

Abstract
To study the mechanism of evolution of the human immunodeficiency virus (HIV) protease gene (pro), we analyzed a database of 213 pro sequences isolated from 11 HIV type 1-infected patients who had not been treated with protease inhibitors. Variation in pro is restricted to rare variable bases which are highly diverse and differ in location among individuals; an average variable base appears in about 16% of individuals. The average intrapatient distance per individual variable site, 27%, is similar for synonymous and nonsynonymous sites, although synonymous sites are twice as abundant. The latter observation excludes selection for diversity as an important, permanently acting factor in the evolution of pro and leaves purifying selection as the only kind of selection. Based on this, we developed a model of evolution, both within individuals and along the transmission chain, which explains variable sites as slightly deleterious mutants slowly reverting to the better-fit variant during individual infection. In the case of a single-source transmission, genetic bottlenecks at the moment of transmission effectively suppress selection, allowing mutants to accumulate along the transmission chain to high levels. However, even very rare coinfections from independent sources are, as we show, able to counteract the bottleneck effect. Therefore, there are two possible explanations for the high mutant frequency. First, the frequency of coinfection in the natural host population may be quite low. Alternatively, a strong variation of the best-adapted sequence between individuals could be caused by a combination of an immune response present in early infection and coselection.
Human immunodeficiency virus (HIV) shows a high level of genetic diversity compared to other viruses (45). Genetic variation in antigenically important regions complicates attempts at vaccine development (3, 29, 48, 49), and drug resistance mutations limit the efficacy of treatment with replication inhibitors (6, 25). From another perspective, a pattern of genetic evolution contains important information about biological factors acting on the virus population. In particular, the role of the immune response in HIV infection is difficult to assess experimentally. It remains a subject of intensive debate, and the mechanism of HIV persistence in vivo in the face of an apparently vigorous immune response is still unknown. At the same time, the presence of an at least partly functional immune response can be inferred from the genetic diversity data. The large proportion of nonsynonymous substitutions, e.g., in env reveals a powerful selection for diversity (4, 33, 41), presumably caused by the immune response and by adaptation to different cell types.
The average genetic distances and relative proportions of different types of substitutions vary strongly both between and within different HIV genes, implying that there are different dominant factors of evolution at different loci. The highest degree of diversity, 3 to 5% within an individual (1, 21, 47) and 8 to 17% between individuals from the same geographical location (1, 20, 26), is observed in the variable regions of the env gene. A somewhat smaller but still impressive level of intrapatient diversity has been reported for gag (1 to 2%) (52) and for pol and pro (0.4 to 1%) (22, 31). In pro, synonymous and conservative substitutions dominate overall variation (22), implying a stronger role for purifying selection than in env.
The aim of this work was to reconstruct the mechanism of evolution in a relatively conserved HIV gene, such as pro, by using data on its genetic variation (22). Genetic evolution of a virus population may be affected by a multitude of different factors (19): mutation, selection forces, stochastic factors (random drift), recombination, transmission between individuals, spread of the virus between infected organs, etc. The main difficulty, as is always the case with mathematical theories of real experimental systems, is that it is not known in advance which factors, among many, are of the greatest importance to the behavior of the system. The quality of approximations cannot be evaluated until the analysis is over, and it depends on the parameters one is trying to predict. We thus face a time paradox: on the one hand, a well-defined set of approximations must be introduced in the beginning to make the analysis possible and intuitively clear; on other hand, finding the right set of approximations (the biological model) is the final aim of such research. The only escape from the time paradox that we know of is a dynamic interplay between experiment and theory (38). One starts from a simple model to match several important experimental features. After calculating its predictions, one searches for a contradiction to the experimental data and, when such is found, checks initial assumptions by changing them, one by one, and estimating what had changed in the predictions. This process has to be repeated several times. The resulting model, when all available information is used up, is considered a working model subject to further experimental tests.
In the present work, we applied this strategy as follows. After several attempts, we chose a set of experimental features from the database of Lech et al. (22) that are difficult to explain within a mathematically and biologically consistent model. This allowed us to select a working model (or models) from a large number of possibilities. We found a model of deterministic evolution in an individual under conditions of purifying selection that agrees with several experimental features from those we have chosen and used it as a starting model of the evolution of pro. Next, to describe genetic variation of the initial virus variant in infecting inocula and to predict the average frequency of variable sites in individuals, we had to take transmission between individuals into consideration. We have determined that such a model can explain a high frequency of variable sites in an individual, which is a very important experimental feature. However, this theoretical result turned out to be entirely dependent on our initial assumption that an animal in the natural host population is, in 99% of cases, infected from a single source. Even if coinfection occurred as rarely as in 5% of cases, this would bring the frequency of variable sites many times below the observed value. We did not find independent facts that would confirm such a low frequency of coinfection. Therefore, we tried to find another explanation for the high frequency of variable sites by searching for a weak spot in our approximations. We have estimated (approximately) quantitative contributions from a number of potential factors of HIV evolution to the frequency of the variable sites and found most of these contributions to be very small. The factors we considered included the possibility that the genetic variation was dominated by random drift (39), temporal changes in the virus population size, adaptation of the virus sequence to different cell types, nonuniformity of virus distribution in the body and virus spread between infection sites, the possibility that the coinfection sources are not independent, coselection between loci, and the immune response. Finally, after excluding all apparent possibilities, we could find a plausible explanation, alternative to a very low coinfection rate in the natural host: in the presence of the immune response, the best-fit sequence of virus will differ strongly between individuals due to individual variation in major histocompatibility complex class I (MHC-I) subtypes, and selection for a variant unrecognized by the immune system leads to a cascade of compensatory mutations, both synonymous and nonsynonymous. This mechanism is also seen following the appearance of drug-resistant mutants.
To make the work available to a broad audience, we parallel qualitative arguments in Results and Discussion with mathematical derivations, given in Materials and Methods. We consider in detail four principal models: deterministic evolution in an individual, the same model with transmission between individuals, the same model including coinfection of an infected individual from independent sources, and the model based on variation in MHC-I subtypes. The other, dead-end models and all approximations are discussed.
The reader should keep in mind that the present work is about the coarse-grained picture of evolution of pro in a typical patient; given the complexity of the problem, we did not attempt to explain the diversity of all HIV genes or all individual variations. Neither did we expect that models and predictions obtained in this work were necessarily final and seek to substitute mathematical analysis for experimental data. The hypotheses selected by this work eventually have to be tested experimentally.
MATERIALS AND METHODS
Calculation of genetic distances.
The genetic distance at a chosen base, between or within patients, is mathematically defined as the probability that two sequences differ at a given base, if sampled randomly from the same or two different patients, respectively. This definition is equivalent to the standard definition of the genetic distance as the average number of pairwise differences between two samples of a genome segment, except it deals with a single base and makes sense only after averaging over many such pairs of sequences. Both genetic distances can be expressed in terms of the frequency of substitutions. Let fki be the frequency of substitutions at site i and for patient k, with respect to any chosen reference sequence, such as a database consensus sequence, a best-fit sequence, etc. Then, the intrapatient distance for each patient, Tkiintra, and the interpatient genetic distances for each pair of patients, Tk1k2iinter, are given by the respective expressions
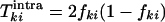 |
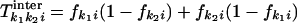 |
1 |
The intrapatient distance, as follows from its definition, has a maximum, Tkiintra = 1/2, at fki = 1/2. The maximum interpatient distance is Tk1k2iinter = 1, which corresponds to either fk1i = 1 and fk2i = 0 or vice versa; the two populations are genetically uniform in opposite alleles. Note that the right-hand sides of equations 1 do not change upon replacement of fki with 1 − fki; i.e., both genetic distances are independent of the choice of consensus, as they should be.
Evolution in an individual.
