

Abstract

The pyrazolo[1,5-a]pyrimidine scaffold is a promising scaffold to develop potent and selective CSNK2 inhibitors with antiviral activity against β-coronaviruses. Herein, we describe the discovery of a 1,2,4-triazole group to substitute a key amide group for CSNK2 binding present in many potent pyrazolo[1,5-a]pyrimidine inhibitors. Crystallographic evidence demonstrates that the 1,2,4-triazole replaces the amide in forming key hydrogen bonds with Lys68 and a water molecule buried in the ATP-binding pocket. This isosteric replacement improves potency and metabolic stability at a cost of solubility. Optimization for potency, solubility, and metabolic stability led to the discovery of the potent and selective CSNK2 inhibitor 53. Despite excellent in vitro metabolic stability, rapid decline in plasma concentration of 53 in vivo was observed and may be attributed to lung accumulation, although in vivo pharmacological effect was not observed. Further optimization of this novel chemotype may validate CSNK2 as an antiviral target in vivo.
Introduction
The COVID-19 pandemic, caused by the β-coronavirus SARS-CoV-2, has resulted in 775 million infections and has claimed 7 million lives to date.1 The rapid emergence and spread of COVID-19 highlight the need for effective treatments against this disease. Other members of the β-coronavirus family have caused previous pandemics or are primed for emergence to cause future pandemics.2,3 While most antiviral drugs currently target viral factors, host-directed therapy is a promising alternative avenue to develop antiviral drugs. The main advantages of host-directed therapy for antivirals are the reduced propensity of the virus to develop resistance, and, importantly, the broad-spectrum ability of such therapeutics to inhibit viral replication of different viruses in the same family.4−6 Developing compounds for host-directed therapy thus has the potential to accelerate drug discovery against β-coronaviruses that emerge in the future, and safeguard humanity against the perils of a future pandemic.
Kinases are among the host proteins hijacked by viruses for viral replication, as phosphorylation events are important for viral replication across various virus genera.4,7 Casein kinase 2 (CSNK2) is a host kinase which plays a key role in replication of β-coronaviruses, including SARS-CoV-2.8 Both CSNK2A1 and CSNK2A2 are highly upregulated upon SARS-CoV-2 infection.9 Both CSNK2A subunits interact with the N protein of SARS-CoV-2, SARS-CoV, and MERS-CoV,10,11 and the N protein is responsible for this upregulation of CSNK2 activity.9 While kinase inhibitors have traditionally been developed for oncology indications, there is an increasing recognition of the role of kinases in other diseases, and a corresponding increase in interest for developing kinase inhibitors for other indications, including infectious diseases.12−14 We and others have previously demonstrated that CSNK2 inhibitors possess antiviral activity against multiple β-coronaviruses including SARS-CoV-2, emphasizing the broad-spectrum antiviral activity of CSNK2 inhibitors.9,15,16 CSNK2 knockdown also demonstrates antiviral effect against the β-coronavirus MHV.15 Efforts have been put into elucidation of the mechanism of CSNK2 inhibition on viral replication. The CSNK2 substrates with increased phosphorylation postinfection included cytoskeletal proteins, and it was proposed that CSNK2 mediates remodeling of the extracellular matrix for viral egress.9 An alternative mechanism of action is the modulation of the stress granule antiviral response.10 We have also previously shown that CSNK2 inhibition disrupts viral entry via the clathrin-mediated endocytosis (CME) pathway.15 It is thus likely that CSNK2 inhibition interferes with these essential viral processes.
Many small molecule CSNK2 inhibitors have been developed and reported in the literature.17,18 Among these, the pyrazolo[1,5-a]pyrimidine series of inhibitors, as exemplified by the CSNK2 chemical probe SGC-CK2-1 (1, Figure 1), has demonstrated good cellular potency and possesses excellent selectivity.19 We have previously reported that both 1 and 2, a related inhibitor also of the pyrazolo[1,5-a]pyrimidine series, inhibit replication of SARS-CoV-2, MHV, and two bat coronaviruses, SHC014-CoV and WIV1–CoV, in vitro.15 However, antiviral activity was only investigated in infected cells in vitro. To further support CSNK2 as a host target for antiviral therapy, our project aims to validate CSNK2 as an antiviral target in vivo, in a mouse model of COVID-19 infection,20 through the development of an in vivo tool molecule. To do so, we sought to better characterize and understand structure–activity relationship (SAR) around the pyrazolo[1,5-a]pyrimidine scaffold to guide lead optimization.
Figure 1.

Exemplar CSNK2 inhibitors of the previously reported pyrazolo[1,5-a]pyrimidine series.
Based on the crystal structure of analogues of the pyrazolo[1,5-a]pyrimidine series bound to CSNK2 (PDB ID: 3U4U,214GUB,225H8B,235H8E,235H8G,236Z83,196Z8419), we noted that the meta-position amide substituent of the aniline ring played a critical role in CSNK2 binding. As exemplified by the cocrystal structure of 1 with CSNK2A1 (PDB ID: 6Z83) (Figure 2, left),19 the amide binds in an s-cis conformation, with the NH of the amide forming a hydrogen bond with the Asp residue of the DWG motif (Asp175 in CSNK2A1), and the carbonyl forming critical hydrogen bonding interactions with the catalytic lysine (Lys68 in CSNK2A1) and a water molecule buried in the ATP-binding pocket. Despite the key interactions the acetamide forms with CSNK2, only limited SAR exploration at this position had been reported in the literature, involving alkylation of the NH of the amide21,22 and replacement with a sulfone or primary alcohol group.21 Herein, we report the successful identification of a 1,2,4-triazole as a bioisosteric replacement for the amide. Co-crystal structures with CSNK2A1 support the binding hypothesis. We identify a lead compound 53 with a balance of potency, solubility, metabolic stability, and low cytotoxicity. Despite the low hepatic clearance measured in vitro, 53 demonstrated a rapid decrease in plasma concentration in vivo, attributed to distribution and accumulation in lungs, although this was insufficient to achieve pharmacological effect. Nevertheless, this successful isosteric replacement afforded an alternative chemotype for investigation of in vitro CSNK2 activity.
Figure 2.

Co-crystal structure of 1 (left, PDB ID: 6Z83)19 and 14 (right, PDB ID: 8P07) with CSNK2A1. Part of the kinase P-loop is hidden for clarity. Water molecules are represented as red spheres. Hydrogen bonds are denoted as yellow dashed lines.
Results and Discussion
Identification of the 1,2,4-Triazole as an Amide Bioisostere
To characterize the SAR in the region of the meta-position amide substituent, we evaluated other substituents with a hydrogen bond acceptor three bond lengths away from the aniline ring, with an objective of recapitulating the same hydrogen bonding interactions the amide makes (Table 1). We first screened our compounds for CSNK2A1 and CSNK2A2 inhibition using the NanoBRET assay.15,19 For compounds of the pyrazolo[1,5-a]pyrimidine series reported previously15,19,24 and here, we did not observe significant selectivity between the two highly homologous CSNK2A subunits. Hence, we elected to focus on CSNK2A2 in the current screening campaign, since this subunit routinely gave a larger dynamic range in the NanoBRET assay. Data for CSNK2A1 inhibition, where collected, is available in Table S1. Additionally, we screened our compounds for inhibition of mouse hepatitis virus (MHV), a β-coronavirus in the same genus as SARS-CoV-2 used as a model for virulence of SARS-CoV-2,25 due to biosafety and technical advantages.
Table 1. SAR at the meta-Position of the Aniline.


Mean of three independent experiments performed in triplicate.
Mean of two independent experiments.
Compounds were separated by chiral chromatography, but stereochemistry was not determined.
As compared to the amide (3), a 5-to-8-fold drop in CSNK2A2 and MHV potency was observed with the sulfonamide 4. Replacement with the sulfone analogue 5 restored potency against both CSNK2A2 and MHV, indicating that the hydrogen bond formed by the NH of the amide was dispensable for CSNK2A2 inhibition. The sulfoximines 6 and 7 were found to be an order of magnitude less potent against CSNK2A2 than the sulfone 5 and correspondingly showed an almost complete loss of activity against MHV, demonstrating that replacement of either oxygen atom of the sulfone with an NH was not tolerated. Replacement of the sulfone with the racemic sulfoxide 8 largely maintained potency, and the phosphine oxide 9 led to a 10-fold drop in potency. Replacement of the amide with alcohol groups (10, 11) proved unsuccessful, with a >25-fold drop in CSNK2A2 and MHV potency. A carboxyl group at this position (12) was also found not to be tolerated by CSNK2A2, and was likewise inactive against MHV. Our attempt to introduce a nitrile as a hydrogen bond acceptor (13) also led to a drastic loss in CSNK2 and MHV potency. As the acetamide was observed to form two hydrogen bonds, one with the catalytic lysine and another with the water molecule, we rationalized that the single hydrogen bond accepting capability of the nitrile was insufficient to recapitulate the same interactions.
Five-membered ring heterocycles are established bioisosteres of the amide bond.26 We hypothesized that CSNK2A2 would similarly accept five-membered ring heterocycles directly attached to the phenyl ring with hydrogen bond acceptors three bond lengths away. To our delight, replacement of the acetamide with a 1,2,4-triazol-4-yl group (14) successfully improved CSNK2A2 activity 4-fold. In concert with the increased CSNK2A2 potency, the potency of 14 in the MHV assay was also improved 4-fold. While the isomeric 1,2,3-triazol-5-yl group (15) had a <3-fold decrease in MHV and CSNK2A2 activity as compared to 14, the 1,2,3-triazol-1-yl group (16) was two orders of magnitude less potent against MHV and CSNK2A2. The sensitivity of CSNK2A2 inhibition to the positions of the nitrogen atoms on the ring suggests that both nitrogen atoms on the 1- and 2-positions of the triazole in 14 form crucial interactions with the kinase.
Introducing a methyl group at the 3-position of the 1,2,4-triazole (17) resulted in decreased activity of CSNK2A2 and MHV, similar to the effects observed upon introducing a methyl group at the equivalent positions in the imidazole 18 and triazole 19. The corresponding imidazolinone 20 was similarly inactive. Together, these analogues demonstrate the steric limitations of the binding pocket around this group. Although, an alternative explanation may be that substituents adjacent to the biaryl bond may reduce the planarity of this biaryl system, preventing efficient interactions with the kinase. The introduction of a methylene linker between the phenyl ring and the triazole (21) also led to a drastic drop in both CSNK2A2 and MHV activity. This suggests that there are tight steric requirements within the kinase’s ATP-binding pocket, and underscores the necessity of positioning the nitrogen atoms on the 1- and 2-positions of the triazole of 14 appropriately for potent binding.
We next explored if the 1,2,4-triazole could be replaced with other heterocycles. The oxadiazole 22, thiazole 23, thiadiazole 24, and tetrazoles 25, 26, and 27 were only weakly active against CSNK2A2 and inactive against MHV. Expanding the ring size to a six-membered ring was shown to be similarly unfavorable as exemplified by the pyridine (28) and pyrazine (29) analogues. These results suggest that the 1,2,4-triazole is indeed a privileged amide bioisostere at this position, in terms of fitting the steric requirement and the optimal positioning of the hydrogen bond acceptors.
To better understand the binding mode of the triazole and the importance of the nitrogen atoms, we obtained a cocrystal structure of CSNK2A1 bound to 14 (Figure 2, right). 14 binds to CSNK2A1 in a very similar manner as compared to the CSNK2 chemical probe SGC-CK2-1 (1).19 Like 1, the pyrazolo[1,5-a]pyrimidine core of 14 binds at the hinge region of the kinase, with the N1 atom and the 7-position exocyclic NH forming key hydrogen bonding interactions with the backbone of Val116. The nitrile group at the 3-position of the pyrazolo[1,5-a]pyrimidine core forms a hydrogen bond with a water molecule buried in the ATP binding pocket. The cocrystal structure with 1 shows the carbonyl group of the amide forming two hydrogen bonding interactions with Lys68 and the buried water molecule. These same two hydrogen bonding interactions were captured by the triazole of 14 using both the 1- and 2-position N atoms separately. Thus, the crystallographic evidence supports our conclusions from the SAR regarding the importance of both the 1- and 2-position N atoms on the triazole, and its suitability as a bioisosteric replacement for the critical amide substituent.21−23
We next decided to investigate how this bioisosteric replacement modified other important properties. Given that metabolic stability is a primary concern for the pyrazolo[1,5-a]pyrimidines,24 we investigated the microsomal and whole hepatocyte stability of compounds 3 and 14 (Table 2). Compared to 3, compound 14 exhibited improved metabolic stability in mouse and human liver microsomes and hepatocytes, providing another advantage of this isosteric replacement, although we noted that the metabolic clearance rates were still high. However, further profiling of compound 14 revealed poor aqueous solubility of 0.47 μg/mL. Despite this, given the excellent potency and improved metabolic stability of the triazole, we decided to maintain this group at the meta-position of the aniline and aimed to enhance aqueous solubility by incorporating further modifications elsewhere on the molecule.
Table 2. Comparison of Metabolic Stability and Solubility of 3 and 14.
| Compound | Mouse liver microsomal stability (% remaining after 30 min)a | Human liver microsomal CLint (mL/min/kg)b | Mouse hepatocyte CLint (mL/min/kg)c | Human hepatocyte CLint (mL/min/kg)c | Kinetic Solubility (μg/mL)d |
|---|---|---|---|---|---|
| 3 | 22 | 25 | 520 | 58 | 2.8 |
| 14 | 74 | 7.6 | 160 | 16 | 0.47 |
Metabolism in MLM measured at 30 min by LC-MS.
Metabolism in HLM quantified by LC-MS over five time points over 1 h, scaled by scaling factors of mass of liver per body weight and microsomal concentration in liver.
Metabolism in hepatocytes quantified by LC-MS over 2 h, scaled by scaling factors of mass of liver per body weight and hepatocyte concentration in liver.
Kinetic solubility is determined from 10 mM DMSO stock solutions in PBS buffered at pH 7.4.
Modifications to Improve Solubility and Metabolic Stability
Based on the above-mentioned crystal structures of the pyrazolo[1,5-a]pyrimidine inhibitors bound to CSNK2, we observed that the 7-position cyclopropylamino substituent points toward the solvent, offering an opportunity to add solubilizing groups at this position (Table 3). Replacement of the cyclopropyl ring with a slightly larger cyclobutyl ring (30) was found to be tolerated by CSNK2A2 and MHV, although this increase in lipophilicity led to a further decrease in solubility of the compound. To our surprise, however, we observed two orders of magnitude decrease in potency against CSNK2A2 and a loss of MHV activity with the oxetane ring (31), a result that contrasts with previous reports of the tolerability of CSNK2 toward the oxetane substituent.21,22 The 3,3-difluorocyclobutyl substituent (32) was similarly disfavored by CSNK2A2 and was inactive against MHV. We next introduced cyclic ethers as solubilizing groups at this position (33 and 34), but both compounds were inactive against CSNK2A2 and MHV. A (1-methyl-1H-pyrazol-4-yl)methyl substituent (35) and a N-acetylpiperidin-4-yl substituent (36) also demonstrated no activity against CSNK2A2 and MHV. Together, these results demonstrated that the steric requirement around this region of the CSNK2 active site is rather strict. We also demonstrated that the compound with a hydroxyethyl group (37) had weak activity against CSNK2A2 and was inactive against MHV. We infer from these results that the introduction of polar heteroatoms in this region could be detrimental for inhibition of CSNK2A2.
Table 3. SAR at the 7-Position of the Pyrazolo[1,5-a]pyrimidine Core.


Mean of three independent experiments performed in triplicate.
Mean of two independent experiments.
Kinetic solubility measurements were carried out in phosphate buffered saline solution (PBS) at pH 7.4 from DMSO stock solutions.
Metabolism in MLM measured at 30 min by LC-MS. n.d. = not determined.
To inform selection of suitable substituents at this position, we surveyed literature for examples of groups successfully incorporated at this position. It has been demonstrated previously that imidazole or pyrazole substituents were tolerated by CSNK2A1 at this position, and pendant solubilizing groups may be successfully attached here without loss of CSNK2A1 potency.22 We initially investigated the imidazole substituents. Despite an improvement in solubility, in contrast with the literature reports, the replacement of the cyclopropyl ring with an N-methyl imidazole (38) resulted in a 17-fold drop in CSNK2A2 potency and a 19-fold drop in MHV potency. Introduction of basic amines on the imidazole (39, 40, 41, 42) led to a further drop in CSNK2A2 potency and a loss of MHV activity. We then turned to the pyrazole substituents. Similarly to the compounds with an imidazole, the compounds containing a pyrazole with pendant amines (43, 44, 45, 46) improved solubility compared to 14. However, we still observed a 5- to 1000-fold drop in CSNK2A2 potency for these compounds, which was accompanied by a ≥25-fold drop in MHV potency. A similar trend was observed for CSNK2A1 potency as well (Table S1). Interestingly, comparing matched pairs of imidazoles and pyrazoles (39 with 43, 40 with 44, 41 with 45), we observed that the pyrazoles were preferred by CSNK2, demonstrating that the exact positioning of the N atoms on this ring leads to significantly different inhibitory effects. The inconsistency of the SAR with the findings by Dowling et al.22 are puzzling, but nevertheless consistent with our previous findings that the cyclopropylamino group is the most optimal 7-position substituent for both CSNK2 and MHV inhibition.15
Selected analogues (30–35, 40–46) were also tested for metabolic stability in mouse liver microsomes. All analogues demonstrated moderate stability with 60–90% remaining after 30 min of incubation. Because these modifications at the 7-position of the pyrazolo[1,5-a]pyrimidine core did not significantly affect metabolic stability, this suggested that the major site of metabolism was unlikely to be at the cyclopropylamine group, in agreement with the metabolite identification study conducted previously for another analogue of this series.24
Having ruled out the feasibility of improving solubility and metabolic stability by attaching solubilizing groups at the 7-position of the pyrazolo[1,5-a]pyrimidine while maintaining cellular potency, we next turned to optimization of the substituents on the aniline ring of the molecule (Table 4). We first explored the effect of introducing fluorine atoms on the aniline ring. Interestingly, the introduction of a fluorine atom at the ortho-position of the aniline ring (47) improved MHV potency while maintaining CSNK2A2 potency, increased stability in mouse liver microsomes, and led to a slight improvement in solubility when compared to 14. However, 47 demonstrated moderate cytotoxicity in A549-ACE2 cells (59% viability at 1 μM, Table 5), and we chose not to proceed further with this compound. Introduction of a fluorine atom at the para-position of the aniline ring (48) maintained CSNK2A2 potency, decreased MHV potency, and improved metabolic stability, although it did not improve solubility compared to 14, demonstrating that a fluorine atom was not favored at this position. Replacement of an aromatic CH with a nitrogen has been recognized as an effective strategy to improve solubility of compounds in multiparameter optimization programs.27 Replacement of the phenyl ring with a pyridine ring with the aromatic N atom at the “ortho-position” (49) or “para-position” (50) both slightly decreased CSNK2A2 and MHV potency. The solubility was indeed slightly improved with 49 but, surprisingly, decreased with 50. The microsomal stability of 49 and 50 was improved compared to 14. Altogether, these four analogues demonstrate that a reduction in electron-rich nature of the aniline ring was a fruitful strategy in reducing metabolic clearance in liver microsomes, presumably by reducing the propensity for cytochrome-P450-mediated oxidation into an arene oxide or a quinone-imine ring.28−30
Table 4. SAR at the para-Position of the Aniline.



Mean of three independent experiments performed in triplicate.
Mean of two independent experiments.
Kinetic solubility measurements were carried out in phosphate buffered saline solution (PBS) at pH 7.4 from DMSO stock solutions.
Metabolism in MLM measured at 30 min by LC-MS. n.d. = not determined.
Table 5. Cytotoxicity of Compounds with Potent Antiviral Activity.
| Compound | A549-ACE2 % viability at 1 μMa | A549-ACE2 % viability at 0.1 μMa |
|---|---|---|
| 14 | 85 | 100 |
| 15 | 85 | 100 |
| 30 | 93 | 102 |
| 47 | 59 | 86 |
| 48 | 99 | 99 |
| 49 | 96 | 106 |
| 50 | 98 | 99 |
| 51 | 87 | 99 |
| 53 | 98b | 101b |
| 55 | 91 | 95 |
| 56 | 76 | 93 |
| 59 | 100 | 98 |
| 60 | 97 | 95 |
| 62 | 99 | 99 |
| 64 | 101 | 96 |
| 66 | 85 | 101 |
| 68 | 102 | 106 |
| 72 | 105 | 105 |
Mean of quadruplicate experiments.
n = 9. n.d. = not determined.
We note that the successful attachment of solubilizing groups containing basic amines at the para-position of the aniline ring has been reported when the meta-position of the aniline ring possesses an acetamide or propionamide group.15,19,23 We hypothesized that this strategy could be similarly employed with our triazole compounds.
As hypothesized, introduction of an N-(2-aminoethyl)-N-methyl group at this position (51) improved solubility drastically. However, an unexpected 58-fold drop in CSNK2A2 potency and a 9-fold drop in MHV potency was observed. We further investigated other solubilizing groups. With an N-(2-aminoethyl)-N-ethyl group at the para-position (53), a modest 2-fold improvement in MHV potency was observed when compared to 51, while the CSNK2A2 potency remained in the micromolar range. Methylation at other positions in this para-position group (55, 56, 57) led to similar MHV and CSNK2A2 potency. Notably, all these analogues maintained excellent solubility. Adding an ethyl group to the aliphatic amine (58) decreased MHV potency drastically, although potency was recovered by the addition of a second ethyl group (59) and maintained good solubility. Submicromolar potency against CSNK2A2 and MHV was obtained with an alcohol (60) or methyl ether (62) in place of the amino group, suggesting that the basic amine may be replaced with neutral substituents. While 60 demonstrated excellent solubility, the solubility of 62 was poor. To investigate the steric requirements in this region, we replaced the pendant amine of 51 with a pyrrolidine (63) or morpholine (64). While this resulted in an improvement in CSNK2A2 potency, a decrease in potency against MHV was observed. The solubility of 64 remained favorable.
We then investigated alternative solubilizing groups with cyclic amines at this position. A basic amine was maintained with a distance of three atoms between the phenyl ring and the amine, in accordance to literature findings about the optimal linker length.23 Both enantiomers with the 3-aminopyrrolidin-1-yl group (65, 66) maintained submicromolar potency against CSNK2A2, but the (S)-enantiomer 66 was 8-fold more potent than the (R)-enantiomer 65 against MHV. Expansion of the ring size by one methylene unit (68) did not significantly improve CSNK2A2 or MHV potency. Methylation of the amine (69) resulted in comparable CSNK2A2 potencies while decreasing the MHV potency 3-fold. The (R)-enantiomer (70) was less potent against CSNK2A2, with similar MHV potency. The solubility of these compounds remained excellent, as expected from the presence of a basic amine.
We next introduced a morpholine ring at this position (72). While we found that this compound possessed good CSNK2A2 and MHV potency as compared to the other analogues with basic amines, the modest increase in solubility as compared to 14 did not justify the 7.5-fold drop in CSNK2A2 potency and 3-fold drop in MHV potency. Changing the morpholine ring to an N-methylpiperazine ring (73) led to a 5-fold drop in potency against CSNK2A2 and an almost 50-fold drop in potency against MHV, while the solubility surprisingly decreased further.
Several analogues with para-position solubilizing groups (51, 53, 55, 60, 62, 64, 66–69, 71, 72) were selected for metabolic stability screening. Except for 64, all analogues exhibited excellent stability in mouse liver microsomes, with 84–100% remaining after 30 min of incubation. This result suggested that the decreased lipophilicity resulting from the introduction of solubilizing groups was beneficial for decreasing hepatic metabolism.
Our results showed that despite improvements in solubility and microsomal stability, introducing solubilizing groups at the para-position decreases both CSNK2A2 and MHV potency. Having established earlier with compound 47 that an ortho-position fluorine atom could improve the MHV potency, we hypothesized that the introduction of this ortho-position fluorine atom could compensate for the lower potency with the solubilizing groups. We thus synthesized five compounds 52, 54, 61, 67, and 71, which are the match-pair analogues of 51, 53, 60, 66, and 70, respectively. However, for all the matched pairs except 71 and 70, we observed a general trend where MHV potency decreased at least 6-fold with the addition of this ortho-position fluorine atom.
We have additionally obtained cocrystal structures of 50 and 53 (Figure 3) with CSNK2A1. Both compounds bind to CSNK2A1 in an almost identical manner as 14. For both compounds, we observed hydrogen bonds between the N1 of the pyrazolo[1,5-a]pyrimidine and the 7-position exocyclic NH with the backbone of Val116, and with both the 1- and 2-position nitrogen atoms of the triazole with the buried water molecule and Lys68, respectively. For 53, the ethyl group points toward the P-loop while the 2-aminoethyl group oriented toward the C-lobe, forming a hydrogen bond with Asn161 and is consistent with a literature crystal structure of the 2-aminoethyl group at this position (PDB ID: 5H8E).23
Figure 3.

Co-crystal structure of 50 (left, PDB ID: 8P06) and 53 (right, PDB ID: 9EZG) with CSNK2A1. Part of the kinase P-loop is hidden for clarity. Water molecules are represented as red spheres. Hydrogen bonds are denoted as yellow dashed lines.
Unlike previously reported for compounds with a meta-position acetamide and propionamide,15,19,23 the addition of a para-position substituent to compounds with a meta-position triazole generally led to a loss of CSNK2A2 and MHV activity. While one possible explanation could be decreased cellular permeability of the more polar compounds, this might also be attributed to disruption of the high degree of coplanarity between the triazole ring and the phenyl ring. A high degree of coplanarity was observed for both 14 and 50, with dihedral angles of 9.6/9.9° and 3.5/4.2°, respectively. As a reference, the measured dihedral angle across the C–N bond between the phenyl ring and the N(H)–C(=O) bond in the crystal structure of CSNK2A1 with 1 was 55.6°. A para-position substituent on the aniline ring would be expected to increase the dihedral angle, deviating from the optimal angle for binding. Because the triazole uses both 1- and 2-position N atoms to form separate hydrogen bonds with Lys68 and the water molecule, and hydrogen bonds are directional interactions sensitive to changes in bond angle, an increase in dihedral angle would be expected to diminish the binding energy of the two hydrogen bonds and preclude efficient binding to CSNK2. We noted that the dihedral angle of 53 in its cocrystal structure with CSNK2A1 was 8.7/15.2°, which was slightly higher than that for 14 and 50. This rationale was also supported by the decrease in potency following introduction of a methyl group at the 3-position of the 1,2,4-triazole (17), at the equivalent positions in the imidazole (18) and triazole (19), and the exocyclic oxygen atom of the imidazolinone (20).
We selected compounds that demonstrated MHV IC50 ≤ 1 μM for evaluation of cytotoxicity in A549-ACE2 cells at 1.0 μM and 0.1 μM inhibitor concentrations using a CellTiter-Glo assay (Table 5). All compounds except 47 and 56 demonstrated ≥85% viability at 1 μM and negligible cytotoxicity at 0.1 μM.
Despite excellent potency against CSNK2 and MHV, low cytotoxicity, and favorable metabolic stability of 14 and 50, their low solubility resulted in significant challenges during chemical synthesis and purification upon reaction scale-up, as well as during formulation development for in vivo studies. As a compromise between potency and aqueous solubility, we selected 53, which had submicromolar potency against both CSNK2 and MHV, and also possessed excellent solubility and exemplary microsomal stability, for further evaluation.
Potency and Selectivity Characterization of Compound 53
To ascertain the potency of 53 in orthogonal assays, we first evaluated 53 against CSNK2A1 and CSNK2A2 in the Eurofins KinaseProfiler assay, an in vitro radiometric enzyme assay performed with [ATP] at the Km of the kinases. 53 was found to be a potent inhibitor of both CSNK2A1 and CSNK2A2, with IC50 values of 1.7 nM and 0.66 nM, respectively (Figure S1). The difference between the potencies as measured by the cellular NanoBRET assay and the in vitro KinaseProfiler assay are likely due to cellular permeability of the inhibitor or due to the higher cellular concentrations of ATP than used in the KinaseProfiler assay. We next assayed 53 in the NanoBRET assay using a modified procedure, where digitonin was added to permeabilize the cells to eliminate the factor of membrane permeability in the assay.31 Under these conditions, a dose-dependent inhibition of CSNK2A2 was also observed, with an IC50 of 80 nM (Figure S2), representing a 4-fold increase in potency as compared to the regular conditions for the NanoBRET. Additionally, when A549-ACE2 cells were treated with 1 or 5 μM of 53, we measured a reduction in phosphorylation levels of the CSNK2 substrate EIF2S232 after 24 h (Figure S3), indicating that 53 inhibits CSNK2 downstream signaling. Altogether, these results confirmed that 53 is a potent CSNK2 inhibitor. Clearly, despite minor permeability limitations, 53 was still sufficiently permeable to achieve efficacy against MHV, and the NanoBRET assay was a good predictor of the cellular activity in the antiviral assay.
Our next step was to evaluate the kinome-wide selectivity of 53. We measured the % occupancy of 53 at 10 μM against 192 human kinases using the NanoBRET K192 assay panel, a selectivity panel that determines target engagement in the cellular context33 (Figure 4). Out of 192 kinases, only two kinases, CSNK2A1 and CSNK2A2, were engaged by 53 with 97% occupancy (Table 6). Eight other kinases (CLK1, CLK2, DAPK2, HIPK4, CLK4, PHKG1, DYRK1A, and CDK7) were identified with % occupancy values between 50–75%, while all other kinases possess <50% occupancy at 10 μM (Table S3). A follow-up dose–response experiment in the NanoBRET assay was conducted for the off-target kinases, which verified that CLK1, CLK2, DAPK2, HIPK4, CLK4, PHKG1, and CDK7 were inhibited only with micromolar IC50s (Table 6). With at least approximately one order of magnitude of potency difference between inhibition of CSNK2 and inhibition of identified off-target kinases, we concluded that 53 will be a selective inhibitor of CSNK2 in cells.
Figure 4.

Selectivity profile of 53 determined at 10 μM using the NanoBRET K192 panel.33 Only CSNK2A1 and CSNK2A2 had 97% occupancy (red). Kinases with 50–75% occupancy are shown in yellow. Kinases with <50% occupancy are shown in green. Kinases not in the NanoBRET K192 panel are shown in gray. Image generated using CORAL.34 No kinases had occupancy >75% except CSNK2A1 and CSNK2A2. Detailed % occupancy values for each kinase are described in Table S3.
Table 6. Selectivity of 53 in the NanoBRET K192 Panel.
| Kinase | NanoBRET K192 % occupancy at 10 μMa | NanoBRET pIC50 |
|---|---|---|
| CSNK2A2 | 97 | 6.5b |
| CSNK2A1 | 97 | n.d. |
| CLK1 | 74 | 5.3b |
| CLK2 | 73 | 5.5c |
| DAPK2 | 66 | 5.6c |
| HIPK4 | 63 | 4.6b |
| CLK4 | 59 | 5.6c |
| PHKG1 | 59 | 5.2c |
| DYRK1A | 56 | n.d.d |
| CDK7 | 50 | 4.8c |
Performed in singlicate.
Mean of two independent experiments.
Determined from one experiment with two dilution curves.
Poor assay window observed but no occupancy observed up to 3 μM. n.d. = not determined.
Having established that 53 is a potent and selective CSNK2A2 inhibitor with good antiviral activity against MHV, we next sought to evaluate it for activity against SARS-CoV-2 in vitro (Figure 5). A dose-dependent inhibition of SARS-CoV-2 replication was observed with an IC50 of 390 nM. No decrease in cell viability by the LDH assay was observed at concentrations below 10 μM, demonstrating that this assay result was not confounded by host cell cytotoxicity. The cytotoxicity results in the LDH assay were also consistent with the CellTiter-Glo assay results.
Figure 5.

(A) Structure of 53. (B) Dose-dependent effect of 53 against SARS-CoV-2 in A549-ACE2 cells and cell viability of A549-ACE2 cells as measured by the LDH assay.
In Vitro ADME and In Vivo PK of Compound 53
Encouraged by these results, we further characterized 53 in in vitro ADME studies (Table 7) and in vivo mice pharmacokinetic experiments (Table 8). As discussed previously, 53 possesses excellent solubility of 56 μg/mL. However, 53 had poor permeability in MDCK cells transfected with MDR1, although it was not susceptible to rapid efflux by MDR1 (efflux ratio of 2.3). Compound 53 was also not highly plasma protein bound in human plasma, with an unbound fraction of 15%. 53 possessed good metabolic stability in mouse and human liver microsomes and whole hepatocytes, a promising finding considering that inhibitors of the pyrazolo[1,5-a]pyrimidine scaffold are known to demonstrate high mouse hepatocyte clearance due to phase I and phase II metabolism, commensurate with rapid metabolism in vivo.24
Table 7. In Vitro ADME Characterization of Compound 53.
| Kinetic Solubilitya (μg/mL) | MDCK-MDR1 Papp A-B/B-A (10–6 cm/s)b | Efflux Ratiob | Human Plasma Protein Binding fu (%)c | Mouse liver microsomal stability (% remaining after 30 min)d | Human liver microsomal CLint (mL/min/kg)e | Mouse hepatocyte CLint (mL/min/kg)f | Human hepatocyte CLint (mL/min/kg)f |
|---|---|---|---|---|---|---|---|
| 56 | 0.16/0.36 | 2.3 | 15 | 92 | <6.8 | <32.0 | <6.9 |
Kinetic solubility is determined from 10 mM DMSO stock solutions in PBS buffered at pH 7.4.
Permeability assay in MDCK cells transfected with human MDR1, in apical-to-basolateral (A-B) and basolateral-to-apical (B-A) directions.
Plasma protein binding measurements performed in duplicate.
Metabolism in MLM measured at 30 min by LC-MS.
Metabolism in HLM quantified by LC-MS over five time points over 1 h, scaled by scaling factors of mass of liver per body weight and microsomal concentration in liver.
Metabolism in hepatocytes quantified by LC-MS over 2 h, scaled by scaling factors of mass of liver per body weight and hepatocyte concentration in liver.
Table 8. In Vivo (5 h) Mice Pharmacokinetic Data for Compound 53a.
| Dose (mg/kg) | Route of Administration | Compartment | t1/2 (h) | CLint (mL/min/kg) | tmax (h) | Cmax (nM) | AUClast (h × nM) | Vdss (L/kg) |
|---|---|---|---|---|---|---|---|---|
| 3 | i.v. | Plasma | 0.91 | 86 | n.d. | n.d. | 1300 | 3.1 |
| 10 | i.p. | Plasma | 0.63 | n.d. | 0.5 | 6300 | 6400 | n.d. |
| 30 | i.p. | Plasma | 0.63 | n.d. | 0.5 | 27000 | 31000 | n.d. |
n.d. = not determined.
In vivo, 53 demonstrates good bioavailability by i.p. dosing at 10 mg/kg or 30 mg/kg (Table 8). However, it showed no bioavailability when dosed at 10 mg/kg p.o. (data not shown), suggesting that intestinal absorption was a key pharmacokinetic challenge moving forward. This is possibly related to its limited cellular permeability. Nevertheless, the i.p. route of administration is a well-recognized and acceptable means of dosing for in vivo proof-of-concept studies,35 and we opted for this route of administration moving forward. Given the excellent in vitro metabolic stability, we were surprised by the short plasma half-life and high apparent intrinsic clearance of 53 when mice were dosed at 3 mg/kg i.v., 10 mg/kg i.p., or 30 mg/kg i.p. The moderate volume of distribution of 3.1 L/kg indicated that this compound distributes evenly throughout blood and tissues. This result was unsurprising, considering the basic character of 53 might predispose it to bind to negatively charged phospholipids.36 We hypothesized that the high apparent intrinsic clearance was due to a rapid drop in plasma concentrations measured during the distribution phase, as opposed to the elimination phase, of the compound. The distribution of 53 into tissues might account for the high apparent clearance. Since the lung concentration is of particular interest for effective treatment of SARS-CoV-2, we decided to investigate the pharmacokinetic parameters of the lung compartment of mice dosed with 10 mg/kg i.p. of 53 (Figure 6). Gratifyingly, we observed that from the 0.5 to 4 h time point, the lung/plasma ratio of 53 increased from 0.6 to 48, before slowly declining to a ratio of 18 by 24 h, supporting our hypothesis that 53 does partition into the lung tissues. The rapid initial increase of the lung/plasma ratio over the first 4 h also supported our hypothesis that the low plasma half-life of the compound measured initially was due to drug distribution rather than metabolism. We measured the plasma half-life of 53 using the data points from 0.5 to 4 h (t1/2 = 0.45 h), and found it to be comparable to the value determined previously (0.63 h). In contrast, the half-life of 53 from 4 to 24 h was much longer, at 8 h in the plasma compartment and 5 h in the lung compartment. These results suggest that the low rate of in vitro metabolism did in fact translate to a reduction in the in vivo metabolic clearance in mice. While the plasma concentration of 53 had dropped below the in vitro SARS-CoV-2 IC50 by 2 h, importantly, the lung concentration of 53 remained above this level for at least 12 h from a single dose, only dropping below this level by 24 h. From these results, we hypothesized that b.i.d. dosing of 10 mg/kg i.p. might be able to maintain an efficacious concentration of this compound at its target tissue.
Figure 6.

(A) Concentration over time of 53 in CD-1 mice, dosed i.p. at 10 mg/kg at t = 0 h. Plasma concentrations (light blue line) and lung concentrations (dark blue line) of the compound were measured at 0.5, 1, 2, 4, 8, 12, and 24 h, and the lung/plasma ratio is calculated (black dashed line). Data points are shown as mean ± s.d. (n = 3). The SARS-CoV-2 IC50 of 53 determined in A549-ACE2 cells is plotted as a yellow dotted line. (B) Pharmacokinetic parameters.
In Vivo Efficacy
We investigated 53 in a prophylactic treatment model for SARS-CoV-2 MA10 infection in mice.20 Briefly, 53 was dosed i.p. at 10 mg/kg every 12 h, with the first dose performed 12 h before viral inoculation. The mice were euthanized 24 h after viral inoculation (36 h after the first dose of 53). The SARS-CoV-2 viral titer measured in mouse lung was similar in mice treated with 53 and mice given a vehicle treatment (Figure 7A). As a biomarker for CSNK2 inhibition, we measured phosphorylation of CSNK2 substrates EIF2S232 and AKT37 in mouse lung 36 h after treatment with 53 dosed at 10 mg/kg i.p. b.i.d. Compared to vehicle controls, no statistically significant reduction in phosphorylation levels of Ser2 of EIF2S2 and Ser129 of AKT were observed (Figure 7B–G). The disconnect between the pharmacokinetics and efficacy of 53 is unexpected. We measured the protein binding levels of 53 in mouse lung homogenate, and found it to be 96.8% bound. As such, the free drug concentration of 53 is likely to be much lower than expected based on the measured total drug concentration, and may not be sufficient to achieve in vivo efficacy. For antivirals, it has been recommended that the in vivo free drug concentration remain above the EC90 levels determined in cellular assays for therapeutic effect.38,39 We acknowledge that the measured lung concentrations represented the organ-wide average, which may not reflect the true concentration in the specific microenvironment of the target, given the heterogeneity in cell types and subcellular compartments present. It was also possible that the concentration of 53 at the target microenvironment of CSNK2 was insufficient to achieve pharmacological effect.
Figure 7.

(A) Viral titer measured in mouse lungs 24 h post inoculation when treated with vehicle or with compound 53 (10 mg/kg i.p. b.i.d.) (n = 5). (B) Western blot for total EIF2S2 expression in mouse lung samples after treatment with vehicle or 53 (10 mg/kg i.p. b.i.d.) (n = 3). (C) Western blot for EIF2S2 (phospho-Ser2) in mouse lung samples after treatment with vehicle or 53 (10 mg/kg i.p. b.i.d.) (n = 3). (D) EIF2S2 (phospho-Ser2) levels normalized to total EIF2S2 levels in mouse lung. (E) Western blot for total AKT expression in mouse lung samples after treatment with vehicle or 53 (10 mg/kg i.p. b.i.d.) (n = 3). (F) Western blot for AKT (phospho-Ser129) in mouse lung samples after treatment with vehicle or 53 (10 mg/kg i.p. b.i.d.) (n = 3). (G) AKT (phospho-Ser129) levels normalized to total AKT levels in mouse lung.
Conclusion
In conclusion, we have successfully identified the 1,2,4-triazole as an appropriate isostere of the amide group. This amide group was previously found to be critical for binding to CSNK2 through its involvement in the hydrogen bonding network with Lys68 and a water molecule buried in the ATP-binding pocket. The 1,2,4-triazole successfully captures these hydrogen bonds using two different nitrogen atoms. This bioisosteric replacement improved on-target potency against CSNK2 and antiviral potency against MHV, and led to favorable metabolic stability profiles. Through a multiparameter optimization campaign, we have characterized SAR at the 7-position of the pyrazolo[1,5-a]pyrimidine and at the para-position of the aniline for this new chemotype. Compound 53 maintained a balance of potency, solubility, metabolic stability, and low cytotoxicity, which are properties favorable for compound progression. Despite the excellent metabolic stability profile in vitro, a rapid decline in plasma levels of 53 was observed in vivo, which may be attributed to the distribution and accumulation into organs such as the lung. Pharmacological efficacy was not achieved in vivo, possibly attributed to insufficient free drug concentrations. Further medicinal chemistry optimization is required to reach in vivo proof of concept. Nevertheless, the SAR described herein further contributes to the future development of improved CSNK2 inhibitors for use in vivo, and represents a significant step forward in the development of antivirals against β-coronaviruses. CSNK2 remains a potential antiviral target. Optimized inhibitors may pharmacologically validate CSNK2 as a target for host-directed therapy, and serve as CSNK2-targeted antiviral therapeutics in the future. Here, we found compound 53 to be a potent and selective CSNK2 inhibitor with well-characterized physiochemical and pharmacokinetic properties both in vitro and in vivo. We envisage that compound 53, a potent, selective, and cell-active inhibitor with a distinct structure from other CSNK2 inhibitors, will find utility in the investigation of CSNK2 activity in a range of biological contexts.
CHEMISTRY
Synthesis of analogues varying the meta-position of the aniline (Scheme 1) was achieved by displacing the 7-chloro substituent on 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) with cyclopropylamine to obtain the common intermediate 75. Buchwald–Hartwig coupling between 75 and anilines 76–101 furnished the desired analogues 3–29, removing a Boc or tBu protecting group where necessary. Aniline coupling partners were purchased where commercially available, and synthesized individually otherwise (Scheme 2). Anilines 79 and 80 were synthesized by an SN2 reaction between benzyl bromide 104 and NaSMe, followed by oxidation using mCPBA to obtain the sulfoxide 106, subsequent Rh(II)-catalyzed oxidation to obtain the Boc-protected sulfaneylidene 107, and finally nitro reduction. An analogous SN2 reaction between benzyl bromide 108 and dimethylphosphine oxide, followed by Boc deprotection afforded aniline 81. Aniline 83 was synthesized by the attack of the ester group of 112 by two equivalents of MeMgBr, with the necessary Cbz protection and deprotection steps for the aniline. The 1H-1,2,3-triazol-5-yl group of aniline 87 was prepared through a copper-catalyzed azide–alkyne “click” reaction between the alkyne of 114 and TMSN3, followed by nitro reduction of the nitro group to afford 87. A base-promoted condensation between the isothiocynate group of 116 and acetohydrazide afforded 117, which was desulfurized by NaNO2 in acetic acid to form 118, and underwent nitro reduction to yield aniline 89. The reaction between isocyanate 119 and 2,2-dimethoxyethan-1-amine afforded the urea 120, which cyclized to the imidazol-2-one 121, and the nitro group was then reduced to the aniline 92. Synthesis of aniline 93 proceeded through a similar SN2 reaction between 1,2,4-triazole and benzyl bromide 104, with a subsequent nitro reduction. The boronate ester of 123 underwent Suzuki coupling reactions with the appropriate aryl bromide to furnish anilines 95, 96, and 101. Finally, the aniline 99 was synthesized by first converting the amino group of 124 to diazonium salt 125, which reacted with TMSCHN2 in a silver(I)-catalyzed [3+2]-cycloaddition to form the tetrazole 126, before reduction of the nitro group to afford 99.
Scheme 1. Synthesis of Analogues Varying the meta-Position of the Aniline 3–29.
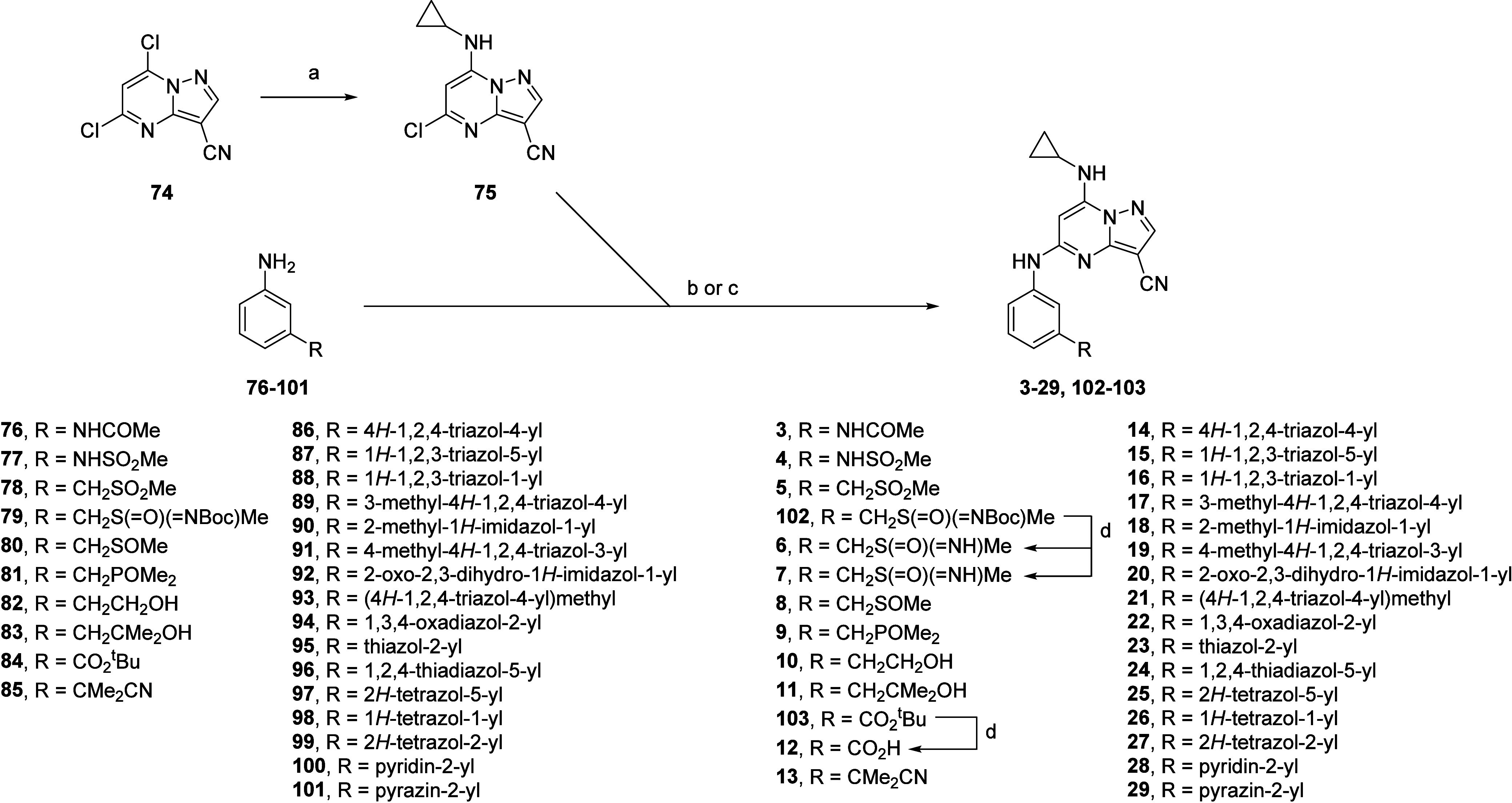
Reagents and conditions: (a) cyclopropylamine, EtOH, 25 °C, 2 h; (b) Pd(OAc)2, BINAP, Cs2CO3 or tBuOLi or tBuOK, dioxane, μW, 130 °C, 0.5 h or 100 °C, 2 h; (c) Brettphos Pd G3, Cs2CO3, dioxane, μW, 130 °C, 0.5 h; (d) TFA, DCM, 25 °C, 2 h.
Scheme 2. Synthesis of Aniline Intermediates 79–81, 83, 87, 89, 92, 93, 95, 96, 99, 101.

