

Abstract
The fluoroalkyl-containing organic molecules are widely used in drug discovery and material science. Herein, we report ligand regulated nickel(0)-catalyzed regiodivergent hydrosilylation of α-(fluoroalkyl)styrenes without defluorination, providing an atom- and step-economical synthesis route of two types of fluoroalkyl substituted silanes with exclusive regioselectivity. The anti-Markovnikov addition products (β-fluoroalkyl substituted silanes) are formed with monodentate phosphine ligand. Noteworthy, the bidentate phosphine ligand promote the generation of the more challenging Markovnikov products (α-fluoroalkyl substituted silanes) with tetrasubstituted saturated carbon centers. This protocol features with easy available starting materials and commercially available nickel catalysis, a wide range of substrates and excellent regioselectivity. The structure divergent products undergo a variety of transformations. Comprehensive mechanistic studies including the inverse kinetic isotope effects demonstrate the regioselectivity controlled by ligand structure through α-CF3 nickel intermediate. DFT calculations reveal a distinctive mechanism involving an open-shell singlet state, which is crucial for generating intricate tetra-substituted Markovnikov products.
Subject terms: Synthetic chemistry methodology, Catalyst synthesis, Catalyst synthesis
The fluoroalkyl-containing organic molecules are widely used in drug discovery and material science. Herein, the authors report ligand regulated nickel(0)-catalyzed regiodivergent hydrosilylation of α-(fluoroalkyl)styrenes without defluorination.
Introduction
Organofluorine compounds have found extensive applications in the area of pharmaceuticals, agrochemicals, and materials science1–4. The incorporation of fluoroalkyl group, particularly the trifluoromethyl group (CF3) into small organic moleculars has a privileged role in drug development and agrochemical industry, which could improve its lipophilicity metabolic stability and bioacitivity1–6. However, compared to the well-developed C(sp2)–CF3 and C(sp)–CF3 bonds formation7–17, the construction of C(sp3)–CF3 bond with transition-metal catalysis still lags behind18–23. Specifically, the efficient and economical synthesis of products with tetrasubstituted carbon centers, which contain C(sp3)–CF3 bonds, is still highly desirable and remains a formidable challenge. One attractive strategy for the synthesis of alkyl-CF3 compounds would be the direct transformation of α-CF3 transition-metal intermediates, but much more challenging due to the thermodynamically favored β-F elimination18–20.
Recently, significant advancements have been made by using the α-(trifluoromethyl)styrenes to synthesize high-value fluorinated organic compounds24–26. While, most of these reports focus on the formation of gem-difluoroalkenes and their derivatives via C–F bond cleavage (Fig. 1A)27. In the last few years, very limited examples to give CF3-containing products using transition-metal catalysts. These include the carbene insertion (Rh or Fe complex), cycloaddition (Pd catalysis), or hydroboration processes (Co or Ir catalysis)28–33. The transformation of α-(trifluoromethyl)styrenes through α-CF3 transition-metal intermediate to give alkyl-CF3 compounds is still extremely rare and remains elusive for the construction of tetrasubstituted carbon centers.
Fig. 1. The transformation of α-(trifluoromethyl)styrenes by transition-metal catalysis.

A Previous strategies for the transformation of α-(trifluoromethyl)styrenes. B The transformation of α-(trifluoromethyl)styrenes with Ni-H species. C The divergent synthesis of CF3-containing silanes without defluorination by Nickel catalysis.
On the other hand, the transition-metal catalyzed hydrosilylation of alkenes is an atom-economic and appealing approach for the preparation of organosilanes, which find broad applications in materials science and medicinal chemistry34–37. Compared to the noble-metal catalysts (such as Pt, Rh, and Pd), the inexpensive and Earth-abundant first-row catalysts (such as Fe, Co, and Ni) have gained great attention over the past few dacades38–50. When terminal alkenes were used in these reactions, the regio control to deliver Markovnikov or anti-Markovnikov addition products is one of the most important issues51–60. The regiodivergent hydrosilylation would be a practical tool for the rapid construction of value-added organosilanes. In 2022, the Lu group developed a Co-catalyzed regiodivergent hydrosilylation of in situ generated α-substituted vinylsilanes for the synthesis of vicinal and geminal bis(silane)s61. With nickel catalysis, linear products were usually favorable, especially for the low reactive 1, 1-disubstituted alkenes45–50,62,63. In 2020, the Norton group reported the insertion of pincer ligated Ni(II)-H species to α-(trifluoromethyl)styrenes with silanes, only delivering the gem-difluoroalkene products through the β-F elimination process (Fig. 1B)64. Herein, we report the ligand-controlled Ni(0)-catalyzed regiodivergent hydrosilylation of α-(fluoroalkyl)styrenes with silanes, enabling the synthesis of two types of fluoroalkylsilanes with excellent regioselectivity. The anti-Markovnikov addition products were formed with monodentate phosphine ligand. In contrast, the bidentate phosphine ligand promoted the Markovnikov products with α-fluoroalkyl-containing tetrasubstituted saturated carbon centers (Fig. 1C). The trifluoromethyl group is similar in size to an isopropyl group and distinct from other alkyl group65,66. So, to overcome the steric hindrance of the trifluoromethyl group and avoid the β-fluorine elimination would be the two key challenges, especially for tetrasubstituted carbon center formation.
