

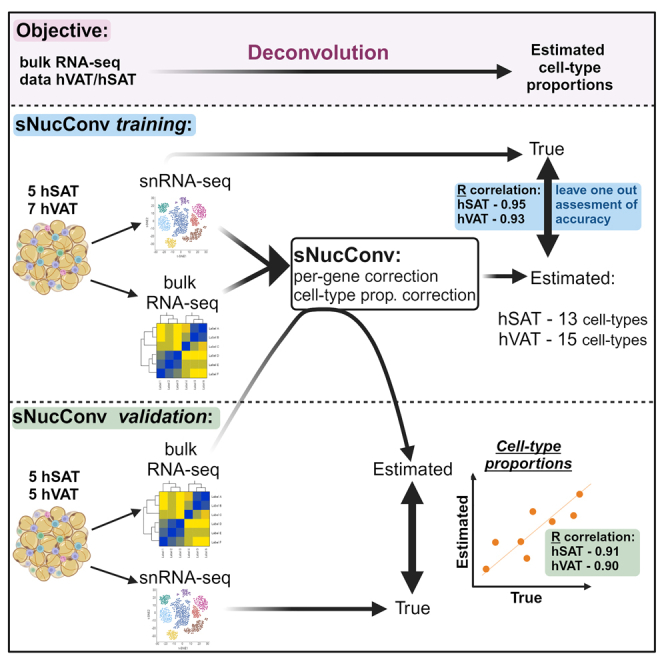
Summary
Deconvolution algorithms mostly rely on single-cell RNA-sequencing (scRNA-seq) data applied onto bulk RNA-sequencing (bulk RNA-seq) to estimate tissues’ cell-type composition, with performance accuracy validated on deposited databases. Adipose tissues’ cellular composition is highly variable, and adipocytes can only be captured by single-nucleus RNA-sequencing (snRNA-seq). Here we report the development of sNucConv, a Scaden deep-learning-based deconvolution tool, trained using 5 hSAT and 7 hVAT snRNA-seq-based data corrected by (i) snRNA-seq/bulk RNA-seq highly correlated genes and (ii) individual cell-type regression models. Applying sNucConv on our bulk RNA-seq data resulted in cell-type proportion estimation of 15 and 13 cell types, with accuracy of R = 0.93 (range: 0.76–0.97) and R = 0.95 (range: 0.92–0.98) for hVAT and hSAT, respectively. This performance level was further validated on an independent set of samples (5 hSAT; 5 hVAT). The resulting model was depot specific, reflecting depot differences in gene expression patterns. Jointly, sNucConv provides proof-of-concept for producing validated deconvolution models for tissues un-amenable to scRNA-seq.
Subject areas: Integrative aspects of cell biology, Biocomputational method, Classification of bioinformatical subject, Transcriptomics, Machine learning
Graphical abstract
Highlights
-
•
Deconvolution algorithms estimate cell-type proportions from bulk RNA-seq data
-
•
sNucConv extends Scaden deep-learning by adding per-gene and cell-type corrections
-
•
Its uniquely high (R∼0.9) accuracy is verified on human adipose single-nucleus RNA-seq
-
•
sNucConv accurately estimates 13 and 15 cell types in hSAT and hVAT, respectively
Integrative aspects of cell biology; Biocomputational method; Classification of bioinformatical subject; Transcriptomics; Machine learning
Introduction
Two seminal papers in 2003 demonstrated that obesity is associated with increased adipose tissue abundance of macrophages, whose presence associates with metabolic dysfunction (insulin resistance).1,2 These papers sparked enormous interest in deciphering the dynamic cell composition of adipose tissue. Indeed, over the following 20 years, most known immune cell types had been identified in adipose tissue and were implicated in linking adipose tissue changes with health risks.3,4 For example, it became clear that it is not only adipose tissue mass per-se but also its cellular composition which importantly differentiates people with obesity and cardiometabolic complications from those with a relatively benign obesity phenotype (arguably termed “healthy obesity”).5 Thus, to understand adipose tissue biology, function, and its interphase with whole-body (patho)physiology, the tissue’s cellular composition must be assessed.6,7
With the advent of single-cell RNA-sequencing (scRNA-seq) technology, novel subtypes of specific cells could be studied in an unprecedentedly unbiased approach. However, adipose tissue poses a unique challenge when trying to apply scRNA-seq: the “adipose cells”–adipocytes—that are the unique functional cell type of this tissue, could not be captured by scRNA-seq. This is because adipocytes are unique in several aspects: they have an exceptionally broad size range, which can vary by > 20-fold; they are buoyant and fragile (due to their high lipid content—85% of the cell volume is composed of triglycerides). Thus, most scRNA-seq platforms, which rely on either microfluidics or the passage of cells through nozzles, disintegrate these cells, particularly those in the larger size range. scRNA-seq analyses of adipose tissue have therefore been confined to analyses only of the non-adipocytes, stromal-vascular cell fraction of the tissue. Indeed, such studies uncovered novel cell subtypes that comprise adipose tissue, underscoring the promise in analyzing adipose tissue at the single-cell level.
More recently, single-nucleus RNA-sequencing (snRNA-seq) was developed8,9 and applied to studying adipose tissue.10,11 snRNA-seq’s major weakness in comparison to scRNA-seq is that the nuclear mRNA repertoire is a fraction of the entire cellular RNA landscape. The latter is dominated by cytoplasmic RNA, while nuclear RNA has higher proportions of “immature” mRNA (i.e., that has more intronic regions) and long non-coding RNAs.8,12 Nevertheless, several studies have demonstrated good correspondence between snRNA-seq and scRNA-seq, suggesting that the latter can reliably capture the transcriptome landscape.8,12,13 On the other hand, snRNA-seq advantages over scRNA-seq have also been noted, in particular, the ability to use flash-frozen (and even formaldehyde-fixed) samples, and avoiding the possible transcriptome changes occurring during tissue disintegration into isolated cells. An atlas of human and mouse adipose tissues’ cellular landscape based on snRNA-seq data was recently reported, highlighting known adipose tissue cell types and novel, unbiasedly identified, subtypes of adipocytes, endothelial cells, and progenitor cells.10
Despite the advances offered by both scRNA-seq and snRNA-seq, their use remains limited due to their high cost and heavy reliance on specific equipment, reagents, and bioinformatics expertise. Deconvolution tools were therefore developed using computational approaches to address these shortcomings, allowing to estimate cell-type composition of tissues from bulk-tissue RNA-sequencing data,14,15,16 also in adipose tissue.17,18,19,20 Yet, most algorithms rely on the gene expression derived from scRNA-seq data. Consequently, the ability of such deconvolution tools to accurately estimate adipose tissue’s cell composition, whose true cell-type proportions can only be studied using snRNA-seq, is questionable.