Reagents and conditions: (a) NaSMe, MeOH, 0–25 °C, 3 h; (b) mCPBA, DCM, 0 °C, 1 h; (c) Rh(OAc)2, MgO, iodobenzene diacetate, tert-butyl carbamate, DCM, 25 °C, 16 h; (d) Fe, NH4Cl, EtOH, H2O, 65–90 °C, 1–3 h; (e) Me2P(=O)H, NaHMDS, THF, −30 °C, 1 h then 25 °C, 12 h; (f) TFA, DCM, 25 °C, 2–10 h; (g) H2, Pd/C, MeOH, 25 °C, 2–12 h; (h) CbzCl, DIPEA, DCM, 25 °C, 16 h; (i) MeMgBr, THF, 25 °C, 16 h; (j) TMSN3, CuI, DMF, MeOH, 100 °C, 12 h; (k) acetohydrazide, DBU, EtOH, 85 °C, 10 h; (l) NaNO2, AcOH, H2O, 0–100 °C, 5 h; (m) SnCl2·H2O, HCl, EtOH, 70 °C, 10 h; (n) NH2CH2CH(OMe)2, DCM, 0–25 °C, 3 h; (o) 4H-1,2,4-triazole, K2CO3, MeCN, 100 °C, 10 h; (p) ArBr, Pd(dppf)Cl2, Cs2CO3, dioxane, H2O, 100 °C, 5 h; (q) tBuONO, HBF4, EtOH, 0–25 °C, 1 h; (r) TMSCHN2, (CF3CO2)Ag, Et3N, THF, −78 °C, 1 h; (s) CsF, MeOH, rt, 0.5 h.
The synthesis of analogues varying the 7-position of the pyrazolo[1,5-a]pyrimidine (Scheme 3) proceeded through an SNAr reaction between 74 and commercially available amines to furnish intermediates 127–134, which reacted with 86 in Buchwald–Hartwig coupling reactions to yield analogues 30–36 and 135, which was converted to 37 by deprotection of the TBDMS group. Synthesis of analogues with an amino-imidazole or amino-pyrazole at the 7-position of the pyrazolo[1,5-a]pyrimidine (Scheme 4) began by functionalizing 4-nitro-1H-imidazole (139) or 3-nitro-1H-pyrazole (140) with the appropriate alkyl halide using an SN2 reaction, followed by functional group interconversion where necessary. The nitro groups of functionalized imidazoles and pyrazoles 136, 144, 146, 153–158 were reduced to an amino group via hydrogenation (137, 147–148, 159–164), which next reacted readily with 74 in an SNAr reaction to form intermediates 138, 149–150, 165–170. Buchwald–Hartwig coupling with 86 furnished the analogues 38–46, after Boc deprotection where necessary.
Scheme 3. Synthesis of Analogues Varying the 7-Position Substituent of the Pyrazolo[1,5-a]pyrimidine 30–37.

Reagents and conditions: (a) RNH2, EtOH, rt, 2 h; (b) 86, Pd(OAc)2, BINAP, Cs2CO3 or tBuOLi, dioxane, μW, 130 °C, 0.5 h; (c) 86, Brettphos Pd G3, Cs2CO3, dioxane, μW, 130 °C, 0.5 h; (d) TFA, DCM, 35 °C, 2 h.
Scheme 4. Synthesis of Analogues Varying the 7-Position Substituent of the Pyrazolo[1,5-a]pyrimidine 38–46.

Reagents and conditions: (a) H2, Pd/C, MeOH or THF, 20–40 °C, 2–12 h; (b) 74, EtOH, 25 °C, 2–5 h; (c) 86, Pd(OAc)2, BINAP, Cs2CO3, dioxane, μW, 130 °C, 0.5–6 h; (d) 2-bromoethanol, K2CO3, MeCN, 60 °C, 10 h; (e) BocNHCH2CH2Br, K2CO3, DMF, 90 °C, 3 h; (f) SOCl2, DCM, DMF, 25 °C, 2 h; (g) MeNH2, K2CO3, NaI, μW, 80 °C, 4 h; (h) Boc2O, K2CO3, THF, 25 °C, 3 h; (i) MeI, NaH, THF, 0–25 °C, 10 h; (j) TFA, DCM, 25–35 °C, 2–6 h; (k) RCH2CH2Cl, K2CO3, MeCN or DMF, 60–120 °C, 4–12 h; (l) RCH2CH2Cl, NaH, DMF, 15 °C, 3.5 h; (m) 86, Brettphos Pd G3, Cs2CO3, dioxane, μW, 130 °C, 4 h.
The synthesis of analogues 47–50 (Scheme 5) started with a condensation between the aniline of 171–174 and 1,2-diformylhydrazine to furnish the 1,2,4-triazole ring of 175–178. The nitro group of 175, 176, and 178 were reduced via hydrogenation to afford anilines 179, 180, and 182 respectively, while 177 underwent an SNAr reaction with ammonia to yield 181. A final Buchwald–Hartwig coupling step with 75 completes the synthesis of 47–50.
Scheme 5. Synthesis of Compounds 47–50.

Reagents and conditions: (a) 1,2-diformylhydrazine, Me3SiCl, Et3N, pyridine, 100 °C, 12 h; (b) for 179, 180, and 182, H2, Pd/C, MeOH, 25 °C, 10 h; (c) for 181, NH3, H2O, 100 °C, 72 h; (d) 75, Brettphos Pd G3, tBuOLi, dioxane, μW, 130 °C, 0.5 h; (e) 75, Pd(OAc)2, BINAP, Cs2CO3 or tBuOLi, dioxane, μW, 130 °C, 0.5 h.
Nonfluorinated analogues varying the para-position of the aniline were prepared starting with 176 (Scheme 6). SNAr reactions with the appropriate amine nucleophile followed by nitro reduction yielded anilines 201–218. Subsequent Buchwald–Hartwig coupling with 75, and Boc or TBDMS deprotection where necessary, completed analogues 51, 53, 55–60, 62–66, 68–70, 72–73.
Scheme 6. Synthesis of Non-fluorinated Analogues Varying the para-Position of the Aniline 51, 53, 55–60, 62–66, 68–70, 72–73.

Reagents and conditions: (a) R-H, K2CO3, MeCN, 100 °C, 10–12 h; (b) H2, Pd/C, MeOH or THF, 25–35 °C, 2–12 h; (c) 75, Pd(OAc)2, BINAP, Cs2CO3, dioxane, μW, 130 °C, 0.5 h; (d) 75, Brettphos Pd G3, Cs2CO3 or tBuOLi, dioxane, μW, 130 °C, 0.5 h; (e) TFA, DCM, 25–35 °C, 1–2 h; (f) for 60, TBAF, THF, 25 °C, 10 h.
Fluorinated analogues varying the para-position of the aniline were synthesized starting with 231 (Scheme 7). SNAr reactions with the appropriate amine nucleophile furnished 232–236 (234 included TBDPS protection of the alcohol group). The anilines were Cbz-protected, before the nitro groups were reduced to an aniline and condensed with 1,2-diformylhydrazine to afford the 1,2,4-triazole-containing compounds 247–251. The Cbz group was then removed via hydrogenation, and coupled with 75 in Buchwald–Hartwig coupling reactions to yield analogues 52, 54, 61, 67, and 71 after Boc or TBDPS deprotection.
Scheme 7. Synthesis of Fluorinated Analogues Varying the para-Position of the Aniline 52, 54, 61, 67, 71.

