

Abstract
Wells’ syndrome is a rare inflammatory skin disease characterized by pruritic erythematous lesions and cutaneous edema, often accompanied by eosinophilia. Parasitic infestations, such as toxocariasis and strongyloidiasis, can serve as triggers. However, Wells’ syndrome associated with toxocariasis and strongyloidiasis has not been reported previously in Indonesia. Herein, we present a case of a 27-year-old male with a chief complaint of recurrent, pruritic, and painful erythematous rash on the right lower leg for 6 months, accompanied by fever and diarrhea. Physical examination showed cutaneous edema with erythematous macules and bullae on the affected leg. Peripheral blood eosinophilia was noted, and the histopathological analysis demonstrated flame figures, confirming the diagnosis of Wells’ syndrome. A stool culture identified Strongyloides stercoralis, confirming strongyloidiasis, and serological testing was positive for toxocariasis immunoglobulin G antibodies. The patient was treated with albendazole 400 mg twice daily for 3 weeks resulted in clinical improvement observed by the 14th day. The diverse clinical features of Wells’ syndrome present a challenge to clinicians in making an accurate diagnosis, which typically hinges on histopathological assessment and identifying flame figures. Therefore, clinicopathological correlation is important to establish an accurate diagnosis.
Keywords: strongyloidiasis, toxocariasis, Wells’ syndrome
Introduction
Wells’ syndrome, also known as eosinophilic cellulitis, is a rare inflammatory skin disease characterized by cutaneous edema and painful or pruritic erythematous lesions, often accompanied by an increased number of eosinophils.1 Less than 200 cases of this uncommon condition have been reported in the medical literature.2 While the pathogenesis of this rare disease is still not fully known, it is considered to result from a hypersensitivity reaction to various endogenous and/or exogenous stimuli, or it may have no identifiable cause.1 Wells’ syndrome is challenging to diagnose due to its variable clinical presentation, which can resemble other skin conditions. Diagnostic assistance can come from blood tests and histopathological examination, which may show hypereosinophilia and eosinophil infiltration with flame-like features in the dermis.1 Additionally, investigating the potential causes of Wells’ syndrome is crucial, including immunization, medication allergies, herpes during pregnancy, mastocytoma, or worm infestation.2
Wells’ syndrome can be triggered by worm infestations such as toxocariasis and strongyloidiasis, among others.3,4 Toxocariasis, caused by Toxocara spp. like Toxocara canis (T. canis) or Toxocara cati (T. cati), is transmitted to humans through the ingestion of worm eggs.4 Strongyloidiasis in humans, primarily caused by Strongyloides stercoralis (S. stercoralis) is another potential trigger.5 Accurate diagnosis of Wells’ syndrome, based on careful observation of risk factors, clinical presentation, and supporting examinations, is crucial for administering the appropriate medication.5 This case report aims to present a rare case of Wells’ syndrome in a patient with both toxocariasis and strongyloidiasis.
Case Illustration
A 27-year-old male was presented to the Internal Medicine Department of Dr. Hasan Sadikin Hospital, Bandung, Indonesia, with a chief complaint of itchy red-brown macules accompanied by cutaneous edema on the patient’s right lower leg. The skin lesions initially manifested 6 months ago as an itchy, nummular-sized erythematous macule, later enlarging to a plaque and evolving into a brownish erythematous macule. Following the doctor’s visit and prescribed oral and topical medications, the skin lesions became hyperpigmented macules. One month later, the patient also experienced fever, chills, arthralgia, and cephalgia. The symptoms resolved after taking paracetamol and antihistamines. However, two itchy, nummular-sized erythematous macules resurfaced at the same location, which later increased in size and combined into a plaque-sized lesion. After receiving oral and topical medications, the skin lesions disappeared. After another month, the patient experienced fever, abdominal pain, and diarrhea, resulting in a weight loss of seven kilograms. During another visit to the Internal Medicine Department, the patient was diagnosed with hypereosinophilia syndrome and strongyloidiasis following the detection of S. stercoralis in a stool sample. The patient was then referred to the Dermatology and Venereology Department after experiencing severe itching, blisters, and cutaneous edema from a brown erythematous macule on the patient’s right lower leg, accompanied by fever and diarrhea that began 5 days ago. The patient regularly consumed raw vegetables and owned a cat.
