

Keywords: anoctamin-1, AKT, RhoA, TGF-β, with-no-lysine (K) kinase
Abstract
Idiopathic pulmonary fibrosis (IPF) is a devastating disease characterized by progressive scarring of the lungs and resulting in deterioration in lung function. Transforming growth factor-β (TGF-β) is one of the most established drivers of fibrotic processes. TGF-β promotes the transformation of tissue fibroblasts to myofibroblasts, a key finding in the pathogenesis of pulmonary fibrosis. We report here that TGF-β robustly upregulates the expression of the calcium-activated chloride channel anoctamin-1 (ANO1) in human lung fibroblasts (HLFs) at mRNA and protein levels. ANO1 is readily detected in fibrotic areas of IPF lungs in the same area with smooth muscle α-actin (SMA)-positive myofibroblasts. TGF-β-induced myofibroblast differentiation (determined by the expression of SMA, collagen-1, and fibronectin) is significantly inhibited by a specific ANO1 inhibitor, T16Ainh-A01, or by siRNA-mediated ANO1 knockdown. T16Ainh-A01 and ANO1 siRNA attenuate profibrotic TGF-β signaling, including activation of RhoA pathway and AKT, without affecting initial Smad2 phosphorylation. Mechanistically, TGF-β treatment of HLFs results in a significant increase in intracellular chloride levels, which is prevented by T16Ainh-A01 or by ANO1 knockdown. The downstream mechanism involves the chloride-sensing “with-no-lysine (K)” kinase (WNK1). WNK1 siRNA significantly attenuates TGF-β-induced myofibroblast differentiation and signaling (RhoA pathway and AKT), whereas the WNK1 kinase inhibitor WNK463 is largely ineffective. Together, these data demonstrate that 1) ANO1 is a TGF-β-inducible chloride channel that contributes to increased intracellular chloride concentration in response to TGF-β; and 2) ANO1 mediates TGF-β-induced myofibroblast differentiation and fibrotic signaling in a manner dependent on WNK1 protein but independent of WNK1 kinase activity.
NEW & NOTEWORTHY This study describes a novel mechanism of differentiation of human lung fibroblasts (HLFs) to myofibroblasts: the key process in the pathogenesis of pulmonary fibrosis. Transforming growth factor-β (TGF-β) drives the expression of calcium-activated chloride channel anoctmin-1 (ANO1) leading to an increase in intracellular levels of chloride. The latter recruits chloride-sensitive with-no-lysine (K) kinase (WNK1) to activate profibrotic RhoA and AKT signaling pathways, possibly through activation of mammalian target of rapamycin complex-2 (mTORC2), altogether promoting myofibroblast differentiation.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive, fatal disease characterized by parenchymal fibrosis and structural distortion of the lungs. Age-adjusted mortality due to pulmonary fibrosis is increasing (1), and it poses a vexing clinical challenge given the lack of effective therapy. IPF is a disorder of abnormal wound healing (2, 3), wherein the initial trigger is an injury to the alveolar epithelial cell, followed by a nonresolving wound-healing response (4–6). Migration of interstitial fibroblasts to sites of injury and their differentiation to myofibroblasts are thought to be critical processes in pathogenesis of pulmonary fibrosis. Myofibroblasts secrete extracellular matrix proteins and profibrotic factors which perpetuate tissue remodeling and fibrosis (7, 8). Myofibroblasts are invariably found in histologic sections of human lung specimens from IPF patients and are thought to be a critical pathogenic mechanism responsible for the progressive nature of IPF.
Transforming growth factor-β (TGF-β) is the most well-established driver of myofibroblast differentiation (9–11). Production of this cytokine has been localized to areas of fibrosis in both experimental pulmonary fibrosis and human disease (9, 12, 13). Lung-targeted overexpression of TGF-β results in the development of lung fibrosis in animal models of IPF (11, 14). Conversely, inhibition of TGF-β, via soluble TGF-β receptors, can inhibit in vivo fibrogenesis (15, 16). TGF-β signaling acts through transmembrane-receptor serine/threonine kinases that phosphorylate Smad transcription factors (Smad2/3), leading to their heteromerization with a common mediator Smad4, nuclear translocation of the Smad2/3/4 complex, and activation of transcription of TGF-β target genes (7, 17, 18). We and others have established that signaling downstream of Smad-dependent gene transcription is critical for myofibroblast differentiation; this includes the TGF-β-induced RhoA/serum response factor (SRF) pathway (19–21) and AKT activation (22–24).
Anoctamine-1 (ANO1), also known as TMEM16A, discovered on gastrointestinal stromal tumors 1) (DOG1), ORAl cancer overexpressed (ORAOV2), and tumor amplified and overexpressed (TAOS-2) was initially found to be overexpressed in a number of cancer tissues and is thought to contribute to cancer cell growth and tumorigenesis (25, 26). Subsequently, ANO1 was identified as a calcium-activated chloride channel (27–29). Reported cellular functions of ANO1 include the control of cancer cell proliferation, cell survival, and migration (25, 26); secretory function in the epithelia (including airways, intestines, salivary glands, renal tubules, and sweat glands) (30); induction of electrical pacemaker activity of interstitial cells of Cajal in gastrointestinal smooth muscles (31), control of acute pain sensation, chronic pain, and anxiety-related behaviors (32, 33); and contribution to contraction of airway and vascular smooth muscles (32). Through an unbiased microarray analysis, we identified ANO1 as one of the top transcripts induced by TGF-β in primary cultured human lung fibroblasts (HLFs), which was not previously recognized. We therefore sought to confirm this finding by appropriate biochemical approaches and to examine the role of ANO1 in control of intracellular chloride homeostasis and myofibroblast differentiation.
MATERIALS AND METHODS
Primary Culture of Human Lung Fibroblasts
Human lung fibroblasts (HLFs) were isolated from human lungs that were rejected for transplantation through the Gift of Hope Organ and Tissue Donor Network. IPF-HLFs were isolated from the lungs of IPF patients shortly after their removal during lung transplantation at the University of Chicago under the Insitutional Review Board Protocol No. 14514 A. Human lung tissue samples were placed in DMEM with antibiotics. Lung tissue was minced to ∼1-mm3 pieces, washed, and plated on 10-cm plates in growth media containing DMEM supplemented with 10% FBS and antibiotics. The medium was changed twice a week. After ∼2 wk, the explanted and amplified fibroblasts were trypsinized, cleared from tissue pieces by sedimentation, and further amplified as passage 1. Unless indicated, cells were grown in 12-well plates at a density of 1 × 105 cells per well in a growth media for 24 h, starved in DMEM containing 0.1% FBS for 48 h (48-h starvation is important to ensure quiescence in all cells), and treated with desired drugs for various times as indicated in the figure legends. Primary cultures were used from passages 3 to 8.
Immunohistochemistry
Nonfibrotic human lungs were obtained from the Gift of Hope Organ and Tissue Donor Network. Fibrotic lungs from IPF patients were obtained shortly after lung transplantation at the University of Chicago under Insitutional Review Board Protocol No. 14514 A. Lung tissue specimens were fixed in formalin, washed in 70% ethanol, and paraffin embedded. Paraffin-embedded tissue was sectioned (5 µm) and stained with control IgG or ANO1 antibodies (Abcam ab53212, 3 µg/ml) by the University of Chicago Immunohistochemistry Core Facility.
Immunofluorescent Microscopy
Paraffin-embedded IPF human lung specimens were sectioned (5 μm) by the University of Chicago Human Tissue Resource Center. Sections were deparaffinized with xylenes and rehydrated with ethanol. They were then permeabilized and blocked with 1% BSA with 0.1% Triton X-100 in PBS for 1 h. Sections were next incubated with primary antibodies for ANO1 (Abcam Ab53212, 3 µg/ml) and smooth muscle actin (SMA; Sigma A2547, 1:100) overnight at 4°C. After being washed with PBS, sections were incubated with secondary goat anti-rabbit IgG conjugated to Alexa Fluor 488 (Invitrogen A-11034, 1:400) and donkey anti-mouse IgG conjugated to Alexa Fluor 647 (Invitrogen A-31571, 1:400) at room temperature for 1 h. After being washed with PBS sections were mounted with mounting medium containing DAPI (Abcam Ab104139). Fluorescence images were acquired on a Zeiss 3i Marianas spinning disk confocal microscope with the ×40/NA 1.3 oil (Plan-Apochromat) objective using Slidebook acquisition software (University of Chicago Integrated Light Microscopy Core). Negative controls included the omission of primary antibodies.
siRNA Transfection
HLFs were plated at a density of 0.8 × 105 cells per well (12-well plates, for Western blotting) or 1.6 × 105 cells per well [6-well plates, for real-time quantitative (q)PCR] and grown for 24 h. Cells were then transfected with 10 nM negative control siRNA or ANO1 siRNA using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions and kept in growth media for an additional 24 h, followed by serum starvation in 0.1% FBS for 48 h and then treatment with TGF-β for desired times.
Real-Time qPCR
Direct-Zol RNA miniprep kit (Zymo Research, Irvin, CA) was used to isolate total RNA following the manufacturer’s protocol. RNA was random primed and reverse transcribed using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer’s protocol. Real-time PCR analysis was performed using iTaq SYBR Green supermix with ROX (Bio-Rad) in a MyIQ single-color real-time PCR detection system (Bio-Rad). The ANO1 primers were as follows: GCAGAGAGGCCGAGTTTCTG (forward) and GCTCAGCCACTTTGGGCTG (reverse).
Western Blotting
Cells were lysed in a buffer containing 8 M deionized urea, 1% SDS, 10% glycerol, 60 mM Tris-HCl, pH 6.8, 0.02% pyronin Y, and 5% β-mercaptoethanol. Lysates were sonicated for 5 s. Samples were then subjected to polyacrylamide gel electrophoresis and Western blotting with desired primary antibodies and corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies and then developed by chemiluminescence reaction. Digital chemiluminescent images below the saturation level were obtained with a LAS-4000 analyzer, and signal intensity was quantified using Multi Gauge software (Fujifilm, Valhalla, NY). Specificity of primary antibodies was validated based on the appropriate molecular weight of target proteins and by the loss of the immunoreactivity in siRNA-mediated knockdown experiments [ANO1 and “with-no-lysine (K)” kinase (WNK1); Supplemental Fig. S1; see https://doi.org/10.6084/m9.figshare.24514975].