Reagents and conditions: (a) R-H, K2CO3, MeCN, 60–100 °C, 10–12 h; (b) for 234, 2-(methylamino)ethan-1-ol, K2CO3, 60 °C, 2 h, then TBDPSCl, imidazole, DMF, 25 °C, 12 h; (c) CbzCl, K2CO3, THF, 25 °C, 2–12 h; (d) Fe, NH4Cl, EtOH, H2O, 80–100 °C, 2–12 h; (e) 1,2-diformylhydrazine, Me3SiCl, Et3N, pyridine, 100 °C, 12 h; (f) H2, Pd/C, MeOH, 25 °C, 2–5 h; (g) 75, Pd(OAc)2, BINAP, Cs2CO3, dioxane, μW, 130 °C, 0.5–6 h; (h) TFA, DCM, 25 °C, 1–3 h; (i) for 61, TBAF, THF, 25 °C, 2 h.
Experimental Section
NanoBRET Assay
Assays were run with a modified version of the previously published protocols.15,19,24 HEK293 cells were cultured at 37 °C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (VWR/Avantor). A transfection complex of DNA at 10 μg/mL was created, consisting of 9 μg/mL carrier DNA (Promega) and 1 μg/mL CSNK2A-NLuc fusion DNA in Opti-MEM without serum (Gibco). FuGENE HD (Promega) was added at 30 μL/mL to form a lipid:DNA complex. The solution was then mixed and incubated at room temperature for 20 min. The transfection complex was mixed with a 20× volume of HEK293 cells in DMEM/FBS to arrive at a final concentration of 200,000 cells/mL, and 100 μL/well was added to a 96-well plate that was incubated overnight at 37 °C and 5% CO2. The following day, the media were removed via aspiration and replaced with 85 μL of Opti-MEM without phenol red. A total of 5 μL per well of 20× NanoBRET Tracer K10 (Promega) at 10 μM for CSNK2A1 or 5 μM for CSNK2A2 in Tracer Dilution Buffer (Promega N291B) was added to all wells, except the “no tracer” control wells. Test compounds (10 mM in DMSO) were diluted 100× in Opti-MEM media to prepare stock solutions and evaluated at 11 concentrations. A total of 10 μL per well of the 10-fold test compound stock solutions (final assay concentration of 0.1% DMSO) were added. For “no compound” and “no tracer” control wells, DMSO in Opti-MEM was added for a final concentration of 1.1% across all wells; 96-well plates containing cells with NanoBRET Tracer K10 and test compounds (100 μL total volume per well) were equilibrated (37 °C/5% CO2) for 2 h. The plates were cooled to room temperature for 15 min. The NanoBRET NanoGlo substrate (Promega) at a ratio of 1:166 to Opti-MEM media in combination with an extracellular NLuc Inhibitor (Promega) diluted at 1:500 (10 μL of 30 mM stock per 5 mL of the Opti-MEM plus substrate) was combined to create a 3× stock solution. A total of 50 μL of the 3× substrate/extracellular NL inhibitor was added to each well. The plates were read within 30 min on a GloMax Discover luminometer (Promega) equipped with a 450 nm BP filter (donor) and 600 nm LP filter (acceptor) using 0.3 s of integration time. Raw milliBRET (mBRET) values were obtained by dividing the acceptor emission values (600 nm) by the donor emission values (450 nm) and multiplying by 1000. Averaged control values were used to represent complete inhibition (no tracer control: Opti-MEM + DMSO only) and no inhibition (tracer only control: no compound, Opti-MEM + DMSO + Tracer K10 only) and were plotted alongside the raw mBRET values. The data was first normalized and then fit using the Sigmoidal 4PL binding curve in Prism Software to determine IC50 values.
NanoBRET Assay in Digitonin-Permeabilized Cells
HEK293 cells were cultured at 37 °C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (VWR/Avantor). A transfection complex of DNA at 10 μg/mL was created, consisting of 9 μg/mL carrier DNA (Promega) and 1 μg/mL CSNK2A2-NLuc fusion DNA in Opti-MEM without serum (Gibco). FuGENE HD (Promega) was added at 30 μL/mL to form a lipid:DNA complex. The solution was then vortexed and incubated at room temperature for 20 min. The transfection complex was mixed with a 20× volume of HEK293 cells in DMEM/FBS to arrive at a final concentration of 200,000 cells/mL, and 100 μL was added to each well in a 96-well plate that was incubated overnight at 37 °C and 5% CO2. The following day, the media was removed via aspiration and replaced with 75 μL of Opti-MEM without phenol red. A total of 5 μL per well of 20× NanoBRET Tracer K10 (Promega) at 5 μM in Tracer Dilution Buffer (Promega N291B) was added to all wells except the “no tracer” control wells. Test compounds (10 mM in DMSO) were diluted 100× in Opti-MEM media to prepare stock solutions and evaluated at 11 concentrations. A total of 10 μL per well of the 10-fold test compound stock solutions (final assay concentration of 0.1% DMSO) was added. For “no compound” and “no tracer” control wells, DMSO in Opti-MEM was added for a final concentration of 1.1% across all wells. A 10× digitonin solution was prepared with Opti-MEM from a 400× stock solution (Promega). 10 μL of the 10× digitonin solution was then added to each well of the 96-well plate (50 μg/mL). The plate, now containing cells with NanoBRET Tracer K10, test compounds, and digitonin (100 μL total volume per well), was then incubated at room temperature for a period no longer than 25 min. The NanoBRET NanoGlo substrate (Promega), at a ratio of 1:166 with Opti-MEM media, in combination with an extracellular NLuc Inhibitor (Promega) diluted at 1:500 (10 μL of 30 mM stock per 5 mL of the Opti-MEM plus substrate) was combined to create a 3× stock solution. A total of 50 μL of the 3× substrate/extracellular NLuc inhibitor was added to each well. The plates were read within 30 min of substrate addition on a GloMax Discover luminometer (Promega) equipped with a 450 nm BP filter (donor) and 600 nm LP filter (acceptor) using 0.3 s of integration time. Raw milliBRET (mBRET) values were obtained by dividing the acceptor emission values (600 nm) by the donor emission values (450 nm) and multiplying by 1000. Averaged control values were used to represent complete inhibition (no tracer control: Opti-MEM + DMSO only) and no inhibition (tracer only control: no compound, Opti-MEM + DMSO + Tracer K10 only). The data was first normalized and then fit using the Sigmoidal 4PL binding curve in Prism Software to determine IC50 values.
In-cell Selectivity Profiling Using NanoBRET K192 Assay
The K192 selectivity assay was run according to the Draft Promega technical manual, NanoBRET Target Engagement K192 Kinase Selectivity System. Reagents were supplied by Promega (Promega NP 4101). For the assay, DNA from the prepared kinase vector panel plates A and B were mixed with Fugene in 96-well plates (Corning 3917) and incubated at room temperature for 30 min. The NanoLuc Low control vector used is pNL1.1.CMV [Nluc/CMV] Vector (Cat.# N1091), and the transfection control vector is NanoLuc-HIPK2 Fusion Vector (Cat.# NV3221).
HEK293 cells were grown to 75–95% confluency in DMEM (Gibco 11995-065) supplemented with FBS (Avantor 97068-085) at 37 °C in 5% RH. On the first day of the assay, cells were harvested and resuspended in Opti-MEM (Gibco 11058-021) supplemented with 1% FBS (Avantor 97068-085) at 2.5 × 105 cells per mL. 60 μL of cell suspension was mixed with 10 μL of prepared DNA (10× concentration) and 30 μL of Fugene (30 μL/mL in Opti-MEM) as outlined by Promega and incubated overnight at 37 °C in a 5% CO2 incubator.
On day two of the assay, 5 μL of 20× K10 tracer was prepared and added at concentrations recommended by Promega. Then, 10 μL of compound 53 at 100 μM in Opti-MEM (diluted from a 10 mM solution in DMSO) was added to the test wells while an equivalent volume of Opti-MEM was added to the high-control wells. Plates were kept at 37 °C in a 5% CO2 incubator for 2 h. After 2 h, plates were allowed to equilibrate to room temperature for 15 min. A solution of 3× Complete Substrate plus Inhibitor Solution was freshly prepared, consisting of a 1:166 dilution of NanoBRET Nano-Glo Substrate plus a 1:500 dilution of Extracellular NanoLuc Inhibitor in Opti-MEM medium without serum or phenol red. 50 μL of the 3× Complete Substrate plus Inhibitor Solution was added to each assay well, including control wells. After 2–3 min, the plate was shaken at 300 rpm for 10 s and the donor emission wavelength (450 nm) and acceptor emission wavelength (610 nm) were measured using the Glomax Discover System.
As a quality check, the donor signal-to-background ratio was calculated for each individual kinase by dividing the mean donor signal for each kinase by the mean donor signal for the signal-to-background control wells. Fractional occupancy for the test drug for each kinase was determined using the following formula:
Where:
Sample = Mean BRET value across all Sample (tracer + compound) wells for an individual kinase.
Top = Mean BRET value across all Top (tracer + vehicle) control wells for an individual kinase.
Bottom = Mean BRET value of NanoLuc control wells (calculated either on a plate-by-plate basis or across the entire experiment).
MHV Assay
DBT cells were cultured at 37 °C in Dulbecco’s modified Eagle medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (Gibco) and penicillin and streptomycin (Sigma-Aldrich). DBT cells were plated in 96-well plates to be 80% confluent at the start of the assay. Test compounds or positive control EIDD-1931 were diluted to 15 μM in DMEM. Serial 4-fold dilutions were made in DMEM, providing a concentration range of 15–0.22 μM. Media were aspirated from the DBT cells, and 100 μL of the diluted test compounds was added to the cells for 1 h at 37 °C. After 1 h, MHV-nLuc5 was added at an MOI of 0.1 in 50 μL of DMEM so that the final concentration of the first dilution of the compound was 10 μM (T = 0). After 10 h, the media were aspirated, and the cells were washed with PBS and lysed with passive lysis buffer (Promega) for 20 min at room temperature. Relative light units (RLUs) were measured by using a luminometer (Promega; GloMax). Triplicate data was analyzed in Graphpad Prism to generate IC50 values. A dose–response of EIDD-1931 was used as a positive control for the assay; each plate also contained a set of wells treated with EIDD-1931 at the IC50 for the assay (1.2 μM).
Kinetic Solubility
Phosphate buffered saline (50 mL, PBS, Fisher, pH 7.4) was added to HPLC grade H2O (450 mL) for a total dilution factor of 1:10 and final PBS concentration of 1×. The test compound (6 μL) as a 10 mM DMSO stock solution was combined with the aqueous PBS solution (294 μL) for 50-fold dilution in a Millipore solubility filter plate with a 0.45 μM polycarbonate filter membrane using a Hamilton Starlet liquid handler. The final DMSO concentration was 2.0%, and the maximum theoretical compound concentration was 200 μM. The filter plate was heat-sealed for the duration of a 24 h incubation period. The sample was placed on a rotary shaker (200 rpm) for 24 h at ambient temperature (21.6–22.8 °C) and then vacuum-filtered. All filtrates were injected into a chemiluminescent nitrogen detector for quantification. The equimolar nitrogen response of the detector was calibrated using standards that span the dynamic range of the instrument from 0.08 to 4500 μg/mL nitrogen. The filtrates were quantified with respect to this calibration curve. The calculated solubility values were corrected for background nitrogen present in DMSO and the media used to prepare the samples.
SARS-CoV-2 Assay
Human lung epithelial A549-ACE2 cells were cultured in DMEM containing 10% heat-inactivated FBS, nonessential amino acids, and pen strep. A549-ACE2 cells were seeded at 20,000 cells per well in a 96-well solid black plate 1 day prior to infection. To assay drug effect, cells were pretreated with drug for 1 h and then infected with SARS-CoV-2, with drug maintained during the infection. Then, 2 h after infection, the supernatant was removed, monolayers were rinsed with PBS, and media containing drug was added to each well. At 48 h post infection start, Nanoglo was added to each well as per the manufacturer’s protocol (Promega), and RLUs were measured using a Promega GloMax.
LDH Cytotoxicity Assay
DBT cells were plated to be 80% confluent at the start of the assay. Compounds were diluted as done for the MHV assay and incubated with cells at 37 °C for 1 h. After 1 h, 50 μL of DMEM was added to the cells (T = 0); 45 min before harvest, lysis buffer was added to positive wells. LDH activity in cell-free supernatants was measured at 10 h after infection using the Sigma Tox7 kit as per the manufacturer’s directions. A549-ACE2 cells were seeded at 20,000 cells per well 1 day prior to infection in 96-well plates. Cells were pretreated for 1 h and then mock-infected. Then, 2 h post mock infection, the media was removed, the monolayer was rinsed one time with PBS, and media containing drug was added to each well. Typically, 48 h after mock infection, plates were centrifuged, and an aliquot of the cell culture supernatant was removed. For LDH assays using Sigma Tox7 kit, the clarified supernatant was transferred to a clean plate and assayed following the manufacturer’s protocol.
Cell-Titer-Glo Cytotoxicity Assay
A549-ACE2 cells were maintained in low-glucose DMEM (Gibco) supplemented with 10% FBS, 1% NEAA, and 1% l-glutamine. No antibiotics were used. Cells were plated at 2000 cells/well in 384-well plate (Costar) and incubated overnight (37 °C, 5% CO2) before adding compound. Compounds were added in quadruplicate and incubated for 48 h. DMSO percentage was constant across all concentrations of compound. Cell viability was measured using CellTiter-Glo2 (Promega), and luminescence signal was read on a GloMax plate reader (Promega). Dose response analysis was performed using GraphPad Prism.
MDCK-MDR1 Permeability Assay
50 μL and 25 mL of cell culture medium were added to each well of the Transwell insert and reservoir, respectively. The HTS transwell plates were incubated at 37 °C, 5% CO2 for 1 h before cell seeding. MDCK-MDR1 cells were diluted to 1.56 × 106 cells/mL with culture medium. 50 μL of cell suspension was dispensed into the filter well of the 96-well HTS Transwell plate. Cells were cultivated for 3–8 days in a cell culture incubator at 37 °C, 5% CO2, 95% relative humidity. Cell culture medium was replaced every other day, beginning no later than 24 h after initial plating. Cell monolayer integrity was verified before the assay. Media was removed from the reservoir and each Transwell insert and replaced with prewarmed fresh culture medium. Transepithelial electrical resistance (TEER) across the monolayer was measured using Millicell Epithelial Volt-Ohm measuring system (Millipore, USA), and the plate was returned to the incubator after measurement. TEER was calculated by the following equation: TEER measurement (ohms) × Area of membrane (cm2) = TEER value (ohm·cm2). A well-qualified MDCK-MDR1 monolayer was defined by a TEER value greater than 42 ohm·cm2. Prior to the assay, the MDCK-MDR1 plate was removed from the incubator, washed twice with prewarmed HBSS (10 mM HEPES, pH 7.4), and then incubated at 37 °C for 30 min.
10 mM stock solutions of compound 53 and positive controls metoprolol, prazosin, and imatinib were prepared in DMSO. The stock solutions of test compounds were diluted in DMSO to 0.2 mM and then diluted with HBSS (10 mM HEPES, pH 7.4) to 1 μM working solutions. The final concentration of DMSO in the incubation system was 0.5%. To determine the rate of drug transport in the apical to basolateral direction, 75 μL of 1 μM working solution of test compound was added to the Transwell insert (apical compartment), and the wells in the receiver plate (basolateral compartment) were filled with 235 μL of HBSS (10 mM HEPES, pH 7.4). To determine the rate of drug transport in the basolateral to apical direction, 235 μL of 1 μM working solution of test compound was added to the receiver plate wells (basolateral compartment), and then the Transwell inserts (apical compartment) were filled with 75 μL of HBSS (10 mM HEPES, pH 7.4). The assay was performed in duplicate. Time 0 samples were prepared by transferring 50 μL of 1 μM working solution to wells of the 96-deepwell plate, followed by the addition of 200 μL cold methanol containing appropriate internal standards (100 nM alprazolam, 200 nM labetalol, 200 nM caffeine and 200 nM diclofenac). The plates were incubated at 37 °C for 2 h. At the end of the incubation, 50 μL samples from donor sides (apical compartment for Ap → Bl flux, and basolateral compartment for Bl → Ap) and receiver sides (basolateral compartment for Ap → Bl flux, and apical compartment for Bl → Ap) were transferred to wells of a new 96-well plate, followed by the addition of 4 volumes of cold methanol containing appropriate internal standards (100 nM alprazolam, 200 nM labetalol, 200 nM caffeine, and 200 nM diclofenac). Samples were vortexed for 5 min and then centrifuged at 3,220g for 40 min. An aliquot of 100 μL of the supernatant was mixed with 100 μL of ultrapure water. The samples were analyzed by LC-MS/MS.
To determine the Lucifer yellow leakage after a 2 h transport period, a stock solution of Lucifer yellow was prepared in water and diluted with HBSS (10 mM HEPES, pH 7.4) to reach the final concentration of 100 μM. 100 μL of the Lucifer yellow solution was added to each Transwell insert (apical compartment), followed by filling the wells in the receiver plate (basolateral compartment) with 300 μL of HBSS (10 mM HEPES, pH 7.4). The plates were incubated at 37 °C for 30 min. 80 μL samples were removed directly from the apical and basolateral wells (using the basolateral access holes) and transferred to wells of new 96-well plates. The Lucifer yellow fluorescence (to monitor monolayer integrity) signal was measured in a fluorescence plate reader at 485 nM excitation and 530 nM emission. Lucifer yellow leakage was <1% for all compounds, indicating a well-qualified MDCK-MDR1 monolayer.
The apparent permeability coefficient (Papp), in units of cm/s, was calculated using the following equation:
Where VA is the volume (in mL) in the acceptor well, Area is the surface area of the membrane (0.143 cm2 for Transwell-96 Well Permeable Supports), and Time is the total transport time in seconds.
The efflux ratio was determined using the following equation:
Plasma Protein Binding Assay
Frozen human plasma (BioIVT, MSE483763, stored at −80 °C) was thawed in a 37 °C water bath. Working solutions of compound 53 and positive control compound ketoconazole were prepared in DMSO at a concentration of 200 μM, then spiked into plasma to achieve a final compound concentration of 1 μM. The final concentration of DMSO was 0.5%.
For plasma protein binding analysis, dialysis membranes were soaked in ultrapure water for 60 min to separate strips, then in 20% ethanol for 20 min, and finally in dialysis buffer for 20 min. The dialysis set up was assembled according to the manufacturer’s protocol. Each cell containing 150 μL of plasma sample was dialyzed against an equal volume of dialysis buffer (PBS). The dialysis plate was sealed and incubated in an incubator at 37 °C with 5% CO2 at 100 rpm for 6 h. At the end of incubation, 50 μL of samples from both buffer and plasma chambers were transferred to wells of a 96-well plate. 50 μL of plasma was added to each buffer sample and an equal volume of PBS was supplemented to the collected plasma sample. 400 μL of quench solution (acetonitrile containing internal standards 200 nM labetalol, 100 nM tolbutamide, and 100 nM ketoprofen) was added to precipitate protein and release compounds. Samples were vortexed for 2 min and centrifuged for 30 min at 3,220g. 100 μL aliquots of the supernatant were diluted by 100 μL ultrapure water, and the mixtures were used for LC-MS/MS analysis. The assay was performed in duplicate.
Plasma stability analysis was performed in parallel. 50 μL of spiked plasma sample was transferred to a new plate. The samples are incubated at 37 °C in an incubator with 5% CO2 for 0 and 6 h. At designated time points, 50 μL of PBS was added and mixed thoroughly. 400 μL of quench solution (acetonitrile containing internal standards 200 nM labetalol, 100 nM tolbutamide, and 100 nM ketoprofen) was added to precipitate protein and release compounds. Samples were vortexed for 2 min and centrifuged for 30 min at 3,220g. 100 μL aliquots of the supernatant were diluted by 100 μL ultrapure water, and the mixtures were used for LC-MS/MS analysis. 53 was stable in plasma.
The concentrations of test compounds in the buffer and plasma chambers were determined from peak area ratios. The percentages of bound compound were calculated as follows:
Lung Protein Binding Assay
Frozen mouse lung tissue homogenate (Pharmaron, PH-Mouse-20240318, stored at −80 °C) was thawed in a 37 °C water bath. Working solutions of compound 53 and positive control compound ketoconazole were prepared in DMSO at a concentration of 200 μM, then spiked into lung tissue homogenate to achieve a final compound concentration of 1 μM.
Dialysis membranes were soaked in ultrapure water for 60 min to separate strips, then in 20% ethanol for 20 min, and finally in dialysis buffer for 20 min. The dialysis set up was assembled according to the manufacturer’s protocol. Each cell containing 150 μL of lung tissue homogenate sample was dialyzed against an equal volume of dialysis buffer (100 mM PBS, pH 7.4). The dialysis plate was sealed and incubated in an incubator at 37 °C with 5% CO2 at 100 rpm for 6 h. At the end of incubation, 50 μL of samples from both buffer and lung tissue homogenate were transferred to wells of a 96-well plate. 50 μL of lung tissue homogenate was added to each buffer sample, and an equal volume of PBS was supplemented to the collected lung tissue homogenate sample. 400 μL of precipitation buffer (acetonitrile containing internal standards 200 nM alprazolam, 200 nM labetalol, 200 nM imipramine, and 2 μM ketoplofen) was added to precipitate protein and release compounds. Samples were vortexed for 2 min and centrifuged for 30 min at 3,220g. 100 μL aliquots of the supernatant were diluted by 100 μL of ultrapure water, and the mixtures were used for LC-MS/MS analysis. The assay was performed in duplicate.
The concentrations of test compounds in the buffer and lung tissue homogenate chambers were determined from peak area ratios. The percentages of bound compound were calculated as follows:
Liver Microsomal Stability Assay
Compounds as 10 mM DMSO stock solutions were diluted to 2.5 mM with DMSO and again to 0.5 mM with MeCN to give a final solution containing a 0.5 mM compound in 1:4 DMSO/MeCN. Liver microsomes from male CD-1 mice were sourced from Xenotech (Kansas City, KS). A reaction plate was prepared by adding 691.25 μL and prewarmed (37 °C) microsomal solution (0.63 mg/mL protein and 1.3 mM EDTA in potassium phosphate buffer made by mixing ∼250 mL of 100 mM K2HPO4 with ∼65 mL of KH2PO4 until the buffer reached a pH of 7.4) to an empty well of a 96-well plate and maintained at 37 °C. The diluted 0.5 mM compound (8.75 μL) was added to the microsomal solution in the reaction plate and mixed thoroughly by repeated pipetting to give a final assay concentration of 5.0 μM. The resulting solutions were preincubated for 5 min at 37 °C and then dispensed into T = 0 and incubation plates. For the T = 0 plates, an aliquot (160 μL) of each reaction solution was added to an empty well of a 96-well plate as an exact replicate of the reaction plate. Cold MeOH (4 °C, 400 μL) was added to each well and mixed thoroughly by repeated pipetting. NADPH regeneration solution (40 μL) was added to each well and mixed thoroughly by repeated pipetting. For the T = 30 min incubation plate, NADPH (95 μL) was added to the remaining solution (microsomes + test compound) in each well in the previously prepared reaction plate to initiate the reaction. The plate was sealed and incubated at 37 °C for 30 min. An aliquot (100 μL) was removed from each well at the desired time point and dispensed into a well of a 96-well plate. Cold MeOH (4 °C, 200 μL) was added to quench the reaction. All plates were sealed, vortexed, and centrifuged at 3000 rpm, 4 °C for 15 min, and the supernatants were transferred for analysis by LC-TOFMS. The supernatant (20 μL) was injected onto an AQUASIL C18 column and eluted using a fast-generic gradient program. TOFMS data was acquired using Agilent 6538 Ultra High Accuracy TOF MS in extended dynamic range (m/z 100–1000) using generic MS conditions in positive mode. Following data acquisition, exact mass extraction and peak integration were performed using MassHunter Software (Agilent Technologies). The stability of the compound was calculated as the percent remaining of the unchanged parent at T = 30 min relative to the peak area at T = 0 min.
To determine CLint, aliquots of 50 μL were taken from the reaction solution at 0, 15, 30, 45, and 60 min. The reaction was stopped by the addition of four volumes of cold MeCN with internal standards (100 nM alprazolam, 200 nM imipramine, 200 nM labetalol, and 2 μM ketoprofen). Samples were centrifuged at 3,220g for 40 min, and 90 μL of the supernatant was mixed with 90 μL of ultrapure H2O and then used for LC-MS/MS analysis. Peak areas were determined from extracted ion chromatograms, and the slope value, k, was determined by linear regression of the natural logarithm of the remaining percentage of the parent drug vs incubation time curve. The intrinsic clearance (CLint in μL/min/mg) was calculated using the relationship CLint = kV/N where V is the incubation volume and N is the amount of protein per well.
Hepatocyte Stability Assay
Human cryopreserved hepatocytes were supplied by BioIVT (lot QZW, 10 pooled donors). Mouse cryopreserved hepatocytes were supplied by BioIVT (lot ZPG, pooled male CD-1). Vials of cryopreserved hepatocytes were removed from storage and thawed in a 37 °C water bath with gentle shaking, then the contents were poured into a 50 mL thawing medium conical tube. Vials were centrifuged at 100g for 10 min at room temperature. The thawing medium was aspirated, and hepatocytes were resuspended with a serum-free incubation medium to yield ∼1.5 × 106 cells/mL. Cell viability and density were counted using AO/PI fluorescence staining, and then cells were diluted with a serum-free incubation medium to a working cell density of 0.5 × 106 viable cells/mL. Aliquots of 198 μL of hepatocytes were dispensed into each well of a 96-well noncoated plate. The plate was placed in an incubator for approximately 10 min. Aliquots of 2 μL of the 100 μM test compound in duplicate and positive control were added into the respective wells of the noncoated 96-well plate to start the reaction. The final concentration of the test compound was 1 μM. The plate was placed in an incubator for the designed time points. Contents (25 μL) were transferred and mixed with six volumes (150 μL) of cold MeCN with internal standards (100 nM alprazolam, 200 nM labetalol, 200 nM caffeine, and 200 nM diclofenac) to terminate the reaction at time points of 0, 15, 30, 60, 90, and 120 min. Samples were centrifuged for 45 min at 3,220g, an aliquot of 100 μL of the supernatant was diluted with 100 μL of ultrapure H2O, and the mixture was used for LC-MS/MS analysis. Peak areas were determined from extracted ion chromatograms, and the slope value, k, was determined by linear regression of the natural logarithm of the remaining percentage of the parent drug vs incubation time curve. The intrinsic clearance (CLint in μL/min/106 cells) was calculated using the relationship CLint = kV/N where V is the incubation volume (0.2 mL) and N is the number of hepatocytes per well (0.1 × 106 cells). Scaling factors to convert CLint from μL/min/106 cells to mL/min/kg were 2540 (human hepatocytes) and 11,800 (mouse hepatocytes).
Inhibition of EIF2S2 Phosphorylation In Vitro
A549ACE2 were seeded at 150,000 cells per well on a 6-well plate in DMEM high-glucose containing 10% fetal bovine serum, 1X non-essential amino acids, and 2 mM L-glutamine and allowed to adhere overnight. Cells were treated with 53 (1 μM or 5 μM) or 0.05% DMSO (vehicle control) for 24 hours. Cells were lysed with RIPA buffer and sonicated at 40% for 10 seconds on ice. Lysates were quantified using Pierce Rapid Gold BCA Protein Assay kit and 20 mg protein was run on 4-20% Tris-Glycine gels and transferred to PVDF. Blots were blocked in 5% Milk in 1X TBST (0.1% Tween 20) for 1 hour at room temperature, washed with 1X TBST, and incubated with primary antibodies in 5% BSA in 1X TBST for 10 hours at room temperature; 1:10,000 phospho-EIF2S2 P-S2 (provided by Laszlo Gyenis from the David Litchfield group), 1:200 EIF2S2 (Novus Biologicals, H00008894-M09), 1:5000 GAPDH (Proteintech, 10494-1-AP). Bands were quantified using Fiji and normalized to GAPDH. Phosphorylated protein was then normalized to its corresponding total protein. Plots were generated using GraphPad Prism version 9.4.1 for macOS (N=2).
Pharmacokinetics
In the 5-h PK study, male CD-1 mice (6–8 weeks, 20–30 g) were dosed by intravenous (i.v.), oral (p.o.), or intraperitoneal (i.p.) administration of compound 53. For i.v. administration, a single dose of compound 53 (3 mg/kg) as a formic acid salt was administered as 5 mL/kg of a 0.6 mg/mL solution in NMP/Solutol/PEG-400/normal saline (v/v/v/v, 10:5:30:55) to two mice. For p.o. administration, a single dose of compound 53 (10 mg/kg) was administered as 10 mL/kg of a 1 mg/mL solution in NMP/Solutol/PEG-400/normal saline (v/v/v/v, 10:5:30:55) to two mice. For i.p. administration, a single dose of compound 53 (10 or 30 mg/kg) was administered as 10 mL/kg of a 1 or 3 mg/mL solution in NMP/Solutol/PEG-400/normal saline (v/v/v/v, 10:5:30:55) to two mice in each cohort. The mice had free access to water and food. At 0.5, 1, 3, and 5 h post dose, 0.03 mL of blood was collected from the dorsal metatarsal vein of each mouse. Blood of each sample was transferred into plastic microcentrifuge tubes containing anticoagulant of EDTA-K2. Blood samples were centrifuged at 4,000g for 5 min at 4 °C to obtain plasma. The samples were stored in a freezer at −75 ± 15 °C prior to analysis. Plasma samples from the two mice of each cohort and each time point were pooled together for analysis.
In the 24-h PK study, male CD-1 mice (6–8 weeks, 20–30 g) were dosed by intraperitoneal (i.p.) administration of compound 53 (10 mg/kg) as a formic acid salt by administration of 10 mL/kg of a 1 mg/mL solution in NMP/Solutol/PEG-400/normal saline (v/v/v/v, 10:5:30:55) to 21 mice. The mice had free access to water and food. At 0.5, 1, 2, 4, 8, 12, and 24 h post dose, 0.15 mL of blood was collected by cardiac puncture from three mice at each time point. Blood of each sample was transferred into plastic microcentrifuge tubes containing anticoagulant of EDTA-K2 and mixed well with anticoagulant, then placed on ice prior to centrifugation at 4000g for 5 min at 4 °C to obtain plasma. The samples were stored in a freezer at −75 ± 15 °C prior to analysis. The three mice at each time point were anaesthetized by a rising concentration of CO2 and lung samples collected. Lung samples were quick frozen in an ice box and stored at −75 ± 15 °C. Prior to analysis, all lung samples were weighed and homogenized with phosphate buffered saline (PBS) by lung weight (g) to buffer volume (mL) ratio of 1:3. The final compound concentration was calculated by multiplying the detected concentration by the dilution factor of 4.
Concentrations of the test compound in the plasma samples were determined using a Shimadzu LC-MS/MS system with a LC-40D X3 CN pump, DGU-405 degasser, CBM-40 CN system controller, SIL-40C X3CN autosampler, CTO-40C CN column oven with a HALO 160A ES-C18, (2.7 μm, 2.1 × 50 mm) column, and AB API 5500+ MS instrument. The mobile phase was 5–95% MeCN in H2O, with 0.1% formic acid. The desired serial concentrations of working solutions were achieved by diluting stock solution of analyte with 50% acetonitrile in water solution. 10 μL of working solutions (1, 2, 5, 10, 50, 100, 500, 1000, and 5000 ng/mL) were added to 10 μL of blank plasma or lung homogenate to achieve calibration standards of 1, 2, 5, 10, 50, 100, 500, and 1000 ng/mL in a total volume of 20 μL. Five quality control samples at 2, 5, 10, 100, and 4000 ng/mL were prepared independently of those used for the calibration curves in the same manner. 20 μL of standards, 20 μL of QC samples, and 20 μL of unknown samples (10 μL of plasma or lung homogenate with 10 μL of blank solution) were added to 200 μL of acetonitrile containing internal standard mixture for precipitating protein, respectively. Then the samples were vortexed for 30 s. After centrifugation at 4 °C, 4000 rpm for 15 min, the supernatant was diluted with ultrapure water at a ratio of 1:2 (v/v, 1:2), then 20 μL of diluted supernatant was injected into the LC/MS/MS system for quantitative analysis. PK parameters were calculated from the mean plasma concentration versus time by a noncompartmental model using WinNonlin 8.3 (Phoenix).
In Vivo SARS-CoV-2 Efficacy
All mouse studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill.
CD-1 mice (female, 8–10 week old) received compound 53 dosed 10 mg/kg i.p. in vehicle (NMP/Solutol/PEG-400/normal saline, v/v/v/v, 10:5:30:55). Mice were dosed every 12 h for a total of 36 h (3 doses). Twelve hours after the first dose, mice were anesthetized with 50 mg/kg ketamine + 5 mg/kg xylazine and intranasally infected with 1 × 104 plaque forming units (pfu) of mouse adapted coronavirus, SARS-CoV-2 MA10,20,40 contained in a 50 μL volume which was pipetted into the nares of each mouse. Post challenge, mice were monitored, weighed, and scored for clinical signs and euthanized at 24 h postinfection. Mice were euthanized by an overdose of isoflurane anesthesia (Baxter), blood was collected by cardiocentesis, and lung lobes collected for downstream analysis.
Infectious viral loads were measured by plaque assay. One day prior to assay, Vero cells were seeded at 2 × 105 cells per well in 12-well plates. Titers were measured from superior and middle lung lobes that were homogenized in 0.5 mL of media (DMEM + 5% FBS + 1% l-glutamine) at 6000 rpm for 40 s using a Roche MagNA Lyser homogenizer. Cell debris was removed by centrifugation for 1 min at full speed. 50 μL of the supernatant of the clarified homogenate was added to 450 μL of dilution media (DMEM + 5% FBS + 1% l-glutamine media). Homogenates were used to create 10-fold serial dilutions (10–1 to 10–6). Approximately 200 μL of each dilution was pipetted onto the previously plated Vero cells and incubated at 37 °C. To ensure even distribution across each well, the plates were rocked every 15 min. After 1 h, 2 mL of overlay (50:50 mixture of 2.5% carboxymethylcellulose and 2X alpha MEM containing 6% FBS + 2% penicillin/streptomycin + 2% l-glutamine + 2% HEPES) was added to each well. After incubation for 4 days in 37 °C, 5% CO2, an equal volume of 4% paraformaldehyde was added to each well and the cells were allowed to fix overnight. The fixative was removed, wells were rinsed with water to remove residual overlay, and 0.25% crystal violet was added to each well. Visible plaques were counted and averaged between two technical replicate wells and used to calculate plaque forming units (pfu) per lung tissue. The limit of detection (LOD) for the assay was determined to be 12.5 pfu/lung tissue, and samples that yielded no plaques were assigned a value of 6.25, half of the LOD.
In Vivo CSNK2 Inhibition
All mouse studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill.
CD-1 mice (female, 8–10 week old) received compound 53 dosed 10 mg/kg i.p. in vehicle (NMP/Solutol/PEG-400/normal saline (v/v/v/v, 10:5:30:55). Mice were dosed every 12 h for a total of 36 h (3 doses). At 36 h after the first dose, mice were euthanized by an overdose of isoflurane (Baxter), blood was collected by cardiac puncture, and lung lobes were collected for downstream processing and analysis. Recovered blood was placed in EDTA tubes, spun at 5000g in a microcentrifuge, and the recovered plasma transferred to a clean tube and frozen at −80 °C until analysis. The left lung lobe was added to a 2 mL O-ring skirted tube containing 750 μL of 1X PBS containing Phos Stop (per manufacturers recommendation, Roche) and glass beads. Lungs were homogenized for 60 s at 6000 rpm in a Roche Magnalyzer. Homogenates were centrifuged for 5 min at 10,000 rpm, and 500 μL of the homogenate was transferred to a clean tube and frozen at −80 °C until analysis.
Halt Protease Inhibitor cocktail (ThermoFisher, 78429) was added to mouse lung homogenates prior to sonication at 40% for 10 s on ice. Samples were quantified using Piece Rapid Gold BCA Protein Assay kit (ThermoFisher, A53226), and 50 μg of protein was run on 4–20% Tris-Glycine gels and transferred to PVDF. Blots were blocked in 5% Milk in 1X TBST (0.1% Tween 20) for 1 h at room temperature, washed with 1X TBST, and incubated with primary antibodies in 5% BSA in 1X TBST for 10 h at room temperature; 1:10,000 phospho-EIF2S2 P-S2 (provided by Laszlo Gyenis from the David Litchfield group), 1:200 EIF2S2 (Novus Biologicals, H00008894-M09), 1:1000 phospho-AKT Ser129 (Cell Signaling Technology, 13461), 1:1000 AKT (Cell Signaling Technology, 2920), 1:1000 GAPDH (Proteintech, 10494-1-AP), 1:1000 Transferrin (Proteintech, 17435-1-AP). Bands were quantified using Fiji41 and normalized to GAPDH. Phosphorylated protein was then normalized to its corresponding total protein. GraphPad Prism version 9.4.1 for macOS was used to plot averages (N = 3 for each treatment) and assess statistical significance using an unpaired t test with Welch’s correction.
Crystallography
CSNK2A1 expression and purification were performed as described previously.19,42,43 Briefly, transformed BL21(DE3) cells were grown in Terrific Broth medium containing 50 mg/mL kanamycin. Protein expression was induced at an OD600 of 2 by using 0.5 mM isopropyl-thio-galactopyranoside (IPTG) at 18 °C for 12 h. Cells expressing His6-tagged CSNK2A1 were lysed in lysis buffer containing 50 mM HEPES pH 7.5, 500 mM NaCl, 25 mM imidazole, 5% glycerol, and 0.5 mM Tris(2 carboxyethyl)phosphine (TCEP) by sonication. After centrifugation, the supernatant was loaded onto a Nickel-Sepharose column equilibrated with 30 mL lysis buffer. The column was washed with 60 mL of lysis buffer. Proteins were eluted by an imidazole step gradient (50, 100, 200, and 300 mM). Fractions containing protein were pooled together and dialyzed overnight using 1 L of final buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 0.5 mM TCEP) at 4 °C. Additionally, TEV protease was added (protein:TEV 1:20 molar ratio) to remove the tag. The next day, the protein solution was loaded onto Nickel-Sepharose column beads again to remove the TEV protease and cleaved Tag. The combined flow through fraction and the wash fraction (25 mM imidazole) containing the protein were concentrated to approximately 4–5 mL and loaded onto Superdex 75 16/60 Hi-Load gel filtration column equilibrated with final buffer. The protein was concentrated to approximately 9 mg/mL.
CSNK2A1 was crystallized using the sitting-drop vapor diffusion method by mixing protein (9 mg/mL) and well solutions in 2:1, 1:1, and 1:2 ratios. The reservoir solution contained 0.2 M ammonia sulfate, 0.1 M bis-tris pH 5.5, and between 23% and 26% (v/v) PEG 3350. Complex structures were achieved by soaking grown apo crystals for at least 24 h with the desired inhibitor dissolved in reservoir solution. Final concentration of the inhibitor was approximately 0.5 mM.
Diffraction data were collected at beamline X06SA (Villigen, CH) at a wavelength of 1.0 Å at 100 K. The reservoir solution supplemented with 20% ethylene glycol was used as cryoprotectant. Data were processed using XDS44 and scaled with AIMLESS.45 The PDB structure with the accession code 6Z83(19) was used as an initial search MR model using the program MOLREP.46 The final model was built manually using Coot47 and refined with REFMAC5.48 Data collection and refinement statistics are summarized in Table S2.
General Chemistry Methods
All chemical reagents were commercially available except those whose synthesis is described below. All reaction mixtures and column eluents were monitored via analytical thin-layer chromatography (TLC) performed on precoated fluorescent silica gel plates, 200 μm with an F254 indicator; visualization was accomplished by UV light (254/365 nm). LC-MS measurements were determined on Shimadzu LC-AB + LCMS-2020, Shimadzu LC-AD + LCMS-2020, Shimadzu LC-AD xR + LCMS-2020, or Agilent 1200 + Infinitylab LC/MSD instruments. Purity was determined by HPLC measurement using a Shimadzu LC-20 + LCMS-2020 instrument fitted with an Agilent PoroShell 120 EC-C18 column (45 °C, 2.7 μm, 3.0 × 50 mm); 8 min chromatography ran 0.037% TFA in water/MeCN (19:1) (solvent A), 0.018% TFA/MeCN (solvent B), and gradient 0–60% (solvent B) over 6.0 min, held at 60% for 1.0 min, and returned to 0% (solvent B) for 1.0 min at a flow rate of 1.0 mL/min; 4 min chromatography ran 0.037% TFA in water/MeCN (19:1) (solvent A), 0.018% TFA/MeCN (solvent B), and gradient 10–80% (solvent B) over 3.0 min, held at 80% for 0.5 min, and returned to 0% (solvent B) for 0.5 min at a flow rate of 1.0 mL/min. All final compounds were >95% pure unless otherwise stated. Nuclear magnetic resonance (NMR) spectra were obtained on Bruker Avance Neo 400 MHz, Bruker Avance Neo 500 MHz, and Bruker Avance 850 MHz instruments. Chemical shifts are reported in parts per million (ppm, δ), with residual solvent peaks referenced as the internal standard. Coupling constants are reported in Hz. Spin multiplicities are described as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), p (pentet), and m (multiplet). Data were processed using MestReNova.
Experimental Procedures in Schemes 1 and 2
5-Chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (5.00 g, 23.47 mmol, 1 eq) in EtOH (30 mL) was added cyclopropylamine (12.00 g, 211.24 mmol, 14 mL, 9 eq) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered and the solid was washed with EtOH (4 mL × 2). 75 (5.47 g, 23.03 mmol, 98.1% yield) was obtained as a yellow solid. 1H NMR (400 MHz, MeOD-d4) δ 8.36 (s, 1H), 6.61 (s, 1H), 2.76 (m, 1H), 1.04–0.93 (m, 2H), 0.83–0.74 (m, 2H). LCMS tR = 0.500 min in 1 min chromatography, Chromolith Flash RP-18e,25-3 mm, MS ESI calcd. for C10H9ClN5+ [M + H]+m/z 234.05, found 234.0.
N-(3-((3-Cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)phenyl)acetamide (3)
To a solution of N-(3-aminophenyl)acetamide (76) (100 mg, 665.88 μmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (124 mg, 532.70 μmol) in dioxane (4 mL) was added Cs2CO3 (651 mg, 2.00 mmol), BINAP (62 mg, 99.88 μmol), and Pd(OAc)2 (22 mg, 99.88 μmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The residue was purified by flash silica gel chromatography (eluent of 0–4%, MeOH/DCM) to give the product. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water(NH4HCO3)-ACN]; B%: 16–56%, 36 min). 3 (45.7 mg, 19.7% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.93 (s, 1H), 9.66 (s, 1H), 8.35 (s, 1H), 8.21 (s, 1H), 7.85–7.75 (m, 2H), 7.24 (t, J = 8.0 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 6.06 (s, 1H), 2.59 (tt, J = 7.0, 3.7 Hz, 1H), 2.05 (s, 3H), 0.84–0.77 (m, 2H), 0.74–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.32, 156.97, 150.86, 148.31, 145.11, 140.51, 139.65, 128.91, 114.87, 114.52, 113.26, 110.48, 76.45, 76.42, 24.07, 23.32, 6.55. HPLC tR = 3.332 min in 8 min chromatography, purity 100.0%. LCMS tR = 1.741 min in 4 min chromatography, MS ESI calcd. for C18H18N7O+ [M + H]+ 348.16, found 348.1.
N-(3-((3-Cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)phenyl)methanesulfonamide (4)
To a solution of N-(3-aminophenyl)methanesulfonamide (77) (200 mg, 1.07 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (201 mg, 0.86 mmol) in dioxane (8 mL) was added Cs2CO3 (1.40 g, 4.30 mmol), BINAP (100 mg, 0.16 mmol), and Pd(OAc)2 (36 mg, 0.16 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 40 min. The mixture was filtered, and the filtrate was concentrated to dryness. The residue was purified by silica gel chromatography (eluent of 0% ∼ 6%, MeOH/DCM) and then purified by prep-HPLC (column: Welch SiO2 10 μm, 250 × 200 mm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 20–60%, 15 min). The residue was further purified by prep-HPLC (column: Welch Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 16–56%, 30 min). 4 (80 mg, 19.4% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 9.12 (br s, 1H), 8.37 (s, 1H), 8.27 (s, 1H), 7.65 (dd, J = 8.2, 2.0 Hz, 1H), 7.50 (t, J = 2.1 Hz, 1H), 7.28 (t, J = 8.1 Hz, 1H), 6.82 (dd, J = 8.1, 2.1 Hz, 1H), 6.02 (s, 1H), 3.04 (s, 3H), 2.60 (tt, J = 7.0, 3.6 Hz, 1H), 0.86–0.78 (m, 2H), 0.78–0.65 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.78, 150.75, 148.35, 145.18, 141.05, 138.76, 129.54, 114.95, 114.81, 113.88, 110.64, 76.60, 76.58, 39.38, 23.33, 6.56. HPLC tR = 3.953 min in 8 min chromatography, purity 99.6%. LCMS tR = 1.780 min in 4 min chromatography, MS ESI calcd. for C17H18N7O2S+ [M + H]+ 384.12, found 384.3.
7-(Cyclopropylamino)-5-((3-((methylsulfonyl)methyl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (5)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (100 mg, 0.43 mmol) and 3-((methylsulfonyl)methyl)aniline (78) (79 mg, 0.43 mmol) in dioxane (2 mL) was added Cs2CO3 (418 mg, 1.28 mmol), BINAP (40 mg, 0.06 mmol), and Pd(OAc)2 (14 mg, 0.06 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h, then concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–3%, MeOH/DCM). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 26–56%, 10 min). 5 (38.5 mg, 23.1% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 8.37 (s, 1H), 8.27 (s, 1H), 7.91 (d, J = 7.7 Hz, 1H), 7.60 (t, J = 1.9 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.06 (d, J = 7.6 Hz, 1H), 6.03 (s, 1H), 4.46 (s, 2H), 2.95 (s, 3H), 2.65–2.57 (m, 1H), 0.85–0.78 (m, 2H), 0.75–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.84, 150.81, 148.40, 145.12, 140.36, 129.68, 128.99, 124.89, 121.91, 119.58, 114.80, 76.54, 76.40, 59.65, 23.33, 6.54. HPLC tR = 4.118 min in 8 min chromatography, purity 98.6%. LCMS tR = 2.028 min in 4 min chromatography, MS ESI calcd. for C18H19N6O2S+ [M + H]+ 383.13, found 383.3.
Methyl(3-nitrobenzyl)sulfane (105)
