

Abstract
INTRODUCTION
Sleep is crucial for memory consolidation and the clearance of toxic proteins associated with Alzheimer's disease (AD). We examined the association between sleep characteristics and imaging biomarkers of early amyloid beta (Aβ) and tau pathology as well as neurodegeneration in brain regions known to be affected in the incipient stages of AD.
METHODS
Thirty‐nine cognitively unimpaired (CU) participants of the Harvard Aging Brain Study underwent at‐home polysomnography as well as tau positron emission tomography (flortaucipir‐PET), amyloid PET (Pittsburgh compound B [PiB]‐PET), and magnetic resonance imaging–derived assessment of cortical thickness (CT).
RESULTS
Increased N1 sleep was associated with a higher tau PET signal (β = 0.009, p = 0.001) and lower CT in the temporal composite region of interest (β = –0.017, p = 0.007). Decreased slow‐wave sleep (SWS) was associated with higher tau burden in the temporal composite (β = –0.008, p = 0.005) and lower CT (β = 0.008, p = 0.002), even after controlling for global PiB‐PET.
DISCUSSION
In CU older adults, lower SWS and higher N1 sleep were associated with higher tau burden and lower CT in brain regions associated with early tau deposition and vulnerable to AD‐related neurodegeneration through mechanisms dissociable from amyloid deposition.
Highlights
We report the results of an observational study, which leveraged ‐a well‐characterized cohort of healthy aging (Harvard Aging Brain Study) by adding in‐home full polysomnograms.
By adding at‐home polysomnograms to this unique and deeply phenotyped cohort, we examined variations in sleep architecture that are associated with Alzheimer's disease (AD) pathologic changes.
Our results confirmed the association of sleep changes with early tau and cortical neurodegenerative changes that were independent of amyloid.
The results will be of importance in monitoring sleep‐related variations in relation to the natural history of AD pathology and in designing sleep‐focused clinical trials.
Keywords: amyloid positron emission tomography, neurodegeneration, neuroimaging, slow‐wave sleep, tau pet
1. BACKGROUND
The accumulation of Alzheimer's disease (AD)–related pathologies can predate its clinical manifestations by years and even decades. 1 This long preclinical phase of AD represents a critical window for early disease detection, interventions with disease‐modifying therapies, and the mitigation of potentially modifiable risk factors for the emergence of cognitive decline. Both the duration and quality of sleep have been associated with the risk of developing AD, 2 , 3 , 4 placing intense focus on sleep disturbance as both an AD biomarker and a potential target to delay the emergence of cognitive decline in older adults. 5 , 6 , 7 , 8
Brain areas regulating sleep are found in the basal forebrain (BF), hypothalamus, thalamus, midbrain, pons, and brainstem, which are also regions that regulate arousal and circadian rhythms. 9 Many of these regions are affected by early tau pathology during pretangle stages (stage 0 Braak), prior to cortical tau or amyloid beta (Aβ) peptide deposition. Research in animals and cognitively unimpaired (CU) humans suggests variations in time spent in each sleep stage, circadian rhythm variability, and total sleep time may be linked to the process of Aβ production and clearance and to the hyperphosphorylation and promotion of tau protein aggregation. 10 , 11 , 12 , 13 , 14 , 15 The early and profound involvement of brainstem nuclei with tau may partially account for the bidirectional relationship between sleep disruption and AD pathology, whereby sleep disruption due to early brainstem tau pathology results in decreased clearance and/or increased production of cortical tau and Aβ, and subsequently induces further accumulation of protein aggregates and neurodegeneration. 16 , 17 , 18 , 19 While the correlational link between sleep and cognition in AD is well established, critical gaps remain.
In contrast to Aβ deposition, which occurs across many cortical regions, tau accumulation occurs in anatomically more circumscribed patterns, and appears to correlate better with regional neurodegeneration and cognitive decline. 20 , 21 , 22 Most research has linked sleep disturbances to amyloid and tau levels in cerebrospinal fluid. 23 , 24 , 25 The advent of tau positron emission tomography (PET) tracers has made it possible to measure tau pathology distribution patterns in vivo and link them to Braak stages thereby providing a framework for reliably identifying sleep disturbances based on the earliest stages of AD pathology. 26 However, data on relationships between polysomnography data and tau PET remain extremely limited. 27
The Harvard Aging Brain Study (HABS) is a longitudinal study of cognitive aging and preclinical AD, consisting of a deeply phenotyped cohort of older (60–90 years) individuals who were cognitively normal at study entry. HABS participants are closely followed with cognitive and clinical measures, magnetic resonance imaging (MRI), and both amyloid and tau PET imaging. 28 By adding at‐home polysomnograms (PSGs) to this unique cohort, we examined how variations in sleep architecture are associated with early changes in Aβ and tau pathology in older adults. A better understanding of sleep changes in relation to emerging amyloid and tau pathology as well as cortical neurodegenerative changes will be a key in designing clinical trials targeting the earliest stages of AD prior to the development of cognitive impairment.
