

Abstract
Unleashing anti-tumor T cell activity by checkpoint inhibitor immunotherapy is effective in cancer patients but clinical responses are limited. Cytokine signaling through the JAK/STAT pathway correlates with checkpoint immunotherapy resistance. We report a phase I clinical trial of the JAK inhibitor ruxolitinib with anti-PD-1 antibody nivolumab in Hodgkin lymphoma patients relapsed or refractory following checkpoint inhibitor immunotherapy. The combination yielded a best overall response rate of 53% (10/19). Ruxolitinib significantly reduced neutrophil-to-lymphocyte ratios and percentages of myeloid suppressor cells but increased numbers of cytokine-producing T cells. Ruxolitinib rescued the function of exhausted T cells and enhanced the efficacy of immune checkpoint blockade in pre-clinical solid tumor and lymphoma models. This synergy was characterized by a switch from suppressive to immunostimulatory myeloid cells which enhanced T cell division.
One-Sentence Summary:
Ruxolitinib reshapes myeloid immunity to synergize with checkpoint inhibitor immunotherapy in Hodgkin lymphoma patients.
Immune control of cancer and response to immunotherapy are hampered by diverse, partially redundant, adaptive immunosuppressive effects mediated by both cancer and non-cancer cells. Suppressive myeloid cells are present in many tumor types, cause lymphocyte dysfunction and poor response to checkpoint inhibitor immunotherapy (1, 2). The pan immune suppressive state generated during persistent infection closely resembles that observed in cancer (3, 4) including T cell exhaustion and suppressive myeloid cells shared between cancer and persistent viral infection, which led to fundamental discoveries such as T cell reactivation by PD-1 blockade in the lymphocytic choriomeningitis virus (LCMV) Clone 13 model (5–7).
Targeting of myeloid derived suppressor cells (MDSCs) and tumor associated macrophages enhances response to checkpoint inhibitors in cancer models. Interestingly, suppressive activity may be associated with stimulus-dependent states in all myeloid lineages (8, 9). The upstream stimuli that drive suppressive programming of myeloid cells offer a promising therapeutic opportunity. Multiple signal transducer of activators of transcription (STAT) transcription factors and the JAK1/2-dependent cytokines G-CSF, GM-CSF and IL-6 are implicated in suppressive programming of MDSCs (9–11). Therapeutics specifically targeting these cells have yet to gain clinical approval (12) and conflicting effects of Janus kinase (JAK) inhibitors on MDSCs have been reported (13–15).
The JAK/STAT pathway plays a central role in activating transcriptional programs in responses to dozens of soluble mediators including cytokines, growth factors and interferons (16). Several small molecule JAK inhibitors have been approved in the clinic and are predominantly used to treat myeloproliferative and autoimmune diseases (17). Despite known genetic links between JAK mutations and cancer, mainstream use of JAK inhibitors for cancer treatment has been impeded by JAK inhibitors’ immune suppressive properties. Here we demonstrate that rather than suppress essential anti-tumor immunity, small molecule JAK inhibition synergizes with checkpoint blockade immunotherapy to reshape the myeloid cell compartment and enhance NK and T cell responses.
Results
JAK inhibitors rescue function of exhausted T cells
We developed a screening strategy to discover small molecules that rescue T cell exhaustion utilizing the LCMV Clone 13 (Cl-13) model of immune suppression (18). Having screened the ReFrame collection of ~12,000 biologically active compounds using the assay, we tested if specific biomolecular targets were overrepresented in the set of hit compounds. Using annotated compound-gene interactions curated by the Drug-Gene Interaction Database (19), we found an enrichment of JAK inhibitors (JAKi) among the hits (Fig. S1A–C). These inhibitor compounds span multiple chemotypes, arguing against a common off-target (Fig. S1D). The top 5 JAKi by z-score were re-obtained and their activity re-tested at a range of concentrations (Fig. S1E). Although all compounds showed some activity in the validation assay, only BMS911543 and ruxolitinib could match or surpass the effect of aPDL1 (Fig. S1E). We further validated the activity of ruxolitinib by assessing IFN-γ protein using intracellular staining in place of the YFP reporter in the Cl13 based assay (Fig. 1A, S1F). Testing commercially available kinase inhibitors revealed that compounds inhibiting JAK1 and/or JAK2 were among the most potent in increasing the frequency of YFP+ CD8 T cells (Fig. S1G–J).
Fig. 1. JAK1/2 inhibitors rescue proliferation of cytokine-producing CD8 T cells without impairing viral control.
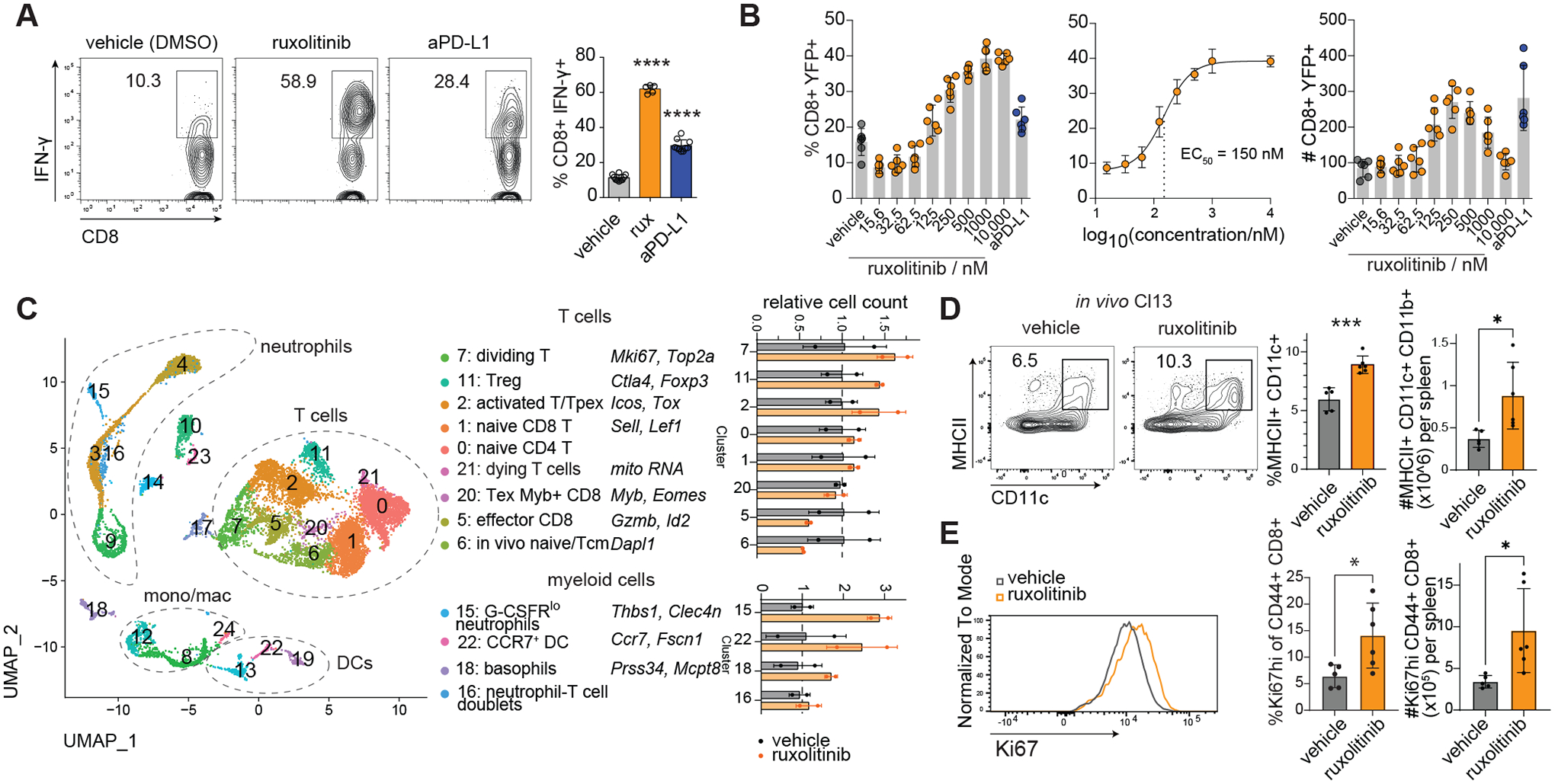
(A-C) In vitro Cl13: splenocytes from LCMV Cl13 infected IFNγ-IRES-YFP mice were harvested at 15 days post infection and cultured in the presence of LCMV peptides for 5 days (more details see Materials and Methods). (A) Validation assay: effect of ruxolitinib and anti-PD-L1 treatment on endogenous IFN-γ labeled by intracellular staining, splenocyte culture conditions equivalent to primary assay with B6 mice substituted for YFP-IFN-γ mice. (B) Dose response and EC50 calculation of ruxolitinib in enhancing %YFP+ of CD8 T cells in 5-day culture assay. (C) CITE-seq of splenocyte cultures treated with ruxolitinib or vehicle, cells analyzed at d0 and d4 of treatment, dot plot shows relative number of cells in each cluster in ruxolitinib vs vehicle treated wells, taking into account the total number of cells recovered. (D-E) In vivo Cl13: B6 mice were infected with Cl13 and treated with vehicle or ruxolitinib by daily gavage: (D) Flow cytometric assessment of splenic DCs, (E) splenic CD44+ CD8 T cells from Cl13 infected mice treated with ruxolitinib or vehicle at 10 dpi. For myeloid clusters, only clusters with >=10 cells per sample in the ruxolitinib group were quantified. Experiments were performed once (C) or 2–4 times (A-B, D-E) and representative results shown. Bars show standard deviation. Statistical comparison of experimental groups was performed using one-way ANOVA with Dunnett’s post test (A), unpaired Student’s t-test (D-E).; Treg, T regulatory cell; DC, dendritic cell; Tcm, T central memory cell; Tex, T exhausted cell; Tpex, T progenitor exhausted cell; *, p ≤ 0.05; ***, p ≤ 0.001; ****, p ≤ 0.0001.
We focused on ruxolitinib, the first JAKi to receive clinical approval (20, 21) and currently in clinical use for indications including myelofibrosis and graft vs host disease (20, 21). Compound titration revealed a sigmoidal dose response to ruxolitinib in the frequency of YFP-IFN-γ+ CD8 T cells (EC50 ~ 150 nM) (Fig. 1B), the number of YFP-IFN-γ+ CD8 T cells showing a bell shape dose response peaking around 250 nM (Fig. 1B). Ruxolitinib was superior to anti-PD-L1 in enhancing the percentage and total number of IFN-γ-producing CD8 T cells as detected by intracellular cytokine staining (Fig. 1B) as well as increasing IFN-γ protein levels in splenocyte cultures (Fig. S2A). The most dramatically reduced cytokine in the culture supernatant was the myeloid chemokine CCL2, suggesting ruxolitinib treatment more broadly impacts the immune compartment (Fig. S2B), although no differences in the intracellular content of these cytokines were detected in the ruxolitinib vs vehicle treated groups (Fig. S2C). To better understand the changes in non-T cells, we performed a comprehensive profiling of cells in this assay by CITE-seq (22). Comparison of CD45+ splenocytes with ruxolitinib or vehicle indicated that T cells increased but most myeloid populations decreased overall by cluster share by day 4 of culture (Fig. 1C, S2D–E). Ruxolitinib increased the percentage and total number of dividing T cells but also caused a ~2-fold increase in the percentage of CD11b− dendritic cells (Fig. 1C, S2F). Further, the cytokines increased by ruxolitinib in the culture supernatant such as GM-CSF, CCL3 and CCL4 were most highly expressed at the mRNA level in clusters enhanced by ruxolitinib, namely dividing T cells and 2 myeloid clusters (Fig. S2G).
We tested whether ruxolitinib compromised effector functions of CD8 T cells following viral infection in vivo. Mice infected with Cl13 and receiving daily ruxolitinib exhibited significantly increased total numbers of GP276-specific splenic CD8 T cells (Fig. S3A) and of IFN-γ-producing GP276-reactive CD8 T cells than vehicle-treated controls (Fig. S3B). Further examination of splenocytes from these mice by CITE-seq and flow cytometry revealed that ruxolitinib increased the percentage and total number of dendritic cells (Fig. 1D, S3C–D) (23). Plasma levels of G-CSF were reduced at d5 post infection (Fig. S3E). Additionally, GP33-specific P14 CD8 T cells isolated from Cl-13 infected mice showed reduced proliferation when stimulated with anti-CD3/CD28 in the presence of ruxolitinib, suggesting that ruxolitinib’s capacity to induce expansion of LCMV-specific T cell expansion in a mixed culture system involves other cellular targets (Fig. S3F). However, viral loads in plasma and organs were unaffected by ruxolitinib treatment at all timepoints examined, suggesting antiviral T cell responses are sufficiently preserved following ruxolitinib treatment early during persistent LCMV infection (Fig. S4A–B); in contrast, treatment with IFNAR-blocking antibody (anti-IFNAR) before Cl13 infection raised viral titers significantly and equivalent percentages of infected splenocytes were seen in ruxolitinib and vehicle in contrast to anti-IFNAR1 treated mice, suggesting that ruxolitinib does not fully inhibit IFNAR signaling (Fig. S4B–C)(24, 25). Treatment of ruxolitinib also increased the percentage and total number of proliferative CD8, CD4 T and NK cells as determined by Ki67 expression (Fig. 1E, S4D). We wondered whether the increase in T cells could be partly attributed to reduced cell death. GP33-specific transgenic CD8 T cells (P14) were adoptively transferred to congenic hosts, infected with Cl13 and treated with ruxolitinib or vehicle, then splenic P14 cells analyzed. The percentage of apoptotic P14 cells was reduced more than 3-fold compared to vehicle treated mice (Fig. S4E). We also observed increased levels of the anti-apoptotic protein Bcl-XL in CD8 T cells (Fig. S4F).
Ruxolitinib enhances lymphocyte function and efficacy of checkpoint blockade immunotherapy in pre-clinical cancer models
We hypothesized that the T cell enhancing effects of ruxolitinib in a persistent viral infection model may be beneficial in enhancing checkpoint blockade immunotherapy in cancer. Mice received MC38 tumor cells and were treated with suboptimal doses of anti-PD-1+anti-CTLA4 immune checkpoint inhibitors (ICI) or controls as described previously (26) and also received ruxolitinib or vehicle starting 2 days after the first ICI dose (Fig. 2A). In the absence of immunotherapy, ruxolitinib had a marginal effect on tumor growth, however ruxolitinib in combination with ICI (ruxolitinib+ICI) significantly reduced tumor growth compared to ICI alone; once daily 30 mg/kg ruxolitinib was the most efficacious dose (Fig. 2A, S5A). Similar results were observed in the B cell lymphoma model A20 and the lung cancer model LLC1 (Fig. 2B, Fig. S5B–C). Although it has been shown that ruxolitinib can sensitize anti-PD-1-resistant melanoma cells to anti-CTLA4 treatment (27), the MC38 and A20 cell lines were not pre-treated with checkpoint inhibitors and do not exhibit prior resistance to anti-PD-1.