To a solution of 1-(bromomethyl)-3-nitrobenzene (104) (2 g, 9.26 mmol) in MeOH (20 mL) was added NaSMe (510 mg, 7.28 mmol, 463.64 μL) at 0 °C. The mixture was stirred at 25 °C for 3 h. The resulting mixture was extracted with EtOAc (10 mL × 3). The combined organic phase was washed with water (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated. 105 (1.5 g, 8.19 mmol, 88.4% yield) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 8.20–8.04 (m, 2H), 7.76 (d, J = 7.6 Hz, 1H), 7.65–7.57 (m, 1H), 3.78 (s, 2H), 1.95 (s, 3H).
1-((Methylsulfinyl)methyl)-3-nitrobenzene (106)
To a solution of methyl(3-nitrobenzyl)sulfane (105) (1.3 g, 7.10 mmol) in DCM (15 mL) was added mCPBA (1.44 g, 7.10 mmol, 85% purity) at 0 °C. The mixture was stirred at 0 °C for 1 h. NaHCO3 (5 mL) was added to the reaction mixture. The resulting mixture was extracted with DCM (10 mL × 3). The combined organic phase was washed with brine (10 mL) and NaHCO3 (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was used in the next step without purification. 106 (1.32 g, 6.63 mmol, 93.4% yield) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 8.27–8.18 (m, 2H), 7.79–7.75 (m, 1H), 7.73–7.65 (m, 1H), 4.35 (d, J = 12.8 Hz, 1H), 4.11 (d, J = 12.8 Hz, 1H), 2.50 (s, 3H).
tert-Butyl N-[methyl-[(3-nitrophenyl)methyl]-oxo-λ6-sulfanylidene]carbamate (107)
To a solution of 1-((methylsulfinyl)methyl)-3-nitrobenzene (106) (1.00 g, 5.02 mmol) in DCM (120 mL) was added MgO (809 mg, 20.1 mmol), iodobenzene diacetate (2.43 g, 7.53 mmol), and rhodium(II) acetate (222 mg, 502 μmol). The mixture was combined with tert-butyl carbamate (1.18 g, 10.0 mmol) and stirred at 25 °C for 16 h. The reaction mixture was filtered, and concentrated under reduced pressure to give a residue. The residue was purified by flash silica gel chromatography (eluent of 0–15% methanol/dichloromethane). 107 (836 mg, 2.66 mmol, 53.0% yield) was obtained as a brown oil. LCMS tR = 0.398 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C9H11N2O5S+ [M + 2H-tBu]+ 259.04, found 259.0.
tert-Butyl ((3-aminobenzyl)(methyl)(oxo)-λ6-sulfaneylidene)carbamate (79)
To a solution of tert-butyl N-[methyl-[(3-nitrophenyl)methyl]-oxo-λ6-sulfanylidene]carbamate (107) (736 mg, 2.34 mmol) in EtOH (30 mL) and H2O (7.5 mL) was added Fe (1.31 g, 23.4 mmol) and NH4Cl (501 mg, 9.37 mmol). The mixture was stirred at 65 °C for 1 h. The reaction mixture was filtered, and concentrated under reduced pressure to give a residue. 79 (491 mg, 1.73 mmol, 73.7% yield) was obtained as a brown oil. LCMS tR = 0.242 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C13H21N2O3S+ [M + H]+ 285.13, found 591.2.
tert-Butyl ((3-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)benzyl)(methyl)(oxo)-λ6-sulfaneylidene)carbamate (102)
To a solution of tert-butyl ((3-aminobenzyl)(methyl)(oxo)-λ6-sulfaneylidene)carbamate (79) (500 mg, 1.76 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (411 mg, 1.76 mmol) in 1,4-dioxane (15 mL) was added BINAP (164 mg, 264 μmol), Pd(OAc)2 (59.2 mg, 264 μmol), and Cs2CO3 (2.29 g, 7.03 mmol). The reaction mixture was heated in a microwave reactor at 100 °C for 2 h. The reaction mixture was filtered, and concentrated under reduced pressure to give a residue. The residue was purified by flash silica gel chromatography (eluent of 0–70% ethyl acetate/petroleum ether). 102 (436 mg, 905 μmol, 51.5% yield) was obtained as a red oil. LCMS tR = 0.453 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C18H20N7OS+ [M + 2H-Boc]+ 382.14, found 382.0.
7-(Cyclopropylamino)-5-[3-[(methylsulfonimidoyl)methyl]anilino]pyrazolo[1,5-a]pyrimidine-3-carbonitrile (6 and 7)
To a solution of tert-butyl ((3-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)benzyl)(methyl)(oxo)-λ6-sulfaneylidene)carbamate (102) (436 mg, 905 μmol) in DCM (10 mL) was added TFA (9.08 g, 79.7 mmol, 5.92 mL). The mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered, and concentrated under reduced pressure to give a residue. The residue was purified by prep-HPLC (column: Phenomenex Luna C18 150 × 25 mm × 10 μm; mobile phase: [water (FA)-ACN]; B%: 13–43%, 10 min). The product was separated by SFC (column: DAICEL CHIRALPAK AS (250 mm × 30 mm, 10 μm); mobile phase: [0.1% NH3H2O IPA]; B%: 40–40%, A5.8; 70 min) to give Peak 1 as 6 (53.2 mg, 28.3% yield) and Peak 2 as 7 (52.2 mg, 29.3% yield) both as white solids. 6:1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.36 (s, 1H), 8.25 (s, 1H), 7.81 (d, J = 8.2 Hz, 1H), 7.66 (s, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.02 (s, 1H), 4.39 (s, 2H), 2.87 (s, 3H), 2.65–2.57 (m, 1H), 0.85–0.78 (m, 2H), 0.75–0.68 (m, 2H). LCMS tR = 0.338 min in 0.8 min chromatography, 5–95AB, MS ESI calcd. for C18H20N7OS+ [M + H]+ 382.14, found 382.0. HPLC tR = 1.428 min in 4 min chromatography, 10–80AB, purity 96.9%. Chiral SFC: ee% = 100%. 7:1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.36 (s, 1H), 8.25 (d, J = 1.9 Hz, 1H), 7.81 (dd, J = 8.1, 2.2 Hz, 1H), 7.66 (t, J = 1.9 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 6.03 (s, 1H), 4.37 (s, 2H), 2.85 (s, 3H), 2.65–2.57 (m, 1H), 0.84–0.79 (m, 2H), 0.74–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.85, 150.83, 148.36, 145.07, 140.16, 131.19, 128.76, 124.96, 122.00, 119.41, 114.80, 76.47, 76.36, 62.40, 40.89, 23.32, 6.52. LCMS tR = 0.343 min in 0.8 min chromatography, 5–95AB, MS ESI calcd. For C18H20N7OS+ [M + H]+ 382.14, found 382.0. HPLC tR = 1.436 min in 4 min chromatography, 10–80AB, purity 96.9%. Chiral SFC: ee% = 100%.
3-((Methylsulfinyl)methyl)aniline (80)
A mixture of 1-((methylsulfinyl)methyl)-3-nitrobenzene (106) (200 mg, 1.00 mmol), NH4Cl (268.49 mg, 5.02 mmol), and Fe (280.33 mg, 5.02 mmol) in EtOH (5 mL) and H2O (5 mL) was degassed and purged with N2 three times, and then the mixture was stirred at 90 °C for 2 h under N2 atmosphere. The reaction mixture was filtered. The filtrate was concentrated. The residue was dissolved in water (4 mL). The resulting suspension was extracted with EtOAc (10 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated. The product was used in the next step without further purification. 80 (100 mg, 590.86 μmol, 58.9% yield) was obtained as a colorless oil. 1H NMR (400 MHz, chloroform-d) δ 7.15–7.10 (m, 1H), 6.65–6.60 (m, 3H), 4.05–3.96 (m, 1H), 3.80 (d, J = 12.8 Hz, 1H), 2.46 (s, 3H)
7-(Cyclopropylamino)-5-((3-((methylsulfinyl)methyl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (8)
To a solution of 3-((methylsulfinyl)methyl)aniline (80) (100 mg, 590.86 μmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (110.45 mg, 472.69 μmol) in dioxane (2 mL) was added BINAP (55.19 mg, 88.63 μmol), Pd(OAc)2 (19.90 mg, 88.63 μmol), and Cs2CO3 (577.55 mg, 1.77 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 2% MeOH/DCM) and prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water(NH4HCO3)-ACN]; gradient: 12–52% B over 1 min). 8 (26.5 mg, 70.57 μmol, 12.3% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.36 (s, 1H), 8.26 (d, J = 1.9 Hz, 1H), 7.82 (dd, J = 8.2, 2.1 Hz, 1H), 7.57 (t, J = 1.9 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 6.97 (dt, J = 7.6, 1.3 Hz, 1H), 6.02 (s, 1H), 4.09 (d, J = 12.7 Hz, 1H), 3.94 (d, J = 12.7 Hz, 1H), 2.64–2.57 (m, 1H), 2.51 (s, 3H), 0.85–0.78 (m, 2H), 0.74–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.83, 150.84, 148.38, 145.09, 140.34, 131.72, 128.90, 124.20, 121.33, 118.96, 114.77, 76.52, 76.38, 58.86, 37.41, 23.32, 6.54. HPLC tR = 3.857 min in 8 min chromatography, purity 97.5%. LCMS tR = 2.572 min in 4.0 min chromatography, 10–80AB, LCMS ESI calcd. for C18H19N6OS+ [M + H]+ 367.13, found 367.1.
tert-Butyl (3-((dimethylphosphoryl)methyl)phenyl)carbamate (109)
A solution of dimethylphosphine oxide (1.36 g, 17.47 mmol) in THF (10 mL) was degassed and purged with N2 three times. Then NaHMDS (1 M, 17.47 mL) was added at −30 °C under N2 atmosphere. The mixture was stirred at −30 °C for 1 h. Then a solution of tert-butyl (3-(bromomethyl)phenyl)carbamate (108) (1 g, 3.49 mmol) was added dropwise, and the mixture was stirred at 25 °C for 12 h under N2 atmosphere. The reaction was quenched with H2O (8 mL) dropwise. The resulting mixture was extracted with DCM (30 mL × 3). The combined organic phase was washed with brine (15 mL) and water (15 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was used in the next step without further purification. 109 (1.3 g, crude) was obtained as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 7.39–7.35 (m, 1H), 7.30–7.14 (m, 2H), 6.86–6.81 (m, 1H), 3.07 (d, J = 14.8 Hz, 2H), 1.47–1.45 (m, 9H), 1.35–1.33 (m, 3H), 1.31–1.30 (m, 3H). LCMS tR = 0.500 min in 1.0 min chromatography, 5–100AB, LCMS ESI calcd. for C14H23NO3P+ [M + H]+ 284.14 found 284.2.
(3-Aminobenzyl)dimethylphosphine oxide (81)
To a solution of tert-butyl (3-((dimethylphosphoryl)methyl)phenyl)carbamate (109) (1.3 g, 4.59 mmol) in DCM (10 mL) was added TFA (4.07 g, 35.72 mmol, 2 mL). The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated directly. The reaction mixture was poured into aq. NaHCO3 (15 mL) slowly. The resulting mixture was then adjusted to pH ∼8 by aq. NaHCO3. The resulting mixture was extracted with DCM (30 mL × 3). The combined organic phase was washed with brine (15 mL) and water (15 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was used in the next step directly. 81 (400 mg, crude) was obtained as a brown oil. 1H NMR (400 MHz, DMSO-d6) δ 6.94 (t, J = 7.6 Hz, 1H), 6.46–6.36 (m, 3H), 5.39–5.22 (m, 1H), 5.20–5.08 (m, 1H), 2.95 (d, J = 14.8 Hz, 2H), 1.33–1.32 (m, 3H), 1.30–1.29 (m, 3H).
7-(Cyclopropylamino)-5-((3-((dimethylphosphoryl)methyl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (9)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 1.09 mmol) and (3-aminobenzyl)dimethylphosphine oxide (81) (204 mg, 873.43 μmol) in dioxane (3 mL) was added BINAP (101 mg, 163.77 μmol), Pd(OAc)2 (36 mg, 163.77 μmol), and Cs2CO3 (1.07 g, 3.28 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in microwave at 130 °C for 0.5 h. After being cooled to 25 °C, the reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 19%,EtOAC/PE) and prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water(NH4HCO3)-ACN]; gradient: 10–50% B over 32 min). 9 (31 mg, 81.50 μmol, 7.8% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.35 (s, 1H), 8.22 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.56 (q, J = 2.1 Hz, 1H), 7.28 (t, J = 7.8 Hz, 1H), 7.04–6.83 (m, 1H), 6.01 (s, 1H), 3.12 (d, J = 15.1 Hz, 2H), 2.64–2.57 (m, 1H), 1.37 (d, J = 12.8 Hz, 6H), 0.84–0.78 (m, 2H), 0.75–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.90, 150.86, 148.31, 145.08, 140.22 (d, J = 2.7 Hz), 134.08 (d, J = 7.9 Hz), 128.70 (d, J = 2.3 Hz), 123.74 (d, J = 5.0 Hz), 120.76 (d, J = 4.8 Hz), 117.63 (d, J = 3.0 Hz), 114.85, 76.47, 76.43, 38.77, 23.32, 15.66 (d, J = 68.2 Hz), 6.55. HPLC tR = 2.118 min in 8 min chromatography, purity 99.2%. LCMS tR = 1.279 min in 4.0 min chromatography, 10–80AB, LCMS ESI calcd. for C19H22N6OP+ [M + H]+ 381.16, found 381.2.
7-(Cyclopropylamino)-5-((3-(2-hydroxyethyl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (10)
To a solution of 2-(3-aminophenyl)ethan-1-ol (82) (150 mg, 1.09 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (204 mg, 874.77 μmol) in dioxane (3 mL) was added Cs2CO3 (1.07 g, 3.28 mmol), BrettPhos Pd G3 (148 mg, 164.02 μmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. No workup was required. The residue was purified by flash silica gel chromatography (eluent of 0–4% MeOH in DCM). The residue was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water(NH4HCO3)-ACN]; gradient: 18–58% B over 1 min). 10 (24.1 mg, 70.35 μmol, 9.41% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.34 (s, 1H), 8.21 (d, J = 1.9 Hz, 1H), 7.60 (dd, J = 8.2, 2.2 Hz, 1H), 7.56 (t, J = 1.9 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 6.87 (d, J = 8.0 Hz, 1H), 6.00 (s, 1H), 4.65 (t, J = 5.2 Hz, 1H), 3.64 (td, J = 7.1, 5.1 Hz, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.63–2.55 (m, 1H), 0.85–0.78 (m, 2H), 0.75–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.92, 150.94, 148.31, 145.02, 140.13, 140.05, 128.52, 122.97, 120.16, 117.29, 114.82, 76.40, 76.26, 62.08, 23.31, 6.55. HPLC tR = 3.019 min in 8 min chromatography, purity 97.6%. LCMS tR = 2.800 min in 4.0 min chromatography, 10–80AB, LCMS ESI calcd. for C18H19N6O+ [M + H]+ 335.16, found 335.5.
Methyl 2-(3-aminophenyl)acetate (111)
To a solution of methyl 2-(3-nitrophenyl)acetate (110) (1 g, 5.12 mmol) in MeOH (10 mL) was added Pd/C (1.00 g, 939.67 μmol, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 (15 psi) at 25 °C and stirred for 12 h. The reaction mixture was filtered. The filtrate was concentrated directly. 111 (756 mg, crude) was obtained as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 6.98–6.91 (m, 1H), 6.47–6.41 (m, 2H), 6.37 (d, J = 7.2 Hz, 1H), 5.04 (s, 2H), 3.59 (s, 3H), 3.46 (s, 2H).
Methyl 2-(3-(((benzyloxy)carbonyl)amino)phenyl)acetate (112)
To a solution of methyl 2-(3-aminophenyl)acetate (111) (656 mg, 3.97 mmol) in DCM (20 mL) was added DIPEA (769.9 mg, 5.96 mmol, 1.04 mL) at 0 °C, then CbzCl (812.9 mg, 4.77 mmol, 680.30 μL) was added to the mixture slowly. The mixture was stirred at 25 °C for 16 h. The reaction mixture was poured into sat. aq. NaHCO3 (20 mL). The resulting mixture was extracted with DCM (15 mL × 2). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was purified by flash silica gel chromatography (ISCO; 20g, Sepa Flash Silica Flash Column, eluent of 0–14% EA/PE @ 35 mL/min). 112 (1.1 g, 3.67 mmol, 92.54% yield) was obtained as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 7.44–7.30 (m, 7H), 7.22 (t, J = 7.6 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 5.15 (s, 2H), 3.65–3.58 (m, 5H).
Benzyl (3-(2-hydroxy-2-methylpropyl)phenyl)carbamate (113)
To a solution of methyl 2-(3-(((benzyloxy)carbonyl)amino)phenyl)acetate (112) (1.1 g, 3.67 mmol) in THF (20 mL) was added MeMgBr (3 M in THF, 6.12 mL) at 0 °C. The mixture was stirred at 25 °C for 16 h. The reaction was quenched with aq. NH4Cl (10 mL) dropwise, then the mixture was extracted with DCM (15 mL × 2). The combined organic phase was dried over Na2SO4, filtered, and concentrated to give the crude product. The residue was purified by flash silica gel chromatography (ISCO; 12g, Sepa Flash Silica Flash Column, eluent of 0–20% EA/PE @ 35 mL/min). 113 (880 mg, 2.94 mmol, 79.99% yield) was obtained as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 7.48–7.25 (m, 7H), 7.15 (t, J = 7.6 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 5.14 (s, 2H), 4.29 (s, 1H), 2.59 (s, 2H), 1.05 (s, 6H).
1-(3-Aminophenyl)-2-methylpropan-2-ol (83)
To a solution of benzyl (3-(2-hydroxy-2-methylpropyl)phenyl)carbamate (113) (330 mg, 1.10 mmol) in MeOH (5 mL) was added Pd/C (400 mg, 375.87 μmol, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 (15 psi) at 25 °C and stirred for 2 h. The reaction mixture was filtered. The filtrate was concentrated directly. 83 (165 mg, crude) was obtained as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 6.87 (t, J = 7.6 Hz, 1H), 6.48–6.27 (m, 3H), 4.85 (s, 2H), 4.21 (s, 1H), 2.48 (s, 2H), 1.04 (s, 6H).
7-(Cyclopropylamino)-5-((3-(2-hydroxy-2-methylpropyl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (11)
To a solution of 1-(3-aminophenyl)-2-methylpropan-2-ol (83) (150 mg, 907.82 μmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (212.1 mg, 907.82 μmol) in dioxane (4 mL) was added BINAP (84.8 mg, 136.17 μmol), Pd(OAc)2 (30.6 mg, 136.17 μmol), and Cs2CO3 (887.3 mg, 2.72 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in microwave at 130 °C for 0.5 h. The reaction mixture was concentrated. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 4% MeOH/DCM) and prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water(NH4HCO3)-ACN]; gradient: 22–62% B over 32 min). 11 (22.9 mg, 63.19 μmol, 6.96% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.33 (s, 1H), 8.19 (br s, 1H), 7.61 (dd, J = 7.8, 1.8 Hz, 1H), 7.53 (t, J = 2.0 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.87 (dt, J = 7.5, 1.3 Hz, 1H), 6.00 (s, 1H), 4.32 (s, 1H), 2.64 (s, 2H), 2.59 (tt, J = 6.9, 3.6 Hz, 1H), 1.10 (s, 6H), 0.83–0.77 (m, 2H), 0.73–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.97, 150.89, 148.26, 145.01, 139.53, 139.46, 127.89, 124.67, 121.83, 117.34, 114.75, 76.36, 76.23, 69.46, 49.87, 29.24, 23.30, 6.53. HPLC tR = 4.287 min in 8 min chromatography, purity 98.5%. LCMS tR = 2.106 min in 4 min chromatography, MS ESI calcd for C20H23N6O+ [M + H]+ 363.19, found 363.2.
tert-Butyl 3-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)benzoate (103)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol) and tert-butyl 3-aminobenzoate (84) (192 mg, 0.94 mmol) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol), BINAP (80 mg, 0.13 mmol), and Pd(OAc)2 (29 mg, 0.13 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–2%, MeOH/DCM). 103 (400 mg, 95.4%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.39 (s, 1H), 8.32 (d, J = 2.0 Hz, 2H), 8.03 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.46 (t, J = 8.0 Hz, 1H), 6.01 (s, 1H), 2.62 (s, 1H), 1.57 (s, 9H), 0.84–0.81 (m, 2H), 0.75–0.74 (m, 2H). LCMS tR = 0.575 min in 1 min chromatography, MS ESI calcd. for C21H23N6O2+ [M + H]+ 391.19, found 391.1.
3-((3-Cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)benzoic acid (12)
To a solution of tert-butyl 3-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)benzoate (103) (100 mg, 0.26 mmol) in DCM (3 mL) was added TFA (2.15 g, 18.87 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 0–30%, 14 min). Then the impure product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 0–30%, 14 min). 12 (8.3 mg, 39.7%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.37 (s, 1H), 8.28 (d, J = 1.8 Hz, 1H), 8.24 (s, 1H), 8.12 (dd, J = 8.2, 2.3 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 6.01 (s, 1H), 2.63–2.57 (m, 1H), 0.87–0.79 (m, 2H), 0.75–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.64, 156.78, 150.77, 148.45, 145.22, 140.55, 128.93, 123.11, 122.89, 120.18, 114.72, 76.75, 76.66, 23.38, 6.60. HPLC tR = 3.776 min in 8 min chromatography, purity 98.9%. LCMS tR = 2.114 min in 4 min chromatography, MS ESI calcd. for C17H15N6O2+ [M + H]+ 335.13, found 335.3.
5-((3-(2-Cyanopropan-2-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (13)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.85 mmol, 1 eq) and 2-(3-aminophenyl)-2-methylpropanenitrile (85) (164 mg, 1.03 mmol, 1.2 eq) in dioxane (5 mL) was added Cs2CO3 (836 mg, 2.57 mmol, 3 eq), BINAP (79 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (28 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The solid was washed with DCM (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (0.05% NH3H2O + 10 mM NH4HCO3)-ACN]; B%: 39–79%, 10 min). 13 (10 mg, 0.03 mol, 3.2% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.36 (s, 1H), 8.27 (s, 1H), 8.13 (t, J = 2.1 Hz, 1H), 7.64 (ddd, J = 8.1, 2.1, 1.0 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 7.16 (ddd, J = 7.8, 2.1, 1.0 Hz, 1H), 6.00 (s, 1H), 2.61 (tt, J = 6.9, 3.6 Hz, 1H), 1.72 (s, 6H), 0.87–0.78 (m, 2H), 0.77–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.71, 150.80, 148.39, 144.98, 142.05, 140.89, 129.28, 124.71, 118.79, 118.41, 116.07, 114.62, 76.75, 76.56, 36.60, 28.30, 23.32, 6.55. HPLC tR = 3.407 min in 8 min chromatography, Xtimate C18 2.1 × 30 mm 3 μm, purity 98.9%. LCMS tR = 1.665 min in 4 min chromatography, Xtimate C18, 3 μm, 2.1 × 30 mm, MS ESI calcd. for C20H20N7+ [M + H]+ 358.18, found 358.4.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (14)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.85 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (164 mg, 1.03 mmol, 1.2 eq) in dioxane (5 mL) was added Cs2CO3 (836 mg, 2.57 mmol, 3 eq), BINAP (79 mg, 0.12 mmol, 0.15 eq), and Pd(OAc)2 (28 mg, 0.12 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The solid was washed with DCM (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (0.05% NH3H2O + 10 mM NH4HCO3)-ACN]; B%: 24–54%, 10 min). 14 (11.2 mg, 0.03 mmol, 3.46% yield) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 9.02 (s, 2H), 8.40 (s, 1H), 8.38 (t, J = 2.3 Hz, 1H), 8.36 (br s, 1H), 7.62 (dd, J = 8.2, 2.0 Hz, 1H), 7.51 (t, J = 8.1 Hz, 1H), 7.31 (dd, J = 8.0, 2.2 Hz, 1H), 6.03 (s, 1H), 2.63 (tt, J = 6.9, 3.6 Hz, 1H), 0.87–0.80 (m, 2H), 0.76–0.71 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.58, 150.80, 148.57, 145.04, 141.90, 141.25, 134.22, 130.35, 118.51, 114.75, 114.55, 111.76, 76.97, 76.75, 23.36, 6.59. HPLC tR = 2.371 min in 8 min chromatography, Xtimate C18 2.1 × 30 mm, 3 μm, purity 94.4%. LCMS tR = 1.602 min in 4 min chromatography, Xtimate C18, 3 μm, 2.1 × 30 mm, MS ESI calcd. for C18H16N9+ [M + H]+ 358.15, found 358.0.
5-(3-Nitrophenyl)-1H-1,2,3-triazole (115)
To a solution of 1-ethynyl-3-nitrobenzene (114) (1.00 g, 6.08 mmol) and CuI (65 mg, 0.34 mmol) in DMF (9 mL) and MeOH (1 mL) was added TMSN3 (1.17 g, 10.20 mmol) under N2. The mixture was stirred at 100 °C for 12 h. Water (30 mL) was added to the residue. The resulting suspension was extracted with DCM (50 mL × 2). The combined organic phase was washed with brine (30 mL), dried over anhydrous with Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0–31%, EtOAc/PE). 115 (1.70 g, crude) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.66–8.62 (m, 2H), 8.32 (d, J = 7.6 Hz, 1H), 8.19 (dd, J = 1.6, 8.4 Hz, 1H), 7.76 (t, J = 8.0 Hz, 1H).
3-(1H-1,2,3-Triazol-5-yl)aniline (87)
To a solution of 5-(3-nitrophenyl)-1H-1,2,3-triazole (115). (686 mg, 3.61 mmol) in EtOH (20 mL) was added Fe (1.01 g, 18.04 mmol). A solution of NH4Cl (579 mg, 10.82 mmol) in H2O (5 mL) was added to the mixture. The mixture was stirred at 80 °C for 2 h. The reaction mixture was diluted with MeOH (20 mL) and filtered via a Celite pad. The pad was washed with MeOH (15 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O)-ACN]; B%: 0–15%, 10 min). 87 (552 mg, 93.0%) was obtained as a yellow solid. LCMS tR = 0.413 min in 4 min chromatography, MS ESI calcd. for C8H9N4+ [M + H]+ 161.08, found 161.1.
5-((3-(4H-1,2,3-Triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (15)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol) and 3-(1H-1,2,3-triazol-5-yl)aniline (87) (137 mg, 0.86 mmol) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol), BINAP (80 mg, 0.13 mmol), and Pd(OAc)2 (29 mg, 0.13 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–10% MeOH/DCM) to give the product. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 21–51%, 10 min). 15 (16.2 mg, 5.2%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 15.09 (br s, 1H), 9.82 (s, 1H), 8.43 (d, J = 2.5 Hz, 1H), 8.38 (s, 1H), 8.28 (d, J = 1.8 Hz, 1H), 8.23 (s, 1H), 7.70 (dd, J = 8.1, 2.2 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.9 Hz, 1H), 6.04 (s, 1H), 2.65–2.57 (m, 1H), 0.85–0.79 (m, 2H), 0.76–0.71 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.85, 150.93, 148.42, 145.03, 141.02, 131.04, 129.43, 119.31, 118.99, 118.96, 116.49, 116.40, 114.88, 76.68, 76.59, 23.34, 6.59. HPLC tR = 1.985 min in 4 min chromatography, purity 94.8%. LCMS tR = 1.655 min in 4 min chromatography, MS ESI calcd. for C18H16N9+ [M + H]+ 358.15, found 357.9.
5-((3-(1H-1,2,3-Triazol-1-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (16)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol, 1 eq) and 3-(1H-1,2,3-triazol-1-yl)aniline (88) (137 mg, 0.86 mmol, 1 eq) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol, 3 eq), BINAP (80 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (29 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The filter cake was washed with DCM (3 mL × 2), and the combined filtrate was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–11%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Waters Torus 2-PIC 150 × 19 mm × 5 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 20–35%, 6 min). 16 (11.6 mg, 0.03 mmol, 8.64% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.70 (s, 1H), 8.49 (t, J = 2.1 Hz, 1H), 8.39 (s, 1H), 8.35 (s, 1H), 7.99 (d, J = 1.1 Hz, 1H), 7.84 (dt, J = 8.2, 1.6 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.53–7.48 (m, 1H), 6.04 (s, 1H), 2.66–2.59 (m, 1H), 0.88–0.80 (m, 2H), 0.77–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.64, 150.74, 148.55, 145.14, 141.78, 137.02, 134.44, 130.24, 122.93, 118.87, 114.69, 113.42, 110.70, 76.97, 76.83, 23.38, 6.59. HPLC tR = 4.145 min in 8 min chromatography, purity 99.0%. LCMS tR = 1.729 min in 4 min chromatography, MS ESI calcd. for C18H16N9+ [M + H]+ 358.15, found 358.3.
5-Methyl-4-(3-nitrophenyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (117)
To a solution of 1-isothiocyanato-3-nitrobenzene (116) (2.00 g, 11.1 mmol, 1 eq) in EtOH (25 mL) was added acetohydrazide (822 mg, 11.1 mmol, 1 eq) and DBU (404 mg, 2.65 mmol). Then the reaction mixture was stirred at 85 °C for 10 h. The reaction mixture was diluted with saturated NH4Cl (20 mL) and extracted with EtOAc (10 mL × 3). The combined organic phase was washed with brine (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 117 (900 mg, 3.01 mmol, 27.12% yield) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 13.77 (br s, 1H), 8.43 (t, J = 2.0 Hz, 1H), 8.40–8.36 (m, 1H), 7.99–7.93 (m, 1H), 7.90–7.84 (m, 1H), 2.15 (s, 3H). LCMS tR = 0.418 min in 1 min chromatography, MS ESI calcd. for C9H9N4O2S+ [M + H]+ 237.04, found 237.0.
3-Methyl-4-(3-nitrophenyl)-4H-1,2,4-triazole (118)
To a solution of 5-methyl-4-(3-nitrophenyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (117) (700 mg, 2.96 mmol, 1 eq) in acetic acid (10 mL) was added NaNO2 (1.39 g, 12.18 mmol, 2 eq) dissolved in H2O (5 mL) dropwise at 0 °C. Then the reaction mixture was stirred at 100 °C for 5 h. The reaction mixture was concentrated in vacuo. Water (10 mL) was added to the residue. The solution was adjusted to pH = 9 with 1 M aq. NaOH. The mixture was extracted with DCM: MeOH = 10:1 (11 mL × 3). The combined organic phase was washed with brine (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 118 (627 mg, 2.32 mmol, 78.15% yield) was obtained as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.43 (t, J = 2.0 Hz, 1H), 8.39–8.36 (m, 1H), 8.03–8.01 (m, 1H), 7.91–7.85 (m, 1H), 2.39 (s, 3H). LCMS tR = 0.382 min in 1 min chromatography, MS ESI calcd. for C9H9N4O2+ [M + H]+ 205.07, found 205.0.
3-(3-Methyl-4H-1,2,4-triazol-4-yl)aniline (89)
To a solution of 3-Methyl-4-(3-nitrophenyl)-4H-1,2,4-triazole (118) (990 mg, 4.85 mmol, 1 eq) in EtOH (10 mL) was added SnCl2·H2O (4.92 g, 21.82 mmol, 4.5 eq) dissolved in EtOH (10 mL) and HCl (12 mol/L, 1.5 mL) dropwise at 0 °C. Then the reaction mixture was stirred at 70 °C for 10 h. The reaction mixture was cooled down to 0 °C and adjusted to pH 11–12 with 50% aq. NaOH. Then the mixture was filtered and the solid was washed with EtOH (10 mL). The filtrate was diluted with brine (10 mL) and extracted with DCM (20 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 89 (789 mg, 2.81 mmol, 57.92% yield) was obtained as a brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (s, 1H), 7.17 (t, J = 8.0 Hz, 1H), 6.69–6.65 (m, 1H), 6.58–6.50 (m, 2H), 5.50 (s, 2H), 2.31 (s, 3H). LCMS tR = 0.404 min in 4 min chromatography, MS ESI calcd. for C9H11N4+ [M + H]+ 175.10, found 175.1.
7-(Cyclopropylamino)-5-((3-(3-methyl-4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (17)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (300 mg, 1.28 mmol, 1 eq) and 3-(3-methyl-4H-1,2,4-triazol-4-yl)aniline (89) (268 mg, 1.54 mmol, 1.2 eq) in dioxane (5 mL) was added Cs2CO3 (1.25 g, 3.85 mmol, 3 eq) and Brettphos Pd G3 (128 mg, 0.141 mmol, 0.1 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The solid was washed with DCM (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Luna 30 × 30 mm × 10 μm + YMC AQ 100 × 30 × 10 μm; mobile phase: [water (0.05%NH3H2O)-ACN]; B%: 30–60%, 20 min). 17 (37.6 mg, 0.1 mmol, 7.84% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.98 (s, 1H), 8.68 (s, 1H), 8.39 (s, 1H), 8.36 (s, 1H), 8.17 (t, J = 2.1 Hz, 1H), 7.65 (ddd, J = 8.3, 2.2, 1.0 Hz, 1H), 7.52 (t, J = 8.1 Hz, 1H), 7.13 (ddd, J = 7.8, 2.1, 0.9 Hz, 1H), 6.02 (s, 1H), 2.63 (tt, J = 6.8, 3.6 Hz, 1H), 2.46 (s, 3H), 0.89–0.80 (m, 2H), 0.80–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.57, 150.65, 149.69, 148.49, 145.17, 143.65, 141.56, 134.35, 130.10, 119.07, 118.10, 115.22, 114.64, 76.92, 76.86, 23.35, 10.88, 6.56. HPLC tR = 3.429 min in 8 min chromatography, Xtimate C18 2.1 × 30 mm 3 μm, purity 98.7%. LCMS tR = 1.401 min in 4 min chromatography, Xtimate C18, 3 μm, 2.1 × 30 mm, MS ESI calcd. for C19H18N9+ [M + H]+ 372.17, found 372.3.
7-(Cyclopropylamino)-5-((3-(2-methyl-1H-imidazol-1-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (18)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.85 mmol, 1 eq) and 3-(2-methyl-1H-imidazol-1-yl)aniline (90) (177 mg, 1.03 mmol, 1.2 eq) in dioxane (5 mL) was added Cs2CO3 (836 mg, 2.57 mmol, 3 eq), BINAP (79 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (28 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The solid was washed with DCM (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (0.05% NH3H2O + 10 mM NH4HCO3)-ACN]; B%: 31–61%, 10 min). 18 (24 mg, 0.06 mmol, 7.57% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.38 (s, 1H), 8.33 (s, 1H), 8.13 (t, J = 2.1 Hz, 1H), 7.59 (dd, J = 8.1, 2.1 Hz, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.29 (d, J = 1.4 Hz, 1H), 7.05 (dd, J = 7.8, 2.1 Hz, 1H), 6.91 (d, J = 1.4 Hz, 1H), 6.01 (s, 1H), 2.62 (tt, J = 6.9, 3.6 Hz, 1H), 2.40 (s, 3H), 0.87–0.80 (m, 2H), 0.76–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.61, 150.67, 148.44, 145.16, 143.51, 141.32, 137.93, 129.79, 127.28, 120.55, 118.07, 115.42, 114.64, 76.86, 76.82, 23.34, 14.01, 6.55. HPLC tR = 1.964 min in 8 min chromatography, Xtimate C18 2.1 × 30 mm 3 μm, purity 99.4%. LCMS tR = 1.442 min in 4 min chromatography, Xtimate C18, 3 μm, 2.1 × 30 mm, MS ESI calcd. for C20H19N8+ [M + H]+ 371.17, found 371.0.
7-(Cyclopropylamino)-5-((3-(4-methyl-4H-1,2,4-triazol-3-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (19)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.85 mmol, 1 eq) and 3-(4-methyl-4H-1,2,4-triazol-3-yl)aniline (91) (156 mg, 0.89 mmol, 1.05 eq) in dioxane (5 mL) was added Cs2CO3 (836 mg, 2.57 mmol, 3 eq), BINAP (79 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (28 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The solid was washed with DCM (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (0.05% NH3H2O + 10 mM NH4HCO3)-ACN]; B%: 15–55%, 11 min). 19 (26.4 mg, 0.07 mmol, 8.2% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.60 (s, 1H), 8.38 (s, 1H), 8.34–8.26 (m, 2H), 7.76 (dt, J = 8.3, 1.4 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.42 (dt, J = 7.7, 1.4 Hz, 1H), 6.03 (s, 1H), 3.86 (s, 3H), 2.62 (tt, J = 6.9, 3.6 Hz, 1H), 0.87–0.80 (m, 2H), 0.76–0.71 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.73, 152.98, 150.75, 148.40, 146.24, 145.17, 140.59, 129.39, 127.39, 122.01, 120.23, 118.57, 114.81, 76.79, 76.69, 32.52, 23.34, 6.56. HPLC tR = 2.462 min in 8 min chromatography, Xtimate C18 2.1 × 30 mm 3 μm, purity 99.3%. LCMS tR = 1.527 min in 4 min chromatography, Xtimate C18, 3 μm, 2.1 × 30 mm, MS ESI calcd. for C19H18N9+ [M + H]+ 372.17, found 372.0.
1-(2,2-Dimethoxyethyl)-3-(3-nitrophenyl)urea (120)
To a solution of 2,2-dimethoxyethan-1-amine (704 mg, 6.70 mmol, 1.1 eq) in DCM (5 mL) was added 1-isocyanato-3-nitrobenzene (119) (1.00 g, 6.09 mmol, 1 eq) dissolved in DCM (5 mL) dropwise at 0 °C. Then the reaction mixture was stirred at 0 to 25 °C for 3 h. The reaction mixture was concentrated in vacuo and carried forward without further purification. 120 (1.64 g, crude) was obtained as a light-yellow solid.
1-(3-Nitrophenyl)-1,3-dihydro-2H-imidazol-2-one (121)
To a solution of 1-(2,2-dimethoxyethyl)-3-(3-nitrophenyl)urea (120) (1.64 g, 6.09 mmol, 1 eq) in DCM (10 mL) was added TFA (1.39 g, 12.18 mmol, 2 eq) dissolved in DCM (5 mL) dropwise. Then the reaction mixture was stirred at 25 °C for 10 h. The reaction mixture was concentrated in vacuo. 121 (1.20 g, crude) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 8.82 (t, J = 2.0 Hz, 1H), 8.15 (dd, J = 1.2, 8.4 Hz, 1H), 8.05 (dd, J = 2.0, 8.4 Hz, 1H), 7.72 (t, J = 8.0 Hz, 1H), 7.20 (dd, J = 2.0, 3.2 Hz, 1H), 6.70 (t, J = 2.8 Hz, 1H). LCMS tR = 0.425 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C9H8N3O3+ [M + H]+ 206.06, found 206.0.
1-(3-Aminophenyl)-1,3-dihydro-2H-imidazol-2-one (92)
To a solution of 1-(3-nitrophenyl)-1,3-dihydro-2H-imidazol-2-one (121) (450 mg, 2.19 mmol, 1 eq) in EtOH (4 mL) was added SnCl2·H2O (1.24 g, 6.58 mmol, 3 eq) dissolved in EtOH (3 mL) and HCl (12 mol/L, 1 mL) dropwise at 0 °C. Then the reaction mixture was stirred at 70 °C for 10 h. The reaction mixture was cooled down to 0 °C and adjusted to pH 11–12 with 50% aq. NaOH. Then the mixture was filtered and the solid was washed with EtOH (5 mL). The combined filtrate was concentrated in vacuo. 92 (360 mg, 1.26 mmol, 57.59% yield) was obtained as a white solid. LCMS tR = 0.243 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C9H10N3O+ [M + H]+ 176.08, found 176.1.
7-(Cyclopropylamino)-5-((3-(2-oxo-2,3-dihydro-1H-imidazol-1-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (20)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.85 mmol, 1 eq) and 1-(3-aminophenyl)-1,3-dihydro-2H-imidazol-2-one (92) (157 mg, 0.89 mmol, 1.05 eq) in dioxane (5 mL) was added Cs2CO3 (836 mg, 2.57 mmol, 3 eq) and Brettphos Pd G3 (78 mg, 0.08 mmol, 0.1 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was filtered. The solid was washed with DCM (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Luna 30 × 30 mm × 10 μm + YMC AQ 100 × 30 × 10 μm; mobile phase: [water (0.05%NH3H2O)-ACN]; B%: 30–90%, 20 min). 20 (10 mg, 0.02 mmol, 3.0% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (d, J = 2.0 Hz, 1H), 8.61 (s, 1H), 7.86 (s, 1H), 7.53 (d, J = 3.4 Hz, 1H), 7.19 (d, J = 3.4 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H), 6.97 (t, J = 2.2 Hz, 1H), 6.78–6.70 (m, 1H), 6.56–6.46 (m, 1H), 5.33 (br s, 2H), 2.76–2.68 (m, 1H), 0.91–0.85 (m, 2H), 0.80–0.75 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.58, 149.94, 149.58, 149.51, 149.48, 146.45, 136.94, 129.46, 114.12, 113.20, 111.91, 108.98, 107.84, 107.26, 78.55, 78.29, 23.63, 6.51. HPLC tR = 2.204 min in 8 min chromatography, Xtimate C18 2.1 × 30 mm 3 μm, purity 95.9%. LCMS tR = 1.529 min in 4 min chromatography, Xtimate C18, 3 μm, 2.1 × 30 mm, MS ESI calcd. for C19H17N8O+ [M + H]+ 373.15, found 373.0.
4-(3-Nitrobenzyl)-4H-1,2,4-triazole (122)
To a mixture of 1-(bromomethyl)-3-nitrobenzene (104) (1.00 g, 4.63 mmol) and 4H-1,2,4-triazole (352 mg, 5.09 mmol) in MeCN (12 mL) was added K2CO3 (1.92 g, 13.89 mmol). Then the mixture was stirred at 100 °C for 10 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. Water (45 mL) was added to the residue. The resulting mixture was extracted with EtOAc (20 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 122 (1.70 g, 70.9%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.20–8.18 (m, 2H), 8.03 (s, 1H), 7.75–7.73 (m, 1H), 7.69–7.67 (m, 1H), 5.60 (s, 2H). LCMS tR = 0.417 min in 1 min chromatography, MS ESI calcd. for C9H9N4O2+ [M + H]+ 205.07, found 205.0.
3-((4H-1,2,4-Triazol-4-yl)methyl)aniline (93)
A solution of 4-(3-nitrobenzyl)-4H-1,2,4-triazole (122) (200 mg, 0.86 mmol) in EtOH (8 mL) was stirred at 25 °C. Then a solution of NH4Cl (739 mg, 13.81 mmol) in H2O (1 mL) was added dropwise, and Fe (1.54 g, 27.62 mmol) was added to the reaction mixture at 25 °C. The mixture was degassed and purged with N2 and stirred at 80 °C for 3 h. The reaction mixture was diluted with MeOH (20 mL) and filtered via a Celite pad. The pad was washed with MeOH (15 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Ultimate XB-CN 250 × 50 × 10 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 15–35%, 9 min). 93 (432 mg, 43%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.95 (s, 1H), 6.97 (t, J = 7.6 Hz, 1H), 6.53–6.50 (m, 1H), 6.44–6.41 (m, 2H), 5.24 (s, 2H). LCMS tR = 0.172 min in 1 min chromatography, MS ESI calcd. for C9H11N4+ [M + H]+ 175.10, found 175.1.
5-((3-((4H-1,2,4-Triazol-4-yl)methyl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (21)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (320 mg, 1.37 mmol) and 3-((4H-1,2,4-triazol-4-yl)methyl)aniline (93) (358 mg, 2.05 mmol) in dioxane (3 mL) was added Cs2CO3 (1.34 g, 4.11 mmol), BINAP (128 mg, 0.25 mmol), and Pd(OAc)2 (46 mg, 0.25 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–7%, MeOH/DCM). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 23–53%, 10 min). 21 (133.1 mg, 26.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.66 (s, 1H), 8.36 (s, 1H), 8.25 (s, 1H), 8.00 (s, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.50 (t, J = 1.9 Hz, 1H), 7.33 (t, J = 7.9 Hz, 1H), 6.93 (d, J = 8.0 Hz, 1H), 5.98 (s, 1H), 5.41 (s, 2H), 2.65–2.55 (m, 1H), 0.84–0.77 (m, 2H), 0.75–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.76, 151.78, 150.79, 148.35, 145.08, 144.27, 140.65, 136.90, 129.09, 121.43, 118.84, 118.46, 114.76, 76.59, 76.41, 52.20, 23.30, 6.52. HPLC tR = 3.712 min in 8 min chromatography, purity 99.5%. LCMS tR = 1.494 min in 4 min chromatography, MS ESI calcd. for C19H18N9+ [M + H]+ 372.17, found 372.3.
5-((3-(1,3,4-Oxadiazol-2-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (22)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol, 1 eq) and 3-(1,3,4-oxadiazol-2-yl)aniline (94) (138 mg, 0.86 mmol, 1 eq) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol, 3 eq), BINAP (80 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (29 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–10%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Phenomenex Luna 30 × 30 mm × 10 μm + YMC AQ 100 × 30 × 10 μm; mobile phase: [water (NH3H2O)-ACN]; B%: 40–80%, 17 min). 22 (34.8 mg, 0.09 mmol) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.98 (s, 1H), 9.35 (s, 1H), 8.51 (t, J = 2.0 Hz, 1H), 8.40 (s, 1H), 8.35 (s, 1H), 8.11–8.04 (m, 1H), 7.66 (dt, J = 7.8, 1.4 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 6.03 (s, 1H), 2.66–2.59 (m, 1H), 0.87–0.81 (m, 2H), 0.77–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 163.75, 156.59, 154.49, 150.64, 148.49, 145.20, 141.35, 129.98, 123.67, 122.19, 119.97, 116.88, 114.55, 76.93, 76.78, 23.33, 6.55. HPLC tR = 4.115 min in 8 min chromatography, purity 98.9%. LCMS tR = 1.686 min in 4 min chromatography, MS ESI calcd. for C18H15N8O+ [M + H]+ 359.14, found 359.3.
3-(Thiazol-2-yl)aniline (95)
A mixture of 2-bromothiazole (500 mg, 3.05 mmol, 1 eq), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (123) (1.34 g, 6.10 mmol, 2 eq), Pd(dppf)Cl2 (299 mg, 0.36 mmol, 0.12 eq), and Cs2CO3 (2.98 g, 9.15 mmol, 3 eq) in dioxane (8 mL) and water (2 mL) was degassed and purged with N2 three times at 25 °C. Then the mixture was stirred at 100 °C for 5 h under N2 atmosphere. The reaction mixture was filtered and the solid was washed with DCM (6 mL × 2). The combined filtrate was concentrated in vacuo. The impure product was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (0.05%NH3H2O)-ACN]; B%: 20–50%, 10 min). 95 (221 mg, 1.23 mmol) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.86 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 3.2 Hz, 1H), 7.19 (d, J = 1.6 Hz, 1H), 7.14–7.06 (m, 2H), 6.65 (dd, J = 0.8, 6.8 Hz, 1H), 5.35 (s, 2H). LCMS tR = 0.384 min in 1 min chromatography, MS ESI calcd. for C9H9N2S+ [M + H]+ 177.05, found 177.0.
7-(Cyclopropylamino)-5-((3-(thiazol-2-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (23)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol, 1 eq) and 3-(thiazol-2-yl)aniline (95) (151 mg, 0.86 mmol, 1 eq) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol, 3 eq), BINAP (80 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (29 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–9%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O)-ACN]; B%: 40–70%, 10 min). 23 (13.1 mg, 0.06 mmol) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (s, 1H), 8.61 (s, 1H), 8.38 (s, 1H), 8.29 (s, 1H), 7.93 (t, J = 2.8 Hz, 1H), 7.87–7.76 (m, 2H), 7.62 (d, J = 7.7 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 6.05 (s, 1H), 2.65–2.57 (m, 1H), 0.87–0.79 (m, 2H), 0.76–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.50, 156.77, 150.72, 148.44, 145.19, 143.91, 141.32, 133.61, 129.70, 120.52, 120.43, 119.40, 116.82, 114.68, 76.95, 76.86, 23.40, 6.64. HPLC tR = 4.747 min in 8 min chromatography, purity 99.5%. LCMS tR = 2.013 min in 4 min chromatography, MS ESI calcd. for C19H16N7S+ [M + H]+ 374.12, found 374.3.
3-(1,2,4-Thiadiazol-5-yl)aniline (96)
A mixture of 5-bromo-1,2,4-thiadiazole (500 mg, 3.03 mmol), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (123) (1.33 g, 6.06 mmol), Pd(dppf)Cl2 (297 mg, 0.36 mmol), and Cs2CO3 (2.96 g, 9.09 mmol) in dioxane (5 mL) and water (2 mL) was degassed and purged with N2 three times at 25 °C. Then the mixture was stirred at 100 °C for 5 h under N2 atmosphere. The reaction mixture was filtered and the solid was washed with DCM (6 mL × 2). The combined filtrate was concentrated in vacuo. The impure product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 12–42%, 10 min). 96 (206 mg, 37.3%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (s, 1H), 7.24–7.22 (m, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.15–7.13 (m, 1H), 6.78 (d, J = 8.0 Hz, 1H), 5.50 (s, 2H). LCMS tR = 0.393 min in 1 min chromatography, MS ESI calcd. for C8H8N3S+ [M + H]+ 178.04, found 178.0.
5-((3-(1,2,4-Thiadiazol-5-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (24)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol) and 3-(1,2,4-thiadiazol-5-yl)aniline (96) (136 mg, 0.77 mmol) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol), BINAP (80 mg, 0.13 mmol), and Pd(OAc)2 (29 mg, 0.13 mmol) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–6%, MeOH/DCM). The product was purified by prep-HPLC (column: Waters Torus 2-PIC 150 × 19 mm × 5 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 5–30%, 15 min). Then the impure product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 36–66%, 10 min). 24 (12 mg, 1.9%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.97 (s, 1H), 8.85 (t, J = 1.9 Hz, 1H), 8.41 (s, 1H), 8.32 (br s, 1H), 7.84 (dd, J = 8.1, 2.2 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 7.9 Hz, 1H), 6.03 (s, 1H), 2.63 (tt, J = 6.9, 3.6 Hz, 1H), 0.87–0.81 (m, 2H), 0.77–0.72 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 188.00, 164.35, 156.61, 150.59, 148.48, 145.15, 141.61, 130.12, 130.07, 122.40, 120.09, 117.65, 114.54, 77.04, 76.85, 23.35, 6.56. HPLC tR = 4.670 min in 8 min chromatography, purity 99.5%. LCMS tR = 1.847 min in 4 min chromatography, MS ESI calcd. for C18H15N8S+ [M + H]+ 375.11, found 375.3.
5-((3-(2H-Tetrazol-5-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (25)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (150 mg, 0.64 mmol) and 3-(2H-tetrazol-5-yl)aniline (97) (103 mg, 0.64 mmol) in dioxane (3 mL) was added tBuOK (216 mg, 1.93 mmol), BINAP (60 mg, 0.10 mmol), and Pd(OAc)2 (22 mg, 0.10 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–98%, MeOH/DCM). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 10–40%, 10 min). The residue was further purified by SFC (column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [0.1%NH3H2O IPA]; B%: 35–35%, 60 min). 25 (5.5 mg, 2.3%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.90 (s, 1H), 8.38 (s, 1H), 8.30 (s, 1H), 8.24 (t, J = 1.9 Hz, 1H), 8.12 (d, J = 7.4 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 6.05 (s, 1H), 2.64–2.59 (m, 1H), 0.86–0.80 (m, 3H), 0.76–0.71 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.77, 156.79, 150.77, 148.46, 145.25, 141.09, 129.71, 123.84, 120.93, 120.32, 117.55, 114.76, 76.74, 76.67, 23.35, 6.60. HPLC tR = 1.884 min in 4 min chromatography, purity 99.0%. LCMS tR = 2.239 min in 7 min chromatography, MS ESI calcd. for C17H15N10+ [M + H]+ 359.15, found 359.3.
5-((3-(1H-Tetrazol-1-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (26)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (100 mg, 0.43 mmol) and 3-(1H-tetrazol-1-yl)aniline (98) (69 mg, 0.43 mmol) in dioxane (3 mL) was added Cs2CO3 (418 mg, 1.28 mmol) and Brettphos Pd G3 (39 mg, 0.04 mmol) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. Water (50 mL) was added to the reaction mixture. The mixture was extracted with DCM (80 mL × 2), and the combined organic phase was washed with water (90 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The impure product was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 25–65%, 10 min). 26 (15.7 mg, 2.5%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 10.02 (s, 1H), 8.52 (t, J = 2.1 Hz, 1H), 8.40 (s, 1H), 8.37 (s, 1H), 7.89 (ddd, J = 8.3, 2.2, 1.0 Hz, 1H), 7.60 (t, J = 8.1 Hz, 1H), 7.51 (ddd, J = 8.0, 2.2, 1.0 Hz, 1H), 6.07 (s, 1H), 2.63 (tt, J = 6.9, 3.6 Hz, 1H), 0.88–0.80 (m, 2H), 0.77–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.59, 150.66, 148.58, 145.20, 142.13, 142.01, 134.07, 130.42, 119.79, 114.60, 114.09, 111.35, 77.07, 76.93, 23.39, 6.59. HPLC tR = 4.096 min in 8 min chromatography, purity 98.1%. LCMS tR = 1.615 min in 4 min chromatography, MS ESI calcd. for C17H15N10+ [M + H]+ 359.15, found 359.3.
3-Nitrobenzenediazonium tetrafluoroborate (125)
To a solution of 3-nitroaniline (124) (500 mg, 3.62 mmol) in EtOH (5 mL) and concentrated HF-BF3 solution (1.32 g, 7.24 mmol, 48%) was added tert-butyl nitrite (747 mg, 7.24 mmol) at 0 °C. The mixture was stirred at 25 °C for 1 h. The reaction mixture was diluted with PE (10 mL). The mixture was filtered and the solid was washed with PE (5 mL × 2). The solid was dried in vacuo. The crude product was used in the next step directly. 125 (850 mg, 99.1%) was obtained as a light orange solid.
2-(3-Nitrophenyl)-2H-tetrazole (126)