Results and discussion
Reaction investigations
To pursue the goal, we initiated our studies using α-(trifluoromethyl)styrene 1a and Ph2SiH2 2a as model substrates in the presence of Nickel catalysis (Table 1, for more details in Supplementary Table 1). The anti-Markovnikov addition product 3a could be obtained in 18% yield by Ni(cod)2 catalysis without the addition of any ligand (entry 1), and 17% yield with >20:1 L:B regioselectivity when PPh3 was used as ligand at 30 °C (entry 2). Notably, altering the Ni/PPh3 equivalent ratio from 1:2 to 1:1 resulted in a notable increase in the yield of 3a to 96% with >20:1 L:B value (entry 3). This 1:1 ratio might be beneficial for the generation of a highly reactive nickel catalyst. We then investigated other commercial available ligands (such as PCy3, Bpy, 1, 10-phenanthroline, L1, L2, and L3), and found that all of these ligands gave anti-Markovnikov addition product 3a or the recovery of starting materials (entries 4–9). Noteworthy, when bidentate phosphine ligand BINAP was used, the more challenging Markovnikov product 4a was obtained with >20:1 B:L regioselectivity, and no by-products from β-F elimination were observed in the process (entries 10–13). The amount of BINAP in the system was significantly important for the efficiency, and 4a could be obtained in 96% yield with >20:1 B:L ratio (entry 11). When the equivalent of BINAP ligand was slightly lower than Nickel, higher efficiency was observed (entry 12), indicating the equivalent of BINAP is significant for maintaining high efficiency with an appropriate Nickel:BINAP ratio. This emphasizes the importance of optimal balance for generating an efficiently active nickel catalyst. Then, we tested the solvent effect, and found that toluene gave the best result (entries 14–19). The ee of 4a could be obtained in 45% value when chiral ligand (R)-BINAP was used under the reaction conditions of entry 11. Some other chiral ligands and solvent effects were also tested, but no better result was obtained (for more details in Supplementary Table 2). Low temperature and one equivalent of Ph2SiH2 led to diminished yield of desired products (entries 20–22). Control experiments showed that the nickel catalysis were essential for the transformation under the conditions of entry 3 or entry 11 (Supplementary Table 1 for details).
Table 1.
Optimization of reaction conditionsa
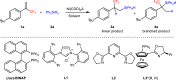 | ||||||
|---|---|---|---|---|---|---|
| Entry | L | L (%) | Solvent | T/(oC) | Yield (%) | L:B (3a:4a) |
| 1 | — | — | toluene | 30 | 18 | >20:1 |
| 2 | PPh3 | 10 mol% | toluene | 30 | 17 | >20:1 |
| 3 | PPh3 | 5 mol% | toluene | 30 | 96 | >20:1 |
| 4 | PCy3 | 5 mol% | toluene | 30 | 38 | >20:1 |
| 5 | BPY | 5 mol% | toluene | 30 | <5 | — |
| 6 | 1,10-phen | 5 mol% | toluene | 30 | <5 | 7:1 |
| 7b | L1 | 5 mol% | toluene | 30 | 81 | >20:1 |
| 8 | L2 | 5 mol% | toluene | 30 | 20 | >20:1 |
| 9 | L3 | 5 mol% | toluene | 30 | trace | — |
| 10 | BINAP | 8 mol% | toluene | 30 | NR | — |
| 11 | BINAP | 5 mol% | toluene | 30 | 96 | >1:20 |
| 12 | BINAP | 4.7 mol% | toluene | 30 | 96 | >1:20 |
| 13 | BINAP | 2.5 mol% | toluene | 30 | 90 | 1:9 |
| 14 | BINAP | 5 mol% | DMA | 30 | trace | — |
| 15 | BINAP | 5 mol% | MeOtBu | 30 | 59 | >1:20 |
| 16 | BINAP | 5 mol% | dioxane | 30 | 95 | >1:20 |
| 17 | BINAP | 5 mol% | DCE | 30 | NR | — |
| 18 | BINAP | 5 mol% | THF | 30 | <5 | — |
| 19 | BINAP | 5 mol% | EtOH | 30 | NR | — |
| 20 | BINAP | 5 mol% | toluene | 0 | NR | — |
| 21c | BINAP | 5 mol% | toluene | 30 | 60 | >1:20 |
| 22c | PPh3 | 5 mol% | toluene | 30 | 81 | >20:1 |
aReaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), Ni(cod)2 (5 mol%), L (x mol%), 30 oC in solvent (2.0 mL) under argon, 24 h, L:B value was determined by 19F NMR, isolated yield.
bCsOAc (20 mol%).
c2a (0.2 mmol).
Substrates scope studies
With the optimized reaction conditions in hand, we next explored the substrate scope of this regiodivergent hydrosilylation reaction. The formation of anti-Markovnikov addition products were investigated first (Fig. 2). A variety of α-(trifluoromethyl)styrenes bearing functional groups such as electron-donating, electron-withdrawing and halide substituents at the para-position of the benzene ring, giving consistently products in 96–99% yield (3a-3h, L:B 7:1 to L:B > 20:1). The reaction can also tolerate reactive groups such as esters (3i, 99% yield and 3ii, 89% yield), amine (3j, 54% yield) and triflate (3k, 42% yield). Other reduction-sensitive functional groups, such as aldehydes, ketones, and imines were not compatible for this hydrosilylation. These meta- substituted α-(trifluoromethyl)styrenes gave the corresponding products in excellent yield with excellent regioselectivity (3l, 94% yield, L:B 17:1 and 3 m, 57% yield, L:B > 20:1). The ortho-substituted alkene gave lower yield of the desired product (3n, 40% yield, L:B > 20:1), potentially attributed to steric effect. Other (hetero)aryl-substituted and biaryl alkenes gave the corresponding products 3o-3t in 96–99% yield. The structure of 3 s was confirmed by X-ray crystallography (CCDC 2214736). The NHC ligand L1 was employed for the electron-rich (hetero)aryl-substituted alkenes to give linear products 3u-3x in 56–99% yield and excellent regioselectivity. Pleasingly, the N-heterocycles containing alkenes such as quinoline and indole also reacted smoothly to provide corresponding products (3y, 55% yield, L:B = 9:1 and 3z, 89% yield, L:B = 18:1). High activity was realized for most substrates. Other fluoroalkyl groups (perfluoroethyl, perfluoropropyl, and difluoroethyl) substituted alkenes also underwent this hydrosilylation to produce β-fluoroalkyl substituted silanes in accepted yield (3aa-3ac, 54–80% yield) and excellent regioselectivity (L:B > 20:1). The reaction also tolerated for PhMeSiH2 and gave the anti-Markovnikov product 3ad in 79% yield but with 1:1 dr ratio of the relative stereochemistry between CF3-containing carbon and Si center. However, the reaction of (3-(trifluoromethyl)but-3-en-1-yl)benzene, Ph3SiH, PhSiH3 all failed to give the desired product and starting materials were recovered even at elevated temperature.