Fig. 2. Ruxolitinib enhances the efficacy of checkpoint blockade cancer immunotherapy.
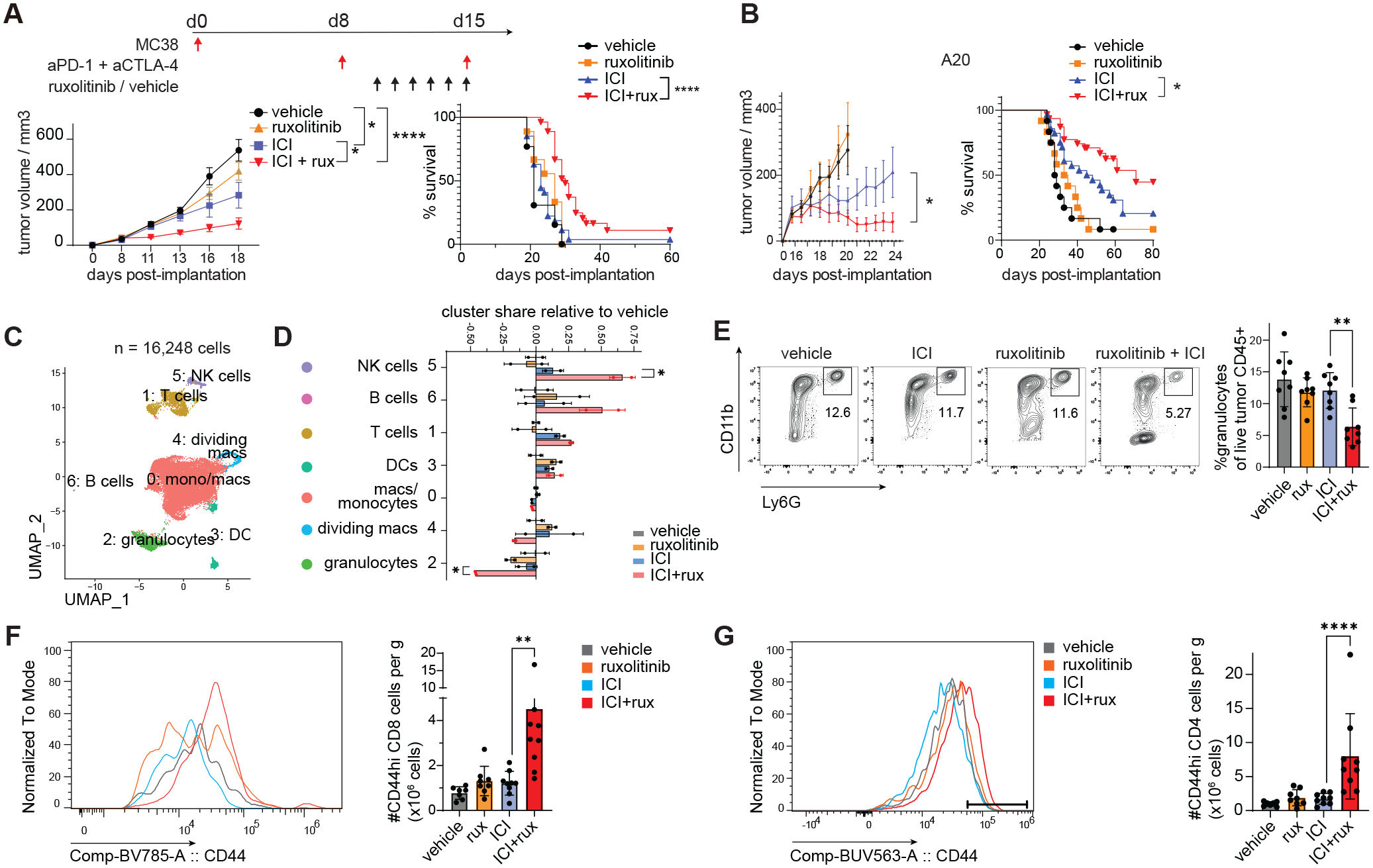
(A, C-G) Mice were implanted with MC38 tumor cells and treated as described in the experimental scheme in 4 experimental groups, tumor measurements were performed every 2–3 days after tumors became palpable. (B) Wild type BALB/c mice implanted with A20 tumor cells were treated with ICI or isotype control when palpable and ruxolitinib or vehicle daily starting 2 days after the first ICI injection. (C-D) Dimensionality reduced map of MC38 tumor-infiltrating cells, treatment groups as described in A; bar graph shows the share of sample cells in each cluster relative to the mean share in vehicle treated samples. Clusters were color-coded as C0 = red, C1 = brown, C2 = green, C3 = turquoise, C4 = sky blue, C5 = purple, C6 = soft maroon. (E-G) Tumor-infiltrating CD45+ cells in mice treated as in 2A were analyzed by flow cytometry at d18 post implantation; (E) flow cytometric quantification of granulocytes. (F-G) Flow cytometric quantification of CD44hi CD8 T (F) and CD4 T cells (G). Statistical comparison of experimental groups was performed by two way-ANOVA (A-B), one-way ANOVA and Sidak’s post-test (E-G), two-tailed Student’s t-test (D). Experiments were performed 2–5 times and pooled (A-B, survival analysis) or representative results shown (C-G). Treatment groups were color-coded as vehicle = grey, ruxolitinib = orange, ICI = blue, ICI+ruxuxolitinib = red. Bars represent standard deviation (D-G) or s.e.m (A-B); anti-PD-1, anti-PD-1; ICI, immune checkpoint blockade; NK, natural killer cell; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
To understand the basis of this beneficial effect we isolated MC38 tumor-infiltrating cells from anti-PD-1+anti-CTLA4 treated mice and analyzed single-cell transcriptomes and protein markers using CITE-seq (22). Low-resolution clustering identified a prominent cluster of monocytes, macrophages and monocytic dendritic cells, and clusters of T cells, granulocytes, dendritic cells, dividing macrophages, NK cells and B cells (Fig. 2C, S5D–E). Mice receiving ruxolitinib with ICI showed a significantly higher percentage of lymphocytes and a lower percentage of granulocytes of CD45+ cells compared to ICI alone, effects validated by flow cytometry and immunohistochemistry (Fig. 2D–E, S5F). Tumors of mice treated with ICI+ruxolitinib contained significantly higher numbers of CD44-high T and NK cells (Fig. 2F–G, S5G). There was a significant increase in the percentage and total number of dividing Ki67+ NK cells in ICI+ruxolitinib vs ICI treated mice (Fig. S5H). Functionally, we observed significantly increased degranulation of T cells comparing ICI+ruxolitinib vs ICI treatment, suggesting ruxolitinib may increase their degranulation capacity (Fig. S5I).
We depleted CD8 T cells or NK cells immediately prior to treatment with ICI, ruxolitinib or combination therapy. As expected, depletion of CD8 T cells in ICI treated mice accelerated tumor growth and the addition of ruxolitinib to ICI did not improve tumor control in depleted mice (Fig. S6A–B). NK cell depletion in ICI+ruxolitinib treated mice eliminated the therapeutic benefit with respect to ICI alone (Fig. S6C–E), suggesting both CD8 T cells and NK cells are required for the efficacy of ICI+ruxolitinib.
Ruxolitinib causes reprogramming of granulocytic cells
We further investigated the modulation of tumor-infiltrating myeloid cells by ICI+ruxolitinib. Granulocytes exhibited significant reductions per gram of tumor in ICI+ruxolitinib vs ICI treated mice (Fig. S7A). Ruxolitinib also reduced the percentage of splenic neutrophils and the neutrophil-to-lymphocyte ratio in LCMV infected mice, indicating the effect of ruxolitinib on neutrophils is not exclusive to cancer (Fig. S7B–C). Granulocytic cells include a population of polymorphonuclear myeloid derived suppressor cells (PMN-MDSCs) which share surface markers with neutrophils (28). To assess the contribution of granulocytic myeloid cells to ruxolitinib’s enhancement of ICI efficacy, Ly6G+ cells were depleted using an anti-Ly6G depleting antibody (aLy6G) prior to ICI, ruxolitinib or ICI+ruxolitinib treatment and depletion was maintained throughout the treatment period. Depletion of Ly6G+ cells in ICI-treated mice significantly reduced tumor growth and weight (Fig. 3A, S7D–E). Tumor-infiltrating myeloid cells were subjected to a suppression assay: Gr1+ cells were isolated from MC38 tumor bearing mice and tested for their ability to suppress IL-2-driven proliferation and IFN-g production of NK cells. As expected, increasing doses of Gr1+ cells reduced the percentage and total number of IFNg+ NK cells, confirming their suppressive abilities (Fig. S7F). Consistently, splenic granulocytes expressed arginase in MC38 tumor-bearing mice and ruxolitinib nearly ablated arginase 1 (ARG1) protein levels in granulocytes both when administered alone or in conjunction with ICI, suggesting ruxolitinib not only modulates granulocyte numbers but also expression of suppressive markers (Fig. S7G). Tumor-infiltrating granulocytes in ICI+ruxolitinib vs ICI treated mice showed significantly reduced expression of MDSC-associated markers Csf1, Cxcl2, Ifitm1, Il1b, S100a8, S100a9 and Wfdc17 (Fig. 3B, S7H) and exhibited higher expression of MHC class II molecules and Cd74 in ICI+ruxolitinib treated mice, suggesting they acquired transcriptomic features of immune stimulatory antigen presenting cells (Fig. 3B–C, S7H). Moreover, the induction of MHC-II expression in granulocytes was also observed in ruxolitinib-treated mice infected with Cl13 (Fig. S7I). We performed an in vitro T cell proliferation assay using splenic granulocytes from MC38 tumor bearing mice treated with vehicle, ruxolitinib, ICI or ICI+ruxolitinib. Remarkably, granulocytes from ruxolitinib and ICI+ruxolitinib treated mice enhanced T cell division and the total number of T cells over 3 days of stimulation with anti-CD3/CD28 to a significantly greater extent than vehicle or ICI derived granulocytes, respectively (Fig. 3D, S7J).
Fig. 3. Ruxolitinib reprograms tumor-infiltrating myeloid cells to enhance lymphocyte proliferation.
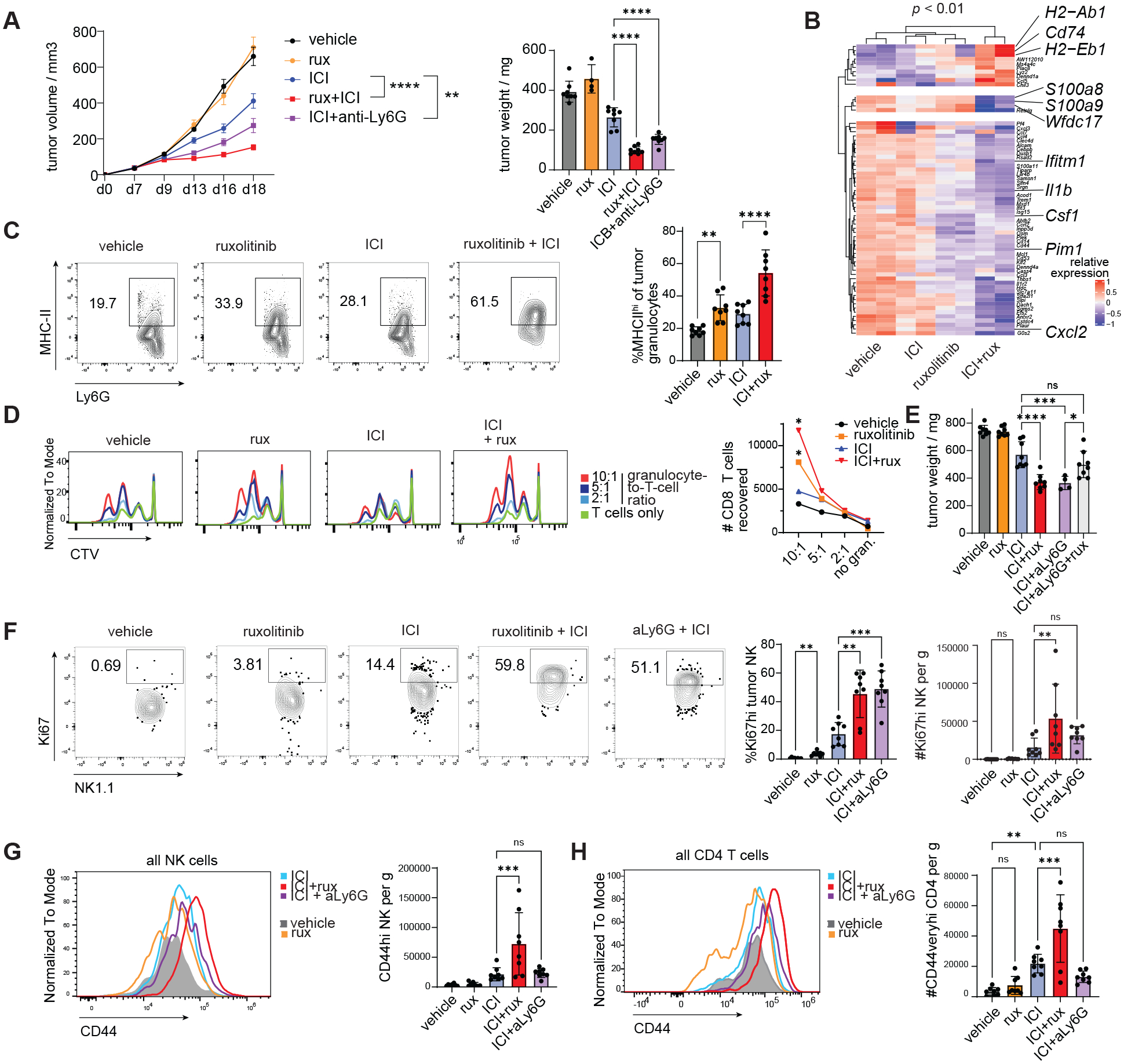
(A) MC38 tumor bearing mice were treated with Ly6G-depleting antibody (aLy6G) or isotype control 2 days prior to treatment with ICI/isotype and ruxolitinib/vehicle, then every 2 days until end of ruxolitinib/vehicle treatment, tumor volume measured every 2–4 days (A) and tumors analyzed at d18 post implantation. (B) Genes differentially expressed between ICI+ruxolitinib treated and ICI treated mice in tumor-infiltrating granulocytes analyzed by scRNAseq. (C) Flow cytometry analysis of tumor-infiltrating granulocytes at d18 post implantation, treatment groups as described in Figure 2A. (D) Splenic granulocytes (CD11b+ Ly6Ghi Ly6Cint) from mice treated as described in Fig. 2A were sorted and mixed with purified, CTV-labeled T cells at the ratios indicated and cell mixtures subjected to anti-CD3/CD28 stimulation, flow cytometry analysis at d3 of assay. (E) MC38 tumor bearing mice were treated as described in Figure 3A with the additional experimental group treated with anti-Ly6G + ICI + ruxolitinib, tumors analyzed 2d after treatment cessation. (F-H) MC38 tumor bearing mice were treated as in Figure 3A and tumor-infiltrating cells analyzed 2d after treatment cessation. Experiments were performed 2–3 times with 6–8 mice per group and representative results shown; scRNAseq was performed once with 2 mice per group. Bars show s.e.m. (A) or standard deviation (all other panels). Statistical comparison of treatment groups was performed using two-way ANOVA (A left panel), one-way ANOVA with Sidak’s post-test (A right panel, C, E-H), or DESeq2 Wald test (B); ICI, immune checkpoint blockade; CTV, cell trace violet; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
To determine the contribution of these granulocytes to the synergy between ICI and ruxolitinib, we depleted Ly6G+ cells during ICI/ICI+ruxolitinib treatment and assessed tumor growth. Treatment with anti-Ly6G (aLy6G) alone had no effect on tumor growth (Fig. S7K). Whereas ruxolitinib+ICI and aLy6G+ICI groups showed significantly reduced tumor weight compared to ICI alone, ruxolitinib provided no additional benefit to aLy6G+ICI treated mice compared to ruxolitinib+ICI or aLy6G+ICI groups (Fig. 3E).
