

SUMMARY
Tumors develop by invoking a supportive environment characterized by aberrant angiogenesis and infiltration of tumor-associated macrophages (TAMs). Whether and how TAMs can be reprogrammed to correct the angiogenic microenvironment are unknown. Here we show that in a transgenic model of breast cancer, the tumor parenchyma-localized TAMs had low activity of the metabolic regulator mTORC1. Depletion of TSC1 in TAMs activated mTORC1 signaling and inhibited tumor growth independent of the adaptive immune system. Instead, TSC1-deficient TAMs relocated to a perivascular niche, depleted PROCR-expressing endovascular endothelial progenitor cells, and rectified the hyperpermeable blood vasculature, causing tumor tissue hypoxia and cancer cell death. TSC1-deficient TAMs were metabolically active and effectively eliminated PROCR-expressing endothelial cells in cell competition experiments. Thus, TAMs can be reprogrammed through the TSC-mTORC1 signaling axis to outcompete endothelial progenitor cells and suppress tumor neoangiogenesis, which defines an innate immune tumor suppression pathway that may be exploited for cancer environment immunotherapy.
Keywords: Tumor-Associated Macrophage, Endothelial Progenitor Cell, Cell Competition, TSC, mTORC1
eTOC Blurb
Tumor neoangiogenesis promotes carcinoma development. Do et al. examine whether such a cancer hallmark can be rectified by tumor-associated macrophages (TAMs) and find that deletion of the mTORC1 inhibitor TSC1 relocates TAMs to perivascular regions, which outcompetes PROCR-expressing endothelial progenitor cells and inhibits tumor angiogenesis, causing starvation-triggered cancer cell death.
INTRODUCTION
Cancer arises from outgrowth of malignant cells and poses a unique challenge to the immune system. Akin to infectious microbes, malignant cells undergo excessive expansion with induction of cytotoxic T lymphocyte typifying a cancer cell-directed immunosurveillance mechanism. Notwithstanding, the host impact of cancer cells is more intimate than that of invading pathogens with cancer pathogenicity dependent on a supportive environment resembling a non-healing wound,1 and helper T cell-mediated tissue healing responses can be mobilized as a cancer environment-directed host defense mechanism.2,3
In addition to adaptive lymphocytes, myeloid cells abundantly infiltrate tumors with tumor-associated macrophages (TAMs) constituting a dominant population.4–6 TAMs can function as antigen-presenting cells and promote dysfunction/exhaustion of tumor-reactive cytotoxic T lymphocytes.7,8 Furthermore, TAMs may support cancer progression by modulating the tumor stroma. Of note, despite acquiring oncogenic mutations that allow sustained proliferation and death evasion, cancer cells cannot grow without a supporting vasculature that supplies oxygen and nutrients.9 Indeed, an angiogenic switch is essential for converting a tumor from the benign to the malignant state.10 Infiltration of macrophages is correlated with vessel network development and tumor progression, and TAMs promote the angiogenic switch.11 In injury models, macrophages undergo progressive phenotypical adaptation from an inflammatory and angiogenic state to an anti-inflammatory and pro-resolving state to restore tissue homeostasis.12,13 Whether such a functional adaption can be elicited in TAMs to influence neoangiogenesis and tumor tissue healing is undefined.
Mammalian/mechanistic target of rapamycin complex 1 (mTORC1) is a metabolic regulator that controls growth, differentiation, and fitness of multiple cell lineages.14 Depletion of growth factors, oxygen or energy represses mTORC1 signaling via the tuberous sclerosis complex (TSC) made of TSC1, TSC2, and TBC1D7, which functions as a GTPase-activating protein for the mTORC1 activator Rheb.15,16 The TSC-mTORC1 signaling axis regulates terminal myeloid differentiation17,18 and polarization of bone marrow-derived macrophages.19–23 TSC1 depletion also impairs survival and migration while enhancing the phagocytic capacities of peritoneal macrophages.24 In addition, mice with myeloid cell-specific deletion of Tsc1 or Tsc2 are more sensitive to septic shock and granulomatous disease but resistant to asthma and mycobacterial infection.21,25,26 Nonetheless, whether TSC-mTORC1 signaling controls macrophage functions in the tumor setting is poorly understood.
Herein, in a transgenic model of breast cancer, we report that the tumor parenchyma-localized TAMs exhibited low mTORC1 activity. Depletion of TSC1 in TAMs caused mTORC1 hyperactivation and suppressed cancer progression independent of adaptive lymphocytes. Instead, metabolically active TSC1-deficient TAMs were redistributed around the intratumoral blood vasculature, outcompeted endothelial progenitor cells, and inhibited sprouting angiogenesis and blood vessel permeability, leading to tumor tissue hypoxia and cancer cell death. Thus, an effective tumor microenvironment-directed innate immune cancer defense response can be elicited through TAM reprogramming.
RESULT
Macrophage subsets adopt distinct tissue localizations and mTORC1 activities in tumor
Solid tumors are abundantly populated by macrophages.4,5 In an MMTV-PyMT (PyMT) transgenic model of breast cancer, we previously identified two distinct populations of macrophages termed TAMs and mammary tissue macrophages (MTMs).7 TAMs accumulate and become the dominant leukocyte population infiltrating late-stage tumors, while MTMs prominent in virgin mammary glands dwindle.7 Both macrophage subsets can arise from Ly6C+ inflammatory monocytes and are characterized by differential expression of the cell adhesion molecule Vcam1, the scavenger receptor CD206, and the integrin molecule CD11b.7 Specifically, TAMs express a high level of Vcam1 and a low level of CD11b while MTMs express high levels of CD206 and CD11b (Figure S1).
Recent studies have revealed that phenotypically distinct monocyte-derived macrophages reside in unique sub-tissular niches across organs.27 Notably, while CD206hi MTMs were preferentially localized in peritumoral stroma region, Vcam1hi TAMs were abundant in tumor parenchyma in close association with cancer cells (Figure 1A). In addition, TAMs had dendrite-like projections and were smaller in size than MTMs (Figure 1B). Cell size is controlled by mTORC1, a pivotal regulator of cell metabolism and growth.14 Indeed, TAMs had lower levels of the mTORC1 signaling target phosphorylated S6 protein (pS6) than MTMs (Figure 1B).
Figure 1. mTORC1 signaling in TAMs is lower than that in MTMs in mammary tumors.

(A) Representative images (left) and quantification (right) of localization of Vcam1hi tumor-associated macrophages (TAMs) and CD206hi mammary tissue macrophages (MTMs) in PyMT tumor tissue. F4/80+ macrophages are indicated by yellow arrowheads. E-Cadherin+ region is defined as tumor and E-Cadherin− region is counted as stroma. Data are pooled from two independent experiments with 5 randomly selected tumor regions for quantification. Scale bars: 200 μm, 50 μm, and 50 μm.
(B) Representative images of Vcam1 (red), CD206 (cyan), F4/80 (white), pS6 (green), and DAPI staining in Vcam1hi TAMs and CD206hi MTMs in PyMT tumor tissue (left). Contour of individual macrophages are traced by dashed line. Quantification of average cell area and pS6 intensity of Vcam1hi TAMs (n = 36) and CD206hi MTMs (n = 52) (right). Scale bars: 5 μm and 5 μm.
Unpaired t-test, two-tailed, ***: p < 0.001, ****: p < 0.0001. Data are shown as mean ± SEM.
See also Figure S1.
TSC1 deficiency alters TAM differentiation in association with tumor suppression
mTORC1 integrates growth factor, oxygen, and energy signals through TSC that functions as a repressor of the mTORC1-activating small GTPase Rheb.14 To determine whether TSC accounted for low mTORC1 activity in TAMs, we crossed mice with a conditional allele of Tsc1 (Tsc1fl) to CD11cCre transgenic mice that effectively target TAMs, but not MTMs or monocytes, in PyMT mice7 (Figure S2A). TAMs from CD11cCreTsc1fl/flPyMT mice had a bigger cell size and lost dendrite-like projections in association with higher levels of mTORC1 targets pS6 and phosphorylated eukaryotic translation initiation factor 4E-binding protein 1 (p4E-BP1) (Figures 2A and S2B). To investigate whether mTORC1 hyperactivation in TAMs caused their phenotypic change, we crossed CD11cCreTsc1fl/flPyMT mice with a conditional allele of Rptor (Rptorfl) encoding regulatory-associated protein of mTOR (RAPTOR), an obligate component of mTORC1. Indeed, the depletion of RAPTOR diminished pS6 levels and reversed the cell growth phenotype of TSC1-deficient TAMs (Figure 2A). Importantly, CD11cCreTsc1fl/flPyMT mice, but not CD11cCreTsc1fl/flRptorfl/flPyMT mice grew tumors more slowly than their littermate controls (Figure 2B). As CD11cCre targets additional hematopoietic cell lineages such as dendritic cells (DCs), we also used a CD64iCre mouse strain28 to deplete TSC1 in macrophages, which resulted in similar tumor inhibition to that observed in CD11cCreTsc1fl/flPyMT mice (Figure S2C). Altogether, these findings demonstrate that TAMs adopt a mTORC1-low state through processes dependent on TSC1, and ectopic activation of mTORC1 in TAMs is associated with tumor suppression.
Figure 2. Loss of TSC1 results in mTORC1-dependent TAM reprogramming and tumor repression.

(A) Representative images of Vcam1 (red), F4/80 (white), pS6 (green), and DAPI staining of Vcam1hi TAMs in Tsc1fl/flPyMT, CD11cCreTsc1fl/flPyMT, CD11cCreRptorfl/flPyMT and CD11cCreTsc1fl/flRptorfl/flPyMT tumor tissues (left). Contour of individual macrophages are traced by dashed line. Quantification of average cell area and pS6 intensity of Vcam1high TAMs (right). Number of Vcam1high TAMs quantified for each genotype: Tsc1fl/flPyMT (n = 38), CD11cCreTsc1fl/flPyMT (n = 34), CD11cCreRptorfl/flPyMT (n = 51) and CD11cCreTsc1fl/flRptorfl/flPyMT (n = 45). Scale bars: 5 μm, 5 μm, 5 μm, and 5 μm.
(B) Total tumor burden of Tsc1fl/flPyMT (n = 48), CD11cCreTsc1fl/flPyMT (n = 56), CD11cCreRptorfl/flPyMT (n = 8) and CD11cCreTsc1fl/flRptorfl/flPyMT (n = 17) mice.
Unpaired t-test, two-tailed, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, and n.s. = not significant. Data are shown as mean ± SEM. T-test in (B) is calculated based on endpoint tumor burden.
See also Figure S2.
TSC1 depletion in TAMs causes cancer cell death independent of adaptive immunity
Tumor suppression can be caused by increased cancer cell death and/or decreased cancer cell proliferation. To delineate the underlying mechanisms of tumor suppression in CD11cCreTsc1fl/flPyMT mice, we assessed cancer cell proliferation and death by the expression of Ki67 and cleaved Caspase 3 (CC3), respectively. While Ki67 expression was comparable in cancer cells from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice, an approximate 6-fold increase of CC3+ cancer cells were observed in CD11cCreTsc1fl/flPyMT mice (Figure 3A).
Figure 3. Increased cancer cell death and diminished tumor growth in mice with TSC1-deficient TAMs are independent of the adaptive immune system.