A solution of 3-nitrobenzenediazonium tetrafluoroborate (125) (850 mg, 3.59 mmol) and silver trifluoroacetate (951 mg, 4.31 mmol) in THF (10 mL) was cooled down to −78 °C. Then triethylamine (545 mg, 5.38 mmol) was added to the reaction mixture dropwise. After 10 min, TMSCHN2 (2 M, 1.97 mL) was added to the reaction mixture dropwise. Then the reaction mixture was stirred at −78 °C for 1 h and the mixture was warmed slowly to 25 °C. A solution of CsF (1.09 g, 7.18 mmol) in MeOH (10 mL) was added to the reaction mixture dropwise. Then the reaction mixture was stirred at 25 °C for 0.5 h. The reaction mixture was diluted with EtOAc (25 mL) and brine (10 mL). The organic phase was separated, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was used in the next step directly. 126 (600 mg, crude) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 1H), 8.80 (t, J = 2.0 Hz, 1H), 8.59–8.57 (m, 1H), 8.47–8.45 (m, 1H), 7.99 (t, J = 8.4 Hz, 1H).
3-(2H-Tetrazol-2-yl)aniline (99)
To a solution of 2-(3-nitrophenyl)-2H-tetrazole (126) (600 mg, 3.14 mmol) in EtOH (8 mL) was added NH4Cl (504 mg, 9.42 mmol) which was dissolved in H2O (2 mL). Then Fe (1.05 g, 18.83 mmol) was added to the mixture at 25 °C. Then the mixture was degassed, purged with N2, and stirred at 80 °C for 3 h. The reaction mixture was diluted with MeOH (10 mL) and filtered. The solid was washed with MeOH (10 mL × 2). The combined filtrate was concentrated in vacuo. The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 15–45%, 10 min). 99 (153 mg, 48.4%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 7.30 (s, 1H), 7.28–7.24 (m, 1H), 7.19–7.17 (m, 1H), 6.73 (d, J = 7.6 Hz, 1H), 5.70 (s, 2H). LCMS tR = 0.381 min in 1 min chromatography, MS ESI calcd. for C7H8N5+ [M + H]+ 162.08, found 162.1.
5-((3-(2H-Tetrazol-2-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (27)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (180 mg, 0.77 mmol) and 3-(2H-tetrazol-2-yl)aniline (99) (124 mg, 0.77 mmol) in dioxane (4 mL) was added Cs2CO3 (753 mg, 2.31 mmol), BINAP (72 mg, 0.12 mmol), and Pd(OAc)2 (29 mg, 0.12 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 4% MeOH/DCM) to give the product. The impure product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 37–67%, 10 min). 27 (35.8 mg, 12.9%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.25 (s, 1H), 8.72 (t, J = 2.2 Hz, 1H), 8.41 (s, 1H), 8.37 (s, 1H), 7.99–7.95 (m, 1H), 7.76–7.69 (m, 1H), 7.62 (t, J = 8.1 Hz, 1H), 6.05 (s, 1H), 2.66–2.58 (m, 1H), 0.88–0.82 (m, 2H), 0.77–0.72 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.52, 153.81, 150.54, 148.54, 145.28, 141.94, 136.52, 130.47, 120.04, 114.47, 112.88, 110.02, 77.09, 76.94, 23.35, 6.56. HPLC tR = 4.643 min in 8 min chromatography, purity 99.7%. LCMS tR = 1.856 min in 4 min chromatography, MS ESI calcd. for C17H15N10+ [M + H]+ 359.15, found 359.4.
7-(Cyclopropylamino)-5-((3-(pyridin-2-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (28)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol, 1 eq) and 3-(pyridin-2-yl)aniline (100) (146 mg, 0.86 mmol, 1 eq) in dioxane (3 mL) was added Cs2CO3 (837 mg, 2.57 mmol, 3 eq), BINAP (80 mg, 0.13 mmol, 0.15 eq), and Pd(OAc)2 (29 mg, 0.13 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–10%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Phenomenex Luna 30 × 30 mm × 10 μm + YMC AQ 100 × 30 × 10 μm; mobile phase: [water (NH3H2O)-ACN]; B%: 45–85%, 17 min). 28 (13.4 mg, 0.06 mmol) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.72 (t, J = 2.0 Hz, 1H), 8.67 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.38 (s, 1H), 8.27 (d, J = 1.8 Hz, 1H), 7.97 (dt, J = 8.2, 1.1 Hz, 1H), 7.86 (td, J = 7.7, 1.9 Hz, 1H), 7.80 (ddd, J = 8.0, 2.3, 1.0 Hz, 1H), 7.74 (dt, J = 8.1, 1.2 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.36 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 6.05 (s, 1H), 2.65–2.57 (m, 1H), 0.86–0.80 (m, 2H), 0.76–0.71 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.87, 155.96, 150.90, 149.52, 148.40, 145.01, 140.96, 139.17, 137.22, 129.19, 122.70, 120.11, 119.71, 117.52, 114.85, 76.71, 76.61, 23.35, 6.58. HPLC tR = 3.540 min in 8 min chromatography, purity 99.0%. LCMS tR = 1.384 min in 4 min chromatography, MS ESI calcd. for C21H18N7+ [M + H]+ 368.16, found 368.3.
3-(Pyrazin-2-yl) aniline (101)
A mixture of 2-bromopyrazine (1.00 g, 6.29 mmol), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (123) (2.76 g, 12.58 mmol), Pd(dppf)Cl2 (616 mg, 0.75 mmol), and Cs2CO3 (6.15 g, 18.87 mmol) in dioxane (10 mL) and water (3 mL) was degassed and purged with N2 three times at 25 °C. Then the mixture was stirred at 100 °C for 5 h under N2 atmosphere. The reaction mixture was filtered and the solid was washed with DCM (10 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Ultimate XB-NH2 250 mm × 100 mm × 10 μm; mobile phase: [heptane-EtOH (0.1% NH3H2O)]; B%: 5–40%, 12 min). The impure product was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O)-ACN]; B%: 10–40%, 10 min). 101 (627 mg, 57.1%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.68–8.67 (m, 1H), 8.56 (d, J = 2.4 Hz, 1H), 7.37 (s, 1H), 7.27–7.25 (m, 1H), 7.20–7.18 (m, 1H), 6.72 (d, J = 2.0 Hz, 1H), 5.33 (s, 2H). LCMS tR = 0.323 min in 1 min chromatography, MS ESI calcd. for C10H10N3+ [M + H]+ 172.09, found 172.0.
7-(Cyclopropylamino)-5-((3-(pyrazin-2-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (29)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (200 mg, 0.86 mmol) and 3-(pyrazin-2-yl) aniline (101) (147 mg, 0.86 mmol) in dioxane (4 mL) was added Cs2CO3 (837 mg, 2.57 mmol), BINAP (80 mg, 0.13 mmol), and Pd(OAc)2 (29 mg, 0.13 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 49% ∼ 67%, EtOAC/PE) to give the product. The impure product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 37–67%, 10 min). 29 (16.9 mg, 7.3%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, 1H), 9.23 (d, J = 1.5 Hz, 1H), 8.73 (dd, J = 2.5, 1.5 Hz, 1H), 8.68 (t, J = 2.0 Hz, 1H), 8.63 (d, J = 2.5 Hz, 1H), 8.38 (s, 1H), 8.29 (d, J = 1.8 Hz, 1H), 7.93–7.87 (m, 1H), 7.83–7.76 (m, 1H), 7.51 (t, J = 7.9 Hz, 1H), 6.05 (s, 1H), 2.66–2.57 (m, 1H), 0.86–0.79 (m, 2H), 0.77–0.72 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.78, 151.40, 150.81, 148.41, 145.06, 144.29, 143.50, 141.79, 141.18, 136.39, 129.50, 120.55, 120.24, 117.47, 114.71, 76.76, 76.63, 23.33, 6.55. HPLC tR = 4.595 min in 8 min chromatography, purity 96.7%. LCMS tR = 1.835 min in 4 min chromatography, MS ESI calcd. for C20H17N8+ [M + H]+ 369.16, found 369.4.
Experimental Procedures in Scheme 3
5-Chloro-7-(cyclobutylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (127)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (350 mg, 1.64 mmol) in EtOH (5 mL) was added cyclobutylamine (234 mg, 3.29 mmol) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The crude compound was used in the next step. 127 (314 mg) was obtained as a light yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.14 (d, J = 6.0 Hz, 1H), 8.67 (s, 1H), 6.50 (s, 1H), 4.39–4.17 (m, 1H), 2.40–2.18 (m, 4H), 1.79–1.57 (m, 2H). LCMS tR = 0.519 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C11H11ClN5+ [M + H]+ 248.07, found 248.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-(cyclobutylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (30)
To a solution of 5-chloro-7-(cyclobutylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (127) (105 mg, 0.42 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (68 mg, 0.42 mmol) in dioxane (3 mL) was added tBuOLi (102 mg, 1.27 mmol), BINAP (40 mg, 0.06 mmol), and Pd(OAc)2 (14 mg, 0.06 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–2%, MeOH/DCM). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 26–56%, 11 min). 30 (30 mg, 19.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 9.02 (s, 2H), 8.42 (s, 1H), 8.34 (d, J = 1.9 Hz, 1H), 8.26 (d, J = 6.2 Hz, 1H), 7.60 (dd, J = 8.1, 2.0 Hz, 1H), 7.51 (t, J = 8.1 Hz, 1H), 7.31 (dd, J = 7.9, 2.1 Hz, 1H), 5.67 (s, 1H), 4.02 (h, J = 7.8 Hz, 1H), 2.40–2.29 (m, 2H), 2.29–2.16 (m, 2H), 1.85–1.71 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.59, 150.85, 146.18, 144.94, 141.78, 141.18, 134.18, 130.29, 118.46, 114.64, 114.53, 111.75, 77.02, 75.90, 46.95, 29.06, 14.94. HPLC tR = 2.721 min in 8 min chromatography, purity 99.2%. LCMS tR = 1.659 min in 4 min chromatography, MS ESI calcd. for C19H18N9+ [M + H]+ 372.17, found 372.3.
5-Chloro-7-(oxetan-3-ylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (128)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (2.00 g, 9.39 mmol, 1 eq) in EtOH (20 mL) was added oxetan-3-amine (1.10 g, 15.05 mmol, 14.6 mL, 1.6 eq) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered and the solid was washed with EtOH (15 mL × 2). The residue was purified by flash silica gel chromatography (eluent 0–4%, MeOH in DCM). 128 (1.80 g, 6.7 mmol, 71.33% yield) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (br s, 1H), 8.71 (s, 1H), 6.48 (s, 1H), 5.09–4.95 (m, 1H), 4.87–4.72 (m, 4H). LCMS tR = 0.424 min in 1 min chromatography, MS ESI calcd. for C10H9ClN5O+ [M + H]+ 250.05, found 250.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-(oxetan-3-ylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (31)
To a solution of 5-chloro-7-(oxetan-3-ylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (128) (100 mg, 0.40 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (64 mg, 0.40 mmol, 1 eq) in dioxane (5 mL) was added Cs2CO3 (391 mg, 1.20 mmol, 3 eq), BINAP (37 mg, 0.06 mmol, 0.15 eq), and Pd(OAc)2 (13 mg, 0.06 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The impure product was purified by prep-HPLC (column: Phenomenex Luna 30 × 30 mm × 10 μm + YMC AQ 100 × 30 × 10 μm; mobile phase: [water (0.05%NH3H2O)-ACN]; B%: 25–55%, 20 min). 31 (4.3 mg, 0.01 mmol, 2.77% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 9.02 (s, 2H), 8.71 (d, J = 3.5 Hz, 1H), 8.46 (s, 1H), 8.32 (t, J = 2.1 Hz, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 5.52 (s, 1H), 4.88–4.80 (m, 2H), 4.81–4.73 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 156.53, 150.91, 146.22, 145.15, 141.65, 141.21, 134.21, 130.35, 118.57, 114.71, 114.58, 111.84, 77.18, 76.00, 75.83, 46.46. HPLC tR = 3.232 min in 8 min chromatography, purity 96.4%. LCMS tR = 1.200 min in 4 min chromatography, MS ESI calcd. for C18H16N9O+ [M + H]+ 374.15, found 374.3.
5-Chloro-7-((3,3-difluorocyclobutyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (129)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (200 mg, 0.94 mmol, 1 eq) in EtOH (5 mL) was added 3,3-difluorocyclobutan-1-amine (404 mg, 2.82 mmol, 3 eq, HCl) and Et3N (304 mg, 3 mmol, 0.42 mL, 3.2 eq) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered and the solid was washed with EtOH (4 mL × 2). 129 (158 mg, 0.55 mmol, 58.49% yield) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.33 (br s, 1H), 8.71 (s, 1H), 6.66 (s, 1H), 4.35–4.16 (m, 1H), 3.20–2.89 (m, 4H). LCMS tR = 0.511 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C11H9ClF2N5+ [M + H]+ 284.05, found 284.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((3,3-difluorocyclobutyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (32)
To a solution of 5-chloro-7-((3,3-difluorocyclobutyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (129) (200 mg, 0.70 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (112 mg, 0.70 mmol, 1 eq) in dioxane (5 mL) was added tBuOLi (169 mg, 2.12 mmol, 3 eq), BINAP (65 mg, 0.10 mmol, 0.15 eq), and Pd(OAc)2 (23 mg, 0.10 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0% ∼ 5%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 26–56%, 10 min). 32 (5.5 mg, 0.01 mmol, 1.9% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 9.03 (s, 2H), 8.50 (s, 1H), 8.45 (s, 1H), 8.34 (t, J = 2.2 Hz, 1H), 7.60 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.32 (ddd, J = 7.9, 2.2, 1.0 Hz, 1H), 5.69 (s, 1H), 4.02 (qd, J = 8.6, 7.7, 4.8 Hz, 1H), 3.14–2.90 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 156.59, 150.89, 146.60, 145.16, 141.70, 141.27, 134.26, 130.42, 119.42 (dd, J = 282.5, 268.2 Hz), 118.55, 114.73, 114.65, 111.79, 77.24, 76.55, 41.50 (t, J = 22.5 Hz), 36.58 (dd, J = 19.3, 6.2 Hz). HPLC tR = 3.938 min in 8 min chromatography, purity 99.1%. LCMS tR = 2.161 min in 4 min chromatography, MS ESI calcd. for C19H16F2N9+ [M + H]+ 408.15, found 408.3.
5-Chloro-7-(((tetrahydrofuran-3-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (130)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (500 mg, 2.35 mmol) in EtOH (5 mL) was added (tetrahydrofuran-3-yl)methanamine (475 mg, 4.69 mmol) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered. The filter cake was washed with EtOH (4 mL × 2). The crude compound was used in next step. 130 (830 mg, crude) was obtained as a light yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.04 (s, 1H), 8.66 (s, 1H), 6.69 (s, 1H), 3.82–3.72 (m, 1H), 3.72–3.65 (m, 1H), 3.63–3.59 (m, 1H), 3.52–3.43 (m, 3H), 2.70–2.56 (m, 1H), 2.14–1.86 (m, 1H), 1.73–1.52 (m, 1H). LCMS tR = 0.466 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C12H13ClN5O+ [M + H]+ 278.08, found 278.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-(((tetrahydrofuran-3-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (33)
To a solution of 5-chloro-7-(((tetrahydrofuran-3-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (130) (200 mg, 0.72 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (115 mg, 0.72 mmol, 1 eq) in dioxane (5 mL) was added tBuOLi (172 mg, 2.16 mmol, 3 eq), BINAP (67 mg, 0.10 mmol, 0.15 eq), and Pd(OAc)2 (24 mg, 0.10 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–11%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 26–56%, 10 min). 33 (14.2 mg, 0.03 mmol, 4.8% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 8.17 (s, 2H), 7.57 (s, 1H), 7.50 (t, J = 2.1 Hz, 1H), 7.41 (t, J = 6.1 Hz, 1H), 6.75 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 6.66 (t, J = 8.1 Hz, 1H), 6.46 (ddd, J = 7.9, 2.2, 1.0 Hz, 1H), 4.93 (s, 1H), 2.94 (td, J = 8.1, 5.7 Hz, 1H), 2.84 (dd, J = 8.7, 6.8 Hz, 1H), 2.78 (td, J = 8.2, 6.7 Hz, 1H), 2.70 (dd, J = 8.6, 4.7 Hz, 1H), 2.42 (t, J = 6.6 Hz, 2H), 1.82 (dtd, J = 14.2, 7.1, 6.5, 3.6 Hz, 1H), 1.13 (dtd, J = 12.3, 8.0, 5.7 Hz, 1H), 0.88–0.75 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 156.71, 150.85, 147.33, 145.09, 141.88, 141.27, 134.24, 130.38, 118.48, 114.74, 114.55, 111.72, 77.11, 75.18, 70.40, 66.81, 44.45, 37.56, 29.39. HPLC tR = 3.500 min in 8 min chromatography, purity 97.8%. LCMS tR = 1.913 min in 4 min chromatography, MS ESI calcd. for C20H20N9O+ [M + H]+ 402.18, found 402.3.
5-Chloro-7-(((tetrahydro-2H-pyran-4-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (131)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (200 mg, 0.94 mmol) in EtOH (5 mL) was added (tetrahydro-2H-pyran-4-yl)methanamine (216 mg, 1.88 mmol) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The crude compound was used in the next step. 131 (273 mg) was obtained as a light yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.05–8.88 (m, 1H), 8.65 (s, 1H), 6.68 (s, 1H), 3.93–3.76 (m, 2H), 3.35–3.31 (m, 2H), 3.27–3.19 (m, 2H), 2.02–1.86 (m, 1H), 1.70–1.52 (m, 2H), 1.32–1.13 (m, 2H). LCMS tR = 0.484 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C13H15ClN5O+ [M + H]+ 292.10, found 292.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-(((tetrahydro-2H-pyran-4-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (34)
To a solution of 5-chloro-7-(((tetrahydro-2H-pyran-4-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (131) (200 mg, 0.68 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (109 mg, 0.68 mmol, 1 eq) in dioxane (5 mL) was added tBuOLi (164 mg, 2.06 mmol, 3 eq), BINAP (64 mg, 0.10 mmol, 0.15 eq), and Pd(OAc)2 (23 mg, 0.10 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–11%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 26–56%, 10 min). 34 (11.1 mg, 0.02 mmol, 3.6% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.18 (s, 2H), 7.57 (s, 1H), 7.49 (t, J = 2.1 Hz, 1H), 7.34 (t, J = 6.1 Hz, 1H), 6.76 (ddd, J = 8.2, 2.1, 1.0 Hz, 1H), 6.66 (t, J = 8.1 Hz, 1H), 6.46 (ddd, J = 7.9, 2.2, 1.0 Hz, 1H), 4.92 (s, 1H), 3.01 (ddd, J = 11.5, 4.5, 1.9 Hz, 2H), 2.42 (td, J = 11.7, 2.0 Hz, 2H), 2.34 (t, J = 6.5 Hz, 2H), 1.12 (ddp, J = 11.2, 7.3, 3.5 Hz, 1H), 0.86–0.72 (m, 2H), 0.40 (qd, J = 12.0, 4.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.68, 150.83, 147.44, 145.08, 141.89, 141.26, 134.23, 130.38, 118.45, 114.75, 114.51, 111.69, 77.06, 75.31, 66.65, 47.30, 33.78, 30.49. HPLC tR = 3.644 min in 8 min chromatography, purity 92.3%. LCMS tR = 2.160 min in 4 min chromatography, MS ESI calcd. for C21H22N9O+ [M + H]+ 416.19, found 416.0.
5-Chloro-7-(((1-methyl-1H-pyrazol-4-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (132)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (200 mg, 0.94 mmol) in EtOH (5 mL) was added (1-methyl-1H-pyrazol-4-yl)methanamine (209 mg, 1.88 mmol) dropwise at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The crude compound was used in the next step. 132 (293 mg) was obtained as a light yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.66 (s, 1H), 7.81–7.63 (m, 1H), 7.55–7.39 (m, 1H), 6.64 (s, 1H), 4.49 (d, J = 6.4 Hz, 2H), 3.82 (s, 1H), 3.77 (s, 3H). LCMS tR = 0.447 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C12H11ClN7+ [M + H]+ 288.08, found 288.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-(((1-methyl-1H-pyrazol-4-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (35)
To a solution of 5-chloro-7-(((1-methyl-1H-pyrazol-4-yl)methyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (132) (200 mg, 0.70 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (111 mg, 0. 70 mmol, 1 eq) in dioxane (5 mL) was added tBuOLi (167 mg, 2.09 mmol, 3 eq), BINAP (65 mg, 0.1 mmol, 0.15 eq), and Pd(OAc)2 (23 mg, 0.10 mmol, 0.15 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–5%, MeOH/DCM). Then the impure product was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 26–56%, 10 min). The residue was purified by SFC (column: DAICEL CHIRALCEL OJ (250 mm × 30 mm, 10 μm); mobile phase: [0.1%NH3H2O EtOH]; B%: 60–60%, 60 min). The impure product was purified by prep-HPLC (column: Phenomenex Gemini-NX C18 75 × 30 mm × 3 μm; mobile phase: [water (10 mM NH4HCO3)-ACN]; B%: 18–48%, 10 min). 35 (13.1 mg, 0.06 mmol) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 9.01 (s, 2H), 8.45 (s, 1H), 8.41 (s, 1H), 8.31 (t, J = 2.1 Hz, 1H), 7.64 (s, 1H), 7.60 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 7.50 (t, J = 8.1 Hz, 1H), 7.45–7.43 (m, 1H), 7.30 (ddd, J = 7.9, 2.2, 1.0 Hz, 1H), 5.75 (s, 1H), 4.37 (s, 2H), 3.78 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 156.56, 150.77, 147.11, 145.09, 141.81, 141.24, 137.82, 134.21, 130.33, 129.34, 118.50, 117.15, 114.70, 114.57, 111.83, 77.05, 75.69, 38.52, 36.05. HPLC tR = 3.389 min in 8 min chromatography, purity 96.2%. LCMS tR = 1.285 min in 4 min chromatography, MS ESI calcd. for C20H18N11+ [M + H]+ 412.17, found 412.3.
7-((1-Acetylpiperidin-4-yl)amino)-5-chloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (133)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (220 mg, 1.03 mmol, 1 eq) in EtOH (5 mL) was added 1-(4-(aminomethyl)piperidin-1-yl)ethan-1-one (734 mg, 5.16 mmol, 5 eq) at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered and the solid was washed with EtOH (4 mL × 2). 133 (300 mg, 0.90 mmol, 87.16% yield) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.57–7.78 (m, 1H), 6.79 (s, 1H), 4.47–4.41 (m, 1H), 3.98 (br t, J = 10.8 Hz, 1H), 3.87 (br d, J = 13.2 Hz, 1H), 3.15 (t, J = 12.8 Hz, 1H), 2.63 (t, J = 12.4 Hz, 1H), 2.01 (s, 3H), 1.94–1.80 (m, 2H), 1.75–1.49 (m, 2H). LCMS tR = 0.447 min in 1 min chromatography, Chromolith Flash RP-18, 5 μm, 3.0 × 25 mm, MS ESI calcd. for C14H16ClN6O+ [M + H]+ 319.11, found 319.1.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-acetylpiperidin-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (36)
To a solution of 7-((1-acetylpiperidin-4-yl)amino)-5-chloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (133) (200 mg, 0.63 mmol, 1 eq) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (101 mg, 0.63 mmol, 1 eq) in dioxane (3 mL) was added Cs2CO3 (613 mg, 1.88 mmol, 3 eq) and Brettphos Pd G3 (57 mg, 0.06 mmol, 0.1 eq) at 25 °C. The mixture was degassed and purged with N2. Then the mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent: 0–10%, MeOH/DCM). The impure product was purified by prep-HPLC (column: Waters Torus 2-PIC 150 × 19 mm × 5 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 5–60%, 15 min). 36 (46.6 mg, 0.10 mmol, 16.07% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 9.02 (s, 2H), 8.42 (s, 1H), 8.37 (t, J = 2.1 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.59 (dd, J = 8.2, 2.0 Hz, 1H), 7.50 (t, J = 8.1 Hz, 1H), 7.31 (dd, J = 7.9, 2.2 Hz, 1H), 5.85 (s, 1H), 4.44 (d, J = 13.2 Hz, 1H), 3.91 (d, J = 13.7 Hz, 1H), 3.66 (dtt, J = 11.6, 8.2, 4.2 Hz, 1H), 3.19–3.07 (m, 1H), 2.64 (ddd, J = 14.7, 12.6, 2.8 Hz, 1H), 2.03 (s, 3H), 1.96 (t, J = 14.4 Hz, 2H), 1.69 (qd, J = 12.3, 4.1 Hz, 1H), 1.56 (qd, J = 12.5, 4.3 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.10, 156.70, 150.94, 146.33, 144.96, 141.84, 141.21, 134.20, 130.31, 118.42, 114.63, 114.50, 111.67, 77.12, 75.62, 49.58, 44.70, 31.09, 30.34, 28.98, 21.31. HPLC tR = 3.429 min in 8 min chromatography, purity 95.75%. LCMS tR = 1.482 min in 4 min chromatography, MS ESI calcd. for C22H23N10O+ [M + H]+ 443.21, found 443.0.
7-((2-((tert-Butyldimethylsilyl)oxy)ethyl)amino)-5-chloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (134)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (500 mg, 2.35 mmol) in EtOH (5 mL) was added 2-((tert-butyldimethylsilyl)oxy)ethan-1-amine (412 mg, 2.35 mmol) at 25 °C. Then the mixture was stirred at 25 °C for 2 h. The residue was purified by flash silica gel chromatography (eluent of DCM) to give the product. 134 (756 mg, 48.9%) was obtained as a white solid. LCMS tR = 0.753 min in 1.5 min chromatography, MS ESI calcd. for C15H23ClN5OSi+ [M + H]+ 352.14, found 352.0.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((2-((tert-butyldimethylsilyl)oxy)ethyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (135)
To a solution of 7-((2-((tert-butyldimethylsilyl)oxy)ethyl)amino)-5-chloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (134) (250 mg, 0.39 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (125 mg, 0.78 mmol) in dioxane (5 mL) was added Cs2CO3 (382 mg, 1.17 mmol), BINAP (36 mg, 0.06 mmol), and Pd(OAc)2 (13 mg, 0.06 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Ultimate XB-CN 250 × 50 × 10 μm; mobile phase: [heptane-EtOH(0.1%NH3H2O)]; B%: 10–50%, 15 min). 135 (227 mg, 34.2%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 9.02 (s, 2H), 8.42 (s, 1H), 8.34 (s, 1H), 7.91 (t, J = 6.0 Hz, 1H), 7.62–7.56 (m, 1H), 7.50 (t, J = 8.1 Hz, 1H), 7.33–7.29 (m, 1H), 5.83 (s, 1H), 3.82 (t, J = 5.6 Hz, 2H), 3.45–3.39 (m, 2H), 0.80 (s, 9H), 0.03 (s, 6H). LCMS tR = 1.633 min in 1 min chromatography, MS ESI calcd. for C23H30N9OSi+ [M + H]+ 476.23, found 476.2.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((2-hydroxyethyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (37)
To a solution of 5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-((2-((tert-butyldimethylsilyl)oxy)ethyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (135) (100 mg, 0.21 mmol) in DCM (8 mL) was added TFA (2 mL) at 25 °C. The mixture was stirred at 35 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: C18-6 100 × 30 mm × 5 μm; mobile phase: [water (FA)-ACN]; B%: 0–60%, 15 min). 37 (45.6 mg, 58.7%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 9.02 (s, 2H), 8.42 (s, 1H), 8.34 (t, J = 2.1 Hz, 1H), 7.87 (t, J = 6.0 Hz, 1H), 7.66–7.56 (m, 1H), 7.51 (t, J = 8.1 Hz, 1H), 7.31 (dd, J = 8.2, 2.2 Hz, 1H), 5.82 (s, 1H), 4.97 (t, J = 5.5 Hz, 1H), 3.66 (q, J = 5.7 Hz, 2H), 3.37 (q, J = 5.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.69, 150.71, 147.58, 145.03, 141.88, 141.24, 134.20, 130.30, 118.52, 114.69, 114.51, 111.75, 76.98, 75.35, 59.03, 44.24. HPLC tR = 3.021 min in 8 min chromatography, purity 93.3%. LCMS tR = 1.719 min in 4 min chromatography, MS ESI calcd. for C17H16N9O+ [M + H]+ 362.15, found 362.2.
Experimental Procedures in Scheme 4
1-Methyl-1H-imidazol-4-amine (137)
To a solution of 1-methyl-4-nitro-1H-imidazole (136) (312 mg, 2.45 mmol) in MeOH (5 mL) was added Pd/C (0.42 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (50 psi) at 25 °C for 3 h. The reaction mixture was filtered. The filter cake was washed with MeOH (5 mL × 3), and then the combined filtrate was concentrated. 137 (238 mg, 99.8%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 7.34–7.28 (m, 1H), 7.08–7.02 (br s, 1H), 3.56–3.51 (m, 2H), 3.46 (s, 3H). LCMS tR = 0.142 min in 1.5 min chromatography, MS ESI calcd. for C8H15N6+ [2M + H]+ 195.14, found 195.1.
5-Chloro-7-((1-methyl-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (138)
To a solution of 1-methyl-1H-imidazol-4-amine (137) (210 mg, 2.16 mmol) in EtOH (5 mL) was added 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (450 mg, 2.11 mmol) under N2 atmosphere. The reaction mixture was stirred under at 25 °C for 5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 78%, EtOAc/PE) to give the product. 138 (520 mg, 44.4%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.98–8.92 (m, 1H), 8.78 (s, 1H), 8.04 (s, 1H), 7.98–7.89 (m, 1H), 7.28–7.26 (m, 1H), 3.73 (s, 3H). LCMS tR = 0.714 min in 2 min chromatography, MS ESI calcd. for C11H9ClN7+ [M + H]+ 274.06, found 273.9.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-methyl-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (38)
To a solution of 5-chloro-7-((1-methyl-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (138) (300 mg, 1.10 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (176 mg, 1.10 mmol) in dioxane (5 mL) was added Cs2CO3 (1.07 g, 3.29 mmol), BINAP (102 mg, 0.16 mmol), and Pd(OAc)2 (37.0 mg, 0.16 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (MeOH) to give a crude product. The crude product was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (FA)-ACN]; B%: 0–38%, 25 min). 38 (15.0 mg, 3.30%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 2H), 9.02 (s, 2H), 8.49 (s, 1H), 8.39 (t, J = 2.1 Hz, 1H), 7.65–7.58 (m, 2H), 7.50 (t, J = 8.1 Hz, 1H), 7.30 (dd, J = 7.9, 2.2 Hz, 1H), 7.07 (d, J = 1.5 Hz, 1H), 6.79 (s, 1H), 3.68 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 145.02, 144.91, 141.92, 141.24, 136.88, 135.15, 134.15, 130.28, 118.56, 118.54, 114.67, 114.55, 111.81, 110.65, 78.85, 77.22, 33.40. HPLC tR = 2.779 min in 8 min chromatography, purity 95.5%. LCMS tR = 1.446 min in 4 min chromatography, MS ESI calcd. for C19H16N11+ [M + H]+ 398.16, found 398.4.
2-(4-Nitro-1H-imidazol-1-yl)ethanol (141)
To a solution of 4-nitro-1H-imidazole (139) (3.00 g, 26.53 mmol) and 2-bromoethanol (3.98 g, 31.84 mmol) in MeCN (30 mL) was added K2CO3 (11.0 g, 79.59 mmol). Then the reaction mixture was stirred at 60 °C for 10 h. The reaction mixture was filtered. The filtrate was concentrated. 141 (4.55 g, crude) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.40–8.35 (m, 1H), 7.83–7.80 (m, 1H), 4.11 (t, J = 5.2 Hz, 2H), 3.70 (t, J = 5.2 Hz, 2H), 3.39 (s, 1H).
1-(2-Chloroethyl)-4-nitro-1H-imidazole (142)
To a solution of 2-(4-nitro-1H-imidazol-1-yl)ethanol (141) (1.50 g, 9.55 mmol) in DCM (20 mL) was added SOCl2 (3.14 g, 28.64 mmol) and DMF (7 mg, 95.46 μmol). The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated directly. 142 (1.56 g, 93.1%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.51–8.45 (m, 1H), 7.94–7.90 (m, 1H), 4.46 (t, J = 5.6 Hz, 2H), 4.08–4.04 (m, 2H).
N-Methyl-2-(4-nitro-1H-imidazol-1-yl)ethanamine (143)
To a solution of 1-(2-chloroethyl)-4-nitro-1H-imidazole (142) (1.50 g, 8.54 mmol) and methylamine (1.33 g, 42.72 mmol) in MeCN (8 mL) was added K2CO3 (7.08 g, 51.26 mmol) at 25 °C. Then NaI (1.54 g, 10.25 mmol) was added to the mixture at 0 °C, and the reaction mixture was heated in a microwave reactor at 80 °C for 4 h. The mixture was concentrated in vacuo. 143 (1.49 g, 13.0%) was obtained as a black brown solid. LCMS tR = 0.107 min in 1 min chromatography, MS ESI calcd. for C6H11N4O2+ [M + H]+ 171.09, found 171.0.
tert-Butyl methyl(2-(4-nitro-1H-imidazol-1-yl)ethyl)carbamate (144)
To a solution of N-methyl-2-(4-nitro-1H-imidazol-1-yl)ethanamine (143) (1.48 g, 8.70 mmol) in THF (10 mL) was added K2CO3 (3.61 g, 26.09 mmol) in H2O (5 mL) at 25 °C. Then Boc2O (3.80 g, 17.39 mmol) was added to the mixture and the resulting mixture was stirred at 25 °C for 3 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–80%, EtOAc/PE). 144 (586 mg, 21.8%) was obtained as a black solid. 1H NMR (400 MHz, DMSO-d6) δ 8.38–8.29 (m, 1H), 7.83–7.78 (m, 1H), 4.19 (t, J = 5.2 Hz, 2H), 3.57 (t, J = 5.2 Hz, 2H), 2.79 (s, 3H), 1.30–1.11 (m, 9H). LCMS tR = 0.766 min in 2 min chromatography, MS ESI calcd. for C11H19N4O4+ [M + H]+ 271.14, found 271.0.
tert-Butyl (2-(4-amino-1H-imidazol-1-yl)ethyl)(methyl)carbamate (147)
To a solution of tert-butyl methyl(2-(4-nitro-1H-imidazol-1-yl)ethyl)carbamate (144) (350 mg, 1.29 mmol) in THF (10 mL) was added Pd/C (0.35 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times and it was stirred under H2 (50psi) at 40 °C for 12 h. The reaction mixture was filtered. The filter cake was washed with MeOH (10 mL × 3), and then the combined filtrate was concentrated. 147 (283 mg, 61.9%) was obtained as a brown oil. 1H NMR (400 MHz, DMSO-d6) δ 7.04 (s, 1H), 6.23–5.98 (m, 1H), 3.17 (s, 2H), 2.69–2.62 (m, 7H), 1.33–1.31 (m, 9H). LCMS tR = 0.825 min in 2 min chromatography, MS ESI calcd. for C11H21N4O2+ [M + H]+ 241.17, found 241.0.
tert-Butyl (2-(4-((5-chloro-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-imidazol-1-yl)ethyl)(methyl)carbamate (149)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (220 mg, 1.03 mmol) in EtOH (10 mL) was added tert-butyl (2-(4-amino-1H-imidazol-1-yl)ethyl)(methyl)carbamate (147) (273 mg, 1.14 mmol) under N2 atmosphere. The reaction mixture was stirred under N2 at 25 °C for 5 h. The reaction mixture was concentrated in vacuo. 149 (586 mg, 21.8%) was obtained as a black solid. 1H NMR (400 MHz, DMSO-d6) δ 11.30–10.75 (m, 1H), 8.94 (s, 1H), 8.81–8.73 (m, 1H), 8.04 (s, 1H), 7.85–7.64 (m, 1H), 4.20–4.09 (m, 2H), 2.85–2.69 (m, 5H), 1.39–1.30 (m, 9H). LCMS tR = 1.301 min in 2 min chromatography, MS ESI calcd. for C18H22ClN8O2+ [M + H]+ 417.15, found 417.1.
tert-Butyl (2-(4-((5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-imidazol-1-yl)ethyl)(methyl)carbamate (151)
To a solution of tert-butyl (2-(4-((5-chloro-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-imidazol-1-yl)ethyl)(methyl)carbamate (149) (300 mg, 0.72 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (115 mg, 0.72 mmol) in dioxane (5 mL) was added Cs2CO3 (703 mg, 2.16 mmol), BINAP (67 mg, 0.11 mmol), and Pd(OAc)2 (24 mg, 0.11 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 4 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 4–5%, MeOH/DCM). 151 (397 mg, 69.1%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.07–10.02 (m, 1H), 9.03 (s, 2H), 8.49 (s, 1H), 8.41–8.36 (m, 1H), 7.65–7.61 (m, 1H), 7.60–7.55 (m, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.32–7.29 (m, 1H), 5.48 (s, 1H), 4.14–4.09 (m, 2H), 3.543.47 (m, 2H), 2.75–2.69 (m, 3H), 1.27 (s, 9H). LCMS tR = 0.797 min in 1.5 min chromatography, MS ESI calcd. for C26H29N12O2+ [M + H]+ 541.25, found 541.2.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-(2-(methylamino)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (39)
To a mixture of tert-butyl (2-(4-((5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-imidazol-1-yl)ethyl)(methyl)carbamate (151) (170 mg, 0.31 mmol) in DCM (3 mL) was added TFA (3 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (FA)-ACN]; B%: 0–26%, 25 min). 39 (40.0 mg, 27.8%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.02 (s, 2H), 8.49 (s, 1H), 8.40 (t, J = 2.2 Hz, 1H), 8.20 (s, 1H), 7.67 (s, 1H), 7.63 (dd, J = 7.6, 2.0 Hz, 1H), 7.50 (t, J = 8.1 Hz, 1H), 7.31 (dd, J = 8.0, 2.2 Hz, 1H), 7.15 (d, J = 1.5 Hz, 1H), 6.90 (s, 1H), 4.16 (t, J = 6.1 Hz, 2H), 3.05 (t, J = 6.2 Hz, 2H), 2.42 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.55, 156.71, 150.76, 144.99, 144.70, 141.92, 141.22, 137.03, 134.74, 134.13, 130.25, 118.56, 114.53, 111.79, 109.33, 79.00, 77.21, 50.03, 44.80, 34.36. HPLC tR = 2.532 min in 8 min chromatography, purity 96.3%. LCMS tR = 1.302 min in 4 min chromatography, MS ESI calcd. for C21H21N12+ [M + H]+ 441.20, found 441.4.
N,N-Dimethyl-2-(4-nitro-1H-imidazol-1-yl)ethanamine (153)
To a solution of 2-chloro-N,N-dimethylethan-1-amine hydrochloride (1.27 g, 8.84 mmol) in MeCN (8 mL) was added K2CO3 (3.67 g, 26.53 mmol) and NaI (1.59 g, 10.61 mmol) at 25 °C. Then 4-nitro-1H-imidazole (139) (1 g, 8.84 mmol) was added to the mixture and the reaction mixture was stirred at 60 °C for 6 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 0–35%, 10 min). 153 (300 mg, 17.9% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 7.85 (s, 1H), 4.15 (t, J = 6.0 Hz, 2H), 2.60 (t, J = 6.0 Hz, 2H), 2.16 (s, 6H). LCMS tR = 0.152 min in 1.5 min chromatography, MS ESI calcd. for C7H13N4O2+ [M + H]+ 185.10, found 185.1.
1-(2-(Dimethylamino)ethyl)-1H-imidazol-4-amine (159)
To a solution of N,N-dimethyl-2-(4-nitro-1H-imidazol-1-yl)ethanamine (153) (250 mg, 1.36 mmol) in MeOH (2 mL) was added Pd/C (100 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 35 °C for 4 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (50 mL × 3), and then the combined filtrate was concentrated to dryness. 159 (224 mg) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.12 (s, 1H), 6.15 (d, J = 1.6 Hz, 1H), 4.08–3.96 (m, 2H), 3.83 (t, J = 6.4 Hz, 2H), 2.14–2.12 (m, 6H).
5-Chloro-7-((1-(2-(dimethylamino)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (165)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (290 mg, 1.36 mmol) in EtOH (5 mL) was added 1-(2-(dimethylamino)ethyl)-1H-imidazol-4-amine (159) (210 mg, 1.36 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was filtered. The filter cake was the target coarse product and was washed with EtOH (4 mL × 2). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 0–38%, 36 min). 165 (80 mg, 17.3%) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.98–10.82 (m, 1H), 8.75 (s, 1H), 7.68 (s, 1H), 7.37 (s, 1H), 7.19 (d, J = 1.6 Hz, 1H), 4.08 (t, J = 6.4 Hz, 2H), 2.57 (t, J = 6.4 Hz, 2H), 2.18 (s, 6H). LCMS tR = 0.560 min in 2.5 min chromatography, MS ESI calcd. for C14H16ClN8+ [M + H]+ 331.12, found 331.1.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-(2-(dimethylamino)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (40)
To a solution of 5-chloro-7-((1-(2-(dimethylamino)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (165) (50 mg, 0.15 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (29 mg, 0.18 mmol) in dioxane (8 mL) was added Cs2CO3 (147 mg, 0.45 mmol), BINAP (14 mg, 0.02 mmol), and Pd(OAc)2 (7 mg, 0.03 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 2 h. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (HCl)-ACN]; B%: 0–40%, 10 min). The residue was purified by prep-HPLC(column: Phenomenex Luna C18 150 × 25 mm × 10 μm; mobile phase: [water(HCl)-ACN]; B%: 0–25%, 30 min). 40 as a HCl salt (8.6 mg, 11.8%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 10.26 (s, 1H), 10.23 (s, 1H), 9.11 (s, 2H), 8.52 (s, 1H), 8.42 (s, 1H), 8.00 (s, 1H), 7.76–7.62 (m, 1H), 7.52 (t, J = 8.1 Hz, 1H), 7.37–7.25 (m, 2H), 6.96 (s, 1H), 4.49 (t, J = 6.6 Hz, 2H), 3.59 (q, J = 5.6 Hz, 2H), 2.82 (d, J = 4.6 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 157.33, 145.83, 142.22, 141.97, 139.90, 135.22, 134.21, 131.70, 130.79, 130.05, 124.39, 121.55, 119.40, 115.62, 112.48, 80.26, 78.10, 55.70, 51.42, 43.00. HPLC tR = 2.637 min in 8 min chromatography, purity 94.5%. LCMS tR = 1.375 min in 4 min chromatography, MS ESI calcd. for C22H23N12+ [M + H]+ 455.22, found 455.4.
4-Nitro-1-(2-(pyrrolidin-1-yl)ethyl)-1H-imidazole (154)
To a solution of 4-nitro-1H-imidazole (139) (1.00 g, 8.84 mmol) and 1-(2-chloroethyl)pyrrolidine (1.50 mg, 8.84 mmol) in DMF (15 mL) was added K2CO3 (3.67 g, 26.53 mmol) at 25 °C. Then the mixture was stirred at 120 °C for 12 h. The reaction mixture was concentrated. Water (50 mL) was added. The resulting mixture was extracted with EtOAc (50 mL × 3). The combined organic phase was washed with brine (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. 154 (425 mg, 20.0%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.42–8.30 (m, 1H), 7.89–7.81 (m, 1H), 4.47–4.12 (m, 2H), 2.82–2.72 (m, 2H), 2.49–2.44 (m, 4H), 1.70–1.60 (m, 4H). LCMS tR = 0.437 min in 2.5 min chromatography, MS ESI calcd. for C9H15N4O2+ [M + H]+ 211.12, found 211.3.
1-(2-(Pyrrolidin-1-yl)ethyl)-1H-imidazol-4-amine (160)
To a solution of 4-nitro-1-(2-(pyrrolidin-1-yl)ethyl)-1H-imidazole (154) (420 mg, 2.00 mmol) in MeOH (8 mL) was added Pd/C (0.33 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 20 °C for 2 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (10 mL × 3), and then the combined filtrate was concentrated to dryness. 160 (412 mg, crude) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 7.35–7.08 (m, 1H), 6.22–5.98 (m, 1H), 4.44–3.80 (m, 4H), 3.17 (s, 1H), 2.67–2.63 (m, 1H), 2.48–2.27 (m, 4H), 1.74–1.56 (m, 4H). LCMS tR = 0.612 min in 1 min chromatography, MS ESI calcd. for C9H17N4+ [M + H]+ 181.14, found 181.1.
5-Chloro-7-((1-(2-(pyrrolidin-1-yl)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (166)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (400 mg, 1.11 mmol) in EtOH (12 mL) was added 1-(2-(pyrrolidin-1-yl)ethyl)-1H-imidazol-4-amine (160) (406 mg, 2.25 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–9%, MeOH/DCM). 166 (185 mg, 24.7%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 7.80 (s, 1H), 7.45 (s, 1H), 7.27 (s, 1H), 4.41 (s, 2H), 3.64–3.46 (m, 4H), 3.17 (s, 1H), 3.08–2.94 (m, 2H), 2.02–1.81 (m, 4H). LCMS tR = 1.039 min in 2.5 min chromatography, MS ESI calcd. for C16H18ClN8+ [M + H]+ 357.13, found 357.2.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-(2-(pyrrolidin-1-yl)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (41)
To a solution of 5-chloro-7-((1-(2-(pyrrolidin-1-yl)ethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (166) (60 mg, 0.17 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (28 mg, 0.17 mmol) in dioxane (3 mL) was added tBuOLi (40 mg, 0.50 mmol), BINAP (16 mg, 0.03 mmol), and Pd(OAc)2 (6 mg, 0.03 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 2 h. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water(NH3H2O+NH4HCO3)-ACN]; B%: 10–50%, 35 min). 41 (6.7 mg, 7.8%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 9.98 (s, 1H), 9.01 (s, 2H), 8.48 (s, 1H), 8.38 (t, J = 2.2 Hz, 1H), 7.67 (d, J = 1.5 Hz, 1H), 7.62 (dd, J = 7.9, 2.1 Hz, 1H), 7.50 (t, J = 8.1 Hz, 1H), 7.30 (dd, J = 7.8, 2.2 Hz, 1H), 7.14 (d, J = 1.5 Hz, 1H), 6.80 (s, 1H), 4.09 (t, J = 6.4 Hz, 2H), 2.76 (t, J = 6.5 Hz, 2H), 1.77–1.61 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 156.69, 150.77, 145.02, 144.88, 141.92, 141.22, 136.58, 134.70, 134.14, 130.27, 118.56, 114.66, 114.53, 111.81, 109.74, 78.84, 77.19, 55.85, 53.47, 45.66, 23.15. HPLC tR = 2.736 min in 8 min chromatography, purity 94.0%. LCMS tR = 1.591 min in 4 min chromatography, MS ESI calcd. for C24H25N12+ [M + H]+ 481.23, found 481.3.
4-(2-(4-Nitro-1H-imidazol-1-yl)ethyl)morpholine (155)
To a solution of 4-nitro-1H-imidazole (139) (2 g, 17.69 mmol) and 4-(2-chloroethyl)morpholine (3.18 g, 21.22 mmol) in MeCN (20 mL) was added K2CO3 (7.33 g, 53.06 mmol) at 25 °C. The mixture was stirred at 100 °C for 5 h. The reaction mixture was concentrated. Water (50 mL) was added. The resulting mixture was extracted with DCM (50 mL × 3). The combined organic phase was washed with brine (30 mL) and water (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Luna C18 250 × 50 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 0–40%, 20 min). 155 (1.2 g, 30.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (d, J = 1.6 Hz, 1H), 7.85 (d, J = 1.2 Hz, 1H), 4.18 (t, J = 6.0 Hz, 2H), 3.57–3.49 (m, 4H), 2.66 (t, J = 6.0 Hz, 2H), 2.46–2.35 (m, 4H). LCMS tR = 0.160 min in 1.5 min chromatography, MS ESI calcd. for C9H15N4O3+ [M + H]+ 227.11, found 227.0.
1-(2-Morpholinoethyl)-1H-imidazol-4-amine (161)
To a solution of 4-(2-(4-nitro-1H-imidazol-1-yl)ethyl)morpholine (155) (1.00 g, 4.42 mmol) in MeOH (15 mL) was added Pd/C (0.84 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 35 °C for 4 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (10 mL × 3), and then the combined filtrate was concentrated to dryness. 161 (870 mg, 99.9%) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.17–7.10 (m, 1H), 6.20–6.15 (m, 1H), 4.33–3.91 (m, 2H), 3.87 (t, J = 6.4 Hz, 2H), 3.56–3.52 (m, 4H), 2.53 (t, J = 6.4 Hz, 2H), 2.40–2.34 (m, 4H). LCMS tR = 0.141 min in 1.5 min chromatography, MS ESI calcd. for C9H17N4O+ [M + H]+ 197.14, found 197.1.
tert-Butyl 5-chloro-7-((1-(2-morpholinoethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (167)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (785 mg, 3.69 mmol) in EtOH (20 mL) was added 1-(2-morpholinoethyl)-1H-imidazol-4-amine (161) (868 mg, 4.42 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 4 h. The reaction mixture was filtered. The filter cake was washed with EtOH (20 mL × 2), and then the combined filtrate cake was concentrated to dryness. 167 (1.4 g, 97.1%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 11.22–10.87 (m, 1H), 8.77 (s, 1H), 7.80 (s, 1H), 7.42 (s, 1H), 7.31–7.25 (m, 1H), 4.48 (s, 2H), 3.84 (s, 4H), 3.47–3.42 (m, 2H), 3.21–2.93 (m, 4H). LCMS tR = 0.673 min in 1.5 min chromatography, MS ESI calcd. for C16H18ClN8O+ [M + H]+ 373.13, found 373.1.
tert-Butyl 5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-((1-(2-morpholinoethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (42)
To a solution of tert-butyl 5-chloro-7-((1-(2-morpholinoethyl)-1H-imidazol-4-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (167) (500 mg, 1.34 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (258 mg, 1.61 mmol) in dioxane (8 mL) was added Cs2CO3 (1.75 g, 5.36 mmol) and Brettphos Pd G3 (182 mg, 0.20 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 4 h. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 78%, MeOH/DCM) to give the product. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 0–90%, 14 min). The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (HCl)-ACN]; B%: 0–34%, 30 min). 42 as a HCl salt (20 mg, 3.0%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.65 (br s, 1H), 10.65 (d, J = 7.2 Hz, 1H), 10.37 (s, 1H), 9.40–9.17 (m, 2H), 8.61–8.53 (m, 1H), 8.53 (s, 1H), 8.48 (t, J = 2.1 Hz, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.65 (s, 1H), 7.54 (t, J = 8.1 Hz, 1H), 7.35 (dd, J = 7.9, 2.2 Hz, 1H), 6.94 (s, 1H), 4.67 (t, J = 6.5 Hz, 2H), 4.11–3.73 (m, 6H), 3.53 (br s, 2H), 3.20 (br s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.89, 150.93, 145.53, 141.80, 141.73, 138.02, 134.75, 133.57, 130.42, 119.22, 119.18, 115.43, 114.53, 112.23, 112.18, 79.98, 77.81, 63.05, 54.62, 51.38, 42.49. HPLC tR = 2.721 min in 8 min chromatography, purity 99.7%. LCMS tR = 1.302 min in 4 min chromatography, MS ESI calcd. for C24H25N12O+ [M + H]+ 497.23, found 497.4.
tert-Butyl (2-(3-nitro-1H-pyrazol-1-yl)ethyl)carbamate (145)
To a solution of 3-nitro-1H-pyrazole (140) (5 g, 44.22 mmol) and tert-butyl (2-bromoethyl)carbamate (11.89 g, 53.06 mmol) in DMF (50 mL) was added K2CO3 (18.33 g, 132.66 mmol) at 25 °C. The mixture was stirred at 90 °C for 3 h. The reaction mixture was poured into ice water (100 mL) slowly. The resulting suspension was filtered. The filter cake was then dried. 145 (7.77 g, 53.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.02–7.92 (m, 1H), 7.13–6.90 (m, 2H), 4.25 (t, J = 6.0 Hz, 2H), 3.41–3.35 (m, 2H), 1.32 (s, 9H). LCMS tR = 0.465 min in 1 min chromatography, MS ESI calcd. for C5H9N4O2+ [M + 2H-Boc]+ 157.07, found 157.0.
tert-Butyl methyl(2-(3-nitro-1H-pyrazol-1-yl)ethyl)carbamate (146)
To a solution of tert-butyl (2-(3-nitro-1H-pyrazol-1-yl)ethyl)carbamate (145) (2 g, 7.80 mmol) in THF (15 mL) was added NaH (468 mg, 60% in mineral oil) at 0 °C for 0.5 h. Then CH3I (1.11 g, 7.80 mmol) was added to the mixture. The mixture was stirred at 25 °C for 10 h. The reaction was quenched with aq. NH4Cl (10 mL) dropwise. Water (50 mL) was added. The resulting mixture was extracted with EtOAc (50 mL × 3). The combined organic phase was washed with brine (50 mL × 2), dried over anhydrous Na2SO4, filtered, and concentrated. 146 (2.13 g, 98.3%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.99 (d, J = 2.4 Hz, 1H), 7.11–6.98 (m, 1H), 4.35 (t, J = 5.6 Hz, 2H), 3.65–3.53 (m, 2H), 2.78–2.68 (m, 3H), 1.32–1.16 (m, 9H). LCMS tR = 1.227 min in 2 min chromatography, MS ESI calcd. for C6H11N4O2+ [M + 2H-Boc]+ 171.09, found 171.3.
tert-Butyl (2-(3-amino-1H-pyrazol-1-yl)ethyl)(methyl)carbamate (148)
To a solution of tert-butyl methyl(2-(3-nitro-1H-pyrazol-1-yl)ethyl)carbamate (146) (2 g, 7.40 mmol) in MeOH (20 mL) was added Pd/C (2.38 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 35 °C for 4 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (20 mL × 3), and then the combined filtrate was concentrated to dryness. 148 (1.56 g, 86.0%) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.20 (s, 1H), 5.37 (d, J = 2.4 Hz, 1H), 4.52 (s, 2H), 3.90 (t, J = 6.0 Hz, 3H), 3.41 (t, J = 6.0 Hz, 2H), 2.59 (s, 3H), 1.42–1.30 (m, 9H). LCMS tR = 0.643 min in 1.5 min chromatography, MS ESI calcd. for C11H21N4O2+ [M + H]+ 241.17, found 241.1.
tert-Butyl (2-(3-((5-chloro-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-pyrazol-1-yl)ethyl)(methyl)carbamate (150)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (1.1 g, 5.16 mmol) in EtOH (20 mL) was added tert-butyl (2-(3-amino-1H-pyrazol-1-yl)ethyl)(methyl)carbamate (148) (1.49 g, 6.20 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 4 h. The reaction mixture was concentrated. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 32%, EtOAC/PE) to give the product. 150 (1.18 g, 45.5%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 11.33–11.12 (m, 1H), 8.79 (s, 1H), 7.77–7.57 (m, 2H), 6.38–6.24 (m, 1H), 4.25 (t, J = 5.6 Hz, 2H), 3.56 (t, J = 5.6 Hz, 2H), 2.72 (s, 3H), 1.33–1.24 (m, 9H) LCMS tR = 1.665 min in 2.5 min chromatography, MS ESI calcd. for C18H22ClN8O2+ [M + H]+ 417.15, found 417.1.