Upon physical examination, cutaneous edema was observed on the right lower leg alongside multiple brownish erythematous macules measuring 10 × 6 cm. The lesions exhibited no warmth, hardness, or pain. Surrounding the brown erythematous macules, numerous areas showed bullae and crusts ranging from 0.5 × 0.5 × 0.1 cm to 1.2 × 1.2 × 0.5 cm in size (Figure 1). Direct microscopic examination of hypopyon bullae fluid using Gram staining revealed a large number of polymorphonuclear (PMN) leukocytes (PMN > 10/large view) and the presence of Gram-positive cocci bacteria. Furthermore, a biopsy was performed on a skin lesion on the right lower leg. Histopathological features revealed no alterations to the epidermis, but there was extensive infiltration of eosinophilic inflammatory cells from the dermal papillae to the subcutaneous fat layer, in addition to perivascular infiltration and interstitial infiltration, including eosinophilic infiltration around the adnexa, some displaying flame figures, indicating Wells’ syndrome (Figure 2). A peripheral blood smear was conducted and showed leukocytosis (19.300/uL) with a significant predominance of eosinophils (57%). Strongyloidiasis was confirmed by the identification of S. stercoralis in a stool culture (Figure 3). The Toxocara immunoglobulin G (IgG) antibody was assessed using the hydatid cyst serology assay method (ELISA, Echinococcus antibody), which typically shows a specificity ranging from 62% to 100%. The result was positive, with an index of 2.04. The patient received a diagnosis of Wells’ syndrome with secondary infection and concurrent strongyloidiasis and toxocariasis.
Figure 1.
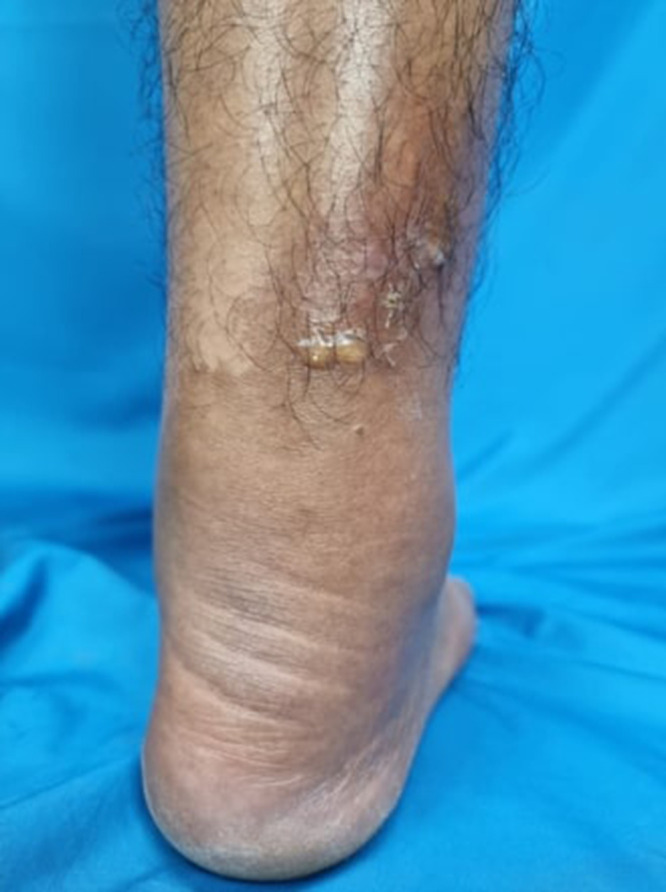
Clinical image of skin lesions before treatments. Multiple brownies erythematous macules with bullae and crusts on the edematous right lower leg.
Figure 2.

Histopathological features of skin lesion. Massive eosinophil inflammatory cell infiltration from the dermal papillae to the subcutaneous fat layer, as well as perivascular and interstitial infiltration with some flame figures (red circle).
Figure 3.