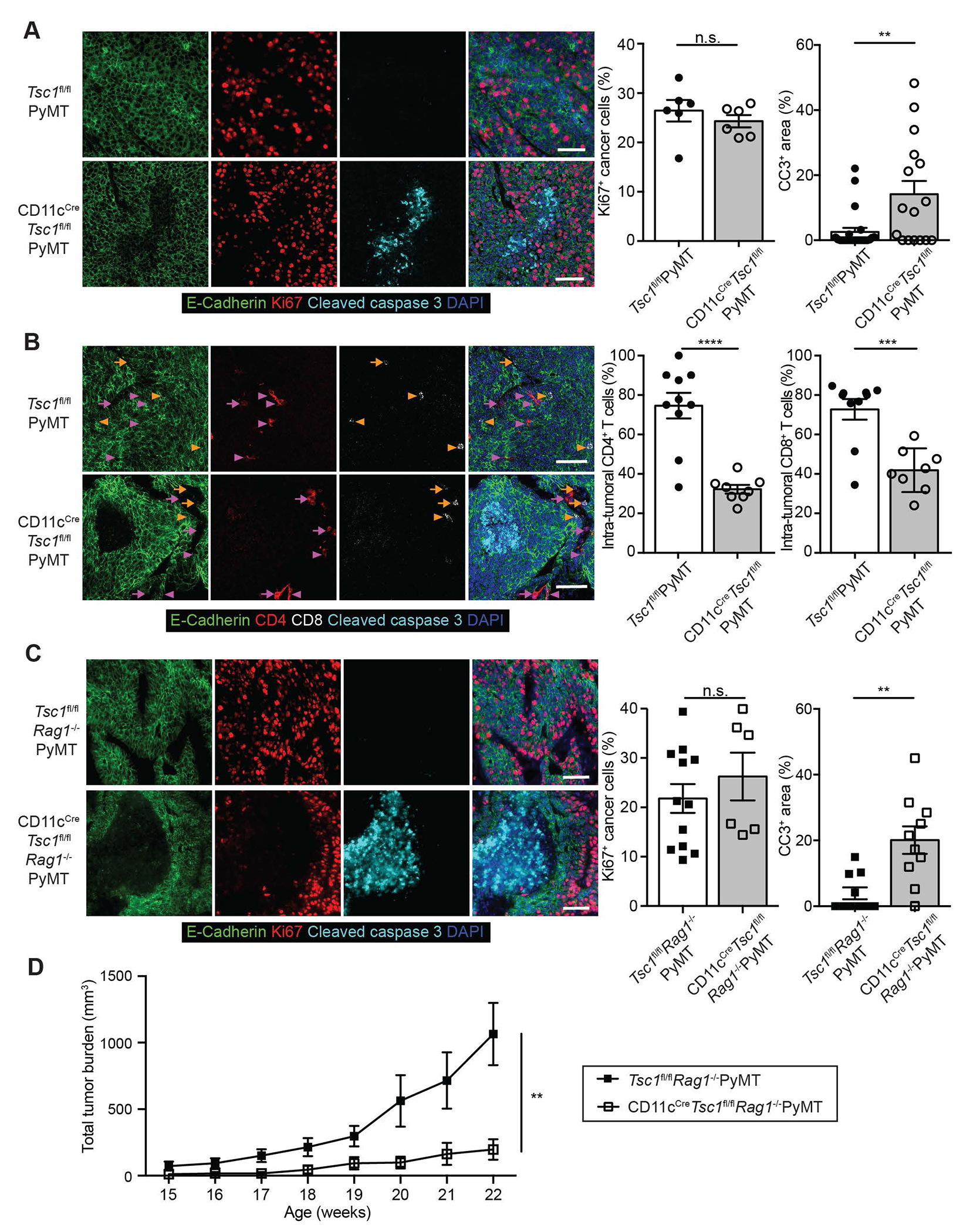
(A) Representative image (left) and quantification (right) of Ki67 and cleaved Caspase 3 (CC3) expression in E-Cadherin+ cancer cells from randomly selected Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT tumor tissues (n = 6–16). Scale bars: 50 μm and 50 μm.
(B) Representative image (left) and quantification (right) of CD4+ and CD8+ T cell localization from randomly selected Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT tumor tissues (n = 8–10). Scale bars: 50 μm and 50 μm. Intra-tumoral and stromal CD4+ T cells are indicated by magenta arrowheads and arrows, respectively. Intra-tumoral and stromal CD8+ T cells are indicated by yellow arrowheads and arrows, respectively.
(C) Representative image (left) and quantification (right) of Ki67+ and CC3+ cancer cells from randomly selected CD11cCreTsc1fl/flRag1−/−PyMT and Tsc1fl/flRag1−/−PyMT tumor regions (n = 6–12). Scale bars: 50 μm and 50 μm.
(D) Total tumor burden of CD11cCreTsc1fl/flRag1−/−PyMT (n = 6) and littermate Tsc1fl/flRag1−/−PyMT controls (n = 7).
Unpaired t-test, two-tailed, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, and n.s. = not significant. Data are shown as mean ± SEM. T-test in (D) is calculated based on endpoint tumor burden.
See also Figure S3.
We have shown that TAMs can function as antigen-presenting cells and regulate cytotoxic T lymphocyte responses in tumor.7,8 In addition, CD11cCre targets DCs that capture tumor-associated antigens to prime T cell responses 29. The proportions and numbers of TAMs, total DCs, XCR1+ type 1 DCs (DC1s) and CD11b+ DC2s in tumors were largely comparable between CD11cCreTsc1fl/flPyMT and Tsc1fl/flPyMT mice (Figures S3A–S3D). To investigate whether TSC1 deficiency might affect myeloid antigen-presenting cell function impacting T cell control of tumor growth, we assessed the abundance and differentiation states of tumor-infiltrating T cells. A 7% increase in total tumor-infiltrating TCRβ+ cells with higher abundance of CD4+Foxp3− conventional T cells was observed in CD11cCreTsc1fl/flPyMT mice (Figure S3E). Nonetheless, similar frequencies of tumor-infiltrating conventional lineages of CD4+ T cells and CD8+ T cells from CD11cCreTsc1fl/flPyMT mice and Tsc1fl/flPyMT mice produced effector cytokines (Figures S3F and S3G). Notably, immunohistochemistry analyses revealed that CD4+ T cells and CD8+ T cells were mostly localized distant from cancer cell nests in CD11cCreTsc1fl/flPyMT mice (Figure 3B), suggesting against direct cancer cell killing by T cells.
To definitively assess T cells’ (lack-of-) function in tumor suppression in CD11cCreTsc1fl/flPyMT mice, we crossed them onto a genetic background deficient in the Rag1 recombinase that supports the development of adaptive lymphocytes. While cancer cell proliferation was comparable between CD11cCreTsc1fl/flRag1−/−PyMT and Tsc1fl/flRag1−/−PyMT mice (Figure 3C), increased numbers of CC3+ cancer cells and diminished tumor growth were preserved in CD11cCreTsc1fl/flRag1−/−PyMT mice (Figures 3C and 3D), ruling out adaptive lymphocytes as effectors of the anti-tumor immune response.
TSC1-deficient TAMs promote vasculature reconfiguration resulting in cancer cell death
To investigate how phenotypically reprogrammed TSC1-deficient TAMs might suppress cancer progression, we examined their localization in the tumor. As expected, MTMs were similarly distributed along the tumor tissue-lining in Tsc1fl/flPyMT mice and CD11cCreTsc1fl/flPyMT mice (Figure 4A). However, in contrast to TAMs from Tsc1fl/flPyMT mice that were intermingled with cancer cells in the tumor parenchyma, TAMs from CD11cCreTsc1fl/flPyMT mice were localized close to the blood vasculature (Figure 4A). Of note, the fraction of TAMs within 5 μm to the nearest vessel was approximately 3-fold higher in CD11cCreTsc1fl/flPyMT mice (Figure 4A), and the TAM re-localization phenotype was preserved in CD11cCreTsc1fl/flRag1−/−PyMT mice (Figure S4A).
Figure 4. Loss of TSC1 triggers TAM repositioning to a perivascular niche in association with vessel reorganization, tumor tissue healing, hypoxia, and cancer cell death.

(A) Representative images of tissue localization of F4/80+Vcam1hi tumor-associated macrophages (TAMs) and F4/80+Vcam1lo/− mammary tissue macrophages (MTMs) in tumor tissues of Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice (top). Orange arrows indicate TAMs or MTMs. Magenta arrows indicate cell death regions with condensed and fragmented chromatin. Quantification of the distance of individual TAMs to their nearest CD31+ vasculature (bottom). Data are representative of three independent experiments with more than 1×103 TAMs quantified in each experiment. Scale bars: 200 μm, 200 μm, 50 μm, 50 μm, 50 μm, and 50 μm.
(B) Representative images of tumor sections from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice with CD31 (red), Fibrinogen (Fg, white), Cleaved Caspase 3 (cyan), E-Cadherin (green), and DAPI staining (left). Isolated CD31 staining is indicated by magenta arrows and extravascular Fg+ events are indicated by orange arrows. Quantification of isolated CD31 staining (top right) and extravascular Fg+ events (bottom right) in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT tumor tissues per 1 mm2 (n = 8-14). Scale bars: 200 μm, 50 μm, 200 μm, and 50 μm.
(C) Representative images (left) and quantification (right) of pimonidazole-positive hypoxic area in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT (n = 10 each). Scale bars: 50 μm and 50 μm.
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05 and ****: p < 0.0001.
See also Figure S4.
The perivascular phenotype of TSC1-deficient TAMs imply that they may regulate the host to endure a growing tumor’s pathological outcomes. In line with the notion that tumors are “wounds that do not heal”,1 blood vessels from tumors of control Tsc1fl/flPyMT mice were disorganized, manifested by abundant isolated CD31 staining (Figure 4B). Furthermore, the vasculature was hyperpermeable with high levels of extravascular deposition of fibrinogen (Figure 4B). In contrast, the microvasculature was more organized in tumors from CD11cCreTsc1fl/flPyMT mice in association with intravascular retention of fibrinogen (Figure 4B). The vasculature structure and permeability phenotypes were preserved in CD11cCreTsc1fl/flRag1−/−PyMT mice (Figure S4B), supporting innate immune vasculature remodeling functions of TSC1-deficient TAMs.
Induction of angiogenesis is a cancer hallmark,10 as neovasculature supplies nutrients and oxygen to promote cancer cell growth and survival.9 Indeed, the extensive sprouting angiogenesis in tumors from Tsc1fl/flPyMT mice was associated with few hypoxic spots (Figure 4C). In contrast, tumors from CD11cCreTsc1fl/flPyMT mice had larger areas positive for the hypoxic probe (Figure 4C). Notably, the ring-shape hypoxic area encircled the cancer cell death region, suggesting that cancer cell death in CD11cCreTsc1fl/flPyMT mice is caused by hypoxia and/or depletion of nutrients. Similarly enhanced hypoxia response and its close association with dying cancer cells were observed in tumors from CD11cCreTsc1fl/flRag1−/−PyMT mice (Figure S4C). These findings demonstrate that TSC1-deficient TAMs promote the reconfiguration of the tumor blood vasculature, causing cancer cell hypoxia and death in distant avascular regions.