tert-Butyl (2-(3-((5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-pyrazol-1-yl)ethyl)(methyl)carbamate (152)
To a solution of tert-butyl (2-(3-((5-chloro-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-pyrazol-1-yl)ethyl)(methyl)carbamate (150) (500 mg, 1.20 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (231 mg, 1.44 mmol) in dioxane (8 mL) was added Cs2CO3 (1.56 g, 4.80 mmol), BINAP (112 mg, 0.18 mmol), and Pd(OAc)2 (40 mg, 0.18 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 4 h. The reaction mixture was concentrated to dryness. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 4%, MeOH/DCM) to give the product. 152 (501 mg, 57.8%) was obtained as a gray solid. 1H NMR (400 MHz, DMSO-d6) δ 10.40–10.29 (m, 1H), 10.11–9.94 (m, 1H), 9.07–8.98 (m, 2H), 8.51 (s, 1H), 8.40–8.31 (m, 1H), 7.68–7.63 (m, 2H), 7.54 (t, J = 8.4 Hz 1H), 7.42–7.29 (m, 2H), 7.27–7.08 (m, 2H), 4.23–4.15 (m, 2H), 3.62–3.52 (m, 2H), 2.66–2.58 (m, 3H), 1.37–1.28 (m, 9H). LCMS tR = 1.572 min in 2.5 min chromatography, MS ESI calcd. for C26H29N12O2+ [M + H]+ 541.25, found 541.2.
tert-Butyl 5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-((1-(2-(methylamino)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (43)
To a solution of tert-butyl (2-(3-((5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)amino)-1H-pyrazol-1-yl)ethyl)(methyl)carbamate (152) (250 mg, 1.20 mmol) in DCM (3 mL) was added TFA (5.98 g 52.44 mmol) at 25 °C. The mixture was stirred at 35 °C for 6 h. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (HCl)-ACN]; B%: 0–90%, 36 min). 43 as a HCl salt (62 mg, 29.3%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (d, J = 4.8 Hz, 1H), 10.44 (s, 1H), 9.47–9.34 (m, 2H), 9.16 (s, 2H), 8.62 (q, J = 2.0 Hz, 1H), 8.52 (s, 1H), 7.90 (dd, J = 8.3, 2.2 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 7.62 (d, J = 1.8 Hz, 1H), 7.56 (t, J = 8.1 Hz, 1H), 7.36 (dd, J = 8.0, 2.2 Hz, 1H), 6.31 (d, J = 2.3 Hz, 1H), 4.46 (t, J = 5.9 Hz, 2H), 3.51 (p, J = 6.0 Hz, 2H), 2.58 (t, J = 5.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 157.11, 150.67, 147.75, 144.90, 142.87, 142.07, 141.52, 133.60, 131.73, 130.21, 119.14, 115.00, 114.60, 97.57, 81.58, 77.53, 48.00, 47.60, 33.10. HPLC tR = 2.964 min in 8 min chromatography, purity 96.3%. LCMS tR = 1.422 min in 4 min chromatography, MS ESI calcd. for C21H21N12+ [M + H]+ 441.20, found 441.4.
N,N-Dimethyl-2-(3-nitro-1H-pyrazol-1-yl)ethan-1-amine (156)
To a solution of 3-nitro-1H-pyrazole (140) (1 g, 8.84 mmol) in DMF (10 mL) was added NaH (530.57 mg, 60% in mineral oil) in 0 °C for 0.5 h, then 2-chloro-N,N-dimethylethan-1-amine hydrochloride (1.27 g, 8.84 mmol) was added and the mixture was stirred at 15 °C for 3.5 h. The reaction was quenched with aq. NH4Cl (50 mL) dropwise. The resulting mixture was extracted with EtOAc (20 mL × 3). The combined organic phase was washed with brine (50 mL) and water (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated. 156 (1.72 g, 92.0%) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (s, 1H), 7.03 (s, 1H), 4.33 (t, J = 6.4 Hz, 2H), 2.73 (s, 2H), 2.18 (s, 6H). LCMS tR = 0.857 min in 2 min chromatography, MS ESI calcd. for C7H13N4O2+ [M + H]+ 185.10, found 185.1.
1-(2-(Dimethylamino)ethyl)-1H-pyrazol-3-amine (162)
To a solution of N,N-dimethyl-2-(3-nitro-1H-pyrazol-1-yl)ethan-1-amine (156) (1.6 g, 8.69 mmol) in MeOH (10 mL) was added Pd/C (1.02 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 35 °C for 2 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (20 mL × 3), and then the combined filtrate was concentrated to dryness. 162 (1.34 g, 100%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.24–7.23 (m, 2H), 5.37–5.36 (m, 2H), 2.88 (s, 1H), 2.73 (s, 1H), 2.59 (t, J = 6.8 Hz, 2H), 2.17 (s, 6H). LCMS tR = 0.118 min in 1 min chromatography, MS ESI calcd. for C7H15N4+ [M + H]+ 155.13, found 155.0.
5-Chloro-7-((1-(2-(dimethylamino)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (168)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (1.05 g, 4.93 mmol) in EtOH (10 mL) was added 1-(2-(dimethylamino)ethyl)-1H-pyrazol-3-amine (162) (912 mg, 5.91 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Ultimate XB-CN 250 × 50 × 10 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 10–45%, 15 min). The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water(NH3H2O+NH4HCO3)-ACN]; B%: 8–48%, 35 min). 168 (266 mg, 11.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 8.79 (s, 1H), 7.77–7.75 (m, 1H), 7.54 (s, 1H), 6.27 (d, J = 2.4 Hz, 1H), 4.22 (t, J = 6.4 Hz, 2H), 2.68 (t, J = 6.4 Hz, 2H), 2.19 (s, 6H). LCMS tR = 1.447 min in 2.5 min chromatography, MS ESI calcd. for C14H16ClN8+ [M + H]+ 331.11, found 331.1.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-(2-(dimethylamino)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (44)
To a solution of 5-chloro-7-((1-(2-(dimethylamino)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (168) (200 mg, 0.60 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (145 mg, 0.91 mmol) in dioxane (8 mL) was added Cs2CO3 (591 mg, 1.81 mmol), BINAP (56 mg, 0.09 mmol), and Pd(OAc)2 (20 mg, 0.09 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 2 h. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water(HCl)-ACN]; B%: 7–37%, 10 min). 44 as a HCl salt (63.5 mg, 22.7%) was obtained as a yellow solid. 1H NMR (400 MHz, D2O) δ 7.94 (s, 2H), 7.78 (s, 1H), 7.31 (s, 1H), 7.16 (s, 1H), 6.50 (s, 1H), 6.30 (s, 1H), 5.99 (s, 1H), 5.94 (s, 1H), 5.51 (s, 1H), 4.19 (s, 2H), 3.53 (s, 2H), 2.82 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 157.15, 157.11, 150.80, 147.70, 145.13, 143.00, 142.18, 141.99, 133.12, 132.21, 130.44, 120.04, 115.57, 114.83, 112.82, 97.85, 77.69, 55.27, 46.49, 42.83. HPLC tR = 2.981 min in 8 min chromatography, purity 98.3%. LCMS tR = 1.528 min in 4 min chromatography, MS ESI calcd. for C22H23N12+ [M + H]+ 455.22, found 455.4.
3-Nitro-1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazole (157)
To a solution of 3-nitro-1H-pyrazole (140) (500 mg, 4.42 mmol) and 1-(2-chloroethyl)pyrrolidine (1.13 g, 6.63 mmol) in MeCN (10 mL) was added K2CO3 (1.83 g, 13.27 mmol) at 25 °C. The mixture was stirred at 120 °C for 4 h. The reaction mixture was concentrated. Water (50 mL) was added. The resulting mixture was extracted with DCM (50 mL × 3). The combined organic phase was washed with brine (30 mL) and water (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. 157 (234 mg, 14.2%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 2.0 Hz, 1H), 7.03 (d, J = 2.0 Hz, 1H), 4.33 (t, J = 6.4 Hz, 2H), 2.85 (t, J = 6.4 Hz, 2H), 2.48–2.45 (m, 4H), 1.66–1.63 (m, 4H). LCMS tR = 1.326 min in 2.5 min chromatography, MS ESI calcd. for C9H15N4O2+ [M + H]+ 211.11, found 211.1.
1-(2-(Pyrrolidin-1-yl)ethyl)-1H-pyrazol-3-amine (163)
To a solution of 3-nitro-1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazole (157) (200 mg, 0.95 mmol) in MeOH (5 mL) was added Pd/C (0.4 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 5 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (10 mL × 3), and then the combined filtrate was concentrated to dryness. 163 (99 mg, 49.3%) was obtained as an off-white oil. 1H NMR (400 MHz, DMSO-d6) δ 7.29 (d, J = 2.0 Hz, 1H), 5.33 (d, J = 2.0 Hz, 1H), 4.48 (s, 2H), 3.90 (t, J = 6.8 Hz, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.44–2.39 (m, 4H), 1.66–1.62 (m, 4H). LCMS tR = 0.131 min in 1 min chromatography, MS ESI calcd. for C9H17N4+ [M + H]+ 181.14, found 181.1.
tert-Butyl 5-chloro-7-((1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (169)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (105 mg, 0.49 mmol) in EtOH (5 mL) was added 1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazol-3-amine (163) (98 mg, 0.54 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 5 h. The reaction mixture was filtered. The filter cake was washed with EtOH (10 mL × 3), and then the combined filtrate cake was concentrated. 169 (178 mg, 93.4%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.39–10.84 (m, 2H), 8.82 (s, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.60 (s, 1H), 6.39 (d, J = 2.4 Hz, 1H), 4.59 (t, J = 6.4 Hz, 2H), 3.63 (t, J = 6.0 Hz, 2H), 3.44 (q, J = 7.0 Hz, 2H), 1.91 (s, 4H), 1.05 (t, J = 7.2 Hz, 2H). LCMS tR = 0.844 min in 2 min chromatography, MS ESI calcd. for C16H18ClN8+ [M + H]+ 357.13, found 357.2.
tert-Butyl 5-((3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-((1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (45)
To a solution of tert-butyl 5-chloro-7-((1-(2-(pyrrolidin-1-yl)ethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (169) (95 mg, 0.27 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (51 mg, 0.32 mmol) in dioxane (5 mL) was added Cs2CO3 (347 mg, 1.06 mmol), BINAP (25 mg, 0.04 mmol), and Pd(OAc)2 (9 mg, 0.04 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 4 h. The residue was purified by flash silica gel chromatography (MeOH) to give the product. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (HCl)-ACN]; B%: 0–90%, 36 min). 45 as a HCl salt (28.3 mg, 21.9%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.12–10.86 (m, 2H), 10.42 (s, 1H), 9.50 (s, 2H), 8.65 (t, J = 2.2 Hz, 1H), 8.51 (s, 1H), 7.98 (dd, J = 8.2, 2.1 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.68–7.47 (m, 2H), 7.36 (dd, J = 8.0, 2.2 Hz, 1H), 6.30 (d, J = 2.3 Hz, 1H), 4.55 (t, J = 6.3 Hz, 2H), 3.91 (q, J = 6.0 Hz, 2H), 3.44 (td, J = 10.4, 5.1 Hz, 2H), 3.14–3.00 (m, 2H), 2.04–1.91 (m, 2H), 1.91–1.74 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 157.16, 150.67, 147.65, 144.89, 142.92, 142.23, 141.74, 133.13, 131.97, 130.15, 119.73, 115.25, 114.63, 112.59, 97.72, 81.37, 77.52, 53.42, 52.83, 47.55, 22.69. HPLC tR = 3.109 min in 8 min chromatography, purity 98.7%. LCMS tR = 1.504 min in 4 min chromatography, MS ESI calcd. for C24H25N12+ [M + H]+ 481.23, found 481.5.
4-(2-(3-Nitro-1H-pyrazol-1-yl)ethyl)morpholine (158)
To a solution of 3-nitro-1H-pyrazole (140) (1.5 g, 13.27 mmol) and 4-(2-chloroethyl)morpholine (2.98 g, 19.90 mmol) in MeCN (20 mL) was added K2CO3 (1.83 g, 13.27 mmol) at 25 °C. The mixture was stirred at 100 °C for 4 h. The reaction mixture was concentrated. Water (50 mL) was added. The resulting mixture was extracted with DCM (50 mL × 3). The combined organic phase was washed with brine (20 mL) and water (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Luna C18 250 × 50 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 0–30%, 15 min). 158 (912 mg, 29.8%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.04 (d, J = 2.4 Hz, 1H), 7.03 (d, J = 2.4 Hz, 1H), 4.35 (t, J = 6.4 Hz, 2H), 3.55–3.50 (m, 4H), 2.74 (t, J = 6.4 Hz, 2H), 2.44–2.38 (m, 4H). LCMS tR = 1.080 min in 2.5 min chromatography, MS ESI calcd. for C9H15N4O3+ [M + H]+ 227.11, found 227.2.
1-(2-Morpholinoethyl)-1H-pyrazol-3-amine (164)
To a solution of 4-(2-(3-nitro-1H-pyrazol-1-yl)ethyl)morpholine (158) (730 mg, 3.23 mmol) in MeOH (15 mL) was added Pd/C (0.84 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 35 °C for 4 h. The reaction mixture was filtered via a Celite pad. The reaction mixture was filtered. The filter cake was washed with MeOH (10 mL × 3), and then the combined filtrate was concentrated to dryness. 164 (815 mg, 99.0%) was obtained as an off-white oil. 1H NMR (400 MHz, DMSO-d6) δ 7.30 (s, 1H), 5.34 (s, 1H), 4.50 (s, 2H), 3.92 (t, J = 6.8 Hz, 2H), 3.53 (t, J = 4.4 Hz, 4H), 2.59 (t, J = 6.8 Hz, 2H), 2.41–2.31 (m, 4H). LCMS tR = 0.733 min in 2.5 min chromatography, MS ESI calcd. for C9H17N4O+ [M + H]+ 197.14, found 197.3.
5-Chloro-7-((1-(2-morpholinoethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (170)
To a solution of 5,7-dichloropyrazolo[1,5-a]pyrimidine-3-carbonitrile (74) (730 mg, 3.43 mmol) in EtOH (20 mL) was added 1-(2-morpholinoethyl)-1H-pyrazol-3-amine (164) (807 mg, 4.11 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 5 h. The reaction mixture was concentrated. The resulting mixture was triturated by H2O (20 mL). The mixture was filtered. The filter cake was washed with EtOH (30 mL × 2) and then dried. 170 (1.34 g, 100.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.27 (s, 2H), 8.81 (s, 1H), 7.85 (s, 1H), 7.57 (s, 1H), 6.36 (s, 1H), 4.94–3.42 (m, 10H), 3.21–2.95 (m, 2H). LCMS tR = 1.452 min in 2.5 min chromatography, MS ESI calcd. for C16H18ClN8O+ [M + H]+ 373.13, found 373.2.
5-((3-(4H-1,2,4-Triazol-4-yl)phenyl)amino)-7-((1-(2-morpholinoethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (46)
To a solution of 5-chloro-7-((1-(2-morpholinoethyl)-1H-pyrazol-3-yl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (170) (300 mg, 0.80 mmol) and 3-(4H-1,2,4-triazol-4-yl)aniline (86) (155 mg, 0.97 mmol) in dioxane (8 mL) was added Cs2CO3 (1.05 g, 3.22 mmol), BINAP (75 mg, 0.12 mmol), and Pd(OAc)2 (27 mg, 0.12 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 6 h. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 48–68%, 36 min). The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (HCl)-ACN]; B%: 0–50%, 10 min). 46 as a HCl salt (26.3 mg, 3.2%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 10.74 (s, 1H), 10.43 (s, 1H), 9.09 (s, 2H), 8.54 (d, J = 18.1 Hz, 2H), 7.91–7.79 (m, 2H), 7.54 (t, J = 8.2 Hz, 1H), 7.46 (s, 1H), 7.33 (d, J = 8.0 Hz, 1H), 6.32 (s, 1H), 4.60 (t, J = 6.6 Hz, 2H), 4.02–3.90 (m, 2H), 3.87–3.74 (m, 4H), 3.44 (d, J = 16.9 Hz, 2H), 3.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 157.08, 150.73, 147.75, 144.94, 143.02, 142.00, 141.32, 134.03, 131.93, 130.23, 118.79, 114.79, 114.59, 112.00, 97.94, 81.21, 77.52, 63.19, 54.86, 51.59, 45.77. HPLC tR = 3.013 min in 8 min chromatography, purity 96.4%. LCMS tR = 1.477 min in 4 min chromatography, MS ESI calcd. for C24H25N12O+ [M + H]+ 497.23, found 497.5.
Experimental Procedures in Scheme 5
4-(4-Fluoro-3-nitrophenyl)-4H-1,2,4-triazole (175)
To a solution of 4-fluoro-3-nitroaniline (171) (1.00 g, 6.41 mmol) and 1,2-diformylhydrazine (1.69 g, 19.22 mmol) in pyridine (10 mL) was added Et3N (4.54 g, 44.84 mmol) and Me3SiCl (10.44 g, 96.05 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated in vacuo. Brine (50 mL) was added. The resulting mixture was extracted with DCM (50 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was purified by flash silica gel chromatography (eluent of 0–3%, MeOH/DCM). 175 (667 mg, 49.7%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.22 (s, 2H), 8.61–8.56 (m, 1H), 8.22–8.16 (m, 1H), 7.89–7.82 (m, 1H). LCMS tR = 0.364 min in 1 min chromatography, MS ESI calcd. for C8H6FN4O2+ [M + H]+ 209.05, found 208.9.
2-Fluoro-5-(4H-1,2,4-triazol-4-yl)aniline (179)
To a solution of 4-(4-fluoro-3-nitrophenyl)-4H-1,2,4-triazole (175) (300 mg, 3.28 mmol) in MeOH (10 mL) was added Pd/C (0.60 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (15 mL × 3). The combined filtrate was concentrated in vacuo. 179 (252 mg, 80.0%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 2H), 7.13–7.07 (m, 1H), 6.87 (dd, J = 2.8, 7.6 Hz, 1H), 6.70–6.65 (m, 1H), 5.50 (s, 2H). LCMS tR = 0.644 min in 1 min chromatography, MS ESI calcd. for C8H8FN4+ [M + H]+ 197.07, found 197.0.
7-(Cyclopropylamino)-5-((2-fluoro-5-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (47)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (250 mg, 1.40 mmol) and 2-fluoro-5-(4H-1,2,4-triazol-4-yl)aniline (179) (300 mg, 1.28 mmol) in dioxane (3 mL) was added Brettphos Pd G3 (175 mg, 0.19 mmol) and tBuOLi (308 mg, 3.85 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of MeOH). The crude product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 10–50%, 28 min). The residue was purified by SFC (column: DAICEL CHIRALPAK IC (250 mm × 30 mm, 10 μm); mobile phase: [0.1%NH3H2O-ETOH]; B%: 60–60%, 90 min). 47 (35.6 mg, 55.6%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.68 (s, 1H), 8.99 (s, 2H), 8.80 (d, J = 6.9 Hz, 1H), 8.40 (s, 2H), 7.51 (t, J = 9.8 Hz, 1H), 7.41 (dt, J = 8.4, 3.5 Hz, 1H), 6.34 (s, 1H), 2.67–2.57 (m, 1H), 0.87–0.78 (m, 2H), 0.78–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.66, 152.33 (d, J = 245.5 Hz), 150.60, 148.71, 145.03, 141.33, 129.99 (d, J = 3.4 Hz), 128.95 (d, J = 12.5 Hz), 116.50 (d, J = 21.8 Hz), 116.07, 115.99, 114.64, 77.11, 76.98, 23.42, 6.58. HPLC tR = 3.582 min in 8 min chromatography, purity 98.5%. LCMS tR = 1.978 min in 4 min chromatography, MS ESI calcd. for C18H15FN9+ [M + H]+ 376.14, found 376.3.
4-(2-Fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176)
To a solution of 2-fluoro-5-nitroaniline (172) (5.00 g, 32.03 mmol) and 1,2-diformylhydrazine (8.46 g, 96.08 mmol) in pyridine (15 mL) was added Et3N (22.69 g, 224.19 mmol, 31 mL) and Me3SiCl (52.19 g, 480.42 mmol, 61 mL) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 1% MeOH/DCM). 176 (4.92g, 73.8%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (dd, J = 2.8, 6.4 Hz, 1H), 8.61 (br d, J = 4.4 Hz, 1H), 8.46–8.40 (m, 1H), 7.87–7.82 (m, 1H), 7.48–7.43 (m, 1H). LCMS tR = 0.358 min in 4 min chromatography, MS ESI calcd. for C8H6FN4O2+ [M + H]+ 209.05, found 209.0.
4-Fluoro-3-(4H-1,2,4-triazol-4-yl)aniline (180)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (1 g, 4.80 mmol) in MeOH (3 mL) was added Pd/C (220 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (400 mL). The combined filtrate was concentrated in vacuo. 180 (157 mg, 17.1%) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (d, J = 1.6 Hz, 2H), 7.15 (dd, J = 8.8, 10.4 Hz, 1H), 6.75–6.60 (m, 2H), 6.14–5.87 (m, 2H). LCMS tR = 0.229 min in 1 min chromatography, MS ESI calcd. for C8H8FN4+ [M + H]+ 179.07, found 179.1.
7-(Cyclopropylamino)-5-((4-fluoro-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (48)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (150 mg, 0.64 mmol) and 4-fluoro-3-(4H-1,2,4-triazol-4-yl)aniline (180) (158 mg, 0.64 mmol) in dioxane (2 mL) was added Cs2CO3 (628 mg, 1.93 mmol), BINAP (60 mg, 0.10 mmol), and Pd(OAc)2 (22 mg, 0.10 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–4%, MeOH/DCM). The crude product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 23–53%, 10 min). 48 (14.3 mg, 10.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 8.93 (d, J = 1.5 Hz, 2H), 8.39 (s, 1H), 8.37 (s, 1H), 8.26 (dd, J = 6.9, 2.7 Hz, 1H), 7.73 (ddd, J = 9.1, 4.2, 2.7 Hz, 1H), 7.53 (dd, J = 10.3, 9.1 Hz, 1H), 5.98 (s, 1H), 2.62 (tt, J = 6.9, 3.6 Hz, 1H), 0.86–0.80 (m, 2H), 0.75–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.44, 150.69, 149.33 (d, J = 244.2 Hz), 148.53, 145.05, 142.64 (d, J = 2.4 Hz), 137.59 (d, J = 2.7 Hz), 121.53 (d, J = 12.9 Hz), 120.56 (d, J = 7.2 Hz), 117.18 (d, J = 20.8 Hz), 116.14, 114.57, 76.91, 76.48, 23.33, 6.53. HPLC tR = 3.904 min in 8 min chromatography, purity 96.6%. LCMS tR = 2.095 min in 4 min chromatography, MS ESI calcd. for C18H15FN9+ [M + H]+ 376.14, found 376.3.
2-Fluoro-4-(4H-1,2,4-triazol-4-yl)pyridine (177)
To a solution of 2-fluoropyridin-4-amine (173) (300 mg, 2.68 mmol) and 1,2-diformylhydrazine (707 mg, 8.03 mmol) in pyridine (5 mL) was added Me3SiCl (4.36 g, 40.14 mmol) and Et3N (1.90 g, 18.73 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 2%, MeOH/DCM). 177 (312 mg, 69.7%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.40 (s, 2H), 8.43 (d, J = 5.6 Hz, 1H), 7.87–7.81 (m, 1H), 7.78–7.75 (m, 1H). LCMS tR = 0.465 min in 2.5 min chromatography, MS ESI calcd. for C7H6FN4+ [M + H]+ 165.06, found 165.2.
4-(4H-1,2,4-Triazol-4-yl)pyridin-2-amine (181)
A mixture of 2-fluoro-4-(4H-1,2,4-triazol-4-yl)pyridine (177) (1.00 g, 6.09 mmol) in concentrated aqueous NH3 (15 mL, 25–28% v/v) was degassed and purged with N2 three times, and then the mixture was stirred at 100 °C for 72 h under N2 atmosphere. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (FA)-ACN]; B%: 0–25%, 25 min). 181 (80 mg, 7.85%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.15 (s, 2H), 8.14 (s, 1H), 8.04 (d, J = 5.6 Hz, 1H), 6.85 (dd, J = 2.0, 5.6 Hz, 1H), 6.65–6.62 (m, 1H), 6.33 (s, 2H). LCMS tR = 0.242 min in 2.5 min chromatography, MS ESI calcd. for C7H8N5+ [M + H]+ 162.08, found 162.1.
5-((4-(4H-1,2,4-Triazol-4-yl)pyridin-2-yl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (49)
To a solution of 4-(4H-1,2,4-triazol-4-yl)pyridin-2-amine (181) (50 mg, 0.31 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (40 mg, 0.17 mmol) in dioxane (3 mL) was added tBuOLi (41 mg, 0.51 mmol), BINAP (16 mg, 0.03 mmol), and Pd(OAc)2 (6 mg, 0.03 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Boston Green ODS 150 × 30 mm × 5 μm; mobile phase: [water (NH4HCO3)-MeOH]; B%: 30–90%, 35 min). 49 (18 mg, 28.9%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 9.16 (s, 2H), 8.57 (d, J = 2.1 Hz, 1H), 8.53 (d, J = 1.8 Hz, 1H), 8.47 (d, J = 5.6 Hz, 1H), 8.46 (s, 1H), 7.43 (dd, J = 5.5, 2.1 Hz, 1H), 6.81 (s, 1H), 2.66–2.59 (m, 1H), 0.90–0.83 (m, 2H), 0.77–0.70 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.72, 154.91, 150.53, 149.92, 148.94, 145.18, 141.65, 140.62, 114.68, 109.02, 103.71, 77.76, 77.03, 23.50, 6.57. HPLC tR = 3.059 min in 8 min chromatography, purity 98.5%. LCMS tR = 1.739 min in 4 min chromatography, MS ESI calcd. for C17H15N10+ [M + H]+ 359.15, found 359.2.
4-Nitro-2-(4H-1,2,4-triazol-4-yl)pyridine (178)
To a solution of 4-nitropyridin-2-amine (174) (800 mg, 5.75 mmol) and 1,2-diformylhydrazine (1.52 g, 17.25 mmol) in pyridine (8 mL) was added Et3N (4.07 g, 40.26 mmol) and Me3SiCl (9.37 g, 86.26 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–4%, MeOH/DCM). 178 (904 mg, 27.2%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 2H), 8.95 (d, J = 5.2 Hz, 1H), 8.77 (d, J = 1.6 Hz, 1H), 8.25 (s, 1H).
2-(4H-1,2,4-Triazol-4-yl)pyridin-4-amine (182)
To a solution of 4-nitro-2-(4H-1,2,4-triazol-4-yl)pyridine (178) (900 mg, 4.71 mmol) in MeOH (5 mL) was added Pd/C (500 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (50 mL). The combined filtrate was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–16%, MeOH/DCM). 182 (241 mg, 29.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 2H), 7.93 (d, J = 6.0 Hz, 1H), 6.74 (d, J = 2.0 Hz, 1H), 6.54–6.51 (m, 3H).
5-((2-(4H-1,2,4-Triazol-4-yl)pyridin-4-yl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (50)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (100 mg, 0.43 mmol) and 2-(4H-1,2,4-triazol-4-yl)pyridin-4-amine (182) (69 mg, 0.43 mmol) in dioxane (3 mL) was added tBuOLi (103 mg, 1.28 mmol, BINAP (40 mg, 0.06 mmol), and Pd(OAc)2 (14 mg, 0.06 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–6%, MeOH/DCM). Then the impure product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 22–52%, 10 min). 50 (22.6 mg, 4.8%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 9.10 (s, 2H), 8.62 (s, 1H), 8.54–8.47 (m, 2H), 8.37 (d, J = 5.7 Hz, 1H), 7.64–7.51 (m, 1H), 6.13 (s, 1H), 2.74–2.62 (m, 1H), 0.93–0.80 (m, 2H), 0.80–0.70 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 156.18, 150.42, 149.96, 149.39, 149.00, 147.18, 145.30, 140.14, 114.48, 112.63, 102.37, 77.78, 77.72, 23.41, 6.55. HPLC tR = 3.999 min in 8 min chromatography, purity 98.4%. LCMS tR = 1.383 min in 4 min chromatography, MS ESI calcd. for C17H15N10+ [M + H]+ 359.15, found 359.4.
Experimental Procedures in Schemes 6 and 7
tert-Butyl (2-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (183)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (200 mg, 0.96 mmol) and tert-butyl (2-(methylamino)ethyl)carbamate (251 mg, 1.44 mmol) in MeCN (5 mL) was added K2CO3 (531 mg, 3.84 mmol) at 25 °C. The mixture was stirred at 100 °C for 10 h under N2 atmosphere. The reaction mixture was concentrated. Water (30 mL) was added. The resulting mixture was extracted with DCM (20 mL × 3). The combined organic phase was washed with brine (20 mL) and water (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 2%, MeOH/DCM) to give the product. 183 (235 mg, 56.6%) was obtained as a black brown oil. LCMS tR = 1.368 min in 2.5 min chromatography, MS ESI calcd. for C16H23N6O4+ [M + H]+ 363.18, found 363.1.
tert-Butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (201)
To a solution of tert-butyl (2-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (183) (235 mg, 0.65 mmol) in MeOH (5 mL) was added Pd/C (0.14 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 2 h. The reaction mixture was filtered. The filter cake was washed with MeOH (30 mL). The combined filtrate was concentrated in vacuo. 201 (189 mg, 84.5%) was obtained as a black brown oil. LCMS tR = 0.912 min in 2.5 min chromatography, MS ESI calcd. for C16H25N6O2+ [M + H]+ 333.20, found 333.2.
tert-Butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (219)
To a solution of tert-butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (201) (90 mg, 0.27 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (32 mg, 0.14 mmol) in dioxane (3 mL) was added Cs2CO3 (265 mg, 0.81 mmol), BINAP (25 mg, 0.04 mmol), and Pd(OAc)2 (9 mg, 0.04 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–3%, MeOH/DCM). 219 (180 mg, 75.9%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.69 (s, 2H), 8.36 (s, 1H), 7.92–7.87 (m, 1H), 7.72–7.66 (m, 1H), 7.35–7.29 (m, 1H), 7.11–7.08 (m, 1H), 5.75 (s, 1H), 3.39–3.38 (m, 1H), 3.34–3.31 (m, 3H), 2.94–2.92 (m, 1H), 2.82–2.79 (m, 2H), 2.72–2.67 (m, 2H), 1.35 (s, 9H), 0.86–0.78 (m, 2H), 0.75–0.69 (m, 2H). LCMS tR = 0.913 min in 1.5 min chromatography, MS ESI calcd. for C26H32N11O2+ [M + H]+ 530.27, found 530.3.
5-((4-((2-Aminoethyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (51)
To a solution of tert-butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (219) (135 mg, 0.25 mmol) in DCM (5 mL) was added TFA (1 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep. HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 18–48%, 11 min). 51 (16.8 mg, 14.3%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.86 (s, 2H), 8.36 (s, 1H), 8.32 (s, 1H), 7.88 (d, J = 2.5 Hz, 1H), 7.71 (dd, J = 8.8, 2.6 Hz, 1H), 7.35 (d, J = 8.9 Hz, 1H), 5.96 (s, 1H), 3.01–2.76 (m, 2H), 2.71 (t, J = 6.6 Hz, 2H), 2.61 (tt, J = 6.8, 3.6 Hz, 1H), 2.43 (s, 3H), 0.85–0.78 (m, 2H), 0.75–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.53, 150.85, 148.39, 145.00, 143.01, 141.54, 136.21, 128.40, 122.62, 120.32, 117.15, 114.69, 76.65, 76.35, 58.11, 41.42, 39.02, 23.32, 6.53. HPLC tR = 3.157 min in 8 min chromatography, purity 92.9%. LCMS tR = 1.768 min in 4 min chromatography, MS ESI calcd. for C21H24N11+ [M + H]+ 430.22, found 430.4.
tert-Butyl (2-((4-amino-5-fluoro-2-nitrophenyl)(methyl)amino)ethyl)carbamate (232)
To a solution of 2,4-difluoro-5-nitroaniline (231) (1 g, 5.74 mmol) and tert-butyl (2-(methylamino)ethyl)carbamate (1.20 g, 6.89 mmol) in MeCN (15 mL) was added K2CO3 (2.38 g, 17.23 mmol). Then the reaction mixture was stirred at 60 °C for 12 h. The reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0–4%, MeOH/DCM). 232 (1.75 g, 83.0%) was obtained as a red solid. 1H NMR (400 MHz, DMSO-d6) δ 7.21–7.07 (m, 2H), 6.69–6.60 (m, 1H), 5.35 (s, 2H), 3.01–2.94 (m, 2H), 2.93–2.87 (m, 2H), 2.63 (s, 3H), 1.33 (s, 9H). LCMS tR = 1.295 min in 1 min chromatography, MS ESI calcd. for C14H22FN4O4+ [M + H]+ 329.16, found 329.0.
tert-Butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)(methyl)amino)ethyl)carbamate (237)
To a solution of tert-butyl (2-((4-amino-5-fluoro-2-nitrophenyl)(methyl)amino)ethyl)carbamate (232) (1.6 g, 4.87 mmol) in THF (20 mL) was added K2CO3 (2.02 g, 14.62 mmol) and CbzCl (1.25 g, 7.31 mmol) at 25 °C under N2. The mixture was stirred at 25 °C for 12 h. The reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0–23%, EtOAc/PE). 237 (1.93 g, 84.9%) was obtained as a red oil. 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H), 8.19–7.97 (m, 1H), 7.44–7.31 (m, 6H), 7.09 (d, J = 13.2 Hz, 1H), 6.78 (t, J = 5.2 Hz, 1H), 5.15 (s, 2H), 3.15–3.09 (m, 4H), 2.77 (s, 3H), 1.31 (s, 9H). LCMS tR = 0.621 min in 1 min chromatography, MS ESI calcd. for C22H28FN4O6+ [M + H]+ 463.20, found 463.2.
tert-Butyl (2-((2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)(methyl)amino)ethyl)carbamate (242)
To a solution of tert-butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)(methyl)amino)ethyl)carbamate (237) (1.70 g, 3.68 mmol) in EtOH (15 mL) was added NH4Cl (590 mg, 11.03 mmol) dissolved in H2O (3 mL) and Fe (616 mg, 11.03 mmol) at 25 °C. Then the mixture was stirred at 100 °C under N2 atmosphere for 2 h. The reaction mixture was filtered and the solid was washed with MeOH (100 mL). The combined filtrate was concentrated in vacuo. The residue was purified by prep. HPLC (column: Welch SiO2 10 μm, 250 × 300 mm; mobile phase: [heptane-EtOH(0.1%NH3H2O)]; B%: 10–40%, 10 min). 242 (418 mg, 24.7%) was obtained as a gray oil. 1H NMR (400 MHz, DMSO-d6) δ 7.45–7.31 (m, 6H), 6.91–6.75 (m, 3H), 5.11 (s, 2H), 3.42 (s, 3H), 3.19–3.15 (m, 2H), 3.08–2.99 (m, 2H), 2.81–2.74 (m, 2H), 1.37 (s, 9H). LCMS tR = 0.867 min in 1.5 min chromatography, MS ESI calcd. for C22H30FN4O4+ [M + H]+ 433.22, found 433.1.
tert-Butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (247)
To a solution of tert-butyl (2-((2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)(methyl)amino)ethyl)carbamate (242) (390 mg, 0.90 mmol) and 1,2-diformylhydrazine (237 mg, 2.7 mmol) in pyridine (5 mL) was added Et3N (639 mg, 6.31 mmol) and Me3SiCl (1.47 g, 13.53 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated. Saturated NaHCO3 (50 mL) solution was added. The resulting mixture was extracted with EtOAc (30 mL × 3). The combined organic phase was washed with brine (30 mL) and water (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. 247 (350 mg, 63.5%) was obtained as a black brown gum. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 2H), 7.44–7.30 (m, 7H), 5.14 (s, 2H), 3.35 (s, 2H), 2.96–2.87 (m, 2H), 2.77–2.69 (m, 2H), 2.43 (s, 3H), 1.33 (s, 9H). LCMS tR = 0.525 min in 1 min chromatography, MS ESI calcd. for C24H30FN6O4+ [M + H]+ 485.23, found 485.1.
tert-Butyl (2-((4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (252)
To a solution of tert-butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (247) (320 mg, 0.66 mmol) in MeOH (10 mL) was added Pd/C (0.4 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 3 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (10 mL × 3). The combine filtrate was concentrated in vacuo. The residue was purified by prep. HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (NH4HCO3)-ACN]; B%: 8–48%, 36 min). 252 (80 mg, 33.1%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (s, 2H), 7.10 (d, J = 12.8 Hz, 1H), 6.74–6.64 (m, 2H), 5.24 (s, 2H), 2.85–2.78 (m, 2H), 2.68–2.64 (m, 2H), 2.33 (s, 3H), 1.36–1.33 (m, 9H). LCMS tR = 0.449 min in 1 min chromatography, MS ESI calcd. for C16H24FN6O2+ [M + H]+ 351.19, found 351.1.
tert-Butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (257)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (70 mg, 0.20 mmol) and tert-butyl (2-((4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (252) (42 mg, 0.18 mmol) in dioxane (5 mL) was added Cs2CO3 (195 mg, 0.60 mmol), BINAP (19 mg, 0.03 mmol), and Pd(OAc)2 (7 mg, 0.03 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 6 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–3% MeOH/DCM). 257 (100 mg, 38.3%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.74–8.66 (m, 4H), 8.35 (s, 1H), 8.32 (s, 1H), 8.24–8.18 (m, 1H), 6.18 (s, 1H), 2.78–2.74 (m, 2H), 2.69–2.65 (m, 2H), 2.33 (s, 3H), 1.35 (s, 9H), 1.26–1.22 (m, 1H), 0.83–0.79(m, 2H), 0.73–0.70 (m, 2H). LCMS tR = 0.880 min in 4 min chromatography, MS ESI calcd. for C26H31FN11O2+ [M + H]+ 548.26, found 548.2.
5-[4-[2-Aminoethyl(methyl)amino]-2-fluoro-5-(1,2,4-triazol-4-yl)anilino]-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (52)
A mixture of tert-butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)carbamate (257) (80 mg, 0.15 mmol) in DCM (2 mL) was added TFA (1 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 3 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep. HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (HCl)-ACN]; B%: 2–42%, 36 min). 52 as a HCl salt (13 mg, 19.0%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 9.38 (s, 2H), 8.37 (s, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.20–8.01 (m, 3H), 7.44 (d, J = 12.5 Hz, 1H), 6.22 (s, 1H), 3.09 (q, J = 6.2 Hz, 2H), 2.92–2.77 (m, 2H), 2.66–2.56 (m, 1H), 2.41 (s, 3H), 0.86–0.79 (m, 2H), 0.75–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.85, 154.47 (d, J = 250.2 Hz), 150.71, 148.62, 145.10, 143.56, 143.05 (d, J = 8.0 Hz), 123.41 (d, J = 13.1 Hz), 122.79 (d, J = 2.9 Hz), 122.57 (d, J = 3.1 Hz), 114.76, 109.75 (d, J = 22.4 Hz), 76.67, 76.62, 51.44, 41.50, 35.71, 23.46, 6.62. HPLC tR = 3.056 min in 8 min chromatography, purity 95.8%. LCMS tR = 1.430 min in 4 min chromatography, MS ESI calcd. for C21H23FN11+ [M + H]+ 448.21, found 448.4.
tert-Butyl (2-(ethyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (184)
To a mixture of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (600 mg, 2.88 mmol) and tert-butyl (2-(ethylamino)ethyl)carbamate (814.0 mg, 4.32 mmol) in MeCN (20 mL) was added K2CO3 (1.59 g, 11.53 mmol). The reaction was heated to 100 °C and stirred for 12 h. After being cooled to 25 °C, the reaction mixture was concentrated to give a crude product. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 5%, MeOH/DCM). 184 (630 mg, 1.12 mmol, 38.96% yield) was obtained as a brown oil. LCMS tR = 0.556 min in 1 min chromatography, MS ESI calcd. for C17H25N6O4+ [M + H]+ 377.19, found 377.2.
tert-Butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (202)
To a solution of tert-butyl (2-(ethyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (184) (630 mg, 1.67 mmol) in MeOH (3 mL) and THF (3 mL) was added Pd/C (310 mg, 291.30 μmol, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 12 h. The reaction mixture was filtered. The filter cake was washed with MeOH (10 mL × 3), then the filtrate was concentrated. 202 (500 mg, 1.44 mmol, 86.23% yield) was obtained as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (s, 2H), 7.09 (d, J = 8.8 Hz, 1H), 6.56–6.48 (m, 1H), 5.24 (s, 2H), 2.87–2.79 (m, 2H), 2.79–2.73 (m, 2H), 2.58 (q, J = 7.2 Hz, 2H), 1.35 (s, 9H), 0.69 (t, J = 7.2 Hz, 3H).
tert-Butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (220)
To a solution of tert-butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (202) (330 mg, 952.58 μmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (178.1 mg, 762.07 μmol) in dioxane (5 mL) was added BINAP (89 mg, 142.89 μmol), Pd(OAc)2 (32.1 mg, 142.89 μmol), and Cs2CO3 (931.1 mg, 2.86 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated. The residue was purified by prep-HPLC (column: Welch Ultimate XB-NH2 250 × 50 × 10 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; gradient: 15–45% B over 15 min). 220 (600 mg, 937.71 μmol, 98.44% yield) was obtained as a white solid. LCMS tR = 0.585 min in 1 min chromatography, MS ESI calcd. for C27H34N11O2+ [M + H]+ 544.29, found 544.4.
5-((4-((2-Aminoethyl)(ethyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (53)
To a solution of tert-butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (220) (600 mg, 1.10 mmol) in DCM (2 mL) was added TFA (1.54 g, 13.46 mmol, 1 mL). The mixture was stirred at 25 °C for 1 h. The reaction mixture was concentrated directly. The residue was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water(FA)-ACN]; gradient: 0–34% B over 25 min). 53 as a formate salt (132 mg, 291.85 μmol, 72.31% yield) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.84 (s, 2H), 8.42 (s, 1H), 8.35 (s, 1H), 7.94 (d, J = 2.5 Hz, 1H), 7.78 (dd, J = 8.8, 2.6 Hz, 1H), 7.37 (d, J = 8.8 Hz, 1H), 6.00 (s, 1H), 3.02 (t, J = 7.1 Hz, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.66 (q, J = 7.1 Hz, 2H), 2.61 (tt, J = 6.9, 3.6 Hz, 1H), 0.85–0.80 (m, 2H), 0.77 (t, J = 7.1 Hz, 3H), 0.74–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.80, 156.57, 150.82, 148.40, 145.01, 143.05, 138.34, 137.00, 129.50, 123.94, 119.94, 117.25, 114.72, 76.73, 76.57, 49.96, 47.61, 36.58, 23.38, 11.40, 6.57. HPLC tR = 3.181 min in 8 min chromatography, purity 96.1%. LCMS tR = 1.302 min in 4 min chromatography, MS ESI calcd. for C22H26N11+ [M + H]+ 444.24, found 444.3.
tert-Butyl (2-((4-amino-5-fluoro-2-nitrophenyl)(ethyl)amino)ethyl)carbamate (233)
To a solution of 2,4-difluoro-5-nitroaniline (231) (1.35 g, 7.75 mmol) and tert-butyl (2-(ethylamino)ethyl)carbamate (1.46 g, 7.75 mmol) in MeCN (6 mL) was added K2CO3 (3.22 g, 23.26 mmol). Then the reaction mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0–21%, EtOAc/PE). 233 (2.08 g, 52.8%) was obtained as a red solid. 1H NMR (400 MHz, DMSO-d6) δ 7.22 (d, J = 12.8 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 6.62–6.53 (m, 1H), 5.77–5.68 (m, 1H), 5.50 (s, 2H), 2.93–2.85 (m, 5H), 1.34 (s, 9H), 0.86 (t, J = 7.2 Hz, 3H). LCMS tR = 0.523 min in 1 min chromatography, MS ESI calcd. for C15H24FN4O4+ [M + H]+ 343.18, found 343.0.
tert-Butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)(ethyl)amino)ethyl)carbamate (238)
To a solution of tert-butyl (2-((4-amino-5-fluoro-2-nitrophenyl)(ethyl)amino)ethyl)carbamate (233) (2.00 g, 4.87 mmol) in THF (20 mL) was added K2CO3 (1.61 g, 11.68 mmol) and CbzCl (1.49 g, 8.76 mmol) at 25 °C under N2. The mixture was stirred at 25 °C for 3 h. The residue was washed with water (180 mL) and extracted with DCM (200 mL × 2). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–21%, EtOAc/PE). 238 (2.26 g, 72.4%) was obtained as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.15–7.98 (m, 1H), 7.46–7.30 (m, 5H), 7.23 (d, J = 12.8 Hz, 1H), 6.73–6.62 (m, 1H), 5.25–5.10 (m, 2H), 3.20–3.16 (m, 2H), 3.13 (d, J = 7.2 Hz, 1H), 3.08–3.05 (m, 1H), 3.04–2.97 (m, 2H), 1.32 (s, 9H), 1.00 (t, J = 7.2 Hz, 3H). LCMS tR = 0.767 min in 1 min chromatography, MS ESI calcd. for C23H30FN4O6+ [M + H]+ 477.21, found 477.0.
tert-Butyl (2-((2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)(ethyl)amino)ethyl)carbamate (243)
To a solution of tert-butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)(ethyl)amino)ethyl)carbamate (238) (2.00 g, 4.20 mmol) in EtOH (30 mL) was added NH4Cl (2.25 g, 41.97 mmol) dissolved in H2O (15 mL) and Fe (1.17 g, 20.99 mmol) at 25 °C. Then the mixture was stirred at 100 °C under N2 atmosphere for 12 h. The reaction mixture was filtered and the solid was washed with EtOH (50 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Ultimate XB-CN 250 × 50 × 10 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 25–49%, 9 min). 243 (960 mg, 48.4%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 7.43–7.32 (m, 5H), 6.93–6.73 (m, 3H), 5.12 (s, 2H), 4.76 (s, 2H), 2.97–2.89 (m, 2H), 2.88–2.80 (m, 4H), 1.36 (s, 9H), 0.87 (t, J = 6.8 Hz, 3H). LCMS tR = 0.882 min in 1.5 min chromatography, MS ESI calcd. for C23H32FN4O4+ [M + H]+ 447.24, found 447.2.
tert-Butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (248)
To a solution of tert-butyl (2-((2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)(ethyl)amino)ethyl)carbamate (243) (830 mg, 1.86 mmol) and 1,2-diformylhydrazine (491 mg, 5.58 mmol) in pyridine (3 mL) was added Et3N (1.32 g, 13.01 mmol) at 25 °C. The mixture was stirred at 100 °C for 30 min, then Me3SiCl (3.03 g, 27.88 mmol) was added to the mixture. The mixture was stirred at 100 °C for 11.5 h. The reaction mixture was concentrated. The residue was purified by prep. HPLC (column: Welch Ultimate XB-CN 250 × 50 × 10 μm; mobile phase: [heptane-EtOH (0.1%NH3H2O)]; B%: 10–50%, 15 min). 248 (200 mg, 19.1%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.72 (s, 2H), 7.59 (d, J = 8.0 Hz, 1H), 7.44–7.38 (m, 4H), 7.37–7.33 (m, 1H), 7.22 (d, J = 12.4 Hz, 1H), 6.81–6.74 (s, 1H), 5.15 (s, 2H), 2.94–2.89 (m, 2H), 2.88–2.81 (m, 2H), 2.64 (d, J = 7.2 Hz, 2H), 1.36–1.32 (m, 9H), 0.75 (t, J = 7.2 Hz, 3H). LCMS tR = 1.047 min in 2.5 min chromatography, MS ESI calcd. for C25H32FN6O4+ [M + H]+ 499.25, found 499.4.
tert-Butyl (2-((4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (253)
To a solution of tert-butyl (2-((4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (248) (135 mg, 0.27 mmol) in MeOH (10 mL) was added Pd/C (0.11 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 2 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (5 mL × 3). The combine filtrate was concentrated in vacuo. 253 (125 mg, crude) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 2H), 7.11 (d, J = 12.8 Hz, 1H), 6.75–6.88 (m, 2H), 5.27 (s, 2H), 2.85–2.81 (m, 2H), 2.78–2.72 (m, 2H), 2.57 (d, J = 7.2 Hz, 2H), 1.35 (s, 9H), 0.70 (t, J = 7.2 Hz, 3H). LCMS tR = 0.572 min in 1 min chromatography, MS ESI calcd. for C17H26FN6O2+ [M + H]+ 365.21, found 365.0.
tert-Butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (258)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (110 mg, 0.30 mmol) and tert-butyl (2-((4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (253) (56 mg, 0.24 mmol) in dioxane (4 mL) was added Cs2CO3 (295 mg, 0.91 mmol), BINAP (28 mg, 0.05 mmol), and Pd(OAc)2 (10 mg, 0.05 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–2%, MeOH/DCM). 258 (140 mg, 75.0%) was obtained as a black solid. 1H NMR (400 MHz, DMSO-d6) δ 9.53–9.47 (m, 1H), 8.82–8.77 (m, 1H), 8.72 (s, 2H), 8.36 (s, 1H), 8.32 (s, 1H), 8.29–8.24 (m, 1H), 7.34–7.26 (m, 2H), 3.17 (s, 1H), 3.16 (s, 1H), 2.96–2.93 (m, 2H), 2.89–2.85 (m, 2H), 2.70–2.68 (m, 1H), 1.37 (s, 3H), 1.35 (s, 9H), 0.80–0.78 (m, 2H), 0.73–0.71 (m, 2H). LCMS tR = 0.807 min in 2 min chromatography, MS ESI calcd. for C27H33FN11O2+ [M + H]+ 562.28, found 562.3.
5-[4-[2-Aminoethyl(ethyl)amino]-2-fluoro-5-(1,2,4-triazol-4-yl)anilino]-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (54)
To a mixture of tert-butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)(ethyl)amino)ethyl)carbamate (258) (110 mg, 0.19 mmol) in DCM (2 mL) was added TFA (2 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 3 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (HCl)-ACN]; B%: 4–44%, 36 min). The residue was purified by SFC (column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [0.1% NH3H2O EtOH]; B%: 45–45%, 60 min). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (HCl)-ACN]; B%: 0–38%, 36 min). 54 as a HCl salt (6.5 mg, 27.0%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.93–8.75 (m, 2H), 8.37 (s, 2H), 8.30 (dd, J = 8.4, 2.5 Hz, 1H), 7.74 (s, 3H), 7.39 (dd, J = 12.7, 2.5 Hz, 1H), 6.22 (d, J = 1.9 Hz, 1H), 3.07 (q, J = 8.0, 6.1 Hz, 2H), 2.86–2.74 (m, 2H), 2.67 (q, J = 7.0 Hz, 2H), 2.63–2.56 (m, 1H), 0.87–0.74 (m, 5H), 0.74–0.69 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 156.73, 153.54 (d, J = 248.3 Hz), 150.65, 148.57, 145.05, 143.12, 140.35 (d, J = 7.6 Hz), 124.53, 123.40 (d, J = 12.7 Hz), 122.04, 114.64, 110.48 (d, J = 21.8 Hz), 76.67, 76.65, 48.01, 47.23, 35.95, 23.40, 11.24, 6.56. HPLC tR = 3.308 min in 8 min chromatography, purity 96.7%. LCMS tR = 5.514 min in 10 min chromatography, MS ESI calcd. for C22H25FN11+ [M + H]+ 462.23, found 462.2.
(R)-tert-Butyl (1-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)propan-2-yl)carbamate (185)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (600 mg, 2.02 mmol) and tert-butyl (R)-(1-(methylamino)propan-2-yl)carbamate (380 mg, 2.02 mmol) in MeCN (7 mL) was added K2CO3 (836 mg, 6.05 mmol) at 25 °C. The mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–8% MeOH/DCM). 185 (700 mg, 65.4%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 2H), 8.19–8.16 (m, 2H), 7.24–7.21 (m, 1H), 6.71 (d, J = 9.2 Hz, 1H), 3.73–3.70 (m, 1H), 2.86–2.84 (m, 1H), 2.75–2.72 (m, 1H), 2.65 (s, 3H), 1.31 (s, 9H), 0.88 (d, J = 6.8 Hz, 3H).
(R)-tert-Butyl (1-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (203)
To a solution of (R)-tert-butyl (1-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)propan-2-yl)carbamate (185) (686 mg, 1.82 mmol) in MeOH (8 mL) was added Pd/C (500 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (50 mL). The combined filtrate was concentrated in vacuo. 203 (570 mg, 51.4%) was obtained as a purple oil. 1H NMR (400 MHz, DMSO-d6) δ 8.71–8.67 (m, 2H), 7.08 (d, J = 8.4 Hz, 1H), 6.65–6.63 (m, 2H), 6.51 (d, J = 2.4 Hz, 1H), 5.22 (s, 2H), 3.48–3.41 (m, 1H), 2.72–2.66 (m, 1H), 2.56–2.52 (m, 1H), 2.26 (s, 3H), 1.37–1.36 (m, 9H), 0.81 (d, J = 6.4 Hz, 3H). LCMS tR = 0.916 min in 2.5 min chromatography, MS ESI calcd. for C17H27N6O2+ [M + H]+ 347.22, found 347.0.
(R)-tert-Butyl (1-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (221)
To a solution of (R)-tert-butyl (1-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (203) (265 mg, 0.73 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (170 mg, 0.73 mmol) in dioxane (3 mL) was added Cs2CO3 (711 mg, 2.18 mmol), BINAP (68 mg, 0.11 mmol), and Pd(OAc)2 (25 mg, 0.11 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–6%, MeOH/DCM). 221 (553 mg, 36.4%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, 1H), 8.80–8.75 (m, 2H), 8.37–8.34 (m, 1H), 8.29 (s, 1H), 7.90 (d, J = 2.4 Hz, 1H), 7.68 (dd, J = 2.0, 8.8 Hz, 1H), 7.30 (d, J = 9.0 Hz, 1H), 5.96 (s, 1H), 3.65–3.53 (m, 1H), 2.75–2.70 (m, 2H), 2.63–2.58 (m, 1H), 2.36 (s, 3H), 1.36 (s, 10H), 0.90 (d, J = 6.4 Hz, 3H), 0.82–0.80 (m, 2H), 0.72 (d, J = 3.2 Hz, 2H). LCMS tR = 1.375 min in 2.5 min chromatography, MS ESI calcd. for C27H34N11O2+ [M + H]+ 544.29, found 544.3.
(R)-5-((4-((2-Aminopropyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (55)
To a solution of (R)-tert-butyl (1-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (221) (550 mg, 1.01 mmol) in DCM (5 mL) was added TFA (2.5 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 30–60%, 10 min). The crude product was purified by SFC (column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [0.1%NH3H2O ETOH]; B%: 35–35%, 60 min). 55 (25.5 mg, 62.5%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H), 8.86 (s, 2H), 8.36 (s, 1H), 7.88 (d, J = 2.5 Hz, 1H), 7.73 (dd, J = 8.9, 2.6 Hz, 1H), 7.37 (d, J = 8.8 Hz, 1H), 5.96 (s, 1H), 2.84–2.71 (m, 1H), 2.65–2.55 (m, 2H), 2.47 (d, J = 8.3 Hz, 1H), 2.42 (s, 3H), 0.86–0.77 (m, 5H), 0.76–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.53, 150.82, 148.38, 145.00, 143.11, 142.00, 136.35, 128.48, 122.93, 120.45, 117.18, 114.68, 76.67, 76.37, 64.17, 44.29, 42.23, 23.32, 21.86, 6.54. HPLC tR = 2.999 min in 8 min chromatography, purity 99.1%. LCMS tR = 1.110 min in 4 min chromatography, MS ESI calcd. for C22H26N11+ [M + H]+ 444.24, found 444.4.