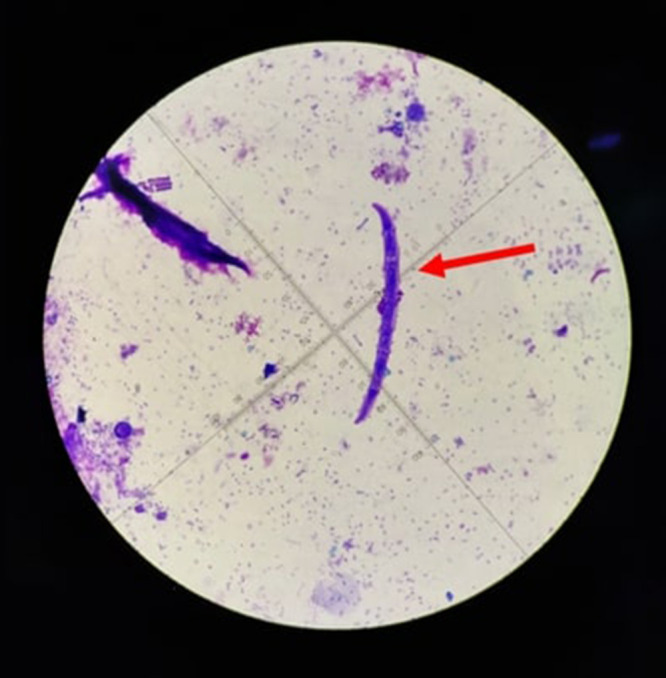
Microscopic examination of a patient’s stool sample. An appearance of the S. stercoralis (red arrow).
The patient received oral medication including 400 mg of albendazole twice daily for 3 weeks, 300 mg of clindamycin for 7 days, 500 mg of mefenamic acid three times daily, and 10 mg of cetirizine. After 2 weeks of treatment, the skin lesions on the right lower leg began to dry out and darken, appearing as hyperpigmented macules with scales (Figure 4). Follow-up laboratory examination results showed improvements and the clinical condition continued to improve throughout the course of the albendazole treatment, with no recurrences observed within 29 days.
Figure 4.

Clinical image of skin lesions after treatments. Hyperpigmented macules with scales on the right lower leg.
Discussion
Wells’ syndrome is an uncommon inflammatory skin disease characterized by polymorphic skin lesions.2 It occurs more frequently in women compared to men, with an incidence ratio of 7:5, typically between the ages of 20 and 50 years.6 The exact etiology remains unknown; however, it is hypothesized to be a hypersensitivity reaction to several endogenous and external stimuli, including insect bites, fungal infections, parasitic infections, and drug hypersensitivity.1,2 In cases of Wells’ syndrome associated with parasitic infestation, stool and serological examinations are essential to identify worm infestation.3 For toxocariasis, serological examination currently employs T. canis excretory-secretory antigen to detect specific IgG antibodies using enzyme-linked immunosorbent assay (ELISA).7 In this case report, a 27-year-old male revealed IgG antibodies to T. cati, confirming the diagnosis of toxocariasis.
Toxocariasis is a zoonotic disease caused by Toxocara nematodes, such as the dog roundworm (T. canis) and the cat roundworm (T. cati). The worms develop into adults in the small intestine and expel their eggs into the environment through feces, with dogs and cats serving as the primary hosts.4 After ingestion, the eggs hatch in the small intestine and the larvae penetrate the intestinal wall to enter the bloodstream.4 Toxocara sp. infestation leads to an inflammatory reaction with diverse clinical signs depending on the organ affected, including skin abnormalities.7
Through history-taking, it was determined that the patient frequently consumed raw vegetables and owned cats, both of which are risk factors. The presence of S. stercoralis larvae in stool samples led to the diagnosis of strongyloidiasis. Strongyloides spp. infects approximately 30–100 million individuals globally, predominantly in tropical and subtropical regions. S. stercoralis is the most frequent cause of strongyloidiasis in humans, potentially resulting in malnutrition and stunted growth. This parasite induces various gastrointestinal symptoms, including nausea, vomiting, flatulence, or diarrhea.8 S. stercoralis is a large nematode notable for its ability to develop into free-living, infectious larvae from excrement. Infection occurs when humans or animals ingest S. stercoralis eggs, leading to infestation. The eggs travel through the duodenum, enter the bloodstream, and eventually hatch into larvae. These larvae proceed to migrate to the liver through the portal vein before entering the lungs. Within approximately 20 days, the larvae enter the digestive tract and mature into adult worms, particularly in the small intestine.9 In this patient, it was considered that S. stercoralis infection originated from the fecal-oral route.