TSC1-deficient TAMs exhibit a pro-resolving macrophage gene expression profile
To investigate how TSC1 deficiency alters TAM differentiation and function, we performed single-cell RNA-sequencing experiments on Vcam1-expressing TAMs isolated from Tsc1fl/flPyMT mice and CD11cCreTsc1fl/flPyMT mice. After sequencing quality control, 1932 wild-type TAMs and 1425 TSC1-deficient TAMs were roughly segregated into five clusters in dimension reduction by uniform manifold approximation and projection (UMAP) and hierarchical clustering analyses (Figure 5A). Although both wild-type TAMs and TSC1-deficient TAMs expressed high levels of macrophage lineage genes including Adgre1, Spi1, and Csf1r (Figure S5A), they exhibited a largely nonoverlapping aggregation pattern in UMAP (Figure 5A). These observations suggest that the depletion of TSC1 reprograms TAMs to a distinct state of cell differentiation.
Figure 5. TSC1-deficient TAMs exhibit an anti-inflammatory and pro-resolving cell differentiation state.

(A) Uniform manifold approximation and projection (UMAP) of single cells in two-dimensions and clustered into 5 communities using the Leiden community detection (left). UMAP projection of single cells in two-dimensions in which tumor-associated macrophages (TAMs) from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT were colored blue and purple respectively (middle). The fraction of TSC1-deficient TAMs and wild-type TAMs in each cluster (right).
(B) Matrix plot showing the expression of representative genes related to different biological processes in cluster 1, cluster 2, and cluster 4. (*: genes encoding lysosomal proteins. See also Table S3.)
See also Figure S5.
In line with previous studies demonstrating the high proliferation potential of TAMs,7 cluster 3 was composed of mitotic cells characterized by high expression of cell cycle-related genes including Birc5 and Mki67 (Figure S5B and Table S1). Both wild-type TAMs and TSC1-deficient TAMs were abundant in cluster 3, suggesting that TSC1 deficiency does not substantially affect TAM proliferation (Figure 5A). In contrast, clusters 1 and 2, and clusters 4 and 5 were dominant by wild-type TAMs and TSC1-deficient TAMs, respectively (Figure 5A). Notably, TAMs in cluster 5 were characterized by enrichment of the T cell lineage transcripts such as Cd3e and Cd3g (Figure S5B and Table S2). As CD3 proteins were undetectable in TAMs (data not shown), these transcripts were likely acquired through TAM engulfment of dying T cells. The higher proportion of TSC1-deficient TAMs (98%) than wild-type TAMs (2%) in cluster 5 (Figure 5A) suggests more active engulfment, in agreement with previous reports that TSC1 deficiency promotes macrophage phagocytosis.24
To define how TSC1 deficiency affects TAM differentiation, we systematically compared the transcriptome of TAMs in clusters 1, 2, and 4 (Table S3). Cluster 1 and cluster 2 had 99% and 98% wild-type TAMs, respectively (Figure 5A) and expressed high levels of transcripts of interferon-stimulated genes such as Ifitm2 and Ifitm3 as well as antigen presentation such as H2-Aa and H2-Ab1 (Figures 5B, S5B, and S5C), which was in line with higher levels of major histocompatibility class II (MHCII) protein expression in wild-type TAMs (Figure S6A). While cluster 1 had few selectively enriched transcripts (Table S3), cluster 2 had the highest expression of transcripts of MAPK and NF-κB signaling pathways including Map2k3, Nfkbiz, and Tnfaip3 as well as transcription factors encoded by immediate early genes including Fos, Jun, and Nr4a1 (Figures 5B and S5B). Moreover, several cytokine genes including Il1b, Tnf, and Vegfa that promote inflammation and angiogenesis were enriched in cluster 2 (Figures 5B and S5B). These findings reveal that the predominant wild-type TAM clusters 1 and 2 exhibit features of mature antigen-presenting cells, with cluster 2 further manifested in a pro-inflammatory and pro-angiogenic differentiation state.
Cluster 4 constituted exclusively of TSC1-deficient TAMs and was characterized by high expression of cell metabolism genes (Figures 5A, S5B, and S5D). Transcripts involved in several catabolic processes, including glyceride and sphingolipid degradation by Gdpd1 and Psap as well as fatty acid beta-oxidation by Acaa2 and Echs1 were enriched (Figure 5B). Genes including Glb1 and Hexa that promote degradation of carbohydrates and their derivatives were also highly expressed (Figure 5B). So were transcripts including Arsg and Ifi30 that degrade sulfur-containing compounds and genes including Ctsd and Sidt2 that catabolize proteins and nucleic acids (Figure 5B). Notably, lysosomal hydrolases (marked by an asterisk) made up most of these degradative enzymes that enable catabolism of a broad class of biomolecules (Figures 5B and S5D). Furthermore, transcripts involved in a number of anabolic processes including fatty acid, isoprenoid, and cholesterol biosynthesis by Hacd4 and Mvk as well as N-linked and O-linked glycosylation by Alg14 and Galnt6 were up-regulated (Figure 5B). Genes including Gsr and Gss that catalyze the biosynthesis and reduction of the sulfur-containing glutathione as well as Gpx3 and Mgst1 that promote glutathione conjugation to electrophilic compounds were also highly expressed (Figure 5B), which may collectively detoxify carcinogens, environmental toxins, and oxidative byproducts.30 Together, these observations suggest that TSC1-deficient TAMs reprogram their metabolic states with prominent features of lysosomal catabolism and glutathione metabolism to fend off biotic and xenobiotic challenges.
In addition to lysosomal enzyme genes, a number of transcripts involved in lysosomal organization such as ATP6v0c and Lamp1 were up-regulated in cluster 4 (Figures 5B and 6A). Genes including Gas6 and Mertk that promote macrophage recognition of apoptotic cells as well as genes including Pikfyve and Sh3gl1 that support phagolysosome maturation and trafficking were also highly expressed (Figure 5B), suggesting enhanced efferocytosis capabilities of TSC1-deficient TAMs. Indeed, ex vivo phagocytosis assay revealed that TSC1-deficient TAMs were highly effective in engulfing apoptotic cells than wild-type TAMs (Figure 6B). Besides being the principal ‘recycling’ organelle, lysosome participates in cellular signaling with a crucial role in mTORC1 activation. Of note, genes encoding the lysosomal components of nutrient mTORC1 signaling such as Lamtor1 and Rragd and cytosolic amino-acid sensing such as Castor2 and Fnip2 were enriched in cluster 4 (Figure 5B), which may work in concert with TSC1 deficiency to regulate mTORC1 signaling in TAMs. Lysosome-mediated clearance of engulfed targets including apoptotic cells promotes tissue homeostasis and suppresses inflammation.31 Notably, transcripts such as C300lf and Nlrc3 that encode plasma membrane and cytosolic anti-inflammatory regulators as well as transcripts such as Il18bp and Igf1 that encode anti-inflammatory and tissue repair factors were also enriched in cluster 4 (Figure 5B). Thus, in contrast to wild-type TAMs manifested in an inflammatory and angiogenic differentiation state, TSC1-deficient TAMs exhibit enhanced lysosomal scavenger function in association with metabolic reprogramming.
Figure 6. TSC1-deficient TAMs display enhanced efferocytosis and mitochondrial activities.

(A) Representative flow cytometric analysis (left) and quantification (right) of CD107a (LAMP1) expression in TAMs from Tsc1fl/flPyMT (n = 7) and CD11cCreTsc1fl/flPyMT (n = 6) mice.
(B) Representative flow cytometric analysis (left) and quantification (right) of CellTrace Violet (CTV)-labelled apoptotic cell engulfment in TAMs from Tsc1fl/flPyMT (n = 3) and CD11cCreTsc1fl/flPyMT (n = 3) mice.
(C) Representative flow cytometric analysis (left) and quantification (right) of mitochondrial mass (MitoTracker Green FM, Y axis) and mitochondrial potential (MitoTracker Red CMXRos, X axis) in TAMs from Tsc1fl/flPyMT (n = 7) and CD11cCreTsc1fl/flPyMT (n = 7) mice.
(D) Representative flow cytometric analysis (left) and quantification (right) of mitochondrial ROS (MitoSOX Red) production in TAMs from Tsc1fl/flPyMT (n = 9) and CD11cCreTsc1fl/flPyMT (n = 9) mice.
(E) Representative analysis (left) and quantification (right) of extracellular acidification rate (ECAR) of TAMs from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Sequential chemical treatments are indicated (Oligo, oligomycin; 2-DG, 2-Deoxy-D-glucose).
(F) Representative analysis (left) and quantification (right) of oxygen consumption rate (OCR) of TAMs from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Sequential chemical treatments are indicated (Oligo, oligomycin; FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone; Ant, antimycin A; Rot, rotenone).
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, and n.s. = not significant.
See also Figure S6.
Perivascular localization of TSC1-deficient TAMs tracks with high mitochondrial activities
To investigate what might drive TSC1-deficient TAMs to redistribute along the tumor blood vasculature, we examined the expression of chemokine receptors CCR2 and CX3CR1 that have been implicated in monocyte/TAM recruitment and control of tumor angiogenesis in response to CCL2 and CX3CL1.32 Compared to monocytes, TAMs expressed lower levels of CCR2 but high levels of CX3CR1 (Figure S6B). Yet, CCR2 and CX3CR1 were expressed at similar levels between wild-type TAMs and TSC1-deficient TAMs (Figure S6B), suggesting that CCR2- or CX3CR1-mediated chemotaxis unlikely accounts for the perivascular localization of TSC1-deficient TAMs.
Aside from chemotaxis, TSC1-deficient TAMs might preferentially adapt to the perivascular region by cell-intrinsic properties. As TSC1-deficient TAMs exhibited a gene expression profile characterized by high metabolic activities (Figures 5B and S5D), they might selectively thrive in the perivascular sub-tissular niche due to their need of access to abundant nutrients. Of note, TSC1-deficient TAMs had higher mitochondrial mass, and showed higher mitochondrial potential as well as higher level reactive oxygen species production than wild-type TAMs (Figures 6C and 6D). Moreover, seahorse experiments revealed that while TSC1-deficient TAMs had moderately enhanced glycolysis (Figure 6E), they displayed much elevated basal, maximal, and spared mitochondrial oxygen consumption rates (Figure 6F). In vitro culture experiments revealed that TSC1-deficient TAMs had a higher rate of apoptosis marked by CC3 expression than wild-type TAMs under a hypoxic condition (Figure S6C). Those observations demonstrate that TSC1-deficient TAMs are metabolically reprogrammed with enhanced mitochondrial respiration, which may promote their accumulation in the high-oxygen perivascular region.
TSC1-deficient TAMs outcompete endovascular endothelial progenitor cells
To investigate how metabolically active TSC1-deficient TAMs affected tumor angiogenesis, we examined the endothelial cell compartment. Recent studies have revealed heterogeneous populations of mouse endothelial cells with phenotypically distinct progenitor cell subsets driving neovascularization in adult tissues.33 In mammary glands, Protein C receptor (PROCR) expression marks a subset of endovascular endothelial progenitor cells with long-term clonal expansion potential and high vessel reconstitution efficiency in transplantation experiments.34 While comparably low frequencies of PROCR+ endothelial cells were detected in peritumor regions of Tsc1fl/flPyMT mice and CD11cCreTsc1fl/flPyMT mice (Figure S7A), a higher fraction of PROCR+ endothelial cells populated the tumor parenchyma of Tsc1fl/flPyMT mice than that of CD11cCreTsc1fl/flPyMT mice (Figure 7A). Reduced numbers of intratumoral PROCR+ endothelial cells in CD11cCreTsc1fl/flPyMT mice were associated with enhanced apoptosis marked by CC3 staining (Figure 7A), whereas endothelial cell death was not different in peritumoral regions of Tsc1fl/flPyMT mice and CD11cCreTsc1fl/flPyMT mice (Figure S7A). These observations suggest that diminished sprouting angiogenesis in CD11cCreTsc1fl/flPyMT mice could be due to depletion of intratumoral endothelial progenitor cells.