Fig. 2. Substrate scope for β-fluoroalkyl substituted silanes.

a Reaction conditions A: [a] 1 (0.2 mmol), [Si]-H 2 (0.4 mmol), Ni(cod)2 (5 mol%), PPh3 (5 mol%), 30 °C, in toluene (2.0 mL) under argon, 24 h, L:B value was determined by 19F NMR, isolated yield. [b] 60 °C. [c] L1 (5 mol%) and CsOAc (20 mol%) were used instead of PPh3.
We then explored the generality of the more challenging Markovnikov addition product systems. The substrate scope was found to be broad. As shown in Fig. 3, the reaction exhibited compatibility with various functional groups at the para-position of the benzene ring and gave the tetrasubstituted saturated silanes with excellent regioselectivity (4a-4h, 86–99% yield, B:L > 20:1). The reduction-sensitive functional groups such as aldehydes, ketones, imines, and amines proved to be incompatible. The hydrosilylation of alkene 1ii, featuring with a carboxylic ester substituent at the benzene ring, resulted in a low yield of the desired product (4ii). For the triflate-substituted alkenes, the desired hydrosilylation product 4k was obtained in 38% yield. The meta-substituted substrate also worked smoothly (4l, 99% yield, B:L > 20:1 and 4m, 31% yield, B:L = 9:1), while the ortho-substituted alkene with a methyl group led to the recovery of starting materials (4n), even at elevated temperature due to steric effect. Other (hetero)aryl-substituted and biaryl α-(trifluoromethyl)styrenes were also tolerated, giving the corresponding products in excellent yield and regioselectivity (4o-4u, 83–99% yield, B:L 5:1 to B:L > 20:1). The structure of 4t was confirmed by X-ray crystallography (CCDC 2214737). Other (hetero)aryl-substituted alkenes such as furan, thiophene, and indole were also compatible (4v, 54% yield, 4x, 56% yield, and 4z, 45% yield). We also briefly investigated alkenes with other fluoroalkyl substituents instead of CF3 group (4aa, 46% yield, B:L > 20:1 and 4ab, 40% yield, B:L > 20:1). The reaction of PhMeSiH2 gave the hydrosilylation product, but with poor regio- and diastereoselectivity (4ad, 51% yield, B:L (4ad:3ad) = 1:2.3, dr (3ad) = 1:1, dr (4ad) = 1:1). The (3-(trifluoromethyl)but-3-en-1-yl)benzene, Ph3SiH and PhSiH3 failed to give the Markovnikov addition products.
Fig. 3. Substrate scope for α-fluoroalkyl substituted silanes.

aReaction conditions B: [a] 1 (0.2 mmol), 2 (0.4 mmol), Ni(cod)2 (5 mol%), rac-BINAP (5.0 mol%), 30 °C, solvent (2.0 mL), argon, 24 h, L:B value was determined by 19F NMR, isolated yield. [b] rac-BINAP (4.7 mol%). [c] 60 °C.
Synthetic applications
The CF3-containing silane products from this divergent hydrosilylation could undergo a series of transformations (Fig. 4). These reactions were successfully scaled up to 2.0 mmol without the erosion of yield and selectivity under reaction conditions A or conditions B (Fig. 4A). The gem-difluoroalkenes are useful synthetic intermediates and present interesting bioactive properties67. When TBAF was employed, the silane 4r could be easily transformed into the gem-difluoroalkene 5. The silane 3r could be oxidized to alcohol 6 in 89% yield through the Fleming-Tamao oxidation process. Additionally, the silane 4r reacted with styrene through the dehydrogenative process to give product 7 in 75% yield, and oxidized to monohydroxysilane 8 in 85% yield. The visible-light-mediated C–Si bond cleavage for the conjugate addition of 4r with activated alkenes was realized, yielding the valuable CF3-containing products with a quaternary carbon center (9, 54% yield and 10, 56% yield) (Fig. 4B).
Fig. 4. Scale-up reaction and derivatization.

A Large scale experiments. B Derivatizations.