Given the importance of characterizing adipose tissue cellular composition, we generated a dataset comprising of human subcutaneous and visceral adipose tissue samples (hSAT and hVAT, respectively), each analyzed in parallel by both bulk RNA-seq and snRNA-seq. We tested the ability to infer cell-type composition from bulk RNA-seq using available deconvolution tools and modified an existing deep-learning deconvolution algorithm in order to generate a validated deconvolution method for human VAT and SAT, effectively bridging the gap between cytoplasmic and nuclear RNA repertoires.
Results
To develop a deconvolution tool that would reliably estimate cell-type composition of human adipose tissue from bulk RNA-seq, we generated an initial sample dataset consisting of 7 hVAT samples and 5 hSAT samples. Each of the 12 adipose tissue biopsies was analyzed in parallel by both snRNA-seq and bulk RNA-seq (Figure 1A1). An additional set of 5 hVAT and 5 hSAT samples were also sequenced and served as a validation sample set. Patients’ basic characteristics, and Cell Ranger-based quality control (QC) parameters of each sample are shown in Tables S1A, S1B, S2A, and S2B, for the initial and validation sets, respectively.
Figure 1.

sNucConv workflow
sNucConv is a Scaden-based algorithm developed for estimating cell-type proportions from bulk RNA-seq data while training on snRNA-seq, rather than scRNA-seq, data.
(A–D) denote the 4 stages of sNucConv, which include: (A) the conversion of snRNA-seq dataset into pseudo-bulk training set with per-gene correction, (B) generating a Scaden-based prediction model, (C) per cell-type regression model generation (D) and correction to obtain the final sNucConv deconvolution output.
(E) sNucConv was trained on the initial samples set (7 hVAT and 5 hSAT) and applied onto an independent validation samples set (5 hVAT and 5 hSAT).
Recent snRNA-seq studies consolidated (harmonized) the sequencing results obtained from the two adipose tissue (anatomically distinct) depots—hVAT and hSAT.10 Yet, these two adipose tissue depots express depot-specific genes and differ in their gene expression profiles.21,22,23 Consistently, bulk RNA-seq principal component analysis (PCA) reveals that the global transcriptome of hSAT and hVAT (of the initial dataset) are in distinctly separable clusters (Figure 2A). Moreover, a multitude of studies demonstrate differences in the function and contribution of the two tissue depots to whole-body physiology (reviewed by Bodis K. and Roden M.; Tchernof A. and Despres J.P.24,25). We therefore considered the various deconvolution tools separately for hVAT and hSAT. Results of initial sample set (7 hVAT and 5 hSAT) snRNA-seq analysis are presented in Figures 2B–2E, S1, and S2, based on processing and cell-type annotation procedures detailed in STAR Methods.
Figure 2.

Bulk RNA-seq and single-nucleus RNA-sequencing (snRNA-seq) analysis of human adipose tissues
(A) Principal component analysis (PCA) of bulk RNA-seq analyses of human visceral and subcutaneous adipose tissues—initial sample set (hVAT and hSAT, n = 7 and 5 samples, respectively).
(B–E) snRNA-seq analyses of the same hVAT and hSAT as in (A), (64,729 and 37,357 nuclei, respectively, of the initial sample set as in (A)), performed using 10x genomics platform. (B and C) UMAP projection uncovering 15 and 13 uniquely colored annotated clusters of nuclei of hVAT and hSAT, respectively. (D and E) Cell-type annotation of nuclei clusters based on single marker genes, as recently used by Emont M.P.10 in hVAT and hSAT, respectively. ASPC1.APC, adipose stem progenitor cells 1. adipocyte progenitor cells; ASPC2.ASC, adipose stem progenitor cells 2. adipose-derived stem cells; SMC, smooth muscle cells.
Performance of existing deconvolution tools applied onto bulk RNA-seq of human adipose tissue using snRNA-seq data
In order to assess the performance (i.e., accuracy) of existing deconvolution tools in correctly estimating cell-type proportions from bulk RNA-seq of hSAT and hVAT, we took advantage of our unique paired dataset of snRNA-seq and bulk RNA-seq. We tested three tools, two of which utilize cell-type-specific gene expression: MuSiC14 (which was also used by Emont M.P. et al.10) and SCDC.16 The third tool—Scaden,15 uses deep neural network ensemble training. We compared the accuracy of each of the three tools in the initial sample set, as detailed in STAR Methods, using a leave-one-out approach: one sample at a time was used as a test sample, while all other samples were used for training. The tools’ accuracy was assessed by evaluating the median R-correlation coefficient between the true, snRNA-seq-derived cell types’ proportions (Figures 3A and 3C for hVAT and hSAT, respectively) and the proportions estimated by each deconvolution tool. The three deconvolution algorithms estimated the relative proportions of the 15 hVAT and 13 hSAT different cell types with low accuracy, indicated by a median correlation coefficient of |R| = 0.1 (Figures 3B and 3D, respectively). Interestingly, all tools exhibited both positive and negative correlations between true and estimated cell-type proportions, particularly in hVAT.
Figure 3.

Performance of cell-type estimation by common deconvolution tools analyzing human visceral and subcutaneous adipose tissue bulk RNA-seq data
(A and C) Cell-type proportions in the initial sample set of 7 hVAT and 5 hSAT samples as assessed by snRNA-seq, respectively. Shown are the proportions of the 15 and 13 cell types, respectively.
(B and D) Accuracy of estimated cell-type proportions achieved by 3 deconvolution tools: MuSiC and SCDC are deconvolution algorithms that utilize cell-type-specific gene expression and Scaden uses a deep neural network ensemble training approach. Boxplot showing each test-case R correlation coefficient between the true proportions (snRNA-seq results) and the tools’ estimated proportions for every sample in the initial sample set (using leave-one-out training methodology, as detailed in STAR Methods).