To identify which antitumor immune responses were invigorated by granulocyte depletion or by ruxolitinib treatment, tumor-infiltrating cells were analyzed by flow cytometry after treatment cessation. The effect of aLy6G+ICI on Ki67 NK cells phenocopied the effect of ruxolitinib+ICI with a >2 fold increase in the percentage of Ki67+ NK cells in these groups compared to ICI alone (Fig. 3F). In contrast, the increased expression of CD44 on NK and T cells was specific to the ICI+ruxolinitinib group, suggesting ruxolitinib has granulocyte-independent effects (Fig. 3G–H). The expression levels of CD44 on CD8 T cells was also enhanced in ICI+ruxolitinib compared to other groups but the total number of CD44hi CD8 T cells was not significantly increased by ICI+ruxolitinib or ICI+aLy6G compared to ICI (Fig. S7L). Treatment with ICI+ruxolitinib+aLy6G resulted in no increase in the number of Ki67+ NK cells and CD44hi CD4 T cells per g of tumor compared to ICI alone, consistent with the poorer tumor control observed (Fig. 3E, S7M).
Ruxolitinib reduces suppressive gene expression in myeloid cells of the bone marrow, blood and tumor
We asked whether the effect of ruxolitinib on tumor-infiltrating myeloid cells had conserved patterns across different tumor types. CITE-seq data of tumor-infiltrating myeloid cells from 3 different mouse tumor types treated with ICI+ruxolitinib or ICI were integrated and subjected to clustering. High diversity was found in the monocytic/macrophage cluster whereas only one granulocyte cluster emerged at that resolution (Fig. 4A–B, S8A). The percentage of myeloid cells in Clusters 0 (ARG1+ mac/mono cells) and 3 (granulocytic cluster) was reduced in ICI+ruxolitinib compared to ICI treated mice in all three tumor models (Fig. 4C, S8B), as was the loss of MDSC markers and gain of MHC gene expression in granulocytes (Fig. S8C). Cells in Cluster 2, expressing markers of monocyte derived dendritic cells (moDC), increased in frequency in all 3 tumor types and the ISG-expressing moDC Cluster 8 increased in the LLC1 and MC38 models but not EL4, consistent with a reduction in IFN-γ signaling and comparable tumor growth in the ICI and ICI+ruxolitinib groups in this model (Fig. 4C, S8D–E). To further understand the phenotypic characteristics of these clusters, we compared markers of human monocytes and macrophages in the MoMac-VERSE (29). Cluster 0 expressed multiple markers such as Spp1, Fabp5 and Apoe which corresponded to the TREM2+ macrophage #3 cluster of the MoMac-VERSE, the cluster most significantly and consistently increased in human cancers compared to non-diseased tissue of all MoMac-VERSE clusters (Fig. 4C, S8F) (29). Furthermore, Cluster 0 and 3 expressed the highest levels of MDSC markers of all myeloid clusters (Fig. S8G). The increase of MHC-II-expressing Ly6Chi monocytic cells was also observed by flow cytometry (Fig. S8H). Flow cytometry also confirmed a switch from TREM2+ MHC− macrophages to TREM2− MHC+ in ICI+ruxolitinib compared to ICI (Fig. S8I). Furthermore, ruxolitinib treatment also increased MHC-II expression and reduced S100a8 expression in splenic monocytes, and switch from TREM2+ MHC− to TREM2− MHC+ macrophages in the persistent viral infection model (Fig. S8J–K). Reductions in CD163+ Ly6Chi cells were observed in the blood of A20 and MC38 tumor bearing ICI+ruxolitinib treated mice and blood CD163+ cells correlated with tumor weight (Fig. S8L–N). These data reveal a broad ruxolitinib-induced shift from suppressive gene expression to MHC expression in granulocytic, monocytic and macrophage populations.
Fig. 4. Systemic myeloid reprogramming by ruxolitinib results in blocking G-CSF.
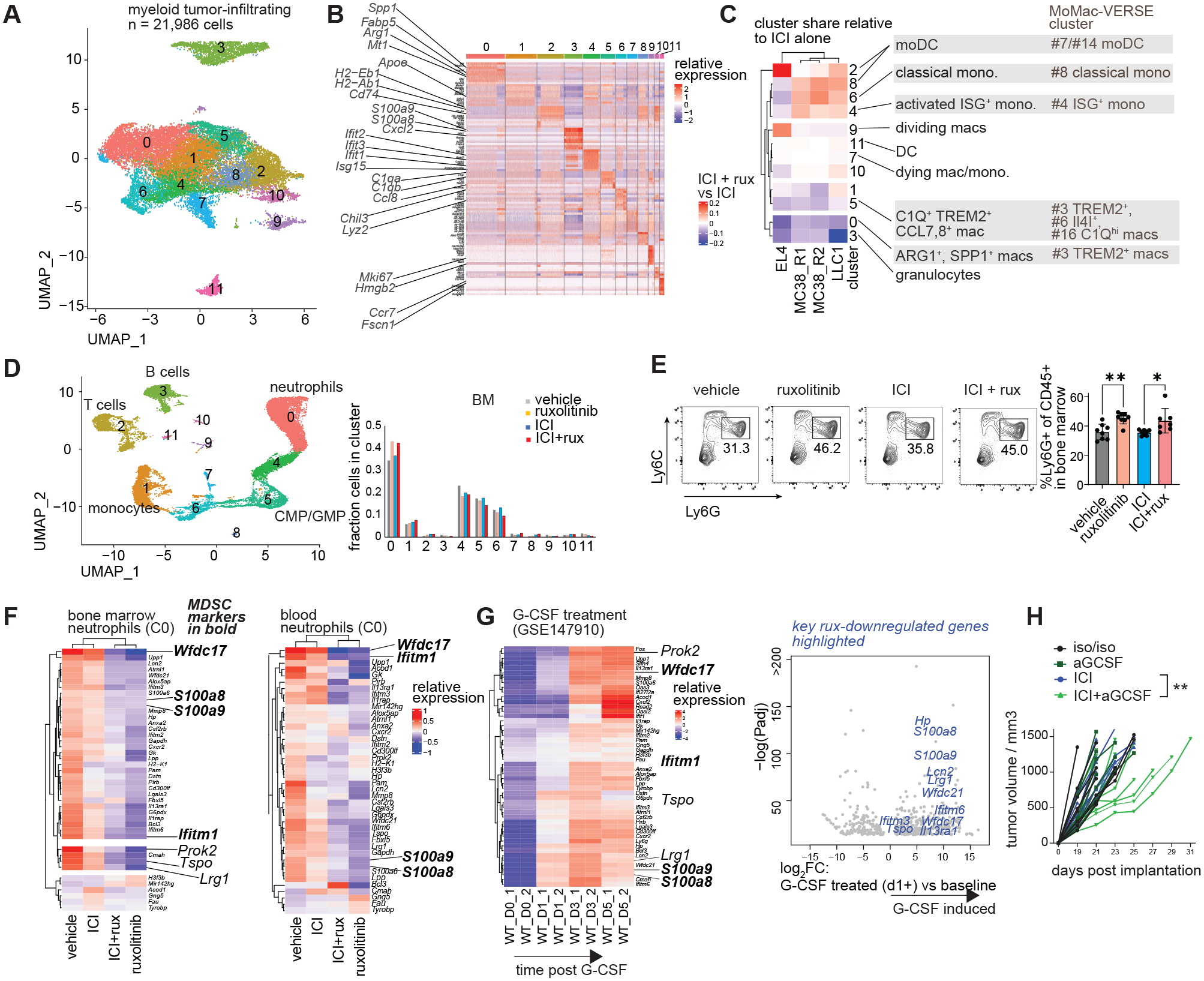
(A-C) Integrated CITE-seq analysis of tumor-infiltrating myeloid cells from EL4, LLC1 and MC38 tumors: (A) dimensionality-reduced plot and clustering of integrated dataset; (B) mRNA markers of 11 clusters. (C) Difference in relative cluster share between ICI+ruxolitinib treated mice and respective ICI treated controls, right: MoMac-VERSE (29) clusters most closely aligned with the observed clusters based on marker genes. (D-F) Bone marrow and blood from mice treated as in Figure 2A were analyzed by CITE-seq and flow cytometry: (D) dimensionality reduced map and clustering of combined dataset. (E) Flow cytometric analysis of bone marrow granulocytes; (F) relative expression of differentially expressed genes between ruxolitinib treated and vehicle treated mice in cluster 0 (differentiated neutrophils), MDSC markers shown in bold italic. (G) Expression of ruxolitinib-regulated genes in myeloid cells cultured in the presence of G-CSF for 0–5 days (GSE147910), MDSC markers in bold italic, right: volcano plot of G-CSF regulated genes at d1-5 vs d0, ruxolitinib-inhibited genes highlighted in blue. (H) MC38 tumor bearing mice were treated with low-dose G-CSF neutralizing antibody or isotype control every 3 days once palpable and ICI or isotype control once palpable and 7 days later, tumor size measured every 2 days; DEG, differentially expressed gene; moDC, monocyte-derived dendritic cell, mac, macrophage; ICI, immune checkpoint blockade. CITE-seq experiments were performed once and integrated data shown; experiments depicted in panels E and H were performed 2–3 times and representative results shown. Bars show standard deviation (E). Statistical comparison of experimental groups was performed using one-way ANOVA with Sidak’s post-test (E), DESeq2 Wald test (F-G) or two-way ANOVA (H); *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Suppressive activity in myeloid cells can be acquired due to excessive stimulation by soluble factors including the JAK-signaling cytokines G-CSF and GM-CSF (9, 30, 31). We asked whether ruxolitinib interfered with the suppressive programming of myeloid cells in the tumor and/or during their development in the bone marrow. Tumor bearing mice were treated as described in Fig. 2A and their peripheral blood and bone marrow CD45+ cells were analyzed by single-cell transcriptome sequencing (Fig. 4D, S9A–B). Blood monocytes from ICI+ruxolitinib treated mice express lower levels of suppressive markers Arg2, S100a8 and S100a9 (Fig. S9C). There was a significant increase in the percentage of differentiated neutrophils in the bone marrow of ruxolitinib and ICI+ruxolitinib treated mice compared to respective controls, validated by flow cytometry, whereas immature neutrophils were underrepresented in these mice based on Neutrotime gene classification (Fig. 4D–E, S9D) (32). Previously identified MDSC markers including Wfdc17, S100a8, S100a9 and Ifitm1 appeared downregulated in the neutrophils of ruxolitinib and ICI+ruxolitinib treated mice (Fig. 4F) (9, 33). These MDSC markers were reduced in both bone marrow and blood neutrophils. There was a dramatic reduction in the expression of myeloid migration genes Lrg1, Prok2 and Tspo in neutrophils of ruxolitinib treated mice (Fig. 4F) (34–37). Ruxolitinib did not significantly reduce the transcriptomic program associated with normal neutrophil differentiation as defined by high Neutrotime score, suggesting ruxolitinib does not prevent neutrophil differentiation (Fig. S9E) (32).
To identify upstream signals targeted by ruxolitinib which lead to the observed gene expression changes, we analyzed the predicted ligand activity in BM neutrophils using Cytosig (Fig. S10A) (38). Several MDSC related ligands showed dominant activity: EGF, IL-17A, IL-36, NO, G-CSF and TRAIL. Of these, only G-CSF uses canonical JAK1 or JAK2 signaling. To determine whether G-CSF activity played a role in inducing ruxolitinib-downregulated genes, the temporal effect of G-CSF on myeloid cell transcriptomes was analyzed. Strikingly, virtually all ruxolitinib-downregulated genes were induced by G-CSF, including genes such as Wfdc21, Wfdc17 and Ifitm6 which were among the most highly induced genes by G-CSF based on fold change and p value (Fig. 4G). Plasma levels of G-CSF were not significantly affected post-ICI+ruxolitinib (Fig. S10B). We treated MC38 tumor-bearing mice with the G-CSF neutralizing antibody (aGCSF) with or without ICI. aGCSF treatment reduced the expression of multiple G-CSF targets including Wfdc17 in bone marrow neutrophils (Fig. S10C). The combination of ICI and aGCSF significantly delayed tumor growth, consistent with the notion that G-CSF exerts immune suppressive effects in this tumor model (Fig. 4H, S10D). As expected, GCSF blockade reduced the percentage of tumor-infiltrating granulocytes and caused other cellular changes partially resembling the effects of ICI+rux (Fig. S10E).
Given that ruxolitinib has been reported to modulate melanoma cell-intrinsic resistance to anti-PD-1 (27), we wondered whether cancer cell-intrinsic JAK1 or JAK2 were required for the effects observed above. To address this, JAK1 and JAK2 deficient MC38 cells generated previously (39) were injected in B6 hosts and treated as described in Fig. 2A, then tumor infiltrating cells analyzed by flow cytometry. Although growth of Jak1−/− MC38 was impaired in vivo as previously reported (Fig. S11A) (39), we observed an increase in the percentage of monocyte-derived dendritic cells in tumors of ICI+ruxolitinib vs ICI treated mice by single-cell transcriptomics (Fig. S11B). Monocytes also expressed higher transcript levels of MHC-II in ICI+ruxolitinib vs ICI treated tumors (Fig. S11C–D). Similar to wt MC38, tumor-infiltrating macrophages in Jak1−/− MC38 tumors exhibited lower expression of TREM2 and higher expression of MHC-II (Fig. S11E). Granulocytic and monocytic tumor-infiltrating populations also expressed higher MHC-II in Jak2−/− MC38 tumors (Fig. S11F). These intradermally implanted Jak2−/− MC38 tumors exhibited slower growth and ICI treatment was ineffective in further reducing tumor growth (Fig. S11G). Although tumor-intrinsic JAK inhibition effects cannot be ruled out, these data demonstrate that ruxolitinib’s myeloid reprogramming effect is at least partially independent of tumor cell-intrinsic JAK1 and JAK2 signaling.