Figure 7. TSC1-deficient TAMs outcompete intratumoral endothelial progenitor cells.
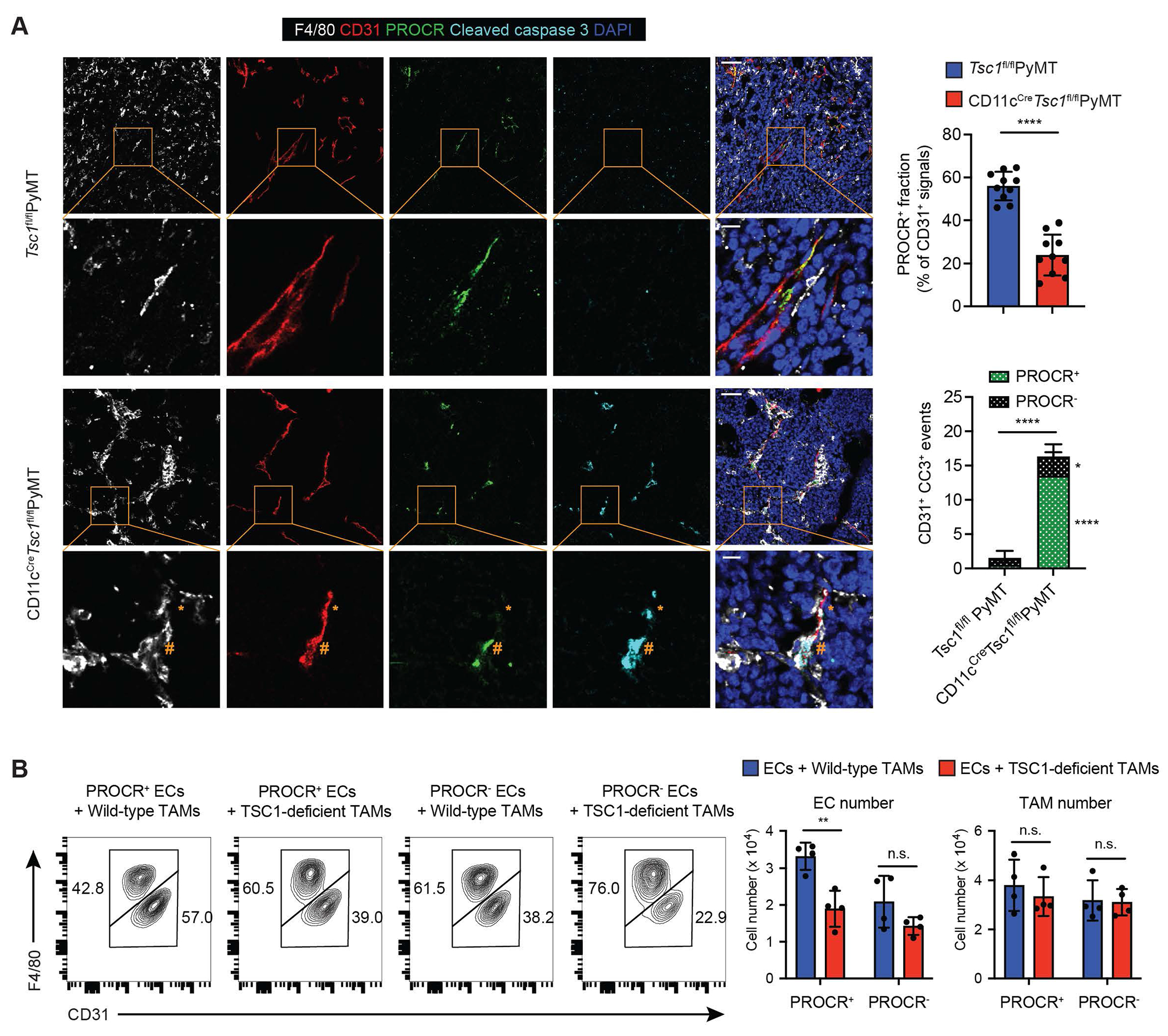
(A) Representative images of tumor sections from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice with F4/80 (white), CD31 (red), protein C receptor (PROCR, green), cleaved caspase 3 (CC3, cyan), and DAPI staining (left). Quantification of PROCR+ fraction among CD31+ endothelial cells by Manders’ colocalization analysis (top right) as well as CD31+CC3+ events differentiated by PROCR expression (bottom right) in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT tumor tissue sections (n = 10). PROCR+CD31+CC3+ events are marked by hashtag, and PROCR−CD31+CC3+ events are marked by asterisk in images from CD11cCreTsc1fl/flPyMT tumors. Scale bars: 50 μm, 10 μm, 50 μm, and 10 μm.
(B) PROCR+ or PROCR− endothelial cells (ECs) were co-cultured with either wild-type or TSC1-deficient TAMs for 3 days. Representative flow cytometric analysis of ECs and TAMs (left) as well as their numbers (right) were shown.
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05, **: p < 0.01, ****: p < 0.0001, and n.s. = not significant.
See also Figure S7.
Three-dimensional imaging experiments revealed close association between TSC1-deficient TAMs and PROCR+ endothelial cells that underwent apoptosis (Videos S1–S5), suggesting that endothelial progenitor cell death might be triggered by cell competition, an evolutionarily conserved cell surveillance program in which comparatively less fit cells are eliminated by their more robust neighbors.35 Of note, depletion of TSC2 activates mTORC1 and creates super-competitive epiblasts during development.36 mTORC1-regulated cell competition has also been demonstrated between different cell lineages. Under a low-protein diet condition, highly phagocytic TAMs gain mTORC1 signaling and outcompete metabolically active MYC-overexpressing cancer cells28. In a SCENITH (single cell energetic metabolism by profiling translation inhibition) assay, PROCR+ endothelial cells were more metabolically active than PROCR− endothelial cells, which was largely dependent on mitochondrial oxidative phosphorylation-mediated energetic metabolism (Figure S7B). In support of their high metabolic activity and progenitor phenotype, PROCR+ endothelial cells more readily expanded than PROCR− endothelial cells in vitro (Figure S7C). These observations are in agreement with previous findings that proliferative endothelial cells manifest high level mitochondrial respiration, which drives neoangiogenesis in tumors.37,38 To investigate whether TAM might directly regulate endothelial cell expansion, we co-cultured PROCR+ or PROCR− endothelial cells with wild-type or TSC1-deficient TAMs. While TAM recovery was largely comparable, recovery of PROCR+ endothelial cells was more substantially reduced than recovery of PROCR− endothelial cells in co-culture experiments with TSC1-deficient TAMs relative to those with wild-type TAMs (Figures 7B). These findings demonstrate that highly metabolically active TSC1-deficent TAMs can outcompete metabolically active endothelial progenitor cells in association with diminished angiogenesis and tumor growth in CD11cCreTsc1fl/flPyMT mice.
DISCUSSION
Tumor development requires a supportive microenvironment characterized by neoangiogenesis and chronic immune filtration. Using a murine model of breast cancer, we found that angiogenic TAMs could be reprogrammed via TSC-mTORC1 signaling to inhibit cancer progression. Depletion of TSC1 led to mTORC1-mediated tumor suppression independent of adaptive lymphocytes. Instead, TSC1-deficient TAMs were redistributed from the tumor epithelium to a perivascular niche and manifested a pro-resolving cell differentiation phenotype. TSC1-deficient TAMs outcompeted intratumoral endothelial progenitor cells, rectified the tortuous and hyperpermeable blood vasculature, and triggered tumor tissue hypoxia and starvation-induced cancer cell death. These observations, together with the finding of Th cell-mediated vasculature remodeling,2,3 reveal that both innate and adaptive immune systems can be mobilized to inhibit cancer progression by targeting the tumor microenvironment.
Heterogeneous populations of macrophages reside in sub-tissular niches in support of tissue functions,39,40 which can be maladapted to foster tumor development.41 TAMs and MTMs, the two major populations of macrophages in the PyMT breast cancer model,7 are localized in the tumor parenchyma and the peritumoral stroma region, respectively. MTMs are phenotypically similar to interstitial tumor monocytes/macrophages characterized by high expression of the angiopoietin receptor Tie2 and CD206,42–44 as well as stromal macrophages that dominate the macrophage landscape in virgin mammary glands.45 In contrast, TAMs resemble ductal macrophages that reside in an intra-epithelial niche and rapidly expand during pregnancy and lactation45 in accordance with expansion of the vasculature.46 Vasculature permeability is also increased during lactation, which is dependent on VEGF produced by epithelial and stromal cells.47,48 Notably, ductal macrophages from lactating mammary gland exhibit a gene expression profile similar to that of cluster 2 TAMs with high expression of pro-angiogenic transcripts including VEGF.45 These findings suggest that TAMs may co-opt a physiological vasculature regulatory function of ductal macrophages to promote pathologic tumor development.
An important finding of our study is that TAMs exhibit a TSC1-dependent mTORC1-low state, which is required to maintain the pro-tumor functions of TAMs. As a critical sensor and integrator of multiple growth conditions, the TSC complex may convey signals such as insufficient growth factors, low oxygen levels, and ATP depletion in the tumor parenchyma to inhibit mTORC1 signaling in TAMs. It is plausible that cancer cell proliferation demands an excessive oxygen supply, resulting in a dynamic low oxygen state in the tumor parenchyma. The inhibition of mTORC1 induced by the lack of oxygen likely promotes TAM expression of VEGF, facilitating tumor angiogenesis to resolve hypoxia. Of note, macrophages in hypoxic regions of the tumor express higher levels of Redd1, a negative regulator of mTORC1, than macrophages in normoxic regions.49 However, Redd1 deficiency does not affect macrophage differentiation to the same extent as with TSC1 ablation,49 implying additional Redd1-independent TSC1-dependent mechanisms of mTORC1 repression in TAMs. Future studies will unravel the constellation of tumor microenvironment-associated sensing pathways that converge on the TSC complex to suppress mTORC1 signaling in TAMs.
Ectopic mTORC1 activation via TSC1 loss redistributed TAMs from intratumoral cancer cell regions to territories around the blood vasculature. This took place despite similar expression of chemokine receptors CCR2 and CX3CR1 between wild-type and TSC1-deficient TAMs, suggesting against differential chemotaxis as a cause of TAM redistribution. Of note, TSC1-deficient TAMs had big cell size and high level of oxygen consumption rate and were susceptible to apoptosis under a hypoxic condition, raising the possibility that abundant nutrients and oxygen around the blood vasculature might be uniquely compatible with the high metabolic state of TSC1-deficient TAMs and thus drive their perivascular localization. Future studies to investigate whether mitochondrial respiration is required to maintain TSC1-deficient TAMs around the blood vasculature can be carried out to test this hypothesis. In addition, change of localization could affect TAM differentiation. Future research will reveal to what extent, the differential gene expression program and different functional properties between wild-type TAMs and TSC1-deficient TAMs are caused by Tsc1 deletion and/or change of the microenvironment.