The initial set of deterministic equations describing the evolution over time of two variants of a virus has the form
 |
2 |
 |
3 |
Here n1 and n2 are the numbers of mutant and wild-type proviruses, respectively; κ1 and κ2 are the replication coefficients; trep is the replication cycle time; and μf and μr are the forward and reverse mutations rates, respectively. We assume that trep is the same for the two variants and that cell death is a random Poisson process. Neither assumption is important for a long-term selection. The selection coefficient s is defined as the relative difference in fitness, s = (κ2/κ1) − 1, and is assumed to be constant and to fulfill the double inequality μf(r) ≪ s ≪ 1. The ratio κ2/κ1 agrees with the standard experimental definition of the relative fitness measured by comparing the exponential growth rates, at a low multiplicity of infection, of the virus variant under study and the reference variant. Using the additional condition that the total population size is constant, n1 + n2 = N, as is the case during the asymptomatic stage of HIV infection (see the section on Verifying approximations), equations 2 and 3 can be replaced by the single equation
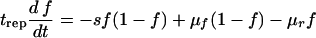 |
4 |
where f ≡ n1/N is the mutant frequency. Equation 4 is nonlinear since the replication coefficients, κi, must depend on the mutant frequency, f. It is implied that κi depends on the number of available target cells, which adjusts to keep the total provirus population constant. Although the clearance rate of infected cells is assumed to be constant and to be equal for the two genetic variants, the same equation, equation 4, can be shown to follow in a more general case as well, except that the definition of the selection coefficient must include the ratio of the clearance rates. Solving equation 4 for the arbitrary initial condition f(0) = f0, we obtain
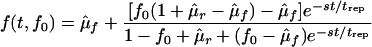 |
5 |
where μ̂f(r) ≡ μf(r)/s and error of the order of (μf(r)/s)2 is allowed in the right-hand side. Two particular initial conditions in equation 5 yield reversion and accumulation dependences: frev(t) = f(t,1) and facc(t) = f(t,0). Plots of frev(t) and facc(t) for a realistic set of parameters are given below (see Fig. 4). We define the reversion time, t50, as given by the equation frev(t50) = 0.5; one obtains t50 = (trep/s)ln(s/μr). In the limit of large t, the mutant frequency converges at the steady-state value, feq = μf/s ≪1.
FIG. 4.

Time dependence of the mutant frequency at different initial populations: purely mutant (reversion) (thicker line in upper panel), purely wild type (accumulation) (lower panel), or 50% mutant (growth competition) (dashed line). The thinner curve shows the intrapatient genetic distance T(t) during reversion. The horizontal bar is the time interval during which the base would be classified as variable (5 to 95%). Values shown in the upper left corner correspond to parameters as follows: the forward (wild type → mutant) and reverse mutation rates, μf and μr, corresponding to either A or C wild type; the replication cycle time, trep; and selection coefficient, s. The reversion half-time, t50, is given by the equation t50 = (trep/s)ln(s/μr).
Chain of single-clone transmission.
Let us assume that each individual infects the next individual at time t* after the moment of his/her own infection with a single genetic variant. Let fn* be the probability that the virus variant which infects person n is mutant. The expected value of the mutant frequency in person n at time t is given by
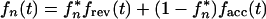 |
6 |
The probability fn* is equal to the expectation value of the mutant frequency in the source of infection at the time of transmission, as given by
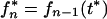 |
7 |
Together, equations 6 and 7 fully describe the evolution of a base along the chain of infection. If transmission occurs late, t* > t50 (t* − t50 ≫ trep/s), probability fn* can be shown to converge, after a few transmissions, to the individual steady-stage value μf/s. If transmission occurs prior to the reversion time, t* < t50, fn* changes slowly with n, and equations 6 and 7 can be simplified to the differential equation
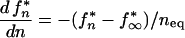 |
8 |
where
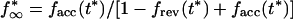 |
9 |
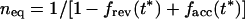 |
10 |
Solving equation 8 at the arbitrary initial condition f0*, we obtain
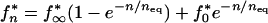 |
11 |
As follows from equation 11, the probability of occurrence of a mutant base converges, after approximately neq transmissions, to a “chain steady-state” value, fn* → f∞*. The dependence of parameters f∞* and neq on the transmission time t*, which is exponentially fast, is shown below (see Fig. 5a and b). In the range of transmission times trep/s < t* < t50, the two parameters are simply related, f∞* = μfneq/s.
FIG. 5.

(a) Probability of a mutant base being in the inoculum at the chain steady state, f*∞, as a function of the transmission time, t∗. Solid line, strictly single-clone infection; dashed line, coinfection from two sources with probability q = 0.05. (b) Number of cycles required to reach the chain steady state, neq, as a function of transmission time, t∗. (c) Probability of a mutant base for a very short transmission time, t∗ → 0, versus the probability of dual infection, q. Upper and lower curves correspond to wild-type A/C and G/T, respectively. (d) The same probability versus s, for a fixed value q = 0.01. trep = 2 days; μf = 4 · 10−5, μr = 4 · 10−6 for wild type A/C and vice versa for G/T.
Coinfection from independent sources.
Let us assume that a pair of genomes is transmitted from two different persons or animals with a probability q and that a single genome is transmitted with a probability 1 − q. Equation 6 is replaced by
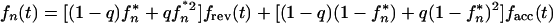 |
12 |
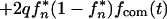 |
where fcom(t) is the growth competition curve defined as f(t,1/2) in equation 5 (the dashed line in Fig. 4) and fn* meets recursive equation 7. Equation 12 assumes that the two infection sources are epidemiologically distant and, therefore, statistically independent. The chain steady-state value, f∞*, is given by the smaller of two zeros of the quadratic equation
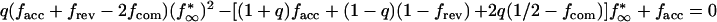 |
13 |
where facc, frev, and fcom are taken at t = t*. The dependence f∞* versus t*, q = 0.05, is shown below (see dashed line in Fig. 5a). The maximum probability of a mutant base, f∞*(t* = 0), is plotted below versus parameter q (see Fig. 5c). At intermediate values of q, such that μf + μr ≪ q ≤ 1, we obtain the asymptotics f∞* (t* = 0) = 2μf/qs. As expected, at 1/q ≈ neq, this expression matches the formula f∞* = μfneq/s obtained above for a singel-clone transmission.
The above derivation assumed that if coinfection with two virus sequences of similar fitness occurs, the steady-state virus population after the acute infection phase consists of equal proportions of the two variants. We also considered a random initial composition of between 0 and 100% and found out that such a change of model does not affect finf* at q → 0 and causes an increase in finf*(q) at larger values of q by a factor of 1.5 (the upper curve in Fig. 5c shifts upward). Consequently, the value of q estimated for HIV below increases ≃50%.
Coinfection from the same source.
Let us assume that transmission always occurs from a single source and that one and two virus variants are transmitted with probabilities 1 − q and q, respectively. (Similar considerations apply to the case of two different sources which are very close epidemiologically.) When two variants are transmitted, the initial steady-state population is assumed to be 50% mutant. In this case, the two sequences sampled are not statistically independent and equation 12 does not apply. Instead, one can write two separate equations for expectation values of the mutant frequency, fn(t), and of its square, yn(t), respectively:
 |
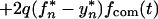 |
and
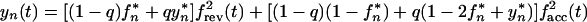 |
14 |
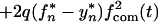 |
where frev(t), facc(t), and fcom(t) are given by equation 5 with f0 = 1, 0, and 1/2, respectively; parameters fn* and yn*, are both defined by recursive equation 7. The chain steady-state values, f∞* and y∞*, are found from equation 14 and the usual steady-state conditions fn* = fn−1* = f∞* and yn* = yn−1* = y∞*. The general expressions are cumbersome, and we give them only for the limit t* → 0 in which f∞* is maximum. In this case, the dependence on q cancels out, and we get the same result as in the case of single-clone transmission (cf. equation 9 at t* → 0):
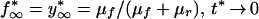 |
15 |
Value of q predicted for HIV.
We consider positions at which the wild type is A or C since such positions are predicted to dominate variable sites (see Fig. 5c). The average observable frequency of a variable base, f∞exp, is given by the general expression
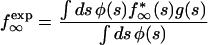 |
16 |
where ϕ(s) is the distribution density of s over different bases, and f∞*(s) is the probability of a mutant base being inserted for wild-type A or C (see Fig. 5c and d). The limits of the integrals in s implied in equation 16 are the boundaries of the variable zone in s, which will be specified below. The factor g(s) is the fraction of patients tested during the time interval in which a base with selection coefficient s is observably variable (see Fig. 3), given by
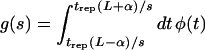 |
17 |
where φ(t) is the distribution density of the time of testing among patients, L = ln(s/μr) = 7.8 (s = 1%), and coefficient α ≈ 1 depends on the boundaries of the interval in f when the base is considered variable. At an average sample size of 20 clones, a site with 0.05 < f < 0.95 is observably diverse, which yields α ≈ 3. Assuming that φ(t) is constant in the interval between 1.25 and 8.75 years, and treating L as a large parameter, from equation 17 we obtain
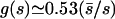 |
18 |
if 0.57s̄ < s <4s̄, and nearly zero otherwise. Here s̄ is defined as given by the equation trepL/s̄=t̄=5 years. The interval in s specified in equation 18 sets the limits of the integrals in equation 16. Approximating the distribution density ϕ(s) by a constant in this interval of s, and using the function f∞*(s) calculated from equation 13, we find that equation 16 matches the observed value of variable sites, f∞exp = 0.16, at q ≈ 0.085.