Combination of ruxolitinib with nivolumab show clinical efficacy in relapsed or refractory Hodgkin lymphoma patients
To investigate whether ruxolitinib combined with checkpoint blockade immunotherapy can enhance antitumor immune responses in humans, we launched an investigator-initiated clinical trial of ruxolitinib with nivolumab in Hodgkin lymphoma patients who previously failed checkpoint inhibitor therapy. In a Phase I open-label trial, eligible patients received ruxolitinib orally using a dose escalation protocol (Materials and methods) followed by nivolumab at day 8 and then every 4 weeks (480 mg intravenously) for 2 years or until progression (NCT03681561). Blood samples were collected pre-treatment, 1 week after the start of ruxolitinib treatment and post-nivolumab at the start of the 2nd cycle, allowing for the effects of ruxolitinib alone and ruxolitinib with nivolumab to be evaluated (Fig. 5A). Of the 21 enrolled patients, 19 were evaluable for response and 18 had viable PBMC samples pre- and post-ruxolitinib available for analysis (Fig. S12 / Table S1). There were no dose limiting toxicities and adverse events were infrequent (Table S2). Patients were a median 3.4 years from the initial diagnosis (range 0.9–16.7 years), 89% had stage III-IV disease, all received prior ICI and progressed (73%) or experienced stable disease/mixed response (27%) (Table S1). Median follow-up was 16.8 months (range 2.8–28 months). Of 19 treated patients, 6 patients exhibited complete metabolic responses, 4 partial remission according to the Lymphoma Response to Immunomodulatory Therapy Criteria (LYRIC) (40) with a best overall response rate of 52% (Fig. 5B). One additional patient had stable disease and 1 patient experienced an indeterminate response and subsequently received autologous stem cell transplantation. The median duration of response was 12.5 months (range 3.7–20.4 months). At 2 years, progression-free survival was 46% (95% confidence interval 9–77%) and overall survival (OS) was 87% (95% CI 58–97%).
Fig. 5. Ruxolitinib and nivolumab show efficacy in patients with relapsed or refractory classical Hodgkin lymphoma in a phase I clinical trial.
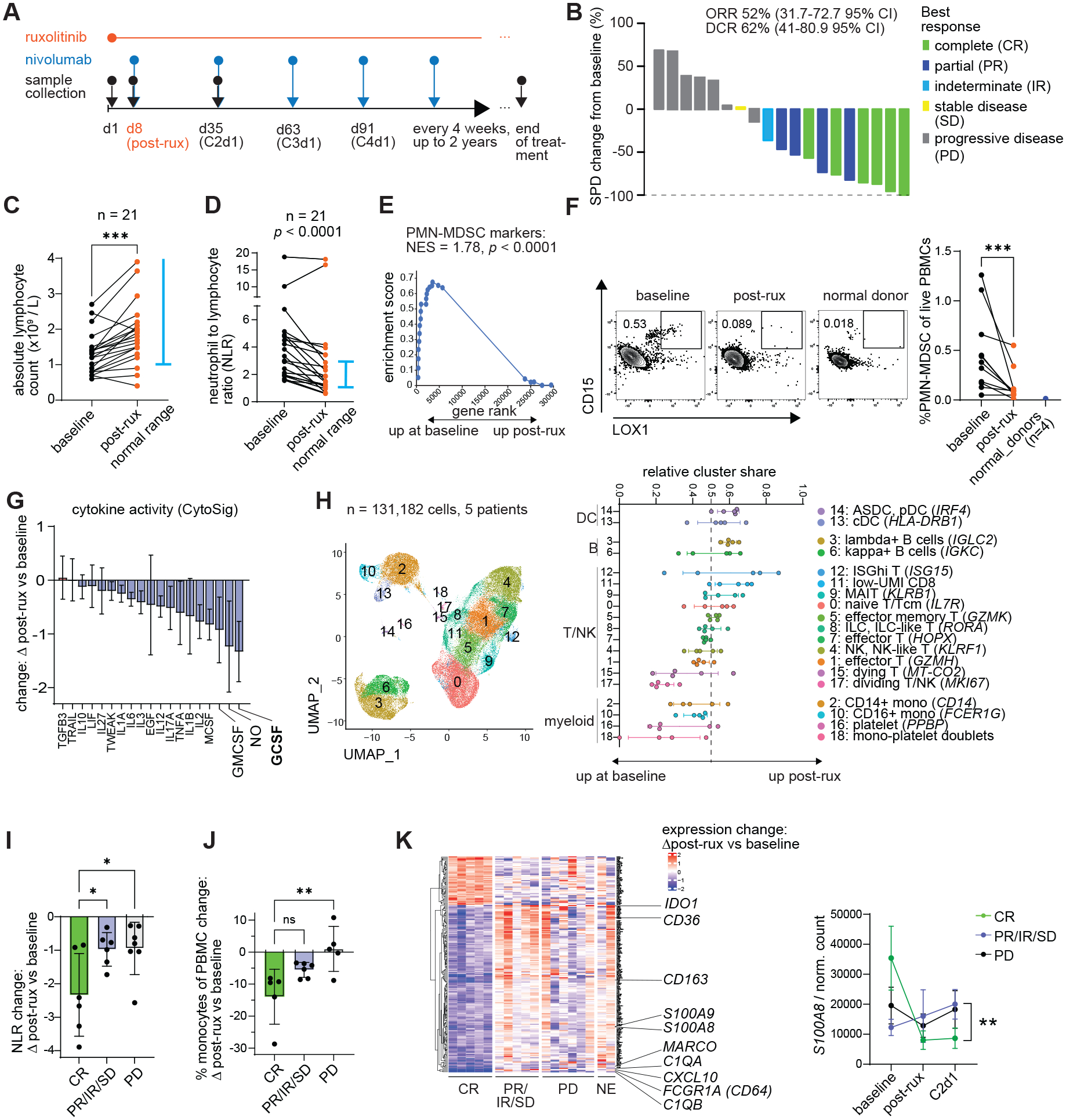
(A) Treatment and sample collection schedule. (B) Waterfall plot depicting tumor burden change as quantified by the sum of the product of diameters (SPD) change from baseline and best response as evaluated by LYRIC criteria. (C-K) Peripheral blood samples were collected at baseline and 8 days post-ruxolitinib treatment (C1d8), subjected to hematologic analysis and mononuclear cells isolated for further analysis: (C) absolute lymphocyte counts established by hematologic analyzer. (D) Neutrophil-to-lymphocyte ratio calculated as absolute neutrophil count divided by absolute lymphocyte count. (E) Change in the expression of PMN-MDSC markers by Veglia et al. as quantified by GSEA in bulk RNA-seq data. (F) Flow cytometric quantification of LOX1+ PMN-MDSCs in patients and normal donors. (G) Relative change in cytokine transcriptomic scores after ruxolitinib compared to baseline, MDSC associated cytokines highlighted. (H) Single-cell RNA-seq analysis of live PBMCs from 5 patients pre- and post-ruxolitinib: integrated dimensionality reduction and clustering and relative change in cluster frequencies post-ruxolitinib. (I) Change in NLR grouped by best clinical response. (J) Change in CIBERSORTx-estimated monocyte percentage grouped by best response. (K) Genes whose change in expression differs significantly between responders and non-responders, quantified by bulk RNA-seq; C2d1 is a timepoint after the first nivolumab dose. DEG, differentially expressed gene; ASDC, Axl+ Siglec6+ dendritic cells; GSEA, gene set enrichment analysis; CR, complete response; IR, indeterminate response; PD; progressive disease; PR, partial response; SD, stable disease; ORR, overall response rate; NES, normalized enrichment score; DC, dendritic cell; ILC; innate lymphoid cells; NK, natural killer cell; RBC, red blood cell; prog., progenitor cell; C1d8, Cycle 1 day 8; C2d1, Cycle 2 day 1; FDR, false discovery rate; MAIT, mucosal associated invariant T cell. Bars show standard deviation. Statistical comparison of experimental groups was performed using ratio paired Student’s t-test (C-D, F), GSEA test (E), one-way ANOVA with Sidak’s post-test (I-J), one-way ANOVA (K left) or two-way ANOVA (K right) (89).
Bulk RNA-sequencing of PBMCs, followed by a computational deconvolution of cell types using a large reference single-cell CITE-seq PBMC dataset (GSE164378), revealed that lymphocytes and dendritic cells increased in percentage accompanied by a sharp decrease in myeloid progenitors and monocytes (Fig. S13, S 14A–D). Concordantly, absolute lymphocyte counts significantly increased following ruxolitinib treatment (Fig. 5C). Mapping differentially expressed genes onto the PBMC reference dataset showed that whereas ruxolitinib-upregulated genes were distributed relatively evenly across B cells, NK cells and CD8 T cells, ruxolitinib-downregulated genes were particularly enriched in CD14+ monocytes (Fig. S14E). Notably, the percentages of cytokine-producing CD8 T cells and CD4 T cells were maintained and the absolute number of cytokine-producing CD8 T cells significantly increased post-treatment with ruxolitinib compared to baseline (Fig. S14F).
The neutrophil-to-lymphocyte ratio (NLR), clinically known to correlate with MDSC frequency and poor prognosis across cancer types including Hodgkin lymphoma (41–44), showed a significant reduction post-ruxolitinib (Fig. 5D, S15A). Assessing the expression of published MDSC markers (9) revealed a statistically significant reduction of their expression after ruxolitinib (Fig. 5E, S15B). Concordantly, the percentage of PMN-MDSCs in blood was sharply reduced after ruxolitinib and no PMN-MDSCs were detected in normal donor controls (Fig. 5F). The expression of G-CSF target genes was significantly downregulated after ruxolitinib treatment (Fig. 5G). Transcription factors STAT1, STAT3 and STAT6 play a role in MDSC programming and signal downstream of JAK1 and JAK2 (9). Of these, STAT3 signals downstream of G-CSF. Gene set enrichment analysis of annotated STAT targets showed a significant reduction in STAT3 target genes post ruxolitinib (Fig. S15C).
To obtain independent verification of the effects of ruxolitinib on PBMCs, a representative subset of 5 patients was subjected to single-cell RNA-seq analysis at baseline and post-ruxolitinib. In agreement with previous results, the relative proportion of DCs, B cells and certain T cell subsets increased in most patients after ruxolitinib (Fig. 5H, S15D). In contrast, the percentage of classical and non-classical monocytes decreased in most patients consistent with bulk RNA-seq and flow cytometry (Fig. 5H). At the transcriptome level, the monocytic cluster showed a statistically significant reduction in MDSC marker genes (Fig. S15E).
We next evaluated differences between responders and non-responders. There was no clear relationship between clinical response and ruxolitinib dose although the low number of patients treated with less than 20 mg bid prevents a formal comparison (Fig. S16A). Complete responders exhibited a significantly greater reduction in the NLR and % of monocytes after ruxolitinib than non-responders and non-evaluable patients (Fig. 5I–J, S16B). NLR levels remained stable after nivolumab treatment compared to post-ruxolitinib (Fig. S16C–D). A global assessment of transcripts modulated differently by ruxolitinib in response groups revealed a monocyte-enriched cluster more strongly downregulated in complete responders than other groups (Fig. 5K). This cluster contained ISGs but also important myeloid and MDSC related genes (Fig. 5K). For example, CD163 and S100A8/9 were more significantly downregulated in the complete responders than other patient groups and the level remained low even after nivolumab treatment (Fig. 5K, S16E). Single-cell RNA-seq data showed decreases of Cluster 3 genes even specifically in the classical monocytes on a per-cell basis (Fig. S16F–G). The single-cell data suggests that ruxolitinib not only reduced the percentage of monocytes in peripheral blood but also reduces the expression of specific genes within monocytes.
Myeloid suppressor cells are increasingly recognized to influence the response to cancer immunotherapy. However, therapies rationally targeting myeloid cells remain in clinical development. JAK inhibitors target pathways directly involved in the suppressive programming of MDSCs (10), however JAK inhibitors have been reported to promote (13, 14, 45) or restrain MDSC function or migration depending on the disease model (46, 47). Moreover, JAK inhibitors exert direct effects on cancer cells (27), dendritic cells (48), NK cells (49), T cells (50) and other cell types (23), suggesting a context-dependent overall impact of therapeutic JAK inhibition. Our results suggest that in the presence of checkpoint inhibitors, ruxolitinib reduces suppressive gene expression in granulocytes, monocytes and macrophages while increasing MHC expression in these cells. Given the important role of JAK-STAT signaling in antigen presentation and T cell recruitment, complete ablation of JAK1/2 activity is unlikely to improve antitumor immunity or ICI response (39, 51). Notably, at the doses tested, T cell function and total numbers were preserved in mice as well as Hodgkin lymphoma patients. This is supported by observations in other studies in which T cell function was preserved at therapeutic doses of JAK inhibitors including preserved graft-versus-tumor effect in ruxolitinib-treated graft-versus-host disease (52, 53). Previously, reports showed PD-1 inhibition can be effective in individual patients treated with ruxolitinib (54, 55) despite the general immunosuppressive effects of ruxolitinib (56, 57). Therefore, identifying patients likely to benefit from JAK inhibitors will be critical for its application to cancer therapy.
To underscore the synergy of JAK inhibition with ICI, we report the results of a phase 1 trial combining ruxolitinib with nivolumab in cHL patients who relapsed or are refractory (R/R) to ICI. Disease control rate (64%) and CR rate (31.5%) with a median duration of response over 1 year are remarkable in this setting and suggests novel mechanism of enhancing immunologic anti-tumor activity. Favorable response rate of this combination contrasts with the limited efficacy of ruxolitinib monotherapy in R/R cHL (58–60).
Classic Hodgkin lymphoma is characterized by frequent alteration of chromosome 9p and exhibit JAK2 and CD274 (PD-L1) copy number changes (61–63). Unexpectedly, the observed efficacy of inhibiting both pathways is strongly correlated with changes in myeloid cells. Ruxolitinib therapy for 7 days yielded reduction of PB MDSC in R/R cHL patients. Ruxolitinib therapy for 7 days yields reduction of PB MDSC and lowered the expression of MDSC-associated S100A8, S100A9 and of CD163 in PB monocytes in R/R cHL patients. CD163 is a gene associated with tumor-associated macrophages and its expression has been associated with high-risk HL (64, 65). G-CSF induces S100A8 and S100A9 expression and has recently been shown to enhance MDSCs in cancer patients, suggesting blockade of G-CSF may contribute to ruxolitinib’s efficacy (11). Our LCMV data and clinical and correlative findings in cHL together suggest that the observed myeloid cell changes are independent of cancer cell-intrinsic JAK signaling.
Given the occurrence of suppressive myeloid cells in various tumor types (66) and adverse interference with immunotherapy, it is possible that JAK inhibition could improve checkpoint responses in other malignancies. Immune modulating effect of ruxolitinib when combined with ICI in Hodgkin lymphoma can abrogate a negative prognostic relevance of NLR for PFS (43, 67) and negative prognostic relevance of monocytes for nivolumab response. However, NLR is also associated with anti-PD-1 response in other tumor types including melanoma and lung cancer (68, 69). Indeed, combination therapy of the JAK inhibitor itacitinib with the anti-PD-1 antibody pembrolizumab in non-small cell lung cancer showed an encouraging 67% overall response rate in a Phase II clinical trial published jointly with this article (70). The itacitinib with pembrolizumab study reported favorable effects of JAK inhibition on CD8 T cell plasticity, suggesting that in addition to reprogramming myeloid cells and increasing T cell proliferation, JAK inhibitors may also enhance the ability of CD8 T cells to differentiate into more functional subsets. Our data highlight that ruxolitinib yields a synergistic effect with checkpoint blockade in cancer immunotherapy and this therapeutic combination offers an avenue to explore in future clinical trials designed to overcomes resistance to and/or enhance ICI therapy.