Wells’ syndrome manifests with burning and itching at the site of the skin lesions, which is followed by cutaneous edema and erythema.1,2 The skin lesions can appear as annular plaques or nodules in some cases. Lesions may progress to edematous plaques and develop bullae.1 Over time, the skin lesions gradually transition from bright red to brownish-red to a greyish-green tint that resembles morphea. Wells’ syndrome may present with unusual clinical features such as hemorrhagic papules, vesicles, and bullae. The skin lesions can be either single or multiple and may occur in any part of the body, although they commonly affect the extremities, followed by the trunk region.1 Wells’ syndrome is characterized by a subfebrile fever as the primary systemic symptom.2 These findings were consistent with the clinical presentation of the patient.
The pathophysiology of toxocariasis and Wells’ syndrome is thought to involve dysregulation of eosinophilia in the tissues.10,11 A study reported a higher percentage of CD3+ and CD4+ T cells in patients with Wells’ syndrome. These cells release significant amounts of interleukin 5 (IL-5), causing eosinophilia in both blood and tissues. In the dermis, eosinophils undergo degranulation, resulting in edema and inflammation.2
There are diagnostic criteria for Wells’ syndrome, which include major and minor criteria. The diagnostic standards consist of major criteria: 1) lesion types such as plaque-type, annular-granuloma-like, urticaria-like, papulovesicular, bullous, papulonodular, fixed-drug eruption-like; 2) the disease typically follows a relapsing, remitting pattern; 3) no evidence of systemic involvement; and 4) histologically: eosinophilic infiltrates are observed without vasculitis. Additionally, the minor criteria include: 1) the presence of flame figures; 2) histological evidence of granulomatous changes; 3) peripheral eosinophilia, which is not persistent and does not exceed >1500/μL; 4) identification of triggering factor (eg, drug). Based on these criteria, the diagnosis of Wells’ syndrome can be established if at least two major criteria and one minor criterion are met.10 In our case report, the diagnosis of Wells’ syndrome was established by reporting three major criteria and three minor criteria as follows: multiple brownish erythematous macules with bullae, a relapsing pattern of skin lesions, and eosinophilic infiltrates observed in histopathological analysis. Additionally, all three minor criteria were also met.
The histological profile of Wells’ syndrome varies depending on the stage of the skin lesion. Early stages exhibit cutaneous edema and infiltration of degranulated eosinophils in the dermis.2 In the subacute phase, flame figures appear in the intermediate to deep dermis, consisting of a central core of collagen surrounded by infiltrates of histiocytes and eosinophils.2 As the lesion progresses, eosinophils diminish and are replaced by phagocytic granulomas composed of histiocyte cells and giant cells.1,2 This case report demonstrated eosinophil infiltration extending from the papillary dermis to the subcutaneous fat, along with perivascular and interstitial infiltration featuring flame figures, suggesting Wells’ syndrome.
In managing etiology or triggers, the primary treatment for Wells’ syndrome typically involves corticosteroids or dapsone. Corticosteroids are the preferred drug of choice for all neutrophilic or eosinophilic skin disorders. Prednisone is prescribed at a dosage of 0.5 to 1 mg/kg/day, with gradual tapering regimen. Dapsone may also be prescribed for individuals with Wells’ syndrome, with dosages ranging from 50 mg to 200 mg daily, particularly when there is moderate inflammation or intolerance to corticosteroids.1,12 Antihistamines, especially hydroxyzine, are commonly administered at doses ranging from 50 mg to 100 mg as an alternative to corticosteroids, which may be avoided due to their potential side effects. The principal treatment for Wells’ syndrome associated with toxocariasis and strongyloidiasis involves anthelmintic medications such as albendazole, mebendazole, and thiabendazole.3,4,9 These medications aim to ameliorate clinical conditions and mitigate cellular damage caused by larval migration to various organs.13 Albendazole, administered at a dosage of 400 mg twice daily for 5 days, constitutes the first-line therapy for toxocariasis and strongyloidiasis.9 In the case presented, the patient received albendazole medication for 7 days at the same dosage and continued for up to 3 weeks. On the 16th day of observation, clinical improvement was evident with reductions in the size and alteration in color of the skin lesions.