The recent human adipose tissue cell atlas estimated cell-type proportions by deconvolution of 331 subcutaneous adipose tissue bulk RNA-seq samples using snRNA-seq data as reference.10 Yet, the estimate was for five major cell-type merged groups: (i) “vascular cells” included endothelial, lymphatic endothelial, pericytes, and smooth muscle cells; (ii) “myeloid-immune cells” included macrophages, monocytes, dendritic cells, mast cells, and neutrophils; (iii) “lymphoid-immune cells” included B cells, NK cells, and T cells; (iv) “adipocytes”; and (v) “adipose stem and progenitor cells” (ASPCs). We therefore assessed if the performance of the 3 deconvolution tools could be improved by decreasing the resolution of cell-type clustering. For this, we generated “medium” and “low resolution” cell-type merged groups from the 15 and 13 cell types identified by our snRNA-seq in hVAT and hSAT, respectively. Medium cell-type resolution included 10 and 8 cell-type groups, respectively, and low cell-type resolution included 5 cell-type groups, as in Emont et al.10 + 1 group named “other” (Figures 4A and 4B). Decreasing the number of cell-type groups indeed improved the performance of the deconvolution tools, as indicated by an increased coefficient of correlation. While hVAT did not show improved performance (higher correlation coefficient) in the middle resolution, it did exhibit improved performance in the low resolution (Figure 4C). In hSAT, the correlation coefficient increased gradually when estimating a lower number of cell type groups (Figure 4D). Overall, the best performance was achieved when using Scaden to estimate hVAT samples at low cell-type resolution. Nevertheless, the coefficient of correlation even for this analysis was 0.61. We also utilized CIBERSORTx that was previously used on GTEX and TwinUK hSAT samples to identify several cell types.18 Different from the aforementioned deconvolution tools, CIBERSORTx was used in that study with expression signatures of cultured specific cell types to estimate their abundance in bulk RNA-seq datasets. Yet, this analysis also yielded similar performance to that achieved by the 3 other deconvolution tools (data not shown). Collectively, the tested deconvolution algorithms exhibit low reliability in estimating the full set of snRNA-seq-identified cell types in hVAT and hSAT. They provide a somewhat improved, but still low, capacity to estimate merged cell-type groups. This performance level of existing tools prompted us to develop an improved deconvolution tool for tissues like human adipose tissue, whose true cell-type composition necessitates snRNA-seq, rather than scRNA-seq, analysis.
Figure 4.

Performance of existing deconvolution algorithms and of sNucConv at various resolutions of cell types/group
(A and B) We merged the “high cell-type resolution” of 15 and 13 specific hVAT and hSAT cell types, respectively, to generate medium and low resolution of cell-type groups, consisting of 10 and 6 cell-type groups, respectively, for hVAT, and 8 and 6, respectively, for hSAT.
(C and D) The accuracy of cell-type proportions estimation at the three cell-type/group resolution levels is shown for Scaden, MuSiC, and SCDC, as in Figure 3, for hVAT and hSAT of the initial sample set, respectively, using leave-one-out approach.
(E and F) Accuracy of cell-type composition estimation by sNucConv—a Scaden-based deconvolution tool developed to train on snRNA-seq data (as illustrated in Figure 1). Boxplot showing each test-case R correlation coefficient between the true proportions (snRNA-seq results) and the tools’ estimated proportions for every sample of the initial sample set using leave-one-out approach (as in C and D). In addition, sNucConv trained on the initial sample set was applied on the bulk-RNA-seq of the independent validation sample set (5 hVAT and 5 hSAT (as illustrated in Figure 1E)). The last bar on the right of the dashed vertical line depicts the R correlation coefficient between the true proportions (snRNA-seq results) and the tools’ estimated proportions in the validation sample set, tested on the high cell-type resolution.
sNucConv methodology
We hypothesized that the poor performance of the existing deconvolution tools stems from their reliance on scRNA-seq as reference data for training. Both scRNA-seq (training dataset) and bulk RNA-seq largely represent a common source of the RNA pool, i.e., mature/cytoplasmic mRNA, in contrast to the nuclear RNA pool, which is distinct.12 Additionally, to accurately estimate cell proportions of adipose tissue, the adipocytes must be also considered. This necessitates reliance on snRNA-seq data, and thus—on the nuclear RNA pool, for the algorithms’ training. We aimed to implement a new, snRNA-seq tailored method (sNucConv) that will adjust for these differences in order to better fit the snRNA-seq dataset to the bulk RNA-seq dataset. The fitted (corrected) dataset, which better represents true bulk RNA-seq samples, can then serve for training deep-learning algorithms. We chose to use the deconvolution deep-learning-based algorithm Scaden as our core training and prediction method on which we implemented our sNucConv methodology.
Per-gene regression model
We took advantage of our paired snRNA-seq and bulk RNA-seq samples from the same donors to bridge the gap between nuclei-based RNA and cytoplasmic mature mRNAs. We set to discover genes whose expression is highly correlated between the nuclei and the cytoplasm (Figure 1A2). These genes enabled us to transform our snRNA-seq data to become more comparable to single-cell data (Figure 1A3,4). First, we simulated “pseudo bulk RNA-seq” samples based on the snRNA-seq dataset by per-gene averaging of randomly sampled 10,000 cells, repeated 30 times, while reflecting the sample’s true cell-type proportions. The final gene counts for each pseudo-bulk sample were then normalized to counts per million (CPM). The specific combination of paired true bulk RNA-seq (also normalized to CPM) and simulated pseudo-bulk samples having true proportions enabled us to look for genes that behave in a correlated manner. A linear regression model was generated for each highly correlated gene (|R|>0.6, Spearman) (Figure 1A2).
Pseudo-bulk simulation by Scaden using random proportions, gene filtration, and correction for sn/bulk correlated genes
In order to create a training dataset that resembles bulk RNA-seq samples, Scaden was used to generate 1,000 simulated pseudo-bulk samples with random (but known) proportions (Figure 1A3). Each simulation was based on 1,000 cells selected from a pooled snRNA-seq dataset comprised of all samples from the same depot. The gene counts of the 1,000 cells were summed up to create a pseudo-bulk sample. The generated simulated pseudo-bulk samples with random proportions were first normalized to CPM and then filtered to include only the highly correlated genes (from Figure 1A2). The normalized gene counts were then corrected, each using the specific gene’s regression model calculated from the paired sn-bulk analysis, to yield “corrected pseudo-bulk RNA-seq” samples (Figure 1A4).
Deep-learning training using corrected pseudo-bulk samples and computing a per cell-type regression model
Using the 1,000 corrected pseudo-bulk RNA-seq dataset, we allowed Scaden to train, while knowing each pseudo-bulk’s (randomly selected) cell-type proportions (Figure 1B). Scaden applied its deep-learning algorithms (training step) to all “pseudo-bulk samples” and finally generated three prediction models. These models were subsequently applied to estimate cell-type proportions of the true bulk RNA-seq samples (that were also used during the training step) (Figure 1C1). To clarify, this constitutes an additional training step and not the final proportion estimation (presented in Figures 1D and 1E). Next, we calculated the correlation between the true proportions (based on the snRNA-seq samples) and Scaden’s estimated proportions (Figure 1C2). We noted that the cell types differed in the strength of correlation between the true snRNA-seq-based and Scaden’s estimated cell-type proportions. Thus, the model could be further improved by adjusting the final predictions. For this, we generated a cell-type-specific regression model (termed “per-cell type regression model”) for each of the cell types (Figure 1C2). Cell types with a calculated R2 correlation coefficient above a certain cutoff (default: R2 > 0.8) were used to adjust (correct) the predicted proportions of our test bulk RNA-seq data. We chose to correct only for the highly correlated cell types, since they present a consistent error of the original Scaden-based model. This ensured that proportions of cell types with high confidence were estimated more accurately while minimizing the overall estimation error.