Mechanistic studies
We then performed several experiments to gain some insights into this regiodivergent hydrosilylation reaction mechanism (Fig. 5). The addition of 1, 1-diphenylethylene or BHT did not affect the yield and regioselectivity either under conditions A or conditions B. These results indicated a Chalk-Harrod reaction pathway rather than a radical pathway (Fig. 5A). EPR experiments also support these findings (for more details in Supplementary Figs. 19–22). When the CF3 group was replaced by the Me or iPr group, only linear product (11 or 12) was obtained no matter under the reaction conditions A or conditions B. These results indicated the trifluoromethyl group serves as a unique σ-electron-withdrawing group and is crucial to regioselectivity for the formation of Markovnikov products 4 (Fig. 5B). Moreover, the cyclopropyl-containing alkene gave the ring-opening product 13 under the conditions A. This regioselectivity is contrary to Me or iPr group substituted alkenes, probably due to the favorable β-C elimination of cyclopropyl substituent, indicating the reversible NiH insertion and β-H elimination (Fig. 5B). The formation of product 13 suggested the NiH insertion pathway rather than the Ni–Si insertion pathway. The deuterium-labeling experiments were performed to investigate the source of hydrogen using the deuterated silane Ph2SiD2. The deuterated product 3d-dn was obtained under the Reaction conditions A, showing the H/D exchanged for proton in the alkene with that in Ph2SiD2 (Fig. 5C). These results indicate that the Ni-D species (from Ph2SiD2) inserted into the alkene 1d and then β-H elimination gave NiH species when PPh3 was used as ligand. While, when BINAP was used as a ligand, the deuterated product 4d-dn was obtained under the Reaction conditions B with 93% deuterium incorporation at the silicon atom, and only 8% H/D exchange occurred in the methyl group. This result indicates that the β-H elimination process is unlikely to proceed with the BINAP ligand. In addition, the parallel KIE studies with BINAP ligand revealed a KIE value of 1.1, suggesting the Si–H bond cleavage is not the turn-over limiting step (Fig. 5D). While, when PPh3 was used as ligand, an unexpected inverse kinetic isotope effect was observed (KIE = 0.4), probably resulting from the generation of 1r-d. These results indicate that the NiH insertion and β-H elimination processes occur before the C–Si bond formation.
Fig. 5. Mechanistic studies.

A Radical-probe experiments. B The effect of trifluoromethyl group in the terminal alkene. C Deuterium-labeling experiments. D Parallel KIE studies.
DFT studies
To elucidate the origin of this unexpectedly excellent regioselectivity in the hydrosilylation of fluoroalkyl-alkenes51–54,68. we performed the density functional theory (DFT) calculations (Figs. 6, 7). We first investigated the detailed mechanism of Ni/BINAP system, which delivered the more challenging Markovnikov product. As shown in Fig. 6, the BINAP-bound-Ni(0) species INT1 was set to the relative zero energy. The Ni(0) species undergo oxidative addition of [Si]–H through the transition state TS1, with a calculated free energy barrier of 1.1 kcal mol−1. This process results in the formation of intermediate INT2 with a slight exergonicity of 4.6 kcal mol−1. Then, the α-(trifluoromethyl)styrene 1b coordinates to the NiH species INT2 and undergoes [1, 2]-insertion, giving intermediate INT4 through the transition state TS3 with an activation barrier of 16.8 kcal mol−1. Alternatively, the NiH [2, 1]-insertion results in the formation of intermediate INT3 through the transition state TS2 with a higher activation barrier of 22.8 kcal mol−1. The lower energy barrier observed in the [1, 2]-insertion compared to [2, 1]-insertion is attributed to the σ-withdrawing effect of the trifluoromethyl group. The subsequent C(sp3)–Si reductive elimination from intermediate INT4 occurs through the transition state TS4, resulting in an overall activation-free energy barrier of 29.2 kcal mol−1. After detailed investigation, we found that the open-shell singlet species INT4-oss through spin-state changes from closed-shell singlet species INT4 would undergo a lower overall activation barrier of 20.7 kcal mol−1 via transition state TS4-oss (which is lower than TS2). Finally, ligand exchange with Ph2SiH2 2a release the Markovnikov product 3b and the active catalytic species INT1 to complete the catalytic cycle. This unique open-shell singlet reaction mechanism is attributed to the d-d orbital transformation in the nickel center69 and steric repulsion between BINAP with the trifluoromethyl group70.
Fig. 6. DFT Studies for Markovnikov product 4.

DFT calculation for the Ni/BINAP system to generate Markovnikov product 4.
Fig. 7. DFT Studies for anti-Markovnikov product 3.

DFT calculations for the Ni/PPh3 system to generate anti-Markovnikov product 3.
Subsequently, a DFT calculation was elucidated on the reaction mechanism of the Ni/PPh3 system. As shown in Fig. 7, the monodentate phosphine ligand PPh3 bound-Ni(0) species INT5 was set to the relative zero energy. The oxidative addition of [Si]–H to the nickel center via transition state TS5 with a low energy barrier of 4.0 kcal mol−1, giving the intermediate INT6. The α-(trifluoromethyl)styrene 1b coordinates to the nickel center and gives INT7, which undergoes [2, 1]-insertion to give the intermediate INT8 through transition state TS6 with an activation barrier of 1.4 kcal mol−1. The following C(sp3)–Si reductive elimination from intermediate INT8 occurs through transition state TS7 with a very low energy barrier of 1.8 kcal mol−1. This process lead to the formation of the final anti-Markovnikov product 3b and the regeneration of active catalytic species INT5 through the coordination of another Ph2SiH2 2a. Alternatively, the Ni-H undergoes [1, 2]-insertion resulting in the formation of intermediate INT9 through transition state TS6’. However, the subsequent C(sp3)–Si reductive elimination from INT9 via transition state TS8 exhibits a high energy barrier of 18.5 kcal mol−1. The high selectivity of the generation of anti-Markovnikov product can be attributed to the elevated energy barrier for C(sp3)–Si reductive elimination energy barrier. DFT calculations of the open-shell singlet transition state TS8-oss reveal that the energy barrier for C(sp3)–Si reductive elimination in Ni/PPh3 system does not decrease but increase to 30.5 kcal mol−1. We also exclude the reaction pathway with two PPh3 ligand bound-Ni(0) species (for more details in Supplementary Fig. 40, and entry 2 vs 3 in Table 1). These DFT calculation results are also consistent with our experimental results.