Measurements of Intracellular Chloride
Intracellular chloride levels ([Cl−]i) were determined using steady-state 36Cl− accumulation, which is proportional to [Cl−]i and can be converted to cytosolic concentration values if the precise volume of intracellular water is known (34, 35). HLFs were replated into six-well plates, grown in DMEM-FBS, and subsequently differentiated and treated as described above. On the day of assay, cells were additionally pretreated with ion transport inhibitors (concentrations and timing are specified in results). 36Cl− accumulation was initiated by adding to each well 1/10 volume of low-FBS assay medium containing 0.5 µCi/ml Na36Cl and 10 mM HEPES (final concentrations). HEPES was supplemented to minimize alkalinization during temporary removal of cells from the CO2 incubator.36Cl− accumulation was carried out for 30 min at 37°C in the CO2 incubator. Radioisotope uptake was terminated by aspirating 36Cl−-containing medium and four immediate washes with 2 mL of ice-cold medium containing (in mM) 300 mannitol, 3 MgSO4, and 10 HEPES (pH 7.4). After the final wash, HLFs were lysed in 1 mL of solution containing 2% SDS plus 8 mM EDTA. Half of each cell lysate sample was used for [36Cl] detection, and the other half was used to determine protein levels in individual wells. 36Cl− in each sample was counted with a TriCarb 4910 scintillation counter (Perkin Elmer, Waltham, MA) after addition of Ecoscint A scintillation cocktail (National Diagnostics, Atlanta, GA). The [Cl−]i values were quantified by normalizing 36Cl− counts to specific 36Cl− activity and either protein levels or cell numbers in each individual well (see results).
Materials
TGF-β was from EMD Millipore (GF111). The following antibodies for Western blotting were from Millipore-Sigma: smooth muscle α-actin (A5228, 10,000×), β-actin (A5441, 10,000×), α-tubulin (T6074, 10,000×), and myosin light chain (MLC; M4401, 1,000×). Antibodies against human collagen-1A1 (sc-28657, 1,000×) and AKT (sc-8312, 1,000×) were from Santa Cruz Biotechnology. Antibodies against ANO1 (14476, 1,000×), Smad2 (L1603, 1,000×), phospho-Smad2-Ser465/467 (138D4, 1,000×), phospho-MLC-Ser19 (3671, 1,000×), and phospho-AKT-Ser473 (9271, 1,000×) were from Cell Signaling Technology. Secondary HRP-conjugated antibodies for Western blotting (3,000×) were from Millipore Sigma (40-139-32ML, anti-rabbit IgG; 40-125-32ML, anti-mouse IgG). For immunohistochemistry and immunofluorescent microscopy, ANO1 antibodies were from Abcam (ab53212, 3 µg/ml final concentration), and SMA antibodies were from Millipore-Sigma (A5228, 100×). The T16Ainh-A01 inhibitor was from EMD Millipore (613551). SB-431542 was from Cayman Chemical (13031), bumetanide (B3023) and 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS, 309795) were from Millipore Sigma. The WNK inhibitor WNK463 (HY-100626HY) was from MedChemExpress. ANO1 siRNA (1027417), WNK1 siRNA (1027417), and AllStars negative control siRNA (1027281) were from Qiagen. Radiolabeled 36Cl (Na36Cl, 0.1 mCi/ml, 16 mCi/g Cl) was from American Radiolabeled Chemicals.
Statistical Analysis
All data are presented as mean values ± SD. Normally distributed data were analyzed using Student t test or one-way ANOVA with the Tukey’s honest significant difference test post hoc correction for multiple comparisons, as appropriate. Normalized-to-controls and nonnormally distributed data were analyzed using the Kruskal-Wallis nonparametric test followed by Dunn’s post hoc correction for multiple comparisons. For data sets presented in Figs. 4, 5, 6, 7, and 8, we performed additional preplanned analyses exploring the effect of ANO1 inhibitor and ANO1 siRNA in TGF-β-treated cells. The latter analysis was conducted using a one-population t test with values compared to 100%. Values of P < 0.05 were considered statistically significant. All statistical analyses were performed in Prism v. 9.5 (GraphPad Software).
Figure 4.
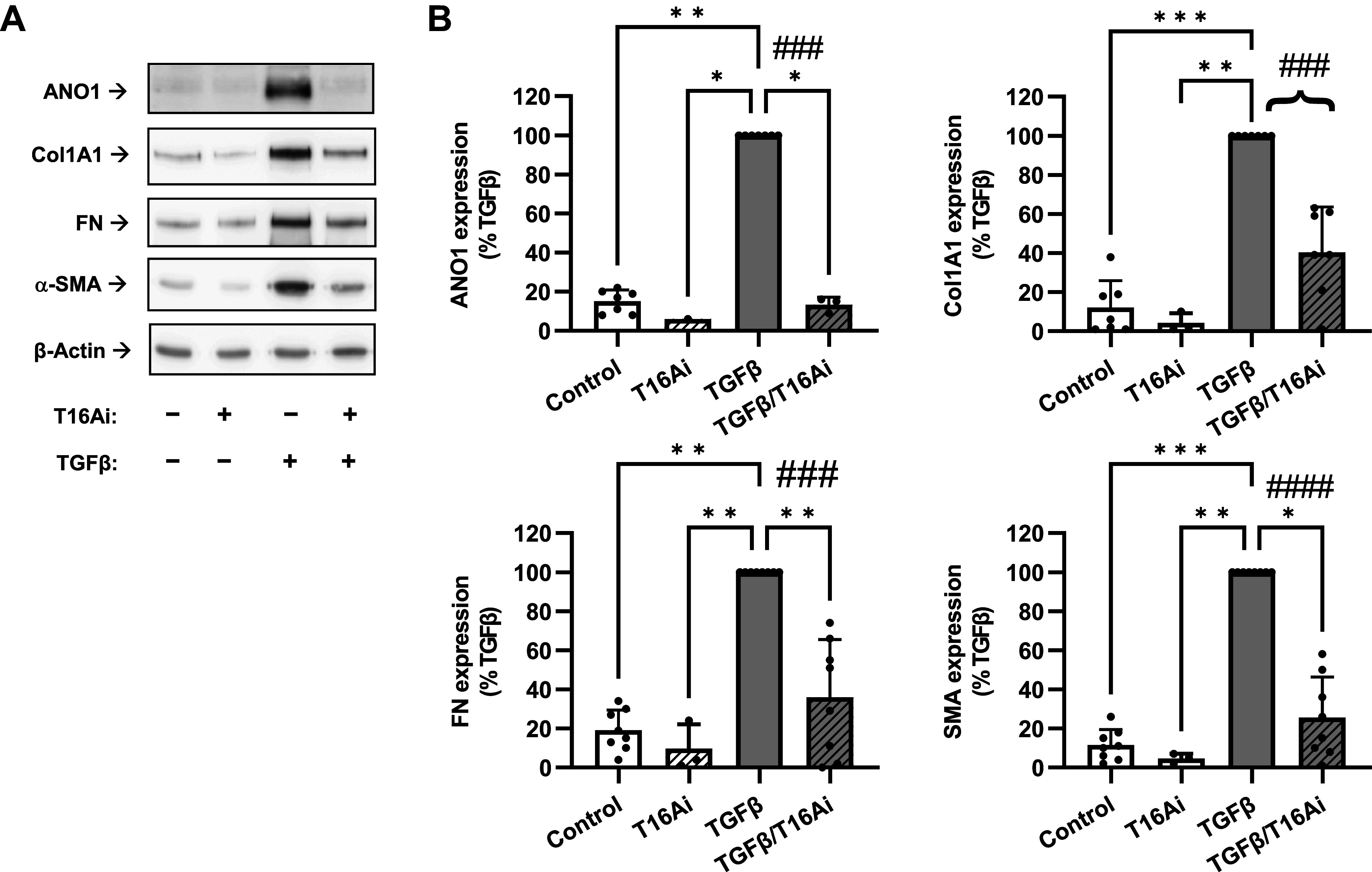
Attenuation of transforming growth factor-β (TGF-β)-induced myofibroblast differentiation by the anoctamin 1 (ANO1) inhibitor T16Ainh-A01. A: representative images of Western blot analyses of the effect of the pharmacological inhibitor of ANO1 T16Ainh-A01 (T16Ai) on myofibroblast differentiation. Serum-starved (48 h) human lung fibroblasts were pretreated with 30 µM T16Ai or vehicle control for 1 h, followed by treatment with TGF-β (1 ng/mL) for 48 h. Cell lysates were then probed for immunoreactivity of ANO1, collagen 1A1 (Col1A1), fibronectin (FN), and smooth muscle α-actin (SMA). B: quantification of ANO1, Col1A1, FN, and SMA immunoreactivities. Data are mean values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, vs. TGF-β group, Kruskal-Wallis test with Dunn’s post hoc correction. Braces, ###P < 0.001; ####P < 0.0001, are the results of preplanned comparisons of TGF-β effect with and without ANO1 inhibitor, one-population t test.
Figure 5.
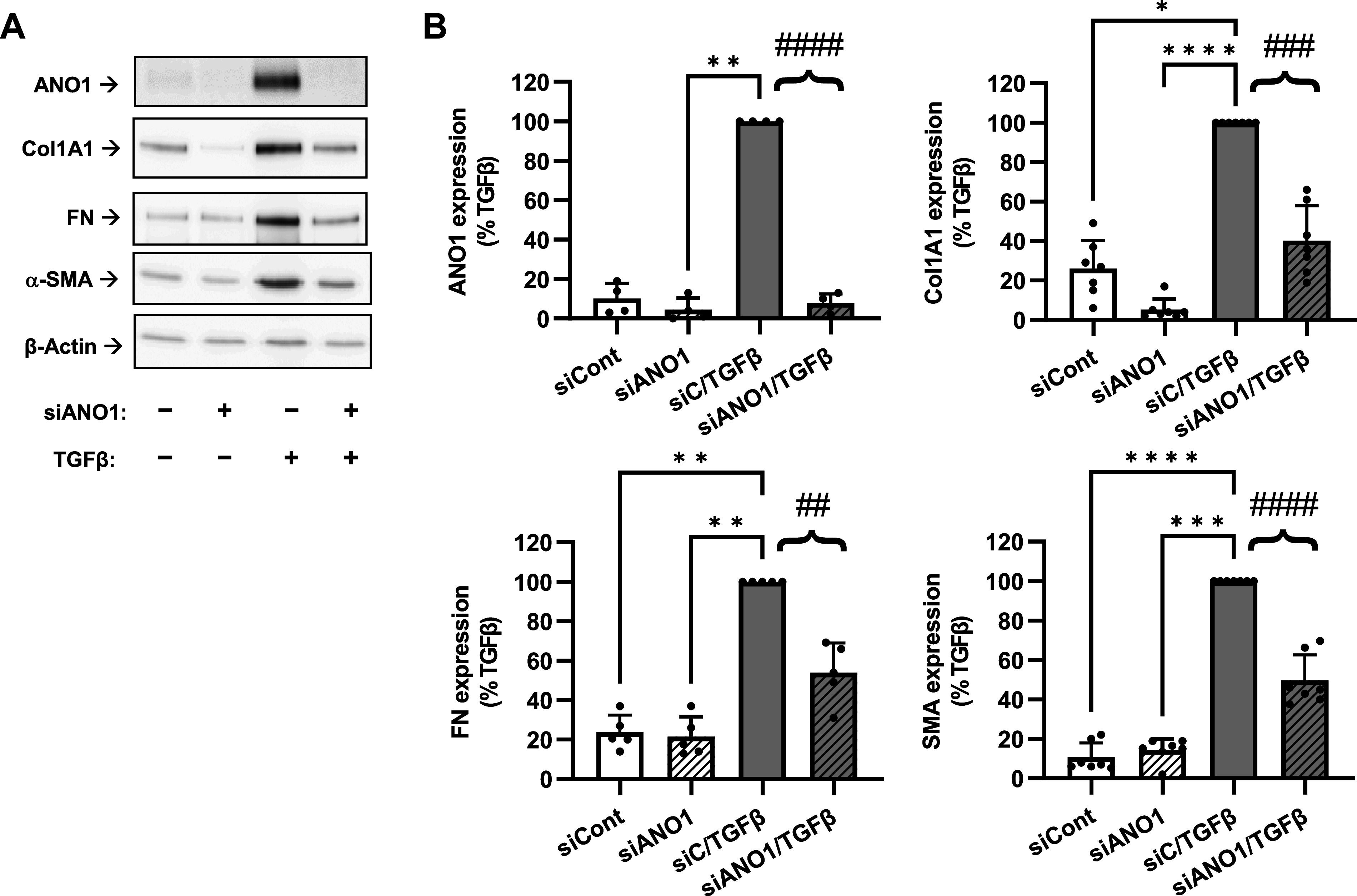
Inhibition of transforming growth factor-β (TGF-β)-induced myofibroblast differentiation by anoctamin 1 (ANO1) knockdown. A: representative images of Western blot analyses of the effect of siRNA targeting ANO1 (siANO1) on myofibroblast differentiation. Human lung fibroblasts were transfected with siRNA targeting ANO1 (siANO1) or control RNA (siCont) for 24 h, serum starved for 48 h, and treated with TGF-β or vehicle control for 48 h. Cell lysates were then probed for immunoreactivity of ANO1, collagen 1A1 (Col1A1), fibronectin (FN), and smooth muscle α-actin (SMA). B: quantification of ANO1, Col1A1, FN, and SMA immunoreactivity. Data are mean values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, vs. TGF-β group, Kruskal-Wallis test with Dunn’s post hoc correction. Braces, ##P < 0.01; ###P < 0.001; ####P < 0.0001, are the results of preplanned comparisons of TGF-β effect in cells treated with either control siRNA (siC) or siANO1, one-population t test.
