

Abstract
Effective cancer immunotherapy is usually blocked by immunosuppressive factors in the tumour microenvironment, resulting in tumour promotion, metastasis and recurrence. Here we combine lipid nanoparticle–mRNA formulations and dendritic cell therapy (named CATCH) to boost the cancer–immunity cycle via progressive steps to overcome the immunosuppressive tumour microenvironment. Multiple types of sugar-alcohol-derived lipid nanoparticles are conceived to modulate the cancer–immunity cycle. First, one type of lipid nanoparticle containing CD40 ligand mRNA induces robust immunogenic cell death in tumoural tissues, leading to the release of tumour-associated antigens and the expression of CD40 ligand. Next, dendritic cells engineered by another type of lipid nanoparticle encapsulating CD40 mRNA are adoptively transferred, which are then activated by the CD40 ligand molecules in tumoural tissues. This promotes the secretion of multiple cytokines and chemokines, and the upregulation of co-stimulatory molecules on dendritic cells, which are crucial for reprogramming the tumour microenvironment and priming the T-cell responses. After dendritic cells present tumour-associated antigens to T cells, all the above stepwise events contribute to boosting a potent tumour-specific T-cell immunity that eradicates established tumours, suppresses distal lesions and prevents tumour rechallenge.
Effective antitumour immunity depends on appropriately activating a series of responses in the cancer–immunity cycle (CIC)1. First, dying cancer cells release tumour-associated antigens (TAAs) that are captured and processed by antigen-presenting cells (APCs) such as dendritic cells (DCs)1. Next, APCs present the antigens to T cells, leading to the priming of effector T-cell responses against TAAs1. These activated effector T cells infiltrate into tumoural tissues and execute the killing of cancer cells1. Last, dead cancer cells further release antigens to increase the breadth and depth of the immune responses in the CIC1. However, these stepwise events are usually dampened by the immunosuppressive tumour microenvironment (TME), resulting in uncontrollable tumour growth, metastasis and recurrence1–12. For example, DCs are one type of pivotal intermediator for the development of antitumour immunity in the CIC1–5,11,12. Nevertheless, their functions are usually restrained by the immunosuppressive TME by reducing their recruitment, infiltration and maturation1–12. Although the adoptive transfer of engineered DCs has provided important safety and immunogenicity data from many clinical trials, the unsatisfactory clinical responses indicate that combining DC therapy with other therapies may be necessary to overcome the immunosuppressive TME3,4,13–16. For example, some chemotherapeutic drugs have shown synergistic effects with DC therapy in clinical trials14,15. These chemotherapeutic drugs enable to induce immunogenic cell death (ICD), leading to the emission of damage-associated molecular patterns and TAAs14,15,17,18. Additionally, appropriate stimulation to DCs is also important to elicit their antitumour potency3,4,13,19–21. CD40 and CD40 ligand (CD40L) are a pair of co-stimulatory molecules that belong to the tumour necrosis factor/tumour necrosis factor receptor family22,23. The interaction between CD40 on DCs and CD40L on CD4+ T cells enable to license DC maturation, which promotes the secretion of proinflammatory cytokines and the upregulation of co-stimulatory molecules20–23. These play central roles in triggering CD8+ T cells to develop cytotoxicity and memory responses20–23. Several CD40 agonist antibodies (CD40 Ab) have been investigated in clinical trials22,23. Overall, CD40 Ab monotherapy has yielded modest tumour response rates and shown dose-limiting toxicity22–24. This has resulted in the exploration of multiple CD40 Ab combination therapies in clinical trials22,23,25–28. Based on these findings, we seek to leverage lipid nanoparticle (LNP)–mRNA formulations to integrate ICD effects in tumoural tissues and CD40-CD40L activation in DCs, providing a unique combination treatment for DC therapy. Specifically, we utilized LNPs containing CD40L mRNA (CD40L-LNPs) to induce ICD and CD40L expression in tumoural tissues. Then, we performed intratumoural (i.t.) adoptive transfer of CD40-overexpressed bone-marrow-derived dendritic cells (CD40-BMDCs) that could be stimulated by interactions with CD40L molecules. The activated DCs may benefit TAA presentation and trigger effector T-cell responses against cancer cells, leading to sufficient priming of the CIC.
Results
Synthesis and formulation of sugar-alcohol-derived LNPs for mRNA delivery
We designed and synthesized three sets of sugar-alcohol-derived ionizable lipids (DIS, DIM and LIS) with sorbitol, mannitol and L-sorbitol as the precursors, respectively (Fig. 1a,b)29. Their structures were confirmed by 1H nuclear magnetic resonance and mass spectroscopy (Supplementary Methods). Next, we formulated LNPs containing firefly luciferase (FLuc) mRNA (FLuc-LNPs) and characterized their sizes, surface charges and mRNA encapsulation efficiency (Supplementary Fig. 1a,b). Among all these LNPs, DIM7 and LIS10 showed higher mRNA delivery efficacy than other LNPs, namely, Lipofectamine 3000 (Lipo 3K) and electroporation (Electro; Fig. 2a). To further optimize the formulations of DIM7 and LIS10, we performed orthogonal screening assays based on an L16 (4)4 orthogonal table (Supplementary Fig. 1c). According to the luminescence intensity of the 16 orthogonal formulations of DIM7 or LIS10 (Fig. 2c,e), we profiled the effect of each lipid component with different molar ratios on mRNA delivery (Fig. 2b,d) and predicted several optimal formulations (Fig. 2f). Then, we validated their mRNA delivery efficacy by comparing with the corresponding top formulations from orthogonal screening assays, namely, DIM7M and LIS10G (Fig. 2c,e). For DIM7, the optimal formulation DIM7S resulted in a 2.0-fold (P < 0.001) and 3.5-fold (P < 0.001) higher luminescence signal than the top orthogonal formulation DIM7M and the initial formulation DIM7, respectively (Fig. 2c). For LIS10, the optimal formulation LIS10W resulted in a 1.2-fold (P < 0.0001) stronger luminescence intensity compared with the top orthogonal formulation LIS10G and a 2.3-fold (P < 0.0001) higher luminescence signal than the initial formulation LIS10 (Fig. 2e). DIM7S and LIS10W LNPs showed similar characteristics. Their particle sizes were about 110 nm with polydispersity index (PDI) < 0.2 (Fig. 2g,i). Their encapsulation efficiency of mRNA was around 90% and they were slightly positively charged (Fig. 2g,i). Moreover, they both exhibited spherical morphology when visualized by cryogenic transmission electron microscopy (cryo-TEM) (Fig. 2h,j and Supplementary Fig. 2d,e). As a result, we chose formulation DIM7S for the ex vivo delivery of mRNA into BMDCs in the following studies. With the same mRNA concentration, formulation DIM7S was 10-fold, 30-fold, 20-fold, 4-fold and 3-fold superior in mRNA delivery than Lipo 3K, Electro, ALC-0315, MC3 and SM-102, respectively (Supplementary Fig. 2f). Furthermore, DIM7S LNPs encapsulating CD40 mRNA (CD40-DIM7S) led to about 60–80% CD40 expression in BMDCs from 6 to 24 h (Supplementary Fig. 2g). To further understand mRNA delivery in BMDCs, we studied the cellular uptake, endocytic pathways and endosomal escape of the optimal formulation, namely, DIM7S, and the control formulation, that is, SM-102. Using Alexa Fluor 647-labelled RNA, we observed over 1.5-fold cellular uptake of DIM7S LNPs than SM-102 LNPs in BMDCs (Supplementary Fig. 2h). Next, we studied the endocytic pathways with CPZ, EIPA and MβCD, which are inhibitors to clathrin-mediated endocytosis, micropinocytosis and caveolae-mediated endocytosis, respectively30,31. The concentration of each inhibitor resulted in about 90% cell viability (Supplementary Fig. 2i). For DIM7S LNPs, MβCD dramatically reduced about 98% cellular uptake (Supplementary Fig. 2j), indicating a major role of caveolae-mediated endocytosis. For SM-102 LNPs, CPZ and MβCD obviously inhibited about 60% and 95% cellular uptake, respectively (Supplementary Fig. 2j). This suggested that SM-102 was mainly internalized through clathrin-mediated and caveolae-mediated endocytic pathways in BMDCs. To evaluate the endosomal escape ability, we analysed the co-localization between formulations and endosomes/lysosomes. Compared with SM-102 LNPs, DIM7S LNPs exhibited a lower Pearson’s correlation coefficient with endosomes and lysosomes (Supplementary Fig. 2k–m). Reflecting the data of luciferase expression and cellular uptake, these results suggested a stronger endosomal escape ability of formulation DIM7S than formulation SM-102.