In summary, we have developed ligand-controlled regiodivergent nickel(0)-catalyzed hydrosilylation of α-(fluoroalkyl)styrenes for the direct generation of fluoroalkyl-containing silanes in good to excellent yield. This approach provides a versatile platform for the selective formation of both α-trifluoromethylsilanes and β-trifluoromethylsilanes, utilizing only two readily available ligands. Mechanistic studies, including DFT calculations, have revealed that the trifluoromethyl group and the structure of the ligand significantly influence the regioselectivity. The distinctive spin-state changes from closed-shell singlet species to open-shell singlet are believed to form unusual tetrasubstituted saturated Markovnikov products. This study contributed to the rapid and divergent synthesis of C(sp3)–Rf (Rf = fluoroalkyl) containing compounds, and provided insights for further development of regio-controlled hydrosilylation involving steric hindrance alkenes.
Methods
General procedure for the generation of anti-Markovnikov addition product 3
General procedure A: Ni(cod)2 (2.8 mg, 0.01 mmol) and PPh3 (2.6 mg, 0.01 mmol) in toluene (2.0 mL) were charged into a 25 mL pressure tube under argon. The mixture was stirred for 30 min at room temperature, followed by the addition of Ph2SiH2 (78 mg, 0.4 mmol), after stirred for 20 min, α-(fluoroalkyl)styrenes 1 (0.2 mmol, 1.0 equiv) was added to the reaction mixture. The reaction tube was then sealed and placed in an oil bath at 30 °C. After stirred for 24 h, the reaction mixture was filtered through a pad of celite, eluted with ethyl acetate, concentrated and purified by silica gel chromatography (PE) to give the indicated product 3.
General procedure for the generation of Markovnikov addition product 4
General procedure B: Ni(cod)2 (2.8 mg, 0.01 mmol) and rac-BINAP (6.2 mg, 0.01 mmol) in toluene (2.0 mL) were charged into a 25 mL pressure tube under argon. The mixture was stirred for 30 min at room temperature, followed by addition of Ph2SiH2 (78 mg, 0.4 mmol), after stirred for 20 min, α-(fluoroalkyl)styrenes 1 (0.2 mmol, 1.0 equiv) was added to the reaction mixture. The reaction tube was then sealed and placed in an oil bath at 30 °C. After stirred for 24 h, the reaction mixture was filtered through a pad of celite, eluted with ethyl acetate, concentrated and purified by silica gel chromatography (PE) to give the indicated product 4.
More details and characterization of the products are available in Supplementary Information.
Supplementary information
Source data
Acknowledgements
This work is supported by the NSFC (Nos. 82130103 (J.C.), U1804283 (J.C.), 21801067 (D.B.)), the Central Plains Scholars and Scientists Studio Fund (2018002 (J.C.)), and the Project funded by the Natural Science Foundation of Henan (242300421351 (D.B.), 202300410225 (H.W.), 222102310562 (H.W.)) and Henan Postdoctoral Science Foundation (202103087 (H.W.)). We also thank the financial support from Henan Key Laboratory of Organic Functional Molecules and Drug Innovation.
Author contributions
D.B. initiated the project, designed and directed the project, completed product characterizations, and wrote the manuscript; L.C. and F.W. did some experiments and some analysis of products; K.Z. and Y.Q. did DFT calculation.; G.X., and J.C. supported the project and wrote the manuscript. J.C. also directed the project.
Peer review
Peer review information
Nature Communications thanks Genping Huang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files, and also available from the corresponding author. The NMR, experimental procedures and characterization for all products and mechanism studies are shown in Supplementary Information files. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2214736 (3 s), CCDC 2214737 (4t). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dachang Bai, Email: baidachang@htu.edu.cn.
Junbiao Chang, Email: changjunbiao@zzu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-50743-w.
References
- 1.Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science317, 1881–1886 (2007). 10.1126/science.1131943 [DOI] [PubMed] [Google Scholar]
- 2.Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev.37, 320–330 (2008). 10.1039/B610213C [DOI] [PubMed] [Google Scholar]
- 3.Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications. 2nd edn (Wiley-VCH, 2013).
- 4.Wang, J. et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001−2011). Chem. Rev.114, 2432–2506 (2014). 10.1021/cr4002879 [DOI] [PubMed] [Google Scholar]
- 5.Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of biosisosteres for drug design. J. Med. Chem.61, 5822–5880 (2018). 10.1021/acs.jmedchem.7b01788 [DOI] [PubMed] [Google Scholar]
- 6.Mei, H. B. et al. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett.31, 2401–2413 (2020). 10.1016/j.cclet.2020.03.050 [DOI] [Google Scholar]
- 7.Chu, L. L. & Qing, F. L. Copper-mediated arobic oxidative trifluoromethylation of terminal alkynes with Me3SiCF3. J. Am. Chem. Soc.132, 7262–7263 (2010). 10.1021/ja102175w [DOI] [PubMed] [Google Scholar]
- 8.Liang, T., Neumann, C. N. & Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed.52, 8214–8264 (2013). 10.1002/anie.201206566 [DOI] [PubMed] [Google Scholar]
- 9.Alonso, C., Marigorta, E. M., Rubiales, G. & Palacios, F. Carbon trifluoromethylation reactions of hydrocarbon derivatives and heteroarenes. Chem. Rev.115, 1847–1935 (2015). 10.1021/cr500368h [DOI] [PubMed] [Google Scholar]
- 10.Liu, X., Xu, C., Wang, M. & Liu, Q. Trifluoromethyltrimethylsilane: nucleophilic trifluoromethylation and beyond. Chem. Rev.115, 683–730 (2015). 10.1021/cr400473a [DOI] [PubMed] [Google Scholar]