FIG. 3.

Model of evolution of a nucleotide along the chain of infected individuals under purifying selection. The numbers at the top denote cycles of transmission. X signs denote mutants. A mutant base appears spontaneously in person n, who infects person A with the mutant and person B with the wild-type variant. Person A passes the mutant down the chain to person C, after which his/her own virus population slowly reverts to the wild type. In person D, who is stably coinfected with a pair of sequences polymorphous at that base, selection clears the mutant virus rapidly.
RESULTS
Two types of selection.
There are several groups of factors which can shape the evolution and diversity of a virus genome at a given locus: mutation, random drift (13, 50), selection, transmission between individuals, and virus spread between different infection sites within an individual. Transmission and spread between infection sites create genetic bottlenecks, which modify the action of other factors, creating founder effects due to random sampling from the infecting population.
Before proceeding, we will define the terminology that we use for different types of selection. We divide all selective forces acting on a separate locus into two components: purifying selection and selection for diversity. Purifying selection exists due to the fact that a certain genetic variant, referred to here as the wild type, is better fit than other variants. The virus fitness is usually defined in experiments as a relative parameter, the ratio of the exponential growth rate of the virus variant to that of a reference variant, when measured at a low multiplicity of infection in culture. For our purposes, it is more convenient to characterize selection by the selection coefficient, s, defined as the relative fitness minus 1, i.e., to the relative difference in the corresponding growth rates. If this type of selection is the only factor of evolution present, the population will eventually become uniform in the better-fit variant.
Selection for diversity comes in two types. The first is time-dependent selection, existing due to temporal changes in the conditions of virus growth, e.g., the appearance of a functional immune response to a specific epitope. The second type of selection for diversity is the ecological niche effect: different host cells may favor different wild-type virus sequences. If this type of selection dominates, the population will gradually assume a constant, albeit highly diverse, genetic composition, consisting of a mixture of viruses adapted to replicate in different cell types.
Note that we use the terms “purifying selection” and “selection for diversity,” as is conventional in population biology, to describe external conditions acting on a population, as opposed to a state or dynamic of the population. This means, in particular, that the genetic diversity of a population can increase transiently in the absence of selection for diversity, e.g., when a wild-type sequence is being fixed in the population under the influence of purifying selection (see below).
Sequence diversity in pro.
To obtain a more detailed picture of genetic variation in pro, we analyzed the database of sequences described by Lech et al. (22). Sequences were obtained at a single time point from protease-inhibitor-naive patients. The 265 clones of proviral DNA were derived from peripheral blood mononuclear cells of 13 individuals by 70 cycles of nested PCR amplification, and the consensus sequence for each patient’s virus was obtained. Of the 13 patients, two (06 and 12) had stop codons in their consensus sequences and were excluded from further analysis. For the remaining 11 patients, 6% of the sequences contained deletions, insertions, or stop codons; these sequences were also filtered out, since the present work addresses point mutations, which do not render the virus nonviable. The remaining database comprised 213 clones of proviral DNA. We determined the common consensus sequence by noting the most frequent variant for each base. The consensus sequence starts from the conserved sequence CCTCAgATCACTCTT (PQITL), where g shows a variable base, and is 297 bases long. It is identical to the subtype B consensus in the LANL database (19a). Of the total number of substitutions, 25% were transversions with respect to the consensus sequence. To simplify further analysis, we ignored transversions, replacing them by the consensus sequence. As a result, each base was represented by two genetic variants: either A/G or C/T. (A theory taking into consideration both types of mutation would have to contain more parameters, namely, the forward and reverse mutation rates for transversions, which are very low and known with poor accuracy. This would complicate the theory without much gain in information.) Finally, to partially correct for errors accumulated during PCR, we ignored mutations present in a single copy in the entire database. These sporadic mutations occurred with a frequency of 0.06% per base, with respect to the consensus, and were distinctly overrepresented in nonsynonymous changes relative to mutations appearing two or more times (Fig. 1; Table 1). This value, as well as the very small number of mutations which appear exactly twice in the database, is consistent with a PCR error rate of about 1 per 105 bases per cycle, which does not exceed reported values (2, 44).
FIG. 1.

Intrapatient (a) and interpatient (b) genetic distances, averaged over patients, at different positions in pro. The upper and lower histograms in each figure correspond to synonymous and nonsynonymous sites, respectively. Dots on the upper horizontal line in panel a show the positions of sporadic mutations (seen only once in the data set).
TABLE 1.
Genetic diversity in pro
| Mutation type | Average intrapatient distance, T (%)a
|
No. of variable sites | ||
|---|---|---|---|---|
| Individual variable siteb | Any variable sitec | Any sited | ||
| Synonymous | 29.5 | 4.7 | 0.49 | 31 |
| Nonsynonymous | 21.9 | 4.3 | 0.23 | 16 |
| Synonymous and nonsynonymous | 27.5 | 4.5 | 0.72 | 47 |
| Sporadic synonymouse | 0.05 | 16 | ||
| Sporadic nonsynonymouse | 0.07 | 23 | ||
| Sporadic synonymous and nonsynonymouse | 0.12 | 39 | ||
Intrapatient genetic distance in pro, T, averaged over one of three groups of bases in each infected individual and then averaged over individuals.
Bases which are variable in a given individual only.
Bases which are variable in any individual.
All bases in pro.
Substitutions present in a single copy per database and excluded from the first three rows as PCR error suspects. Note the difference in relative number of synonymous sites between sporadic and nonsporadic substitutions.
For the database thus filtered, we classified each base as either variable or conserved. For each variable site and each patient, we determined the frequency of substitutions with respect to the database consensus. We also determined, for each patient, the average genetic distance at a base as the proportion of sequence pairs (randomly sampled from the virus population) which differ at that base. This definition is equivalent to the standard definition of the genetic distance as the average number of pairwise differences, except it applies to a separate base rather than a long genomic segment. We determined the interpatient genetic distance in a similar way, except that the two sequences of a pair were sampled from different patients. By definition, the intrapatient genetic distance varies between 0 and 1/2 (0 and 50%), corresponding to a uniform population and a 50:50 mixture, respectively. The interpatient distance is 0 when the two virus populations consist uniformly of the same genetic variant, and it is 1 (100%) when the two virus populations are composed entirely of opposite genetic variants. The interpatient distance, as one can show, cannot be smaller than the average of the two intrapatient distances.
Note that although genetic distances can be mathematically expressed in terms of substitution frequencies, as used for calculations of genetic distances in Materials and Methods, the genetic distances do not depend on the choice of reference sequence (which, in our case, is the subtype B consensus, the same as the database consensus). This invariance with respect to reference sequence makes the intrapatient genetic distance a more adequate parameter for the comparison of theory and experiment than the substitution frequency. Indeed, as we argue below, the best-fit sequence and the consensus sequence do not necessarily coincide, making the choice of a proper reference sequence less than obvious.
Figure 1 shows both kinds of distances averaged over patients (or pairs of patients) versus the position of a base in pro. For individual patients, the intrapatient distances at different variable sites are shown in a gray-scale diagram in Fig. 2. Table 1 shows the intrapatient genetic distance averaged twice: first over one of three groups of sites for each patient separately (sites which are variable in the patient, sites which are variable in any patient, and all sites in pro), and then over all patients. Since multiple substitutions within the same codon were rare and did not generally affect the net diversity, we were able to classify the mutations at each variable base as either synonymous or nonsynonymous. (When two or more substitutions with respect to the consensus did occur in the same codon of the same sequence, we counted these substitutions as if they occurred in different sequences.) Table 1 shows separate data for the two types of sites. The principal conclusions from this material are as follows. (i) The bulk of variation within an individual is contributed by rare sites. In all individuals combined, only 47 of the 297 total bases were variable. (ii) An average variable site occurs in approximately 16% of patients (2 of 11). (iii) A typical variable site is highly diverse; the intrapatient genetic distance is 27% per variable site. (iv) Synonymous and nonsynonymous variable sites are similarly diverse. (v) However, synonymous sites are twice as frequent; if variable sites were chosen at random, the latter ratio would be inverted.
FIG. 2.

Gray-scale diagram of intrapatient genetic distance, Tki, in different patients at different variable sites in pro. The intrapatient genetic distance is indicated by the degree of shading, as shown on the scale on the right. Letters and numbers under the diagram show consensus nucleotides and positions in pro, respectively.