(S)-tert-Butyl (1-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)propan-2-yl)carbamate (186)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (450 mg, 1.95 mmol) and tert-butyl (S)-(1-(methylamino)propan-2-yl)carbamate (366 mg, 1.95 mmol) in MeCN (5 mL) was added K2CO3 (807 mg, 5.84 mmol) at 25 °C. The mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–15% MeOH/DCM). 186 (590 mg, 69.9%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 2H), 8.21–8.15 (m, 2H), 7.24–7.19 (m, 1H), 6.72 (d, J = 8.8 Hz, 1H), 3.78–3.65 (m, 2H), 2.86 (dd, J = 4.8, 14.0 Hz, 1H), 2.65 (s, 3H), 1.31 (s, 9H), 0.88 (d, J = 6.8 Hz, 3H). LCMS tR = 0.476 min in 1 min chromatography, MS ESI calcd. for C17H25N6O4+ [M + H]+ 377.19, found 377.1.
(S)-tert-Butyl (1-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (204)
To a solution of (S)-tert-butyl (1-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)propan-2-yl)carbamate (186) (560 mg, 1.49 mmol) in MeOH (6 mL) was added Pd/C (360 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (100 mL). The residue was purified by flash silica gel chromatography (eluent of 0–9%, MeOH/DCM). 204 (253 mg, 44.3%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 2H), 7.09 (d, J = 8.8 Hz, 1H), 6.69–6.59 (m, 2H), 6.51 (d, J = 2.8 Hz, 1H), 5.20 (s, 2H), 3.44 (q, J = 6.8 Hz, 1H), 2.73–2.64 (m, 1H), 2.55 (dd, J = 6.4, 12.4 Hz, 1H), 2.26 (s, 3H), 1.36 (s, 9H), 0.81 (d, J = 6.4 Hz, 3H). LCMS tR = 0.407 min in 1 min chromatography, MS ESI calcd. for C17H27N6O2+ [M + H]+ 347.22, found 347.1.
(S)-tert-Butyl (1-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (222)
To a solution of (S)-tert-butyl (1-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (204) (204 mg, 0.59 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (125 mg, 0.53 mmol) in dioxane (3 mL) was added Cs2CO3 (523 mg, 1.60 mmol), BINAP (50 mg, 0.08 mmol), and Pd(OAc)2 (18 mg, 0.08 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–13%, MeOH/DCM). 222 (153 mg, 31.9%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.83–8.72 (m, 2H), 8.36 (s, 1H), 8.30 (s, 1H), 7.89 (s, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.30 (d, J = 8.8 Hz, 1H), 6.73 (d, J = 8.4 Hz, 1H), 4.10 (q, J = 5.2 Hz, 1H), 3.67–3.52 (m, 1H), 2.73 (d, J = 6.8 Hz, 2H), 2.61 (s, 1H), 2.35 (s, 3H), 1.36 (s, 9H), 0.90 (d, J = 6.4 Hz, 3H), 0.84–0.78 (m, 2H), 0.72 (d, J = 3.2 Hz, 2H). LCMS tR = 0.533 min in 1 min chromatography, MS ESI calcd. for C27H34N11O2+ [M + H]+ 544.29, found 544.3.
(S)-5-((4-((2-Aminopropyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (56)
To a solution of (S)-tert-butyl (1-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)propan-2-yl)carbamate (222) (149 mg, 0.27 mmol) in DCM (1 mL) was added TFA (0.5 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 20–59%, 9 min). The crude product was purified by SFC (column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [0.1%NH3H2O IPA]; B%: 35–35%, 60 min). 56 (2.3 mg, 26.1%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.84 (s, 2H), 8.37 (s, 1H), 8.33 (s, 1H), 7.92 (s, 1H), 7.88–7.77 (m, 4H), 7.42 (d, J = 9.0 Hz, 1H), 6.03 (s, 1H), 3.18 (dq, J = 14.0, 7.2, 6.7 Hz, 1H), 3.01 (dd, J = 13.1, 6.2 Hz, 1H), 2.80 (dd, J = 12.9, 7.1 Hz, 1H), 2.60 (tt, J = 6.9, 3.6 Hz, 1H), 2.38 (s, 3H), 1.05 (d, J = 6.1 Hz, 3H), 0.86–0.78 (m, 2H), 0.74–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.52, 150.77, 148.36, 145.00, 143.15, 143.10, 140.98, 137.05, 122.89, 120.38, 117.45, 114.66, 76.68, 76.53, 58.72, 44.42, 42.60, 23.33, 16.87, 6.52. HPLC tR = 3.025 min in 8 min chromatography, purity 97.9%. LCMS tR = 1.722 min in 4 min chromatography, MS ESI calcd. for C22H26N11+ [M + H]+ 444.24, found 444.5.
tert-Butyl methyl(2-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (187)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (200 mg, 0.96 mmol) and tert-butyl methyl(2-(methylamino)ethyl)carbamate (271 mg, 1.44 mmol) in MeCN (5 mL) was added K2CO3 (531 mg, 3.84 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 9%, MeOH/DCM) to give the product. 187 (241 mg, 35.6%) was obtained as a black brown oil. LCMS tR = 0.875 min in 1.5 min chromatography, MS ESI calcd. for C17H25N6O4+ [M + H]+ 377.19, found 377.1.
tert-Butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(methyl)carbamate (205)
To a solution of tert-butyl methyl(2-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (187) (241 mg, 0.64 mmol) in MeOH (5 mL) was added Pd/C (0.11 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered. The filter cake was washed with MeOH (20 mL). The combined filtrate was concentrated in vacuo. 205 (219 mg, 84.6%) was obtained as a black brown oil. LCMS tR = 1.035 min in 2.5 min chromatography, MS ESI calcd. for C17H27N6O2+ [M + H]+ 347.22, found 347.1.
tert-Butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(methyl)carbamate (223)
To a solution of tert-butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(methyl)carbamate (205) (100 mg, 0.29 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (34 mg, 0.14 mmol) in dioxane (3 mL) was added Cs2CO3 (282 mg, 0.86 mmol), BINAP (27 mg, 0.04 mmol), and Pd(OAc)2 (10 mg, 0.04 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–5%, MeOH/DCM). 223 (154 mg, 47.7%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.79 (s, 1H), 8.73 (s, 1H), 8.34 (s, 1H), 8.28 (s, 1H), 7.897.84 (m, 1H), 7.73–7.69 (m, 1H), 7.35–7.33 (m, 1H), 5.93 (s, 1H), 5.73 (s, 1H), 3.19–3.06 (m, 4H), 2.66–2.58 (m, 6H), 2.56–2.61 (m, 1H), 1.38–1.31 (m, 9H), 0.80–0.76 (m, 2H), 0.72–0.68 (m, 2H). LCMS tR = 0.965 min in 1.5 min chromatography, MS ESI calcd. for C27H34N11O2+ [M + H]+ 544.29, found 544.3.
7-(Cyclopropylamino)-5-((4-(methyl(2-(methylamino)ethyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (57)
To a solution of tert-butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(methyl)carbamate (223) (120 mg, 0.22 mmol) in DCM (5.1 mL) was added TFA (0.9 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 20–52%, 9 min). 57 (10.8 mg, 10.6%) was obtained as a white solid. 1H NMR (850 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.86 (s, 2H), 8.36 (s, 1H), 8.29 (s, 1H), 7.89 (dd, J = 5.1, 2.6 Hz, 1H), 7.73 (ddd, J = 8.7, 5.8, 2.5 Hz, 1H), 7.37 (d, J = 8.9 Hz, 1H), 5.97 (s, 1H), 2.83 (t, J = 6.7 Hz, 2H), 2.61 (tt, J = 7.4, 3.1 Hz, 1H), 2.52–2.51 (m, 2H), 2.44 (s, 3H), 2.24 (s, 3H), 0.83–0.80 (m, 2H), 0.73–0.71 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 159.67, 153.96, 151.53, 148.14, 146.08, 144.16, 139.55, 131.64, 125.80, 123.37, 120.20, 117.81, 79.81, 79.51, 56.82, 51.19, 44.85, 38.44, 26.46, 9.67. HPLC tR = 1.972 min in 8 min chromatography, purity 96.4%. LCMS tR = 2.695 min in 7 min chromatography, MS ESI calcd. for C22H26N11+ [M + H]+ 444.24, found 444.4.
tert-Butyl ethyl(2-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (188)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (400 mg, 1.92 mmol) and tert-butyl ethyl(2-(methylamino)ethyl)carbamate (583 mg, 2.88 mmol) in MeCN (5 mL) was added K2CO3 (797 mg, 5.77 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. Water (20 mL) was added, and the resulting mixture was extracted with DCM (20 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 188 (331 mg, 31.5%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.94–8.64 (m, 2H), 8.19–8.15 (m, 1H), 7.34–7.5 (m, 1H), 7.30–7.26 (m, 1H), 3.19–3.13 (m, 4H), 3.04–2.92 (m, 3H), 2.73–2.61 (m, 2H), 1.36–1.33 (m, 9H), 0.99–0.90 (m, 3H). LCMS tR = 0.926 min in 1.5 min chromatography, MS ESI calcd. for C18H27N6O4+ [M + H]+ 391.21, found 391.2.
tert-Butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(ethyl)carbamate (206)
To a solution of tert-butyl ethyl(2-(methyl(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)amino)ethyl)carbamate (188) (310 mg, 0.79 mmol) in MeOH (20 mL) was added Pd/C (290 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (20 mL × 3). The combined filtrate was concentrated in vacuo. 206 (195 mg, 46.9%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 2H), 7.15–7.09 (m, 1H), 6.68–6.64 (m, 1H), 6.54–6.50 (m, 1H), 5.24 (s, 2H), 3.21–3.17 (m, 3H), 3.02–2.95 (m, 4H), 2.77–2.66 (m, 3H), 1.39 (s, 9H), 0.96–0.91 (m, 3H). LCMS tR = 1.095 min in 2.5 min chromatography, MS ESI calcd. for C18H29N6O2+ [M + H]+ 361.23, found 361.3.
tert-Butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(ethyl)carbamate (224)
To a solution of tert-butyl (2-((4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(ethyl)carbamate (206) (162 mg, 0.45 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (70 mg, 0.30 mmol) in dioxane (3 mL) was added Cs2CO3 (293 mg, 0.90 mmol), BINAP (28 mg, 0.04 mmol), and Pd(OAc)2 (67 mg, 0.30 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 38–67%, 10 min). 224 (65 mg, 36.8%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.77 (s, 2H), 8.39–8.26 (m, 2H), 7.91–7.85 (m, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.3–7.31 (m, 1H), 5.96 (s, 1H), 3.14–2.94 (m, 4H), 2.86–2.73 (m, 2H), 2.65–2.56 (m, 1H), 2.48–2.42 (m, 3H), 1.35 (s, 9H), 0.94 (t, J = 7.2 Hz, 3H), 0.86–0.77 (m, 2H), 0.77–0.67 (m, 2H). LCMS tR = 0.539 min in 1 min chromatography, MS ESI calcd. for C28H36N11O2+ [M + H]+ 558.30, found 558.4.
7-(Cyclopropylamino)-5-((4-((2-(ethylamino)ethyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (58)
To a solution of tert-butyl (2-((4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)(methyl)amino)ethyl)(ethyl)carbamate (224) (60 mg, 0.11 mmol) in DCM (8.5 mL) was added TFA (1.5 mL) at 25 °C. The mixture was stirred at 35 °C for 2 h. Then the mixture was degassed and purged with N2. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Xtimate C18 100 × 30 mm × 10 μm; mobile phase: [water (FA)-ACN]; B%: 15–35%, 10 min). 58 (35 mg, 70.4%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (d, J = 11.0 Hz, 1H), 8.87 (s, 2H), 8.37 (s, 1H), 8.32 (br s, 2H), 7.92 (t, J = 2.7 Hz, 1H), 7.76 (dd, J = 8.9, 2.6 Hz, 1H), 7.38 (d, J = 8.9 Hz, 1H), 6.00 (d, J = 3.0 Hz, 1H), 2.98 (q, J = 6.5, 5.7 Hz, 2H), 2.80–2.67 (m, 4H), 2.61 (tt, J = 6.7, 3.5 Hz, 1H), 2.42 (s, 3H), 1.08 (td, J = 7.3, 2.7 Hz, 3H), 0.86–0.78 (m, 2H), 0.74–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.53, 150.82, 148.40, 145.01, 143.02, 140.59, 136.79, 128.56, 122.66, 120.22, 117.20, 114.70, 76.70, 76.47, 52.00, 44.24, 42.43, 41.85, 23.34, 12.32, 6.55. HPLC tR = 3.400 min in 8 min chromatography, purity 99.0%. LCMS tR = 1.656 min in 4 min chromatography, MS ESI calcd. for C23H28N11+ [M + H]+ 458.25, found 458.4.
N1,N1-Diethyl-N2-methyl-N2-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)ethane-1,2-diamine (189)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (200 mg, 0.96 mmol) and N1,N1-diethyl-N2-methylethane-1,2-diamine (188 mg, 1.44 mmol) in MeCN (5 mL) was added K2CO3 (531 mg, 3.84 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. Water (30 mL) was added to the residue. The resulting mixture was extracted with DCM (20 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 189 (187 mg, 34.2%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 2H), 8.22–8.14 (m, 2H), 7.29 (d, J = 9.2 Hz, 1H), 2.66 (s, 3H), 2.46–2.42 (m, 4H), 2.36–2.31 (m, 4H), 0.82 (t, J = 7.2 Hz, 6H). LCMS tR = 0.788 min in 2.5 min chromatography, MS ESI calcd. for C15H23N6O2+ [M + H]+ 319.19, found 319.1.
N1-(2-(Diethylamino)ethyl)-N1-methyl-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (207)
To a solution of N1,N1-diethyl-N2-methyl-N2-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)ethane-1,2-diamine (189) (170 mg, 0.53 mmol) in MeOH (5 mL) was added Pd/C (0.11 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 2 h. The reaction mixture was filtered. The filter cake was washed with MeOH (20 mL). The combined filtrate was concentrated in vacuo. 207 (149 mg, 83.1%) was obtained as a black brown oil. LCMS tR = 0.488 min in 2.5 min chromatography, MS ESI calcd. for C15H25N6+ [M + H]+ 289.21, found 289.1.
7-(Cyclopropylamino)-5-((4-((2-(diethylamino)ethyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (59)
To a solution of N1-(2-(diethylamino)ethyl)-N1-methyl-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (207) (145 mg, 0.50 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (117 mg, 0.50 mmol) in dioxane (4 mL) was added Cs2CO3 (491 mg, 1.51 mmol), BINAP (47 mg, 0.07 mmol), and Pd(OAc)2 (16 mg, 0.07 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–12%, MeOH/DCM). The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 42–72%, 10 min). 59 (26 mg, 10.2%) was obtained as a yellow solid. 1H NMR (850 MHz, DMSO-d6) δ 9.80 (s, 1H), 8.92 (s, 2H), 8.36 (s, 1H), 8.29 (d, J = 2.3 Hz, 1H), 7.89 (d, J = 2.6 Hz, 1H), 7.70 (dd, J = 8.8, 2.6 Hz, 1H), 7.35 (d, J = 8.8 Hz, 1H), 5.96 (s, 1H), 2.78 (t, J = 6.9 Hz, 2H), 2.63–2.58 (m, 1H), 2.46 (s, 3H), 2.38–2.33 (m, 4H), 2.30 (t, J = 6.8 Hz, 2H), 0.85 (t, J = 7.1 Hz, 6H), 0.83–0.79 (m, 2H), 0.75–0.70 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 156.54, 150.84, 148.37, 144.98, 142.83, 140.92, 136.15, 128.34, 122.52, 120.08, 116.82, 114.67, 76.64, 76.32, 52.68, 49.97, 46.40, 41.61, 23.31, 11.64, 6.52. HPLC tR = 3.325 min in 8 min chromatography, purity 96.0%. LCMS tR = 1.708 min in 7 min chromatography, MS ESI calcd. for C25H32N11+ [M + H]+ 486.28, found 486.5.
N-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-N-methyl-4-nitro-2-(4H-1,2,4-triazol-4-yl)aniline (190)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (1.00 g, 3.36 mmol) and 2-((tert-butyldimethylsilyl)oxy)-N-methylethan-1-amine (637 mg, 3.36 mmol) in MeCN (10 mL) was added K2CO3 (1.39 g, 10.09 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–8%, MeOH/DCM). 190 (1.24 g, 67.0%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 2H), 8.21–8.18 (m, 1H), 8.18–8.16 (m, 1H), 7.30 (d, J = 9.2 Hz, 1H), 3.58 (t, J = 5.2 Hz, 2H), 2.91 (t, J = 5.2 Hz, 2H), 2.72 (s, 3H), 0.76 (s, 9H), 0.00 (s, 6H). LCMS tR = 0.560 min in 1 min chromatography, MS ESI calcd. for C17H28N5O3Si+ [M + H]+ 378.20, found 378.1.
N1-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-N1-methyl-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (208)
To a solution of N-(2-((tert-butyldimethylsilyl)oxy)ethyl)-N-methyl-4-nitro-2-(4H-1,2,4-triazol-4-yl)aniline (190) (1.20 g, 3.18 mmol) in MeOH (12 mL) was added Pd/C (950 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (60 mL). The crude compound was used in the next step directly. 208 (1.15 g, 48.9%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 2H), 7.09 (d, J = 8.8 Hz, 1H), 6.65–6.62 (m, 1H), 6.54 (d, J = 2.8 Hz, 1H), 5.22 (s, 2H), 3.45 (t, J = 6.0 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.37 (s, 3H), 0.82 (s, 9H), −0.02 (s, 6H).
5-((4-((2-((tert-Butyldimethylsilyl)oxy)ethyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (225)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (240 mg, 1.03 mmol) and N1-(2-((tert-butyldimethylsilyl)oxy)ethyl)-N1-methyl-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (208) (376 mg, 1.03 mmol) in dioxane (2 mL) was added Cs2CO3 (1.00 g, 1.28 mmol), BINAP (96 mg, 0.15 mmol), and Pd(OAc)2 (35 mg, 0.15 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–5%, MeOH/DCM). 225 (1.00 g, 56.8%) was obtained as a gray solid. 1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.87 (s, 2H), 8.37 (s, 1H), 8.31 (s, 1H), 7.92 (d, J = 2.0 Hz, 1H), 7.71–7.68 (m, 1H), 7.35 (d, J = 8.8 Hz, 1H), 5.97 (s, 1H), 3.54 (t, J = 5.6 Hz, 2H), 2.84 (t, J = 6.0 Hz, 2H), 2.62 (d, J = 2.8 Hz, 1H), 2.50 (s, 3H), 0.83 (s, 11H), 0.74 (d, J = 2.8 Hz, 2H), 0.00 (s, 6H). LCMS tR = 1.683 min in 2.5 min chromatography, MS ESI calcd. for C27H37N10OSi+ [M + H]+ 545.29, found 545.4.
7-(Cyclopropylamino)-5-((4-((2-hydroxyethyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (60)
To a solution of 5-((4-((2-((tert-butyldimethylsilyl)oxy)ethyl)(methyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (225) (200 mg, 0.37 mmol) in THF (1 mL) was added TBAF (0.4 mL, 1 M in THF) dropwise at 25 °C. The mixture was stirred at 25 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–14%, MeOH/DCM). The crude product was purified by prep-HPLC (column: Xtimate C18 100 × 30 mm × 10 μm; mobile phase: [water (FA)-ACN]; B%: 25–55%, 10 min). 60 (11 mg, 12.5%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H), 8.89 (s, 2H), 8.36 (s, 1H), 8.28 (s, 1H), 7.90 (d, J = 2.6 Hz, 1H), 7.69 (dd, J = 8.8, 2.5 Hz, 1H), 7.34 (d, J = 8.8 Hz, 1H), 5.97 (s, 1H), 4.57 (s, 1H), 3.37 (d, J = 6.1 Hz, 2H), 2.83 (t, J = 5.9 Hz, 2H), 2.66–2.55 (m, J = 3.3 Hz, 1H), 2.43 (s, 3H), 0.86–0.75 (m, 2H), 0.78–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.54, 150.85, 148.37, 144.96, 142.80, 140.98, 136.16, 128.32, 122.68, 120.14, 116.86, 114.69, 76.64, 76.35, 58.30, 56.82, 41.76, 23.32, 6.53. HPLC tR = 3.467 min in 8 min chromatography, purity 98.8%. LCMS tR = 1.365 min in 4 min chromatography, MS ESI calcd. for C21H23N10O+ [M + H]+ 431.21, found 431.4.
2-((4-Amino-5-fluoro-2-nitrophenyl)(methyl)amino)ethan-1-ol (262)
To a solution of 2,4-difluoro-5-nitroaniline (231) (5.00 g, 28.7 mmol) and 2-(methylamino)ethan-1-ol (4.31 g, 57.4 mmol) in DMF (20 mL) was added K2CO3 (11.9 g, 86.2 mmol). Then the reaction mixture was stirred at 60 °C for 2 h. The reaction mixture was concentrated, and sat. aqueous NaCl (100 mL) was added. The resulting mixture was extracted with EtOAc (20 mL × 3). The combined organic phase was washed with water (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0–30%, EtOAc/PE). 262 (3.24 g, 98.1%) was obtained as a red oil. 1H NMR (400 MHz, DMSO-d6) δ 7.02 (d, J = 9.2 Hz, 1H), 6.95 (d, J = 13.2 Hz, 1H), 5.12 (s, 2H), 4.35 (t, J = 5.2 Hz, 1H), 3.30 (q, J = 6.4 Hz, 2H), 2.79 (t, J = 6.4 Hz, 2H), 2.50 (s, 3H). LCMS tR = 0.289 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C9H13FN3O3+ [M + H]+ 230.09, found 230.2.
N1-(2-((tert-Butyldiphenylsilyl)oxy)ethyl)-5-fluoro-N1-methyl-2-nitrobenzene-1,4-diamine (234)
A solution of 2-((4-amino-5-fluoro-2-nitrophenyl)(methyl)amino)ethan-1-ol (262) (3.24 g, 14.1 mmol), TBDPS-Cl (3.89 g, 14.1 mmol), and imidazole (2.12 g, 31.1 mmol) in DMF (11 mL) was stirred at 25 °C for 12 h. The reaction mixture was diluted with brine (50 mL) and extracted with CH2Cl2 (50 mL). The combined organic phase was washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0–20%, EtOAc/PE). 234 (3.44 g, 98.5%) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.56–7.52 (m, 4H), 7.45–7.37 (m, 6H), 7.18 (d, J = 9.1 Hz, 1H), 7.11 (d, J = 13.2 Hz, 1H), 5.29 (s, 2H), 3.66 (t, J = 5.6 Hz, 2H), 3.11 (t, J = 5.6 Hz, 2H), 2.68 (s, 3H), 0.91 (s, 9H). LCMS tR = 0.577 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C25H31FN3O3Si+ [M + H]+ 468.21, found 468.6.
Benzyl (4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluoro-5-nitrophenyl)carbamate (239)
To a solution of N1-(2-((tert-butyldiphenylsilyl)oxy)ethyl)-5-fluoro-N1-methyl-2-nitrobenzene-1,4-diamine (234) (1.76 g, 3.75 mmol) in THF (25.0 mL) was added K2CO3 (1.56 g, 11.3 mmol). Then Cbz-Cl (960 mg, 5.63 mmol) was added to the mixture at 0 °C. Then the mixture was stirred at 25 °C for 12 h. The reaction mixture was concentrated under reduced pressure to remove solvent to give a residue. The crude compound was used in the next step without further purification. 239 (2.20 g, 92.7%) was obtained as a red oil. 1H NMR (400 MHz, DMSO-d6) δ 7.52–7.50 (m, 3H), 7.44–7.36 (m, 10H), 7.34 (br d, J = 6.8 Hz, 1H), 7.31 (d, J = 4.4 Hz, 2H), 7.17 (d, J = 13.6 Hz, 1H), 5.15 (s, 2H), 4.49 (s, 1H), 3.74 (t, J = 5.2 Hz, 2H), 3.34–3.31 (m, 2H), 2.80 (s, 3H), 0.89 (s, 9H). LCMS tR = 0.629 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C33H37FN3O5Si+ [M + H]+ 602.25, found 602.7.
Benzyl (5-amino-4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluorophenyl)carbamate (244)
To a solution of benzyl (4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluoro-5-nitrophenyl)carbamate (239) (2.10 g, 3.49 mmol) in EtOH (25.0 mL) was added NH4Cl (1.12 g, 20.9 mmol) dissolved in H2O (12.0 mL) and Fe (585 mg, 10.5 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 12 h. The reaction mixture was filtered and the solid was washed with MeOH (10.0 mL × 3). The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 25%, EtOAc/PE) to give the product. 244 (2.03 g, 97.9%) was obtained as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.75–7.60 (m, 4H), 7.50–7.31 (m, 11H), 6.84 (br d, J = 7.2 Hz, 1H), 6.77 (d, J = 12.0 Hz, 1H), 5.12 (s, 2H), 4.88 (s, 2H), 3.72 (br t, J = 5.2 Hz, 2H), 2.97–2.92 (m, 2H), 2.56 (s, 3H), 1.17 (t, J = 7.2 Hz, 1H), 0.99 (s, 9H). LCMS tR = 0.565 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C33H39FN3O3Si+ [M + H]+ 572.27, found 572.7.
Benzyl (4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluoro-5-(4H-1,2,4-triazol-4-yl)phenyl)carbamate (249)
To a solution of benzyl (5-amino-4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluorophenyl)carbamate (244) (2.03 g, 3.55 mmol) and 1,2-diformylhydrazine (1.56 g, 17.8 mmol) in pyridine (18 mL) was added Et3N (2.51 g, 24.9 mmol) and Me3SiCl (5.79 g, 53.3 mmol) at 25 °C. Then the reaction mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated under reduced pressure to remove solvent to give a residue. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 5%, MeOH/DCM) to give the product. 249 (1.42 g, 98.6%) was obtained as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 2H), 7.60–7.55 (m, 1H), 7.54–7.51 (m, 4H), 7.46–7.44 (m, 1H), 7.44–7.42 (m, 3H), 7.41 (s, 4H), 7.39 (d, J = 2.4 Hz, 2H), 7.36 (br d, J = 3.6 Hz, 1H), 7.16 (d, J = 12.4 Hz, 1H), 5.15 (s, 2H), 3.54 (br t, J = 5.2 Hz, 2H), 2.82 (br t, J = 5.2 Hz, 2H), 2.53 (s, 3H), 1.17 (s, 1H), 0.93 (s, 9H). LCMS tR = 0.588 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C35H39FN5O3Si+ [M + H]+ 624.28, found 624.7.
N1-(2-((tert-Butyldiphenylsilyl)oxy)ethyl)-5-fluoro-N1-methyl-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (254)
To a solution of benzyl (4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluoro-5-(4H-1,2,4-triazol-4-yl)phenyl)carbamate (249) (800 mg, 1.28 mmol) in MeOH (15 mL) was added Pd/C (800 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (20 psi) at 25 °C for 4 h. The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to remove solvent to give a residue. The crude compound was used into the next step without further purification. 254 (600 mg, 96.5%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.54 (d, J = 1.6 Hz, 2H), 7.52 (d, J = 1.6 Hz, 2H), 7.46–7.39 (m, 6H), 7.08 (d, J = 12.8 Hz, 1H), 6.74 (d, J = 9.2 Hz, 1H), 5.23 (s, 2H), 3.50 (t, J = 5.6 Hz, 2H), 3.18–3.17 (m, 1H), 3.17–3.16 (m, 1H), 2.80 (t, J = 5.6 Hz, 2H), 2.41 (s, 3H), 0.95 (s, 9H). LCMS tR = 0.540 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C27H33FN5OSi+ [M + H]+ 490.24, found 490.6.
5-((4-((2-((tert-Butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluoro-5-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (259)
To a solution of N1-(2-((tert-butyldiphenylsilyl)oxy)ethyl)-5-fluoro-N1-methyl-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (254) (600 mg, 1.34 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (313 mg, 1.34 mmol) in dioxane (12.0 mL) was added Cs2CO3 (1.74 g, 5.35 mmol), BINAP (125 mg, 201 μmol), and Pd(OAc)2 (45.1 mg, 201 μmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–44%, EtOAc/PE). 259 (500 mg, 60.1%) was obtained as a yellow oil. LCMS tR = 0.597 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C37H40FN10OSi+ [M + H]+ 687.31, found 687.8.
7-(Cyclopropylamino)-5-[2-fluoro-4-[2-hydroxyethyl(methyl)amino]-5-(1,2,4-triazol-4-yl)anilino]pyrazolo[1,5-a]pyrimidine-3-carbonitrile (61)
To a mixture of 5-((4-((2-((tert-butyldiphenylsilyl)oxy)ethyl)(methyl)amino)-2-fluoro-5-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (259) (200 mg, 291 μmol) in THF (5 mL) was added TBAF (1.5 mL, 1 mol/L in THF) at 0 °C. The reaction mixture was stirred at 25 °C for 2 h. The resulting mixture was quenched by water (10 mL), extracted with EtOAc (20 mL × 3). The mixture was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Luna C18 150 × 25 mm × 10 μm; mobile phase: [water (FA)-ACN]; B%: 20–50%, 8 min). 61 (8.00 mg) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.45 (s, 1H), 8.81 (s, 2H), 8.35 (s, 1H), 8.30 (d, J = 1.8 Hz, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 12.8 Hz, 1H), 6.15 (s, 1H), 4.58 (t, J = 5.1 Hz, 1H), 3.37 (q, J = 6.0 Hz, 2H), 2.81 (t, J = 5.8 Hz, 2H), 2.64–2.54 (m, 1H), 2.48 (s, 3H), 0.85–0.78 (m, 2H), 0.73–0.68 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −121.98. 13C NMR (214 MHz, DMSO-d6) δ 160.09, 157.00 (d, J = 248.0 Hz), 153.89, 151.66, 148.15, 146.60 (d, J = 8.8 Hz), 146.08, 126.26, 125.10, 125.09 (d, J = 24.9 Hz), 117.84, 112.17 (d, J = 21.8 Hz), 79.65, 79.50, 61.30, 59.44, 44.03, 26.50, 9.68. HPLC tR = 1.694 min in 4 min chromatography, purity 95.7%. LCMS tR = 0.384 min in 0.8 min chromatography, 5–95AB, LCMS ESI calcd. for C21H22FN10O+ [M + H]+ 449.20, found 449.4.
N-(2-Methoxyethyl)-4-nitro-2-(4H-1,2,4-triazol-4-yl)aniline (191)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (1.00 g, 4.80 mmol) and 2-methoxyethan-1-amine (361 mg, 4.80 mmol) in MeCN (10 mL) was added K2CO3 (1.99 g, 14.41 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–8%, MeOH/DCM). 191 (2.18 g, 54.3%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 2H), 8.20–8.17 (m, 1H), 8.01 (d, J = 2.8 Hz, 1H), 7.01 (d, J = 9.6 Hz, 1H), 6.65 (t, J = 5.6 Hz, 1H), 3.45–3.43 (m, 2H), 3.40–3.36 (m, 2H), 3.24 (s, 3H). LCMS tR = 0.411 min in 1 min chromatography, MS ESI calcd. for C11H14N5O3+ [M + H]+ 264.11, found 264.0.
N1-(2-Methoxyethyl)-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (209)
To a solution of N-(2-methoxyethyl)-4-nitro-2-(4H-1,2,4-triazol-4-yl)aniline (191) (863 mg, 3.28 mmol) in MeOH (8 mL) was added Pd/C (720 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (50 mL). The combined filtrate was concentrated in vacuo. 209 (637 mg, 77.8%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 2H), 6.72–6.69 (m, 1H), 6.65–6.63 (m, 1H), 6.41 (d, J = 2.4 Hz, 1H), 4.74 (s, 2H), 3.92 (t, J = 6.0 Hz, 1H), 3.37–3.36 (m, 2H), 3.20 (s, 3H), 3.04–2.99 (m, 2H).
7-(Cyclopropylamino)-5-((4-((2-methoxyethyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (62)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (150 mg, 0.64 mmol) and N1-(2-methoxyethyl)-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (209) (158 mg, 0.64 mmol) in dioxane (2 mL) was added Cs2CO3 (628 mg, 1.93 mmol), BINAP (60 mg, 0.10 mmol), and Pd(OAc)2 (22 mg, 0.10 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by silica gel chromatography (eluent of 0–5%, MeOH/DCM). The crude product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 24–47%, 8 min). 62 (79.5 mg, 17.1%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.70 (s, 2H), 8.31 (s, 1H), 8.18 (d, J = 1.8 Hz, 1H), 7.61 (dd, J = 8.9, 2.5 Hz, 1H), 7.53 (d, J = 2.5 Hz, 1H), 6.91 (d, J = 8.9 Hz, 1H), 5.88 (s, 1H), 4.73 (t, J = 5.8 Hz, 1H), 3.44 (t, J = 5.9 Hz, 2H), 3.24 (s, 3H), 3.20 (q, J = 5.9 Hz, 2H), 2.62–2.54 (m, 1H), 0.83–0.76 (m, 2H), 0.73–0.66 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.89, 151.05, 148.19, 144.90, 143.42, 139.13, 129.87, 122.71, 119.65, 119.57, 114.84, 112.74, 76.18, 75.69, 70.36, 58.04, 42.63, 23.28, 6.53. HPLC tR = 3.585 min in 8 min chromatography, purity 99.8%. LCMS tR = 1.426 min in 4 min chromatography, MS ESI calcd. for C21H23N10O+ [M + H]+ 431.21, found 431.3.
N-Methyl-4-nitro-N-(2-(pyrrolidin-1-yl)ethyl)-2-(4H-1,2,4-triazol-4-yl)aniline (192)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (300 mg, 1.44 mmol) and N-methyl-2-(pyrrolidin-1-yl)ethan-1-amine (185 mg, 1.44 mmol) in MeCN (5 mL) was added K2CO3 (598 mg, 4.32 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. Water (150 mL) was added to the reaction mixture. The mixture was extracted with EtOAc (200 mL × 2), and the combined organic phase was washed with brine (200 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–21%, MeOH/DCM). 192 (400 mg, 79.7%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 2H), 8.24–8.15 (m, 2H), 7.32 (d, J = 9.2 Hz, 1H), 3.16 (s, 3H), 2.61 (s, 4H), 2.45 (s, 4H), 1.65 (s, 4H). LCMS tR = 0.783 min in 2.5 min chromatography, MS ESI calcd. for C15H21N6O2+ [M + H]+ 317.17, found 317.0.
N1-Methyl-N1-(2-(pyrrolidin-1-yl)ethyl)-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (210)
To a solution of N-methyl-4-nitro-N-(2-(pyrrolidin-1-yl)ethyl)-2-(4H-1,2,4-triazol-4-yl)aniline (192) (400 mg, 1.26 mmol) in MeOH (3 mL) was added Pd/C (220 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (200 mL). The combined filtrate was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–6%, MeOH/DCM). 210 (290 mg, 77.1%) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 2H), 7.11 (d, J = 8.8 Hz, 1H), 6.64 (dd, J = 2.4, 8.8 Hz, 1H), 6.52 (d, J = 2.4 Hz, 1H), 5.23 (s, 2H), 3.16 (s, 2H), 2.75 (t, J = 6.8 Hz, 2H), 2.36 (s, 3H), 2.33 (s, 4H), 1.63 (s, 4H). LCMS tR = 0.132 min in 1 min chromatography, MS ESI calcd. for C15H23N6+ [M + H]+ 287.20, found 287.2.
7-(Cyclopropylamino)-5-((4-(methyl(2-(pyrrolidin-1-yl)ethyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (63)
To a solution of N1-methyl-N1-(2-(pyrrolidin-1-yl)ethyl)-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (210) (196 mg, 0.68 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (160 mg, 0.68 mmol) in dioxane (5 mL) was added tBuOLi (165 mg, 2.05 mmol) and Brettphos Pd G3 (62 mg, 0.07 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–15%, MeOH/DCM). The crude product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 30–62%, 9 min). 63 (11.2 mg, 6.0%) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.90 (s, 2H), 8.36 (s, 1H), 8.30 (d, J = 1.8 Hz, 1H), 7.89 (d, J = 2.5 Hz, 1H), 7.70 (dd, J = 8.8, 2.6 Hz, 1H), 7.36 (d, J = 8.9 Hz, 1H), 5.96 (s, 1H), 2.82 (t, J = 7.0 Hz, 2H), 2.65–2.57 (m, 1H), 2.46 (s, 3H), 2.38–2.27 (m, 6H), 1.68–1.58 (m, 4H), 0.86–0.79 (m, 2H), 0.75–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.54, 150.84, 148.38, 145.01, 142.87, 140.92, 136.27, 128.48, 122.53, 120.13, 116.92, 114.70, 76.66, 76.35, 53.57, 53.40, 52.89, 41.68, 23.32, 23.07, 6.54. HPLC tR = 3.061 min in 8 min chromatography, purity 99.2%. LCMS tR = 1.732 min in 4 min chromatography, MS ESI calcd. for C25H30N11+ [M + H]+ 484.27, found 484.5.
N-Methyl-N-(2-morpholinoethyl)-4-nitro-2-(4H-1,2,4-triazol-4-yl)aniline (193)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (300 mg, 1.30 mmol) and N-methyl-2-morpholinoethan-1-amine (187 mg, 1.30 mmol) in MeCN (5 mL) was added K2CO3 (538 mg, 3.89 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–5%, MeOH/DCM). 193 (266 mg, 55.8%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.95 (s, 2H), 8.22–8.17 (m, 2H), 7.32 (d, J = 9.2 Hz, 1H), 3.45 (t, J = 4.4 Hz, 4H), 2.99 (t, J = 6.4 Hz, 2H), 2.66 (s, 3H), 2.33 (t, J = 6.4 Hz, 2H), 2.26 (s, 4H). LCMS tR = 0.332 min in 1 min chromatography, MS ESI calcd. for C15H21N6O3+ [M + H]+ 333.17, found 333.1.
N1-Methyl-N1-(2-morpholinoethyl)-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (211)
To a solution of N-methyl-N-(2-morpholinoethyl)-4-nitro-2-(4H-1,2,4-triazol-4-yl)aniline (193) (266 mg, 0.80 mmol) in MeOH (12 mL) was added Pd/C (200 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (50 mL). The combined filtrate was concentrated in vacuo. 211 (248 mg, 99.5%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 2H), 7.12 (d, J = 8.8 Hz, 1H), 6.65–6.53 (m, 2H), 5.23 (s, 2H), 3.52–3.45 (m, 4H), 2.77–2.74 (m, 2H), 2.36 (s, 3H), 2.22 (s, 4H), 2.14 (t, J = 6.4 Hz, 2H). LCMS tR = 0.132 min in 1 min chromatography, MS ESI calcd. for C15H23N6O+ [M + H]+ 303.19, found 303.2.
7-(Cyclopropylamino)-5-((4-(methyl(2-morpholinoethyl)amino)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (64)
To a solution of N1-methyl-N1-(2-morpholinoethyl)-2-(4H-1,2,4-triazol-4-yl)benzene-1,4-diamine (211) (207 mg, 0.69 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (160 mg, 0.69 mmol) in dioxane (2 mL) was added Cs2CO3 (669 mg, 2.05 mmol), BINAP (64 mg, 0.10 mmol), and Pd(OAc)2 (23 mg, 0.10 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–7%, MeOH/DCM). The crude product was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 21–50%, 10 min). 64 (31.3 mg, 17.1%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.94 (s, 2H), 8.36 (s, 1H), 8.29 (s, 1H), 7.91 (d, J = 2.6 Hz, 1H), 7.70 (dd, J = 8.8, 2.6 Hz, 1H), 7.36 (d, J = 8.9 Hz, 1H), 5.96 (s, 1H), 3.51 (t, J = 4.6 Hz, 4H), 2.83 (t, J = 6.7 Hz, 2H), 2.65–2.57 (m, 1H), 2.46 (s, 3H), 2.31–2.18 (m, 6H), 0.85–0.78 (m, 2H), 0.74–0.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.54, 150.84, 148.38, 144.99, 142.89, 140.89, 136.31, 128.43, 122.63, 120.10, 116.81, 114.70, 76.67, 76.36, 66.15, 55.53, 53.28, 51.35, 41.69, 23.33, 6.55. HPLC tR = 3.016 min in 8 min chromatography, purity 99.6%. LCMS tR = 1.665 min in 4 min chromatography, MS ESI calcd. for C25H30N11O+ [M + H]+ 500.26, found 500.5.
tert-Butyl (R)-(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (194)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (900 mg, 4.32 mmol) and tert-butyl (R)-pyrrolidin-3-ylcarbamate (966 mg, 5.19 mmol) in MeCN (12 mL) was added K2CO3 (1.79 g, 12.97 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. Water (150 mL) was added to the reaction mixture. The mixture was extracted with EtOAc (180 mL × 2), and the combined organic phase was washed with brine (250 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 194 (1.22 g, 60.1%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 2H), 8.17 (dd, J = 2.8, 9.6 Hz, 1H), 8.06 (d, J = 2.8 Hz, 1H), 7.18 (d, J = 6.4 Hz, 1H), 4.00 (s, 1H), 3.82 (s, 1H), 2.70–2.59 (m, 4H), 1.93 (dt, J = 5.6, 12.8 Hz, 2H), 1.37 (s, 9H). LCMS tR = 0.481 min in 1 min chromatography, MS ESI calcd. for C17H23N6O4+ [M + H]+ 375.18, found 375.1.
tert-Butyl (R)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (212)
To a solution of tert-butyl (R)-(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (194) (1.2 g, 3.21 mmol) in MeOH (12 mL) was added Pd/C (600 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (200 mL). The combined filtrate was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–8%, MeOH/DCM). 212 (385 mg, 33.9%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 2H), 7.04 (d, J = 7.2 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 6.62 (dd, J = 2.4, 8.8 Hz, 1H), 6.48 (d, J = 2.4 Hz, 1H), 4.99 (s, 2H), 3.97–3.85 (m, 1H), 2.83 (dd, J = 6.8, 9.2 Hz, 1H), 2.72–2.63 (m, 1H), 2.58–2.51 (m, 1H), 2.46 (dd, J = 5.2, 8.8 Hz, 1H), 2.00–1.89 (m, 1H), 1.59 (qd, J = 6.4, 12.8 Hz, 1H), 1.37 (s, 9H). LCMS tR = 0.404 min in 1 min chromatography, MS ESI calcd. for C17H25N6O2+ [M + H]+ 345.20, found 345.2.
tert-Butyl (R)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (226)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (190 mg, 0.81 mmol) and tert-butyl (R)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (212) (336 mg, 0.96 mmol) in dioxane (5 mL) was added Cs2CO3 (795 mg, 2.44 mmol), BINAP (76 mg, 0.12 mmol), and Pd(OAc)2 (27 mg, 0.12 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–4%, MeOH/DCM). 226 (207 mg, 21.9%) was obtained as a yellow solid. LCMS tR = 0.524 min in 1 min chromatography, MS ESI calcd. for C27H32N11O2+ [M + H]+ 542.27, found 542.2.
(R)-5-((4-(3-Aminopyrrolidin-1-yl)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (65)
To a solution of tert-butyl (R)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (226) (200 mg, 0.37 mmol) in DCM (3 mL) was added TFA (3.10 g, 27.21 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 20–63%, 10 min). Then the impure product was purified by SFC (column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [0.1%NH3H2O ETOH]; B%: 45–45%, 60 min). 65 (13.6 mg, 17.6%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.76 (s, 2H), 8.32 (s, 1H), 8.22 (br s, 1H), 7.72–7.61 (m, 2H), 6.98 (d, J = 8.8 Hz, 1H), 5.90 (s, 1H), 3.38–3.31 (m, 1H), 2.94 (q, J = 7.7 Hz, 1H), 2.86 (dd, J = 9.2, 6.0 Hz, 1H), 2.76 (q, J = 7.6 Hz, 1H), 2.58 (tt, J = 7.1, 3.7 Hz, 1H), 2.42 (dd, J = 9.2, 4.9 Hz, 1H), 1.88 (dq, J = 16.0, 9.7, 8.2 Hz, 1H), 1.49 (dq, J = 12.8, 6.5 Hz, 1H), 0.86–0.76 (m, 2H), 0.73–0.63 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.72, 150.98, 148.23, 144.92, 144.15, 140.40, 131.62, 121.78, 121.39, 116.50, 114.77, 76.31, 75.88, 58.07, 50.66, 47.88, 34.09, 23.29, 6.52. HPLC tR = 2.870 min in 8 min chromatography, purity 98.5%. LCMS tR = 1.597 min in 4 min chromatography, MS ESI calcd. for C22H24N11+ [M + H]+ 442.22, found 442.4.
tert-Butyl (S)-(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (195)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (1 g, 4.80 mmol) and tert-butyl (S)-pyrrolidin-3-ylcarbamate (895 mg, 4.80 mmol) in MeCN (12 mL) was added K2CO3 (1.99 g, 14.41 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. Water (150 mL) was added to the reaction mixture. The mixture was extracted with EtOAc (180 mL × 2), and the combined organic phase was washed with brine (250 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 195 (1.22 g, 67.8%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 2H), 8.16 (dd, J = 2.4, 9.6 Hz, 1H), 8.04 (d, J = 2.8 Hz, 1H), 7.18 (d, J = 6.4 Hz, 1H), 2.59–2.43 (m, 2H), 2.00–1.71 (m, 4H), 1.48 (ddd, J = 5.2, 7.6, 18.0 Hz, 2H), 1.36 (s, 9H). LCMS tR = 0.753 min in 1.5 min chromatography, MS ESI calcd. for C17H23N6O4+ [M + H]+ 375.18, found 375.1.
tert-Butyl (S)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (213)
To a solution of tert-butyl (S)-(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (195) (1.20 g, 3.21 mmol) in MeOH (12 mL) was added Pd/C (600 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (200 mL). The combined filtrate was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–6%, MeOH/DCM). 213 (430 mg, 37.8%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.75 (s, 2H), 7.06 (d, J = 7.2 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 6.62 (dd, J = 2.4, 8.8 Hz, 1H), 6.48 (d, J = 2.4 Hz, 1H), 5.00 (s, 2H), 3.96–3.85 (m, 1H), 2.83 (dd, J = 6.8, 9.2 Hz, 1H), 2.72–2.63 (m, 1H), 2.57–2.51 (m, 1H), 2.45 (dd, J = 5.2, 9.2 Hz, 1H), 2.00–1.87 (m, 1H), 1.63–1.54 (m, 1H), 1.36 (s, 9H). LCMS tR = 0.399 min in 1 min chromatography, MS ESI calcd. for C17H25N6O2+ [M + H]+ 345.20, found 345.1.
tert-Butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (227)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (260 mg, 1.11 mmol) and tert-butyl (S)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (213) (383 mg, 1.11 mmol) in dioxane (5 mL) was added Cs2CO3 (1.09 g, 3.34 mmol), BINAP (104 mg, 0.17 mmol), and Pd(OAc)2 (37 mg, 0.17 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–5%, MeOH/DCM). 227 (440 mg, 67.1%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.66 (s, 1H), 8.79 (s, 2H), 8.33 (s, 1H), 8.25 (s, 1H), 7.70–7.61 (m, 2H), 7.12 (d, J = 7.1 Hz, 1H), 7.00 (d, J = 9.2 Hz, 1H), 5.90 (s, 1H), 4.03–3.89 (m, 1H), 2.96 (dd, J = 6.8, 9.2 Hz, 1H), 2.90–2.83 (m, 1H), 2.76–2.69 (m, 1H), 2.62–2.54 (m, 2H), 1.94 (td, J = 6.0, 12.4 Hz, 1H), 1.66 (qd, J = 6.4, 12.4 Hz, 1H), 1.37 (s, 9H), 0.84–0.77 (m, 2H), 0.73–0.67 (m, 2H). LCMS tR = 0.511 min in 1 min chromatography, MS ESI calcd. for C27H32N11O2+ [M + H]+ 542.27, found 542.2.
(S)-5-((4-(3-Aminopyrrolidin-1-yl)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (66)
To a solution of tert-butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (227) (400 mg, 0.74 mmol) in DCM (3 mL) was added TFA (6.21 g, 54.43 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 20–50%, 10 min). 66 (101.7 mg, 35.5%) was obtained as a gray solid. 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.78 (s, 2H), 8.32 (s, 1H), 8.22 (s, 1H), 7.74–7.58 (m, 2H), 7.00 (d, J = 8.9 Hz, 1H), 5.91 (s, 1H), 3.42 (p, J = 5.6 Hz, 1H), 3.01–2.85 (m, 2H), 2.76 (q, J = 7.6 Hz, 1H), 2.58 (tt, J = 7.0, 3.6 Hz, 1H), 2.48–2.45 (m, 1H), 1.93 (dq, J = 13.2, 6.8 Hz, 1H), 1.55 (dq, J = 12.7, 6.5 Hz, 1H), 0.86–0.76 (m, 2H), 0.74–0.66 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.70, 150.96, 148.24, 144.92, 144.06, 140.06, 131.97, 121.70, 121.68, 119.80, 116.75, 114.76, 76.34, 75.94, 57.20, 50.36, 33.19, 23.29, 6.52. HPLC tR = 2.874 min in 8 min chromatography, purity 96.6%. LCMS tR = 1.570 min in 4 min chromatography, MS ESI calcd. for C22H24N11+ [M + H]+ 442.22, found 442.4.
tert-Butyl (S)-(1-(4-amino-5-fluoro-2-nitrophenyl)pyrrolidin-3-yl)carbamate (235)
To a solution of 2,4-difluoro-5-nitroaniline (231) (2.00 g, 11.49 mmol) and tert-butyl (S)-pyrrolidin-3-ylcarbamate (2.57 g, 13.78 mmol) in MeCN (30 mL) was added K2CO3 (4.76 g, 34.46 mmol). Then the reaction mixture was stirred at 60 °C for 12 h. The reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0–26%, EtOAc/PE). 235 (2.5 g, 59.47%) was obtained as a red solid. 1H NMR (400 MHz, DMSO-d6) δ 7.29–7.23 (m, 1H), 7.15 (br d, J = 5.6 Hz, 1H), 6.86–6.76 (m, 1H), 4.95 (s, 2H), 3.32 (s, 1H), 3.26–3.18 (m, 1H), 3.16–3.07 (m, 2H), 2.81–2.74 (m, 1H), 2.09–1.99 (m, 1H), 1.90–1.79 (m, 1H), 1.37 (s, 9H). LCMS tR = 1.274 min in 2 min chromatography, MS ESI calcd. for C15H22FN4O4+ [M + H]+ 341.15, found 340.9.
tert-Butyl (S)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)pyrrolidin-3-yl)carbamate (240)
To a solution of tert-butyl (S)-(1-(4-amino-5-fluoro-2-nitrophenyl)pyrrolidin-3-yl)carbamate (235) (2.5 g, 7.35 mmol) in THF (30 mL) was added K2CO3 (3.05 g, 22.04 mmol). Then CbzCl (1.88 g, 11.02 mmol) was added to the mixture at 0 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated directly. The reaction mixture was concentrated. Saturated aqueous NH4Cl (15 mL) solution was added. The resulting mixture was extracted with EtOAc (50 mL × 3). The combined organic phase was washed with brine (15 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0–31%, EtOAc/PE). 240 (3.4 g, 86.0%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.99 (s, 1H), 7.50–7.31 (m, 5H), 7.26–7.21 (m, 1H), 6.98–6.87 (m, 1H), 5.14 (s, 2H), 4.12–4.00 (m, 1H), 3.34–3.29 (m, 1H), 3.28–3.16 (m, 2H), 2.88–2.77 (m, 1H), 2.15–2.02 (m, 1H), 1.96–1.83 (m, 1H), 1.37 (s, 9H). LCMS tR = 1.581 min in 2 min chromatography, MS ESI calcd. for C23H28FN4O6+ [M + H]+ 475.20, found 474.9.
tert-Butyl (S)-(1-(2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)pyrrolidin-3-yl)carbamate (245)
To a solution of tert-butyl (S)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)pyrrolidin-3-yl)carbamate (240) (3.40 g, 7.17 mmol) in EtOH (30 mL) was added NH4Cl (1.15 g, 21.50 mmol) dissolved in H2O (6 mL) and Fe (2.40 g, 42.99 mmol) at 25 °C. Then the mixture was stirred at 80 °C under N2 atmosphere for 12 h. The reaction mixture was filtered and the solid was washed with MeOH (150 mL × 2). The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 37%, EtOAc/PE) to give the product. 245 (1.9 g, 51.3%) was obtained as a purple solid. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (s, 1H), 7.45–7.28 (m, 5H), 7.22–7.15 (m, 1H), 6.83–6.73 (m, 1H), 6.68–6.61 (m, 1H), 5.10 (s, 2H), 4.68 (s, 2H), 4.09–4.04 (m, 1H), 3.15–2.99 (m, 2H), 2.84–2.69 (m, 2H), 2.23–2.09 (m, 1H), 1.70–1.61 (m, 1H), 1.39 (s, 9H). LCMS tR = 0.721 min in 1 min chromatography, MS ESI calcd. for C23H30FN4O4+ [M + H]+ 445.22, found 445.1.
tert-Butyl (S)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (250)
To a solution of tert-butyl (S)-(1-(2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)pyrrolidin-3-yl)carbamate (245) (1.00 g, 2.25 mmol) and 1,2-diformylhydrazine (991 mg, 11.25 mmol) in pyridine (5 mL) was added Et3N (1.59 g, 15.75 mmol). The mixture was stirred at 100 °C for 30 min, then Me3SiCl (3.67 g, 33.75 mmol, 4.28 mL) was added to the mixture. The mixture was stirred at 100 °C for 12 h. Water (5 mL) was added to the residue. The resulting mixture was extracted with EtOAc (20 mL × 3). The combined organic phase was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 86%, EtOAc/PE) to give the product. 250 (210 mg, 14.4%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 2H), 7.43–7.28 (m, 6H), 7.12–7.06 (m, 1H), 6.83–6.73 (m, 1H), 5.11 (s, 2H), 4.00–3.89 (m, 1H), 2.98–2.86 (m, 2H), 2.81–2.73 (m, 1H), 2.57–2.52 (m, 1H), 1.73–1.62 (m, 1H), 1.36 (s, 9H). LCMS tR = 0.676 min in 1 min chromatography, MS ESI calcd. for C25H30FN6O4+ [M + H]+ 497.23, found 441.1.
tert-Butyl (S)-(1-(4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (255)
To a solution of tert-butyl (S)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (250) (210 mg, 422.93 μmol) in MeOH (5 mL) was added Pd/C (0.1 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 2 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (100 mL). The combined filtrate was concentrated in vacuo. 255 (100 mg, 46.9%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 2H), 7.08–7.00 (m, 1H), 6.83–6.76 (m, 1H), 6.70–6.62 (m, 1H), 4.94 (s, 2H), 3.95–3.85 (m, 1H), 2.88–2.79 (m, 1H), 2.78–2.68 (m, 1H), 2.63–2.54 (m, 1H), 2.46–2.39 (m, 1H), 1.98–1.88 (m, 1H), 1.64–1.55 (m, 1H), 1.36 (s, 9H). LCMS tR = 0.452 min in 1 min chromatography, MS ESI calcd. for C17H24FN6O2+ [M + H]+ 363.19, found 363.0.
tert-Butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (260)
To a solution of tert-butyl (S)-(1-(4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (255) (100 mg, 275.94 μmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (52 mg, 220.75 μmol) in dioxane (3 mL) was added Cs2CO3 (270 mg, 827.81 μmol), BINAP (26.0 mg, 41.39 μmol), and Pd(OAc)2 (10.0 mg, 41.39 μmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–76%, EtOAc/PE). 260 (85 mg, 15.9%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.97–7.95 (m, 2H), 7.90 (s, 2H), 7.65 (s, 3H), 5.76 (s, 2H), 3.98–3.87 (m, 1H), 3.02–2.91 (m, 2H), 2.85–2.74 (m, 2H), 2.69–2.64 (m, 1H), 1.92–1.88 (m, 2H), 1.37–1.36 (m, 9H), 0.92–0.57 (m, 4H). LCMS tR = 0.559 min in 1 min chromatography, MS ESI calcd. for C22H23FN11+ [M + 2H-Boc]+ 460.21, found 460.2.
(S)-5-((4-(3-Aminopyrrolidin-1-yl)-2-fluoro-5-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (67)