- 11.Feng, Z., Xiao, Y. L. & Zhang, X. Transition-metal (Cu, Pd, Ni)-catalyzed difluoroalkylation via cross-coupling with difluoroalkylhalides. Acc. Chem. Res.51, 2264–2278 (2018). 10.1021/acs.accounts.8b00230 [DOI] [PubMed] [Google Scholar]
- 12.Ni, C. F., Hu, M. Y. & Hu, J. B. Good partnership between sulfur and fluorine: sulfur-based fluorination and fluoroalkylation reagents for organic synthesis. Chem. Rev.115, 765–825 (2015). 10.1021/cr5002386 [DOI] [PubMed] [Google Scholar]
- 13.Yang, X., Wu, T., Phipps, R. J. & Toste, F. D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev.115, 826–870 (2015). 10.1021/cr500277b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qing, F. L. et al. A fruitful decade of organofluorine chemistry: new reagents and reactions. CCS Chem.4, 2518–2549 (2022). 10.31635/ccschem.022.202201935 [DOI] [Google Scholar]
- 15.Zhang, C. P. et al. Copper-mediated trifluoromethylation of heteroaromatic compounds by trifluoromethyl sulfonium salts. Angew. Chem. Int. Ed.50, 1896–1900 (2011). 10.1002/anie.201006823 [DOI] [PubMed] [Google Scholar]
- 16.Bai, D. C., Wang, X. L., Zheng, G. F. & Li, X. W. Redox-divergent synthesis of fluoroalkylated pyridines and 2-pyridones through Cu-catalyzed N-O cleavage of oxime acetates. Angew. Chem. Int. Ed.57, 6633–6637 (2018). 10.1002/anie.201802311 [DOI] [PubMed] [Google Scholar]
- 17.Wang, X. et al. Controllable single and double difluoromethylene insertions into C-Cu bonds: copper-mediated tetrafluoroethylation and hexafluoropropylation of aryl iodides with TMSCF2H and TMSCF2Br. J. Am. Chem. Soc.144, 12202–12211 (2022). 10.1021/jacs.2c03104 [DOI] [PubMed] [Google Scholar]
- 18.Hu, M., Ni, C. & Hu, J. Copper-mediated trifluoromethylation of α-diazo esters with TMSCF3: the important role of water as a promoter. J. Am. Chem. Soc.134, 15257–15260 (2012). 10.1021/ja307058c [DOI] [PubMed] [Google Scholar]
- 19.Liang, Y. & Fu, G. C. Stereoconvergent Negishi arylations of racemic secondary alkyl electrophiles: differentiating between a CF3 and an alkyl group. J. Am. Chem. Soc.137, 9523–9526 (2015). 10.1021/jacs.5b04725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang, W., Hu, M., Wan, X. & Shen, Q. Facilitating the transmetalation step with aryl-zincates in nickel-catalyzed enantioselective arylation of secondary benzylic halides. J. Am. Chem. Soc.141, 11446–11451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin, T. Y. et al. Design and synthesis of TY-phos and application in palladium-catalyzed enantioselective fluoroarylation of gem-difluoroalkenes. Angew. Chem. Int. Ed.59, 22957–22962 (2020). 10.1002/anie.202008262 [DOI] [PubMed] [Google Scholar]
- 22.Huang, S. et al. Regio- and enantioselective umpolung gemdifluoroallylation of hydrazones via palladium catalysis enabled by N-heterocyclic carbene ligand. Nat. Commun.12, 6551 (2021). 10.1038/s41467-021-26667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai, D. C. et al. Highly regio- and enantioselective hydrosilylation of gem-difluoroalkenes by nickel catalysis. Angew. Chem. Int. Ed.61, e202114918 (2022). 10.1002/anie.202114918 [DOI] [PubMed] [Google Scholar]
- 24.Tian, F. T., Yan, G. B. & Yu, J. Recent advances in the synthesis and applications of α-(trifluoromethyl)styrenes in organic synthesis. Chem. Commun.55, 13486–13505 (2019). 10.1039/C9CC06465F [DOI] [PubMed] [Google Scholar]
- 25.Lu, X. et al. Nickel-catalyzed allylic defluorinative alkylation of trifluoromethyl alkenes with reductive decarboxylation of redox-active esters. Chem. Sci.10, 809–814 (2019). 10.1039/C8SC04335C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen, F. L., Xu, X. F., He, Y. L., Huang, G. P. & Zhu, S. L. NiH-catalyzed migratory defluorinative olefin cross-coupling: trifluoromethyl-substituted alkenes as acceptor olefins to form gem-difluoroalkenes. Angew. Chem. Int. Ed.59, 5398–5402 (2020). 10.1002/anie.201915840 [DOI] [PubMed] [Google Scholar]
- 27.Deng, Y. P., He, J. J., Cao, S. & Qian, X. H. Advances in cycloaddition and hydroaddition reaction of α-(trifluoromethyl)styrenes without defluorination: an alternative approach to CF3-containing compounds. Chin. Chem. Lett.33, 2363–2371 (2022). 10.1016/j.cclet.2021.11.049 [DOI] [Google Scholar]
- 28.Huang, W. S. et al. General catalytic enantioselective access to monohalomethyl and trifluoromethyl cyclopropanes. Chem. Eur. J.24, 10339–10343 (2018). 10.1002/chem.201802685 [DOI] [PubMed] [Google Scholar]
- 29.Hock, K. J., Spitzner, R. & Koenigs, R. M. Towards nitrile-substituted cyclopropanes – a slow-release protocol for safe and scalable applications of diazo acetonitrile. Green. Chem.19, 2118–2122 (2017). 10.1039/C7GC00602K [DOI] [Google Scholar]
- 30.Trost, B. M. & Debien, L. Palladium-catalyzed trimethylenemethane cycloaddition of olefins activated by the σ-electron-withdrawing trifluoromethyl group. J. Am. Chem. Soc.137, 11606–11609 (2015). 10.1021/jacs.5b07573 [DOI] [PubMed] [Google Scholar]
- 31.Magre, M., Biosca, M., Pámies, O. & Diéguez, M. Filling the gaps in the challenging asymmetric hydroboration of 1,1-disubstituted alkenes with simple phosphite-based phosphinooxazoline iridium catalysts. Chem. Cat. Chem.7, 114–120 (2015). [Google Scholar]
- 32.Hu, M., Tan, B. B. & Ge, S. Z. Enantioselective cobalt-catalyzed hydroboration of fluoroalkyl-substituted alkenes to access chiral fluoroalkylboronates. J. Am. Chem. Soc.144, 15333–15338 (2022). 10.1021/jacs.2c06488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu, C. et al. Nickel-catalyzed anti-Markovnikov hydroalkylation of trifluoromethylalkenes. ACS Catal.12, 9410–9417 (2022). 10.1021/acscatal.2c02052 [DOI] [Google Scholar]
- 34.Brook, M. A. Silicon in Organic, Organometallic and Polymer Chemistry (Wiley, 2000).