2. METHODS
2.1. Participants
The study was approved by the Mass General Brigham Institutional Review Board and all study participants signed informed consents. All participants were recruited from the HABS cohort. As previously described, HABS participants undergo comprehensive cognitive assessments, MRI, and tau and Aβ PET imaging. 28 For the purpose of this study, HABS participants were invited to also undergo at‐home polysomnography, as described below. At study entry, all participants were CU (Clinical Dementia Rating [CDR] = 0, 29 Mini‐Mental State Examination [MMSE] score > 25, 30 performance within 1.5 standard deviations [SDs] of age and education‐adjusted norms on the Logical Memory Delayed Recall, 31 without current clinical depression [Geriatric Depression Scale < 11]). 32 Key exclusion criteria included a history of severe medical, neurological, and psychiatric condition, as well as clinically significant sleep‐related disorders. All participants with available amyloid PET, tau PET, MRI, and PSG data were included in the analyses (n = 39).
2.2. PET data acquisition and analysis
Amyloid PET was acquired using previously described methods. 28 In brief, administration of 10 to 15 mCi 11C‐Pittsburgh compound B (PiB) was followed by a 60 minute acquisition (63 parallel planes: axial field of view: 15.2 cm; in‐plane resolution: 4.1 mm full‐width at half maximum; slice width: 2.4 mm), using a 39‐frame dynamic protocol: 8 × 15s, 4 × 60s, and 27 × 120s. PET data were reconstructed and corrected for scatter, attenuation, and randoms using vendor‐supplied software. We calculated the distribution volume ratio (DVR) for PiB‐PET based on the Logan graphical analysis technique, 33 using the FreeSurfer‐defined cerebellar gray reference region of interest (ROI). 34 As in our prior publications, PiB retention was summarized using an aggregate cortical ROI including frontal, lateral temporal, and retrosplenial cortices. 35
Tau PET imaging was performed using [18F]flortaucipir tau PET tracer using previously described procedures. 35 , 36 Regional values for tau PET were calculated based on FreeSurfer‐defined cortical ROIs, using cerebellar gray as a reference for calculating standardized uptake value ratios (SUVRs). The prespecified region for analysis was a composite ROI comprised of the bilateral entorhinal cortex, inferior temporal cortex, middle temporal region, and fusiform regions. 37 Follow‐up exploratory analyses examined the broader regional changes across individual FreeSurfer‐defined ROIs. 35 , 38 All PET data were partial volume corrected (PVC) using the symmetric geometric transform matrix approach. 39
2.3. MRI data acquisition and analysis
MRI scans were performed at the Massachusetts General Hospital Athinoula A. Martinos Center for Biomedical Imaging using a 3T Siemens Trio TIM scanner fitted with a 12‐channel head coil. T1‐weighted volumetric magnetization‐prepared rapid‐acquisition gradient‐echo (MP‐RAGE) images were collected (repetition time/echo time/inversion time = 6400/2.8/900 ms, flip angle = 8°, voxel size = 1 × 1 × 1.2 mm3). Image segmentation and ROI labeling were performed using FreeSurfer version 6.0.
We calculated cortical thickness (CT) in FreeSurfer‐defined ROIs (averaged across right and left hemispheres). As with tau PET data, a composite of temporal regions, comprised of the bilateral entorhinal cortex, inferior temporal cortex, middle temporal region, and fusiform regions, was used as the prespecified ROI for analysis. Follow‐up exploratory analyses using individual ROIs were also performed. 37 , 40
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional sources (e.g., PubMed) as well as research presented at conferences. Previous studies demonstrated reduced slow‐wave sleep (SWS) being linked with higher amyloid burden in the preclinical stages of Alzheimer's disease (AD), yet less is known about the relationship between sleep architecture including SWS and early regional tau and cortical correlates of neurodegeneration.
Interpretation: In cognitively unimpaired older adults, lower amounts of SWS and increased amounts of N1 sleep are associated with higher tau accumulation and lower cortical thickness in brain areas prone to early tau build‐up and susceptible to AD, independent of amyloid deposition.
Future directions: This study illuminates the emergence of sleep changes early in the course of AD and the prospect of sleep‐based therapeutic and preventative strategies during the preclinical stages of AD. Further research is warranted to explore the mechanisms and time course of this relationship in larger and more diverse longitudinal cohorts.