Figure 6.
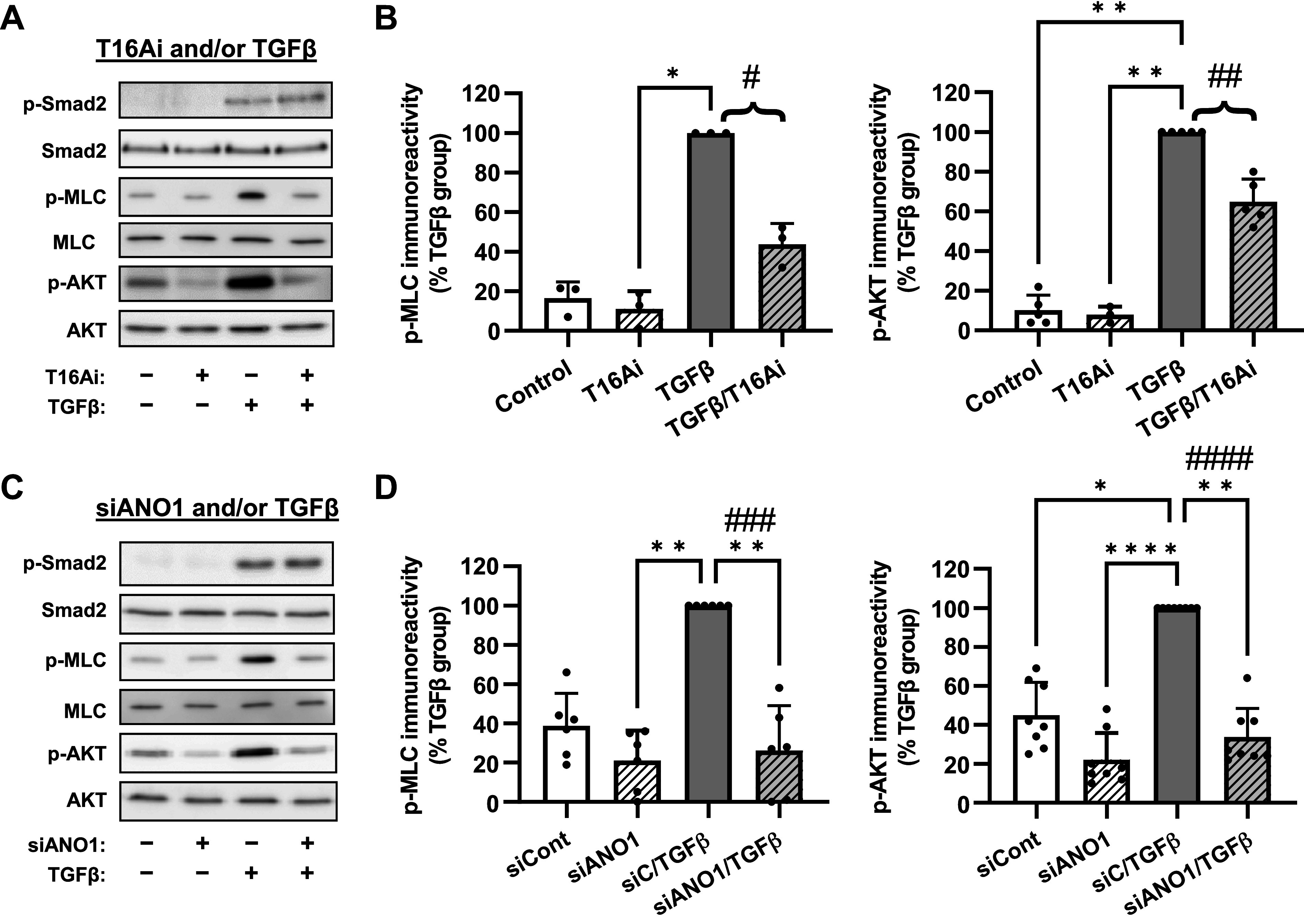
Anoctamin 1 (ANO1) contributes to transforming growth factor-β (TGF-β)-induced RhoA and AKT signaling without affecting Smad2 phosphorylation. A: representative images of Western blot analyses of the pharmacological effect of the ANO1 inhibitor T16Ainh-A01 (T16Ai) on intracellular signaling pathways in human lung fibroblasts (HLFs). Serum-starved (48 h) cells were pretreated for 1 h with T16Ai, followed by treatment with TGF-β (1 ng/mL) for either 30 min (p-Smad2 and Smad2) or 48 h [phosphorylated-myosin light chain (p-MLC), MLC, p-AKT, AKT). B: quantification of p-MLC and p-AKT immunoreactivities in T16Ai-treated cells. Data represent the mean immunoreactivity values ± SD normalized to TGF-β group within the same experiment. *P < 0.05, **P < 0.01, vs. TGF-β group, Kruskal-Wallis test with Dunn’s post hoc correction. Braces, #P < 0.05; ##P < 0.01, are the results of preplanned comparisons of the TGF-β effect in cells treated with or without T16Ai, one population t test. C: representative images of Western blot analyses of the effects of the siRNA targeting ANO1 (siANO1) on intracellular signaling pathways in HLFs. HLF cells were transfected with the siRNA targeting ANO1 (siANO1) or control RNA (siCont) for 24 h, serum starved for 48 h, and treated with TGF-β (1 ng/mL) for either 30 min (p-Smad2 and Smad2), or for 48 h (p-MLC, MLC, p-AKT, and AKT). D: quantification of p-MLC and p-AKT immunoreactivities in siCont and siANO1-treated cells. Data represent mean immunoreactivity values ± SD normalized to β-actin loading control and TGF-β group within the same experiment. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, vs. TGF-β group, Kruskal-Wallis test with Dunn’s post hoc correction. Braces, ###P < 0.001; ####P < 0.0001, are the results of preplanned comparisons of the TGF-β effect in cells treated with control siRNA (siC) or siANO1, one population t test.
Figure 7.
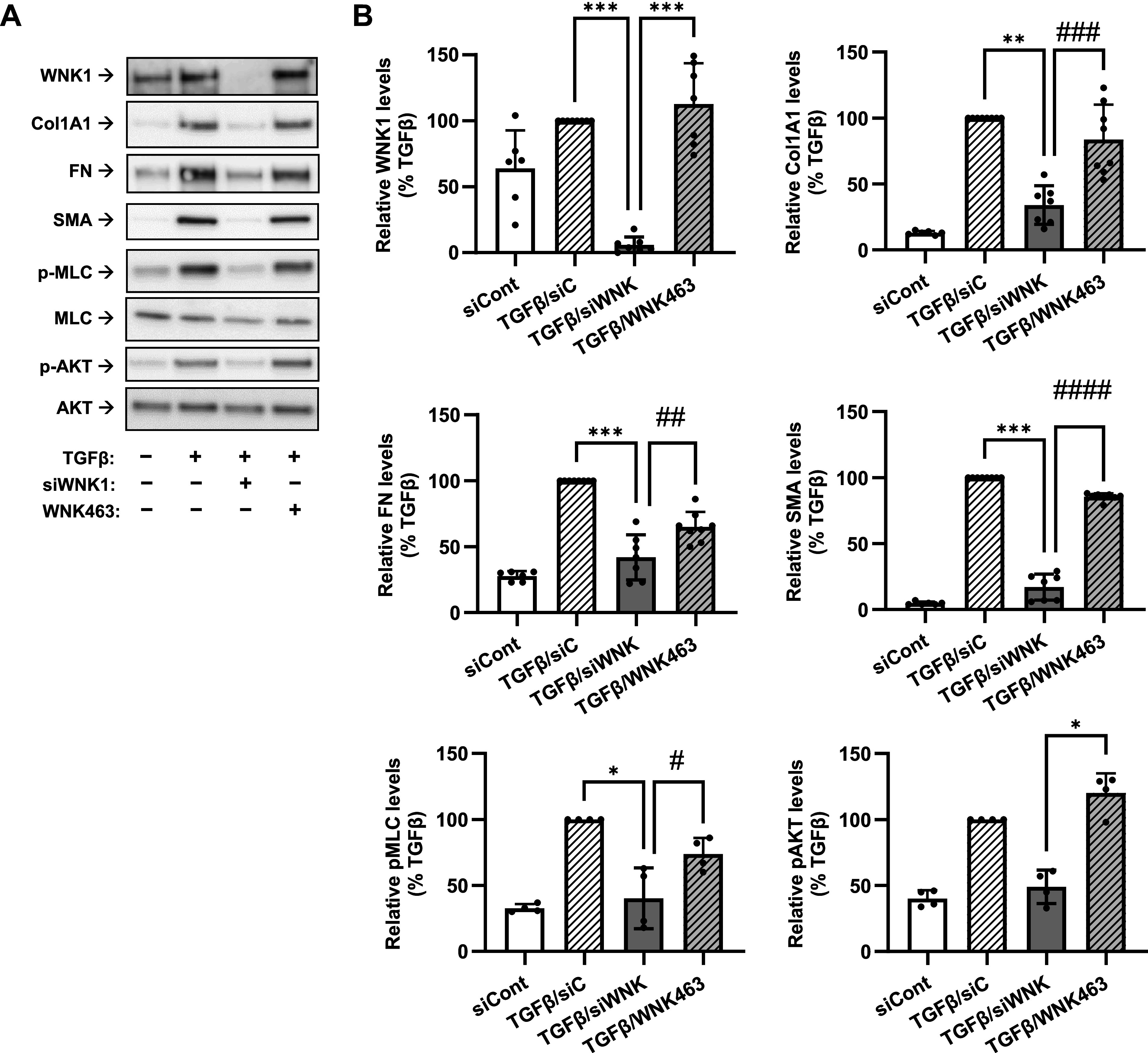
With-no-lysine (K) kinase (WNK1) mediates transforming growth factor-β (TGF-β)-induced myofibroblast differentiation in a kinase-independent manner. A: representative images of Western blot analyses of the effects of siRNA targeting WNK1 (siWNK1) and the WNK kinase inhibitor WNK463 on myofibroblast markers and intracellular signaling pathways in human lung fibroblasts (HLFs). HLFs were transfected with siRNA targeting anoctamin 1 (siANO1) or control RNA (siCont) for 24 h, serum starved for 48 h, pretreated with or without WNK1 inhibitor WNK463 (10 µM) for 1 h, followed by treatment with TGF-β (1 ng/mL) or vehicle control for 48 h. Cell lysates were then probed for the immunoreactivity of WNK1, collagen 1A1 (Col1A1), fibronectin (FN), smooth muscle α-actin (SMA), phosphorylated-myosin light chain (p-MLC), MLC, p-AKT, and AKT. B: quantification of the immunoreactivity of WNK, Col1A1, FN, SMA, p-MLC, and p-AKT. Data are mean immunoreactivity values ± SD normalized to β-actin loading control and TGF-β group within the same experiment. *P < 0.05, **P < 0.01, ***P < 0.001, vs. TGF-β group, Kruskal-Wallis test with Dunn’s post hoc correction. #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001; results of preplanned comparisons of the TGF-β effect in cells treated with siWNK1 or the WNK1 inhibitor WNK463, t test.