Fig. 1 |. Chemical synthesis of sugar-alcohol-derived ionizable lipids.
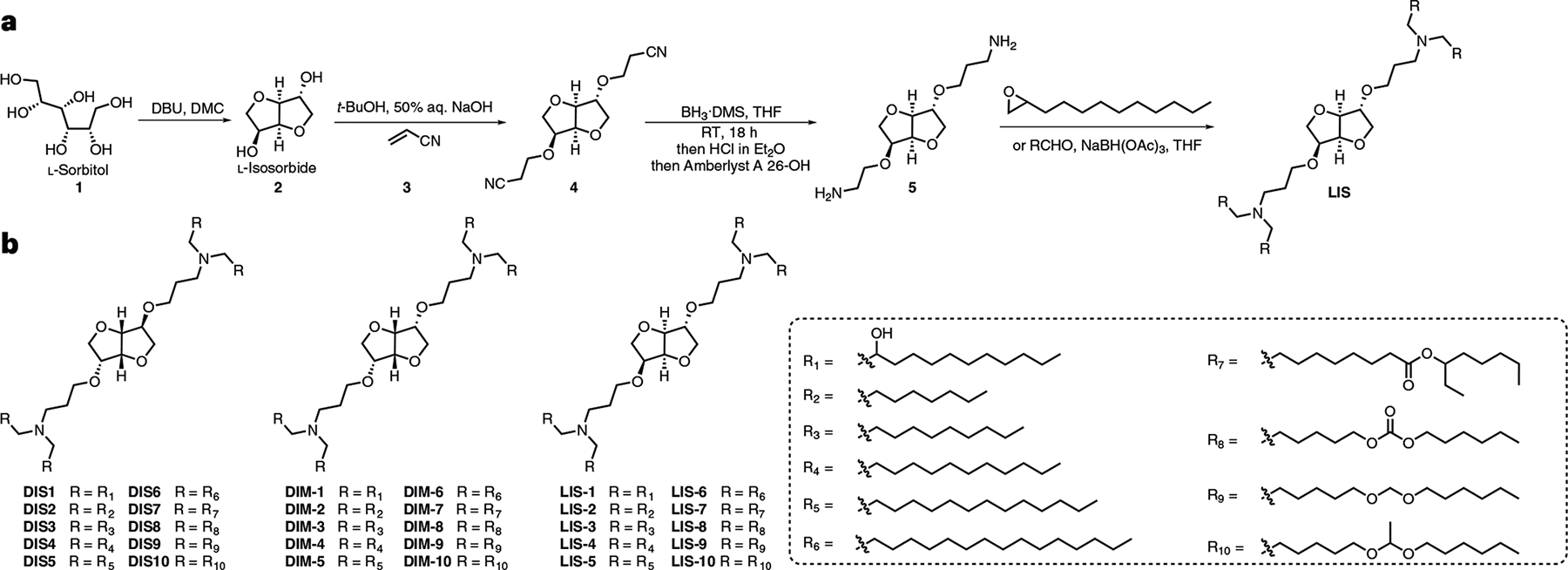
a, A representative synthesis route for sugar-alcohol-derived ionizable lipids. b, Structures of sugar-alcohol-derived ionizable lipids (DIS, DIM and LIS series).
Fig. 2 |. Screening, optimization and characterization of LNP–mRNA formulations.
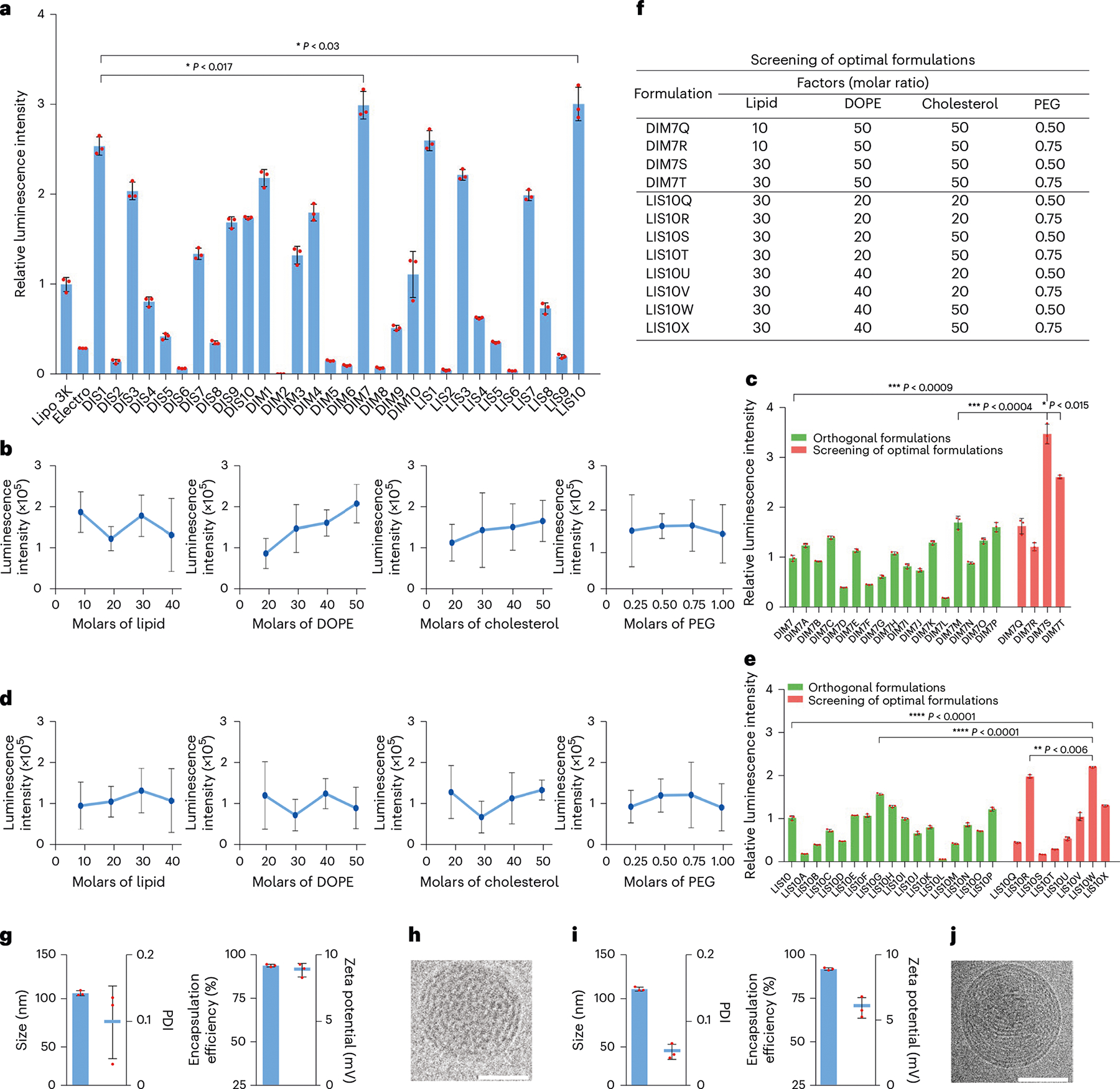
a, mRNA delivery efficacy of DIS, DIM and LIS LNPs in BMDCs. b, Effects of each lipid component at different molar ratios of DIM7 LNPs on mRNA delivery. c, Relative luminescence intensity of DIM7 LNPs’ orthogonal formulations and optimal formulations. d, Effects of each lipid component at different molar ratios of LIS10 LNPs on mRNA delivery. e, Relative luminescence intensity of LIS10 LNPs’ orthogonal formulations and optimal formulations. f, Table for optimal formulations. g, Characterization of DIM7S LNPs, including size, PDI, encapsulation efficiency and zeta potential. h, Cryo-TEM image of DIM7S LNPs. Scale bar, 50 nm. i, Characterization of LIS10W LNPs, including size, PDI, encapsulation efficiency and zeta potential. j, Cryo-TEM image of LIS10W LNPs. Scale bar, 50 nm. Data in h and j are representative images from n = 3 independent experiments. Data in a–e, g and i are from n = 3 biologically independent samples and are presented as mean ± standard deviation (s.d.). Statistical significance was analysed by the two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Combination of CD40-BMDCs and CD40L-LNPs (CATCH regimen) eliminates established tumours, suppresses distal lesions and prevents tumour rechallenge
The activation of DCs is an essential step for priming T-cell responses in the CIC1. Thus, we studied whether CD40 overexpression facilitates BMDC activation in the presence of CD40 Ab. Compared with phosphate-buffered saline (PBS), CD40 Ab and CD40-DIM7S treatments, CD40-DIM7S+CD40 Ab stimulated higher levels of DC activation markers (Fig. 3a) To test whether the activation was due to DC40-CD40L interactions, we utilized CD40−/− BMDCs to perform the activation assay. First, CD40-DIM7S resulted in over 90% CD40 expression in CD40−/− BMDCs (Supplementary Fig. 2a,b). Next, CD40L-B16F10 or FLuc-B16F10 cells were co-cultured with CD40−/− BMDCs or CD40-BMDCs. Compared with the PBS group, CD40L-B16F10 cells did not obviously activate CD40−/− BMDCs until the CD40−/− BMDCs were treated by CD40-DIM7S (Supplementary Fig. 2c,d). Moreover, FLuc-B16F10 cells showed limited stimulation on CD40-BMDCs (Supplementary Fig. 2c,d). These results indicated that CD40-BMDCs were mainly activated by CD40-CD40L interactions. To evaluate whether the enhanced activation of DCs improves the in vivo antitumour effects, we performed the i.t. injection of multiple treatments in a B16F10 melanoma mouse model (Fig. 3b). Compared with all other treatments, CD40-BMDCs+CD40 Ab suppressed tumour growth by 5- to 15-fold (23 d post-inoculation; Fig. 3c and Supplementary Fig. 2e) and extended the overall survival time (Fig. 3d). Indeed, CD40-DIM7S can in situ engineer DCs to increase the expression of CD40 (Supplementary Fig. 3a). However, CD40-DIM7S+CD40 Ab exhibited limited therapeutic effects compared with CD40-BMDCs+CD40 Ab (Fig. 3c,d). Similarly, BMDCs+CD40 Ab supplemented DCs in tumoural tissues, but the antitumour potency was still weaker than CD40-BMDCs+CD40 Ab (Fig. 3c,d). Collectively, these results revealed the importance of adoptive DC transfer, CD40 overexpression and CD40 stimulation for the antitumour effects of the CD40-BMDCs+CD40 Ab treatment.
Fig. 3 |. DC activation, LNP-induced ICD and therapeutic effects on primary tumour and rechallenged tumour.
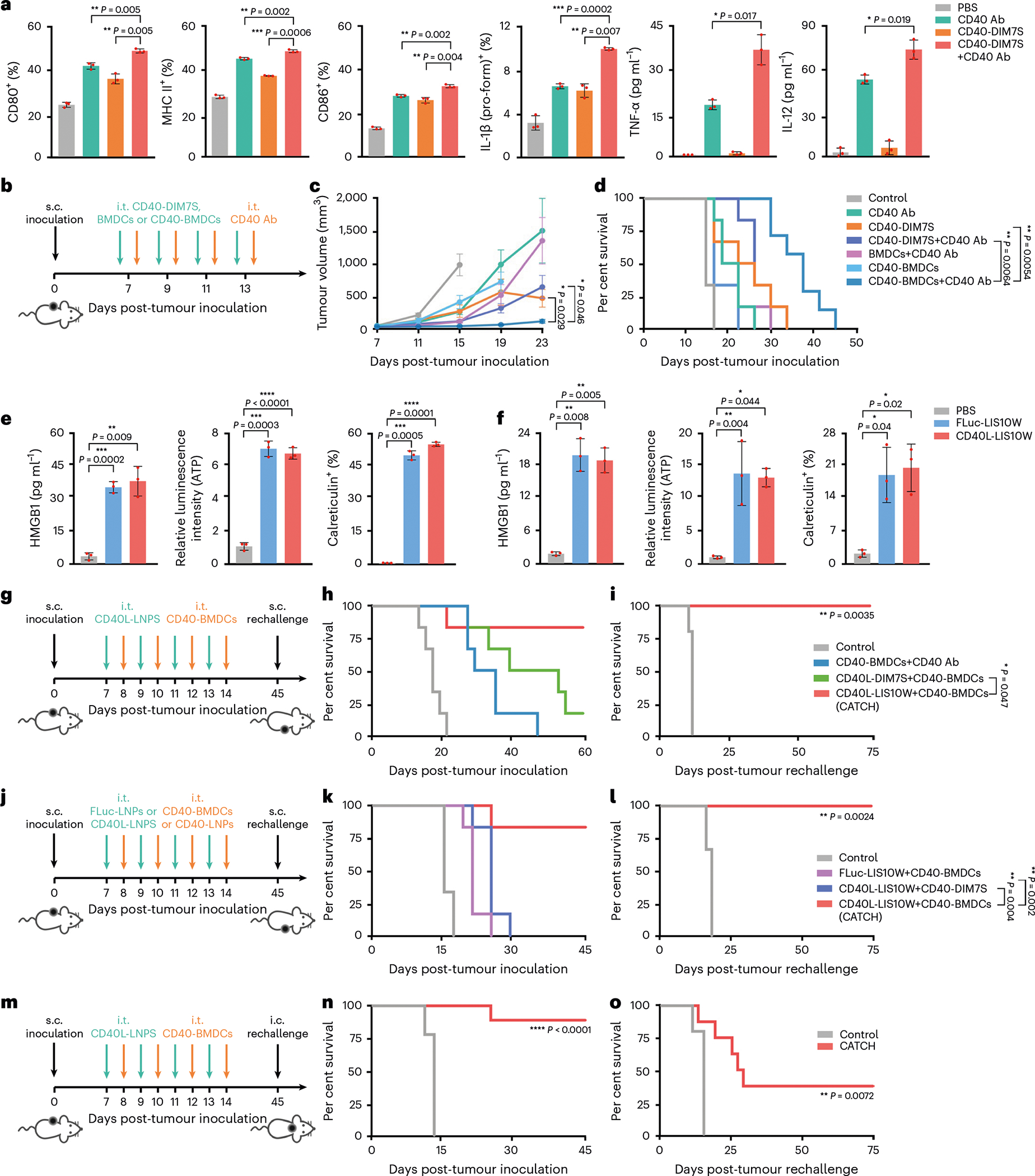