2.4. Polysomnography methods
One night of polysomnography was performed using a Compumedics Somte II device. The montage consisted of six electroencephalogram (EEG) leads (F3, F4, C3, C4, O1, O2, sampling frequency 256 Hz) bilateral electrooculogram (EOG), submental electromyogram (EMG), bilateral anterior tibialis EMG, and standard electrocardiogram (ECG) electrodes. Sensors recommended by the American Academy of Sleep Medicine (AASM) for assessment of sleep‐disordered breathing included: nasal/oral airflow (thermistor), nasal pressure (Validyne transducer), chest plus abdominal wall motion (inductance plethysmography), and finger pulse oxygen saturation. PSG scoring was performed by a registered sleep technologist in accordance with the AASM scoring manual. 41 Main PSG features extracted included total sleep time (TST), sleep efficiency (SE), wake after sleep onset (WASO, minutes), as well as duration and percentage of N1, N2, N3, and rapid eye movement (REM) sleep. Arousals were scored according to the AASM manual scoring criteria, which require an abrupt shift of EEG frequency including alpha, theta, and/or frequencies greater than 16 Hz (but not spindles) that last at least 3 seconds with 10 seconds of stable sleep preceding. Apnea–hypopnea index (AHI) was calculated as apneas and hypopneas with ≥ 4% oxygen desaturation per hour of sleep.
2.5. Statistical analyses
Statistical analyses were performed using R version 4.0.5 (http://www.r‐project.org/).
Pearson r values were calculated for associations between the PSG sleep variables and tau PET ROIs, CT ROIs, as well as age and arousal index (AI). To study the relationship between sleep parameters and temporal lobe tau burden, and temporal lobe CT, we created separate general linear models (GLMs) with tau ROI, CT ROI as dependent variables, and PSG‐derived sleep features as independent predictors while controlling for age and sex. To determine the extent to which the effects of poor sleep were dependent on amyloid pathology, we computed additional models adjusted for global cortical amyloid burden (PiB‐PET SUVR). To account for the presence of nocturnal arousals we performed sensitivity analyses on the significant models correcting by AI.
3. RESULTS
A total of 39 participants were included in the main analyses. The study population consisted of CU, highly educated (16.5 ± 2.75 years of education) adults with a mean ± SD age at tau PET of 74.6 ± 8.7 years, of which 61.5% were females (Table 1). The average total sleep time was 351 ± 54 minutes with a mean sleep efficiency of 73.6 ± 11.6% and WASO of 109 ± 1.0 minutes. Sleep stage distributions and further data from polysomnography are summarized in Table 1.
TABLE 1.
Demographic and PSG characteristics.
| N = 39 | |
|---|---|
| Age (years) | 74.6 (8.69) |
| Sex: female | 24.0 (61.5%) |
| Education (years) | 16.5 (2.75) |
| Total sleep time (minutes) | 351 (54.0) |
| Sleep efficiency (%) | 73.6 (11.6) |
| Wake after sleep onset (minutes) | 109 (71.0) |
| N1 (%) | 17.0 (9.54) |
| N1 (min) | 58.9 (31.8) |
| N2 (%) | 56.1 (10.9) |
| N2 (min) | 196 (46.0) |
| N3 (%) | 11.4 (10.0) |
| N3 (minutes) | 40.2 (35.0) |
| REM (%) | 15.5 (7.68) |
| Time REM (minutes) | 55.6 (30.0) |
| Arousal index (events/hour) | 16.4 (8.44) |
| Apnea‐hypopnea index (events/hour) | 9.0 (9.10) |
Notes: Data are presented as mean ± SD or n (%). N1(%) = percentage of sleep period spent in stage 1 sleep; N2(%) = percentage of sleep period spent in stage 2 sleep; N3(%) = percentage of sleep period spent in N3 sleep; REM (%) = percentage of sleep period spent in REM sleep.
Abbreviations: AHI, apnea–hypopnea index (calculated as apneas and hypopneas with ≥ 4% oxygen desaturation per hour of sleep); PSG, polysomnogram; REM, rapid eye movement; SD, standard deviation.
Analysis of sex differences (Table S1 in supporting information) revealed that males spent more time in N1 sleep measured in minutes (p = 0.028) and percentage (p = 0.0380). Conversely, females spent more time in N3 sleep (p < 0.001, minutes and percentage). Males had a higher AHI compared to females (p = 0.011).