Random drift, in the case of a small effective population size, could have played a significant role in the pattern of genetic diversity observed. However, in other work (39), we tested the pro sequences for a characteristic linkage disequilibrium effect between pairs of sites, a phenomenon expected if the effective virus population is small. The negative result we obtained shows that stochastic effects do not greatly influence the evolution of separate bases in an individual quasi-steady-state HIV infection. The preponderance of synonymous substitutions (observation v in the previous paragraph) suggests that selection for diversity is not a dominant factor of evolution in pro during most of the asymptomatic phase of infection. Therefore, we will treat the evolution of pro in an individual as approximately deterministic and mostly controlled by purifying selection.
Our starting idea of HIV evolution, to explain the high level of diversity at individual variable sites and the random difference between infected individuals, is illustrated in Fig. 3. Slightly deleterious pro mutants emerge spontaneously in some individuals and are passed from one individual to the next, down the chain of transmission. Sequences are sampled randomly during transmission, so that the infecting genome may or may not contain a mutation. In a person who is infected with a mutunt virus (person A in Fig. 3), the population gradually reverts to wild type. During the reversion process, when there are comparable quantities of mutant and wild type, the base will be very diverse. We will analyze this process in two parts. First we consider evolution in an individual, and then we include transmission.
Model 1: evolution in an individual.
Neglecting transversions (which we do in both theory and experiment), a base can assume one of two variants, either A/C or G/T. One of the two variants (by definition the wild type) is better fit, in the sense defined above. We assume that the wild-type sequence is the same for all individuals and all cell types involved. (This and other assumptions are discussed in detail in Discussion.) Each base is characterized by the relative difference in fitness between the two variants (the selection coefficient, s) and the mutation rate (μ). The best estimate of the HIV mutation rate is μ = 4 · 10−5 for A→G and C→T transitions, and 5 to 10 times lower for the opposite transitions (24). The selection coefficient, s, varies strongly among different bases and is almost never known in vivo; therefore, it will be treated as a fitting parameter (see its formal definition in Materials and Methods).
To complete the model, we have to make an assumption about the initial genetic composition at the chosen site. It is known that the virus population early (a few weeks) postinfection can be either genetically diverse or uniform, depending on the genomic region. For example, the V3 and V4 regions of env, which are highly diverse late in infection, are uniform early in infection (9, 18, 23, 53), while the region of gag coding for the p17 matrix protein can have a level of diversity early in infection comparable to that exhibited late in infection (53). This effect probably reflects transmission of multiple clones which can coexist on the time scale of a few weeks due to relatively weak selection conditions for p17 (53). Since we are not aware of analogous data that could clarify transmission and early selection conditions in pro, we will consider all possibilities. In this and the following section we consider a single-clone transmission. Subsequently, we will investigate effects of multiple-clone transmission, including that from the same and different sources.
For the moment, let us assume that the initial, postseroconversion virus population is either 100% mutant or 100% wild type with respect to a chosen base. Suppose it is 100% mutant. In the course of a persistent infection involving numerous infection cycles, wild-type variants will emerge due to mutation. Since the wild type, by definition, is selected for and the mutant is selected against, the population will gradually revert to an almost entirely wild-type population, as shown in the upper panel of Fig. 4. The resulting steady-state population will have a small proportion of mutants due to the balance between selection and mutations (7). We denote t50 as the time it takes to reach a 50% composition; t50 is inversely proportional to the selection coefficient and directly proportional to the time per replication cycle (see Materials and Methods). The latter time can be approximated as the average duration of the productive phase of an infected cell, approximately 2 days (16, 17, 34, 46). If a base is observed near time t50, it will display a high degree of diversity. It is reasonable to assume that the mean sampling time is about 5 years, which is somewhat more than one-half of the average duration of infection (42). This estimate yields a selection coefficient s = 0.008; for a patient tested earlier or later, the respective fitting value of s will be higher or lower. Figure 4 shows the case in which A or C is wild type; when G or T is wild type, reversion occurs somewhat faster due to the higher reverse mutation rate, and we have a slightly lower fitting value, s = 0.005.
The reversion model explains almost all experimental features discussed in the previous section. (i) Variable bases are rare in the sequence, since such a base must have a very small selection coefficient, on the order of or less than 0.01. (ii) Variable bases differ among individuals, since an individual may be randomly infected with a virus that is either mutant or wild type at a particular base. Also, since different individuals are tested at different times, most of them are sampled outside the narrow time interval in which reversion of a particular base can be observed (Fig. 4). (iii) An average variable base in an individual is very diverse, since it is in the middle of reversion. (iv) For the same reason, synonymous and nonsynonymous variable sites have approximately equal diversities per site. (v) Synonymous variable sites are more abundant (Table 1), implying that synonymous substitutions tend to have smaller s values. Note that according to the model, positions of variable sites are not fixed but depend on the time of observation.
According to this model, the consensus sequence of the entire data set for all patients can be either mutant or wild type at a variable site. A base with a selection coefficient somewhat smaller than 0.008 and wild type A or C will tend to be observed before it has completed its reversion in patients who were infected with a virus that is mutant at that base (for a 5-year observation point). Therefore, if such patients are sufficiently frequent, the majority of samples at the time of observation will be mutant at that base. This example illustrates our previous comment, that wild type and consensus sequences, even if determined for a very large group of patients, do not necessarily coincide. (Alternatively, the wild-type sequence may vary among individuals; this possibility is discussed later.)
It is tempting to use the fact that each variable site is expected to evolve in the same direction in different patients as an experimental test of the reversion model. Suppose we compare virus variants from two patients that share a few variable sites. Since one of the patients has been infected longer than the other, the mutant frequency is expected to differ from the first to the second patient in the same direction for all variable sites. Unfortunately, to use this prediction as a test, one would have to know the wild-type sequence at these bases, which, as we just discussed, is unknown. This is also why we have used the pairwise genetic distance instead of the substitution frequency: the former does not change if all sequences mutant at a base are replaced by sequences wild type at the base, and vice versa. In the database studied, the intrapatient distance increases at some bases shared by both patients and decreases at others (Fig. 2). This does not contradict the reversion model in which the genetic distance at a base is expected to have a maximum in the middle of reversion (Fig. 4). We once hoped that comparing more than two patients at once might allow prediction of wild types and, therefore, directions of reversion for separate bases. Using a Boolean algorithm written for this purpose, we determined that the average proportion of patients sharing the same variable site is too low for useful analysis (results not shown). Determining wild types at different sites would require two sequence sets obtained, in the same group of patients, at two sufficiently distant time points. We are not aware of such data for pro.
Model 2: chain of single-clone transmission.
In the absolute sense, the average proportion of patients sharing the same variable site (the frequency of variable sites in individuals) is still quite high, 16%, contributing to the high average intrapatient distance in pro. Can the reversion model explain such a high value? To answer this question, we have to explicitly consider the evolution of the virus along the chain of infection (Fig. 3). Suppose that each individual infects the next individual at a fixed time since the moment of his/her own infection with, as we still assume, a single genetic variant, either mutant or wild type at a particular base. A predictable parameter, related to the frequency of variable sites in an individual, is the probability that a base is mutant in the inoculum for person n in the transmission chain.
As our calculation shows (see Materials and Methods), if the transmission time is shorter than the reversion time (5 years, for the bases of interest), the probability of a mutant being in the inoculum will grow with every passage from individual to individual, until it saturates at a constant value, which can be described as a steady state in the transmission chain sense (Fig. 5a) (see Materials and Methods). This value is not equal to the individual steady-state mutant frequency; rather, it is much higher. In fact, for very small transmission time intervals, and if the forward mutation rate is much higher than the reverse mutation rate, the probability of a mutant being in the inoculum can eventually approach 100%.
On an intuitive level, the predicted strong accumulation of mutants is a combined effect of the transmission bottleneck, which causes an initial population to be genetically uniform at a base, and of the short passage time. Selection does not act on a uniformly mutant population; its effect becomes important after mutations create a sufficiently large wild-type subpopulation. Hence, quick passage of the virus to the next person in the chain, before such a subpopulation can be created, effectively suppresses selection against mutants.
As we have found (see Materials and Methods), the number of transmission intervals required to reach the steady state in the transmission chain and the level to which mutants accumulate are proportional. To explain the high level of mutants observed in the inoculum, the transmission interval has to be rather short, ∼1 year or less (Fig. 5a). In this case, hundreds of transmission cycles will be required. Thus, the main contributor to the occurrence of mutants in the inoculum is the virus evolution occurring before the start of the HIV pandemic, which includes the human endemic and enzootic infection in the original (animal) host. Although the transmission time and other parameters may differ between human and animal hosts, this difference is not expected to affect these conclusions seriously.