a, Expression of DC activation markers, including CD80, MHC II, CD86, IL-1β (pro-form), TNF-α and IL-12. b, Schematic of the treatment regimen in the B16F10 tumour model. c,d, Tumour volumes (c) and mouse survival (d) over time; n = 7 and 6 for CD40-BMDCs+CD40 Ab group and other groups, respectively. e,f, LNP-induced ICD markers in vitro (e) and in vivo (f), including extracellular HMGB1, extracellular ATP and cellular surface calreticulin. g, Schematic of the treatment regimen in the B16F10 tumour model. h, Mouse survival over time; n = 6. i, Survival of responder mice in the CATCH group post-s.c. tumour rechallenge; n = 5 and 6 for control group and CATCH group, respectively. j, Schematic of the treatment regimen in the B16F10 tumour model. k, Mouse survival over time; n = 6. l, Survival of responder mice in the CATCH group post-s.c. tumour rechallenge; n = 5 and 6 for the control group and CATCH group, respectively. m, Schematic of the treatment regimen in the B16F10-Luc2 tumour model. n, Mouse survival over time; n = 9. o, Survival of responder mice in the CATCH group post-i.c. tumour rechallenge; n = 5 and 8 for the control group and CATCH group, respectively. Data in a, e and f are from n = 3 biologically independent samples and are presented as mean ± s.d. Data in c are presented as mean ± standard error of the mean (s.e.m.). Statistical significance in a, c, e and f was analysed by the two-tailed Student’s t-test. Statistical significance in d, h, i, k, l, n and o was analysed by the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Although CD40-BMDCs+CD40 Ab showed promising antitumour effects, the incomplete eradication of tumour burden indicated the necessity of further boosting the CIC. Cancer cell death, as the initial step in the CIC, enables the release of TAAs that are presented by DCs and prime tumour-specific T-cell responses1,17,18,32. Therefore, we hypothesized that ICD induced by LNPs may promote the antitumour effects of CD40-BMDCs. We also speculated that the LNP-mediated expression of CD40L in tumoural tissues could activate the adoptively transferred CD40-BMDCs. As a result, the combination of ICD induced by CD40L-LNPs and adoptive CD40-BMDC transfer may synergistically proceed the CIC and thereby eliminate tumoural lesions. To test these hypotheses, we evaluated the cytotoxicity of DIM7S and LIS10W containing CD40L mRNA (CD40L-DIM7S and CD40L-LIS10W, respectively) in B16F10 cells. Although both CD40L-DIM7S and CD40L-LIS10W resulted in nearly 100% CD40L expression in B16F10 cells (Supplementary Fig. 3b), LIS10W LNPs induced more potent cytotoxicity (~85%) of B16F10 cells than DIM7S LNPs (~50%) after 18 h treatment (Supplementary Fig. 3c). The highest CD40L expression induced by CD40L-LIS10W was observed at the 12 and 18 h time points (Supplementary Fig. 3d). Moreover, apoptosis was the main cell death form induced by LIS10W LNPs (Supplementary Fig. 3e–g) and over 50% CD40L+ cells showed apoptosis at 18 h (Supplementary Fig. 3f,g). Thus, LIS10W was selected to evaluate the ability to induce ICD markers in B16F10 cells, including extracellular high mobility group box 1 (HMGB1), extracellular ATP and cellular surface calreticulin17,18,32. Compared with PBS treatment, FLuc-LIS10W or CD40L-LIS10W induced about 10-fold, 7-fold and 45-fold higher levels of cell extracellular HMGB1, extracellular ATP and cellular surface calreticulin in vitro, respectively (Fig. 3e). Consistently, obvious ICD effects induced by FLuc-LIS10W or CD40L-LIS10W were observed in B16F10 melanoma tissues in vivo (Fig. 3f), suggesting that LIS10W LNPs rather than the mRNA cargo contributed to the ICD effects. Because the expression profile of CD40L in tumoural tissues may affect its bioactivity, we studied its biodistribution at a cellular level. A single injection of CD40L-LIS10W led to about 15% of CD40L+ B16F10 cells in tumoural tissues (Supplementary Fig. 4a). In addition, immune cells in tumoural tissues, including macrophages, DCs, CD4+ T cells and CD8+ T cells, exhibited about 1.5- to 2.0-fold increased expression of CD40L (Supplementary Fig. 4b). The expression of CD40L in both cancer cells and immune cells may contribute to CD40-BMDC activation (Supplementary Fig. 4c,d)33,34. The in vivo function of CD40-BMDCs could be affected by their viability, and thus, we evaluated the cytotoxicity of DIM7S LNPs in BMDCs. The treatment with DIM7S LNPs resulted in ~50% viability in melanoma cells and ~90% in BMDCs (Supplementary Figs. 3c and 4e). Moreover, DIM7S LNPs did not induce obvious apoptosis or necrosis in BMDCs from 6 to 24 h (Supplementary Fig. 4f). These data indicated that DIM7S LNPs induced limited cytotoxicity in BMDCs. The differential cytotoxicity may be attributed to the stronger cellular uptake of DIM7S LNPs in melanoma cells than BMDCs (Supplementary Fig. 4g). With the same cell number and mRNA concentration, the luciferase expression in melanoma cells was 18-fold and 30-fold higher than that in BMDCs at 6 and 12 h, respectively (Supplementary Fig. 4h). This suggested that DIM7S LNPs enabled more effective endosome membrane disturbance in melanoma cells than BMDCs. Collectively, the higher cytotoxicity of DIM7S LNPs in melanoma cells than BMDCs may be caused by the stronger capacity of cellular uptake and membrane disturbance35,36.