3.1. Associations between sleep architecture and AD pathology
We observed that a higher percentage of time spent in N1 sleep was significantly associated with greater tau PET signal in the temporal composite region (β = 0.009, p = 0.001) while a lower percentage of N3 sleep was significantly associated with greater tau burden in the same region (β = −0.008, p = 0.005). Follow‐up exploratory analyses in other temporal regions revealed a significant association between a higher percentage of N1 sleep and tau PET signal in the entorhinal cortex (β = 0.016, p = 0.007), and the inferior temporal cortex (β = 0.008, p = 0.002). Lower percentage of N3 sleep was significantly associated with greater tau burden in the entorhinal cortex (β = −0.012, p = 0.035) and inferior temporal cortex (β = −0.007, p = 0.010; Figure 1). No significant associations were observed between tau PET signal and total sleep time, sleep efficiency, or WASO (Table S2 in supporting information), as well as the percentage of N2 and REM sleep and tau PET signal in the temporal composite region (Table 2).
FIGURE 1.
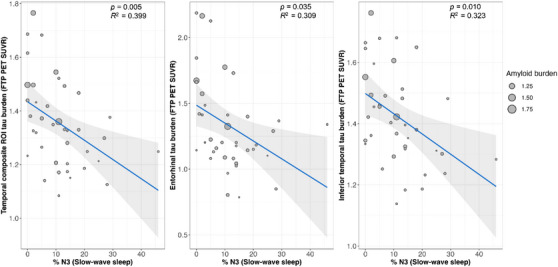
Temporal lobe tau PET signal is higher in individuals with less time spent in N3 sleep. Tau PET signal in the temporal composite ROI was negatively correlated with the percentage of time each participant spent in N3 sleep (left). Similar results were seen in exploratory analyses of individual temporal ROIs, including the entorhinal cortex (middle) and inferior temporal cortex (right). The X axis represents %N3 sleep (slow‐wave sleep), where higher values indicate a higher proportion of N3 sleep. The Y axis represents tau PET signal. FTP, flortaucipir; PET, positron emission tomography; ROI, region of interest; SUVR, standardized uptake value ratio.
TABLE 2.
Associations between regional tau burden and sleep macroarchitecture features.
| Temporal composite ROI tau burden | ||||||
|---|---|---|---|---|---|---|
| Predictor | β coef. | SE | t statistic | p‐value | CI low | CI high |
| % time N1 | 0.009 | 0.002 | 3.782 | 0.001 | 0.005 | 0.014 |
| % time N2 | 0.001 | 0.002 | 0.318 | 0.753 | −0.004 | 0.005 |
| % time N3 | −0.008 | 0.002 | −3.030 | 0.005 | −0.012 | −0.003 |
| % time REM | −0.004 | 0.004 | −1.051 | 0.301 | −0.012 | 0.004 |
| Entorhinal cortex tau burden | ||||||
|---|---|---|---|---|---|---|
| Predictor | β coef. | SE | t statistic | p‐value | CI low | CI high |
| % time N1 | 0.016 | 0.006 | 2.881 | 0.007 | 0.005 | 0.027 |
| % time N2 | 0.001 | 0.005 | 0.143 | 0.887 | −0.009 | 0.010 |
| % time N3 | −0.012 | 0.006 | −2.199 | 0.035 | −0.023 | −0.001 |
| % time REM | −0.007 | 0.008 | −0.784 | 0.438 | −0.023 | 0.010 |
| Inferior temporal cortex tau burden | ||||||
|---|---|---|---|---|---|---|
| Predictor | β coef. | SE | t statistic | p‐value | CI low | CI high |
| % time N1 | 0.008 | 0.003 | 3.353 | 0.002 | 0.003 | 0.013 |
| % time N2 | 0.000 | 0.002 | 0.193 | 0.848 | −0.004 | 0.005 |
| % time N3 | −0.007 | 0.002 | −2.725 | 0.010 | −0.012 | −0.002 |
| % time REM | −0.003 | 0.004 | −0.838 | 0.408 | −0.011 | 0.004 |
Notes: p value uncorrected for multiple comparisons, t statistic – t value. Linear regression models were corrected for age and sex. Temporal composite ROI composed of entorhinal, inferior temporal, middle temporal, and fusiform.
Abbreviations: CI, confidence interval; REM, rapid eye movement; ROI, region of interest; SE, standard error.
There was no association between any sleep stages and global amyloid PET signal, though a trend was observed with N3 sleep percentage being negatively correlated with amyloid burden (β = −0.006, p = 0.067; Table S3 in supporting information).
We also tested whether tau PET associations were dissociable from global amyloid PET and found that adjustment for cortical amyloid burden did not alter the associations between percentages of N1 and N3 sleep with tau in the temporal composite ROI (N1[β = 0.009, p = 0.002]; N3[β = −0.007, p = 0.016]; Table S4 in supporting information).