Figure 8.
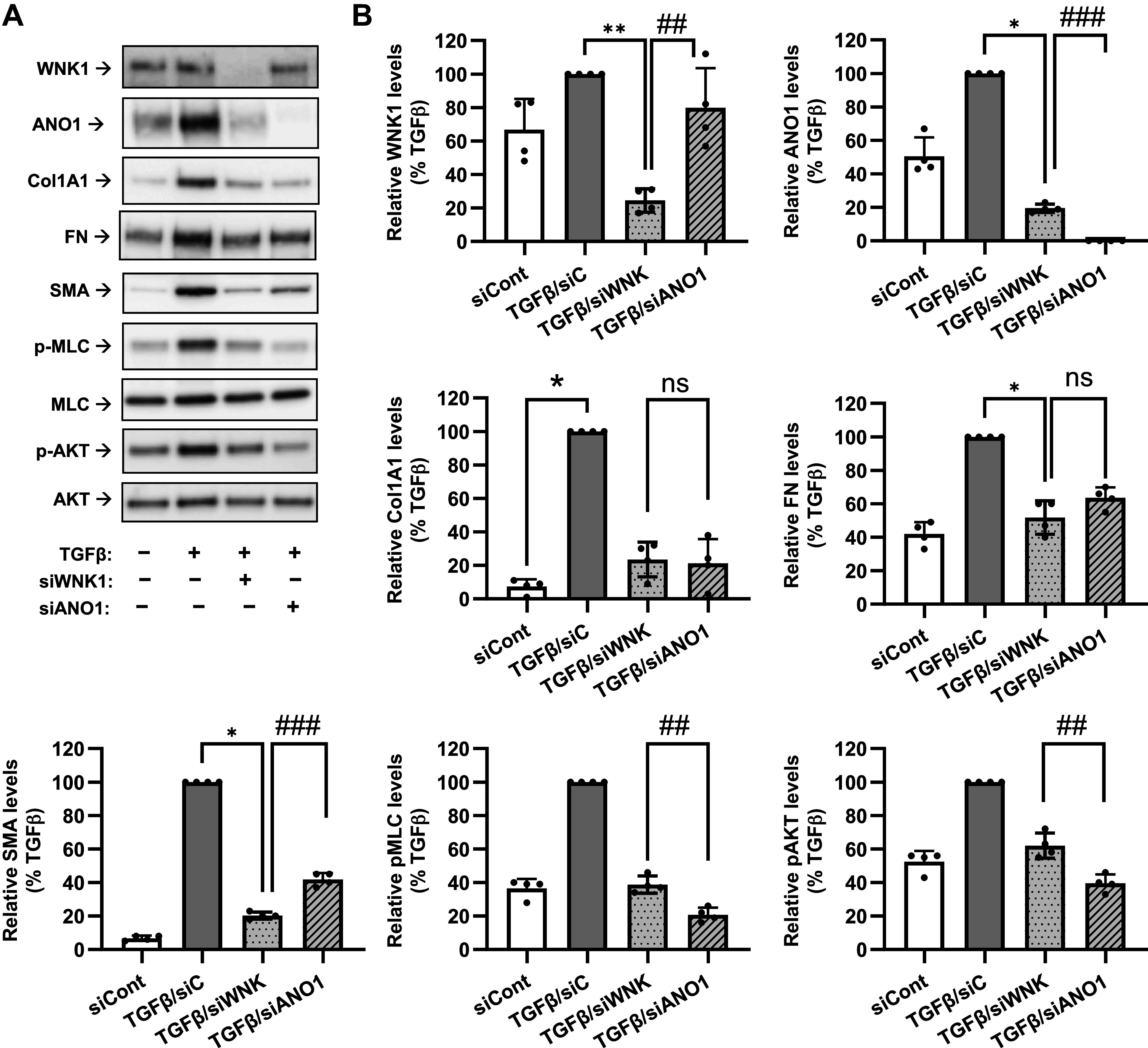
Relative contributions of with-no-lysine (K) kinase (WNK1) and anoctamin 1 (ANO1) to transforming growth factor-β (TGF-β)-induced myofibroblast differentiation and signaling in human lung fibroblasts isolated from the idiopathic pulmonary fibrosis lungs (IPF-HLFs). A: representative images of Western blot analyses of the effects of siRNA targeting WNK1 (siWNK1) or ANO1 (siANO1) on myofibroblast differentiation and intracellular signaling in IPF-HLFs. Cells were transfected with siRNA targeting WNK1 (siWNK1), ANO1 (siANO1) or control RNA (siCont) for 24 h, serum starved for 48 h, followed by treatment with TGF-β (1 ng/mL) or vehicle control for 48 h. Cell lysates were then probed for the immunoreactivity of WNK1, collagen 1A1 (Col1A1), fibronectin (FN), smooth muscle α-actin (SMA), p-MLC, MLC, p-AKT, and AKT. B: quantification of the immunoreactivity of WNK, Col1A1, FN, SMA, p-MLC, and p-AKT. Data are mean values ± SD normalized to TGF-β group within the same experiment. *P < 0.05, **P < 0.01, vs. TGF-β group, Kruskal-Wallis test with Dunn’s post hoc correction. #P < 0.05, ##P < 0.01, ###P < 0.001; results of preplanned comparisons of the TGF-β effect in cells treated with or without T16Ai, one population t test.
RESULTS
Expression of ANO1 in Human Lung Myofibroblasts in Cell Culture and in Human Lung Fibrotic Tissues
Treatment of primary cultured human lung fibroblasts (HLFs) with TGF-β resulted in a robust accumulation of ANO1 expression at mRNA and protein levels, as assessed by real-time qPCR and Western blotting (Fig. 1, A and B). This effect was mediated by the TGF-β receptor II (TGFBRII), as it was inhibited by the specific TGFBRII kinase inhibitor SB-431542 (Fig. 1C). Given that myofibroblasts are significant contributors to tissue scaring during fibrotic processes, we then assessed the expression of ANO1 in lung explants from patients with idiopathic pulmonary fibrosis (IPF) as compared to nonfibrotic lungs. As shown in Fig. 2A, ANO1 expression in nonfibrotic (NF) lungs was largely detected in smooth muscle. In the IPF lungs, ANO1 expression was clearly observed in fibrotic areas with a patchy appearance (Fig. 2A). Double immunofluorescent staining demonstrated a specific ANO1 signal in IPF tissue, which was localized to the same areas as the SMA-expressing myofibroblasts (Fig. 2B).
Figure 1.