Materials and Methods:
Experimental design
Analysis of blood samples collected in a clinical trial was performed using all samples available collected at predetermined timepoints. Sample size for preclinical tumor studies was selected to detect an effect size of at least 1.5 as measured by tumor weight.
Clinical trial design
Patients were enrolled in a Phase I/II multi center, open-label, dose escalation/dose expansion study to evaluate the safety and tolerability of ruxolitinib when combined with nivolumab in patients with relapsed/refractory Hodgkin lymphoma (NCT03681561). Trial was approved by FDA (IND number 136877; VB is holder) and each institution’s Investigational Review Board (TSRI #IRB-19-7408 and #IRB-21-7803; UMN IRB #STUDY00001341) and conducted through the Big Ten Cancer Center Consortium. All patients signed the informed consent and were treated according to Declaration of Helsinki. Eligible patients had to exhibit refractory, relapsed or stable disease (less than partial remission) to prior checkpoint inhibitor treatment. Three dose levels of ruxolitinib were tested: 10 mg orally twice a day (bid), 15 mg bid, and 20 mg bid. The dose escalation used the continual reassessment method (CRM) with 2 patients per dose level 1 and 2. Planned duration of therapy was 2 years. Primary objective was to find the maximum tolerated dose (MTD) of ruxolitinib when given with nivolumab and characterize the safety and tolerability of this combination. Primary objective of expansion phase was to evaluate best overall response rate (ORR) and best overall disease control rate (DCR).
Clinical responses were evaluated by positron emission tomography (PET/CT) or CT scan at 1, 3, 6, 9, 12, 18 and 24 months using the Lugano criteria adapted to immune-based therapy, the Lymphoma Response to Immunomodulatory Therapy Criteria (LYRIC)(40). LYRIC is identical to the Lugano criteria for CR and PR, however it uses an expanded definition of indeterminate response (IR) to taken into account “tumor flare” and “pseudo-progression” both associated with immunomodulatory drugs. For patients with IR the subsequent scan determined CR, PR, SD or PD. There was 1 patient who had resolution of all disease with suspected hypermetabolic tumor flare in a new isolated area and subsequently underwent stem cell transplantation with IR disease status. To be assigned a status of CR or PR, changes in tumor measurements must be confirmed by repeat assessments performed no less than four weeks after the criteria for response are first met. Duration of overall response: the period measured from the time that measurement criteria are met for CR or PR (whichever status is recorded first) until the date that progressive disease is objectively documented. The objective response rate is the proportion of all patients with confirmed PR or CR according to LYRIC, from the start of treatment until disease progression/recurrence. The disease control rate is the proportion of all patients with stable disease (SD) for 8 weeks, or PR or CR according to LYRIC. Non-evaluable subjects received less than 1 month of prescribed therapy. Additional information on trial design is available on clinicaltrials.gov.
Human samples
We analyzed blood samples from patients with HL treated in the clinical trial of ruxolitinib with nivolumab (NCT03681561). Pre-planned blood collection for correlative endpoints occurred at baseline, 1 week of ruxolitinib therapy and 4 weeks of combined therapy of ruxolitinib with nivolumab. Peripheral blood mononuclear cells were isolated and cryopreserved according to standard protocols. Normal peripheral blood mononuclear cells were obtained through the TSRI Normal Blood Donor Services program (TSRI IRB #15-6710). Details on the patients whose samples were available for analysis in each experiment are given in Table S3.
Animal studies
Mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) of The Scripps Research Institute (TSRI IACUC #09-0098 and #15-0017) and performed in accordance with its guidelines. P14 mice were generated by Pircher et al. and are available as Jackson Laboratory stock 37394 (71). Wild type C57BL/6J mice were obtained from an institutional breeding colony derived from Jackson breeders (Jackson #000664). Wild type BALB/cByJ mice were obtained from Jackson (#001026). Sample sizes for mouse experiments were estimated using statistical power data from pilot experiments. Cohort expansion was performed for experiments with measurable differences between experimental groups that were initially statistically underpowered to establish significance. No samples were excluded from analysis.
Virus growth and titration
LCMV Clone 13 was passaged on BHK-21 cells as previously reported (72). Serum was isolated by low-speed centrifugation to remove red and white blood cells. 10 μl of serum was used to perform 10-fold serial dilutions and quantified by focus forming assay on VeroE6 cells as previously described (73). BHK-21 and VeroE6 were provided by the Oldstone laboratory.
Kinase inhibitor library screen
A library of kinase inhibitors was obtained from a commercial source (cat. HY-L008, MedChemExpress, Monmouth Junction, NJ). Spleens from LCMV-CL13 infected IFN-γ-YFP mice were isolated at day 15 p.i. and single cell suspensions prepared. Following depletion of B cells, 2×105 cells were seeded in duplicate onto 96-well plate wells pre-spotted with DMSO or library compounds at 1000 nM, 500 nM, 250 nM and 100 nM final concentration. Following a 5-day incubation period, the viability dye 7-AAD was added to each well (1:50 dilution) and plates were rested for 15 minutes. Cells were then analyzed on a ZE5 flow cytometer (Bio-Rad, Hercules, CA). Z scores were calculated for the frequency of YFP+ 7AAD− cells in compound wells compared to DMSO control wells.
Compound target gene enrichment analysis
Plate list of compounds in the ReFrame library (74) was filtered for duplicates such that only a single plate well per unique molecule was included. Compounds with autofluorescence in the YFP channel were removed by applying the threshold raw %YFP− CD8 T cells > 25. To obtain drug gene interactions, ChEMBL IDs were first retrieved using the ChEMBL API (Plate list of compounds in the ReFrame library (74) was filtered for duplicates such that only a single plate well per unique molecule was included. Compounds with autofluorescence in the YFP channel were removed by applying the threshold raw %YFP− CD8 T cells > 25. To obtain drug gene interactions, ChEMBL IDs were first retrieved using the ChEMBL API (https://chembl.gitbook.io/chembl-interface-documentation/web-services/chembl-data-web-services), then the Drug Gene Interaction Database queried using ChEMBL IDs (19). JAK inhibitors were defined as compounds with a curated interaction with JAK1, JAK2 or JAK3. Fisher’s exact test was used to assess enrichment of JAK inhibitors among hit compounds.
T cell proliferation and intracellular cytokine detection assays
Splenocytes from LCMV-CL13 infected IFN- γ-YFP mice were labeled with CellTrace Violet (CTV) and seeded onto round-bottom 96-well plates at 2×105 cells/well in complete T cell media (10% FBS, L-glutamine, Pen/Strep, NEAA, Sodium Pyruvate, HEPES, β-mercaptoethanol) supplemented with a cocktail of all known immunodominant LCMV CD8-specific (LCMV-GP33-41, LCMV-GP276-286 and LCMV-NP396-404) and CD4-specific (LCMV-GP61-80) epitope peptides (75, 76). Cells were treated with either DMSO, 250 nM ruxolitinib phosphate (LC Laboratories, Woburn, MA; #R-6688), 50 μg/ml anti-IFN-γ (BioXCell, # BE0055) or 25 μg/ml anti-PD-L1 (BioXCell, #BE0101). Brefeldin A (Thermo Fisher, #B7450) was added at a 1:500 dilution to each well 6 h prior to cell isolation to block T cell-specific cytokine secretion. Surface antigens were stained for 30 minutes on ice, cell fixed and permeabilized using the Transcription Factor Staining Buffer Set (Thermo Fisher, #00-5523-00). Permeabilized cells were then stained for intracellular antigens. To track GP-specific CD8 T cells in vitro, 1,000 congenic Thy1.1+ CD8+ T cells (P14) from TCR tg mice that recognize the LCMV-GP33-41 epitope (71) were engrafted into IFN-γ-YFP mice 1 day prior to LCMV infection.
Extracellular cytokine detection assay
Supernatants from in vitro IFN-γ-YFP cultures were isolated at day 3 and 5 post-ruxolitinib treatment. To measure IFN-γ protein, supernatants were diluted at 1:4 and measured by Bio-Plex Pro mouse IFN-γ immunoassay (Bio-Rad, #171G5017M) according to manufacturer’s instructions.
Tumor models
7–8 week-old C57BL/6J (B6) mice were implanted intradermally in the right flank with 1–5×105 cells (MC38 and knockout MC38 cell lines) or subcutaneously (s.c.) in the right flank with 1×106 (EL4) or 2.5×105 cells (LLC1). For A20 tumor experiments, 7–8-week-old BALB/cByJ mice were implanted subcutaneously in the right flank with A20 4×106 cells. For MC38 and LLC1 tumor-infiltrating lymphocyte analysis, mice were treated with anti-PD1 and anti-CTLA4 (ICI) when tumors were palpable and 1 week later (~d8 and d15 for MC38 tumors) and 30 mg/kg ruxolitinib phosphate by oral gavage (LC Laboratories cat #R-6688) 2 days after the first ICI treatment and until the second ICI treatment. A20 tumor bearing mice were treated as described in Figure 2B: once palpable, mice received ICI then daily ruxolitinib starting 2 days after the first ICI treatment. Mice were randomized at the start of treatment to prevent baseline size bias between groups. Investigators were not blinded to the mouse treatment groups.
For TIL isolation, 1–2 days post 2nd ICI treatment, tumors were excised, manually dissociated, incubated with collagenase, hyaluronidase and DNase (STEMCELL Technologies cat #07912, 100-0762) according to manufacturer’s instructions and mashed through 100 μm filters. TILs were then stained with surface antibodies and analyzed by flow cytometry. For intranuclear staining, cells were fixed overnight with FOXP3 Fixation/Permeabilization reagent (eBioscience, catalog# 00-5523-00) and stained with nuclear protein targeting antibodies the following day. For blood analysis, blood was isolated using the retroorbital route and red blood cells lysed twice using Lonza ACK Lysing buffer (Lonza, Basel, Switzerland), then subjected to staining as described above. For bone marrow analysis, femur bone marrow was isolated and stained as described above. For splenocyte analysis, spleens were manually dissociated and mashed through 100 μm filters, then stained as described above. Ruxolitinib phosphate treatment was performed using daily oral gavage, concentration 30 mg/kg unless specified otherwise in text. Immune checkpoint inhibitor treatment was performed at the specified timepoint using anti-PD1 (50 μg/mouse, Leinco #P362) and anti-CTLA4 (50 μg/mouse, bioXcell #BE0164) or isotype controls (mouse IgG2b and rat IgG2a) by intraperitoneal injection. For cell depletion experiments, CD8 T cells, NK cells and granulocytes were depleted using anti-CD8 (Leinco cat #C2442), anti-NK1.1 (bioXcell #BE0036) and anti-Ly6G (Leinco #L280) antibodies, respectively, or respective control antibodies (rat IgG2b, mouse IgG2a, rat IgG2a) using intraperitoneal injection 1 day before ICI treatment and every 2 days thereafter until end of ICI treatment. On-treatment depletion efficiency was verified by flow cytometry of peripheral blood cells.
Cell lines were obtained from ATCC (EL4: #TIB-39, A20: #TIB-208) and Kerafast (MC38 #ENH204-FP). LLC1 cell line was a kind gift from Dr Linda Sherman. Jak1−/− and Jak2−/− MC38 cells were kindly provided by Dr Antoni Ribas (39) and authenticated by bulk RNA-seq which detected frameshift mutations at the Jak1 and Jak2 loci, respectively. The growth of intradermally implanted Jak1−/− and Jak2−/− MC38 cells in vivo was significantly slower compared to wt MC38 cells (Fig. S11A, S11G). Therefore, the tumors required additional days of in vivo growth before palpable and were smaller at harvest. Cellular analysis was performed 2 days after the second ICI treatment, equivalent to wt MC38 tumor analysis.
Myeloid cell functional assays
To test the ability of MDSCs to suppress NK cell proliferation and cytokine production, Gr1+ cells from MC38 tumors were isolated using the CD11b+ Gr1+ magnetic isolation kit (STEMCELL Technologies cat #19867) and incubated with NK cells isolated using the mouse NK cell isolation kit (STEMCELL Technologies cat #19855) from tumor-free wild type B6 mice in the indicated ratios in the presence of IL-2 as described previously (77). To test the ability of granulocytes to stimulate T cell proliferation, T cells were isolated from tumor-free wild type B6 mice using the mouse CD8 T cell isolation kit (STEMCELL Technologies cat #19853), labeled with Cell Trace Violet (Thermo Fisher cat #C34557) according to the manufacturer’s specifications; live CD11b+ Ly6G+ Ly6Clo were isolated from spleens of mice treated with ICI, ICI+rux, ruxolitinib or vehicle and incubated with the indicated ratios of T cells in anti-CD3/CD28 coated plates for 3 days, then analyzed by flow cytometry.
Immunohistochemistry
Freshly isolated tumor tissue was fixed using Bouin’s solution (Sigma Aldrich #HT10132) and subjected to tissue processing for paraffin-embedded sectioning according to standard protocols. Immunohistochemistry staining was performed using anti-NK1.1 antibody and 3,3’ Diaminobenzidine (DAB) Substrate Kit.
High-throughput screen for compounds that rescue T cell exhaustion
An assay using a YFP-IFNγ reporter strain was developed and scaled to high throughput format as described previously (18). Briefly, B cell depleted splenocytes were prepared from LCMV-CL13 infected IFN-γ-YFP mice at day 15 p.i. Next, 5×104 cells were seeded onto 384-well plate wells pre-spotted with DMSO or ReFrame compounds at 5 μM final concentration. Following a 5-day incubation period, the viability dye 7-AAD was added to each well (1:50 dilution) and plates were rested for 15 minutes. Following a 5-day incubation period, the viability dye 7-AAD was added to each well (1:50 dilution) and plates were rested for 15 minutes.
Cell-free IC50 values were derived from the sources below.
Flow cytometry
Surface staining: Single cell suspensions were incubated with 5% normal rat serum and rat anti-mouse CD16/CD32 antibody (BD Biosciences clone 2.4G2, RRID AB_394656) then stained with fluorophore-conjugated antibodies, washed and analyzed by flow cytometry without fixation. Intracellular staining: spleens from Cl13 infected mice were collected and dissociated manually. Red blood cells were lysed using Lonza ACK Lysing buffer (Lonza, Basel, Switzerland, #BP10-548E). Splenocytes were stained with LCMV-specific gp-33 tetramer and surface antibodies then fixed overnight with FOXP3 Fixation/Permeabilization reagent (eBioscience, catalog #00-5523-00) and stained with intranuclear antibodies the following day. Stained cells were analyzed using the 5-laser Cytek Aurora (Cytek Biosciences, Fremont, CA). Gating schemes for human and mouse cell populations are given in Figures S13 and S17, respectively.