Despite well-established functions of angiogenesis in cancer,10 the cellular mechanisms of tumor neoangiogenesis remain incompletely understood. Hematopoietic origin of endothelial progenitor cells was suggested in earlier research,50 which was disputed by later studies with cell lineage tracing and parabiosis experiments in tumor-bearing mice.51 In fact, de novo vascularization in adult tissues has recently been shown to be mediated by tissue-resident endovascular endothelial progenitor cells.33 Of note, PROCR expression marks an endovascular endothelial progenitor cell population in multiple tissues including mammary glands, the skin, and retina.34 In mammary glands, PROCR-based lineage tracing is more prevalent during the pubertal period than adult age in regions of epithelial outgrowth towards the fat pad.34 These observation are in line with our finding that the frequency of PROCR-expressing endothelial cells was higher in the epithelial tumor bed than in the peritumoral region enriched for adipocytes, and support the conclusion that neovascularization in high turnover tissues such as the tumor parenchyma is driven by endovascular endothelial progenitor cells.
Perivascular localization of TSC1-deficient TAMs cooccurred with depletion of PROCR-expressing endothelial cells and suppression of neoangiogenesis, causing tumor tissue hypoxia and starvation-induced cancer cell death. The anti-angiogenic and tumor suppressor function of TSC1-deficient TAMs was distinct from a previous report of a pro-angiogenic role of TSC-deficient monocytes/macrophages in a xenograft tumor model.52 Much of the pro-angiogenic function was demonstrated with monocytes/macrophages stimulated by LPS,52 the relevance of which in tumor was unclear. Notably, TSC1-deficient TAMs inhibited expansion of PROCR+ endothelial cells in co-culture experiments, supporting a cell competition mechanism of endothelial progenitor cell depletion. Cell competition defines a social role of metazoan cell interaction in which growth or death outcomes of neighboring cells are controlled by their relative fitness. The best characterized cell competition occurs among cells of the same lineage, mostly among stem cell populations as a mode of homocellular interaction to promote the selection of fitter progenitors in support of organismal development and tissue regeneration.35 Our finding of TAM outcompeting endothelial cells, together with the finding of TAM outcompeting cancer cells,28 defines a novel type of heterocellular cell competition engaged as innate immune tumor suppression mechanisms.
How perivascular TSC1-deficient TAMs deplete endothelial progenitor cells remains to be elucidated. The cell competition outcome between epiblast pluripotent stem cells is dictated by their metabolic state with ‘winner’ cells exhibiting higher anabolic capacity associated with higher mTORC1 activation.36 Furthermore, ‘winner’ pluripotent stem cells engulf dying ‘loser’ cells as a possible means to boost their metabolic profile.53 Notably, TSC1-deficient TAMs manifest not only high mTORC1 signaling but also high phagocytic activities that may outcompete endothelial progenitor cells for engulfment-dependent nutrient acquisition. In addition, as endothelial cell macropinocytosis is triggered by VEGF signaling54 and can promote tumor-associated vessel tube formation downstream of integrin signaling55, competing engulfment may suppress sprouting angiogenesis and lead to the demise of endothelial progenitor stem cells. In addition to engulfment-regulated processes, TSC1-deficient TAMs may outcompete endothelial progenitor cells for local survival/differentiation signals such as those of the Notch pathway7,56 and/or generate local toxic signals such as reactive oxygen species to trigger their non-autonomous cell death.
In summary, our studies demonstrate that the tumor parenchyma-localized TAMs are in close association with cancer cells and manifest an inflammatory and pro-angiogenic cell differentiation state. Ectopic mTORC1 activation via TSC1 loss results in TAM relocation in close proximity to the tumor blood vasculature and suppresses tumor development independent of the adaptive immune system. Instead, highly metabolically active TSC1-deficient TAMs adopt a pro-resolving gene expression profile and rectify the tumor blood vasculature abnormality by outcompeting endothelial progenitor cells. As the TAM response in conserved in human malignancies,57 the TAM TSC-mTORC1 pathway may be targeted to harness the novel innate immune anti-tumor mechanism for cancer environment immunotherapy.
LIMITATIONS OF THE STUDY
While we show that TSC1-deficient tumor-associated macrophages (TAMs) effectively inhibit MMTV-PyMT mammary tumor development in association with depletion of PROCR-expressing endothelial cells, we could not directly prove that diminished PROCR+ endothelial cells accounted for the suppression of tumor neoangiogenesis. The progenitor/stem cell property of PROCR-expressing endothelial cells in tumors was inferred from their metabolic phenotype ex vivo and growth phenotype in vitro, and we could not ascertain it, for instance, by performing cell lineage-tracing experiments in vivo as previously done during mammary gland development. Furthermore, although phenotypically similar TAMs to those in MMTV-PyMT tumors have been reported in human breast cancer patients, whether human TAMs can be similarly reprogrammed via the TSC-mTORC1 pathway to suppress tumor neoangiogenesis remains to be determined.
STAR METHODS
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Ming O. Li (lim@mskcc.org). This study did not generate new unique reagents. The raw sequencing data that support the findings of this study are available from the lead contact upon reasonable request.
Animal Experimental Model Details
Tsc1fl/fl, Rptorfl/fl, Rag1−/− mice were purchased from Jackson Laboratory. CD11cCre and MMTV-PyMT mice on C57BL/6 background was previously described.7 CD64iCre (Fcgr1iCre) mice were generated in the M.O.L laboratory and has been recently reported.28 In all experiments, female littermate controls were used when possible. Mice were maintained under specific pathogen-free conditions, and animal experimentation was conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center.
Method Details
Tumor measurement.
Tumors were measured regularly with a caliper starting at 12 weeks of age. Tumor volume was calculated using the equation (L×W2) x (π/6) where “L”=length and “W”=width. Individual tumor volumes were added together to calculate total tumor burden. The endpoint for tumor experiment is when total volume reached 3,000 mm3 or one tumor reached 2,000 mm3. Investigators were blinded to genotypes of mice during measurements.
Immune cell isolation from tissues.
Tumor-infiltrating leukocytes were isolated from size-matched tumors as follows. Tumor tissues were prepared by mechanical disruption followed by 1 h treatment with 280 U/ml Collagenase Type 3 (Worthington) and 4 μg/ml DNase I (Sigma) at 37 °C. After the digestion steps, the cell suspension was filtered through a 70-μM cell strainer, layered in a 44% and 66% Percoll gradient (Sigma), and centrifuged at 1900 rpm for 30 mins without brake at 4 °C. Cells at the interface were collected and analyzed by flow cytometry.
Flow cytometry.
Cell surface staining was performed by incubating cells with specific antibodies for 30 mins on ice in the presence of 2.4G2 mAb to block FcγR binding. For intracellular antibodies, Tonbo transcription factor kit was used to fix and permeabilize cells according to the manufacturer’s instruction. To determine cytokine expression, isolated cells were stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (Sigma), 1 mM ionomycin (Sigma) and GolgiStop (BD Biosciences) for 4 h prior to staining. For all stains, dead cells were excluded from analysis by means of Ghost Dye (Tonbo) or DAPI stain. All samples were acquired and analyzed with LSRII flow cytometer (Becton Dickson) and FlowJo software (TreeStar).
Immunofluorescence microscopy.
Tumors were isolated from PyMT mice and fixed in 4% paraformaldehyde overnight at 4 °C. Tumors were then transferred to a 30% sucrose solution at 4 °C overnight. Samples were embedded in OCT and stored at −80 °C before sectioning. Tumors were cut into 20-μm sections using a cryostat. Sections were rehydrated in PBS for 10 mins and blocked for 1 h with normal donkey serum at room temperature (RT). Primary antibodies were stained for either 3.5 h at RT or overnight at 4 °C. Secondary antibodies were stained for 1 h at RT. Slides were mounted with ProLong Gold Antifade Mountant (Invitrogen). Slides were scanned with Panoramic Digital Slide Scanners (3DHISTECH LTD) and Leica SP5 confocal. Random tumor fields were selected for quantification.
Hypoxia detection.
Tumor-bearing mice were i.p. injected with 60 mg/kg pimonidazole hydrochloride. Mice were sacrificed after 1 h and tumors were collected for immunostaining. Tumor sections were stained with Hypoxyprobe-Pab27 kit (Hypoxyprobe) according to the manufacturer’s instruction.
Cell sorting.
After gating on morphology and singlets, TAMs were gated as live CD45+SiglecF−B220−Ly6G−Ly6C−TCRβ−F4/80+Vcam1+ and were sorted from tumors of Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Cell sorting was conducted on Aria II (Becton Dickson).
Single cell RNA sequencing.
From each sample, around 10,000 FACS sorted tumor macrophages (1,000 cells/ul suspension) from one Tsc1fl/flPyMT mouse and one CD11cCreTsc1fl/flPyMT mouse were suspended in 1x PBS (calcium and magnesium free) containing 0.04% BSA (A2153, SIGMA). Cell suspension was used as input in 10x chromium controller system (10x Genomics Inc., product code 120223) and cell barcoding was performed by gel beads in emulsion (GEMs) in assembly chip. GEM RT reaction was performed in thermocycler (53 °C for 45 mins, 85 °C for 5 mins, 4 °C hold overnight). SPRIselect dynabeads (B23318, Beckman Coulter) were used for GEM recovery post RT. 2-50 ng of DNA was used for target enrichment. cDNA amplification, fragmentation, end repair and a tailing preparation were performed as per manual instructions. High sensitivity DNA chips and Agilent 2100 bioanalyzer (Agilent Technologies) was used for gene expression library profile quality control and quantification at several steps. Quality control was performed twice before sequencing. Two barcoded scRNA samples were pooled together before sequencing. scRNA libraries were sequenced on NovaSeq 6000 S1 with sequencing depth of approximately 300 million reads.
TAM phagocytosis assay.
PY8119 breast cancer cells were labelled by CellTrace Violet (CTV) and subsequently treated with 200 μM H2O2 overnight to induce apoptosis. Wild-type or TSC1-deficient TAMs were purified by cell sorting as described above and plated in 24-well plate (4×105 cells/well) for 2 h to allow TAMs to adhere to the plates. Subsequently, CTV-labelled apoptotic cells (1×106 cells/well) were added to TAMs for 6 h. Cells were washed twice by PBS and were harvested to stain with anti-F4/80 and anti-Vcam1 antibodies. Flow cytometry analyses were performed to identify TAMs that had engulfed apoptotic cells.
Mitochondrial mass, potential, and reactive oxygen species (ROS) measurement.
Tumor-infiltrating leukocytes were purified from Tsc1fl/flPyMT and CD11creTsc1fl/flPyMT mice as described above. TAMs were stained with MitoTracker Green FM to measure mitochondrial mass, MitoTracker Red CMXRos to measure mitochondrial potential, and MitoSOX Red to measure mitochondrial ROS following the manufacturer’ instructions.
Seahorse assay.