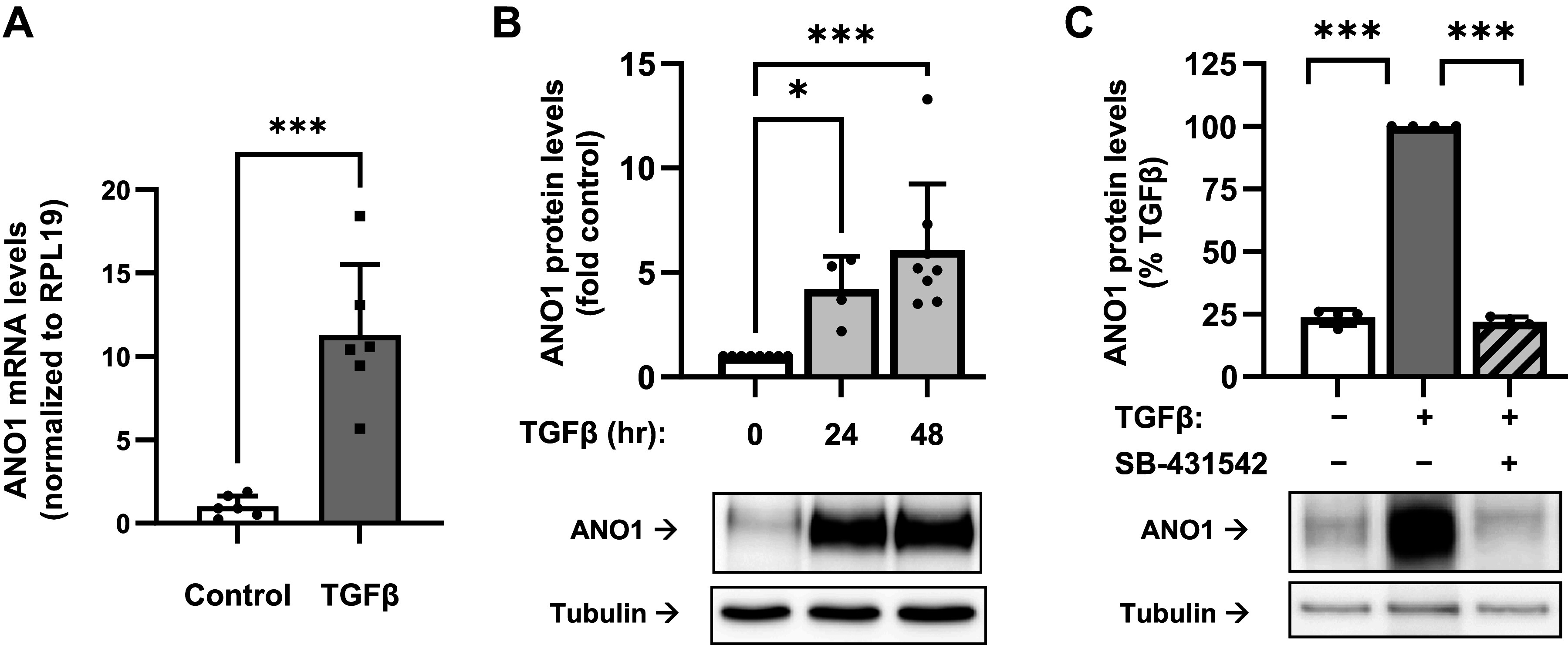
Transforming growth factor-β (TGF-β)-induced expression of anoctamin 1 (ANO1) in human lung fibroblasts. Human lung fibroblasts were serum starved in 0.1% FBS for 48 h, followed by treatment with TGF-β (1 ng/mL) for 24 or 48 h, as indicated. A: ANO1 mRNA expression after 24-h treatment with TGF-β as determined by quantitative RT-PCR. Data are mean values ± SD. ***P < 0.001, t test. B: Western blot analysis and quantitation of ANO1 protein expression in response TGF-β. Data are mean values normalized to control cells ± SD. *P < 0.05, ***P < 0.001, Kruskal-Wallis test with Dunn’s correction for multiple comparisons. C: effect of the TGF-β receptor II (TGFBRII) inhibitor SB-431542 (5 µM, 30-min preincubation) on TGF-β-induced ANO1 protein. expression (48 h). Shown are representative images of ANO1 immunoreactivity and quantification. Data are mean immunoreactivity values normalized to TGF-β-treated cells ± SD. ***P < 0.001, one-population t test with Bonferroni correction for multiple comparisons.
Figure 2.
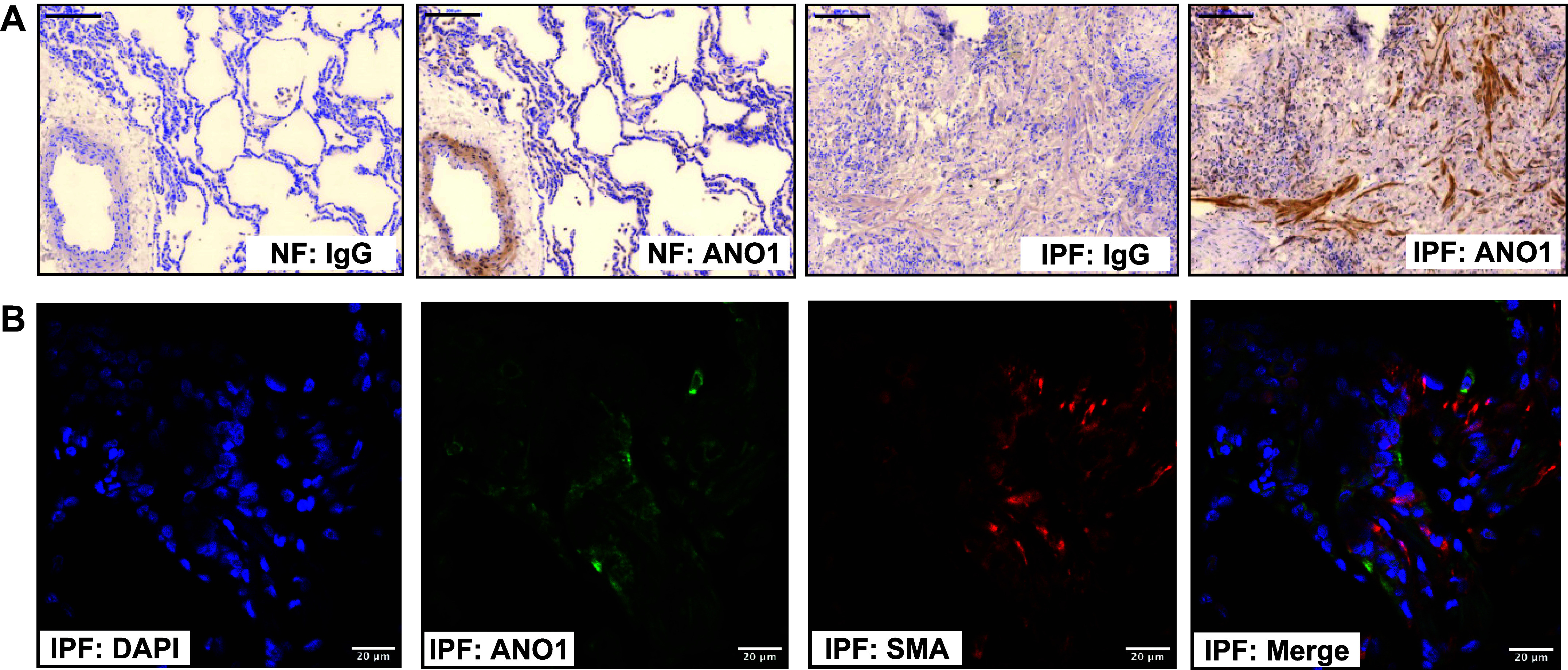
Anoctamin 1 (ANO1) protein expression in fibrotic areas of human lungs with idiopathic pulmonary fibrosis (IPF). A: representative immunohistochemical images of nonfibrotic (NF) lungs and fibrotic areas of the IPF lungs. Sequential sections of the same lung were probed with either ANO1 antibody or IgG control. Scale bars = 200 μm. B: high-magnification immunofluorescence images of section from an IPF lung, stained for ANO1 (green) and smooth muscle a-actin (SMA; red), and counterstained with DAPI (blue). Scale bars = 20 μm. Shown are representative images of lung sections from 2 IPF individuals. Note that we did not expect stains precisely overlapping because of membrane localization of ANO1 and cytosolic localization of SMA.
Effect of TGF-β on Intracellular Chloride Levels and the Role of ANO1 in HLFs
Given that ANO1 is a chloride channel, we next asked whether TGF-β affects intracellular chloride homeostasis in an ANO1-dependent manner. To assess intracellular chloride levels ([Cl−]i), we measured the steady-state accumulation of 36Cl− inside the cells. At equilibrium, 36Cl− accumulation is directly proportional to [Cl−]i and can be converted to cytosolic concentration values if the precise volume of intracellular water is known (34, 35). To determine appropriate conditions for measuring steady-state 36Cl− accumulation, we first assessed the kinetics of 36Cl− uptake in HLFs. As shown in Fig. 3A, intracellular 36Cl− levels reached equilibrium after ∼10–15 min. The fast equilibration was determined by 36Cl− uptake through several ion transport systems. Addition of bumetanide, an inhibitor of the Na+-K+-2Cl− cotransporter (NKCC1), reduced steady-state chloride accumulation by ∼25% (n = 4, P < 0.05, data not shown). When bumetanide was combined with the anion exchanger blocker DIDS, 36Cl− uptake values were reduced by >90% and failed to saturate within 30 min (Fig. 3A). These results indicate that the main transport system that equilibrates 36Cl− across the plasma membrane of HLFs is the DIDS-sensitive chloride-bicarbonate exchanger, with additional contributions from NKCC1 and possibly other DIDS-sensitive Cl− channels.
Figure 3.
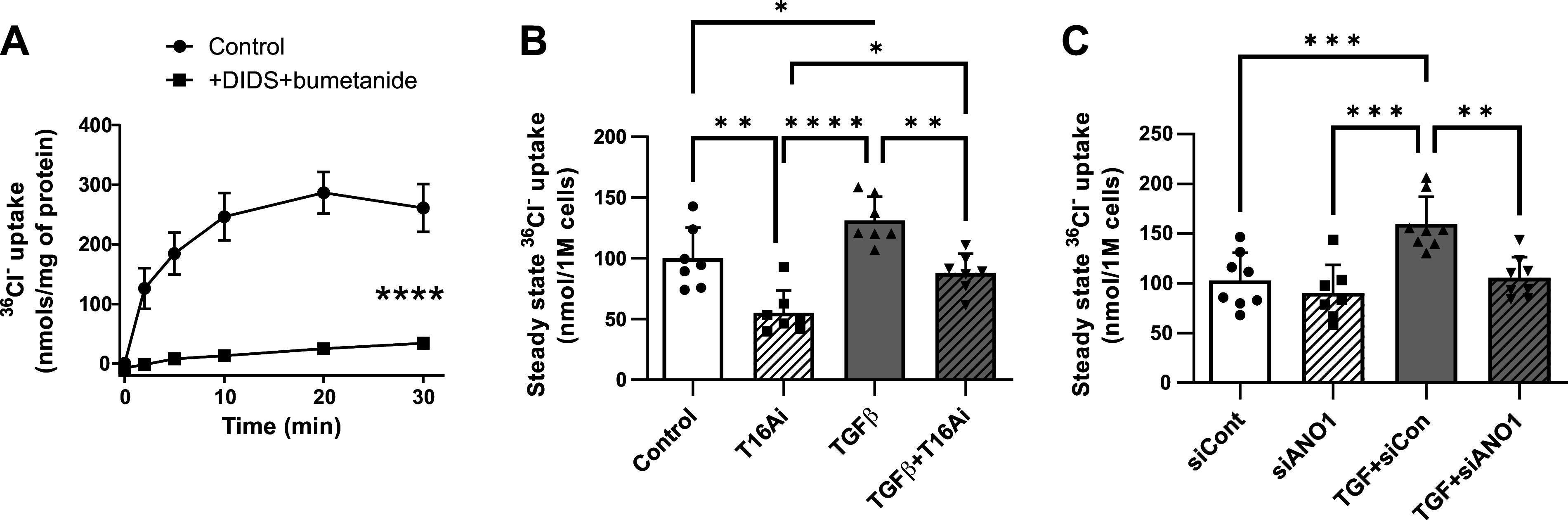
Effect of transforming growth factor-β (TGF-β) and the role of anoctamin 1 (ANO1) in control of intracellular Cl− levels in human lung fibroblasts (HLFs). A: kinetics of 36Cl− accumulation in HLFs. Cl− uptake was measured as radiotracer accumulation after adding 36Cl− into cell culture medium. Serum-starved HLF cells were pretreated for 30 min with either vehicle or a combination of 1 mM DIDS plus 20 µM bumetanide. The same inhibitors were present in transport assay media. Data are mean values ± SD in 3 experiments. ****P < 0.0001, repeated measures ANOVA; inhibitors vs. control. B: effect of the ANO1 inhibitor T16Ainh-A01 (T16Ai) on intracellular Cl− levels in control and TGF-β-treated HLFs. Intracellular Cl− levels were measured as the steady-state 36Cl− accumulation and normalized to the number of cells per well as described in materials and methods. T16Ainh-A01 (30 µM) was applied 1 h before and was present during 36Cl− transport assay. Data represent mean values ± SD from 7 independent experiments in 2 HLF passages. *P < 0.01, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey post hoc correction for multiple comparisons. C: the effect of siRNA-mediated ANO1 knockdown on intracellular Cl− levels in control and TGF-β-treated primary human lung fibroblasts. HLF cells were transfected with the siRNA targeting ANO1 (siANO1) or the negative control RNA (siCont) for 24 h, serum starved for 48 h, and treated with TGF-β or vehicle control for 48 h. The steady-state 36Cl− accumulation was measured and normalized as in B. Data represent mean values ± SD of 8 independent experiments performed in 2 HLF passages. **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey post hoc correction for multiple comparisons.