Cell type proportion estimation on test data with cell-type proportions adjustment
The final cell-type proportion estimation from true bulk RNA-seq data (Figure 1D) included: (i) applying the prediction models generated by Scaden on the test bulk RNA-seq sample (which was not part of the training process) to produce estimated cell-type proportions and (ii) adjusting the proportions using the “per-cell-type regression model”. Consequently, the proportions adjustment of only highly correlated cell types shifted the total sum of all cell-type proportions from 1. Therefore, shifting was then required for the non-adjusted proportions.
sNucConv outperformed existing deconvolution tools
The performance of sNucConv in estimating cell-type proportions in bulk RNA-seq samples is shown in Figures 4E and 4F; Figure S3. First utilizing a leave-one-out approach on the initial sample set, sNucConv achieved median absolute correlations of 0.954, 0.900, and 0.932 in estimating 6, 10, and 15 hVAT cell types/groups, respectively, and 0.924, 0.918, and 0.953 in estimating 6, 8, and 13 cell types/groups, respectively, in hSAT. In comparison to the second-highest performing tool in hVAT—Scaden—which exhibited median absolute correlations of 0.474, 0.046, and 0.103 for 6, 10, and 15 hVAT cell types/groups, respectively, sNucConv predictions outperformed Scaden. In hSAT, the second highest performing tool was MuSiC, achieving median absolute correlations of 0.524, 0.315, and 0.141 for 6, 8, and 13 cell-types/groups, respectively, again, markedly lower than sNucConv’s performance. Notably, sNucConv was the only tool to estimate cell-types with consistent accuracy, producing only positive correlations in all cell type/groups for both depots. A similar trend was observed assessing the performance of the different deconvolution tools using root-mean-square error (RMSE, data not shown) as a performance accuracy measure. To complement our leave-one-out approach and address the concern of data overfitting, we further verified the accuracy of sNucConv by training the model on the initial sample set (7 hVAT and 5 hSAT), and applying it on the bulk RNA-seq of the independent validation sample set (5 hVAT and 5 hSAT). Results of the cell-type proportion estimates were then correlated to the true cell type proportions obtained from the same samples analyzed by snRNA-seq. To first determine the robustness of the model in this training-validation setting, we performed rarefaction analysis, wherein the model was trained based on an increasing number of samples of the initial sample set. Training sNucConv on from 4 up to 7 hVAT, or from 3 up to 5 hSAT samples of the initial sample set (we considered all possible combinations of samples), achieved a similarly high accuracy, with nearly all individual samples exhibiting true-to-estimated cell-type proportion correlation exceeding 0.8 (Figure S4). This underscores the ability of sNucConv to be trained on a relatively small number of bulk/snRNA-seq sample pairs. When all initial sample set were used to train sNucConv and applying it onto the validation sample set, the coefficient of correlation was similar to the performance of the model using the leave-one-out approach (Figures 4E and 4F, bar right to the vertical line). On the validation set, sNucConv achieved median absolute correlations of 0.91 and 0.90 for hVAT and hSAT, respectively. These results highlight the accuracy and robustness of sNucConv as a deconvolution algorithm to deduce cell-type proportions from bulk RNA-seq of hSAT and hVAT.
Discussion
Deconvolution algorithms have been developed to estimate cell-type composition of tissues from their bulk RNA-seq data. However, existing deconvolution tools largely rely on cell-specific RNA markers derived from scRNA-seq. Moreover, rarely are the estimations obtained by such deconvolution tools experimentally validated by results of same samples analyzed in parallel by both bulk RNA-seq and snRNA-seq to assess the estimated-to-true correlation. Here, we experimentally analyzed same samples of two independent sets totaling 22 samples, from two different adipose tissue depots—hSAT and hVAT—all sequenced by both bulk and snRNA-seq. Our results demonstrate the following main points.
-
•
Most existing deconvolution tools perform poorly in estimating cell-type proportions when trained on snRNA-seq data (instead of scRNA-seq) and applying it to bulk RNA-seq.
-
•
This likely reflects the known differences in the RNA repertoire between the nuclear compartment and the whole cell. The latter is dominated by cytoplasmic RNA, while nuclear RNA includes larger proportions of not fully processed RNA molecules and long non-coding RNAs.12
-
•
Thus, we herein show that training a deconvolution algorithm on a subset of genes whose expression patterns correlate well between the snRNA-seq and bulk RNA-seq, along with cell proportion correction, can greatly improve the estimated-to-true correlation coefficient.
-
•
Rarefaction analysis suggests that training sNucConv on 3–4 samples may be sufficient to obtain performance accuracy that exceeds that of most existing tools.
Comparing sNucConv to other deconvolution tools (Table S3) highlights important strengths of this new tool: although seemingly relying on a limited number of samples, the actual number of samples assessed in parallel by both bulk and snRNA-seq (total of 22 samples) exceed other studies, as is the total number of nuclei (or single cells). This is a major strength, as it enables direct assessment of estimated to true cell type proportions. Moreover, our rarefaction analysis demonstrates the stability of the model when trained on 3–4 samples. An additional significant strength of sNucConv is its apparent relative independence of the resolution (i.e., number of cell types/groups) it estimates from bulk RNA-seq data. Indeed, sNucConv enables accurate estimation of 13 and 15 cell types in hSAT and hVAT, respectively, more than other deconvolution tools applied onto human adipose tissue (Table S3). Lastly, for human adipose tissue deconvolution, sNucConv is a validated, “ready to use” tool for analyzing human adipose bulk RNA-seq data without pre-use training on a sample set analyzed also by snRNA-seq.
Our bioinformatic approach to develop sNucConv includes several elements that were similarly used in19 to generate Bisque, which was also tested using a smaller number of samples sequenced by both bulk and snRNA-seq and applied to estimate hSAT cell-type proportions. sNucConv extends Bisque further by relying on a larger set of samples, estimated performance by both leave-one-out and independent training and validation sets, and by providing a ready to use tool on both hSAT and hVAT bulk RNA-seq data: in sNucConv, we apply the transformation to the snRNA-seq-based pseudo-bulk samples during the training process to achieve a one-to-one relationship favoring the bulk data, while reducing any need for modifications prior to using sNucConv. In addition, sNucConv does not rely on gene expression profiles (GEPs), but rather it is based on deep neural networks (DNNs), with the potential to create optimal features for cell deconvolution, without relying on complex generation of GEPs.