Model 3: coinfection.
Thus, if a single clone of virus is transmitted each time, any base with a small selection coefficient will eventually become highly diverse. (The selection coefficient has to be less than the inverse of the number of virus replication cycles between two transmissions [Fig. 5a]) Is this model a realistic explanation for the large number of variable sites in pro? To answer this question, we reexamined the approximations that were incorporated in the model and found that the theoretical explanation critically depends on the assumption that a recipient is always infected from a single source. As we show below, even very rare coinfections from two or more independent sources within a short time interval can prevent accumulation of mutants and sharply decrease the number of variable sites.
Uniformity of the initial virus population is the factor that, as we have shown, suppresses selection in the case of single-clone transmission. A uniformly mutant population gives rise to a new uniformly mutant population in the next individual (person C in Fig. 3), which is then passed further along the chain until a rare sampling of the wild type is obtained for an inoculum. As a result, a typical transmission chain is expected to consist of long series of individuals infected with the same initial variant: mutant - mutant -…-mutant - mutant - wild type - wild type -…-wild type - wild type - mutant - mutant -…- mutant - mutant, and so on. Suppose, now, that an individual (patient D in Fig. 3) is infected, before passing the virus further, with two sequences from different sources, one from a mutant series and another from a wild-type series. Suppose, also, that the two transmission events with viruses of very similar fitness occur within a short time interval (weeks), so that the virus transmitted first does not have a significant advantage in establishing infection with respect to the virus transmitted second. (This assumption is indirectly supported by the short transmission time we had to assume above to explain the accumulation of mutants for single-clone transmission, by the actual observation that the protection effect in simian immunodeficiency virus (SIV)-infected animals is absent during the first few weeks postinfection [51], and by the observation that multiple virus clones can coexist in primary infections [53].) In this case, both genetic variants are well represented in the initial population. Therefore, even weak selection between the two viruses starts to act immediately [fcom (t) in Fig. 4], and there will be less mutant virus to transmit to the next recipient. Such an event can interrupt a long series of “mutant” individuals. Thus, even very rare dual infections can prevent mutant variants from accumulating. This qualitative conclusion is confirmed by direct calculation (see Materials and Methods). The effect of dual infections on diversity turns out to be especially strong for short transmission time intervals, when the level of diversity otherwise would be very large (Fig. 5a). Even if the probability of dual infection is as small as q = 0.05, the mutant frequency in the inoculum is decreased by a factor of 10 compared to the case of exclusively single-clone transmission (Fig. 5c). (Note that the above argument and corresponding calculation make sense only on average. A typical pair of sequences from different sources is also heterozygous at many other sites. The difference in fitness between the two clones is the sum of a random term contributed from the other sites and of a regular term due to the chosen site. The random term has a random sign and vanishes when averaged over many pairs of clones. The same consideration applies to evolution within an individual: although competition between a particular pair of clones depends on the entire configuration of each sequence, evolution of a chosen base can be considered separately, since configurations of other bases average out over many pairs of clones. This is the reason why evolution of a site, in the absence of coselection and in a sufficiently large population, can be considered separately from other sites, even if recombination is absent.)
Thus, the accumulation of mutants to high levels occurs only as long as the spread of infection among the animal or human population is exactly represented by a branching tree, but it may be efficiently suppressed if the branches can sometimes merge together (by coinfection), creating an image of a cluster with loops. The topology of epidemics determines the outcome of virus evolution.
Transmission of multiple sequences from the same source, unlike coinfection from independent sources, does not greatly affect the accumulation of mutants in the transmission chain. We have calculated the frequency of mutants in an inoculum in the model in which either one or two clones are transmitted from a single source (see Materials and Methods). At short transmission time intervals (when the mutant frequency in the inoculum is maximum), coinfection has no effect at all on the frequency. (Such an effect may appear for a very large, in terms of parameter s/μf, number of transmitted clones and at finite transmission times.) The intuitive reason for this is clear from Fig. 3: if person D received two or more genomes from person A, all of them would be mutant. This situation would not differ from that in which a single mutant genome is received from person A, since the initial population established in person D would be mutant in both cases. In mathematical terms, the difference between coinfection from two epidemiologically distant persons and coinfection from the same source is that sequences from two sources are statistically independent (see Materials and Methods) while two sequences sampled from the same person are not. Since each person has an almost uniform initial population, they tend to be either both mutant or both wild type; only rarely is one mutant and the other wild type. Conditions under which two typical individuals in an infected population can be considered as epidemiologically independent are discussed below.
Value of q estimated for HIV.
We now return to the experimental data to find out how small the probability of coinfection, q, must be to explain the high degree of diversity observed in pro. Since, as follows from our calculation, the probability of a mutant base is much less for sites with wild-type G or T (Fig. 5c), most variable bases are expected to have A or C as the wild type. If the test time and the replication cycle time did not vary between patients, the probability of receiving a mutant base in the inoculum would be equal to the observed frequency at which a variable site appears in individuals, which is 16%. Since both times, in fact, vary between patients, the probability of a mutant being in the inoculum must be higher than 16%, because if an infected person is tested too early or too late (compared to an average patient), given a limited sample size (∼20), a reverting base will not be detectably diverse (Fig. 4). The variation in replication time, as follows from statistical analysis of data from 20 patients (17), is relatively small and can be neglected. Still, we have to take into account the probable broad distribution of the test time among patients. The time of progression to AIDS for HIV-infected individuals is almost uniformly distributed between 2 and 14 years (42). We assume that the test time is somewhat past the middle of the infection course and is distributed uniformly as well, between 1.3 and 8.7 years, with the average being 5 years. The distribution density of the selection coefficient around the value s ≈ 0.01 (which is, of course, unknown) also enters our calculation. Fortunately, as we had checked, the final answer happens to be rather insensitive to the shape of the distribution density; as an estimate, we assume a uniform distribution in the region of interest, which happens to be (see Materials and Methods) s = 0.005 to 0.05. Under these approximations, we estimate that the observed experimental frequency of a variable site, 16%, requires a q value of 0.01 or less (see Materials and Methods). At this value of q, the probability of a mutant being in the inoculum is close to 100% if the wild type is A or C and around 10% if it is G or T (Fig. 5c).
The above calculation assumes that if coinfection with two virus sequences occurs, the virus population after the acute infection phase contains 50% of each variant. As the period of time between two infections increases, the symmetry between the two clones may be lost. A random initial composition of between 0 and 100% may be a better approximation in this case. As we have checked, such a change of the model does not affect the mutant frequency in the inoculum at very small values of q and causes an increase in this parameter at larger q values by a constant factor of 1.5 (the upper curve in Fig. 5c shifts upward). As a result, the value of q estimated above increases by 50%, and the final conclusions are not affected.
The above results suggest a surprisingly strong effect of quite rare coinfection events on mutant frequency. Given our assumptions, a probability of coinfection from independent sources of 1% during the evolutionary history of the virus allows for a high observed frequency of variable sites, 16%. A 10-fold-higher probability of coinfection would decrease the latter frequency by a factor of ∼10 (cf. Fig. 5c). On the other hand, lowering q below 1% does not increase the variable-site frequency further above 16%. Indeed, an HIV population has to be, according to the model, in the rare-coinfection limits, when the probability of a mutant being in the inoculum is already close to 100%. The difference between the 16% observed and 100% values is primarily due to fluctuations of the observation time between patients.
DISCUSSION
In this paper, we present and evaluate a model designed to explain the distribution of mutations observed in a relatively conserved region of the HIV genome. In its simplest form, the model describes the accumulation of diversity at individual sites as a consequence of passing of slightly deleterious mutations upon relatively rapid transmission of the virus from individual to individual, accompanied by a slow reversion within infected individuals. As described in more detail below, the model is robust to variation in most of the assumptions we initially made, with one exception. The accumulation of mutations during the chain of transmission is exquisitely sensitive to coinfection of infected individuals prior to further transmission from epidemiologically distant sources. To sustain the observed variability in pro, either the frequency of coinfection must be 1% or less or one of the other assumptions must be changed radically. We present an alternative explanation at the end of this section.
The scenario we describe here is also a possible mechanism for the accumulation of deleterious mutations in frequently passaged viruses, as observed in vitro for vesicular stomatitis virus (5, 10, 11, 43). Another possible explanation (10) is the Muller’s ratchet effect, in which the processes of reversion at different loci interfere with each other. This effect of stochastic evolution theory, which applies to populations that are on the order of the inverse mutation rate, leads to enhanced accumulation of slightly deleterious mutations (12, 14, 30). The testable differences between the more simple effect described in this analysis and Muller’s ratchet are follows. (i) The “simple” ratchet acts on an individual locus, while Muller’s ratchet involves two or more loci and can, therefore, be suppressed if recombination events are frequent (12, 14, 30). (ii) While the Muller’s ratchet effect is restricted to small virus populations (12, 14, 30), the simple ratchet is expected to work even if the average virus population size between transmissions is large. (iii) Simple ratchet implies that the reversion processes at different loci do not have sufficient time to occur, making their interference (Muller’s ratchet) redundant.