Antibodies for flow cytometry
Antibodies were obtained from commercial vendors. Details on antibody clones, fluorophore conjugates and catalog numbers are given below.
| Index | Antibody | Fluorophore | Clone | Catalog# | Vendor |
|---|---|---|---|---|---|
| 1 | Anti-active caspase-3 | PE | C92-605 | 550821 | BD Biosciences |
| 2 | Anti-dog/cynomolgus monkey/human/ mouse/non-human primate/Rat Ki-67 | BUV496 | SolA15 | 364-5698-82 | ThermoFisher |
| 3 | Anti-human CD107a (LAMP-1) | PE | 301107 | W18263B | Biolegend |
| 4 | Anti-human CD107a (LAMP-1) | Pacific Blue | 328624 | W18263B | Biolegend |
| 5 | Anti-human CD11c | PerCP/Cyanine5.5 | 3.9 | 301623 | Biolegend |
| 6 | Anti-human CD123 | APC | 6H6 | 306011 | Biolegend |
| 7 | Anti-human CD14 | BUV805 | 61D3 | 368-0149-42 | ThermoFisher |
| 8 | Anti-human CD15 (SSEA-1) | Alexa Fluor® 700 | HI98 | 301919 | Biolegend |
| 9 | Anti-human CD15 (SSEA-1) | FITC | HI98 | 301903 | Biolegend |
| 10 | Anti-human CD15 (SSEA-1) | Pacific Blue | HI98 | 394703 | Biolegend |
| 11 | Anti-human CD16 | BV711 | 3G8 | 302043 | Biolegend |
| 12 | Anti-human CD161 | BV785 | HP-3G10 | 339930 | Biolegend |
| 13 | Anti-human CD163 | Brilliant Violet 605™ | GHI/61 | 333615 | Biolegend |
| 14 | Anti-human CD19 | BUV496 | SJ25C1 | 364-0198-42 | ThermoFisher |
| 15 | Anti-human CD19 | APC/Cyanine7 | SJ25C1 | 363010 | Biolegend |
| 16 | Anti-human CD197 (CCR7) | PE/Fire™ 810 | G043H7 | 353269 | Biolegend |
| 17 | Anti-human CD1c | Brilliant Violet 650™ | L161 | 331541 | Biolegend |
| 18 | Anti-human CD274 (B7-H1, PD-L1) | PE/Fire™ 810 | 29E.2A3 | 329755 | Biolegend |
| 19 | Anti-Human CD3 | BUV395 | UCHT1 | 563548 | BD Biosciences |
| 20 | Anti-Human CD3 | PerCP/Cyanine5.5 | UCHT1 | 300429 | Biolegend |
| 21 | Anti-human CD33 | PE/Cyanine7 | P67.6 | 366617 | Biolegend |
| 22 | Anti-human CD4 | BV650 | 317435 | OKT4 | Biolegend |
| 23 | Anti-human CD45 | Alexa Fluor® 700 | HI30 | 304023 | Biolegend |
| 24 | Anti-human CD45 | PE/Dazzle™ 594 | HI30 | 304051 | Biolegend |
| 25 | Anti-human CD45 | Alexa Fluor® 700 | HI30 | 304023 | Biolegend |
| 26 | Anti-human CD45 | PE/Dazzle™ 594 | HI30 | 304051 | Biolegend |
| 27 | Anti-human CD56 (NCAM) | PE/Cyanine7 | HCD56 | 362509 | Biolegend |
| 28 | Anti-human CD68 | PerCP/Cyanine5.5 | Y1/82A | 333813 | Biolegend |
| 29 | Anti-human CD8a | Brilliant Ultra Violet™ 563 | RPA-T8 | 365-0088-42 | ThermoFisher |
| 30 | Anti-human CD90 (Thy-1) | BIUV395 | eBio5E10 | 363-0909-42 | ThermoFisher |
| 31 | Anti-human CXCL10 (IP-10) | PE/Cyanine7 | J034D6 | 519507 | Biolegend |
| 32 | Anti-human LOX-1 | BV421 | 15C4 | 358609 | Biolegend |
| 33 | Anti-human/non-human primate/rhesus monkey CD56 (NCAM) | BUV737 | MTULY56 | 367-0566-42 | ThermoFisher |
| 34 | Anti-mouse anti-mouse H-2Kb | Pacific Blue | AF6-88.5 | 116513 | Biolegend |
| 29 | Anti-mouse Bcl-xL | PE | 7B2.5 | MA5-28638 | ThermoFisher |
| 30 | Anti-mouse CD107a (LAMP-1) | PE | 1D4B | 121612 | Biolegend |
| 31 | Anti-mouse CD107a (LAMP-1) | Alexa Fluor® 647 | 1D4B | 121610 | Biolegend |
| 32 | Anti-mouse CD107a (LAMP-1) | BV421 | 1D4B | 121618 | Biolegend |
| 33 | Anti-mouse CD115 (CSF-1R) | APC | AFS98 | 135510 | Biolegend |
| 34 | Anti-mouse CD11b | BUV805 | M1/70 | 368-0112-82 | Thermo Fisher |
| 35 | Anti-mouse CD11c | BV650 | N418 | 117339 | Biolegend |
| 36 | Anti-mouse CD163 | PE/Dazzle™ 594 | S15049I | 155315 | Biolegend |
| 37 | Anti-mouse CD163 | PE/Cyanine7 | S15049I | 155319 | Biolegend |
| 38 | Anti-mouse CD19 | APC/Cyanine7 | 6D5 | 115530 | Biolegend |
| 39 | Anti-mouse CD226 (DNAM-1) | PerCP/Cyanine5.5 | 10E5 | 128813 | Biolegend |
| 40 | Anti-mouse CD244.2 (2B4 B6 Alloantigen) | PE/Cyanine7 | m2B4 (B6)458.1 | 133511 | Biolegend |
| 41 | Anti-mouse CD279 (PD-1) | PE/Fire™ 810 | 29F.1A12 | 135253 | Biolegend |
| 42 | Anti-mouse CD335 (NKp46) | PE | 29A1.4 | 137603 | Biolegend |
| 43 | Anti-mouse CD335 (NKp46) | FITC | 29A1.4 | 137605 | Biolegend |
| 44 | Anti-mouse CD366 (Tim-3) | BV421 | RMT3-23 | 119723 | Biolegend |
| 45 | Anti-human LOX-1 | BV421 | 15C4 | 358609 | Biolegend |
| 46 | Anti-human/non-human primate/rhesus monkey CD56 (NCAM) | BUV737 | MTULY56 | 367-0566-42 | ThermoFisher |
| 47 | Anti-mouse anti-mouse H-2Kb | Pacific Blue | AF6-88.5 | 116513 | Biolegend |
| 48 | Anti-mouse Bcl-xL | PE | 7B2.5 | MA5-28638 | ThermoFisher |
| 49 | Anti-mouse CD107a (LAMP-1) | PE | 1D4B | 121612 | Biolegend |
| 50 | Anti-mouse CD107a (LAMP-1) | Alexa Fluor® 647 | 1D4B | 121610 | Biolegend |
| 51 | Anti-mouse CD366 (Tim-3) | BV785 | RMT3-23 | 119725 | Biolegend |
| 52 | Anti-mouse CD366 (Tim-3) | BV711 | RMT3-23 | 119727 | Biolegend |
| 53 | Anti-mouse CD39 | BUV 805 | 24DMS1 | 368-0391-82 | ThermoFisher |
| 54 | Anti-mouse CD4 | BV421 | GK1.5 | 100437 | Biolegend |
| 55 | Anti-mouse CD4 | BUV737 | GK1.5 | 367-0041-82 | BD Biosciences |
| 56 | Anti-mouse CD4 | BUV395 | GK1.5 | 363-0041-82 | BD Biosciences |
| 57 | Anti-mouse CD4 | BUV805 | RM4-5 | 368-0042-82 | BD Biosciences |
| 58 | Anti-mouse CD4 | BV605 | GK1.5 | 100451 | Biolegend |
| 59 | Anti-mouse CD4 | APC/Cyanine7 | GK1.5 | 100414 | Biolegend |
| 60 | Anti-mouse CD4 | eFluor™ 506 | RM4-5 | 69-0042-82 | Thermo Fisher |
| 61 | Anti-mouse CD43 | PE/Dazzle™ 594 | S11 | 143217 | Biolegend |
| 62 | Anti-mouse CD45 | BUV395 | 30-F11 | 363-0451-82 | ThermoFisher |
| 63 | Anti-mouse CD45 | BUV563 | 30-F11 | 365-0451-82 | ThermoFisher |
| 64 | Anti-mouse CD45.2 | BUV737 | 104 | 612779 | BD Biosciences |
| 65 | Anti-mouse CD45.2 | Alexa Fluor® 700 | 104 | 109822 | Biolegend |
| 66 | Anti-mouse CD49b | APC/Cyanine7 | DX5 | 108905 | Biolegend |
| 67 | Anti-mouse CD49b | BUV563 | HMα2 | 741280 | BD Biosciences |
| 68 | Anti-mouse CD49b | PE/Cyanine7 | DX5 | 108921 | Biolegend |
| 69 | Anti-mouse CD63 | Alexa Fluor® 700 | NVG-2 | 143923 | Biolegend |
| 70 | Anti-mouse CD8a | BV711 | 53-6.7 | 100747 | Biolegend |
| 71 | Anti-mouse CD8a | APC/Cyanine7 | 53-6.7 | 100714 | Biolegend |
| 72 | Anti-mouse CD8a | PE/Cyanine7 | 53-6.7 | 100722 | Biolegend |
| 73 | Anti-mouse CD8a | Alexa Fluor® 700 | 53-6.7 | 100729 | Biolegend |
| 74 | Anti-mouse CD8a | BUV805 | 53-6.7 | 368-0081-82 | ThermoFisher |
| 75 | Anti-mouse CD8a | Pacific Blue | 53-6.7 | 100725 | Biolegend |
| 76 | Anti-mouse CD9 | PE/Dazzle™ 594 | MZ3 | 124821 | Biolegend |
| 77 | Anti-mouse CD90.2 (Thy1.2) | PerCP/Cyanine5.5 | 30-H12 | 105338 | Biolegend |
| 78 | Anti-mouse F4/80 | PerCP/Cyanine5.5 | BM8 | 123127 | Biolegend |
| 79 | Anti-mouse F4/80 | PE/Dazzle™ 594 | BM8 | 123145 | Biolegend |
| 80 | Anti-mouse F4/80 | APC/Cyanine7 | BM8 | 123118 | Biolegend |
| 81 | Anti-mouse GM-CSF | FITC | MP1-22E9 | 505403 | Biolegend |
| 82 | Anti-mouse H-2D b | PE/Cyanine7 | KH95 | 111515 | Biolegend |
| 83 | Anti-mouse I-A/I-E | BV711 | M5/114.15.2 | 107643 | Biolegend |
| 84 | Anti-mouse I-A/I-E | PerCP/Cyanine5.5 | M5/114.15.2 | 107625 | Biolegend |
| 85 | Anti-mouse IFN-γ | Alexa Fluor® 488 | XMG1.2 | 505813 | Biolegend |
| 86 | Anti-mouse IFN-γ | BV421 | XMG1.2 | 505829 | Biolegend |
| 87 | Anti-mouse IL-2 | Alexa Fluor® 488 | JES6-5H4 | 503813 | Biolegend |
| 88 | Anti-mouse IL-2 | BV605 | JES6-5H4 | 503829 | Biolegend |
| 89 | Anti-mouse Ki-67 | PE | 16A8 | 652404 | Biolegend |
| 90 | Anti-mouse Ki-67 | FITC | 16A9 | 652410 | Biolegend |
| 91 | Anti-mouse Ly-6C | APC | HK1.4 | 128015 | Biolegend |
| 92 | Anti-mouse Ly-6C | PerCP/Cyanine5.5 | HK1.4 | 128011 | Biolegend |
| 93 | Anti-mouse Ly-6C | Pacific Blue | HK1.4 | 128013 | Biolegend |
| 94 | Anti-mouse Ly-6G | Alexa Fluor® 488 | 1A8 | 127626 | Biolegend |
| 95 | Anti-mouse Ly-6G | APC/Cyanine7 | 1A8 | 127623 | Biolegend |
| 96 | Anti-mouse Ly-6G | BV421 | 1A8 | 127627 | Biolegend |
| 97 | Anti-mouse Ly-6G | PE/Cyanine7 | 1A8 | 127617 | Biolegend |
| 98 | Anti-mouse Ly-6G | BV605™ | 1A8 | 127639 | Biolegend |
| 99 | Anti-mouse Ly-6G | BV650™ | 1A8 | 127641 | Biolegend |
| 100 | Anti-mouse Ly-6G | PE/Fire™ 810 | 1A8 | 127673 | Biolegend |
| 101 | Anti-mouse Ly49H | PE/Cyanine7 | 144713 | 3D10 | Biolegend |
| 102 | Anti-mouse Ly49H | Alexa Fluor® 647 | 144709 | 3D10 | Biolegend |
| 103 | Anti-mouse Ly6C | BV605 | HK1.4 | 128036 | Biolegend |
| 104 | Anti-mouse Ly6C | PerCP/Cyanine5.5 | HK1.4 | 128011 | Biolegend |
| 105 | Anti-mouse MHC Class II (I-A/I-E) | BUV496 | M5/114.15.2 | 364-5321-82 | ThermoFisher |
| 106 | Anti-mouse NK-1.1 | BUV661 | PK136 | 741477 | BD Biosciences |
| 107 | Anti-mouse NK-1.1 | BUV661 | PK136 | 741477 | BD Biosciences |
| 108 | Anti-mouse NK1.1 | BV711 | PK136 | 108745 | Biolegend |
| 109 | Anti-mouse NK1.1 | APC/Cy7 | PK137 | 108724 | Biolegend |
| 110 | Anti-mouse NK1.1 | BUV496 | PK136 | 364-5941-82 | ThermoFisher |
| 111 | Anti-mouse TCR β chain | Alexa Fluor® 700 | H57-597 | 109223 | Biolegend |
| 112 | Anti-mouse TCR β chain | PE/Cyanine7 | H57-597 | 109221 | Biolegend |
| 113 | Anti-mouse TNF-α | BV605 | MP6-XT22 | 506329 | Biolegend |
| 114 | Anti-mouse TNF-α | APC/Cyanine7 | MP6-XT22 | 506343 | Biolegend |
| 115 | Anti-mouse TNF-α | PerCP/Cyanine5.5 | MP6-XT22 | 506321 | Biolegend |
| 116 | Anti-mouse TREM2 | FITC | 78.18 | MA5-28223 | ThermoFisher |
| 117 | Anti-mouse/human Arginase 1 | BUV805 | A1exF5 | 368-3697-82 | Thermo Fisher |
| 118 | Anti-mouse/human CD11b | BV650 | M1/70 | 101239 | Biolegend |
| 119 | Anti-mouse/human CD11b | PE/Dazzle™ 594 | M1/70 | 101256 | Biolegend |
| 120 | Anti-mouse/human CD11b | PerCP/Cyanine5.5 | M1/70 | 101228 | Biolegend |
| 121 | Anti-mouse/human CD11b | BV421 | M1/70 | 101235 | Biolegend |
| 122 | Anti-mouse/human CD11b | BV650 | M1/70 | 101239 | Biolegend |
| 123 | Anti-mouse/human CD11b | Alexa Fluor® 700 | M1/70 | 101222 | Biolegend |
| 124 | Anti-mouse/human CD44 | PE/Cyanine5 | IM7 | 103009 | Biolegend |
| 125 | Anti-mouse/human CD44 | BV650 | IM7 | 103049 | Biolegend |
| 126 | Anti-mouse/human CD44 | PE/Cyanine7 | IM7 | 103029 | Biolegend |
| 127 | Anti-mouse/human CD44 | BV785 | IM7 | 103059 | Biolegend |
| 128 | Anti-mouse/human CD44 | BUV563 | IM7 | 365-0441-82 | ThermoFisher |
| 129 | Anti-mouse/human KLRG1 (MAFA) | APC/Cyanine7 | 138425 | 2F1/KLRG1 | Biolegend |
| 130 | Anti-mouse/human TCF1/TCF7 | Alexa Fluor® 488 | C63D9 | 6444S | Cell Signaling |
| 131 | Anti-mouse/human TCF1/TCF7 | PE-Cy7 | C63D9 | 90511S | Cell Signaling |
| 132 | Anti-mouse/human TCF1/TCF7 | PE | C63D9 | 14456S | Cell Signaling |
| 133 | Anti-mouse/human TCF1/TCF7 | Alexa Fluor® 700 | C63D9 | 90904S | Cell Signaling |
| 134 | Anti-mouse/rat Bcl-2 | PE/Cyanine7 | BCL/10C4 | 633512 | Biolegend |
Bulk RNA sequencing
For bulk PBMC RNA-seq analysis, RNA was isolated using RNeasy Mini kit (Qiagen, #74104), polyA+ library preparation performed and followed by 2×150 bp NovaSeq 6000 (Illumina) sequencing. For bulk RNA-seq analysis of sorted cells, RNA was isolated using Arcturus Picopure RNA kit (Thermo), converted to cDNA and subjected to amplification followed by 2×150 bp sequencing on the NovaSeq 6000 (Illumina).