The cellular landscape of adipose tissue may be tightly linked to obesity complications in cross-sectional analyses. Furthermore, studies suggest that the abundance of specific cell types in adipose tissue can predict response to anti-obesity treatment.26,27 For example, higher abundance of mast cells in hVAT was shown to predict greater weight-loss response 6 months post-bariatric surgery.28 This proof-of-principle study suggests that information on the cellular composition of adipose tissue could uncover novel predictors of response to treatment, and could thereby help in personalizing anti-obesity therapy. Yet, identifying predictors to therapy requires large cohorts, which is unlikely to be obtainable with snRNA-seq analysis, given its high cost and requirements of still non-standard laboratory equipment. Deconvolution can fill in this missing gap by providing cell-type composition estimates from large cohorts of adipose tissue analyzed by bulk RNA-seq. As a proof-of-concept, we applied sNucConv on a large publicly available GTEx hVAT and hSAT sample set. This analysis revealed that in males, the proportions of dendritic, Pre.Myeloid, ASPC1.APC and adipocytes in hVAT, and monocytes in hSAT, were significantly associated with age groups, consistent with a recent report.29 Thus, applying sNucConv on large sample sets of bulk RNA-seq of hVAT or hSAT, with additional clinical data, could potentially reveal new insights. Finally, our study highlights the need to ensure, experimentally, the robustness and trustworthiness of deconvolution algorithms, before applying their use on large bulk RNA-seq data. Here, this was experimentally achieved by parallel examination of a relatively limited number of same samples analyzed in parallel by bulk and snRNA-seq.
In summary, we hereby present sNucConv, a bioinformatic approach to adopt deconvolution tools that were designed to perform with scRNA-seq data, to enable extracting cell-type composition of tissues necessitating snRNA-seq.
Limitations of the study
Our study and sNucConv have several noteworthy limitations. Clinically, samples included in this study were all from people with obesity (BMI range 34.5–55.0 kg/m2), and the performance of sNucConv when estimating cell-type proportions in adipose tissues of people with lower BMI is unknown. Yet, sNucConv algorithm may be used to assess its accuracy if samples from lean patients are available for parallel analysis by bulk and snRNA-seq. Another possible limitation of sNucConv may be in its high tissue-specificity, as the deconvolution algorithm performed best when applied to the same adipose tissue depot it was trained on: a combined model that trained on both hSAT and hVAT exhibited lower accuracy in estimating cell type proportions from bulk RNA-seq data (median R = 0.77 and 0.86 for hVAT and hSAT, respectively, compared to 0.93 and 0.95, respectively, when the training was on samples from the same depot (Figures 4E and 4F)). However, accuracy level of the model trained on both depots is still acceptable. Additionally, this finding may reflect both the high sensitivity of the tool to unique expression patterns, and the fact that the seemingly similar hSAT and hVAT are in fact distinct, as can be observed even at the bulk RNA-seq PCA analysis (Figure 2A).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| All analyzed data | This study | https://cellxgene.cziscience.com/collections/ba84c7ba-8d8c-4720-a76e-3ee37dc89f0b |
| Original code (sNucConv) | This study | https://github.com/bioinfo-core-BGU/sNuConv |
| Software and algorithms | ||
| R v4.1.1 | R Core Team | https://www.r-project.org/ |
| Scaden | Menden et al. | https://github.com/KevinMenden/scaden |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Liron Levin (levinl@bgu.ac.il).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Data have been deposited at CZ CELLxGENE portal and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code, including sNucConv algorithm, the 2 models, and the harmonized Seurat objects for hSAT and hVAT has been deposited at GitHub and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Twelve hVAT and 10 hSAT samples in total were obtained from an adipose tissue bio-bank established in Beer-Sheva, Israel, and subjected to both bulk RNA-seq and snRNA-seq. The “initial sample set” included 7 and 5 hVAT and hSAT samples, and the remaining 5 and 5 samples, respectively, constituted the “validation sample set”. In the Beer-Sheva human adipose tissue biobank, adipose tissue biopsies are obtained from patients who had signed in advance a written informed consent before undergoing elective abdominal surgeries, and tissues are immediately delivered from the operating room to the lab, where they are frozen at −80°C, as detailed previously.30,31 Ethical approval of the study procedures was obtained by the Helsinki Ethics Committee of Soroka University Medical Center (approval no: 15–0348). Samples included in the present analyses are from consecutive patients with at least 1gr biopsy available, and whose individual basic clinical characteristics are shown in Tables S1A and S1B for the initial and validation sample sets, respectively.
Method details
Bulk RNA sequencing
Total RNA was isolated from hSAT and hVAT samples (150–250 mg) using RNeasy Lipid Tissue Mini Kit (QIAGEN, Germany), according to the manufacturer’s instructions. RNA quality (RINe) was determined by 4150 TapeStation System (Agilent, CA, USA) with RNA ScreenTape (Agilent, CA, USA), while RNA concentration was determined using Qubit 4 Fluorometer (ThermoFisher Scientific, MA, USA) using Qubit RNA Assay Kit. Only samples with RINe>8.0 were further processed. RNA libraries were prepared using KAPA Stranded RNA-seq Kit (Roche, Switzerland), pooled, and subjected to single-end sequencing according to the manufacturer’s protocol (NovaSeq 6000, Illumina, CA, USA). An average of 41.2 million paired-reads per sample were generated. Sequences were quality trimmed and filtered using Trim Galore (v0.4.4) and cutadapt (v1.15). Alignment of the reads to the human genome (GRCh38.100) was performed with STAR (v2.5.2a).32 The number of reads per gene per sample was counted using RSEM (v1.2.28).33 Quality assessment of the process was carried out with FastQC (v0.11.8) and MultiQC (v1.0.dev0).34 After trimming each sample had an average of 40.9M ± 10.9M reads with an average sequence length of 88.5bp.