Freshly purified wild-type or TSC1-deficient TAMs were plated onto Seahorse Xfe-96 microplates (1×105 cells/well) and rested for two hours in DMEM complete medium to allow TAMs to adhere to the plates. Subsequently, the plates were washed and cultured in Seahorse XF base DMEM medium in a non-CO2 incubator for 1 h before Seahorse assays. Specifically, for the glycolysis stress assay, the Seahorse XF base DMEM medium contained 2 mM glutamine, while for the mitochondrial stress assay, the Seahorse XF base DMEM medium contained 2 mM glutamine, 1 mM pyruvate, and 10 mM glucose. Glycolysis-associated parameters were determined by a Seahorse XF Glycolysis Stress Test Kit with three injections: (1) 10 mM glucose; (2) 1 μM oligomycin; and (3) 50 mM 2-Deoxy-D-glucose (2-DG). Oxidative phosphorylation-associated parameters were determined by a Seahorse XF Mito Stress Test Kit with three injections: (1) 1.5 μM oligomycin; (2) 2 μM FCCP, and (3) 0.5 μM rotenone/antimycin A.
TAM and endothelial cell co-culture assay.
TAMs and endothelial cells were isolated from PyMT tumors. 6×104 freshly purified wild-type or TSC1-deficient TAMs were co-cultured with 2×104 PROCR+ or PROCR− tumor-associated endothelial cells for 3 days in IMDM medium containing 10% FBS, 10−4 mM 2-mercaptoethanol, 1% Glutamax, 1% ITS-X (Insulin, transferrin and selectin), 100 ng/mL rmVEGFα, 50 ng/mL rmFGF-basic and 10 ng/mL rmM-CSF. Total cell numbers were counted, and the relative frequencies of TAMs and ECs were determined by flow cytometry.
RNA extraction and quantitative PCR (qPCR).
Freshly purified wild-type or TSC1-deficient TAMs were lysed in TRIzol reagent, and RNA was extracted according to the manufacturer’s instruction. Total RNA was reversed transcribed to cDNA by Maxima First Strand cDNA Synthesis Kit for RT-qPCR. Real-time qPCR was performed in triplicates with SYBR Green Master Mix on a StepOnePlus termal cycler. Primer sequences are listed for the following genes: Tsc1 (forward) 5’-CGGCTCTGGAGGAACACAAT-3’, (reverse) 5’-CTGGCTATGCAGTTGGGTCA-3’; Rplp0 (forward) 5’-CGTCCTGGCATTGTCTGTG-3’, (reverse) 5’-TCTGATTCCTCCGACTCTTCC-3’.
Immunoblotting.
Freshly purified wild-type or TSC1-deficient TAMs or TAMs cultured under a hypoxic condition were lysed in 1xSDS loading buffer and denatured at 95°C for 10 min. Denatured cell lysates were separated by 10% SDS-PAGE and transferred to polyvinylidence fluoride (PVDF) membrane for probing with primary antibodies and HRP-conjugated secondary antibodies. Immobilon Western Chemiluminescent HRP Substrate was used for detection. Relative density of blotting bands was quantified using ImageJ (v2.0.0).
SCENITH (single cell energetic metabolism by profiling translation inhibition) assay.
Modified from a previous study,58 single-cell suspension prepared from PyMT tumors were seeded in 48-well plates at 1×106 cells per well in the RPMI medium containing 10% FBS. Control medium and oligomycin (1 μM) were added to the cells for 15 min. Subsequently, puromycin (10 μg/mL) was added, and incubated for another 45 min. Cells were washed with ice-cold PBS before staining for Live/Dead and surface markers, and then fix/permeabilized for intracellular staining of puromycin. Puromycin intensities were determined by flow cytometry.
Quantification and Statistical Analysis
For scRNAseq analysis, data output from sequencing arrived in the form of as FASTQ files. These files were then processed into count matrices using Bustools (v0.39.3)59 and the accompanying pseudo-aligner Kallisto (v0.46.0).60 Briefly, transcriptome alignment was performed using Kallisto and a generated index from Gencode GRCm38_M22 transcriptome. Of the 400,857,218 reads found in Tsc1fl/flPyMT sample, approximately 82% were pseudo aligned (n = 326,986,504). In the CD11cCreTsc1fl/flPyMT sample, approximately 82% (n = 343,186,512) of the 418,086,280 total reads were pseudo aligned. Barcodes containing fewer than 10 unique molecular identifiers (UMIs) or cells containing counts in 10 or fewer genes were then discarded. Single cell count matrices were then evaluated with ‘knee plots’ to guide empty droplet detection with DropletUtils (v1.8.0).61 This filtered out empty cells with 4256 cells remained in the Tsc1fl/flPyMT sample 3604 cells remained in CD11cCreTsc1fl/flPyMT sample. Single cell analysis, further cell filtering, normalization, and visualization were performed using Scanpy (v1.4.5.post1).62 Several more steps in quality control included cell and gene filtering such that density and scatter plots of UMI counts and genes detected per cell were minimally filtered to obtain a gaussian like distribution, and we retained cells that contained a maximum of 20% mitochondrial gene expression. Cells with 0% Vcam1 expression were also filtered out. After filtering the total number of high-quality cells in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT samples amounted to 1932 and 1425, respectively.
Related to Figures 5 and S5, the log-transformed counts of highly variable genes (HVGs) were used as features for dimensionality reduction and clustering. HVGs were first identified using the method of Seurat.63 This calculates a mean and dispersion measure (variance/mean) for each gene across all single cells, and places these genes into 20 bins based on their average expression. Within each bin the dispersion measure of all genes is z-normalized to identify highly variable expression values compared to genes with similar average expression. These genes were then used by PCA to reveal the main axes of variation and then input to K-nearest neighbors to compute the neighborhood graph. We then used UMAP to embed the neighborhood graph and visually project cells into a two-dimensional space. Clustering was performed using the leiden community detection algorithm.64
Related to Figures 5 and S5 as well as Tables S1–3, single cell differential expression was evaluated by Scanpy’s rank_genes_groups using a Wilcoxon rank sum test and Benjamini-Hochberg for p-value correction. Related to Table S3, cluster 1 enriched genes are transcripts with FDR < 0.01 and LogFC >1 in both 1_vs_2 and 1_vs_4 comparisons. Cluster 2 enriched genes are transcripts with FDR < 0.01 and LogFC <−1 in 1_vs_2 comparison and FDR < 0.01 and LogFC >1 in 2_vs_4 comparison. Clusters 1&2 enriched genes are transcripts with FDR < 0.01 and LogFC >1 in both 1_vs_4 and 2_vs_4 comparisons. Cluster 4 enriched genes are ranscripts with FDR < 0.01 and LogFC <−1 in both 1_vs_4 and 2_vs_4 comparisons. Related to Figure S5D and S5E, KEGG pathway analyses were performed by using David Bioinformatics Resources. Gene lists of wild-type TAMs were from clusters 1 and 2, while gene lists of TSC1-deficient TAMs were from cluster 4.
All experiments were repeated at least three times except measurements of individual tumors. All statistical measurements are presented as the mean values ± SEM. Comparisons between groups were analyzed using unpaired t-tests with GraphPad Prism software. N.s.=not significant, *: p < 0.05, **: p < 0.01, ***: p < 0.001, and ****: p < 0.0001
Data and Code Availability
The datasets supporting the current study are available from the corresponding author on request.
Supplementary Material
Table S1. Differentially expressed transcripts between TAMs from cluster 3 and other clusters
Table S2. Differentially expressed transcripts between TAMs from cluster 5 and other clusters
Table S3. Three-way comparison of transcripts expressed by TAMs from clusters 1, 2, and 4
Video S1. F4/80 staining (white) in a CD11cCreTsc1fl/flPyMT tumor section
Video S2. CD31 staining (red) in a CD11cCreTsc1fl/flPyMT tumor section
Video S3. Protein C receptor (PROCR) staining (green) in a CD11cCreTsc1fl/flPyMT tumor section
Video S4. Cleaved Caspase 3 (CC3) staining (cyan) in a CD11cCreTsc1fl/flPyMT tumor section
Video S5. Merged staining of F4/80, CD31, Protein C receptor (PROCR), cleaved Caspase 3 (CC3), and DAPI in a CD11cCreTsc1fl/flPyMT tumor section
Figure S1. Phenotypic characterization of TAMs and MTMs in virgin mammary glands and mammary tumors
Representative flow cytometric plots of total macrophages as well as tumor-associated macrophage (TAM) and mammary tissue macrophage (MTM) subsets in virgin mammary glands and PyMT tumors: terminally differentiated TAMs are CD45+SiglecF−Ly6C−F4/80+MHCIIhiCD11bloVcam1hiCD206lo/−; MTMs are CD45+SiglecF−Ly6C−F4/80+MHCIIhiCD11bhiVcam1lo/−CD206hi. Data are representative of at least three independent experiments.
Figure S2. TSC1 ablation in TAMs promotes mTORC1 signaling and inhibits tumor growth
(A) mRNA expression of Tsc1 was determined by real time quantitative PCR (qPCR) in TAMs, MTMs, and monocytes from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice.
(B) p4EBP1 and pS6 signals were determined by immunoblotting in TAMs from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Quantification of p4EBP1 and pS6 signals is normalized to that of β-actin.
(C) Total tumor burden of CD64iCreTsc1fl/flPyMT (n = 6) and littermate control Tsc1fl/flPyMT (n = 8) mice.
Figure S3. CD11cCre-mediated TSC1 ablation does not substantially affect expansion of TAMs and dendritic cells or differentiation of T cells
(A and B) TAM percentage (A) and number (B) in Tsc1fl/flPyMT (n = 4) and CD11cCreTsc1fl/flPyMT (n = 5) mice.
(C and D) Percentage (C) and number (D) of total tumor-infiltrating dendritic cells (DCs) as well as XCR1+ DC1 and CD11b+ DC2 subsets in Tsc1fl/flPyMT (n = 9 for percentage and n = 4 for number) and CD11cCreTsc1fl/flPyMT (n = 7 for percentage and n = 5 for number) mice.
(E) Quantification of tumor-infiltrating T cell populations in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Data are representative of more than five independent experiments (n = 6-13).
(F) Quantification of cytokine (IFN-γ, TNF-α, IL-4, and IL-17) production by tumor-infiltrating CD4+ T cells in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice (n = 4-14).
(G) Quantification of cytokine (TNF-α and IFN-γ) production by tumor-infiltrating conventional CD8+ T cells in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice (n = 6-14).
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05, **: p < 0.01, and n.s. = not significant.
Figure S4. Loss of TSC1 triggers TAM repositioning to a perivascular niche as well as vessel reorganization and hypoxia independent of adaptive lymphocytes
(A) Representative images of localization of F4/80+Vcam1hi TAMs in tumor tissues of Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT mice (top). TAMs are indicated by orange arrows. Quantification of the distance of individual TAM to the nearest CD31+ vasculature (bottom). Data are representative of three independent experiments with more than 1×103 TAMs quantified in each experiment. Scale bars: 50 μm, 20 μm, 50 μm, and 20 μm.
(B) Representative images of tumor sections from Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT mice with CD31 (red), Fibrinogen (Fg, white), Cleaved Caspase 3 (cyan), E-Cadherin (green), and DAPI (blue) (left). Isolated CD31 staining is indicated by magenta arrows and extravascular Fg+ events are indicated by orange arrows. Quantification of isolated CD31 staining (top right) and extravascular Fg+ events (bottom right) in randomly selected Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT tumor tissues per 1 mm2 (n = 5-14). Scale bars: 200 μm, 50 μm, 200 μm, and 50 μm.
(C) Representative images (left) and quantification (right) of pimonidazole-positive hypoxic area in randomly selected Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT tumor tissues (n = 13 each). Scale bars: 50 μm and 50 μm.