We next examined the effect of TGF-β and the pharmacological inhibitor of ANO1 T16Ainh-A01 (36) on [Cl−]i in HLFs. As shown in Fig. 3B, 48-h treatment of HLFs with TGF-β resulted in a significant increase in steady-state 36Cl− accumulation in HLFs. Further, T16Ainh-A01 significantly suppressed both basal and TGF-β-induced steady-state 36Cl− accumulation in HLFs (Fig. 3B). We then used siRNA to knock down ANO1 in HLFs. TGF-β increased 36Cl− accumulation in cells treated with control siRNA, and, similar to T16Ainh-A01, ANO1 knockdown completely blunted TGF-β-induced accumulation of intracellular 36Cl− (Fig. 3C). One difference was that, unlike T16Ainh-A01, siANO1 did not affect intracellular 36Cl− levels under the basal conditions (Fig. 3, B and C, and see discussion). Together, these data suggest that ANO1 activity modulates [Cl−]i in HLFs, and TGF-β-induced increase in intracellular Cl− (36Cl−) is fully determined by the increased expression of ANO1.
Role of ANO1 in TGF-β-Induced Myofibroblast Differentiation
Consistent with our prior work and other publications in the field, treatment of HLFs with TGF-β for 48 h resulted in differentiation to myofibroblasts as evidenced by a profound increase in the expression of the extracellular matrix proteins collagen-1 (Col1A1) and fibronectin (FN) and dramatic increase in the myofibroblast marker smooth muscle α-actin (SMA) (Fig. 4). These changes paralleled newly found six- to -sevenfold increases in ANO1 expression (Fig. 4). Unexpectedly, pretreatment of HLFs with the ANO1 inhibitor T16Ainh-A01 blocked the TGF-β-induced increase in ANO1 levels (Fig. 4). More importantly, T16Ainh-A01 blunted the TGF-β-induced upregulation of α-SMA, Col1A1, and FN without affecting levels of the housekeeping β-actin protein. Attenuation of TGF-β-induced ANO1 expression by T16Ainh-A01 (Fig. 4) may suggest that in addition to pharmacological inhibition, long-term T16Ainh-A01 treatment also affects the expression of ANO1 via as yet unknown mechanisms. To corroborate the results with pharmacological blockade of ANO1, we used an siRNA approach. Figure 5 shows efficient ANO1 protein knockdown by ANO1 siRNA and a significant decrease in TGF-β-induced expression of α-SMA, Col1A1, and FN very similar to the effect of T16Ainh-A01.
Control of TGF-β-Induced Signaling by ANO1 in HLFs
We next began exploring the mechanism through which ANO1 controls TGF-β-induced myofibroblast differentiation, starting from established fibrotic pathways. Phosphorylation of Smad2/3 transcription factors by TGF-β receptors is the proximal event in TGF-β signaling. As shown in representative images in Fig. 6 (P-Smad2 blots), a 30-min treatment of HLFs with TGF-β resulted in phosphorylation of Smad2 without affecting total levels of Smad2. Pretreatment of HLFs with the ANO1 inhibitor T16Ainh-A01 had no effect on TGF-β-induced Smad2 phosphorylation (Fig. 6A), suggesting that the initial signaling event is not affected. We next focused on the downstream, delayed signaling processes that are required for myofibroblast differentiation in response to TGF-β. We and others have previously established the critical role of the RhoA pathway in TGF-β-induced myofibroblast differentiation (19–21). We assessed changes in this pathway by measuring the phosphorylation state of myosin light chain (MLC), which is controlled by RhoA-mediated signaling (37) and represents a widely used indirect assay for RhoA pathway activation (38, 39). Treatment of HLFs with TGF-β for 48 h increased MLC phosphorylation more than fivefold, and this effect was significantly inhibited by the pharmacological ANO1 inhibitor T16Ainh-A01, whereas the total MLC levels remained unchanged (Fig. 6, A and B). We also examined the role of ANO1 in the control of AKT phosphorylation in HLFs, as has been reported in other cells (40, 41) albeit not in the context of TGF-β. Treatment of HLFs with TGF-β for 48 h resulted in a significant AKT phosphorylation; and this effect was inhibited by T16Ainh-A01 (Fig. 6, A and B). To corroborate the data obtained with the pharmacological ANO1 inhibitor, we used the ANO1 knockdown approach. Similarly to T16Ainh-A01, ANO1 siRNA had no significant effect on Smad2 phosphorylation, but it completely abolished TGF-β-induced phosphorylation of MLC and AKT (Fig. 6, C and D).
Control of TGF-β-Induced Myofibroblast Differentiation and Signaling by With-No-Lysine (K) Kinase, WNK1
WNK kinases control electrolyte homeostasis and cell volume in renal epithelial cells (42). The WNK1 isoform is ubiquitously expressed, but its function outside of kidney remains poorly understood. It is well established that the kinase activity of WNK1 is activated in response to lowering [Cl−]i and conversely inhibited by increased concentrations of intracellular chloride (43). On the other hand, it was recently reported that increased [Cl−] promotes a scaffolding (kinase activity-independent) function of WNK1 by bringing together the mammalian target of rapamycin complex-2 (mTORC2) with its targets, such as serum/glucocorticoid regulated kinase 1 (SGK1) (44). Therefore, we assessed the role of WNK1 and its kinase activity in TGF-β-induced myofibroblast differentiation and signaling. As shown in Fig. 7, siRNA knockdown of WNK1 significantly reduced TGF-β-induced myofibroblast differentiation (Col1A1, FN, and SMA expression) as well as RhoA (P-MLC) and AKT (P-AKT) activation. In striking contrast, the pan-WNK kinase inhibitor WNK463 was largely ineffective at concentrations (10 μM) that are far above its nanomolar WNK1-kinase inhibitory IC50 (45). Together, these data suggest that WNK1 mediates TGF-β-induced myofibroblast differentiation and RhoA and AKT signaling in a kinase-independent manner.
Control of TGF-β-Induced Myofibroblast Differentiation and Signaling by ANO1 and WNK1 in IPF-HLFs
We further examined the generality of our findings using lung fibroblasts cultured from IPF lung (IPF-HLFs), comparing side-by-side the effects of siWNK1 and siANO1. As shown in Fig. 8, ANO1 was readily detectable under basal conditions in IPF-HLFs, and its expression was further induced by TGF-β. Both siWNK1 and siANO1 strongly inhibited myofibroblast differentiation (expression of Col1A1, FN, and SMA) as well as two relevant signaling pathways (P-MLC, P-AKT). Interestingly, WNK1 knockdown also attenuated the expression of ANO1 in response to TGF-β (Fig. 8). This suggests a potentially novel feed-forward mechanism in TGF-β signaling through WNK1-dependent upregulation of ANO1.
DISCUSSION
This study describes a novel and unexpected mechanism of TGF-β-induced myofibroblast differentiation involving the Ca2+-activated Cl− channel anoctamin-1 (ANO-1). Our principal findings suggest that ANO1 and the chloride-sensitive signaling protein WNK1 play critical roles in the myofibroblast differentiation process.
We discovered that ANO1 is dramatically upregulated during TGF-β-induced human lung myofibroblast differentiation in vitro (Fig. 1). The pathological significance of this discovery is supported by our observation of ANO1 immunoreactivity in human IPF lungs, where it is expressed in the same areas as SMA-positive myofibroblasts (Fig. 2). These findings are novel and have no precedent in the literature. We have previously extensively reviewed the published data on the regulation of ANO1 expression under various conditions and in a variety of cell types (46). ANO1 can be upregulated by several cytokines from the interleukin family (e.g., IL-4, IL-6, IL-13), lysophosphatidic acid, and epidermal growth factor in various cell types. There were several controversial reports on the bidirectional (up or down) regulation of ANO1 expression by serum supplementation or by the G-protein coupled receptor agonist angiotensin II (reviewed in Ref. 46). In this context, the generality of effect of TGF-β on ANO1 expression we identified is yet to be tested in other cell types. The downstream mechanism(s) of TGF-β-induced ANO1 expression in HLFs and other cells also requires further exploration. Nevertheless, a number of important deductions can be made based on the existing literature and findings of the present work (see below).
The most important discovery of this study is that ANO1 expression is a critical, previously unknown step in myofibroblast differentiation. Either pharmacological inhibition or siRNA-mediated knockdown of ANO1 significantly attenuates TGF-β-induced myofibroblast differentiation of HLFs (Figs. 4 and 5, respectively). This conclusion about ANO1 involvement in myofibroblast transformation is supported by the analysis of expression of three independent myofibroblast markers including the extracellular matrix proteins collagen 1A1 and fibronectin and the myofibroblast marker smooth muscle α-actin.