Single-nucleus RNA sequencing
Human adipose tissue samples were analyzed by isolating nuclei, barcoding them using Chromium 10X technology, followed by RNA sequencing using NovaSeq 6000 (Illumina, CA, USA). Briefly, nuclei were isolated from 500 mg of frozen hSAT or hVAT samples with all steps carried out on ice. One mL of ice-cold adipose tissue nuclei lysis buffer (AST: 5 mM PIPES, 80 mM KCl, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630) supplemented with 0.2U/μL of RNAse Protector (Roche, Switzerland), were added to each sample, followed by mincing the frozen tissue using small surgical scissors. Then, small pieces of minced tissue in 1 mL of AST lysis buffer were transferred into 7 mL WHEATON Dounce Tissue Grinder (DWK Life Sciences, Germany) and additional 2 mL of AST were added. Tissues were dissociated to retrieve nuclei by 12 strokes with loose pestle (A), followed by 10 strokes with tight pestle (B). Samples were then incubated for 7 min on ice to ensure maximal nuclei retrieval, filtered through 70 μm SMARTStrainer (Miltenyi Biotech, Germany), and centrifuged (4°C, 5 min, 500 RCF). Supernatants were discarded, and nuclei pellet was resuspended in 2.5 mL of ice-cold PBS 0.5% BSA, supplemented with 0.2U/μL of RNAse Protector, and filtered through 40 μm SmartStrainer, followed by additional centrifugation. Next, 2 mL of supernatant were discarded, and nuclei pellet was resuspended in the remained 0.5 mL of ice-cold PBS 0.5% BSA. Nuclei were counted using LUNA-FL Dual Fluorescence Cell Counter (logos biosystems, South Korea), and 13,000–16,500 nuclei/sample were immediately (i.e., without an additional sorting step) loaded onto 10X Chromium Controller (10X GENOMICS, CA, USA) according to the manufacturer’s protocol (Chromium Next GEM Chip C. Chromium Next GEM Single Cell 3′ Kits v3.1). cDNA and gene expression libraries size fragments were assessed using 4150 TapeStation System (Agilent, CA, USA) with high sensitivity D5000 ScreenTape, while their concentrations were quantified using Qubit 4 Fluorometer using Qubit dsDNA HS Assay Kit (ThermoFisher Scientific, MA, USA). Gene expression libraries were sequenced by NovaSeq 6000 using S1-100 flow cell (Illumina, CA, USA).
Quality control and mapping
Reads were transformed into a raw counts matrix with CellRanger software, which was used as input for CellBender to remove ambient RNA. For downstream analysis, only cell barcodes which were determined to be true cells by both CellRanger and CellBender algorithms were used (with default parameters). The result of CellBender’s counts matrix (including only filtered barcodes) of each sample was analyzed with Seurat v3.0 R package. Our quality control procedure included removal of broken nuclei, i.e., nuclei with less than 200 genes or nuclei that contained more than 20% mitochondrial genes. Parameters of each sample are presented in Tables S2A and S2B, for the initial and validation samples sets, respectively.
Integration and clustering
Counts were normalized (Log-normalization) and scaled using the 3,000 most variable genes. Linear dimensionality reduction via principal component analysis (PCA) and graph-based clustering via k-nearest-neighbors (KNN) were used to group individual cells into subsets. We removed cell clusters with low mean gene count and low expression level (mean read counts less than one standard deviation below the sample’s mean). We then used the DoubletFinder R package to remove possible doublets, and compared to putative further doublet identification by Scrublet (Figure S5). Overall, doublets identification by the two tools partially overlapped, and when we deleted any nucleus identified as doublet by either of the two tools the performance of sNucConv did not materially change, though it did show a trend toward less variability between samples. Immunofluorescence studies of human adipose tissues demonstrated that nuclei with high remaining doublet score are in fact bona-fide cells (not shown). Thus, we recommend careful consideration of the use of doublet elimination tools, and continued our analyses with doublet elimination by DoubletFinder only, given that such acceptable though less stringent doublet elimination strategy did not compromise sNucConv’s performance. Once each sample passed quality control, we integrated separately the 7 hVAT and 5 hSAT of the initial sample set using Harmony, and performed dimensionality reduction and clustering again to obtain the final Uniform Manifold Approximation and Projection (UMAP) plots (Figures 2B and 2C, respectively).
hVAT snRNA-seq annotations
snRNA-seq analysis of the 7 hVAT samples of the initial sample set included, after quality control (QC), the nuclear transcriptome of 64,729 nuclei (an average of 9,247 nuclei per sample). UMAP projection map unbiasedly identified 15 unique clusters (Figure 2B; Figure S1A). Our initial cluster/cell-type annotation relied on single cell-type specific gene markers used in an atlas of the human adipose tissue cellular landscape10(Figure 2D). Adipocytes, adipocyte progenitor/stem cells (ASPC), mesothelial cells, endothelial cells, lymphatic endothelial cells, pericytes, smooth muscle cells (SMC), mast cells, and B-lymphocytes were easily identifiable using these single cell-type specific markers. Myeloid cells (macrophages, monocytes and dendritic cells) shared more than one marker, and lymphoid T and NK cells clustered into a single cluster. In addition to these readily identifiable clusters, two clusters were largely negative to all 15 markers (see further discussion on their annotation below). To support our clustering, we also searched unbiasedly the 10 highest preferentially (compared to all other clusters) expressed genes (heatmap - Figure S1C, list of all 10 genes/cluster - Table S4). This analysis confirmed that indeed the identified clusters exhibited at least 10 differentially expressed genes per cluster when compared to all other clusters. Ten genes were mildly upregulated in one of the two clusters that were negative to all single marker genes used in Figure S1B. Expression levels of these genes were also similarly observed in an adjacent cluster, which was identified as mesothelial cells based on an additional set of 10 genes and expression of Mesothelin (MSLN, Figure S1B). We therefore named this initially unidentified cluster ASPC2.ASC, representing PDGFRA-negative adipocyte stem cells that express several stem cell genes, including SMO, NGR4, and WT1. The myeloid cell clusters overlapped in their up-regulated genes, and a final non-annotated cluster based on single gene markers seemed to express relatively low levels and/or percentage of myeloid markers nuclear RNA, and was therefore named “pre-myeloid”. All samples contributed to each of the cell-type clusters, except for B-cells, to which samples v3387 and v3399 contributed 0 nuclei. The snRNA-seq analysis of the 5 hVAT samples of the validation sample set included the nuclear transcriptome of 67,709 nuclei (an average of 9,315 nuclei per sample).