3.2. Associations between sleep architecture and CT
A higher percentage of N1 sleep was significantly associated with lower CT in the temporal composite ROI (β = −0.017, p = 0.007; β = −0.007, p = 0.011; Table 3) while a lower percentage of N3 sleep was significantly associated with lower CT in the temporal composite ROI (β = 0.008, p = 0.002). Follow‐up exploratory analyses demonstrated similar associations between percent N3 sleep and CT in other temporal lobe ROIs (entorhinal cortex [β = 0.013, p = 0.031]; inferior temporal cortex [β = 0.005, p = 0.023]; see Figure 2). Adjustment for global amyloid burden revealed that associations between percent N3 sleep and CT in the temporal composite ROI remained significant (β = 0.008, p = 0.006; see Table S5 in supporting information). Exploratory regional analyses are summarized in Table S5. Similar to regional tau PET signal analysis, we observed no significant associations between percentage of N2 and REM sleep and temporal lobe CT measures.
TABLE 3.
Associations between regional cortical thickness and sleep macroarchitecture features.
| Temporal composite ROI thickness | ||||||
|---|---|---|---|---|---|---|
| Predictor | β coef. | SE | t statistic | p‐value | CI low | CI high |
| % time N1 | −0.007 | 0.003 | −2.677 | 0.011 | −0.013 | −0.002 |
| % time N2 | −0.001 | 0.002 | −0.618 | 0.541 | −0.006 | 0.003 |
| % time N3 | 0.008 | 0.002 | 3.341 | 0.002 | 0.003 | 0.013 |
| % time REM | 0.001 | 0.004 | 0.213 | 0.833 | −0.007 | 0.009 |
| Entorhinal cortex thickness | ||||||
|---|---|---|---|---|---|---|
| Predictor | β coef. | SE | t statistic | p‐value | CI low | CI high |
| % time N1 | −0.017 | 0.006 | −2.866 | 0.007 | −0.029 | −0.005 |
| % time N2 | 0.002 | 0.005 | 0.298 | 0.767 | −0.009 | 0.012 |
| % time N3 | 0.013 | 0.006 | 2.246 | 0.031 | 0.002 | 0.025 |
| % time REM | −0.001 | 0.009 | −0.067 | 0.947 | −0.018 | 0.017 |
| Inferior temporal cortex thickness | ||||||
|---|---|---|---|---|---|---|
| Predictor | β coef. | SE | t statistic | p‐value | CI low | CI high |
| % time N1 | −0.003 | 0.002 | −1.192 | 0.241 | −0.008 | 0.002 |
| % time N2 | −0.003 | 0.002 | −1.683 | 0.101 | −0.007 | 0.001 |
| % time N3 | 0.005 | 0.002 | 2.381 | 0.023 | 0.001 | 0.010 |
| % time REM | 0.004 | 0.003 | 1.269 | 0.213 | −0.002 | 0.011 |
Notes: P value uncorrected for multiple comparisons, t statistic – t‐value. Linear regression models were corrected for age and sex. Temporal composite ROI composed of entorhinal, inferior temporal, middle temporal, and fusiform.
Abbreviations: CI, confidence interval; REM, rapid eye movement; ROI, region of interest; SE, standard error.
FIGURE 2.
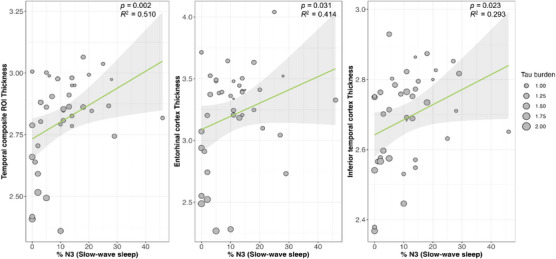
Temporal lobe cortical thickness is lower in individuals with less time spent in N3 sleep. Cortical thickness in the temporal composite region of interest (ROI) was positively correlated with the percentage of time each participant spent in N3 sleep (left). Similar results were seen in exploratory analyses of individual temporal ROIs, including the entorhinal cortex (middle) and inferior temporal cortex (right). The X axis represents %N3 sleep (slow‐wave sleep), where higher values indicate a higher proportion of N3 sleep. The Y axis represents cortical thickness of significant ROIs.
3.3. Sensitivity analyses
We performed separate linear regression analyses accounting for the AHI index to investigate whether the significant increases in N1 and reductions in %N3 (slow‐wave sleep [SWS]) are the result of sleep fragmentation due to sleep‐disordered breathing.
A higher percentage of N1 sleep, as well as a lower percentage of N3, remained significantly associated with tau burden in the temporal composite ROI (%N1[β = 0.010, p = 0.001]; %N3[β = −0.008, p = 0.005]), as well as the entorhinal (%N1[β = 0.017, p = 0.006]; %N3[β = −0.012, p = 0.038]) and inferior temporal cortices tau burden (%N1[β = 0.009, p = 0.001]; %N3[β = –0.007, p = 0.011]), respectively, after correcting for the total AHI index (see Table S6 in supporting information).