Because the most recognized role for ANO1 is in the regulation of chloride transport and homeostasis, we first explored the effect of TGF-β on intracellular Cl− levels ([Cl−]i) and found that this cytokine drives an increase in [Cl−]i in HLFs, in a manner dependent on ANO1 activity and expression. The ANO1 blocker, T16Ainh-A01, strongly reduces intracellular Cl− levels under both basal conditions and in the TGF-β-treated HLFs (Fig. 3B). ANO1 siRNA prevents the effect TGF-β on the intracellular [Cl−]i but does not modify baseline [Cl−]i on its own (Fig. 3C). This partial discrepancy in the effects of ANO1 manipulation on baseline [Cl−]i may be explained by 1) either limited specificity of T16Ainh-A01, or 2) by the differential duration of ANO1 inhibition (acute with T16Ainh-A01 vs. chronic with siRNA). T16Ainh-A01 has off-target effects on the ubiquitously expressed LRRC8 volume-regulated anion channels, with an IC50 value of ∼5 µM (47). It is also possible that acute inhibition of anion channels (ANO1 and/or LRRC8) can reduce intracellular levels of the negatively charged Cl− via hyperpolarization of HLFs. If so, the TGF-β-induced cell transformation would be mediated by changes in membrane potential rather than [Cl−]i. To address this uncertainty and further test [Cl−]i dependency, we examined the long-term effects of DIDS and bumetanide, two inhibitors of electroneutral transporters that reduced [Cl−]i (Fig. 3A), on myofibroblast differentiation. As shown in Supplemental Fig. S2 (see https://doi.org/10.6084/m9.figshare.24514876), bumetanide + DIDS significantly attenuated the effect of TGF-β on FN and SMA expression in HLFs. Paradoxically, bumetanide + DIDS increased basal Col1A1 protein expression and did not inhibit, but rather potentiated, the effect of TGF-β on levels of this protein (Supplemental Fig. S2). This may point to a nonspecific long-term effect of ion transport blockers on HLF cell physiology and deposition of extracellular proteins. However, with the exception of the surprising upregulation of Col1A1 by bumetanide +DIDS, the majority of the experiments employing ANO1 inhibitors (Fig. 4), ANO1 siRNA (Fig. 5), and [Cl−]i manipulation (Supplemental Fig. S2) are consistent with the idea of [Cl−]i-dependent regulation of myofibroblast differentiation.
The effect of TGF-β on ANO1 expression needs to be considered in the context of known mechanisms driving TGF-β-dependent changes in gene expression. First, ANO1 can be a direct target of canonical SMAD signaling, and the ANO1 promoter may have previously unrecognized SMAD-binding elements. Second, ANO1 levels may be indirectly modulated by Smad-dependent gene transcription through regulation of downstream signaling pathways. For example, ANO1 expression is induced by IL-6 (through STAT3 transcription factor) (48) while IL-6 expression is induced by TGF-β also by recruiting the STAT3 pathway (49, 50). Third, ANO1 transcription may be under the control of myocardin/serum response factor (SRF) as seen in vascular smooth muscle cells (51). Given our previous findings that TGF-β promotes the SMAD-dependent activation of the RhoA/SRF pathway in HLFs (20, 21), the TGF-β/SRF-dependent transcription of ANO1 is also plausible.
Findings presented in Fig. 6 indicate that inhibition of ANO1 does not affect the immediate TGF-β signaling involving Smad2 phosphorylation but does block downstream signaling events, including phosphorylation of MLC and AKT (Fig. 6). The net phosphorylation of MLC (controlled by RhoA-dependent inactivation of MLC phosphatase; Ref. 37) has been commonly used for an indirect assessment of RhoA activation (38, 39). Our previous work established a critical role of the RhoA/SRF pathway in TGF-β-induced myofibroblast differentiation (19–21). The present study found complete blockade of MLC phosphorylation by pharmacological inhibition of ANO1 or ANO1 siRNA knockdown (Fig. 6). Based on these observations, we argue that TGF-β-dependent activation of the RhoA/SRF pathway could be one of the mechanisms by which ANO1 controls myofibroblast differentiation. On the other hand, ANO1 transcription has been reported to be driven by SRF (51), potentially suggesting the reciprocal regulation and a positive feed-forward loop mechanism involving both SRF and ANO1. Besides the RhoA/SRF axis, we also found that inhibition and siRNA knockdown of ANO1 both reduce TGF-β-dependent phosphorylation of AKT (Fig. 6). A role of ANO1 in regulation of AKT phosphorylation has been reported in other cells (40, 41) but not in the context of TGF-β stimulation. AKT plays an important role in TGF-β-induced myofibroblast differentiation (24) and may additionally contribute to the profibrotic effects of ANO1.
What is a common denominator among intracellular Cl− levels, TGF-β signaling, and ANO1 expression? Our study uncovers a novel, Cl−-dependent, mechanism for TGF-β-induced myofibroblast differentiation and RhoA and AKT signaling that involves the chloride-sensing protein kinase WNK1. All the effects of TGF-β we observed on myofibroblast differentiation and signaling were inhibited by WNK1 knockdown (Fig. 7). Yet, TGF-β signaling and induction of myofibroblast markers were largely insensitive to the actions of the pan-WNK kinase inhibitor WNK463 (Fig. 7). These new findings are consistent with a recently reported kinase-independent role of WNK1 in activation of mTORC2 signaling under conditions of high intracellular [Cl−] (44). According to that study, binding of Cl− ions to the autoinhibitory domain of WNK1 blocks its kinase activity but promotes WNK1 association with the mTOR complex 2, followed by activation of downstream signaling molecules (44). It is established that mTORC2 promotes AKT autophosphorylation at serine-473 (52), the phosphorylation site we assessed in this study (Figs. 6–8). mTORC2 was also shown to be required for RhoA activity in rat uterine leiomyoma cells and smooth muscle-like lymphangioleiomyomatosis cells (53), in mesenchymal stem cells (54), and in neutrophils (55).
In a separate noteworthy series of experiments, we explored the relative contributions of ANO1 and WNK1 to regulation of myofibroblast markers in HLFs that were isolated from IPF lungs (IPF-HLFs). In IPF fibroblasts, ANO1 expression is readily detectable even in the absence of TGF-β (Fig. 8). Treatment with TGF-β causes additional increases in ANO1 levels; upregulates expression levels of the myofibroblast markers Col1A1, FN, and SMA; and stimulates phosphorylation of MLC and AKT (Fig. 8). Consistent with findings in non-IPF fibroblasts (Figs. 4–7), the effects of TGF-β are strongly reduced or completely inhibited by siRNA targeting either ANO1 or WNK1 (Fig. 8). However, we also found that downregulation of WNK1 reduces the levels of the “upstream” ANO1 (Fig. 8). This latter discovery points to the bidirectional positive interaction between ANO1 and WNK1, which may constitute a feed-forward mechanism in myofibroblast differentiation.
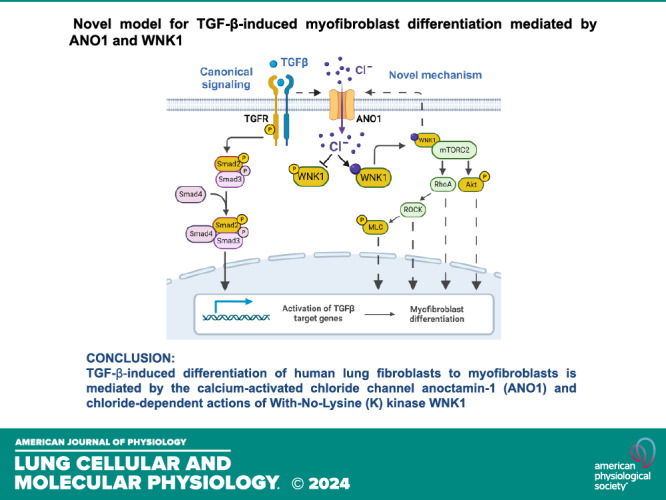
In conclusion, we propose a novel model for myofibroblast differentiation, which is summarized in Fig. 9. Our study uncovered an entirely unexpected role of intracellular Cl− in profibrotic TGF-β signaling. The new hypothetical mechanism incorporates the following signaling steps: 1) TGF-β stimulates expression of ANO1, resulting in an increase in intracellular [Cl−] in HLFs; 2) high intracellular chloride promotes a scaffolding, kinase-independent function of WNK1 and drives its association with mTORC2 and activation of downstream signaling pathways (44); 3) mTORC2 activates AKT and RhoA, all of which drive myofibroblast differentiation (19–21, 24, 56, 57), and P-MLC, activated by RhoA, also promotes myofibroblast differentiation (58, 59) and fibronectin assembly (60) through myosin-dependent cell contraction; and 4) interestingly, ANO1 expression is both a prerequisite for the TGF-β WNK1/mTORC2 signaling and is also dependent on WNK1 expression. This suggests a bidirectional feed-forward mechanism contributing to myofibroblast differentiation. The validity of this model and its relevance to diverse aspects of myofibroblast pathophysiology deserve further exploration, particularly in in vivo models of pulmonary fibrosis.
Figure 9.

Novel model for transforming growth factor-β (TGF-β)-induced myofibroblast differentiation mediated by anoctamin 1 (ANO1) and with-no-lysine (K) kinase (WNK1). Left: illustration of the canonical model of the TGF-β-induced myofibroblast differentiation involving Smad signaling. Right: diagram depicts a novel mechanism that this study suggests. 1) TGF-β upregulates the expression of ANO1 and elevates intracellular [Cl−] ([Cl−]i). 2) Increased [Cl−]i stabilizes autoinhibitory conformation of WNK1 and prompts its association with and activation of mammalian target of rapamycin complex-2 (mTORC2). 3) mTORC2 activates AKT and RhoA pathways. 4) AKT and RhoA promote myofibroblast differentiation via downstream signaling mechanisms, also including phosphorylated-myosin light chain (p-MLC). 5) WNK1 signaling forms a feedforward mechanism for ANO expression. Yellow color represents signaling molecules whose contribution was tested in this study. TGFR, TGF receptor. [Images created with a licensed version of BioRender.com.]
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL MATERIAL
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.24514975.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.24514876.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Award R01 HL149993 (to N.O.D.) and National Institute of Neurological Disorders and Stroke Award R01 NS111943 (to A.A.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.B.R., B.A.M., I.L., A.A.M., and N.O.D. conceived and designed research; E.B.R., S.O., B.A.M., A.S., B.C., I.L., A.A.M., and N.O.D. performed experiments; E.B.R., S.O., B.A.M., A.S., B.C., I.L., A.A.M., and N.O.D. analyzed data; E.B.R., S.O., B.A.M., A.S., B.C., I.L., J.S., G.M.M., Y.F. A.A.M., and N.O.D. interpreted results of experiments; B.A.M., A.A.M., and N.O.D. prepared figures; A.A.M. and N.O.D. drafted manuscript; B.A.M., J.S., G.M.M., Y.F., A.A.M., and N.O.D., edited and revised manuscript; E.B.R., S.O., B.A.M., A.S., B.C., I.L., J.S., G.M.M., Y.F. A.A.M., and N.O.D. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank J. W. Nalwalk for critical reading and helpful suggestions on the manuscript. Graphical abstract and Fig. 9 were created with a licensed version of BioRender.com.