hSAT snRNA-seq annotation
snRNA-seq analysis of the 5 hSAT samples of the initial sample set included the nuclear transcriptome of 37,357 nuclei (an average of 7471 nuclei/sample). UMAP projection map unbiasedly identified 13 unique clusters (Figure 2C). Single cell-type specific marker genes, depicting the same fifteen single marker genes that were used for the hVAT annotation analysis, are shown in a violin plot (Figure 2E). The main 12 cell-type groups (adipocytes, ASPC, endothelial, lymphatic endothelial, pericytes, SMC, mast, B-lymphocytes, macrophages, monocytes, dendritic, lymphoid T and NK cells) that were identified in the hVAT analysis, were also easily identifiable in the hSAT. As reported previously,10 nuclei of cells annotated as mesothelial cells based on the expression of mesothelin were absent in hSAT. In addition, nuclei of cells annotated as PDGFRA-negative adipocyte stem cells (ASPC2.ASC) in the hVAT were also absent in the hSAT. Beyond these readily identifiable clusters, one unique cluster showed co-expression of the endothelial and pericytes marker genes (see further discussion on this annotation below). To support our clustering, we again identified at least 10 preferentially expressed genes in each cluster (heatmap – Figure S2C, list of all 10 genes/cluster - Table S5). The previously mentioned unique cluster again overlapped with the top 10 genes for endothelial and pericytes. However, it also exhibited at least 10 unique markers, confirming that it was not a cluster of doublets composed of two nuclei – of endothelial and pericytes. Intriguingly, these same preferentially expressed genes were also present within the mesothelial cells cluster identified in the hVAT analysis, suggesting that these cells are mesothelial-like, and the cluster was therefore named “Pseudo-Meso”. All samples contributed to each of the cell-type clusters, except for B-cells, to which samples s3387 and s3399, similarly to their corresponding VAT samples, contributed 0 nuclei. The snRNA-seq analysis of the 5 hSAT samples of the validation sample set included the nuclear transcriptome of 49,224 nuclei (an average of 8,136 nuclei per sample).
Deconvolution tool assessment
We first assessed three deconvolution tools (Scaden, MuSiC and SCDC) using a leave-one-out approach. Specifically, for each tool, one sample at a time was used as a test sample, while all other samples were used for training. In each run, the test sample’s estimated proportions by each of the tools were compared to the true, snRNA-seq -derived, proportions, and R Pearson coefficient was used to quantify the correlation accuracy. MuSiC14 requires raw read counts for both bulk RNA-seq and single-cell expression (snRNA-seq, in our case). The prediction was done according to the 'estimation of cell type proportions' protocol in MuSiC’s documentation. Data preparation included the creation of bulk RNA-seq and snRNA-seq ExpressionSets (also known as ESETs), holding the expression data along with sample and feature annotation. Cell-type proportions were estimated with the 'music_prop' function, while using all marker genes that were overlapping in the bulk and snRNA-seq ESETs. All other arguments were set to default. SCDC16 takes ExpressionSet objects with raw read counts as input. The prediction protocol was performed according to SCDC’s vignettes. The single-cell (snRNA-seq, in our case) ESET was first pre-processed, and a quality control procedure was performed to remove cells with questionable cell-type assignments. The processed ESET was evaluated using the 'SCDC_qc' function, with a threshold set to 0.7. The cell-type proportions were then estimated using the 'SCDC_prop_ONE' function and default settings, with the processed ESET used as a reference. Scaden15 requires two input files: (i) cell-type labels file of size (n x 1), where n is the number of cells in the data, and a single 'Celltype' column with cell-type assignments; (ii) count data file of size (n x g), where g is the number of genes and n is the number of cells (nuclei, in our case). These files were then used to create pseudo bulk samples for training using the 'simulate' step, generating 1000 artificial samples from 1000 cells per sample. Scaden was then used to process the training data using the 'process' step, creating a new file for training, which only contains the intersection of genes between the training and the estimation data. The variance cutoff used for the process step was 0.1. Scaden was then trained on three deep neural network models using the 'train' step, each of them trained for 5000 steps. As a final step, the cell-type proportions were predicted using the 'predict' step, by providing Scaden bulk count data with gene counts over samples.
Quantification and statistical analysis
Correlation coefficients and statistical significance were performed with R (v4.1.1). A p-value <0.05 was considered statistically significant. The development of a new deconvolution algorithm (sNucConv) is described in results section 'sNucConv methodology'.
Acknowledgments
This publication is part of the Human Cell Atlas—www.humancellatlas.org/publications/. This study has been made possible in part by CZI grant CZIF2019-002441 and grant DOI https://doi.org/10.37921/206883bpivjy from the Chan Zuckerberg Initiative Foundation. Additional support was provided by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) 209933838: SFB1052 subproject B02 A.R., L.L: “Obesity mechanisms”, and the Israel Science Foundation (ISF-2176/19). The authors would like to acknowledge the help of Daphne Perlman (daphneperlman@gmail.com) in the graphical design of the figures in this article.
Author contributions
Conceptualization, G.S., Y.H., V.C.-C., A.R., and L.L.; methodology, G.S., Y.H., O.L., M.Z.-A., T.H., and P.A.-N.-N.; investigation, G.S., Y.H. A.R., and L.L.; writing – original draft, A.R. and L.L.; writing – review and editing, G.S., Y.H., V.C.-C., T.H., M.B., A.R., and L.L.; funding acquisition, A.R., M.B., and E.Y.-L.; resources, I.F.L., O.D., and I.K.; supervision, A.R. and L.L.
Declaration of interests
The authors declare no competing interests.
Published: June 24, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110368.
Contributor Information
Assaf Rudich, Email: rudich@bgu.ac.il.
Liron Levin, Email: levinl@bgu.ac.il.