As for the CT, significant associations between the percentage of N1 sleep remained significant for the temporal composite (β = −0.007, p = 0.016) and the entorhinal cortex (β = −0.016, p = 0.014; see Table S7 in supporting information). Similarly, a reduced percentage of N3 sleep remained significantly associated with lower CT in the temporal composite ROI (β = 0.008, p = 0.003). Exploratory analyses in the entorhinal and inferior temporal regions remained largely unchanged after adjustment for the AHI (entorhinal cortex [β = 0.013, p = 0.038]; inferior temporal cortex [β = 0.005, p = 0.022]).
4. DISCUSSION
Individual variations in sleep may play an important role in the development of AD pathology, especially early in the course of the disease. Here we observed a significant association among N3, SWS, and N1 sleep with regional tau accumulation in temporal lobe brain regions susceptible to early tau deposition in cognitively healthy older adults. These relationships were also present with respect to CT in the temporal lobe, suggesting that, in particular, individual variations in SWS may play a role in the establishment of early tau tangle pathology in both isocortical and neocortical regions of the temporal lobe, as well as to very early neuronal loss in these same regions. Strikingly, the associations of SWS to tau pathology and CT observed here were independent of amyloid PET signal, indicating that SWS may not be directly involved in the promotion of early amyloid accumulation. In turn, this suggests that interventions on SWS may have added benefit even in individuals receiving potent anti‐amyloid disease‐modifying therapies. Importantly, the associations of SWS with tau PET signal and CT were also independent of sleep‐disordered breathing, suggesting that SWS may have a relatively specific association with these early neurodegenerative changes.
SWS is the deepest phase of non‐REM sleep, characterized by reduced cholinergic activity and low‐frequency delta wave oscillations at 0.5 to 2 Hz. 42 While it is recognized that SWS is a marker of homeostatic sleep pressure, particularly the slow oscillatory activity during SWS, it has been linked to global synaptic downscaling, a process which preserves the overall balance of synaptic strength and helps consolidate long‐term memory representations in the neocortex. 43 , 44 Slow waves originate from specific cortical areas (insula, superior temporal gyri) and tend to spread through the cingulate cortices, typically involving the anterior cingulate gyrus; the middle, medial, and inferior frontal gyri; and the posterior cingulate and precuneus, a process during which they can grow in relative current amplitude as they travel. Although slow oscillations have generally been considered a cortical phenomenon, including local cortical circuits, there are also indications for subcortical structures in particular sleep‐promoting circuitries in the brainstem playing an active modulatory role in regulating the quantity and quality of SWS. 45 , 46
Given the time course of tau deposition pattern, it is possible that early tau accumulations in the brainstem inhibit sleep‐promoting systems necessary for sleep stability and SWS induction.
In contrast to SWS, N1 sleep is considered a transitional sleep stage after wakefulness and is characterized by low‐voltage and fast EEG activity, including theta (4–8 Hz) activity. An increase in N1 sleep is considered a marker of more frequent transitions between sleep stages and lower sleep stability. Notably, this increase has been observed in AD patients and has also been identified as a marker for dementia using an electroencephalography‐based brain age index (BAI) machine‐learning model. 47 , 48
Furthermore, in this study, we also observed a sex differential effect with respect to sleep stages with males exhibiting a greater duration of N1 sleep, while females showed a propensity for a higher N3 sleep percentage. This observation aligns with prior research demonstrating 49 variations in sleep characteristics, particularly in EEG frequency, based on sex in older populations, potentially due to anatomical variations such as differences in skull thickness. 50 , 51
Strengths of this study include the application of multimodal imaging techniques (including quantitative structural MRI and the combination of amyloid and tau PET) allowing for a more comprehensive assessment of structural and functional brain changes that may be associated with sleep disturbances and early cognitive decline. 52 , 53 The use of at‐home polysomnography reduces disruption of an individual's natural sleep patterns compared to laboratory‐based polysomnography, and thereby provides a more ecologically valid and representative measure of individuals’ real sleeping habits.
We also acknowledge several limitations of the present work. While the study was sufficiently powered for the main outcome variables (tau PET, amyloid PET, and CT in the temporal lobe), we lacked sufficient power to properly assess cross‐sectional relationships between sleep measures and cognitive performance (particularly as the sample was CU, limiting the range of cognitive test scores). Additionally, due to the nature of the cross‐sectional design, we were not able to determine the directionality between sleep disturbances and AD pathology. Future larger scale longitudinal studies with longer post‐PSG follow‐up periods are needed to establish the temporal sequence and underlying mechanisms of the relationship between sleep and AD pathology, and cognition.