Single-cell RNA-sequencing
Live single cells were isolated from peripheral blood mononuclear cells using flow cytometry. All samples were subjected to 3’ transcriptome single-cell library preparation using Chromium Single Cell Gene Expression kit version 3.1 (10X Genomics, Pleasanton, CA). For CITE-seq experiments, cells were first stained using TotalSeq B DNA barcoded antibodies (Biolegend) including cell hashing antibodies, then subjected to single-cell transcriptome and barcode library preparation using the Chromium Single Cell Gene Expression kit version 3.1 (10X Genomics, Pleasanton, CA). Libraries were sequenced using NextSeq 2000 or NovaSeq 6000 (Illumina, San Diego, CA) to an average depth of 10,000 reads per cell.
Bulk RNA-seq analysis
Reads were aligned to the mouse genome and genic reads quantified using STAR version 2.7.0f (Dobin et al., 2013) and Ensembl version 101 GRCm38 or GRCh38 genome and transcriptome annotations. Normalization, differential expression analysis, principal component analysis were performed using R package DESeq2 v1.35.0 (Love et al., 2014); heatmaps were constructed using R package ComplexHeatmap v2.12.0. Gene counts for publicly available studies GSE83978, GSE147910 were obtained from the Sequence Read Archive and analyzed using the same pipeline. Analyses were performed using R v4.2.0 (Team, 2008). Digital cytometry was performed using CIBERSORTx (87) in single-cell RNA-seq reference mode. The Seurat large PBMC dataset was used as the reference (88).
Single-cell RNA-seq analysis
Raw sequencing data were demultiplexed using cellranger mkfastq v6.1 (10X Genomics). Cellranger count v6.1 (10X Genomics) was used to identify cells, align cDNA reads, identify hashtag barcode – cell barcode reads, count reads per gene and produce a cell-gene count matrix. Cellranger-associated genome references (GRCh38 and mm10) were used. Filtering of low-quality cells, filtering of doublets, dimensionality reduction, clustering and visualization were performed using Seurat v4.0.1.
Statistical tests
All replicates in this study were biological replicates unless explicitly described otherwise. Statistical comparisons of experimental data were performed using tests appropriate for the type of data and comparison being made. Fisher’s exact test was used to compare the frequency of members of a specific class in two different groups, for example the frequency of JAK inhibitors among hit and non-hit compounds. Student’s two-tailed t-test was used to compare two groups with continuous data in which normality could be reasonably assumed. One-way ANOVA with Dunnett’s post-test was used to compare multiple groups with continuous data with respect to a single control group. One-way ANOVA with Sidak’s multiple comparisons post-test was used to compare selected groups in a multi-group experiments. The log-rank test was used to compare survival between groups. Two-way ANOVA was used to compare the rate of tumor growth between groups. The Coefficient of determination was used to measure correlation. Statistical normalization and differential expression analysis of bulk RNA-seq data was performed using DESeq2 using the default Wald test. DESeq2 was also used to perform differential expression analysis of scRNAseq data with replicates using pseudobulk counts. Student’s ratio paired test was used to assess the change in paired values of continuous variables before and after ruxolitinib treatment. The GSEA test was used to assess significance of gene set expression analysis. Statistical testing was performed using R version 4.2.2, Prism v10 (GraphPad Software, Boston, MA) or GSEA (89).
Supplementary Material
Acknowledgments:
The authors thank the patients, their families and study staff for participating in the clinical study. We also thank The Scripps Research Institute flow cytometry and genomics cores, The UCSD Moores Cancer Center histology core, University of Minnesota Translational Therapy Laboratory, T. Mondala and H. Nguyen for experimental assistance, S. Kaech for advice on data interpretation, A. Ribas for providing MC38 knockout cell lines, D. Lazar for sharing primary blood cells and L. Sherman for sharing LLC1 cells. The clinical trial was managed by The Big Ten Cancer Research Consortium Administrative Headquarters at Hoosier Cancer Research Network, Inc. 7676 Interactive Way, Suite 120, Indianapolis, IN 46278.
Funding:
Cancer Research Institute/Irvington Postdoctoral Fellowship CRI2904 (JZ)
National Institute of Health grant R01AI123210 (JRT)
National Institute of Health grant UL1TR002550 Pilot Award (JRT)
National Institute of Health grant R01AI164744 (JRT)
National Institute of Health grant R21AI141842 (LLL)
Incyte Corporation clinical funding IST-USA-000382 (VB)
Bristol- Myers Squibb clinical funding CA209-9EF (VB)
Footnotes
Data and materials availability:
Mouse transcriptomic studies have been deposited to GEO (GSE209647). Human transcriptomic studies have been deposited to dbGap (phs003601).
References
- 1.Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, Utikal J, Umansky V, Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Frontiers in immunology 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu X, Hogg GD, DeNardo DG, Rethinking immune checkpoint blockade: ‘Beyond the T cell’. Journal for ImmunoTherapy of Cancer 9, e001460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Moldawer LL, Parallels between Cancer and Infectious Disease. New England Journal of Medicine 371, 380–383 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion. Nature reviews. Immunology 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R, Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Norris Brian A., Uebelhoer Luke S., Nakaya Helder I., Price Aryn A., Grakoui A, Pulendran B, Chronic but Not Acute Virus Infection Induces Sustained Expansion of Myeloid Suppressor Cell Numbers that Inhibit Viral-Specific T Cell Immunity. Immunity 38, 309–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snell LM, McGaha TL, Brooks DG, Type I Interferon in Chronic Virus Infection and Cancer. Trends in Immunology 38, 542–557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde S, Leader AM, Merad M, MDSC: Markers, development, states, and unaddressed complexity. Immunity 54, 875–884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veglia F, Sanseviero E, Gabrilovich DI, Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nature Reviews Immunology, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Haas N, de Koning C, Spilgies L, de Vries IJM, Hato SV, Improving cancer immunotherapy by targeting the STATe of MDSCs. OncoImmunology 5, e1196312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Victoire C-R, Naima B, Luisa D, Wang SJ, Lauren KB, Joshua R, Keheler CE, Keri MS, Thomas AA, Leah HB, Peter CE, Nadine JM, Anuj KP, Douglas AR, Benjamin S, Sarah S, Matthew BY, James MC, Kimberly P, Michael D, Kimmie N, Brian MW, Harshabad S, Stephanie KD, G-CSF rescue of FOLFIRINOX-induced neutropenia leads to systemic immune suppression in mice and humans. Journal for ImmunoTherapy of Cancer 11, e006589 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law AMK, Valdes-Mora F, Gallego-Ortega D, Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maenhout SK, Du Four S, Corthals J, Neyns B, Thielemans K, Aerts JL, AZD1480 delays tumor growth in a melanoma model while enhancing the suppressive activity of myeloid-derived suppressor cells. Oncotarget; Vol 5, No 16, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sendo S, Saegusa J, Yamada H, Nishimura K, Morinobu A, Tofacitinib facilitates the expansion of myeloid-derived suppressor cells and ameliorates interstitial lung disease in SKG mice. Arthritis Research & Therapy 21, 184 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao W, Klement JD, Lu C, Ibrahim ML, Liu K, IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. The Journal of Immunology 201, 264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A, The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annual Review of Medicine 66, 311–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka Y, Luo Y, O’Shea JJ, Nakayamada S, Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nature Reviews Rheumatology 18, 133–145 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marro BS*, Zak J*, Zavareh RB, Teijaro JR, Lairson LL, Oldstone MBA, Discovery of Small Molecules for the Reversal of T Cell Exhaustion. Cell Reports 29, 3293–3302.e3293 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, Griffith M, DGIdb 3.0: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Research 46, D1068–D1073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakafi (ruxolitinib) [package insert]. U. S. Food and Drug Administration website. 2019. [Google Scholar]
- 21.Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, Rodgers JD, Haley P, Kantarjian H, Fridman JS, Verstovsek S, Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115, 3109–3117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P, Simultaneous epitope and transcriptome measurement in single cells. Nature Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elli EM, Baratè C, Mendicino F, Palandri F, Palumbo GA, Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Frontiers in Oncology 9, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, Schreiber RD, Carlos de la Torre J, Oldstone MBA, Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling. Science 340, 207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG, Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection. Science 340, 202–207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Ruiz E, Minute L, Otano I, Alvarez M, Ochoa MC, Belsue V, de Andrea C, Rodriguez-Ruiz ME, Perez-Gracia JL, Marquez-Rodas I, Llacer C, Alvarez M, de Luque V, Molina C, Teijeira A, Berraondo P, Melero I, Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 569, 428–432 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, Gangadhar TC, Amaravadi RK, Schuchter LM, Feldman MD, Ishwaran H, Vonderheide RH, Maity A, Wherry EJ, Minn AJ, Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554.e1512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI, Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature Communications 7, 12150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, Khalilnezhad S, Irac SE, Dubuisson A, Chevrier M, Zhang XM, Tam JKC, Lim TKH, Wong RMM, Pai R, Khalil AIS, Chow PKH, Wu SZ, Al-Eryani G, Roden D, Swarbrick A, Chan JKY, Albani S, Derosa L, Zitvogel L, Sharma A, Chen J, Silvin A, Bertoletti A, Blériot C, Dutertre C-A, Ginhoux F, Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 54, 1883–1900.e1885 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, Grassi F, Bronte V, Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. European Journal of Immunology 40, 22–35 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Abrams SI, Waight JD, Identification of a G-CSF-Granulocytic MDSC axis that promotes tumor progression. OncoImmunology 1, 550–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grieshaber-Bouyer R, Radtke FA, Cunin P, Stifano G, Levescot A, Vijaykumar B, Nelson-Maney N, Blaustein RB, Monach PA, Nigrovic PA, Aguilar O, Allan R, Astarita J, Austen KF, Barrett N, Baysoy A, Benoist C, Brown BD, Buechler M, Buenrostro J, Casanova MA, Chowdhary K, Colonna M, Crowl T, Deng T, Desland F, Dhainaut M, Ding J, Dominguez C, Dwyer D, Frascoli M, Gal-Oz S, Goldrath A, Johanson T, Jordan S, Kang J, Kapoor V, Kenigsberg E, Kim J, Kim K. w., Kiner E, Kronenberg M, Lanier L, Laplace C, Lareau C, Leader A, Lee J, Magen A, Maier B, Maslova A, Mathis D, McFarland A, Merad M, Meunier E, Monach PA, Mostafavi S, Muller S, Muus C, Ner-Gaon H, Nguyen Q, Novakovsky G, Nutt S, Omilusik K, Ortiz-Lopez A, Paynich M, Peng V, Potempa M, Pradhan R, Quon S, Ramirez R, Ramanan D, Randolph G, Regev A, Rose SA, Seddu K, Shay T, Shemesh A, Shyer J, Smilie C, Spidale N, Subramanian A, Sylvia K, Tellier J, Turley S, Vijaykumar B, Wagers A, Wang C, Wang PL, Wroblewska A, Yang L, Yim A, Yoshida H, ImmGen C, The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nature Communications 12, 2856 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alshetaiwi H, Pervolarakis N, McIntyre Laura L, Ma D, Nguyen Q, Rath Jan A, Nee K, Hernandez G, Evans K, Torosian L, Silva A, Walsh C, Kessenbrock K, Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Science Immunology 5, eaay6017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu B, Yang L, Song S, Li W, Wang H, Cheng J, LRG1 facilitates corneal fibrotic response by inducing neutrophil chemotaxis via Stat3 signaling in alkali-burned mouse corneas. American Journal of Physiology-Cell Physiology 321, C415–C428 (2021). [DOI] [PubMed] [Google Scholar]
- 35.de Lima CB, Tamura EK, Montero-Melendez T, Palermo-Neto J, Perretti M, Markus RP, Farsky SHP, Actions of translocator protein ligands on neutrophil adhesion and motility induced by G-protein coupled receptor signaling. Biochemical and Biophysical Research Communications 417, 918–923 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Shojaei F, Wu X, Zhong C, Yu L, Liang X-H, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RAD, Meng YG, Ferrara N, Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450, 825–831 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Curtis VF, Wang H, Yang P, McLendon RE, Li X, Zhou Q-Y, Wang X-F, A PK2/Bv8/PROK2 Antagonist Suppresses Tumorigenic Processes by Inhibiting Angiogenesis in Glioma and Blocking Myeloid Cell Infiltration in Pancreatic Cancer. PLOS ONE 8, e54916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang P, Zhang Y, Ru B, Yang Y, Vu T, Paul R, Mirza A, Altan-Bonnet G, Liu L, Ruppin E, Wakefield L, Wucherpfennig KW, Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nature Methods 18, 1181–1191 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torrejon DY, Abril-Rodriguez G, Champhekar AS, Tsoi J, Campbell KM, Kalbasi A, Parisi G, Zaretsky JM, Garcia-Diaz A, Puig-Saus C, Cheung-Lau G, Wohlwender T, Krystofinski P, Vega-Crespo A, Lee CM, Mascaro P, Grasso CS, Berent-Maoz B, Comin-Anduix B, Hu-Lieskovan S, Ribas A, Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discovery 10, 1140–1157 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheson BD, Ansell S, Schwartz L, Gordon LI, Advani R, Jacene HA, Hoos A, Barrington SF, Armand P, Refinement of the Lugano Classification lymphoma response criteria in the era of immunomodulatory therapy. Blood 128, 2489–2496 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Howard R, Kanetsky PA, Egan KM, Exploring the prognostic value of the neutrophil-to-lymphocyte ratio in cancer. Scientific Reports 9, 19673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Templeton AJ, McNamara MG, Šeruga B, Vera-Badillo FE, Aneja P, Ocaña A, Leibowitz-Amit R, Sonpavde G, Knox JJ, Tran B, Tannock IF, Amir E, Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. JNCI: Journal of the National Cancer Institute 106, (2014). [DOI] [PubMed] [Google Scholar]