As we have found, a high frequency of variable sites in HIV could be explained as a combined effect of a transmission bottleneck and frequent passaging between individuals, provided coinfection from independent sources is extremely rare. The rate of superinfection of individuals depends on both virological and epidemiological factors and is hard to estimate, in general. A clue to the rate in modern times is given by the frequency of coinfection with multiple HIV subtypes in geographical regions where no single subtype dominates. Infections with two different HIV type 1 (HIV-1) subtypes (or with HIV-1 and HIV-2) have been directly observed and are estimated to occur in 10 to 15% of such cases (35, 36). This figure does not include reports of intersubtype recombination (15, 40), which imply coinfection at some indeterminate time in the past.
The fact that coinfection is relatively frequent despite the effect of superinfection protection detected in SIV-positive animals (51) suggests either that a narrow time interval between primary infection and establishment of protection exists or that the protection is only partial in a probabilistic sense; data (51) allow for either interpretation. The two-subtype data can be used to estimate the overall frequency of coinfection, q. Assuming that the two subtypes circulate in equal quantities, and taking into account the fact that cases of double infection with the same subtype are not included in the above percentage, we obtain, as a lower bound, q = 0.2 to 0.3, much higher than our prediction of q ≈ 0.01. We have no way of knowing whether this value is representative of conditions in ancient animal transmission, which, according to the model, determine the mutant frequency in an inoculum in the modern-day pandemic; however, there is no obvious barrier to the coinfection rate which would keep its value as low as 1%. Although we cannot dismiss the possibility of a very low coinfection rate in the natural host, we do not know any independent facts to support this model.
Verifying approximations.
The above considerations suggest that we may have to look elsewhere for an explanation for the high frequency of variable sites. To find another possibility, we will reexamine the simplifications common for all models discussed so far.
(i) Ignoring transversions.
We ignored transversions in both experiment and theory. The danger of this approximation is that a double transversion may appear as a transition and thus interfere with our count of transitions. Given the small relative weight of single transversions in the net variation (25%), this effect is expected to be but a relatively small correction to the numbers of transitions, on the order of (0.25)2 (∼0.06).
(ii) Neglecting stochastic effects in steady state.
Stochastic effects include randomness of mutation times and random drift of the genetic composition. The linkage disequilibrium test (39) implies that the effective HIV population is larger than 105 infected cells (P = 0.05) and that, correspondingly, stochastic effects on the evolution of separate bases within an individual are relatively small. In the same work, we considered the effect of recombination on the test results and found that it cannot decrease the lower estimate of the effective population size by more than a factor of ∼2.
(iii) Constant HIV population size.
In fact, in a typical patient, the virus load expands sharply within 1 to 2 weeks and then drops by 1 to 2 orders of magnitude over 2 to 3 months during the acute phase of infection. Then, during the asymptomatic phase of infection, the virus load climbs back slowly, over a scale of a few years, before a large expansion at the final stage, leading to AIDS. In this work, we consider the evolution of bases with a small selection coefficient, which occurs slowly during the asymptomatic phase of infection. The ongoing slow change in the population size is not important for the almost deterministic evolution (which we found is the case), since it does not greatly depend on the population size.
(iv) Same wild type for all cells and individuals.
In fact, HIV adapts to new cell types. The magnitude of this effect on the HIV genome after a thousand replication cycles, the relevant time scale, is hard to infer from the existing literature. It is certain that adaptation on a scale comparable to the fraction of variable sites in HIV pro, 16%, can be caused within 1 year by a very adverse factor, such as an efficient protease inhibitor (8). It could be also forced by the immune response, which we have not considered to this point.
(v) “Well-stirred pot” approximation.
We implicitly replaced the complex topography of virus distribution between and within different lymphoid organs by an imaginary infected organ with well-mixed cells infected with different genetic variants. Direct visualization of two env variants of HIV in the spleen by selective labeling shows that although infected cells are nonuniformly distributed into islands, most of these islands are shared by different variants (37). This result implies that virions mix well between these islands; i.e., a significant part of the de novo infection is due to far-travelling virus particles (38). The conclusion is supported (38) by the large number of virion particles removed daily from peripheral blood of SIV-infected animals (28).
(vi) Independent sources of coinfection.
When calculating the purifying effect of coinfection, we implied that two typical sources of coinfection in the host population are epidemiologically distant, so that the probabilities of receiving mutant bases from the two sources are independent. For epidemiologically close sources, the purifying effect of coinfection is expected to become weaker. Transmission of two clones from the same source is a limiting case of nonindependent coinfection; it does not have a purifying effect (see Materials and Methods). Whether two typical animals in a population are epidemiologically distant depends on the coinfection frequency q and on the size of the animal population. Coinfections randomize the distribution of virus variants between animals and make them more independent. If coinfections are very rare (i.e., q is much less than 1), a mutant base can pass through a long chain of animals without converting to the opposite variant. Then, two animals from even a large population are likely to have the same initial variant. If, however, the value of q is not too small, say ∼0.2 to 0.3, then the two typical animals are statistically independent even in a small herd comprising just a few animals. Therefore, invoking the epidemiological proximity does not really help to explain the high mutant frequency in inocula; one still has to postulate that q is very small.
(vii) Absence of group selection.
The possibility of group selection (as opposed to selection of individual genomes) has been raised (27). However, at this time, there is no compelling evidence that such an effect exists for RNA viruses.
(viii) Neglecting coselection.
This approximation is rather crude for a particular pair of sites. When studying linkage disequilibrium (39) at close pairs of variable sites, we found that some haplotypes are 2.5-fold more frequent than one would expect for independently evolving sites, implying a noticeable coselection effect. However, when obtaining an average pattern of variation in a segment of genome as long as pro, containing over 40 variable sites, contributions from negatively and positively coselected pairs of bases to the net variation are expected to cancel out, at least partially. For example, if coselection is responsible for 50% of the relative fitness of haplotypes at a separate pair of sites, and there are 40 variable sites with a random sign of coselection between each pair, then the effect of coselection on the relative fitness at an average site is expected to be, by the laws of statistics, ∼50%/√39 (∼8%), i.e., a small correction. This argument, however, applies only under certain restrictions (see below).
(ix) Neglecting selection for diversity.
The predominance of synonymous and conservative substitutions in pro in patients at late stages of asymptomatic infection was the reason why we initially neglected selection for diversity. This argument does not imply that immune pressure is completely absent from pro but rather states that its selective pressure is smaller than the effect of purifying selection at a typical observation time.
Let us search for weak spots in these approximations. Note, first, that a few of them have restrictions. First, the wild-type pro sequence could differ between individuals strongly if, despite the predominance of synonymous mutations, the immune function was somehow involved; amino acids serving as binding sites for MHC subtypes expressed in a person are highly individual. Second, coselection could be relevant if the 47 variable sites in pro were somehow dependent on a much smaller number of sites. Third, strong selection for diversity may be present in pro during isolated periods far removed from the typical observation time.
In an attempt to find out how these clues could help to explain the high frequency of mutant bases in inocula, we came up with the following possible scenario.
Model 4: individual variation in wild type due to MHC subtypes.
Suppose that the cytotoxic T-cell (CTL) response in HIV-infected patients is at least partly functional and contributes to clearance of infected cells. Since the CTL response can be directed against epitopes in any part of the HIV genome, the pro gene, in general, is likely to contain some of them. Therefore, the best-adapted type in pro, which we denote here as the wild type, will be one that does not contain CTL epitopes for the MHC-I subtype set of the infected individual. In other words, the wild type will vary between individuals.
In this scenario, soon after infection, pro will rapidly accumulate antigenic escape mutations within the CTL epitopes, abrogating binding of the epitopes by the individual MHC-I subtypes. Immune memory ensures that these anchor switches become permanent. Coselection comes into play at this point: a switch at an anchor residue is expected to redefine best-fit configurations for a number of other bases linked to the residue by coselection. These bases now become mutant and start to revert gradually to the new wild type, just as we described in the beginning (model 1), under a constant, purifying selection. Some of the evolving substitutions will be synonymous, and some will be nonsynonymous.