A mixture of tert-butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)pyrrolidin-3-yl)carbamate (260) (85.0 mg, 151.90 μmol) in DCM (5 mL) was added TFA (1 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 1 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (FA)-ACN]; B%: 0–34%, 25 min). 67 (4.80 mg, 6.12%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 8.76 (s, 2H), 8.32 (s, 1H), 8.24 (s, 1H), 7.81 (d, J = 8.3 Hz, 1H), 6.93 (d, J = 13.5 Hz, 1H), 6.03 (s, 1H), 3.67–3.58 (m, 1H), 3.04–2.95 (m, 2H), 2.88 (q, J = 8.6, 7.9 Hz, 1H), 2.64 (dd, J = 10.3, 4.2 Hz, 1H), 2.60–2.54 (m, 1H), 2.10–1.99 (m, 1H), 1.79–1.63 (m, 1H), 0.85–0.74 (m, 2H), 0.69 (p, J = 4.8, 4.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 159.15 (d, J = 239.8 Hz), 157.38, 150.89, 148.42, 145.01, 144.47, 125.61 (d, J = 3.2 Hz), 116.77, 114.82, 105.61, 105.09, 103.60 (d, J = 24.9 Hz), 76.20, 75.84, 54.44, 49.63, 47.39, 30.63, 23.33, 6.53. HPLC tR = 1.924 min in 8 min chromatography, purity 89.4%. LCMS tR = 1.030 min in 4 min chromatography, MS ESI calcd. for C22H23FN11+ [M + H]+ 460.21, found 460.4.
tert-Butyl (S)-(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (196)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (200 mg, 0.96 mmol) and tert-butyl (S)-piperidin-3-ylcarbamate (289 mg, 1.44 mmol) in MeCN (5 mL) was added K2CO3 (531 mg, 3.84 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. Water (40 mL) was added to the residue. The resulting mixture was extracted with DCM (30 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 196 (498 mg, crude) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.26–8.22 (m, 1H), 7.35 (d, J = 9.0 Hz, 1H), 6.97–6.86 (m, 1H), 6.69–6.62 (m, 1H), 5.76 (s, 1H), 3.39–3.38 (m, 1H), 2.90–2.82 (m, 2H), 2.76–2.68 (m, 2H), 1.78–1.71 (m, 2H), 1.59–1.52 (m, 2H), 1.38 (s, 9H). LCMS tR = 1.293 min in 2.5 min chromatography, MS ESI calcd. for C18H25N6O4+ [M + H]+ 389.19, found 389.0.
tert-Butyl (S)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (214)
To a solution of tert-butyl (S)-(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (196) (214 mg, 0.55 mmol) in MeOH (5 mL) was added Pd/C (0.11 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (10 mL × 3). The combined filtrate was concentrated in vacuo. 214 (198 mg, 79.5%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H), 7.01 (br d, J = 8.6 Hz, 1H), 6.75–6.68 (m, 2H), 6.65–6.60 (m, 1H), 6.55–6.52 (m, 1H), 5.23 (s, 1H), 3.51–3.46 (m, 1H), 3.16 (s, 1H), 2.96–2.84 (m, 2H), 2.79–2.73 (m, 2H), 1.73–1.68 (m, 2H), 1.61–1.53 (m, 2H), 1.37 (s, 9H). LCMS tR = 1.057 min in 2.5 min chromatography, MS ESI calcd. for C18H27N6O2+ [M + H]+ 359.22, found 359.2.
tert-Butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (228)
To a solution of tert-butyl (S)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (214) (190 mg, 0.53 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (74 mg, 0.32 mmol) in dioxane (5 mL) was added Cs2CO3 (518 mg, 1.59 mmol) and Brettphos Pd G3 (74 mg, 0.08 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–1%, MeOH/DCM). 228 (45 mg, 11.1%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.84–8.81 (m, 1H), 8.76–8.70 (m, 1H), 8.39–8.35 (m, 1H), 7.86–7.83 (m, 1H), 7.58–7.52 (m, 1H), 7.30–7.21 (m, 1H), 7.07–7.01 (m, 1H), 6.84–6.77 m, 1H), 5.75 (s, 1H), 3.57 (s, 1H), 3.29–3.26 (m, 2H), 2.98–2.90 (m, 2H), 2.83–2.76 (m, 2H), 1.78–1.74 (m, 2H), 1.67–1.62 (m, 2H), 1.38 (s, 9H), 0.99–0.89 (m, 2H), 0.87–0.82 (m, 2H). LCMS tR = 0.93 min in 1.5 min chromatography, MS ESI calcd. for C28H34N11O2+ [M + H]+ 556.29, found 556.3.
(S)-5-((4-(3-Aminopiperidin-1-yl)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (68)
To a solution of tert-butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (228) (38 mg, 0.07 mmol) in DCM (4 mL) was added TFA (1 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h. Then the mixture was degassed and purged with N2. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 18–58%, 11 min). 68 (11.2 mg, 35.6%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.84 (s, 2H), 8.37 (s, 1H), 8.30 (br s, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.69 (dd, J = 8.8, 2.6 Hz, 1H), 7.27 (d, J = 8.8 Hz, 1H), 5.97 (s, 1H), 2.81 (dd, J = 10.9, 3.8 Hz, 1H), 2.70–2.55 (m, 3H), 2.41 (td, J = 11.1, 2.8 Hz, 1H), 2.26 (dd, J = 10.9, 8.9 Hz, 1H), 1.81–1.70 (m, 1H), 1.61–1.51 (m, 1H), 1.43–1.28 (m, 1H), 1.07–0.94 (m, 1H), 0.91–0.77 (m, 2H), 0.77–0.65 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.55, 150.87, 148.41, 145.04, 142.76, 141.44, 136.39, 128.13, 121.75, 120.27, 116.97, 114.75, 76.68, 76.38, 60.42, 51.87, 47.90, 33.25, 23.82, 23.34, 6.57. HPLC tR = 3.237 min in 8 min chromatography, purity 96.9%. LCMS tR = 1.744 min in 4 min chromatography, MS ESI calcd. for C23H26N11+ [M + H]+ 456.24, found 456.4.
tert-Butyl (S)-methyl(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (197)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (300 mg, 1.44 mmol) and tert-butyl (S)-methyl(piperidin-3-yl)carbamate (340 mg, 1.59 mmol) in MeCN (2 mL) was added K2CO3 (598 mg, 4.32 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–17%, MeOH/DCM). 197 (162 mg, 12.5%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.29–8.25 (m, 1H), 8.01 (d, J = 9.6 Hz, 2H), 7.43 (d, J = 9.6 Hz, 1H), 3.63–3.58 (m, 2H), 3.14–3.11 (m, 1H), 2.92–2.74 (m, 2H), 2.70–2.65 (m, 3H), 1.76–1.74 (m, 4H), 1.42 (s, 9H). LCMS tR = 0.514 min in 1 min chromatography, MS ESI calcd. for C19H27N6O4+ [M + H]+ 403.21, found 403.2.
tert-Butyl (S)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (215)
To a solution of tert-butyl (S)-methyl(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (197) (977 mg, 2.43 mmol) in MeOH (10 mL) was added Pd/C (600 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (8 mL × 2). The combined filtrate was concentrated in vacuo. 215 (803 mg, 76.8%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 2H), 7.08 (d, J = 8.4 Hz, 1H), 6.65–6.62 (m, 1H), 6.54 (d, J = 2.4 Hz, 1H), 5.24 (s, 2H), 4.13 (br s, 1H), 3.73–3.72 (m, 1H), 3.17 (s, 2H), 2.66–2.64 (m, 4H), 1.59 (br d, J = 8.4 Hz, 2H), 1.40 (d, J = 2.0 Hz, 2H), 1.39 (s, 9H). LCMS tR = 1.125 min in 2.5 min chromatography, MS ESI calcd. for C19H29N6O2+ [M + H]+ 373.23, found 373.1.
tert-Butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (229)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (330 mg, 1.41 mmol) and tert-butyl (S)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (215) (368 mg, 0.99 mmol) in dioxane (5 mL) was added Cs2CO3 (1.38g, 4.24 mmol) and Brettphos Pd G3 (128 mg, 0.14 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–4%, MeOH/DCM). 229 (669 mg, 20.7%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H), 8.81 (s, 2H), 8.36–8.35 (m, 1H), 8.30 (s, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.75–7.73 (m, 1H), 7.34 (d, J = 8.8 Hz, 1H), 5.96 (s, 1H), 4.10 (br d, J = 4.8 Hz, 1H), 3.34 (s, 2H), 2.75–2.63 (m, 4H), 2.61–2.58 (m, 2H), 1.62 (br s, 2H), 1.52–1.48 (m, 2H), 1.39 (s, 9H), 0.82–0.79 (m, 2H), 0.78–0.72 (m, 2H). LCMS tR = 1.528 min in 2.5 min chromatography, MS ESI calcd. for C29H36N11O2+ [M + H]+ 570.30, found 570.2.
(S)-7-(Cyclopropylamino)-5-((4-(3-(methylamino)piperidin-1-yl)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (69)
To a solution of tert-butyl (S)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (229) (610 mg, 1.07 mmol) in DCM (6 mL) was added TFA (1.85 g, 16.21 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 5–55%, 10 min). 69 (75.8 mg, 14.3%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.84 (s, 2H), 8.36 (s, 1H), 8.29 (s, 1H), 7.92 (d, J = 2.5 Hz, 1H), 7.69 (dd, J = 8.8, 2.6 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 5.96 (s, 1H), 2.94–2.81 (m, 1H), 2.71–2.64 (m, 1H), 2.61 (tt, J = 6.9, 3.6 Hz, 1H), 2.48–2.42 (m, 1H), 2.38–2.28 (m, 1H), 2.19 (dd, J = 11.0, 9.1 Hz, 1H), 2.13 (s, 3H), 1.83–1.71 (m, 1H), 1.61–1.53 (m, 1H), 1.45–1.32 (m, 1H), 1.07–0.94 (m, 1H), 0.86–0.78 (m, 2H), 0.74–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.55, 150.86, 148.40, 145.01, 142.76, 141.52, 136.28, 127.98, 121.54, 120.25, 117.02, 114.73, 76.67, 76.37, 57.11, 56.01, 51.86, 33.37, 30.25, 23.78, 23.34, 6.56. HPLC tR = 3.252 min in 8 min chromatography, purity 95.4%. LCMS tR = 1.214 min in 4 min chromatography, MS ESI calcd. for C24H28N11+ [M + H]+ 470.25, found 470.4.
tert-Butyl (R)-methyl(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (198)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (300 mg, 1.44 mmol) and tert-butyl (R)-methyl(piperidin-3-yl)carbamate (340 mg, 1.59 mmol) in MeCN (2 mL) was added K2CO3 (598 mg, 4.32 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 10 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–12%, MeOH/DCM). 198 (367 mg, 42.0%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.94 (s, 2H), 8.26–8.25 (m, 2H), 7.42–7.40 (m, 1H), 3.17 (d, J = 4.4 Hz, 1H), 2.93–2.88 (m, 2H), 2.73 (d, J = 2.4 Hz, 2H), 2.68 (br s, 3H), 1.77–1.74 (m, 2H), 1.65–1.59 (m, 2H), 1.40 (s, 9H). LCMS tR = 0.514 min in 1 min chromatography, MS ESI calcd. for C19H27N6O4+ [M + H]+ 403.21, found 403.1.
tert-Butyl (R)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (216)
To a solution of tert-butyl (R)-methyl(1-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (198) (360 mg, 0.89 mmol) in MeOH (3 mL) was added Pd/C (150 mg, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 for 3 h. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (8 mL × 2). The combined filtrate was concentrated in vacuo. 216 (274 mg, 41.5%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 2H), 7.99 (d, J = 10.0 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.54 (d, J = 2.4 Hz, 1H), 5.25 (s, 2H), 3.81–3.64 (m, 2H), 2.73–2.71 (m, 3H), 2.66 (br s, 3H), 1.73 (br d, J = 8.0 Hz, 2H), 1.59 (br d, J = 9.6 Hz, 2H), 1.37 (s, 9H). LCMS tR = 1.182 min in 2.5 min chromatography, ESI calcd. for C19H29N6O2+ [M + H]+ 373.23, found 373.2.
tert-Butyl (R)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (230)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (80 mg, 0.34 mmol) and tert-butyl (R)-(1-(4-amino-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (216) (115 mg, 0.31 mmol) in dioxane (5 mL) was added Cs2CO3 (335 mg, 1.03 mmol) and Brettphos Pd G3 (31 mg, 0.03 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–5%, MeOH/DCM). 230 (189 mg, 29.4%) was obtained as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.80 (s, 2H), 8.36–8.35 (m, 1H), 8.29 (s, 1H), 7.88 (d, J = 2.4 Hz, 1H), 7.76–7.73 (m, 1H), 7.35 (d, J = 8.8 Hz, 1H), 5.96 (s, 1H), 3.85–3.76 (m, 1H), 2.69–2.67 (m, 5H), 2.61–2.59 (m, 2H), 2.46 (br s, 1H), 1.62 (br s, 2H), 1.52–1.49 (m, 1H), 1.40 (s, 9H), 1.23 (s, 1H), 0.82–0.80 (m, 2H), 0.74–0. 72 (m, 2H). LCMS tR = 1.552 min in 2.5 min chromatography, MS ESI calcd. for C29H36N11O2+ [M + H]+ 570.30, found 570.3.
(R)-7-(Cyclopropylamino)-5-((4-(3-(methylamino)piperidin-1-yl)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (70)
To a solution of tert-butyl (R)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (230) (160 mg, 0.28 mmol) in DCM (3 mL) was added TFA (924 mg, 0.01 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex C18 75 × 30 mm × 3 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 5–65%, 12 min). 70 (22.5 mg, 16.5%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.84 (s, 2H), 8.36 (s, 1H), 8.29 (br s, 1H), 7.91 (d, J = 2.5 Hz, 1H), 7.69 (dd, J = 8.8, 2.6 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 5.96 (s, 1H), 2.88 (dd, J = 11.1, 3.6 Hz, 1H), 2.72–2.64 (m, 1H), 2.61 (tt, J = 6.9, 3.6 Hz, 1H), 2.48–2.41 (m, 1H), 2.33 (tt, J = 9.2, 3.7 Hz, 1H), 2.18 (dd, J = 10.9, 9.0 Hz, 1H), 2.13 (s, 3H), 1.86–1.68 (m, 1H), 1.63–1.50 (m, 1H), 1.45–1.31 (m, 1H), 1.09–0.92 (m, 1H), 0.86–0.75 (m, 2H), 0.75–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.54, 150.86, 148.39, 145.00, 142.75, 141.53, 136.27, 127.98, 121.54, 120.25, 117.02, 114.73, 76.66, 76.36, 57.15, 56.02, 51.86, 33.39, 30.28, 23.79, 23.33, 6.56. HPLC tR = 3.310 min in 8 min chromatography, purity 96.9%. LCMS tR = 1.244 min in 4 min chromatography, MS ESI calcd. for C24H28N11+ [M + H]+ 470.25, found 470.4.
tert-Butyl (R)-(1-(4-amino-5-fluoro-2-nitrophenyl)piperidin-3-yl)(methyl)carbamate (236)
To a solution of 2,4-difluoro-5-nitroaniline (231) (2.00 g, 11.49 mmol) and tert-butyl (R)-methyl(piperidin-3-yl)carbamate (2.71 g, 12.64 mmol) in MeCN (20 mL) was added K2CO3 (4.76 g, 34.46 mmol). Then the reaction mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0–18%, EtOAc/PE). 236 (3.70 g, 85.7%) was obtained as a red solid. 1H NMR (400 MHz, DMSO-d6) δ 7.26–7.12 (m, 2H), 5.46 (s, 2H), 4.03–3.84 (m, 1H), 2.94–2.79 (m, 2H), 2.75 (s, 1H), 2.71 (s, 3H), 2.65–2.52 (m, 1H), 1.78–1.67 (m, 2H), 1.60–1.49 (m, 2H), 1.39 (s, 9H). LCMS tR = 0.763 min in 1 min chromatography, MS ESI calcd. for C17H26FN4O4+ [M + H]+ 369.19, found 369.1.
tert-Butyl (R)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)piperidin-3-yl)(methyl)carbamate (241)
To a solution of tert-butyl (R)-(1-(4-amino-5-fluoro-2-nitrophenyl)piperidin-3-yl)carbamate (236) (3.7 g, 10.04 mmol) in THF (30 mL) was added K2CO3 (4.16 g, 30.13 mmol). Then CbzCl (2.57 g, 15.07 mmol) was added to the mixture at 0 °C. The mixture was stirred at 25 °C for 12 h. The reaction mixture was concentrated directly. Saturated NH4Cl (100 mL) solution was added. The resulting mixture was extracted with EtOAc (100 mL × 3). The combined organic phase was washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash silica gel chromatography (eluent of 0–20%, EtOAc/PE). 241 (5.14 g, 99.9%) was obtained as a red oil. 1H NMR (400 MHz, DMSO-d6) δ 7.45–7.21 (m, 8H), 5.16 (s, 2H), 4.05–3.86 (m, 1H), 3.11–3.04 (m, 1H), 3.03–2.97 (m, 1H), 2.94–2.85 (m, 1H), 2.72 (s, 4H), 1.78–1.70 (m, 2H), 1.65–1.56 (m, 2H), 1.40 (s, 9H). LCMS tR = 0.830 min in 1 min chromatography, MS ESI calcd. for C25H32FN4O6+ [M + H]+ 503.23, found 503.2.
tert-Butyl (R)-(1-(2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)piperidin-3-yl)(methyl)carbamate (246)
To a solution of tert-butyl (R)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-nitrophenyl)piperidin-3-yl)carbamate (241) (5.14 g, 10.23 mmol) in EtOH (30 mL) was added NH4Cl (3.28 g, 61.37 mmol) dissolved in H2O (15 mL) and Fe (1.71 g, 30.68 mmol) at 25 °C. Then the mixture was stirred at 100 °C under N2 atmosphere for 12 h. The reaction mixture was filtered and the solid was washed with MeOH (50 mL × 3). The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 11%, EtOAc/PE) to give the product. 246 (2.92 g, 48.1%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 7.43–7.30 (m, 7H), 6.89–6.82 (m, 1H), 6.77 (d, J = 12.4 Hz, 1H), 5.11 (s, 2H), 4.64 (s, 2H), 4.08–3.89 (m, 1H), 2.99–2.92 (m, 1H), 2.73 (s, 3H), 1.82–1.62 (m, 4H), 1.61–1.48 (m, 2H), 1.39 (s, 9H). LCMS tR = 1.651 min in 2.5 min chromatography, MS ESI calcd. for C25H34FN4O4+ [M + H]+ 473.26, found 473.3.
tert-Butyl (R)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (251)
To a solution of tert-butyl (R)-(1-(2-amino-4-(((benzyloxy)carbonyl)amino)-5-fluorophenyl)piperidin-3-yl)carbamate (246) (2.92 g, 6.18 mmol) and compound 2A (2.72 g, 30.90 mmol) in pyridine (20 mL) was added Et3N (4.38 g, 43.25 mmol) and Me3SiCl (10.07 g, 92.69 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated directly. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 35%, EtOAc/PE) to give the product. 251 (1.52 g, 15.8%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.63–9.57 (m, 1H), 8.87 (s, 1H), 8.80 (s, 1H), 7.45–7.38 (m, 7H), 5.15 (s, 3H), 2.71–2.65 (m, 5H), 1.78–1.68 (m, 2H), 1.66–1.52 (m, 4H), 1.43–1.37 (m, 9H). LCMS tR = 2.298 min in 4 min chromatography, MS ESI calcd. for C27H34FN6O4+ [M + H]+ 525.26, found 525.4.
tert-Butyl (R)-(1-(4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (256)
To a solution of tert-butyl (R)-(1-(4-(((benzyloxy)carbonyl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (251) (1.51 g, 2.88 mmol) in MeOH (10 mL) was added Pd/C (1.56 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 25 °C for 2 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (30 mL × 3). The combined filtrate was concentrated in vacuo. 256 (1.49 g, 36.3%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.71–8.68 (m, 1H), 7.12–7.07 (m, 1H), 6.79–6.74 (m, 1H), 5.31–5.24 (m, 2H), 2.83–2.79 (m, 1H), 2.76–2.69 (m, 3H), 2.66 (s, 4H), 2.61–2.58 (m, 1H), 1.62–1.55 (m, 3H), 1.41–1.35 (m, 9H). LCMS tR = 1.294 min in 2 min chromatography, MS ESI calcd. for C19H28FN6O2+ [M + H]+ 391.23, found 391.0.
tert-Butyl (R)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)(methyl)carbamate (261)
To a solution of tert-butyl (R)-(1-(4-amino-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (256) (500 mg, 0.51 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (108 mg, 0.46 mmol) in dioxane (8 mL) was added Cs2CO3 (501 mg, 1.54 mmol), BINAP (48 mg, 0.08 mmol), and Pd(OAc)2 (17 mg, 0.08 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–3%, MeOH/DCM). 261 (180 mg, 41.5%) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 8.77 (s, 1H), 8.35 (s, 1H), 8.33–8.30 (m, 1H), 8.28–8.21 (m, 1H), 7.44–7.23 (m, 3H), 6.18 (s, 1H), 1.79–1.55 (m, 4H), 1.41–1.38 (m, 9H), 1.34 (s, 2H), 0.87–0.77 (m, 2H), 0.75–0.66 (m, 2H). LCMS tR = 0.643 min in 1 min chromatography, MS ESI calcd. for C29H35FN11O2+ [M + H]+ 588.29, found 588.2.
(R)-7-(Cyclopropylamino)-5-((2-fluoro-4-(3-(methylamino)piperidin-1-yl)-5-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (71)
To a mixture of tert-butyl (R)-(1-(4-((3-cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-yl)amino)-5-fluoro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperidin-3-yl)carbamate (261) (180 mg, 0.31 mmol) in DCM (3 mL) was added TFA (3 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 3 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep. HPLC (column: Welch Xtimate C18 150 × 30 mm × 5 μm; mobile phase: [water (FA)-ACN]; B%: 0–34%, 25 min). 71 as a formate salt (40 mg, 25.2%) was obtained as a gray solid. 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H), 8.83 (s, 2H), 8.36 (s, 1H), 8.33 (s, 1H), 8.29 (dd, J = 5.1, 2.4 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 12.5 Hz, 1H), 6.18 (s, 1H), 3.02 (d, J = 11.1 Hz, 1H), 2.71–2.62 (m, 2H), 2.63–2.56 (m, 1H), 2.48–2.36 (m, 2H), 2.30 (s, 3H), 1.89 (d, J = 12.1 Hz, 1H), 1.65–1.54 (m, 1H), 1.38 (q, J = 11.7 Hz, 1H), 1.25–1.12 (m, 1H), 0.86–0.78 (m, 2H), 0.75–0.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.60, 160.31, 156.86, 153.77 (d, J = 247.1 Hz), 150.72, 148.55, 145.03, 142.85, 123.60, 122.94 (d, J = 13.9 Hz), 121.80, 114.70, 108.82 (d, J = 22.5 Hz), 76.58, 76.51, 55.07, 54.23, 51.26, 31.49, 28.08, 23.38, 23.19, 6.55. HPLC tR = 3.324 min in 8 min chromatography, purity 95.2%. LCMS tR = 1.655 min in 4 min chromatography, MS ESI calcd. for C24H27FN11+ [M + H]+ 488.24, found 488.5.
4-(4-Nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)morpholine (199)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (500 mg, 2.40 mmol) and morpholine (313.91 mg, 3.60 mmol) in MeCN (15 mL) was added K2CO3 (996 mg, 7.21 mmol) at 25 °C. Then the mixture was stirred at 100 °C for 12 h. The reaction mixture was concentrated in vacuo. Water (50 mL) was added to the mixture. The residue was extracted with DCM (50 mL × 2). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. 199 (1.16 g, crude) was obtained as a black brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 8.31–8.26 (m, 1H), 8.02 (s, 1H), 7.48–7.32 (m, 1H), 3.60–3.57 (m, 4H), 3.40–3.35 (m, 4H). LCMS tR = 0.921 min in 2.5 min chromatography, MS ESI calcd. for C12H14N5O3+ [M + H]+ 276.11, found 276.2.
4-Morpholino-3-(4H-1,2,4-triazol-4-yl)aniline (217)
To a solution of 4-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)morpholine (199) (750 mg, 2.72 mmol) in MeOH (20 mL) was added Pd/C (0.70 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. Then the reaction mixture was stirred under H2 (15 psi) at 25 °C for 10 h. The reaction mixture was filtered via a Celite pad. The pad was washed with MeOH (15 mL × 3). The residue was purified by flash silica gel chromatography (eluent of 0–6%, MeOH/DCM). 217 (516 mg, 77.2%) was obtained as a black brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.82–8.76 (m, 1H), 8.02 (s, 1H), 7.16–6.94 (m, 1H), 6.67–6.54 (m, 1H), 5.25 (s, 1H), 3.56–3.55 (m, 2H), 3.38–3.38 (m, 2H), 2.75–2.71 (m, 3H), 2.55–2.53 (m, 2H). LCMS tR = 0.593 min in 2.5 min chromatography, MS ESI calcd. for C12H16N5O+ [M + H]+ 246.13, found 246.1.
7-(Cyclopropylamino)-5-((4-morpholino-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (72)
To a solution of 4-morpholino-3-(4H-1,2,4-triazol-4-yl)aniline (217) (140 mg, 0.34 mmol) and 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (40 mg, 0.17 mmol) in dioxane (3 mL) was added Cs2CO3 (167 mg, 0.51 mmol), BINAP (16 mg, 0.03 mmol), and Pd(OAc)2 (6 mg, 0.03 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by flash silica gel chromatography (eluent of 0–3%, MeOH/DCM). 72 (15 mg, 19.8%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 8.89 (s, 2H), 8.37 (s, 1H), 8.31 (d, J = 1.9 Hz, 1H), 7.94 (d, J = 2.5 Hz, 1H), 7.73 (dd, J = 8.8, 2.5 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 5.97 (s, 1H), 3.55 (dd, J = 5.9, 3.2 Hz, 4H), 2.65–2.58 (m, 5H), 0.87–0.79 (m, 2H), 0.75–0.70 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 156.51, 150.81, 148.41, 145.00, 142.75, 140.46, 136.76, 128.09, 121.46, 120.21, 116.99, 114.66, 76.70, 76.40, 66.10, 51.33, 23.32, 6.53. HPLC tR = 4.039 min in 8 min chromatography, purity 97.5%. LCMS tR = 2.172 min in 1 min chromatography, MS ESI calcd. for C22H23N10O+ [M + H]+ 443.21, found 443.1.
1-Methyl-4-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperazine (200)
To a solution of 4-(2-fluoro-5-nitrophenyl)-4H-1,2,4-triazole (176) (1 g, 4.80 mmol) and 1-methylpiperazine (529 mg, 5.28 mmol) in MeCN (10 mL) was added K2CO3 (664 mg, 4.80 mmol) at 25 °C. The mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated in vacuo. Water (30 mL) was added to the residue. The residue was purified by flash silica gel chromatography (eluent of 0% ∼ 3%, MeOH/DCM) to give the product. 200 (918 mg, 59.1%) was obtained as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 2H), 8.30–8.22 (m, 2H), 7.39–7.34 (m, 1H), 2.81–2.75 (m, 4H), 2.33–2.30 (m, 4H), 2.18 (s, 3H). LCMS tR = 0.137 min in 1 min chromatography, MS ESI calcd. for C13H17N6O2+ [M + H]+ 289.14, found 289.1.
4-(4-Methylpiperazin-1-yl)-3-(4H-1,2,4-triazol-4-yl)aniline (218)
To a solution of 1-methyl-4-(4-nitro-2-(4H-1,2,4-triazol-4-yl)phenyl)piperazine (200) (910 mg, 3.16 mmol) in MeOH (10 mL) was added Pd/C (0.82 g, 10% Pd) under N2 atmosphere. The suspension was degassed and purged with H2 three times. The reaction mixture was stirred under H2 (15 psi) at 35 °C for 2 h. The reaction mixture was filtered. The filter cake was washed with MeOH (20 mL × 3). The combined filtrate was concentrated in vacuo. 218 (744 mg, crude) was obtained as a gray solid. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 2H), 7.06–7.01 (m, 1H), 6.66–6.61 (m, 1H), 6.56–6.53(m, 1H), 5.22 (s, 2H), 2.57–2.52 (m, 4H), 2.30–2.19 (m, 4H), 2.12 (s, 3H).
7-(Cyclopropylamino)-5-((4-(4-methylpiperazin-1-yl)-3-(4H-1,2,4-triazol-4-yl)phenyl)amino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (73)
To a solution of 5-chloro-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (75) (300 mg, 1.28 mmol) and 4-(4-methylpiperazin-1-yl)-3-(4H-1,2,4-triazol-4-yl)aniline (218) (498 mg, 1.93 mmol) in dioxane (5 mL) was added K2CO3 (532 mg, 3.85 mmol), BINAP (120 mg, 0.19 mmol), and Pd(OAc)2 (43 mg, 0.19 mmol) at 25 °C. Then the mixture was degassed and purged with N2. The reaction mixture was heated in a microwave reactor at 130 °C for 0.5 h. The reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC (column: Xtimate C18 150 × 40 mm × 10 μm; mobile phase: [water (NH3H2O+NH4HCO3)-ACN]; B%: 20–60%, 10 min). 73 (139 mg, 23.8%) was obtained as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.84 (s, 2H), 8.36 (d, J = 0.9 Hz, 1H), 8.20 (br s, 1H), 7.93 (dd, J = 2.5, 1.1 Hz, 1H), 7.70 (dd, J = 8.8, 2.5 Hz, 1H), 7.29 (dd, J = 8.9, 1.5 Hz, 1H), 5.96 (s, 1H), 2.69–2.56 (m, 5H), 2.28 (br s, 4H), 2.15 (s, 3H), 0.81 (td, J = 7.2, 6.6, 5.0 Hz, 2H), 0.76–0.67 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 156.53, 150.85, 148.40, 145.02, 142.75, 140.74, 136.56, 127.98, 121.38, 120.18, 116.97, 114.74, 76.70, 76.41, 54.64, 50.83, 45.74, 23.34, 6.57. HPLC tR = 2.941 min in 8 min chromatography, purity 99.9%. LCMS tR = 1.642 min in 4 min chromatography, MS ESI calcd. for C23H26N11+ [M + H]+ 456.24, found 456.5.
Acknowledgments
The Structural Genomics Consortium (SGC) is a registered charity (no. 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and United States), Pfizer, and Takeda. Research reported in this publication was supported in part by the NC Biotech Center Institutional support grant 2018-IDG-1030, by the NIH Illuminating the Druggable Genome 1U24DK116204-01, and Department of Defense ALSRP award AL190107. This project was supported by the Rapidly Emerging Antiviral Drug Development Initiative (READDI) at the University of North Carolina at Chapel Hill with funding from the North Carolina Coronavirus State and Local Fiscal Recovery Funds program, appropriated by the North Carolina General Assembly. Additional funding was provided by a grant from Millennium Pharmaceuticals (Takeda). This work was supported by NIH grant S10OD032476 for upgrading the 500 MHz NMR spectrometer in the UNC Eshelman School of Pharmacy NMR Facility. Constructs for NanoBRET measurements of CSNK2A1 and CSNK2A2 were provided by Promega Corporation (Madison, WI). We thank Laszlo Gyenis and David Litchfield for providing us with the EIF2S2 P-S2 antibody. We thank K. Saikatendu Singh (Takeda, San Diego, CA) for facilitating collaborative interactions and constructive criticism throughout the project. WuXi AppTec (Shanghai, China) provided the chemical synthesis support. Analiza, Inc. (Cleveland, OH) performed kinetic solubility and mouse liver microsome studies. Pharmaron (Beijing, China) provided the in vitro and in vivo pharmacokinetic assay support. The kinome tree in Figure 4 was generated using CORAL.34
Glossary
ABBREVIATIONS USED
- AKT
RAC-alpha serine/threonine-protein kinase
- b.i.d.
twice daily
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- BRET
bioluminescence resonance energy transfer
- CME
clathrin-mediated endocytosis
- COVID-19
Coronavirus Disease 2019
- CSNK2
casein kinase II
- CSNK2A1
casein kinase II subunit alpha
- CSNK2A2
casein kinase II subunit alpha’
- DBU
1,8-diazabicyclo(5.4.0)undec-7-ene
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- EIF2S2
eukaryotic translation initiation factor 2 subunit 2
- HLM
human liver microsome
- HPLC
high-performance liquid chromatography
- i.p.
intraperitoneal
- i.v.
intraveneous
- IPTG
isopropyl-thio-galactopyranoside
- LC-MS
liquid chromatography mass spectrometry
- LC-TOFMS
liquid chromatography time-of-flight mass spectrometry
- LDH
lactate dehydrogenase
- LOD
limit of detection
- mBRET
milliBRET
- mCPBA
meta-chloroperoxybenzoic acid
- MDR1
ATP-dependent translocase ABCB1
- MHV
mouse hepatitis virus
- MLM
mouse liver microsome
- MS
mass spectrometry
- NanoBRET
nanoluciferase bioluminescence resonance energy transfer
- NLuc
nanoluciferase
- NMP
N-methyl-2-pyrrolidone
- p.o.
oral administration
- PBS
phosphate-buffered saline
- PFU
plaque forming units
- PK
pharmacokinetics
- PVDF
polyvinylidene difluoride
- RLU
relative light unit
- SAR
structure–activity relationship
- SN2
bimolecular nucleophilic substitution
- SNAr
nucleophilic aromatic substitution
- TBAF
tetra-n-butylammonium fluoride
- TBDMS
tert-butyldimethylsilyl
- TBDPS
tert-butyldiphenylsilyl
- TEER
transepithelial electrical resistance
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00962.
Eurofins KinaseProfiler radiometric enzymatic assay results; NanoBRET in digitonin-permeabilized cells; phosphorylation of EIF2S2 in A549ACE2 cells in vitro; comparison between CSNK2A2 and CSNK2A1 NanoBRET pIC50s; crystallographic refinement statistics; NanoBRET K192 Selectivity Panel results; 1H and 13C NMR spectra for analogues synthesized; HPLC trace for compound 53 (PDF)
Molecular formula strings with associated biochemical and biological data (CSV)
Accession Codes
The crystal structures of CSNK2A1 with compounds 14 (PDB ID: 8P07), 50 (PDB ID: 8P06), and 53 (PDB ID: 9EZG) have been deposited in the PDB.
Author Contributions
# H. W. Ong and X. Yang contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization . WHO COVID-19 Dashboard. https://covid19.who.int/ (accessed 2024–04–22).
- Payne S.Family Coronaviridae. In Viruses; Elsevier, 2017; pp 149–158. [Google Scholar]
- Shereen M. A.; Khan S.; Kazmi A.; Bashir N.; Siddique R. COVID-19 Infection: Emergence, Transmission, and Characteristics of Human Coronaviruses. J. Adv. Res. 2020, 24, 91–98. 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Sharma S.; Kumar R.; Tripathi B. N.; Barua S.; Ly H.; Rouse B. T. Host-Directed Antiviral Therapy. Clin. Microbiol. Rev. 2020, 33, 1–36. 10.1128/CMR.00168-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi D.; Sodani M.; Gupta P. K.; Kulkarni S. Host Directed Therapies: COVID-19 and Beyond. Curr. Res. Pharmacol. Drug Discovery 2021, 2, 100058 10.1016/j.crphar.2021.100058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X.; Li Z. Medicinal Chemistry Strategies toward Host Targeting Antiviral Agents. Med. Res. Rev. 2020, 40 (5), 1519–1557. 10.1002/med.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating J. A.; Striker R. Phosphorylation Events during Viral Infections Provide Potential Therapeutic Targets. Rev. Med. Virol. 2012, 22, 166–181. 10.1002/rmv.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada Meza C. P.; Ruzzene M. Protein Kinase CK2 and SARS-CoV-2: An Expected Interplay Story. Kinases and Phosphatases 2023, 1, 141–150. 10.3390/kinasesphosphatases1020009. [DOI] [Google Scholar]
- Bouhaddou M.; Memon D.; Meyer B.; White K. M.; Rezelj V. V.; Correa Marrero M.; Polacco B. J.; Melnyk J. E.; Ulferts S.; Kaake R. M.; Batra J.; Richards A. L.; Stevenson E.; Gordon D. E.; Rojc A.; Obernier K.; Fabius J. M.; Soucheray M.; Miorin L.; Moreno E.; Koh C.; Tran Q. D.; Hardy A.; Robinot R.; Vallet T.; Nilsson-Payant B. E.; Hernandez-Armenta C.; Dunham A.; Weigang S.; Knerr J.; Modak M.; Quintero D.; Zhou Y.; Dugourd A.; Valdeolivas A.; Patil T.; Li Q.; Hüttenhain R.; Cakir M.; Muralidharan M.; Kim M.; Jang G.; Tutuncuoglu B.; Hiatt J.; Guo J. Z.; Xu J.; Bouhaddou S.; Mathy C. J. P.; Gaulton A.; Manners E. J.; Félix E.; Shi Y.; Goff M.; Lim J. K.; McBride T.; O’Neal M. C.; Cai Y.; Chang J. C. J.; Broadhurst D. J.; Klippsten S.; De wit E.; Leach A. R.; Kortemme T.; Shoichet B.; Ott M.; Saez-Rodriguez J.; TenOever B. R.; Mullins R. D.; Fischer E. R.; Kochs G.; Grosse R.; García-Sastre A.; Vignuzzi M.; Johnson J. R.; Shokat K. M.; Swaney D. L.; Beltrao P.; Krogan N. J. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712. 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; White K. M.; O’Meara M. J.; Rezelj V. V.; Guo J. Z.; Swaney D. L.; Tummino T. A.; Hüttenhain R.; Kaake R. M.; Richards A. L.; Tutuncuoglu B.; Foussard H.; Batra J.; Haas K.; Modak M.; Kim M.; Haas P.; Polacco B. J.; Braberg H.; Fabius J. M.; Eckhardt M.; Soucheray M.; Bennett M. J.; Cakir M.; McGregor M. J.; Li Q.; Meyer B.; Roesch F.; Vallet T.; Mac Kain A.; Miorin L.; Moreno E.; Naing Z. Z. C.; Zhou Y.; Peng S.; Shi Y.; Zhang Z.; Shen W.; Kirby I. T.; Melnyk J. E.; Chorba J. S.; Lou K.; Dai S. A.; Barrio-Hernandez I.; Memon D.; Hernandez-Armenta C.; Lyu J.; Mathy C. J. P.; Perica T.; Pilla K. B.; Ganesan S. J.; Saltzberg D. J.; Rakesh R.; Liu X.; Rosenthal S. B.; Calviello L.; Venkataramanan S.; Liboy-Lugo J.; Lin Y.; Huang X.-P.; Liu Y.; Wankowicz S. A.; Bohn M.; Safari M.; Ugur F. S.; Koh C.; Savar N. S.; Tran Q. D.; Shengjuler D.; Fletcher S. J.; O’Neal M. C.; Cai Y.; Chang J. C. J.; Broadhurst D. J.; Klippsten S.; Sharp P. P.; Wenzell N. A.; Kuzuoglu-Ozturk D.; Wang H.-Y.; Trenker R.; Young J. M.; Cavero D. A.; Hiatt J.; Roth T. L.; Rathore U.; Subramanian A.; Noack J.; Hubert M.; Stroud R. M.; Frankel A. D.; Rosenberg O. S.; Verba K. A.; Agard D. A.; Ott M.; Emerman M.; Jura N.; von Zastrow M.; Verdin E.; Ashworth A.; Schwartz O.; D’Enfert C.; Mukherjee S.; Jacobson M.; Malik H. S.; Fujimori D. G.; Ideker T.; Craik C. S.; Floor S. N.; Fraser J. S.; Gross J. D.; Sali A.; Roth B. L.; Ruggero D.; Taunton J.; Kortemme T.; Beltrao P.; Vignuzzi M.; García-Sastre A.; Shokat K. M.; Shoichet B. K.; Krogan N. J. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Hiatt J.; Bouhaddou M.; Rezelj V. V.; Ulferts S.; Braberg H.; Jureka A. S.; Obernier K.; Guo J. Z.; Batra J.; Kaake R. M.; Weckstein A. R.; Owens T. W.; Gupta M.; Pourmal S.; Titus E. W.; Cakir M.; Soucheray M.; McGregor M.; Cakir Z.; Jang G.; O’Meara M. J.; Tummino T. A.; Zhang Z.; Foussard H.; Rojc A.; Zhou Y.; Kuchenov D.; Hüttenhain R.; Xu J.; Eckhardt M.; Swaney D. L.; Fabius J. M.; Ummadi M.; Tutuncuoglu B.; Rathore U.; Modak M.; Haas P.; Haas K. M.; Naing Z. Z. C.; Pulido E. H.; Shi Y.; Barrio-Hernandez I.; Memon D.; Petsalaki E.; Dunham A.; Marrero M. C.; Burke D.; Koh C.; Vallet T.; Silvas J. A.; Azumaya C. M.; Billesbølle C.; Brilot A. F.; Campbell M. G.; Diallo A.; Dickinson M. S.; Diwanji D.; Herrera N.; Hoppe N.; Kratochvil H. T.; Liu Y.; Merz G. E.; Moritz M.; Nguyen H. C.; Nowotny C.; Puchades C.; Rizo A. N.; Schulze-Gahmen U.; Smith A. M.; Sun M.; Young I. D.; Zhao J.; Asarnow D.; Biel J.; Bowen A.; Braxton J. R.; Chen J.; Chio C. M.; Chio U. S.; Deshpande I.; Doan L.; Faust B.; Flores S.; Jin M.; Kim K.; Lam V. L.; Li F.; Li J.; Li Y.-L.; Li Y.; Liu X.; Lo M.; Lopez K. E.; Melo A. A.; Moss F. R.; Nguyen P.; Paulino J.; Pawar K. I.; Peters J. K.; Pospiech T. H.; Safari M.; Sangwan S.; Schaefer K.; Thomas P. V.; Thwin A. C.; Trenker R.; Tse E.; Tsui T. K. M.; Wang F.; Whitis N.; Yu Z.; Zhang K.; Zhang Y.; Zhou F.; Saltzberg D.; Hodder A. J.; Shun-Shion A. S.; Williams D. M.; White K. M.; Rosales R.; Kehrer T.; Miorin L.; Moreno E.; Patel A. H.; Rihn S.; Khalid M. M.; Vallejo-Gracia A.; Fozouni P.; Simoneau C. R.; Roth T. L.; Wu D.; Karim M. A.; Ghoussaini M.; Dunham I.; Berardi F.; Weigang S.; Chazal M.; Park J.; Logue J.; McGrath M.; Weston S.; Haupt R.; Hastie C. J.; Elliott M.; Brown F.; Burness K. A.; Reid E.; Dorward M.; Johnson C.; Wilkinson S. G.; Geyer A.; Giesel D. M.; Baillie C.; Raggett S.; Leech H.; Toth R.; Goodman N.; Keough K. C.; Lind A. L.; Klesh R. J.; Hemphill K. R.; Carlson-Stevermer J.; Oki J.; Holden K.; Maures T.; Pollard K. S.; Sali A.; Agard D. A.; Cheng Y.; Fraser J. S.; Frost A.; Jura N.; Kortemme T.; Manglik A.; Southworth D. R.; Stroud R. M.; Alessi D. R.; Davies P.; Frieman M. B.; Ideker T.; Abate C.; Jouvenet N.; Kochs G.; Shoichet B.; Ott M.; Palmarini M.; Shokat K. M.; García-Sastre A.; Rassen J. A.; Grosse R.; Rosenberg O. S.; Verba K. A.; Basler C. F.; Vignuzzi M.; Peden A. A.; Beltrao P.; Krogan N. J.; Owens T. W.; Gupta M.; Pourmal S.; Titus E. W.; Azumaya C. M.; Billesbølle C.; Brilot A. F.; Campbell M. G.; Diallo A.; Dickinson M. S.; Diwanji D.; Herrera N.; Hoppe N.; Kratochvil H. T.; Liu Y.; Merz G. E.; Moritz M.; Nguyen H. C.; Nowotny C.; Puchades C.; Rizo A. N.; Schulze-Gahmen U.; Smith A. M.; Sun M.; Young I. D.; Zhao J.; Asarnow D.; Biel J.; Bowen A.; Braxton J. R.; Chen J.; Chio C. M.; Chio U. S.; Deshpande I.; Doan L.; Faust B.; Flores S.; Jin M.; Kim K.; Lam V. L.; Li F.; Li J.; Li Y.-L.; Li Y.; Liu X.; Lo M.; Lopez K. E.; Melo A. A.; Moss F. R.; Nguyen P.; Paulino J.; Pawar K. I.; Peters J. K.; Pospiech T. H.; Safari M.; Sangwan S.; Schaefer K.; Thomas P. V.; Thwin A. C.; Trenker R.; Tse E.; Tsui T. K. M.; Wang F.; Whitis N.; Yu Z.; Zhang K.; Zhang Y.; Zhou F.; Trinidad D.; Agard D. A.; Cheng Y.; Fraser J. S.; Frost A.; Jura N.; Kortemme T.; Manglik A.; Southworth D. R.; Stroud R. M.; Rosenberg O. S.; Verba K. A.; Damas J.; Hughes G. M.; Keough K. C.; Painter C. A.; Persky N. S.; Corbo M.; Kirilenko B.; Hiller M.; Koepfli K.-P.; Kaplow I.; Wirthlin M.; Pfenning A. R.; Zhao H.; Genereux D. P.; Swofford R.; Lind A.; Pollard K. S.; Ryderq O. A.; Nweeia M. T.; Meadows J.; Dong M.; Wallerman O.; Marinescu V.; Lindblad-Toh K.; Ray D. A.; Power S.; Teeling E. C.; Chauhan G.; Li S. X.; Karlsson E. K.; Lewin H. A. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson F. M.; Gray N. S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discovery 2018, 17, 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Xie Z.; Yang X.; Duan Y.; Han J.; Liao C. Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. J. Med. Chem. 2021, 64, 1283–1345. 10.1021/acs.jmedchem.0c01511. [DOI] [PubMed] [Google Scholar]
- García-Cárceles J.; Caballero E.; Gil C.; Martínez A. Kinase Inhibitors as Underexplored Antiviral Agents. J. Med. Chem. 2022, 65, 935–954. 10.1021/acs.jmedchem.1c00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Dickmander R. J.; Bayati A.; Taft-Benz S. A.; Smith J. L.; Wells C. I.; Madden E. A.; Brown J. W.; Lenarcic E. M.; Yount B. L.; Chang E.; Axtman A. D.; Baric R. S.; Heise M. T.; McPherson P. S.; Moorman N. J.; Willson T. M. Host Kinase CSNK2 Is a Target for Inhibition of Pathogenic SARS-like β-Coronaviruses. ACS Chem. Biol. 2022, 17, 1937–1950. 10.1021/acschembio.2c00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón A. C.; Pérez G. V.; Caballero E.; Rosales M.; Aguilar D.; Vázquez-Blomquist D.; Ramos Y.; Rodríguez-Ulloa A.; Falcón V.; Rodríguez-Moltó M. P.; Yang K.; Perera Y.; Perea S. E. Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection. Viruses 2022, 14, 552. 10.3390/v14030552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Wang Y.; Wang J.; Zhou Z.; Cao S.; Zhang J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257–2281. 10.1021/acs.jmedchem.2c01523. [DOI] [PubMed] [Google Scholar]
- Ong H. W.; Drewry D. H.; Axtman A. D. CK2 Chemical Probes: Past, Present, and Future. Kinases and Phosphatases 2023, 1, 288–305. 10.3390/kinasesphosphatases1040017. [DOI] [Google Scholar]
- Wells C. I.; Drewry D. H.; Pickett J. E.; Tjaden A.; Krämer A.; Müller S.; Gyenis L.; Menyhart D.; Litchfield D. W.; Knapp S.; Axtman A. D. Development of a Potent and Selective Chemical Probe for the Pleiotropic Kinase CK2. Cell Chem. Biol. 2021, 28, 546–558. 10.1016/j.chembiol.2020.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist S. R.; Dinnon K. H.; Schäfer A.; Tse L. V.; Okuda K.; Hou Y. J.; West A.; Edwards C. E.; Sanders W.; Fritch E. J.; Gully K. L.; Scobey T.; Brown A. J.; Sheahan T. P.; Moorman N. J.; Boucher R. C.; Gralinski L. E.; Montgomery S. A.; Baric R. S. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 2020, 183, 1070–1085. 10.1016/j.cell.2020.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J. E.; Chuaqui C.; Pontz T. W.; Lyne P. D.; Larsen N. A.; Block M. H.; Chen H.; Su N.; Wu A.; Russell D.; Pollard H.; Lee J. W.; Peng B.; Thakur K.; Ye Q.; Zhang T.; Brassil P.; Racicot V.; Bao L.; Denz C. R.; Cooke E. Potent and Selective Inhibitors of CK2 Kinase Identified through Structure-Guided Hybridization. ACS Med. Chem. Lett. 2012, 3, 278–283. 10.1021/ml200257n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J. E.; Alimzhanov M.; Bao L.; Block M. H.; Chuaqui C.; Cooke E. L.; Denz C. R.; Hird A.; Huang S.; Larsen N. A.; Peng B.; Pontz T. W.; Rivard-Costa C.; Saeh J. C.; Thakur K.; Ye Q.; Zhang T.; Lyne P. D. Structure and Property Based Design of Pyrazolo[1,5-a]Pyrimidine Inhibitors of CK2 Kinase with Activity in Vivo. ACS Med. Chem. Lett. 2013, 4, 800–805. 10.1021/ml400197u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J. E.; Alimzhanov M.; Bao L.; Chuaqui C.; Denz C. R.; Jenkins E.; Larsen N. A.; Lyne P. D.; Pontz T.; Ye Q.; Holdgate G. A.; Snow L.; O’Connell N.; Ferguson A. D. Potent and Selective CK2 Kinase Inhibitors with Effects on Wnt Pathway Signaling in Vivo. ACS Med. Chem. Lett. 2016, 7, 300–305. 10.1021/acsmedchemlett.5b00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Ong H. W.; Dickmander R. J.; Smith J. L.; Brown J. W.; Tao W.; Chang E.; Moorman N. J.; Axtman A. D.; Willson T. M. Optimization of 3-Cyano-7-Cyclopropylamino-Pyrazolo[1,5-a]Pyrimidines toward the Development of an In Vivo Chemical Probe for CSNK2A. ACS Omega 2023, 8, 39546–39561. 10.1021/acsomega.3c05377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner R.; Majjouti M.; Alcazar M.; Mahabir E. Of Mice and Men: The Coronavirus MHV and Mouse Models as a Translational Approach to Understand SARS-CoV-2. Viruses 2020, 12, 880. 10.3390/v12080880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S.; Carmona A. V.; Tiwari A. K.; Trippier P. C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. 10.1021/acs.jmedchem.0c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington L. D.; Aquila B. M.; Choi Y.; Valiulin R. A.; Muegge I. Positional Analogue Scanning: An Effective Strategy for Multiparameter Optimization in Drug Design. J. Med. Chem. 2020, 63, 8956–8976. 10.1021/acs.jmedchem.9b02092. [DOI] [PubMed] [Google Scholar]
- Potęga A. Glutathione-Mediated Conjugation of Anticancer Drugs: An Overview of Reaction Mechanisms and Biological Significance for Drug Detoxification and Bioactivation. Molecules 2022, 27, 5252. 10.3390/molecules27165252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll J. P.; Yadav A. S.; Shah N. R. Role of Glucuronidation and P450 Oxidation in the Bioactivation of Bromfenac. Chem. Res. Toxicol. 2018, 31, 223–230. 10.1021/acs.chemrestox.7b00293. [DOI] [PubMed] [Google Scholar]
- Evison B. J.; Sleebs B. E.; Watson K. G.; Phillips D. R.; Cutts S. M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. 10.1002/med.21364. [DOI] [PubMed] [Google Scholar]
- Vasta J. D.; Corona C. R.; Wilkinson J.; Zimprich C. A.; Hartnett J. R.; Ingold M. R.; Zimmerman K.; Machleidt T.; Kirkland T. A.; Huwiler K. G.; Ohana R. F.; Slater M.; Otto P.; Cong M.; Wells C. I.; Berger B.-T.; Hanke T.; Glas C.; Ding K.; Drewry D. H.; Huber K. V. M.; Willson T. M.; Knapp S.; Müller S.; Meisenheimer P. L.; Fan F.; Wood K. V.; Robers M. B. Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 2018, 25, 206–214. 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V.; Masvidal L.; Cargnello M.; Gyenis L.; McLaughlan S.; Cai Y.; Tenkerian C.; Morita M.; Balanathan P.; Jean-Jean O.; Stambolic V.; Trost M.; Furic L.; Larose L.; Koromilas A. E.; Asano K.; Litchfield D.; Larsson O.; Topisirovic I. MTORC1 and CK2 Coordinate Ternary and EIF4F Complex Assembly. Nat. Commun. 2016, 7, 11127. 10.1038/ncomms11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robers M. B.; Wilkinson J. M.; Vasta J. D.; Berger L. M.; Berger B. T.; Knapp S. Single Tracer-Based Protocol for Broad-Spectrum Kinase Profiling in Live Cells with NanoBRET. STAR Protoc. 2021, 2, 100822 10.1016/j.xpro.2021.100822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz K. S.; Deoudes E. M.; Berginski M. E.; Jimenez-Ruiz I.; Aksoy B. A.; Hammerbacher J.; Gomez S. M.; Phanstiel D. H. Coral: Clear and Customizable Visualization of Human Kinome Data. Cell Syst. 2018, 7, 347–350. 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Shoyaib A.; Archie S. R.; Karamyan V. T. Intraperitoneal Route of Drug Administration: Should It Be Used in Experimental Animal Studies?. Pharm. Res. 2020, 37, 12. 10.1007/s11095-019-2745-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58, 5691–5698. 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- Di Maira G.; Salvi M.; Arrigoni G.; Marin O.; Sarno S.; Brustolon F.; Pinna L. A.; Ruzzene M. Protein Kinase CK2 Phosphorylates and Upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. 10.1038/sj.cdd.4401604. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yang Y.; Grimstein M.; Liu G.; Kitabi E.; Fan J.; Wang Y.; Earp J.; Weaver J. L.; Zhu H.; Liu J.; Reynolds K. S.; Huang S.; Wang Y. Anti–SARS-CoV-2 Repurposing Drug Database: Clinical Pharmacology Considerations. CPT Pharmacometrics Syst. Pharmacol. 2021, 10, 973–982. 10.1002/psp4.12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Delft A.; Hall M. D.; Kwong A. D.; Purcell L. A.; Saikatendu K. S.; Schmitz U.; Tallarico J. A.; Lee A. A. Accelerating Antiviral Drug Discovery: Lessons from COVID-19. Nat. Rev. Drug Discovery 2023, 22, 585–603. 10.1038/s41573-023-00692-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinnon K. H.; Leist S. R.; Schäfer A.; Edwards C. E.; Martinez D. R.; Montgomery S. A.; West A.; Yount B. L.; Hou Y. J.; Adams L. E.; Gully K. L.; Brown A. J.; Huang E.; Bryant M. D.; Choong I. C.; Glenn J. S.; Gralinski L. E.; Sheahan T. P.; Baric R. S. A Mouse-Adapted Model of SARS-CoV-2 to Test COVID-19 Countermeasures. Nature 2020, 586, 560–566. 10.1038/s41586-020-2708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J.-Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Gilbert Z. W.; Krämer A.; Dunford J. E.; Howell S.; Senbabaoglu F.; Wells C. I.; Bashore F. M.; Havener T. M.; Smith J. L.; Hossain M. A.; Oppermann U.; Drewry D. H.; Axtman A. D. Discovery of a Potent and Selective Naphthyridine-Based Chemical Probe for Casein Kinase 2. ACS Med. Chem. Lett. 2023, 14, 432–441. 10.1021/acsmedchemlett.2c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A.; Kurz C. G.; Berger B.-T.; Celik I. E.; Tjaden A.; Greco F. A.; Knapp S.; Hanke T. Optimization of Pyrazolo[1,5-a]Pyrimidines Lead to the Identification of a Highly Selective Casein Kinase 2 Inhibitor. Eur. J. Med. Chem. 2020, 208, 112770 10.1016/j.ejmech.2020.112770. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How Good Are My Data and What Is the Resolution?. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. A.; Vagin A. A.; Murshudov G. N. Model Preparation in MOLREP and Examples of Model Improvement Using X-Ray Data. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 33–39. 10.1107/S0907444907049839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot : Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. REFMAC 5 Dictionary: Organization of Prior Chemical Knowledge and Guidelines for Its Use. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2184–2195. 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.