To evaluate the antitumour effects of the combination of ICD and adoptive CD40-BMDC transfer, CD40-BMDCs were i.t. administrated 18 h after the i.t. injection of CD40L-LIS10W (Fig. 3g). Four doses of this treatment regimen resulted in complete tumour regression in 83% B16F10 tumour-bearing mice (Fig. 3h and Supplementary Fig. 5a), which was obviously higher than CD40-BMDCs+CD40 Ab treatment (0%) and CD40L-DIM7S+CD40-BMDCs treatment (17%). Importantly, all the responder mice in CD40L-LIS10W+CD40-BMDCs group were resistant to B16F10 tumour rechallenge on the opposite flank (Fig. 3i and Supplementary Fig. 5b), indicating the development of antitumour memory. These results highlighted the crucial role of ICD in priming antitumour immunity in the CIC. To further confirm the function of CD40-BMDCs and CD40L-mediated activation in this treatment, we tested another two treatment regimens, including FLuc-LIS10W+CD40-BMDCs and CD40L-LIS10W+CD40-DIM7S (Fig. 3j). Although FLuc-LIS10W (ICD control without CD40L expression) and CD40L-LIS10W showed similar in vivo ICD effects (Fig. 3f), CD40L-LIS10W+CD40-BDMCs dramatically suppressed tumour growth by about 21-fold and 7-fold (19 d post-inoculation) compared with FLuc-LIS10W+CD40-BDMCs and CD40L-LIS10W+CD40-DIM7S, respectively (Supplementary Fig. 5c,d). Furthermore, 83% mice from the CD40L-LIS10W+CD40-BDMCs group survived 45 d after tumour inoculation, whereas none of the mice survived from the other two treatment groups (Fig. 3k). Additionally, on rechallenge with B16F10 cells on the opposite flank, 100% responder mice were resistant to secondary tumour development (Fig. 3l and Supplementary Fig. 5e). These findings implied that ICD treatment alone was not sufficient to eradicate tumoural lesions, emphasizing the importance of DC supplement and activation. Here we name the CD40L-LIS10W+CD40-BDMCs regimen as the CATCH treatment. The long-term benefit of immunotherapy requires antitumour memory against tumour recurrence. Thus, we further validated the development of antitumour memory via an intracranial (i.c.) rechallenged mouse model (Fig. 3m). In this experiment, we used another melanoma cell line (B16F10-Luc2) containing a luciferase reporter, which facilitated monitoring brain tumour growth via bioluminescence imaging. The CATCH treatment achieved about 88% complete regression of the primary subcutaneous (s.c.) tumour (Fig. 3n and Supplementary Fig. 5f). In particular, the survived mice showed markedly delayed tumour growth in the brain (11 d post-inoculation; Supplementary Fig. 5g). Moreover, 38% of the responder mice remained tumour free compared with 0% of naive controls (75 d post-i.c. rechallenge; Fig. 3o). These data showed that a robust antitumour-memory T-cell population was generated from this local therapy.
The efficacy of local immunotherapy in treating metastatic cancer is dependent on the development of a systemic antitumour immunity32,37–39. To evaluate this, B16F10 cells were inoculated on both flanks of each mouse, followed by CATCH treatment on one tumour site (Fig. 4a). The treatment eliminated 80% primary tumour (Supplementary Fig. 5h) and 70% distal tumour (Fig. 4b), and the overall survival rate was around 70% (Fig. 4c). In another two-tumour model (skin + brain tumours), B16F10-Luc2 cells were s.c. and i.c. inoculated (Fig. 4d). The CATCH treatment eradicated 80% of the s.c. tumour (Supplementary Fig. 5i). Meanwhile, the treatment visibly regressed tumour growth in the brain (11 d post-inoculation; Fig. 4e) and remarkably prolonged the overall survival time (Fig. 4f). Altogether, these results indicated that this local treatment resulted in systemic antitumour immunity.
Fig. 4 |. Therapeutic effects on two tumour or immune cell depletion models, and dynamic expression of cytokines and chemokines in tumoural tissues and blood.