In the idealized model suggested above, the immune selection pressure is said to be absent during most of the infection. This is an approximation, as we already discussed. In fact, for this explanation to work, the immune pressure has to be only much smaller than both the effect of purifying selection most of the time and the immune pressure early in infection.
The positions of epitopes for a particular MHC subtype, and the bases linked to them, are expected to overlap only partially among patients. A base variant which is wild type for one patient may therefore be mutant for another patient. This effect could also explain the very high probability of mutant bases in inocula that we inferred from the frequency of variable sites in individuals assuming that the wild-type sequence is identical for all patients and that coinfections are very rare (model 3). In the present model, based on the differences among MHC subtypes between patients, the frequency of variable sites could reflect the degree of overlap between wild types in different patients.
Reversion of substitutions with larger selection coefficients occurs more rapidly. Therefore, in the course of time, on a scale of months to years, the variable zone will shift gradually to sites with smaller selection coefficients, slowing down the tempo of evolution (Fig. 6). There will be a gradual decrease in the relative number of nonsynonymous sites versus the number of synonymous variable sites; synonymous sites, on average, are expected to have smaller selection coefficients. (At the same time, the average intrapatient distance per variable site, regardless of whether it is synonymous, is expected to stay at a constant high value.) Note that the env gene, which may be under immune pressure most of the time, shows the opposite tendency: at early stages of infection, the synonymous-to-nonsynonymous ratio is decreasing (4, 32, 33). We are not aware of whether such sequence databases at multiple time points have been obtained for the pro gene.
FIG. 6.

A working model of evolution in pro. A quick initial switch of dominant epitopes redefines wild type for a number of sites, which then revert to this new wild type at speeds inversely proportional to their selection coefficients. (a) Time dependence of mutant frequency at three sites linked to an epitope. The three selection coefficients after the epitope switch are shown near corresponding curves. (b) Evolution of an individual consensus sequence at the three sites. pt, patient.
The suggested scenario for pro in the absence of antiviral drugs is very similar to the cascade of compensating mutations that follows the appearance of drug resistance mutations observed after protease inhibitor treatment. In one study (8), the changes in the intrapatient nucleotide consensus of pro after 32 to 60 weeks of indinavir therapy comprised 10.4 nucleotide substitutions per patient, of which about 25% were synonymous. The difference in the synonymous/nonsynonymous composition between data (8) and the group of drug-naive patients considered here (66% [Table 1]) is consistent with the difference in time which elapsed after the escape or resistance mutations occurred, around 1 year versus 5 years. According to the model, most nonsynonymous substitutions in the drug-naive patients already finished reversion and are no longer detectably variable given the sample size.
To summarize, we analyzed the genetic variation of the HIV pro gene in a group of patients sampled at a single time point. The pattern of diversity at separate variable sites supports the hypothesis that the major contribution to genetic diversity comes from rare bases slowly evolving from deleterious variants to wild type under purifying selection. The high frequency of variable sites in individual patients suggests that either the mutant bases are due to events of early antigenic escape in pro or coinfection from different sources was a very rare event in the ancient animal host population.
ACKNOWLEDGMENTS
We are grateful to A. Kaplan, who supplied us with a database of protease sequences, and to F. McCutchan, for help with data on multiple infections. We also thank E. Kandel, D. Lazinski, W. Lech, and J. Mullins for useful discussions.
This work was supported by grant R35 CA44385 from the National Cancer Institute. J.M.C. was a research professor of the American Cancer Society.
REFERENCES
- 1.Balfe P, Simmonds P, Ludlam C A, Bishop J O, Leigh Brown A J. Concurrent evolution of human immunodeficiency virus type 1 in patients infected from the same source: rate of sequence change and low frequency of inactivating mutations. J Virol. 1990;64:6221–6233. doi: 10.1128/jvi.64.12.6221-6233.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes W M. The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene. 1992;112:29–35. doi: 10.1016/0378-1119(92)90299-5. [DOI] [PubMed] [Google Scholar]
- 3.Burns D P, Desrosiers R C. Envelope sequence variation, neutralizing antibodies, and primate lentivirus persistence. Curr Top Microbiol Immunol. 1994;188:185–219. doi: 10.1007/978-3-642-78536-8_11. . (Review.) [DOI] [PubMed] [Google Scholar]
- 4.Burns D P W, Desrosiers R C. Selection of genetic variants of simian immunodeficiency virus in persistently infected rhesus monkeys. J Virol. 1991;65:1843–1854. doi: 10.1128/jvi.65.4.1843-1854.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke D K, Duarte E A, Moya A, Elena S F, Domingo E, Holland J. Genetic bottlenecks and population passages cause profound fitness differences in RNA viruses. J Virol. 1993;67:222–228. doi: 10.1128/jvi.67.1.222-228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cleland A, Watson H G, Robertson P, Ludlam C A, Leigh Brown A J. Evolution of zidovudine resistance-associated genotypes in human immunodeficiency virus type 1-infected patients. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12:6–18. doi: 10.1097/00042560-199605010-00002. [DOI] [PubMed] [Google Scholar]
- 7.Coffin J M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–488. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 8.Condra J H, Holder D J, Schleif W A, Blahy O M, Danovich R M, Gabryelski L J, Graham D J, Laird D, Quintero J C, Rhodes A, Robbins H L, Roth E, Shivaprakash M, Yang T, Chodakewitz J A, Deutsch P J, Leavitt R Y, Massari F E, Mellors J W, Squires K E, Steigbigel R T, Teppler H, Emini E A. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J Virol. 1996;70:8270–8276. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delwart E L, Sheppard H W, Walker B D, Goudsmit J, Mullins J I. Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J Virol. 1994;68:6672–6683. doi: 10.1128/jvi.68.10.6672-6683.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duarte E, Clarke D, Moya A, Dombingo E, Holland J. Rapid fitness losses in mammalian RNA virus clones due to Muller’s ratchet. Proc Natl Acad Sci USA. 1992;89:6015–6019. doi: 10.1073/pnas.89.13.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elena S F, Gonzalez-Candelas F, Novella I S, Duarte E A, Clarke D K, Domingo E, Holland J J, Moya A. Evolution of fitness in experimental populations of vesicular stomatitis virus. Genetics. 1996;142:673–679. doi: 10.1093/genetics/142.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–756. doi: 10.1093/genetics/78.2.737. . (Review.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher R A. On the dominance ratio. Proc R Soc Edinb. 1922;42:321–341. [Google Scholar]
- 14.Fisher R A. The genetical theory of natural selection. Oxford, United Kingdom: Clarendon Press; 1958. [Google Scholar]
- 15.Groenink M, Andeweg A C, Fouchier R A M, Broersen S, van der Jagt R C M, Schuitemaker H, de Goede R E Y, Bosch M L, Huisman H G, Tersmette M. Phenotype-associated env gene variation among eight related human immunodeficiency virus type 1 clones: evidence for in vivo recombination and determinants of cytotropism outside the V3 domain. J Virol. 1992;66:6175–6180. doi: 10.1128/jvi.66.10.6175-6180.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haase A T. Population biology of HIV-1 infection: viral and CD4+ T cell demographics in lymphatic tissues. Annu Rev Immunol. 1999;17:625–656. doi: 10.1146/annurev.immunol.17.1.625. [DOI] [PubMed] [Google Scholar]
- 17.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 18.Holmes E C, Zhang L Q, Simmonds P, Ludlam C A, Leigh Brown A J. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci USA. 1992;89:4835–4839. doi: 10.1073/pnas.89.11.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura M. Population genetics, molecular evolution, and the neutral theory. Selected papers. Chicago, Ill: The University of Chicago Press; 1994. [Google Scholar]
- 19a.Korber B, Kuiken C, Foley B, Hahn B, McCutchan F, Mellors J, Sodroski J, editors. Human retroviruses and AIDS, p. I-A-37–I-A-39. Los Alamos, N.Mex: Los Alamos National Laboratory; 1998. http://hiv-web.lanl.gov . [ http://hiv-web.lanl.gov.] .] [Google Scholar]