- 35.Pooni, P. K. & Showell, G. A. Silicon switches of marketed drugs. Mini Rev. Med. Chem.6, 1169–1177 (2006). 10.2174/138955706778560120 [DOI] [PubMed] [Google Scholar]
- 36.Xu, L. W., Li, L., Lai, G. Q. & Jiang, J. X. The recent synthesis and application of silicon-stereogenic silanes: a renewed and significant challenge in asymmetric synthesis. Chem. Soc. Rev.40, 1777–1790 (2011). 10.1039/C0CS00037J [DOI] [PubMed] [Google Scholar]
- 37.Franz, A. K. & Wilson, S. O. Organosilicon molecules with medicinal applications. J. Med. Chem.56, 388–405 (2013). 10.1021/jm3010114 [DOI] [PubMed] [Google Scholar]
- 38.Bart, S. C., Lobkovsky, E. & Chirik, P. J. Preparation and molecular and electronic structures of iron(0) dinitrogen and silane complexes and their application to catalytic hydrogenation and hydrosilation. J. Am. Chem. Soc.126, 13794–13807 (2004). 10.1021/ja046753t [DOI] [PubMed] [Google Scholar]
- 39.Chen, J. H., Cheng, B., Cao, M. Y. & Lu, Z. Iron-catalyzed asymmetric hydrosilylation of 1,1-disubstitutedAlkenes. Angew. Chem. Int. Ed.54, 4661–4664 (2015). 10.1002/anie.201411884 [DOI] [PubMed] [Google Scholar]
- 40.Cheng, B., Liu, W. B. & Lu, Z. Iron-catalyzed highly enantioselective hydrosilylation of unactivated terminal alkenes. J. Am. Chem. Soc.140, 5014–5017 (2018). 10.1021/jacs.8b01638 [DOI] [PubMed] [Google Scholar]
- 41.Hu, M. Y. et al. Iron-catalyzed regiodivergent alkyne hydrosilylation. J. Am. Chem. Soc.142, 16894–16902 (2020). 10.1021/jacs.0c09083 [DOI] [PubMed] [Google Scholar]
- 42.Cheng, B., Lu, P., Zhang, H. Y., Cheng, X. P. & Lu, Z. Highly enantioselective cobalt-catalyzed hydrosilylation of alkenes. J. Am. Chem. Soc.139, 9439–9442 (2017). 10.1021/jacs.7b04137 [DOI] [PubMed] [Google Scholar]
- 43.Wang, C., Teo, W. J. & Ge, S. Z. Cobalt-catalyzed regiodivergent hydrosilylation of vinylarenes and aliphatic alkenes: ligand- and silane-dependent regioselectivities. ACS Catal.7, 855–863 (2017). 10.1021/acscatal.6b02518 [DOI] [Google Scholar]
- 44.Wen, H. N., Wang, K., Zhang, Y. L., Liu, G. X. & Huang, Z. Cobalt-catalyzed regio- and enantioselective Markovnikov 1,2- hydrosilylation of conjugated dienes. ACS Catal.9, 1612–1618 (2019). 10.1021/acscatal.8b04481 [DOI] [Google Scholar]
- 45.Lipschutz, M. I. & Tilley, T. D. Synthesis and reactivity of a conveniently prepared two-coordinate bis(amido) nickel(II) complex. Chem. Commun.48, 7146–7148 (2012). 10.1039/c2cc32974c [DOI] [PubMed] [Google Scholar]
- 46.Buslov, I., Becouse, J., Mazza, S., Montandon-Clerc, M. & Hu, X. Chemoselective alkene hydrosilylation catalyzed by nickel pincer complexes. Angew. Chem. Int. Ed.54, 14523–14526 (2015). 10.1002/anie.201507829 [DOI] [PubMed] [Google Scholar]
- 47.Steiman, T. J. & Uyeda, C. Reversible substrate activation and catalysis at an intact metal− metal bond using a redox-active supporting ligand. J. Am. Chem. Soc.137, 6104–6110 (2015). 10.1021/jacs.5b03092 [DOI] [PubMed] [Google Scholar]
- 48.Buslov, I., Keller, S. C. & Hu, X. L. Alkoxy hydrosilanes as surrogates of gaseous silanes for hydrosilylation of alkenes. Org. Lett.18, 1928–1931 (2016). 10.1021/acs.orglett.6b00792 [DOI] [PubMed] [Google Scholar]
- 49.Pappas, I., Treacy, S. & Chirik, P. J. Alkene hydrosilylation using tertiary silanes with α-diimine nickel catalysts. Redox-active ligands promote a distinct mechanistic pathway from platinum catalysts. ACS Catal.6, 4105–4109 (2016). 10.1021/acscatal.6b01134 [DOI] [Google Scholar]
- 50.Mathew, J. et al. Olefin hydrosilylation catalyzed by cationic nickel(ii) allyl complexes: a non-innocent allyl ligand-assisted mechanism. Chem. Commun.52, 6723–6726 (2016). 10.1039/C6CC01665K [DOI] [PubMed] [Google Scholar]
- 51.Corey, J. Y. Reactions of hydrosilanes with transition metal complexes and characterization of the products. Chem. Rev.111, 863–1071 (2011). 10.1021/cr900359c [DOI] [PubMed] [Google Scholar]
- 52.Nakajima, Y. & Shimada, S. Hydrosilylation reaction of olefins: recent advances and perspectives. RSC Adv.5, 20603–20616 (2015). 10.1039/C4RA17281G [DOI] [Google Scholar]
- 53.Du, X. Y. & Huang, Z. Advances in base-metal-catalyzed alkene hydrosilylation. ACS Catal.7, 1227–1243 (2017). 10.1021/acscatal.6b02990 [DOI] [Google Scholar]
- 54.Sunada, Y. & Nagashima, H. in Organosilicon Chemistry: Novel Approaches and Reactions (eds Hiyama, T. & Oestreich, M.) (WileyVCH, 2019).