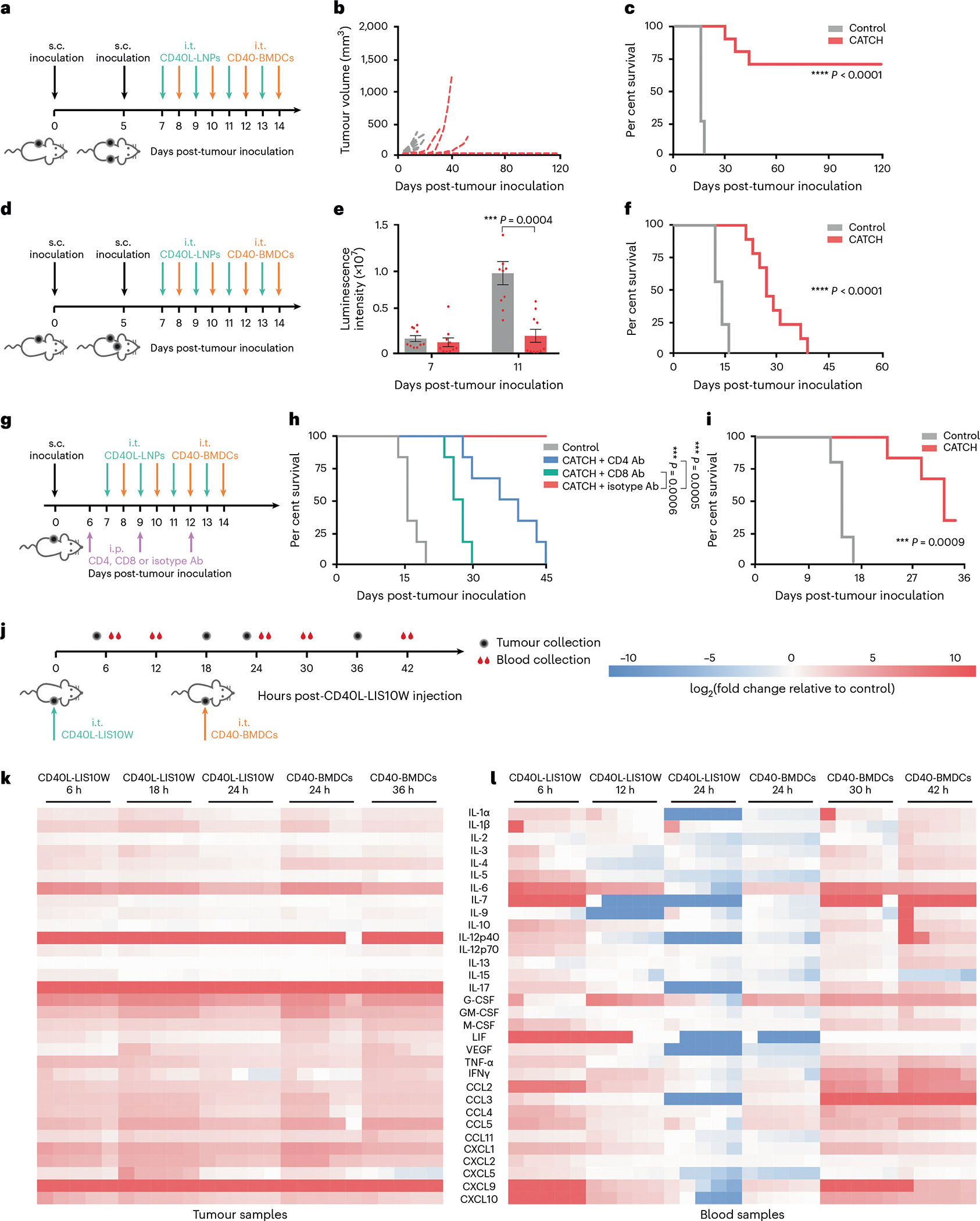
a, Schematic of the treatment regimen in the B16F10 tumour model. b,c, Distal tumour volumes of individual mice (b) and mouse survival (c) over time; n = 8 and 10 for the control group and CATCH group, respectively. d, Schematic of the treatment regimen in the B16F10-Luc2 tumour model. e,f, Brain tumour volumes (e) and mouse survival (f) over time; n = 10. g, Schematic of the treatment regimen in the B16F10 tumour model. h,i, Tumour volumes (h) and mouse survival (i) over time; n = 6. j, Schematic of the treatment regimen and sample collection. k,l, Dynamic expression of cytokines and chemokines in tumoural tissues (k) and blood (l); n = 5 biologically independent mice. Data in e are presented as mean ± s.e.m. Statistical significance in e was analysed by the two-tailed Student’s t-test. Statistical significance in c, f, h and i was analysed by the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
T-cell-mediated cancer cell killing is an important step for eradicating tumoural lesions in the CIC1. To evaluate the roles of CD8+ and CD4+ T cells on therapeutic efficacy, CATCH treatment was performed on mice receiving anti-CD8, anti-CD4 or isotype control Abs (Fig. 4g). Relative to CATCH treatment, the depletion of CD8+ or CD4+ T cells greatly compromised the tumour regression and mouse survival (Fig. 4h and Supplementary Fig. 5j,k). In particular, the depletion of CD8+ T cells led to a more remarkably weakened therapeutic efficacy relative to the depletion of CD4+ T cells (Fig. 4h and Supplementary Fig. 5j,k), suggesting the essential function of CD8+ T cells in this treatment regimen. Because DCs are crucial intermediators for stimulating antitumour T-cell responses, we utilized Batf3−/− mice to examine the antitumour effect of endogenous DCs in the CATCH treatment. Although the treatment improved the survival rate (33%) relative to the control (0%; Fig. 4i and Supplementary Fig. 5l), the therapeutic effect was obviously decreased compared with the survival in wild-type mice (83–88%; Fig. 3h,k,n). These data indicated the important antitumour effects of both endogenous and adoptively transferred DCs in the CATCH treatment.
CATCH regimen reprograms the immunosuppressive TME
To profile the TME changes after the CATCH treatment, we studied the dynamic expression of cytokines and chemokines in mouse melanoma tissues and blood during administration of the first dose (Fig. 4j). In tumoural tissues, CD40L-LIS10W elicited 29 cytokines and chemokines from 6 to 24 h (Fig. 4k and Supplementary Fig. 6). Moreover, their concentrations were further increased after the i.t. administration of CD40-BDMCs (Fig. 4k and Supplementary Fig. 6). In the blood, most of the cytokines and chemokines were upregulated 6 h post-CD40L-LIS10W injection, whereas their concentrations gradually decreased in the following 18 h (Fig. 4l and Supplementary Fig. 7). However, the administration of CD40-BDMCs restimulated the production of many cytokines and chemokines from 24 to 42 h (Fig. 4l and Supplementary Fig. 7). These data implied that CATCH treatment (CD40L-LIS10W+CD40-BDMCs) could better reprogram the immunosuppressive TME and induce stronger systemic immune responses than CD40L-LIS10W alone. The upregulation of these inflammatory cytokines and chemokines may not only enhance the recruitment and activation of immune cells but also facilitate the development of immunological memory. Thus, we profiled the changes in immune cell populations in tumoural tissues after CATCH treatment. In this experiment, CD40-BMDCs were labelled by CellTrace Blue before i.t. injection, which enabled to distinguish the administrated DCs from endogenous DCs. The treatment resulted in a massive influx of conventional type 1 and type 2 dendritic cells (cDC1s and cDC2s, respectively), CD8+ T cells and CD4+ T cells, whereas decreased the recruitment of macrophages and regulatory T (Treg) cells in tumoural tissues (Fig. 5a and Supplementary Fig. 8a–c). Importantly, the treatment enhanced the percentage of antitumour phenotype APCs, including CD80/86+ macrophages and CD80/86+ DCs (Supplementary Fig. 8d). Moreover, the treatment obviously stimulated the expression of Ki-67, IFN-γ/TNF-α and granzyme B in CD8+ T cells (Supplementary Fig. 8e), indicating the generation of cytotoxic T cells. Furthermore, the treatment downregulated the expression of CTLA-4 but not PD-1 on T cells (Supplementary Fig. 8f). Similar to the trend of immune cells in tumoural tissues, the treatment enhanced the trafficking of macrophages, cDC1s, cDC2s, CD8+ T cells and CD4+ T cells in tumour-draining lymph nodes (Fig. 5b and Supplementary Fig. 9a–c). By co-culturing Pmel-1 CD8+ T cells and DCs isolated from tumoural tissues and tumour-draining lymph nodes, we found that the DCs from CATCH-treated mice stimulated higher levels of T-cell activation markers, including Ki-67, granzyme B, IFN-γ and TNF-α compared with the controls (Supplementary Fig. 9d,e), suggesting an enhanced T-cell priming ability. In addition, we examined memory T cells in the spleen and blood from responder mice. Compared with naive controls, the numbers of effector memory and central memory T cells in both spleen and blood dramatically increased (Fig. 5c and Supplementary Fig. 9f,g), indicating the development of long-term antitumour immunity.