Data are representative of three independent experiments. All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05, **: p < 0.01, ***: p < 0.001, and n.s. = not significant.
Figure S5. Enriched transcripts in TAM clusters as well as wild-type and TSC1-deficient TAMs
(A) UMAP projection of single cells in two-dimensions colored by normalized counts of macrophage lineage genes Adgre1 (left), Spi1 (middle), and Csf1r (right) encoding F4/80, PU1.1, and CD115 proteins, respectively.
(B) Violin plot of top 5 most significantly differential genes (ranked by adjusted p-value) for all 5 clusters. See also Tables S1 and S2.
(C and D) Top10 enriched KEGG pathways in wild-type (cluster 1 and 2) (C) and TSC1-deficient (cluster 4) (D) TAMs.
Figure S6. Loss of TSC1 does not affect chemokine receptor expression in TAMs, but enhances apoptosis under a hypoxic condition
(A) Representative flow cytometric analysis (left) and quantification (right) of MHCII expression in TAMs from Tsc1fl/flPyMT (n = 9) and CD11cCreTsc1fl/flPyMT (n = 8) mice.
(B) Representative flow cytometric analysis (left) and quantification (right) of CCR2 (top) and CX3CR1 (bottom) expression in TAMs and monocytes from Tsc1fl/flPyMT (n = 4) and CD11cCreTsc1fl/flPyMT (n = 4) mice.
(C) TAMs from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice were cultured under normoxic or hypoxic (1% oxygen) conditions overnight. Cleaved Caspase 3 signals were determined by immunoblotting. Quantification of cleaved Caspase 3 level is normalized to that of β-actin.
All data are shown as mean ± SEM, unpaired t-test, two-tailed, ****: p < 0.0001 and n.s. = not significant.
Figure S7. Tsc1-deficient TAMs outcompete endothelial progenitor cells in intratumoral regions but not peritumoral regions
(A) Representative peritumoral images of tumor sections from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice with F4/80 (white), CD31 (red), protein C receptor (PROCR, green), cleaved Caspase 3 (CC3, cyan), and DAPI staining (left). Quantification of PROCR+ fraction among CD31+ endothelial cells by Manders’ colocalization analysis (top right) as well as CD31+CC3+ events differentiated by PROCR expression (bottom right) in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT tumor tissue sections (n = 10). Scale bars: 50 μm, 10 μm, 50 μm, and 10 μm.
(B) SCENITH (single cell energetic metabolism by profiling translation inhibition) assay to reveal puromycin incorporation-based protein biosynthesis among PROCR+ and PROCR− endothelial cells treated with phosphate-buffered saline (PBS) or oligomycin.
(C) PROCR+ and PROCR− endothelial cells were isolated from Tsc1fl/flPyMT tumors and cultured for 3 days. Fold change of cell number was determined.
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05 and n.s. = not significant.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies for flow cytometry | ||
| Anti-mouse IL-17a | BD Biosciences | TC11-18H10.1 |
| Anti-mouse CD206 | BioLegend | C068C2 |
| Anti-mouse TNFα | BioLegend | MP6-XT22 |
| Anti-mouse XCR1 | BioLegend | ZET |
| Anti-mouse CD107a (LAMP1) | Cell Signaling | D2D11 |
| Anti-mouse CD11b | eBioscience | M1/70 |
| Anti-mouse CD11c | eBioscience | N418 |
| Anti-mouse CD4 | eBioscience | RM4-5 |
| Anti-mouse CD45 | eBioscience | 30-F11 |
| Anti-mouse CD8α | BioLegend | 53-6.7 |
| Anti-mouse F4/80 | eBioscience | BM8 |
| Anti-mouse Foxp3 | eBioscience | FJK-16s |
| Anti-mouse IFN-γ | eBioscience | XMG1.2 |
| Anti-mouse IL-4 | eBioscience | 11B11 |
| Anti-mouse Ly6C | eBioscience | AL-21 |
| Anti-mouse Ly6G | eBioscience | 1A8 |
| Anti-mouse MHC-II I-A/I-E | eBioscience | M5/114.15.2 |
| Anti-mouse SiglecF | eBioscience | E50-2440 |
| Anti-mouse TCR-β | eBioscience | H57-595 |
| Anti-mouse B220 | Thermo Scientific | RA3-6B2 |
| Anti-mouse NK1.1 | Thermo Scientific | PK136 |
| Anti-mouse Vcam1 | Thermo Scientific | 429 |
| Anti-mouse CCR2 | BioLegend | SA203G11 |
| Anti-mouse CX3CR1 | BioLegend | SA011F11 |
| Anti-mouse CD31 | BioLegend | MEC 13.3 |
| Anti-mouse cleaved Caspase 3 | Cell Signaling | Asp175, D3E9 |
| Anti-mouse Protein C receptor | Thermo Scientific | PA5-47382 |
| Anti-Puromycin | BioLegend | 381508 |
| Ghost Dye | Tonbo Biosciences | 13-0865-T500 13-0863-T500 13-0870-T500 |
| FcR Block (2.4G2 mAB) | Bio X Cell | 25mg |
| Antibodies for immunofluorescence | ||
| Anti-mouse CD206 | Abd Serotec | MR5D3 |
| Anti-mouse CD31 | BioLegend | MEC 13.3 |
| Anti-mouse CD4 | BioLegend | RM4-5 |
| Anti-mouse CD8α | BioLegend | 53-6.7 |
| Anti-mouse fibrinogen | Bio-Rad Laboratories | 4440-8004 |
| Anti-mouse cleaved Caspase3 | Cell Signaling | Asp175, D3E9 |
| Anti-mouse Phospho-S6 (Ser235/236) | Cell Signaling | D57.2.2E |
| Anti-mouse E-Cadherin | eBioscience | DECMA-1 |
| Anti-mouse F4/80 | eBioscience | BM8 |
| Anti-mouse Ki67 | eBioscience | SolA15 |
| Anti-mouse Vcam1 | eBioscience | 429 |
| Anti-mouse Vcam1 | R&D Systems | AF643 |
| Anti-mouse Protein C receptor | Thermo Scientific | PA5-47382 |
| DAPI | Thermo Scientific | D1306 |
| Antibodies for immunoblotting | ||
| Anti-mouse phospho-4EBP1 (Thr37/46) | Cell Signaling | 2855S |
| Anti-mouse phospho-S6 (Ser240/244) | Cell Signaling | 2215S |
| Anti-mouse β-actin | Cell Signaling | 3700S |
| Anti-mouse cleaved Caspase 3 | Cell Signaling | Asp175, D3E9 |
| Chemicals, Peptides and Recombinant Proteins | ||
| Collagenase Type 3 | Worthington Biochemical | LS004183 |
| DNase I | Sigma-Aldrich | |
| Percoll | Sigma-Aldrich | P1644-1L |
| 2-Mercaptoethanol 2-Mercaptoethanol |
Thermo Scientific | 21985023 |
| Glutamax™ Supplement | Thermo Scientific | 35050061 |
| Recombinant murine VEGF165 | PeproTech | 450-32 |
| Recombinant murine FGF-basic | PeproTech | 450-33 |
| Recombinant murine M-CSF | PeproTech | 315-02 |
| Insulin-Transferrin-Selenium-Ethanolamine (ITS-X) (100X) | Thermo Scientific | 51500056 |
| Critical Commercial Assays | ||
| Transcription factor Fix/Perm Kit | Tonbo Biosciences | TNB-0607-KIT |
| Hypoxyprobe-Pab27 kit | Hypoxyprobe | hp12-100kit |
| CellTrace Violet Proliferation Kit | Thermo Scientific | C34557 |
| MitoTracker Green FM | Thermo Scientific | M7514 |
| Mitotracker Red CMXRos | Thermo Scientific | M7512 |
| MitoSOX Red Mitochondrial Superoxide | Thermo Scientific | M36008 |
| Agilent Seahorse XFp Glycolysis Stress Test Kit | Agilent Technology | 103017-100 |
| Agilent Seahorse XF Cell Mito Stress Test Kit | Agilent Technology | 103015-100 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.FVB-Tg(MMTV-PyVT)634Mul/LellJ (PyMT) | Jackson Laboratories | Stock: 021614 |
| Mouse: B6.Cg-Tg(Itgax-cre)1-1Reiz/J(Received from Dr. Boris Reizis Lab) | Jackson Laboratories | Stock: 008068 |
| Mouse: Tsc1tm1Djk/J | Jackson Laboratories | Stock: 005680 |
| Mouse: B6.Cg-Rptortm1.1Dmsa/J | Jackson Laboratories | Stock: 013188 |
| Mouse: B6.129S7-Rag1tm1Mom/J | Jackson Laboratories | Stock: 002216 |
| Software and Algorithms | ||
| GraphPad Prism V.7-9 | GraphPad | |
| FlowJo V.9-10 | TreeStar | |
| FacsDIVA | BD Bioscience | |
| Fiji - ImageJ | ||
| ImarisViewer 10.0.0 | Oxford Instruments | |
Highlights.
Tumor parenchyma-located TAMs exhibit low mTORC1 activity
Depletion of TSC1 in TAMs activates mTORC1 and suppresses tumor growth
TSC1-deficient TAMs relocate to a perivascular niche and inhibit angiogenesis
TSC1-deficient TAMs outcompete PROCR-expressing endothelial progenitor cells
ACKNOWLEDGEMENTS
We thank members of the M.O.L. laboratory for helpful discussions. This work was supported by the National Institutes of Health (F30 AI29273-03 to M.H.D., F31 CA210332 to B.G.N. and R01 CA198280 to M.O.L.), Cancer Research Institute (Irvington Fellow awards to L.J., X.Z., S.L., C.C.), the Howard Hughes Medical Institute (Faculty Scholar Award to M.O.L.), Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center (M.O.L.), the Mazumdar-Shaw Translational Research Initiative in Kidney Cancer, and a Cancer Center Support Grant (P30 CA08748).
Footnotes
DECLARATION OF INTERESTS
MSKCC has filed a patent application (number 63/502054) with the US Patent and Trademark Office directed towards targeting the mTORC1 signaling pathway in macrophages for cancer immunotherapy. M.O.L. is a scientific advisory board member of and holds equity or stock options in Amberstone Biosciences and META Pharmaceuticals.