- 43.Romano A, Parrinello NL, Vetro C, Chiarenza A, Cerchione C, Ippolito M, Palumbo GA, Di Raimondo F, Prognostic meaning of neutrophil to lymphocyte ratio (NLR) and lymphocyte to monocyte ration (LMR) in newly diagnosed Hodgkin lymphoma patients treated upfront with a PET-2 based strategy. Annals of Hematology 97, 1009–1018 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Tavakkoli M, Wilkins CR, Mones JV, Mauro MJ, A novel paradigm between leukocytosis, G-CSF secretion, neutrophil-to-lymphocyte ratio, myeloid-derived suppressor cells, and prognosis in non-small cell lung cancer. Frontiers in Oncology 9, 295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao Y, Wang J, Jiang S, Lyu M, Zhao F, Liu J, Wang M, Pei X, Zhai W, Feng X, Feng S, Han M, Xu Y, Jiang E, JAK1/2 inhibitor ruxolitinib promotes the expansion and suppressive action of polymorphonuclear myeloid-derived suppressor cells via the JAK/STAT and ROS-MAPK/NF-κB signalling pathways in acute graft-versus-host disease. Clinical & Translational Immunology 12, e1441 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon N, Antignani A, Hewitt SM, Gadina M, Alewine C, FitzGerald D, Tofacitinib enhances delivery of antibody-based therapeutics to tumor cells through modulation of inflammatory cells. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajappa P, Cobb WS, Vartanian E, Huang Y, Daly L, Hoffman C, Zhang J, Shen B, Yanowitch R, Garg K, Cisse B, Haddock S, Huse J, Pisapia DJ, Chan TA, Lyden DC, Bromberg JF, Greenfield JP, Malignant Astrocytic Tumor Progression Potentiated by JAK-mediated Recruitment of Myeloid Cells. Clinical Cancer Research 23, 3109–3119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heine A, Held SAE, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, Wolf D, Brossart P, The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 122, 1192 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Schönberg K, Rudolph J, Vonnahme M, Parampalli Yajnanarayana S, Cornez I, Hejazi M, Manser AR, Uhrberg M, Verbeek W, Koschmieder S, Brümmendorf TH, Brossart P, Heine A, Wolf D, JAK Inhibition Impairs NK Cell Function in Myeloproliferative Neoplasms. Cancer Research 75, 2187–2199 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Parampalli Yajnanarayana S, Stübig T, Cornez I, Alchalby H, Schönberg K, Rudolph J, Triviai I, Wolschke C, Heine A, Brossart P, Kröger N, Wolf D, JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. British Journal of Haematology 169, 824–833 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, Parisi G, Azhdam A, Chmielowski B, Cherry G, Seja E, Berent-Maoz B, Shintaku IP, Le DT, Pardoll DM, Diaz LA Jr., Tumeh PC, Graeber TG, Lo RS, Comin-Anduix B, Ribas A, Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discovery 7, 188–201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan B, Shang L, Liu C, Gao J, Zhang F, Xu M, Li L, Sun Z, Li Z, Xu K, PD-1 antibody and ruxolitinib enhances graft-versus-lymphoma effect without increasing acute graft-versus-host disease in mice. American Journal of Transplantation 21, 503–514 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Lu C, Talukder A, Savage NM, Singh N, Liu K, JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. OncoImmunology 6, e1291106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Debureaux PE, Arrondeau J, Bouscary D, Goldwasser F, Nivolumab combined with ruxolitinib: antagonism or synergy? Annals of Oncology 29, 1334–1335 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Nijland M, van Meerten T, Seitz A, Huls G, Kibbelaar R, Visser L, van den Berg A, Diepstra A, Combined PD-1 and JAK1/2 inhibition in refractory primary mediastinal B-cell lymphoma. Annals of Hematology 97, 905–907 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Lussana F, Cattaneo M, Rambaldi A, Squizzato A, Ruxolitinib-associated infections: A systematic review and meta-analysis. American Journal of Hematology 93, 339–347 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Winthrop KL, The emerging safety profile of JAK inhibitors in rheumatic disease. Nature Reviews Rheumatology 13, 234–243 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Eric Van Den N, Marc A, Thomas G, Aspasia S, Corinne H, Amine B, Oumedaly R, Olivier C, Hervé G, Gregor V, Marie-José C, Hélène AP, Marie-Christine C, Romain D, Peter V, Ioanna-Andrea S, Anne SC, Sarah B, Laurent K, Franck M, A phase II study of the oral JAK1/JAK2 inhibitor ruxolitinib in advanced relapsed/refractory Hodgkin lymphoma. Haematologica 103, 840–848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gillessen S, Pluetschow A, Vucinic V, Ostermann H, Kobe C, Bröckelmann PJ, Böll B, Eichenauer DA, Heger J-M, Borchmann S, Fuchs M, Borchmann P, Engert A, von Tresckow B, JAK Inhibition with Ruxolitinib in Relapsed or Refractory Classical Hodgkin Lymphoma: Final Results of A Phase II, Open Label, Multicenter Clinical Trial (JeRiCHO). European Journal of Haematology n/a, (2022). [DOI] [PubMed] [Google Scholar]
- 60.Kim SJ, Yoon DH, Kang HJ, Hong JY, Lee HS, Oh SY, Shin H-J, Kong JH, Yi JH, Sakamoto K, Ko YH, Huh J, Lee S-S, Takeuchi K, Shin D-Y, Suh C, Kim WS, Ruxolitinib shows activity against Hodgkin lymphoma but not primary mediastinal large B-cell lymphoma. BMC Cancer 19, 1080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramchandren R, Domingo-Domènech E, Rueda A, Trněný M, Feldman TA, Lee HJ, Provencio M, Sillaber C, Cohen JB, Savage KJ, Willenbacher W, Ligon AH, Ouyang J, Redd R, Rodig SJ, Shipp MA, Sacchi M, Sumbul A, Armand P, Ansell SM, Nivolumab for Newly Diagnosed Advanced-Stage Classic Hodgkin Lymphoma: Safety and Efficacy in the Phase II CheckMate 205 Study. Journal of Clinical Oncology 37, 1997–2007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, Kutok JL, Shipp MA, Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116, 3268–3277 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roemer MGM, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, Connelly CF, Sun HH, Daadi SE, Freeman GJ, Armand P, Chapuy B, de Jong D, Hoppe RT, Neuberg DS, Rodig SJ, Shipp MA, PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. Journal of Clinical Oncology 34, 2690–2697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koh YW, Park CS, Yoon DH, Suh C, Huh J, CD163 expression was associated with angiogenesis and shortened survival in patients with uniformly treated classical Hodgkin lymphoma. PLoS One 9, e87066 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoon DH, Koh YW, Kang HJ, Kim S, Park CS, Lee SW, Suh C, Huh J, CD68 and CD163 as prognostic factors for Korean patients with Hodgkin lymphoma. Eur J Haematol 88, 292–305 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Kumar V, Patel S, Tcyganov E, Gabrilovich DI, The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends in Immunology 37, 208–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Romano A, Pavoni C, Di Raimondo F, Tarella C, Viviani S, Rossi A, Patti C, Picardi M, Cantonetti M, La Nasa G, Trentin L, Bolis S, Zoli V, Gavarotti P, Corradini P, Cimminiello M, Schiavotto C, Parvis G, Zanotti R, Gini G, Ferreri AJM, Viero P, Chauvie S, Biggi A, Massimo Gianni A, Gallamini A, Rambaldi A, The neutrophil to lymphocyte ratio (NLR) and the presence of large nodal mass are independent predictors of early response: A subanalysis of the prospective phase II PET-2-adapted HD0607 trial. Cancer Medicine 9, 8735–8746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sacdalan DB, Lucero JA, Sacdalan DL, Prognostic utility of baseline neutrophil-to-lymphocyte ratio in patients receiving immune checkpoint inhibitors: a review and meta-analysis. Onco Targets Ther 11, 955–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hwang M, Canzoniero JV, Rosner S, Zhang G, White JR, Belcaid Z, Cherry C, Balan A, Pereira G, Curry A, Niknafs N, Zhang J, Smith KN, Sivapalan L, Chaft JE, Reuss JE, Marrone K, Murray JC, Li QK, Lam V, Levy BP, Hann C, Velculescu VE, Brahmer JR, Forde PM, Seiwert T, Anagnostou V, Peripheral blood immune cell dynamics reflect antitumor immune responses and predict clinical response to immunotherapy. Journal for ImmunoTherapy of Cancer 10, e004688 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathew D, Marmarelis ME, Foley C, Bauml JM, Ye D, Ghinnagow R, Foong Ngiow S, Klapholz M, Jun S, Zhang Z, Zorc R, Davis CW, Diehn M, Giles JR, Huang AC, Wei-Ting H, Zhang NR, Schoenfeld AJ, Langer CJ, Wherry EJ, Minn Andy J., Combined JAK Inhibition and PD-1 Immunotherapy for Non-Small Cell Lung Cancer Patients. In Press, (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM, Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342, 559–561 (1989). [DOI] [PubMed] [Google Scholar]
- 72.Welsh RM, Seedhom MO, Lymphocytic Choriomeningitis Virus (LCMV): Propagation, Quantitation, and Storage. Current Protocols in Microbiology 8, 15A.11.11–15A.11.11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Battegay M, Cooper S, Althage A, Banziger J, Hengartner H, Zinkernagel RM, Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J. Virol. Methods 33, 191–198 (1991). [DOI] [PubMed] [Google Scholar]
- 74.Janes J, Young ME, Chen E, Rogers NH, Burgstaller-Muehlbacher S, Hughes LD, Love MS, Hull MV, Kuhen KL, Woods AK, Joseph SB, Petrassi HM, McNamara CW, Tremblay MS, Su AI, Schultz PG, Chatterjee AK, The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proceedings of the National Academy of Sciences 115, 10750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gairin JE, Mazarguil H, Hudrisier D, Oldstone MB, Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. Journal of Virology 69, 2297 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oxenius A, Bachmann MF, Ashton-Rickardt PG, Tonegawa S, Zinkernagel RM, Hengartner H, Presentation of endogenous viral proteins in association with major histocompatibility complex class II: On the role of intracellular compartmentalization, invariant chain and the TAP transporter system. European Journal of Immunology 25, 3402–3411 (1995). [DOI] [PubMed] [Google Scholar]
- 77.Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, Horn LA, Palena C, Schlom J, Maeda DY, Zebala JA, Clavijo PE, Allen C, Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clinical Cancer Research 26, 1420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, McCurdy SP, Kudlacz EM, Conklyn MJ, Elliott EA, Koslov ER, Fisher MB, Strelevitz TJ, Yoon K, Whipple DA, Sun J, Munchhof MJ, Doty JL, Casavant JM, Blumenkopf TA, Hines M, Brown MF, Lillie BM, Subramanyam C, Shang-Poa C, Milici AJ, Beckius GE, Moyer JD, Su C, Woodworth TG, Gaweco AS, Beals CR, Littman BH, Fisher DA, Smith JF, Zagouras P, Magna HA, Saltarelli MJ, Johnson KS, Nelms LF, Des Etages SG, Hayes LS, Kawabata TT, Finco-Kent D, Baker DL, Larson M, Si M-S, Paniagua R, Higgins J, Holm B, Reitz B, Zhou Y-J, Morris RE, Shea JJ, Borie DC, Prevention of Organ Allograft Rejection by a Specific Janus Kinase 3 Inhibitor. Science 302, 875 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Wu S-C, Li Loretta S., Kopp N, Montero J, Chapuy B, Yoda A, Christie Amanda L., Liu H, Christodoulou A, van Bodegom D, van der Zwet J, Layer Jacob V., Tivey T, Lane Andrew A., Ryan Jeremy A., Ng Samuel Y., DeAngelo Daniel J., Stone Richard M., Steensma D, Wadleigh M, Harris M, Mandon E, Ebel N, Andraos R, Romanet V, Dölemeyer A, Sterker D, Zender M, Rodig Scott J., Murakami M, Hofmann F, Kuo F, Eck Michael J., Silverman Lewis B., Sallan Stephen E., Letai A, Baffert F, Vangrevelinghe E, Radimerski T, Gaul C, Weinstock David M., Activity of the Type II JAK2 Inhibitor CHZ868 in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 28, 29–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M, The JAK2 Inhibitor AZD1480 Potently Blocks Stat3 Signaling and Oncogenesis in Solid Tumors. Cancer Cell 16, 487–497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, Lepescheux L, Christophe T, Conrath K, Vandeghinste N, Vayssiere B, De Vos S, Fletcher S, Brys R, van ‘t Klooster G, Feyen JHM, Menet C, Preclinical Characterization of GLPG0634, a Selective Inhibitor of JAK1, for the Treatment of Inflammatory Diseases. The Journal of Immunology 191, 3568 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Ito M, Yamazaki S, Yamagami K, Kuno M, Morita Y, Okuma K, Nakamura K, Chida N, Inami M, Inoue T, Shirakami S, Higashi Y, A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. Journal of Pharmacological Sciences 133, 25–33 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Gonzales AJ, Bowman JW, Fici GJ, Zhang M, Mann DW, Mitton-Fry M, Oclacitinib (APOQUEL®) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. Journal of Veterinary Pharmacology and Therapeutics 37, 317–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, Finke C, Mak CC, Mesa R, Zhu H, Soll R, Gilliland DG, Tefferi A, TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 21, 1658–1668 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Baffert F, Régnier CH, De Pover A, Pissot-Soldermann C, Tavares GA, Blasco F, Brueggen J, Chène P, Drueckes P, Erdmann D, Furet P, Gerspacher M, Lang M, Ledieu D, Nolan L, Ruetz S, Trappe J, Vangrevelinghe E, Wartmann M, Wyder L, Hofmann F, Radimerski T, Potent and Selective Inhibition of Polycythemia by the Quinoxaline JAK2 Inhibitor NVP-BSK805. Molecular Cancer Therapeutics 9, 1945 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Pardanani A, Roberts AW, Seymour JF, Burbury K, Verstovsek S, Kantarjian HM, Begna K, Yoshitsugu H, Gestone TA, Phillips P, Xing G, Peltz G, Lorenzi MV, Alland L, Woolfson A, Tefferi A, BMS-911543, A Selective JAK2 Inhibitor: A Multicenter Phase 1/2a Study In Myelofibrosis. Blood 122, 664 (2013). [Google Scholar]
- 87.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA, Determining cell type abundance and expression from bulk tissues with digital cytometry. Nature Biotechnology 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse transcriptomic studies have been deposited to GEO (GSE209647). Human transcriptomic studies have been deposited to dbGap (phs003601).