Overall, the present study was able to detect sleep architecture variations that are sensitive to early tau deposition and reduced CT in the temporal lobe independent of amyloid burden. It is important to note that the interpretation of these results is contingent upon the characteristics of the sample population, comprising clinically unimpaired older adults, as well as the relatively modest sample size. Our focus was to examine the very early stages of disease, prior to clear clinical cognitive impairment. However, to clearly distinguish the extent to which our findings are within the scope of normal aging or the beginning stages of the degenerative processes of AD will require further studies, including longitudinal data collection of our own HABS study sample and subsequent investigations involving cohorts with early AD‐related mild cognitive impairment and encompassing larger samples.
Data from previous AD trials and the relatively slow rate of change in AD pathology strongly support the idea that the preclinical stage of disease may be optimal for intervention. Ultimately, studies applying interventions directed at sleep—either independently or in conjunction with amyloid‐lowering therapies—to mitigate the onset of AD‐related cognitive decline will enable a more comprehensive assessment of the potential role of sleep disruption as a modifiable risk factor in the progression of AD.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This research was supported by NIH R01 AG 062667 American Academy of Sleep Medicine, Bridge to Success Award, 171‐BS‐17 HABS: NIH‐P01AG036694.
Stankeviciute L, Chhatwal JP, Levin R, et al. Amyloid beta–independent sleep markers associated with early regional tau burden and cortical thinning. Alzheimer's Dement. 2024;16:e12616. 10.1002/dad2.12616
Laura Stankeviciute and Jasmeer P. Chhatwal contributed equally.
REFERENCES
- 1. Sperling RA, Aisen PS, Beckett LA. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Edinger JD, Glenn DM, Bastian LA, Marsh GR. Slow‐wave sleep and waking cognitive performance II: findings among middle‐aged adults with and without insomnia complaints. Physiol Behav. 2000;70(1‐2):127‐134. [DOI] [PubMed] [Google Scholar]
- 3. Mccann UD, Penetar DM, Shaham Y. Sleep deprivation and impaired cognition. Biol Psychiatry. 1992;31(11):1082‐1097. Possible role of brain catecholamines. [DOI] [PubMed] [Google Scholar]
- 4. Liguori C, Romigi A, Nuccetelli M. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71(12):1498‐1505. [DOI] [PubMed] [Google Scholar]
- 5. Blackwell T, Yaffe K, Ancoli‐Israel S. Poor sleep is associated with impaired cognitive function in older women: the study of osteoporotic fractures. J Gerontol A Biol Sci Med Sci. 2006;61(4):405‐410. [DOI] [PubMed] [Google Scholar]
- 6. Blackwell T, Yaffe K, Ancoli‐Israel S. Associations between sleep architecture and sleep‐disordered breathing and cognition in older community‐dwelling men: the Osteoporotic Fractures in Men Sleep Study. J Am Geriatr Soc. 2011;59(12):2217‐2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song Y, Blackwell T, Yaffe K, et al. Relationships between sleep stages and changes in cognitive function in older men: the MrOS sleep study. Sleep. 2015;38(3):411‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim ASP, Ellison BA, Wang JL. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain. 2014;137(Pt 10):2847‐2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68(6):1023‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ju Yo‐ElS, Ooms SJ, Sutphen C. Slow wave sleep disruption increases cerebrospinal fluid amyloid‐β levels. Brain. 2017;140(8):2104‐2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mander BA, Marks SM, Vogel JW. β‐amyloid disrupts human NREM slow waves and related hippocampus‐dependent memory consolidation. Nat Neurosci. 2015;18(7):1051‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang J‐E, Lim MM, Bateman RJ. Amyloid‐beta dynamics are regulated by orexin and the sleep‐wake cycle. Science. 2009;326(5955):1005‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Varga AW, Wohlleber ME, Giménez S. Reduced slow‐wave sleep is associated with high cerebrospinal fluid abeta42 levels in cognitively normal elderly. Sleep. 2016;39(11):2041‐2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shokri‐Kojori E, Wang G‐J, Wiers CE. beta‐Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A. 2018;115(17):4483‐4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie L, Kang H, Xu Q. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheime’ s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology. 2019;45:104‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holth JK, Patel TK, Holtzman DM. Sleep in Alzheimer's disease—beyond amyloid. Neurobiol Sleep Circadian Rhythms . 2017;2:4‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ju Yo‐ElS, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology–a bidirectional relationship. Nat Rev Neurol . 2014;10(2):115‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354(6315):1004‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Desikan RS, Mcevoy LK, Thompson WK. Amyloid‐beta–associated clinical decline occurs only in the presence of elevated P‐tau. Arch Neurol. 2012;69(6):709‐713. Alzheimer's Disease Neuroimaging Initiative FT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jack CR, Knopman DS, Jagust WJ. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schöll M, Lockhart SN, Schonhaut DR. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Osorio RS, Ayappa I, Mantua J. Interaction between sleep‐disordered breathing and apolipoprotein E genotype on cerebrospinal fluid biomarkers for Alzheimer's disease in cognitively normal elderly individuals. Neurobiol Aging. 2014;35(6):1318‐1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lucey BP, Mccullough A, Landsness EC. Reduced non‐rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Sci Transl Med. 2019;11(474). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu H, Barthelemy NR, Ovod V, et al. Acute sleep loss decreases CSF‐to‐blood clearance of Alzheimer's disease biomarkers. Alzheimers Dement. 2023;19(7):3055‐3064. doi: 10.1002/alz.12930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Braak H, Braak E. Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239‐259. [DOI] [PubMed] [Google Scholar]
- 27. Winer JR, Mander BA, Helfrich RF. Sleep as a potential biomarker of tau and beta‐amyloid burden in the human brain. J Neurosci. 2019;39(32):6315‐6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dagley A, Lapoint M, Huijbers W. Harvard aging brain study: dataset and accessibility. Neuroimage. 2017;144(Pt B):255‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412‐2414. [DOI] [PubMed] [Google Scholar]
- 30. Folstein MF, Folstein SE, Mchugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189‐198. [DOI] [PubMed] [Google Scholar]
- 31. Wechsler D. Wechsler adult intelligence scale‐revised. Psychological Corporation; 1988. [Google Scholar]
- 32. Yesavage JA, Brink TL, Rose TL. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17(1):37‐49. [DOI] [PubMed] [Google Scholar]
- 33. Logan J, Fowler JS, Volkow ND. Graphical analysis of reversible radioligand binding from time‐activity measurements applied to [N‐11C‐methyl]‐(‐)‐cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10(5):740‐747. [DOI] [PubMed] [Google Scholar]
- 34. Becker JA, Hedden T, Carmasin J. Amyloid‐beta associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69(6):1032‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson KA, Schultz A, Betensky RA. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chien DT, Bahri S, Szardenings AK. Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis. 2013;34(2):457‐468. [DOI] [PubMed] [Google Scholar]
- 37. Jack CR, Wiste HJ, Weigand SD. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13(3):205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dani M, Brooks DJ, Edison P. Tau imaging in neurodegenerative diseases. Eur J Nucl Med Mol Imaging. 2016;43(6):1139‐1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39(5):904‐911. [PubMed] [Google Scholar]
- 40. Jack CR, Wiste HJ, Weigand SD. Age‐specific and sex‐specific prevalence of cerebral beta‐amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50‐95 years: a cross‐sectional study. Lancet Neurol. 2017;16(6):435‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iber C, Ancoli‐Israel S, Quan SF, Chesson AL. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology, and Technical Specifications. Westchester, Ill, American Academy of Sleep Medicine., 2007.
- 42. Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137(4):1087‐1106. [DOI] [PubMed] [Google Scholar]
- 43. Achermann P. Mathematical models of sleep regulation. Front Biosci. 2003;8:s683‐693. [DOI] [PubMed] [Google Scholar]
- 44. Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81(1):12‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G. The sleep slow oscillation as a traveling wave. J Neurosci. 2004;24(31):6862‐6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anaclet C, Fuller PM. Brainstem regulation of slow‐wave‐sleep. Curr Opin Neurobiol. 2017;44:139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Ye, Ren R, Yang L. Sleep in Alzheimer's disease: a systematic review and meta‐analysis of polysomnographic findings. Transl Psychiatry. 2022;12(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ye E, Sun H, Leone MJ. Association of sleep electroencephalography‐based brain age index with dementia. JAMA Netw Open. 2020;3(9):e2017357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dijk DJ, Beersma DGM, Bloem GM. Sex differences in the sleep EEG of young adults: visual scoring and spectral analysis. Sleep. 1989;12(6):500‐507. [DOI] [PubMed] [Google Scholar]
- 50. Eggert T, Dorn H, Danker‐Hopfe H. Nocturnal brain activity differs with age and sex: comparisons of sleep EEG power spectra between young and elderly men, and between 60‐80‐year‐old men and women. Nat Sci Sleep. 2021;13:1611‐1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carrier J, Land S, Buysse DJ, Kupfer DJ, Monk TH. The effects of age and gender on sleep EEG power spectral density in the middle years of life (ages 20‐60 years old). Psychophysiology. 2001;38(2):232‐242. [PubMed] [Google Scholar]
- 52. Hanseeuw BJ, Betensky RA, Jacobs HIL. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76(8):915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ossenkoppele R, Rabinovici GD, Smith R. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2018;320(11):1151‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information