- 20.Kuiken C A, Swart G, Baan E, Coutinho R A, van den Hoek J A R, Goudsmit J. Increasing antigenic and genetic diversity of the V3 variable domain of the human immunodeficiency virus envelope protein in the course of the AIDS epidemic. Proc Natl Acad Sci USA. 1993;90:9061–9065. doi: 10.1073/pnas.90.19.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamers S L, Sleasman J W, She J X, Barrie K A, Pomeroy S M, Barrett D J, Goodenow M M. Independent variation and positive selection in env V1 and V2 domains within maternal-infant strains of human immunodeficiency virus type 1 in vivo. J Virol. 1993;67:3951–3960. doi: 10.1128/jvi.67.7.3951-3960.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lech W J, Wang G, Yang Y L, Chee Y, Dorman K, McCrae D, Lazzeroni L C, Erickson J W, Sinsheimer J S, Kaplan A H. In vivo sequence diversity of the protease of human immunodeficiency virus type 1: presence of protease inhibitor-resistant variants in untreated subjects. J Virol. 1996;70:2038–2043. doi: 10.1128/jvi.70.3.2038-2043.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S-L, Schaeker T, Musey L, Shriner D, McElrath M J, Corey L, Mullins J I. Divergent patterns of progression to AIDS after infection from the same source: human immunodeficiency virus type 1 evolution and antiviral responses. J Virol. 1997;71:4284–4295. doi: 10.1128/jvi.71.6.4284-4295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansky L M, Temin H M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayers D L, McCutchan F E, Sandersbuell E E, Merritt L I, Dilworth S, Fowler A K, Marks C A, Ruiz N M, Richman D D, Roberts C R, Burke D S. Characterization of HIV isolates arising after prolonged zidovudine therapy. J Acquir Immune Defic Syndr. 1992;5:749–759. [PubMed] [Google Scholar]
- 26.McCutchan F E, Artenstein A W, Sanders-Buell E, Salminen M O, Carr J K, Mascola J R, Yu X-F, Nelson K E, Khamboonruang C, Schmitt D, Kieny M P, McNeil J G, Burke D S. Diversity of the envelope glycoprotein among human immunodeficiency virus type 1 isolates of clade E from Asia and Africa. J Virol. 1996;70:3331–3338. doi: 10.1128/jvi.70.6.3331-3338.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miralles R, Moya A, Elena S F. Is group selection a factor modulating the virulence of RNA viruses? Genet Res. 1997;69:165–172. doi: 10.1017/s0016672397002735. [DOI] [PubMed] [Google Scholar]
- 28.Mohri H, Bonhoeffer S, Monard S, Perelson A S, Ho D D. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science. 1998;279:1223–1227. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- 29.Moore J P, Cao Y, Leu J, Qin L, Korber B, Ho D D. Inter- and intraclade neutralization of human immunodeficiency virus type 1: genetic clades do not correspond to neutralization serotypes but partially correspond to gp120 antigenic serotypes. J Virol. 1996;70:427–444. doi: 10.1128/jvi.70.1.427-444.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller H J. Some genetic aspects of sex. Am Nat. 1932;66:118. [Google Scholar]
- 31.Nájera I, Holguín A, Quiñones-Mateu M E, Muñoz-Fernández M, Nájera R, López-Galíndez C, Domingo E. pol gene quasispecies of human immunodeficiency virus: mutations associated with drug resistance in virus from patients undergoing no drug therapy. J Virol. 1995;69:23–31. doi: 10.1128/jvi.69.1.23-31.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pang S, Shlesinger Y, Daar E S, Moudgil T, Ho D D, Chen I S. Rapid generation of sequence variation during primary HIV-1 infection. AIDS. 1992;6:453–460. doi: 10.1097/00002030-199205000-00003. [DOI] [PubMed] [Google Scholar]
- 33.Pang S, Vinters H V, Akashi T, O’Brien W A, Chen I S Y. HIV-1 Env sequence variation in brain tissue of patients with AIDS-related neurological disease. J Acquired Immune Defic Syndr. 1991;4:1082–1092. [PubMed] [Google Scholar]
- 34.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. HIV-1 dynamics in vivo: virion clearance rate, infected cell lifespan, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 35.Pfutzner A, Dietrich U, von Eichel U, von Briesen H, Brede H D, Maniar J K, Rubsamen-Waigmann H. HIV-1 and HIV-2 infections in a high-risk population in Bombay, India: evidence for the spread of HIV-2 and presence of a divergent HIV-1 subtype. J Acquir Immune Defic Syndr. 1992;5:972–977. [PubMed] [Google Scholar]
- 36.Pieniazek D, Janini L M, Ramos A, Tanuri A, Schechter M, Peralta J M, Vicente A C, Pieniazek N K, Schochetman G, Rayfield M A. HIV-1 patients may harbor viruses of different phylogenetic subtypes: implications for the evolution of the HIV/AIDS pandemic. Emerg Infect Dis. 1995;1:86–88. doi: 10.3201/eid0103.950303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reinhart T A, Rogan M J, Amedee A M, Murphey-Corb M, Rausch D M, Eiden L E, Haase A T. Tracking members of the simian immunodeficiency virus deltaB670 quasispecies population in vivo at single-cell resolution. J Virol. 1998;72:113–120. doi: 10.1128/jvi.72.1.113-120.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouzine I M, Coffin J M. Interplay between experiment and theory in development of a working model for HIV-1 population dynamics. In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. London, United Kingdom: Academic Press Ltd.; 1999. pp. 225–262. [Google Scholar]
- 39.Rouzine, I. M., and J. M. Coffin. Linkage disequilibrium test implies large effective population of HIV in vivo. Proc. Natl. Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed]
- 40.Sabino E C, Shpaer E G, Morgado M G, Korber B T M, Diaz R S, Bongertz V, Cavalcante S, Galvão-Castro B, Mullins J I, Mayer A. Identification of human immunodeficiency virus type 1 envelope genes recombinant between subtypes B and F in two epidemiologically linked individuals from Brazil. J Virol. 1994;68:6340–6346. doi: 10.1128/jvi.68.10.6340-6346.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmonds P, Balfe P, Ludlam C A, Bishop J O, Leigh Brown A J. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J Virol. 1990;64:5840–5850. doi: 10.1128/jvi.64.12.5840-5850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith M W, Dean M, Carrington M, Winkler C, Huttley G A, Lomb D A, Goedert J J, O’Brien T R, Jacobson L P, Kaslow R, Buchbinder S, Vittinghoff E, Vlahov D, Hoots K, Hilgartner M W, O’Brien S J. Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- 43.Steinhauer D A, de la Torre J C, Meier E, Holland J J. Extreme heterogeneity in populations of vesicular stomatitis virus. J Virol. 1989;63:2072–2080. doi: 10.1128/jvi.63.5.2072-2080.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tindall K R, Kunkel T A. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry. 1988;27:6008–6013. doi: 10.1021/bi00416a027. [DOI] [PubMed] [Google Scholar]
- 45.Wain-Hobson S. The fastest genome evolution ever described: HIV variation in situ. Curr Opin Genet Dev. 1993;3:878–883. doi: 10.1016/0959-437x(93)90008-d. . (Review.) [DOI] [PubMed] [Google Scholar]
- 46.Wei X, Ghosh S, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, Saag M S, Shaw G M. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 47.Wolfs T F W, de Jong J-J, van den Berg H, Tijnagel J M G H, Drone W J A, Goudsmit J. Evolution of sequences encoding the principal neutralization epitope of human immunodeficiency virus 1 is host dependent, rapid, and continuous. Proc Natl Acad Sci USA. 1990;87:9938–9942. doi: 10.1073/pnas.87.24.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolfs T F W, Zwart G, Bakker M, Valk M, Kuiken C L, Gousmit J. Naturally occurring mutations within HIV-1 V3 genomic RNA lead to antigenic variation dependent on a single amino acid substitution. Virology. 1991;185:195–205. doi: 10.1016/0042-6822(91)90767-6. [DOI] [PubMed] [Google Scholar]
- 49.Wolinsky S M, Korber B T M, Neumann A U, Daniels M, Kunstman K J, Whetsell A J, Furtado M R, Cao Y, Ho D D, Safrit J T, Koup R A. Adaptive evolution of human immunodeficiency virus type 1 during the natural course of infection. Science. 1996;272:537–542. doi: 10.1126/science.272.5261.537. [DOI] [PubMed] [Google Scholar]
- 50.Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wyand M S, Manson K H, Garcia-Moll M, Montefiori D, Desrosiers R C. Vaccine protection by a triple deletion mutant of simian immunodeficiency virus. J Virol. 1996;70:3724–3733. doi: 10.1128/jvi.70.6.3724-3733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshimura F K, Diem K, Learn G H, Jr, Riddell S, Corey L. Intrapatient sequence variation of the gag gene of human immunodeficiency virus type 1 plasma virions. J Virol. 1996;70:8879–8887. doi: 10.1128/jvi.70.12.8879-8887.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L Q, MacKenzie P, Cleland A, Holmes E C, Leigh Brown A J, Simmonds P. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol. 1993;67:3345–3356. doi: 10.1128/jvi.67.6.3345-3356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]