Conclusion
This case report represents the first documented instance in Indonesia of Wells’ syndrome associated with toxocariasis and strongyloidiasis. Establishing the diagnosis of Wells’ syndrome is still challenging for clinicians due to the diverse clinical presentations. Furthermore, the identification of flame figures in a histological analysis serves as the primary basis for the diagnosis of Wells’ syndrome; however, these results are not unique to Wells’ syndrome and can be seen in a variety of other skin conditions. Clinicopathological correlation is, therefore, crucial to determining an accurate diagnosis.
Acknowledgments
The authors would like to thank the staff of the Department of Dermatology and Venereology, Faculty of Medicine, Universitas Padjadjaran, Bandung, West Java, Indonesia.
Funding Statement
The authors declare that this study has received no financial support.
Ethic Statement
The study's ethics approval was obtained from the Research Ethics Committee of Dr. Hasan Sadikin General Hospital Bandung with the registry number DP.04.03/D.X.IV.6.5/05/2024.
Consent Statement
The patient has signed the consent forms for the use of case details, images for publication, and scientific purposes. Institutional approval has been obtained to publish the case details.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ujiie H, Shimizu H. Eosinophilic Diseases. In: Kang S, Amagai M, Bruckner AL, Enk AH, Margolis DJ, McMichael AJ, editors. Fitzpatrick’s Dermatology in General Medicine. 9th ed. New York: McGraw-Hill Education; 2019:665–668. [Google Scholar]
- 2.Weins AB, Biedermann T, Weiss T, Weiss JM. Wells-Syndrome. J Dtsch Dermatol Ges. 2016;14(10):989–993. [DOI] [PubMed] [Google Scholar]
- 3.Tan LYC, Wang D, Lee JSS, Bwy H, Lim JHL. Eosinophilic cellulitis secondary to occult strongyloidiasis, case report. AME Med J. 2021;6:9. doi: 10.21037/amj-20-150 [DOI] [Google Scholar]
- 4.Ma G, Holland CV, Wang T, et al. Human toxocariasis. Lancet Infect Dis. 2018;18(1):14–24. doi: 10.1016/S1473-3099(17)30331-6 [DOI] [PubMed] [Google Scholar]
- 5.Sinno H, Lacroix J-P, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells’ syndrome): a case series and literature review. Can J Plast Surg. 2012;20(2):91–97. doi: 10.1177/229255031202000204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caputo R, Marzano AV, Vezzoli P, Lunardon L. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142(9):1157–1161. doi: 10.1001/archderm.142.9.1157 [DOI] [PubMed] [Google Scholar]
- 7.Seo M, Yoon SC. A seroepidemiological survey of toxocariasis among eosinophilia patients in chungcheongnam-do. Korean J Parasitol. 2012;50(3):249–251. doi: 10.3347/kjp.2012.50.3.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Echazú A, Bonanno D, Juarez M, et al. Effect of poor access to water and sanitation as risk factors for soil-transmitted helminth infection: selectiveness by the infective route. PLoS Negl Trop Dis. 2015;9(9):1–14. doi: 10.1371/journal.pntd.0004111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piranavan P, Kalantri P, Pandey D, Bharadwaj HS, Verma A. Strongyloides stercoralis hyperinfection syndrome: a neglected cause of abdominal pain. Cureus. 2020;12(9):e10671. doi: 10.7759/cureus.10671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurni MA, Gerbig AW, Braathen LR, Hunziker T. Toxocariasis and Wells’ syndrome: a causal relationship? Dermatology. 1997;195(4):325–328. doi: 10.1159/000245981 [DOI] [PubMed] [Google Scholar]
- 11.Bassukas ID, Gaitanis G, Zioga A, Boboyianni C, Stergiopoulou C. Febrile ”migrating” eosinophilic cellulitis with hepatosplenomegaly: adult toxocariasis - a case report. Cases J. 2008;1(1):356. doi: 10.1186/1757-1626-1-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinig B, Vojvocic A, Lotti T, Tirant M, Wollina U. Wells syndrome – an Odyssey. Open Access Maced J Med Sci. 2019;7(18):3002–3005. doi: 10.3889/oamjms.2019.572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearson DR, Margolis DJ. Cellulitis and erysipelas. In: Kang S, Amagai M, Bruckner AL, Enk AH, Margolis DJ, McMichael AJ, editors. Fitzpatrick’s Dermatology in General Medicine. 9th ed. New York: McGraw-Hill Education; 2019:2746–2756. [Google Scholar]