Fig. 5 |. Changes in immune cell infiltration, memory T cells and mRNA transcripts.
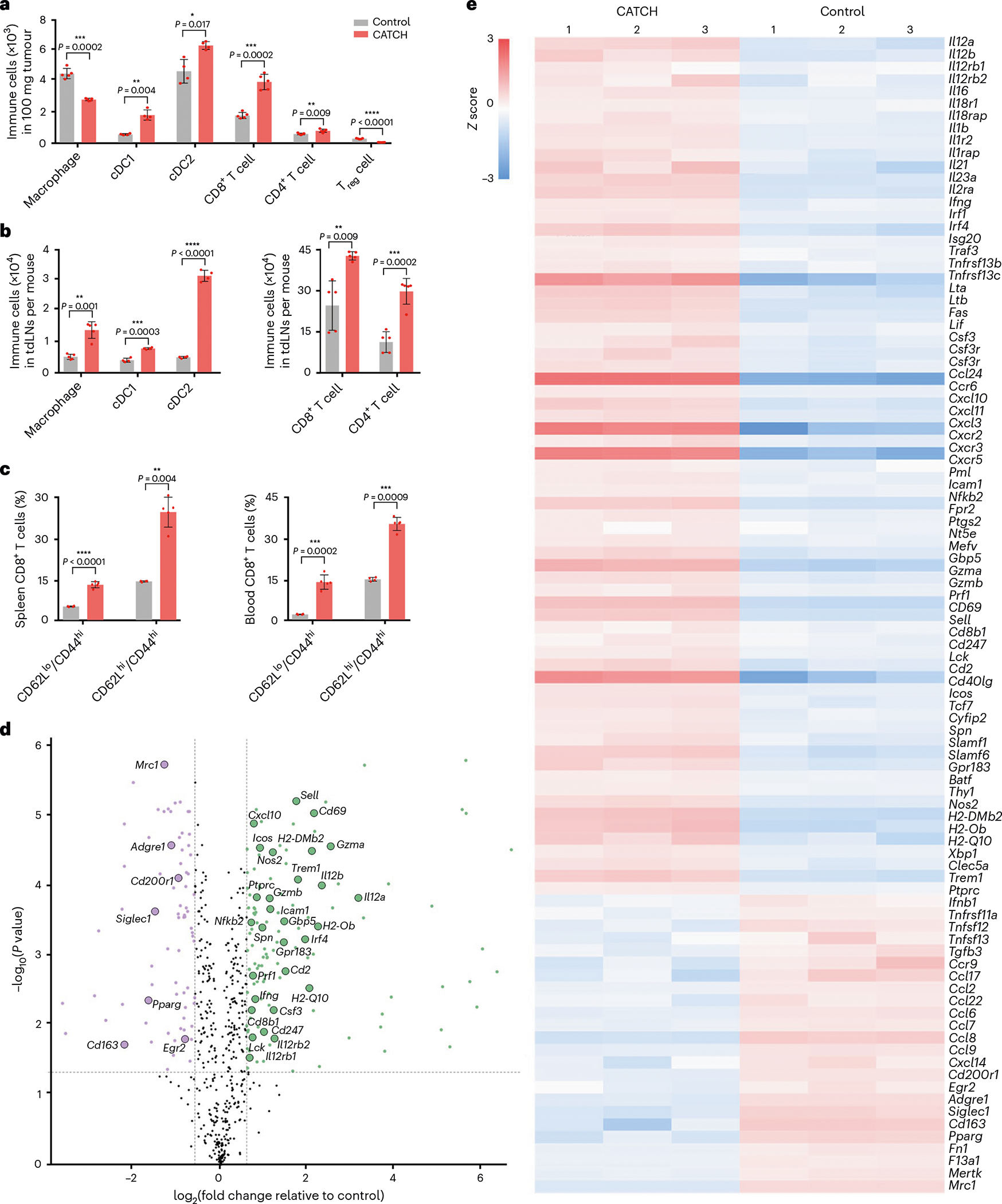
a, Immune cell populations in tumoural tissues; n = 4 and 5 biologically independent mice for DCs and other cells, respectively. b, Immune cell populations in tumour-draining lymph nodes (tdLNs); n = 5 biologically independent mice. c, Percentage of effector memory T cells and central memory T cells in the spleen and blood; n = 5 biologically independent mice. d, Volcano plot of differential mRNA transcripts; n = 3 biologically independent mice. e, Clustering heat map of representative differential mRNA transcripts; n = 3 biologically independent mice. Data in d and e are analysed by nSolver Advanced Analysis. Data in a–c are presented as mean ± s.d. and statistical significance was analysed by the two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We further performed a NanoString assay to quantify the mRNA levels in tumour-infiltrating immune cells after CATCH treatment (Fig. 5d,e). Among them, cytokine- and chemokine-associated transcripts (for example, Il12 and Ifng) and inflammatory-response-associated genes (for example, Icam1 and Nfkb2) were upregulated, which were similar to the results of cytokines and chemokines measured by Luminex assays (Fig. 4k,l). Moreover, consistent with the changes in tumour-infiltrating immune cells (Fig. 5a), the treatment elevated the levels of Ptprc (CD45), Cd247 (CD3 zeta chain) and Cd8b1 (CD8 beta chain), whereas reduced the levels of Adgre1 and Siglec1 (F4/80 and CD169, respectively). In particular, many M2 phenotype macrophage-related mRNAs (for example, Mrc1, Cd163 and Pparg) were downregulated, whereas antitumour phenotype-related mRNAs (for example, Nos2 and Trem1) were upregulated40. Additionally, many transcripts involved in antigen presentation were increased, including MHC II-related mRNAs (H2-DMb2, H2-Ob and H2-Q10)41. These results demonstrated the ability of CATCH treatment to activate tumour-infiltrating APCs for TAA presentation and T-cell priming. Consequently, several T-cell-response-associated transcripts were increased (Fig. 5d,e). For example, T-cell co-stimulatory molecule-related mRNAs (for example, Icos and Cd2) and cytotoxic protein-related mRNAs (for example, Gzma, Gzmb and Prf1) were upregulated42, indicating enhanced cytotoxic T-cell differentiation. More importantly, the levels of Cd69 and Sell (CD62L)—two memory T-cell markers—were elevated43, which correlated with the increased numbers of memory T cells in the spleen and blood from the responder mice (Fig. 5c). Overall, the NanoString data supported the activation of a potent antitumour immunity induced by the CATCH regimen.
Applicability and safety studies of the CATCH regimen
To evaluate the applicability of CATCH treatment, we tested the anti-tumour effects in two other tumour models. The complete response rates in A20 and 4T1 models were 83% and 50%, respectively (Fig. 6a–c and Supplementary Fig. 9h,i). The lower complete response rate of immunotherapy in 4T1 tumour than A20 tumour has been previously reported, which may be attributed to the poor immunogenicity and harsh immunosuppressive TME44,45. In addition, we optimized the treatment regimen to reduce the number of i.t. injections. In this treatment regimen, we performed a total of four i.t. injections, consisting of two doses of CD40L-LIS10W and CD40-BDMCs (Fig. 6d). A similar complete response rate (83%) was observed in this optimized treatment relative to the original eight-time treatment (Fig. 6e and Supplementary Fig. 9j). Meanwhile, two doses of CATCH treatment did not elevate the levels of alanine transaminase (ALT), aspartate transaminase (AST) and blood urea nitrogen (BUN) (Fig. 6f)24. Furthermore, no obvious toxicity was observed in major tissues from the histopathology analyses (Supplementary Fig. 10a). To explore the clinical translatability of the CATCH treatment, we tested whether DIM7S enabled effective CD40 mRNA delivery into human peripheral blood monocyte-derived DCs. CD40-DIM7S induced about 60% CD40 expression in such DCs without obvious cytotoxicity (Fig. 6g and Supplementary Fig. 10b). Additionally, we also evaluated the ability of CD40L-LIS10W to induce ICD effects in A375 human melanoma cells. Similar to the results in mouse B16F10 melanoma cells, the treatment increased CD40L expression, and in the meantime elicited the levels of ICD markers including cell extracellular HMGB1, extracellular ATP and cellular surface calreticulin (Fig. 6h and Supplementary Fig. 10c–e).