- 55.Gao, Y. F., Wang, L. J. & Deng, L. Distinct catalytic performance of cobalt(I)–N-heterocyclic carbene complexes in promoting the reaction of alkene with diphenylsilane: selective 2,1-hydrosilylation, 1,2-hydrosilylation, and hydrogenation of alkene. ACS Catal.8, 9637–9646 (2018). 10.1021/acscatal.8b02513 [DOI] [Google Scholar]
- 56.Agahi, R. et al. Regiodivergent hydrosilylation, hydrogenation, [2π + 2π]-cycloaddition and C–H borylation using counterion activated earth-abundant metal catalysis. Chem. Sci.10, 5079–5084 (2019). 10.1039/C8SC05391J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuai, C. S. et al. Ligand-regulated regiodivergent hydrosilylation of isoprene under iron catalysis. Angew. Chem. Int. Ed.59, 19115–19120 (2020). 10.1002/anie.202007930 [DOI] [PubMed] [Google Scholar]
- 58.Hossain, I. & Schmidt, J. A. Cationic nickel(II)-catalyzed hydrosilylation of alkenes: role of P, N-type ligand scaffold on selectivity and reactivity. Organometallics39, 3441–3451 (2020). 10.1021/acs.organomet.0c00551 [DOI] [Google Scholar]
- 59.Wu, X. Y. et al. Nickel-catalyzed hydrosilylation of terminal alkenes with primary silanes via electrophilic silicon–hydrogen bond activation. Org. Lett.23, 1434–1439 (2021). 10.1021/acs.orglett.1c00111 [DOI] [PubMed] [Google Scholar]
- 60.Docherty, J. H., Dominey, A. P. & Thomas, S. P. Cobalt-catalysed, ligand-controlled regiodivergent alkene hydrosilylation. Asian J. Org. Chem.10, 2379–2384 (2021). 10.1002/ajoc.202100361 [DOI] [Google Scholar]
- 61.Cheng, Z.-Y. et al. Cobalt-catalyzed regiodivergent double hydrosilylation of arylacetylenes. Angew. Chem. Int. Ed.61, e202215029 (2022). [DOI] [PubMed] [Google Scholar]
- 62.Chen, Y. & Zargarian, D. Phenylsilane dehydrocoupling and addition to styrene catalyzed by (R-indenyl)Ni(phosphine)(methyl) complexes. Can. J. Chem.87, 280–287 (2009). 10.1139/v08-132 [DOI] [Google Scholar]
- 63.Junquera, L. B., Puerta, M. C. & Valerga, P. R-Allyl nickel(II) complexes with chelating N-heterocyclic carbenes: synthesis, structural characterization, and catalytic activity. Organometallics31, 2175–2183 (2012). 10.1021/om200937d [DOI] [Google Scholar]
- 64.Yao, C. B., Wang, S., Norton, J. & Hammond, M. Catalyzing the hydrodefluorination of CF3-substituted alkenes by PhSiH3. H• transfer from a nickel hydride. J. Am. Chem. Soc.142, 4793–4799 (2020). 10.1021/jacs.9b13757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bott, G., Field, L. D. & Sternhell, S. Steric effects. a study of a rationally designed system. J. Am. Chem. Soc.102, 5618–5626 (1980). 10.1021/ja00537a036 [DOI] [Google Scholar]
- 66.Furuya, T., Kamlet, A. S. & Ritter, T. Catalysis for fluorination and trifluoromethylation. Nature473, 470–477 (2011). 10.1038/nature10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meanwell, N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem.54, 2529–2591 (2011). 10.1021/jm1013693 [DOI] [PubMed] [Google Scholar]
- 68.Chang, A. S. et al. (NHC)Ni(0)-catalyzed branched-selective alkene hydrosilylation with secondary and tertiary silanes. ACS Catal.12, 11002–11014 (2022). 10.1021/acscatal.2c03580 [DOI] [Google Scholar]
- 69.Zhu, B. & Sakaki, S. C(sp3)−F bond activation and hydrodefluorination of the CF3 group catalyzed by a nickel(II) hydride complex: theoretical insight into the mechanism with a spin-state change and two ion-pair intermediates. ACS Catal.11, 10681–10693 (2021). 10.1021/acscatal.1c02251 [DOI] [Google Scholar]
- 70.Marciniec, B. Catalysis by transition metal complexes of alkene silylation – recent progress and mechanistic implications. Coord. Chem. Rev.249, 2374–2390 (2005). 10.1016/j.ccr.2005.02.025 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files, and also available from the corresponding author. The NMR, experimental procedures and characterization for all products and mechanism studies are shown in Supplementary Information files. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2214736 (3 s), CCDC 2214737 (4t). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided with this paper.