Supplemental information
References
- 1.Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu H., Barnes G.T., Yang Q., Tan G., Yang D., Chou C.J., Sole J., Nichols A., Ross J.S., Tartaglia L.A., Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cinkajzlová A., Mráz M., Haluzík M. Adipose tissue immune cells in obesity, type 2 diabetes mellitus and cardiovascular diseases. J. Endocrinol. 2021;252:R1–R22. doi: 10.1530/JOE-21-0159. [DOI] [PubMed] [Google Scholar]
- 4.Blüher M. Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clin. Sci. (Lond.) 2016;130:1603–1614. doi: 10.1042/CS20160005. [DOI] [PubMed] [Google Scholar]
- 5.Klöting N., Fasshauer M., Dietrich A., Kovacs P., Schon M.R., Kern M., Stumvoll M., Bluher M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010;299:E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 6.Dewal R.S., Wolfrum C. Master of disguise: deconvoluting adipose tissue heterogeneity and its impact on metabolic health. Curr. Opin. Genet. Dev. 2023;81 doi: 10.1016/j.gde.2023.102085. [DOI] [PubMed] [Google Scholar]
- 7.Sun W., Dong H., Balaz M., Slyper M., Drokhlyansky E., Colleluori G., Giordano A., Kovanicova Z., Stefanicka P., Balazova L., et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature. 2020;587:98–102. doi: 10.1038/s41586-020-2856-x. [DOI] [PubMed] [Google Scholar]
- 8.Habib N., Avraham-Davidi I., Basu A., Burks T., Shekhar K., Hofree M., Choudhury S.R., Aguet F., Gelfand E., Ardlie K., et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods. 2017;14:955–958. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habib N., Li Y., Heidenreich M., Swiech L., Avraham-Davidi I., Trombetta J.J., Hession C., Zhang F., Regev A. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353:925–928. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emont M.P., Jacobs C., Essene A.L., Pant D., Tenen D., Colleluori G., Di Vincenzo A., Jørgensen A.M., Dashti H., Stefek A., et al. A single-cell atlas of human and mouse white adipose tissue. Nature. 2022;603:926–933. doi: 10.1038/s41586-022-04518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massier L., Jalkanen J., Elmastas M., Zhong J., Wang T., Nono Nankam P.A., Frendo-Cumbo S., Bäckdahl J., Subramanian N., Sekine T., et al. An integrated single cell and spatial transcriptomic map of human white adipose tissue. Nat. Commun. 2023;14:1438. doi: 10.1038/s41467-023-36983-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta A., Shamsi F., Altemose N., Dorlhiac G.F., Cypess A.M., White A.P., Yosef N., Patti M.E., Tseng Y.H., Streets A. Characterization of transcript enrichment and detection bias in single-nucleus RNA-seq for mapping of distinct human adipocyte lineages. Genome Res. 2022;32:242–257. doi: 10.1101/gr.275509.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakken T.E., Hodge R.D., Miller J.A., Yao Z., Nguyen T.N., Aevermann B., Barkan E., Bertagnolli D., Casper T., Dee N., et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 2018;13 doi: 10.1371/journal.pone.0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Park J., Susztak K., Zhang N.R., Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019;10:380. doi: 10.1038/s41467-018-08023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menden K., Marouf M., Oller S., Dalmia A., Magruder D.S., Kloiber K., Heutink P., Bonn S. Deep learning-based cell composition analysis from tissue expression profiles. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aba2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong M., Thennavan A., Urrutia E., Li Y., Perou C.M., Zou F., Jiang Y. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief. Bioinform. 2021;22:416–427. doi: 10.1093/bib/bbz166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donovan M.K.R., D'Antonio-Chronowska A., D'Antonio M., Frazer K.A. Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants. Nat. Commun. 2020;11:955. doi: 10.1038/s41467-020-14561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glastonbury C.A., Couto Alves A., El-Sayed Moustafa J.S., Small K.S. Cell-Type Heterogeneity in Adipose Tissue Is Associated with Complex Traits and Reveals Disease-Relevant Cell-Specific eQTLs. Am. J. Hum. Genet. 2019;104:1013–1024. doi: 10.1016/j.ajhg.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jew B., Alvarez M., Rahmani E., Miao Z., Ko A., Garske K.M., Sul J.H., Pietiläinen K.H., Pajukanta P., Halperin E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020;11:1971. doi: 10.1038/s41467-020-15816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brotman S.M., Oravilahti A., Rosen J.D., Alvarez M., Heinonen S., van der Kolk B.W., Fernandes Silva L., Perrin H.J., Vadlamudi S., Pylant C., et al. Cell-Type Composition Affects Adipose Gene Expression Associations With Cardiometabolic Traits. Diabetes. 2023;72:1707–1718. doi: 10.2337/db23-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vohl M.C., Sladek R., Robitaille J., Gurd S., Marceau P., Richard D., Hudson T.J., Tchernof A. A survey of genes differentially expressed in subcutaneous and visceral adipose tissue in men. Obes. Res. 2004;12:1217–1222. doi: 10.1038/oby.2004.153. [DOI] [PubMed] [Google Scholar]
- 22.Gesta S., Blüher M., Yamamoto Y., Norris A.W., Berndt J., Kralisch S., Boucher J., Lewis C., Kahn C.R. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc. Natl. Acad. Sci. USA. 2006;103:6676–6681. doi: 10.1073/pnas.0601752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto Y., Gesta S., Lee K.Y., Tran T.T., Saadatirad P., Kahn C.R. Adipose depots possess unique developmental gene signatures. Obesity. 2010;18:872–878. doi: 10.1038/oby.2009.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bódis K., Roden M. Energy metabolism of white adipose tissue and insulin resistance in humans. Eur. J. Clin. Invest. 2018;48 doi: 10.1111/eci.13017. [DOI] [PubMed] [Google Scholar]
- 25.Tchernof A., Després J.P. Pathophysiology of human visceral obesity: an update. Physiol. Rev. 2013;93:359–404. doi: 10.1152/physrev.00033.2011. [DOI] [PubMed] [Google Scholar]
- 26.Pincu Y., Yoel U., Haim Y., Makarenkov N., Maixner N., Shaco-Levy R., Bashan N., Dicker D., Rudich A. Assessing Obesity-Related Adipose Tissue Disease (OrAD) to Improve Precision Medicine for Patients Living With Obesity. Front. Endocrinol. 2022;13 doi: 10.3389/fendo.2022.860799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ledoux S., Boulet N., Belles C., Zakaroff-Girard A., Bernard A., Germain A., Decaunes P., Briot A., Galitzky J., Bouloumié A. Subcutaneous Stromal Cells and Visceral Adipocyte Size Are Determinants of Metabolic Flexibility in Obesity and in Response to Weight Loss Surgery. Cells. 2022;11 doi: 10.3390/cells11223540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein N., Kezerle Y., Gepner Y., Haim Y., Pecht T., Gazit R., Polischuk V., Liberty I.F., Kirshtein B., Shaco-Levy R., et al. Higher Mast Cell Accumulation in Human Adipose Tissues Defines Clinically Favorable Obesity Sub-Phenotypes. Cells. 2020;9 doi: 10.3390/cells9061508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moraes D.d., Mousovich-Neto F., Cury S.S., Oliveira J., Souza J.D.S., Freire P.P., Dal-Pai-Silva M., Mori M.A.d.S., Fernandez G.J., Carvalho R.F. The Transcriptomic Landscape of Age-Induced Changes in Human Visceral Fat and the Predicted Omentum-Liver Connectome in Males. Biomedicines. 2023;11 doi: 10.3390/biomedicines11051446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haim Y., Blüher M., Slutsky N., Goldstein N., Klöting N., Harman-Boehm I., Kirshtein B., Ginsberg D., Gericke M., Guiu Jurado E., et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy. 2015;11:2074–2088. doi: 10.1080/15548627.2015.1094597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maixner N., Pecht T., Haim Y., Chalifa-Caspi V., Goldstein N., Tarnovscki T., Liberty I.F., Kirshtein B., Golan R., Berner O., et al. A TRAIL-TL1A Paracrine Network Involving Adipocytes, Macrophages, and Lymphocytes Induces Adipose Tissue Dysfunction Downstream of E2F1 in Human Obesity. Diabetes. 2020;69:2310–2323. doi: 10.2337/db19-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ewels P., Magnusson M., Lundin S., Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data have been deposited at CZ CELLxGENE portal and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code, including sNucConv algorithm, the 2 models, and the harmonized Seurat objects for hSAT and hVAT has been deposited at GitHub and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.