REFERENCES
- 1. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med 176: 277–284, 2007. doi: 10.1164/rccm.200701-044OC. [DOI] [PubMed] [Google Scholar]
- 2. White ES, Lazar MH, Thannickal VJ. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol 201: 343–354, 2003. doi: 10.1002/path.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol 175: 3–16, 2009. doi: 10.2353/ajpath.2009.081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noble PW, Homer RJ. Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 33: 113–120, 2005. doi: 10.1165/rcmb.F301. [DOI] [PubMed] [Google Scholar]
- 5. Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 3: 3, 2002. doi: 10.1186/rr175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med 55: 395–417, 2004. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 7. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J 18: 816–827, 2004. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 8. Leask A, Holmes A, Abraham DJ. Connective tissue growth factor: a new and important player in the pathogenesis of fibrosis. Curr Rheumatol Rep 4: 136–142, 2002. doi: 10.1007/s11926-002-0009-x. [DOI] [PubMed] [Google Scholar]
- 9. Broekelmann TJ, Limper AH, Colby TV, McDonald JA. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci U S A 88: 6642–6646, 1991. doi: 10.1073/pnas.88.15.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, Huang X, Sheppard D, Heller RA. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci U S A 97: 1778–1783, 2000. doi: 10.1073/pnas.97.4.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Santana A, Saxena B, Noble NA, Gold LI, Marshall BC. Increased expression of transforming growth factor beta isoforms (beta 1, beta 2, beta 3) in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 13: 34–44, 1995. doi: 10.1165/ajrcmb.13.1.7541221. [DOI] [PubMed] [Google Scholar]
- 13. Phan SH, Kunkel SL. Lung cytokine production in bleomycin-induced pulmonary fibrosis. Exp Lung Res 18: 29–43, 1992. doi: 10.3109/01902149209020649. [DOI] [PubMed] [Google Scholar]
- 14. Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem 282: 7723–7732, 2007. doi: 10.1074/jbc.M610764200. [DOI] [PubMed] [Google Scholar]
- 15. Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax 48: 959–966, 1993. doi: 10.1136/thx.48.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Q, Wang Y, Hyde DM, Gotwals PJ, Koteliansky VE, Ryan ST, Giri SN. Reduction of bleomycin induced lung fibrosis by transforming growth factor beta soluble receptor in hamsters. Thorax 54: 805–812, 1999. doi: 10.1136/thx.54.9.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693, 2005. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 18. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425: 577–584, 2003. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 19. Sandbo N, Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res 158: 181–196, 2011. doi: 10.1016/j.trsl.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sandbo N, Kregel S, Taurin S, Bhorade S, Dulin NO. Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-beta. Am J Respir Cell Mol Biol 41: 332–338, 2009. doi: 10.1165/rcmb.2008-0288OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-beta. Am J Physiol Lung Cell Mol Physiol 301: L656–L666, 2011. doi: 10.1152/ajplung.00166.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Conte E, Fruciano M, Fagone E, Gili E, Caraci F, Iemmolo M, Crimi N, Vancheri C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS One 6: e24663, 2011. doi: 10.1371/journal.pone.0024663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fagone E, Conte E, Gili E, Fruciano M, Pistorio MP, Lo Furno D, Giuffrida R, Crimi N, Vancheri C. Resveratrol inhibits transforming growth factor-beta-induced proliferation and differentiation of ex vivo human lung fibroblasts into myofibroblasts through ERK/Akt inhibition and PTEN restoration. Exp Lung Res 37: 162–174, 2011. doi: 10.3109/01902148.2010.524722. [DOI] [PubMed] [Google Scholar]
- 24. Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS One 6: e15909, 2011. doi: 10.1371/journal.pone.0015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bill A, Alex Gaither L. The mechanistic role of the calcium-activated chloride channel ANO1 in tumor growth and signaling. Adv Exp Med Biol 966: 1–14, 2017. doi: 10.1007/5584_2016_201. [DOI] [PubMed] [Google Scholar]
- 26. Crottes D, Jan LY. The multifaceted role of TMEM16A in cancer. Cell Calcium 82: 102050, 2019. doi: 10.1016/j.ceca.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322: 590–594, 2008. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 28. Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134: 1019–1029, 2008. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455: 1210–1215, 2008. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 30. Jang Y, Oh U. Anoctamin 1 in secretory epithelia. Cell Calcium 55: 355–361, 2014. doi: 10.1016/j.ceca.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 31. Sanders KM, Zhu MH, Britton F, Koh SD, Ward SM. Anoctamins and gastrointestinal smooth muscle excitability. Exp Physiol 97: 200–206, 2012. doi: 10.1113/expphysiol.2011.058248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oh U, Jung J. Cellular functions of TMEM16/anoctamin. Pflugers Arch 468: 443–453, 2016. doi: 10.1007/s00424-016-1790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cho CH, Lee S, Kim A, Yarishkin O, Ryoo K, Lee YS, Jung HG, Yang E, Lee DY, Lee B, Kim H, Oh U, Im HI, Hwang EM, Jy P. TMEM16A expression in cholinergic neurons of the medial habenula mediates anxiety-related behaviors. EMBO Rep 21: e48097, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simchowitz L, De Weer P. Chloride movements in human neutrophils. Diffusion, exchange, and active transport. J Gen Physiol 88: 167–194, 1986. doi: 10.1085/jgp.88.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mattes PM, Maloney PC, Littlefield JW. Altered chloride metabolism in cultured cystic fibrosis skin fibroblasts. Proc Natl Acad Sci U S A 84: 3009–3013, 1987. doi: 10.1073/pnas.84.9.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem 286: 2365–2374, 2011. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245–248, 1996. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 38. Birukova AA, Shah AS, Tian Y, Moldobaeva N, Birukov KG. Dual role of vinculin in barrier-disruptive and barrier-enhancing endothelial cell responses. Cell Signal 28: 541–551, 2016. doi: 10.1016/j.cellsig.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Birukova AA, Tian X, Cokic I, Beckham Y, Gardel ML, Birukov KG. Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc Res 87: 50–57, 2013. doi: 10.1016/j.mvr.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa MO, Lilley S, Choudhury H, Gosling M, Wang L, Fitzgerald S, Borawski J, Baffoe J, Labow M, Gaither LA, Bentires-Alj M. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci USA 110: E1026–1034, 2013. doi: 10.1073/pnas.1217072110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Z, Zhang S, Hou F, Zhang C, Gao J, Wang K. Inhibition of Ca(2+) -activated chloride channel ANO1 suppresses ovarian cancer through inactivating PI3K/Akt signaling. Int J Cancer 144: 2215–2226, 2019. doi: 10.1002/ijc.31887. [DOI] [PubMed] [Google Scholar]
- 42. Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 43. Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saha B, Leite-Dellova DCA, Demko J, Sorensen MV, Takagi E, Gleason CE, Shabbir W, Pearce D. WNK1 is a chloride-stimulated scaffold that regulates mTORC2 activity and ion transport. J Cell Sci 135: jcs260313, 2022. doi: 10.1242/jcs.260313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamada K, Park HM, Rigel DF, DiPetrillo K, Whalen EJ, Anisowicz A, , et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol 12: 896–898, 2016. doi: 10.1038/nchembio.2168. [DOI] [PubMed] [Google Scholar]
- 46. Dulin NO. Calcium-activated chloride channel ANO1/TMEM16A: regulation of expression and signaling. Front Physiol 11: 590262, 2020. doi: 10.3389/fphys.2020.590262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Friard J, Tauc M, Cougnon M, Compan V, Duranton C, Rubera I. Comparative effects of chloride channel inhibitors on LRRC8/VRAC-mediated chloride conductance. Front Pharmacol 8: 328, 2017. doi: 10.3389/fphar.2017.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Q, Bai L, Luo S, Wang T, Yang F, Xia J, Wang H, Ma K, Liu M, Wu S, Wang H, Guo S, Sun X, Xiao Q. TMEM16A Ca(2+)-activated Cl(-) channel inhibition ameliorates acute pancreatitis via the IP3R/Ca(2+)/NFkappaB/IL-6 signaling pathway. J Adv Res 23: 25–35, 2020. doi: 10.1016/j.jare.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P, Perruchoud AP, Roth M. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem 274: 12933–12938, 1999. doi: 10.1074/jbc.274.18.12933. [DOI] [PubMed] [Google Scholar]
- 50. Pedroza M, Le TT, Lewis K, Karmouty-Quintana H, To S, George AT, Blackburn MR, Tweardy DJ, Agarwal SK. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J 30: 129–140, 2016. doi: 10.1096/fj.15-273953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang XH, Zheng B, Yang Z, He M, Yue LY, Zhang RN, Zhang M, Zhang W, Zhang X, Wen JK. TMEM16A and myocardin form a positive feedback loop that is disrupted by KLF5 during Ang II-induced vascular remodeling. Hypertension 66: 412–421, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05280. [DOI] [PubMed] [Google Scholar]
- 52. Baffi TR, Newton AC. mTOR regulation of AGC kinases: new twist to an old tail. Mol Pharmacol 101: 213–218, 2022. doi: 10.1124/molpharm.121.000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goncharova EA, Goncharov DA, Li H, Pimtong W, Lu S, Khavin I, Krymskaya VP. mTORC2 is required for proliferation and survival of TSC2-null cells. Mol Cell Biol 31: 2484–2498, 2011. doi: 10.1128/MCB.01061-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thompson WR, Guilluy C, Xie Z, Sen B, Brobst KE, Yen SS, Uzer G, Styner M, Case N, Burridge K, Rubin J. Mechanically activated Fyn utilizes mTORC2 to regulate RhoA and adipogenesis in mesenchymal stem cells. Stem Cells 31: 2528–2537, 2013. doi: 10.1002/stem.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saha S, Town JP, Weiner OD. Mechanosensitive mTORC2 independently coordinates leading and trailing edge polarity programs during neutrophil migration. Mol Biol Cell 34: ar35, 2023. doi: 10.1091/mbc.E22-05-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee YS, Bae Y, Park N, Yoo JC, Cho CH, Ryoo K, Hwang EM, Park JY. Surface expression of the Anoctamin-1 (ANO1) channel is suppressed by protein-protein interactions with beta-COP. Biochem Biophys Res Commun 475: 216–222, 2016. doi: 10.1016/j.bbrc.2016.05.077. [DOI] [PubMed] [Google Scholar]
- 57. Bernard M, Dieude M, Yang B, Hamelin K, Underwood K, Hebert MJ. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy 10: 2193–2207, 2014. doi: 10.4161/15548627.2014.981786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Southern BD, Grove LM, Rahaman SO, Abraham S, Scheraga RG, Niese KA, Sun H, Herzog EL, Liu F, Tschumperlin DJ, Egelhoff TT, Rosenfeld SS, Olman MA. Matrix-driven myosin II mediates the pro-fibrotic fibroblast phenotype. J Biol Chem 291: 6083–6095, 2016. doi: 10.1074/jbc.M115.712380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu YY, Fang CC, Hong CH, Wu CH, Lin YH, Chang KL, Lee CH. Nonmuscle myosin II activation regulates cell proliferation, cell contraction, and myofibroblast differentiation in keloid-derived fibroblasts. Adv Wound Care (New Rochelle) 9: 491–501, 2020. doi: 10.1089/wound.2019.0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Torr EE, Ngam CR, Bernau K, Tomasini-Johansson B, Acton B, Sandbo N. Myofibroblasts exhibit enhanced fibronectin assembly that is intrinsic to their contractile phenotype. J Biol Chem 290: 6951–6961, 2015. doi: 10.1074/jbc.M114.606186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.24514975.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.24514876.
Data Availability Statement
Data will be made available upon reasonable request.