Fig. 6 |. Applicability of CATCH treatment regimen in other tumour models and human-derived cells.
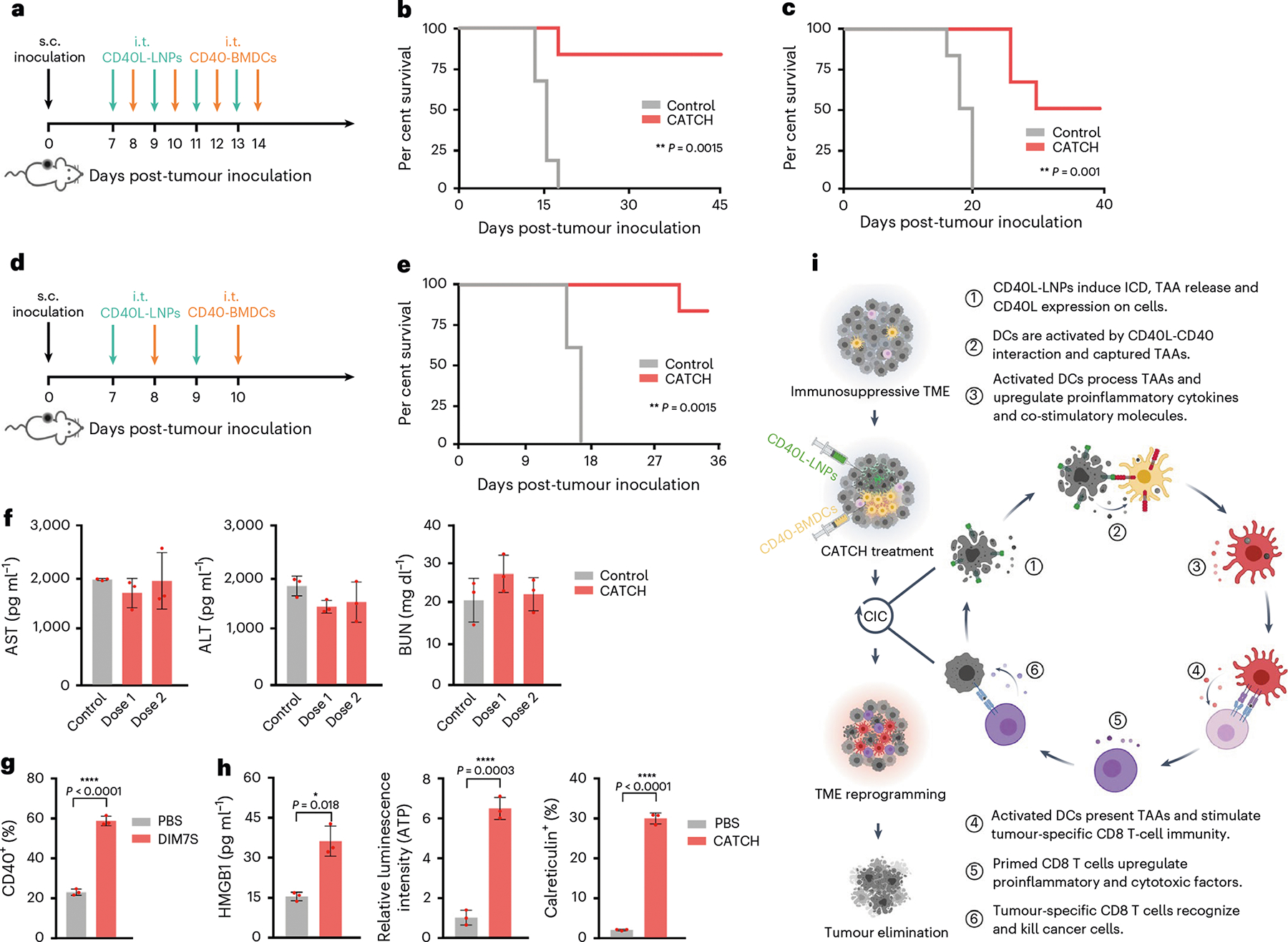
a, Schematic of the treatment regimen in the A20 and 4T1 tumour models. b,c, Mouse survival over time in A20 (b) and 4T1 (c) tumour models; n = 6. d, Schematic of the optimized treatment regimen in the B16F10 tumour model. e, Mouse survival over time; n = 6. f, Liver transaminase and BUN levels in the blood after CATCH treatment in the B16F10 tumour model; n = 3 biologically independent mice. g, CD40 expression mediated by CD40-DIM7S in human peripheral blood monocyte-derived DCs. h, CD40L-LIS10W induced ICD markers in A375 human melanoma cells. i, Illustration of closing the CIC by integrating LNP–mRNA formulations and DC therapy (CATCH). This illustration was created with BioRender.com. Statistical significance in b, c and e was analysed by the log-rank (Mantel–Cox) test. Data in g and h are from n = 3 biologically independent samples, presented as mean ± s.d. Statistical significance was analysed by the two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Conclusion
Collectively (Fig. 6i), CD40L-LIS10W in the CATCH treatment simultaneously induced ICD and CD40L expressions in tumoural tissues, which enabled to activate both endogenous DCs and adoptively transferred CD40-BMDCs, the second component of the CATCH regimen. The activation of DCs enhanced their presentation of TAAs and expression of co-stimulatory molecules, and in the meantime, induced multiple cytokines and chemokines in both tumoural tissues and blood. These immune events contributed to reprogramming the TME and systemic immune responses, which promoted the migration of immune cells into tumoural tissues, the priming of effector T cells and the development of T-cell memory. The primed T cells elicited their antitumour cytotoxicity. As a result, dying cancer cells further release antigens to increase the breadth and depth of the immune responses in the CIC. Overall, closing the CIC by integrating LNP–mRNA formulations and DC therapy (CATCH) is a promising strategy for cancer immunotherapy (Supplementary Discussion).
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41565-023-01453-9.
Methods
mRNA synthesis
The dsDNA of FLuc, mouse CD40 and mouse CD40L were obtained from Integrated DNA Technologies. The pUC19 vector was used for Golden Gate assembly to generate plasmids. The mRNAs of FLuc, mouse CD40 and mouse CD40L were synthesized by an in vitro transcription assay46.
Preparation, characterization and optimization of LNPs
The mRNA LNPs were prepared by mixing an ethanol solution containing ionizable lipids, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol and 1,2-dimyristoyl-rac-glycero-3-methoxypolyet-hylene glycol-2000 (DMG-PEG2000), and a citrate solution containing mRNA via NanoAssemblr (Precision NanoSystems)47,48. The size, PDI and zeta potential were measured by a NanoZS Zetasizer instrument (Malvern). The encapsulation efficiency was detected by a RiboGreen assay47,48. The morphology was observed on Glacios cryo-TEM (Thermo Scientific)31,47. In the initial screening, the LNPs were prepared by the ionizable lipids, DOPE, cholesterol and DMG-PEG2000 at a molar ratio of 20.00:30.00:40.00:0.75, as well as FLuc mRNA with a mass ratio of 10:1 (refs. 31,48). In the optimization study, the LNPs were prepared based on an L16 (4)4 orthogonal table and the predicted formulations31,48. A Lipo 3K/mRNA complex was prepared based on the recommended protocol (Thermo Fisher, L3000015). Electroporation for BMDCs was based on the mouse DC nucleofector (Lonza, VAPA1011). The mRNA delivery efficacy was tested by a luciferase assay using BioTek Cytation 5 plate reader.
CD40 and CD40L expressions
For in vitro assays, 1 × 106 BMDCs or human DCs were seeded into each well of a six-well plate and treated with 2.5 μg CD40-LNPs. At different time points, the cells were stained with the CD40 antibody and analysed by an LSRFortessa flow cytometer (BD Biosciences). For in vivo studies, 6 h post-i.t. injection with Fluc, CD40 or CD40L-LNPs, the tumours were dissociated, and the infiltrating immune cells were isolated by Ficoll-Paque density gradient media. After staining with antibody combinations, the cells were analysed by an LSRFortessa flow cytometer (BD Biosciences).
LNP-induced ICD
ICD markers were detected by the adaptation of previous methods32. The cytotoxicity of LNPs in melanoma cells was examined by an MTT assay. Extracellular ATP was measured by a luminescent ATP detection kit (Abcam, ab113849). The extracellular HMGB1 level was detected by ELISA. Cell surface calreticulin was detected by flow cytometry. Cell apoptosis was detected by a dead-cell apoptosis kit (Invitrogen, V35113).
Luminex analysis of cytokines and chemokines
Mouse tumoural tissues and serum were collected from 0 to 42 h time course post-injection of the first dose of CD40L-LNPs+CD40-BMDCs. Tumoural tissues were frozen in liquid nitrogen and pulverized tissues were extracted with the RIPA lysis buffer (Thermo Scientific, 89900) with protease inhibitors (Thermo Scientific, 87785). The whole blood was collected in tubes containing sodium citrate and serum was collected by centrifugation. The tumoural lysate and serum were stored at −80 °C. Mouse cytokines and chemokines were detected by mouse cytokine/chemokine discovery assay (Eve Technologies)32,47.
Immune cell activation in vivo
After two doses of CD40L-LNPs+CD40-BMDCs, tumoural tissues or tumour-draining lymph nodes were dissociated, and then the immune cells were stained for flow cytometry. In this experiment, CD40-BMDCs were labelled by CellTrace Blue (Invitrogen, C34568) before i.t. injection, which can distinguish the administrated DCs from the endogenous DCs via flow cytometry.
NanoString analysis
After two doses of CD40L-LNPs+CD40-BMDCs, the tumoural tissues were harvested and dissociated. Next, CD45+ immune cells were sorted, and their total RNA transcripts were extracted by an RNeasy Mini Kit (QIAGEN, 74104). The RNA samples were detected via the nCounter Mouse PanCancer Immune Profiling Panel based on the manufacturer’s instructions. Raw NanoString data were normalized and analysed by ROSALIND. Based on the analysis results, 1.5-fold changes with statistical significance of P < 0.05 were used to define the differential RNA transcripts.
Regulatory
All the mouse studies were approved by the Institutional Animal Care and Use Committee at The Ohio State University (2014A00000106) and The Icahn School of Medicine at Mount Sinai (IPROTO202200000134), and complied with local, state and federal regulations.
Supplementary Material
Acknowledgements
Cryo-TEM imaging was performed at the Center for Electron Microscopy and Analysis (CEMAS) at The Ohio State University. The Genomics Shared Resource performed the nCounter NanoString analysis for this study with part support from the National Cancer Institute (grant P30 CA016058). Y.D. acknowledges support from the Maximizing Investigators’ Research Award R35GM144117 from the National Institute of General Medical Sciences as well as the funding from the Icahn School of Medicine at Mount Sinai. X.H. and J.Y. acknowledge support from the Professor Sylvan G. Frank Graduate Fellowship.
Footnotes
Competing interests
Y.D. is a scientific advisory board member of Oncorus Inc., Arbor Biotechnologies and FL85. The other authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Peer review information Nature Nanotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41565-023-01453-9.
Data availability
Source data are provided with this paper. Additional materials for this study are available from the corresponding author upon reasonable request.
References
- 1.Chen DS & Mellman I Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Wculek SK et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Anguille S, Smits EL, Lion E, van Tendeloo VF & Berneman ZN Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 15, e257–e267 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Belderbos RA, Aerts JG & Vroman H Enhancing dendritic cell therapy in solid tumors with immunomodulating conventional treatment. Mol. Ther. Oncolytics 13, 67–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saxena M, van der Burg SH, Melief CJ & Bhardwaj N Therapeutic cancer vaccines. Nat. Rev. Cancer 21, 360–378 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Tang M et al. Toll-like receptor 2 activation promotes tumor dendritic cell dysfunction by regulating IL-6 and IL-10 receptor signaling. Cell Rep. 13, 2851–2864 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Villablanca EJ et al. Tumor-mediated liver X receptor-α activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 16, 98–105 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Veglia F et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 8, 2122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottfried E et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Park MD et al. On the biology and therapeutic modulation of macrophages and dendritic cells in cancer. Annu. Rev. Cancer Biol. 7, 291–311 (2023). [Google Scholar]
- 12.Merad M, Sathe P, Helft J, Miller J & Mortha A The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 31, 563–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harari A, Graciotti M, Bassani-Sternberg M & Kandalaft LE Antitumour dendritic cell vaccination in a priming and boosting approach. Nat. Rev. Drug Discov. 19, 635–652 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Kongsted P et al. Dendritic cell vaccination in combination with docetaxel for patients with metastatic castration-resistant prostate cancer: a randomized phase II study. Cytotherapy 19, 500–513 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Tanyi JL et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 10, eaao5931 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Saxena M, Balan S, Roudko V & Bhardwaj N Towards superior dendritic-cell vaccines for cancer therapy. Nat. Biomed. Eng. 2, 341–346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kroemer G, Galluzzi L, Kepp O & Zitvogel L Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Galluzzi L, Buqué A, Kepp O, Zitvogel L & Kroemer G Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Bhardwaj N et al. Flt3 ligand augments immune responses to anti-DEC-205-NY-ESO-1 vaccine through expansion of dendritic cell subsets. Nat. Cancer 1, 1204–1217 (2020). [DOI] [PubMed] [Google Scholar]
- 20.De Keersmaecker B et al. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 8, e000329 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau SP et al. Dendritic cell vaccination and CD40-agonist combination therapy licenses T cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J. Immunother. Cancer 8, e000772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vonderheide RH CD40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med. 71, 47–58 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Bullock TN CD40 stimulation as a molecular adjuvant for cancer vaccines and other immunotherapies. Cell. Mol. Immunol. 19, 14–22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salomon R et al. Bispecific antibodies increase the therapeutic window of CD40 agonists through selective dendritic cell targeting. Nat. Cancer 3, 287–302 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Li D-K & Wang W Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol. Lett. 20, 176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beatty GL et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 19, 6286–6295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonderheide RH et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2, e23033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak AK et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann. Oncol. 26, 2483–2490 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Aricò F, Tundo P, Maranzana A & Tonachini G Synthesis of five-membered cyclic ethers by reaction of 1,4-diols with dimethyl carbonate. ChemSusChem 5, 1578–1586 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Sahay G, Alakhova DY & Kabanov AV Endocytosis of nanomedicines. J. Control. Release 145, 182–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou X et al. Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nat. Nanotechnol. 15, 41–46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer 1, 882–893 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kikuchi T, Moore MA & Crystal RG Dendritic cells modified to express CD40 ligand elicit therapeutic immunity against preexisting murine tumors. Blood 96, 91–99 (2000). [PubMed] [Google Scholar]
- 34.Hernandez MGH, Shen L & Rock KL CD40-CD40 ligand interaction between dendritic cells and CD8+ T cells is needed to stimulate maximal T cell responses in the absence of CD4+ T cell help. J. Immunol. 178, 2844–2852 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Love SA, Maurer-Jones MA, Thompson JW, Lin Y-S & Haynes CL Assessing nanoparticle toxicity. Annu. Rev. Anal. Chem. 5, 181–205 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Yang W, Wang L, Mettenbrink EM, DeAngelis PL & Wilhelm S Nanoparticle toxicology. Annu. Rev. Pharmacol. Toxicol. 61, 269–289 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Melero I, Castanon E, Alvarez M, Champiat S & Marabelle A Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol. 18, 558–576 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hewitt SL et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 11, eaat9143 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Hotz C et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci. Transl. Med. 13, eabc7804 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jhunjhunwala S, Hammer C & Delamarre L Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 21, 298–312 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Chen L & Flies DB Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mueller SN, Gebhardt T, Carbone FR & Heath WR Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 31, 137–161 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Sagiv-Barfi I et al. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl Acad. Sci. USA 112, E966–E972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ouzounova M et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 8, 14979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 46.Zeng C et al. Leveraging mRNA sequences and nanoparticles to deliver SARS-CoV-2 antigens in vivo. Adv. Mater. 32, 2004452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W et al. Biomimetic nanoparticles deliver mRNAs encoding costimulatory receptors and enhance T cell mediated cancer immunotherapy. Nat. Commun. 12, 7264 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B et al. An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett. 15, 8099–8107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data are provided with this paper. Additional materials for this study are available from the corresponding author upon reasonable request.