REFERENCES
- 1.Dvorak HF Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315, 1650–1659, doi: 10.1056/NEJM198612253152606 (1986). [DOI] [PubMed] [Google Scholar]
- 2.Liu M et al. TGF-beta suppresses type 2 immunity to cancer. Nature 587, 115–120, doi: 10.1038/s41586-020-2836-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li S et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in T(H) cells. Nature 587, 121–125, doi: 10.1038/s41586-020-2850-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noy R & Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61, doi: 10.1016/j.immuni.2014.06.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franklin RA & Li MO Ontogeny of Tumor-associated Macrophages and Its Implication in Cancer Regulation. Trends Cancer 2, 20–34, doi: 10.1016/j.trecan.2015.11.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeNardo DG & Ruffell B Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19, 369–382, doi: 10.1038/s41577-019-0127-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franklin RA et al. The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925, doi: 10.1126/science.1252510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nixon BG et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity 55, 2044–2058 e2045, doi: 10.1016/j.immuni.2022.10.002 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergers G & Benjamin LE Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3, 401–410, doi: 10.1038/nrc1093 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Lin EY et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66, 11238–11246, doi: 10.1158/0008-5472.CAN-06-1278 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Vannella KM & Wynn TA Mechanisms of Organ Injury and Repair by Macrophages. Annu Rev Physiol 79, 593–617, doi: 10.1146/annurev-physiol-022516-034356 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Oishi Y & Manabe I Macrophages in inflammation, repair and regeneration. Int Immunol 30, 511–528, doi: 10.1093/intimm/dxy054 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Saxton RA & Sabatini DM mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371, doi: 10.1016/j.cell.2017.03.035 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Inoki K, Li Y, Xu T & Guan KL Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17, 1829–1834, doi: 10.1101/gad.1110003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tee AR, Manning BD, Roux PP, Cantley LC & Blenis J Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13, 1259–1268, doi: 10.1016/s0960-9822(03)00506-2 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Lee PY et al. The metabolic regulator mTORC1 controls terminal myeloid differentiation. Sci Immunol 2, doi: 10.1126/sciimmunol.aam6641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu S et al. TSC1 regulates osteoclast podosome organization and bone resorption through mTORC1 and Rac1/Cdc42. Cell Death Differ 25, 1549–1566, doi: 10.1038/s41418-017-0049-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan H, O’Brien TF, Zhang P & Zhong XP The role of tuberous sclerosis complex 1 in regulating innate immunity. J Immunol 188, 3658–3666, doi: 10.4049/jimmunol.1102187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byles V et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun 4, 2834, doi: 10.1038/ncomms3834 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu L et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun 5, 4696, doi: 10.1038/ncomms5696 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Yang T et al. TSC1 controls IL-1beta expression in macrophages via mTORC1-dependent C/EBPbeta pathway. Cell Mol Immunol 13, 640–650, doi: 10.1038/cmi.2015.43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Covarrubias AJ et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 5, doi: 10.7554/eLife.11612 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang C et al. Tsc1 is a Critical Regulator of Macrophage Survival and Function. Cell Physiol Biochem 36, 1406–1418, doi: 10.1159/000430306 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Pan H, Zhong XP & Lee S Sustained activation of mTORC1 in macrophages increases AMPKalpha-dependent autophagy to maintain cellular homeostasis. BMC Biochem 17, 14, doi: 10.1186/s12858-016-0069-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linke M et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol 18, 293–302, doi: 10.1038/ni.3655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakarov S et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, doi: 10.1126/science.aau0964 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Zhang X et al. Reprogramming tumour-associated macrophages to outcompete cancer cells. Nature 619, 616–623, doi: 10.1038/s41586-023-06256-5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy TL et al. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol 34, 93–119, doi: 10.1146/annurev-immunol-032713-120204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bansal A & Simon MC Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 217, 2291–2298, doi: 10.1083/jcb.201804161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doran AC, Yurdagul A Jr. & Tabas I Efferocytosis in health and disease. Nat Rev Immunol 20, 254–267, doi: 10.1038/s41577-019-0240-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmall A et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med 191, 437–447, doi: 10.1164/rccm.201406-1137OC (2015). [DOI] [PubMed] [Google Scholar]
- 33.Dight J, Zhao J, Styke C, Khosrotehrani K & Patel J Resident vascular endothelial progenitor definition and function: the age of reckoning. Angiogenesis 25, 15–33, doi: 10.1007/s10456-021-09817-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu QC, Song W, Wang D & Zeng YA Identification of blood vascular endothelial stem cells by the expression of protein C receptor. Cell Res 26, 1079–1098, doi: 10.1038/cr.2016.85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Neerven SM & Vermeulen L Cell competition in development, homeostasis and cancer. Nat Rev Mol Cell Biol 24, 221–236, doi: 10.1038/s41580-022-00538-y (2023). [DOI] [PubMed] [Google Scholar]
- 36.Bowling S et al. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat Commun 9, 1763, doi: 10.1038/s41467-018-04167-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coutelle O et al. Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol Med 6, 624–639, doi: 10.1002/emmm.201303016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiffmann LM et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat Commun 11, 3653, doi: 10.1038/s41467-020-17472-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bleriot C, Chakarov S & Ginhoux F Determinants of Resident Tissue Macrophage Identity and Function. Immunity 52, 957–970, doi: 10.1016/j.immuni.2020.05.014 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Guilliams M, Thierry GR, Bonnardel J & Bajenoff M Establishment and Maintenance of the Macrophage Niche. Immunity 52, 434–451, doi: 10.1016/j.immuni.2020.02.015 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Ji L & Li MO Control of tumor-associated macrophage responses by nutrient acquisition and metabolism. Immunity 56, 14–31, doi: 10.1016/j.immuni.2022.12.003 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pucci F et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 114, 901–914, doi: 10.1182/blood-2009-01-200931 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Lewis CE, Harney AS & Pollard JW The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell 30, 18–25, doi: 10.1016/j.ccell.2016.05.017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laviron M et al. Tumor-associated macrophage heterogeneity is driven by tissue territories in breast cancer. Cell Rep 39, 110865, doi: 10.1016/j.celrep.2022.110865 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Dawson CA et al. Tissue-resident ductal macrophages survey the mammary epithelium and facilitate tissue remodelling. Nat Cell Biol 22, 546–558, doi: 10.1038/s41556-020-0505-0 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Djonov V, Andres AC & Ziemiecki A Vascular remodelling during the normal and malignant life cycle of the mammary gland. Microsc Res Tech 52, 182–189, doi: (2001). [DOI] [PubMed] [Google Scholar]
- 47.Rossiter H et al. Inactivation of VEGF in mammary gland epithelium severely compromises mammary gland development and function. FASEB J 21, 3994–4004, doi: 10.1096/fj.07-8720com (2007). [DOI] [PubMed] [Google Scholar]
- 48.Qiu Y et al. Mammary alveolar development during lactation is inhibited by the endogenous antiangiogenic growth factor isoform, VEGF165b. FASEB J 22, 1104–1112, doi: 10.1096/fj.07-9718com (2008). [DOI] [PubMed] [Google Scholar]
- 49.Wenes M et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab 24, 701–715, doi: 10.1016/j.cmet.2016.09.008 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Asahara T et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967, doi: 10.1126/science.275.5302.964 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Purhonen S et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A 105, 6620–6625, doi: 10.1073/pnas.0710516105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen W et al. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res 72, 1363–1372, doi: 10.1158/0008-5472.CAN-11-2684 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Claveria C, Giovinazzo G, Sierra R & Torres M Myc-driven endogenous cell competition in the early mammalian embryo. Nature 500, 39–44, doi: 10.1038/nature12389 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Basagiannis D et al. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J Cell Sci 129, 4091–4104, doi: 10.1242/jcs.188219 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Bae E et al. Integrin alpha3beta1 promotes vessel formation of glioblastoma-associated endothelial cells through calcium-mediated macropinocytosis and lysosomal exocytosis. Nat Commun 13, 4268, doi: 10.1038/s41467-022-31981-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akil A et al. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front Cell Dev Biol 9, 642352, doi: 10.3389/fcell.2021.642352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nalio Ramos R et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell 185, 1189–1207 e1125, doi: 10.1016/j.cell.2022.02.021 (2022). [DOI] [PubMed] [Google Scholar]
- 58.Arguello RJ et al. SCENITH: A Flow Cytometry-Based Method to Functionally Profile Energy Metabolism with Single-Cell Resolution. Cell Metab 32, 1063–1075 e1067, doi: 10.1016/j.cmet.2020.11.007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melsted P, Ntranos V & Pachter L The barcode, UMI, set format and BUStools. Bioinformatics 35, 4472–4473, doi: 10.1093/bioinformatics/btz279 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527, doi: 10.1038/nbt.3519 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Lun ATL et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 20, 63, doi: 10.1186/s13059-019-1662-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15, doi: 10.1186/s13059-017-1382-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502, doi: 10.1038/nbt.3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9, 5233, doi: 10.1038/s41598-019-41695-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed transcripts between TAMs from cluster 3 and other clusters
Table S2. Differentially expressed transcripts between TAMs from cluster 5 and other clusters
Table S3. Three-way comparison of transcripts expressed by TAMs from clusters 1, 2, and 4
Video S1. F4/80 staining (white) in a CD11cCreTsc1fl/flPyMT tumor section
Video S2. CD31 staining (red) in a CD11cCreTsc1fl/flPyMT tumor section
Video S3. Protein C receptor (PROCR) staining (green) in a CD11cCreTsc1fl/flPyMT tumor section
Video S4. Cleaved Caspase 3 (CC3) staining (cyan) in a CD11cCreTsc1fl/flPyMT tumor section
Video S5. Merged staining of F4/80, CD31, Protein C receptor (PROCR), cleaved Caspase 3 (CC3), and DAPI in a CD11cCreTsc1fl/flPyMT tumor section
Figure S1. Phenotypic characterization of TAMs and MTMs in virgin mammary glands and mammary tumors
Representative flow cytometric plots of total macrophages as well as tumor-associated macrophage (TAM) and mammary tissue macrophage (MTM) subsets in virgin mammary glands and PyMT tumors: terminally differentiated TAMs are CD45+SiglecF−Ly6C−F4/80+MHCIIhiCD11bloVcam1hiCD206lo/−; MTMs are CD45+SiglecF−Ly6C−F4/80+MHCIIhiCD11bhiVcam1lo/−CD206hi. Data are representative of at least three independent experiments.
Figure S2. TSC1 ablation in TAMs promotes mTORC1 signaling and inhibits tumor growth
(A) mRNA expression of Tsc1 was determined by real time quantitative PCR (qPCR) in TAMs, MTMs, and monocytes from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice.
(B) p4EBP1 and pS6 signals were determined by immunoblotting in TAMs from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Quantification of p4EBP1 and pS6 signals is normalized to that of β-actin.
(C) Total tumor burden of CD64iCreTsc1fl/flPyMT (n = 6) and littermate control Tsc1fl/flPyMT (n = 8) mice.
Figure S3. CD11cCre-mediated TSC1 ablation does not substantially affect expansion of TAMs and dendritic cells or differentiation of T cells
(A and B) TAM percentage (A) and number (B) in Tsc1fl/flPyMT (n = 4) and CD11cCreTsc1fl/flPyMT (n = 5) mice.
(C and D) Percentage (C) and number (D) of total tumor-infiltrating dendritic cells (DCs) as well as XCR1+ DC1 and CD11b+ DC2 subsets in Tsc1fl/flPyMT (n = 9 for percentage and n = 4 for number) and CD11cCreTsc1fl/flPyMT (n = 7 for percentage and n = 5 for number) mice.
(E) Quantification of tumor-infiltrating T cell populations in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice. Data are representative of more than five independent experiments (n = 6-13).
(F) Quantification of cytokine (IFN-γ, TNF-α, IL-4, and IL-17) production by tumor-infiltrating CD4+ T cells in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice (n = 4-14).
(G) Quantification of cytokine (TNF-α and IFN-γ) production by tumor-infiltrating conventional CD8+ T cells in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice (n = 6-14).
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05, **: p < 0.01, and n.s. = not significant.
Figure S4. Loss of TSC1 triggers TAM repositioning to a perivascular niche as well as vessel reorganization and hypoxia independent of adaptive lymphocytes
(A) Representative images of localization of F4/80+Vcam1hi TAMs in tumor tissues of Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT mice (top). TAMs are indicated by orange arrows. Quantification of the distance of individual TAM to the nearest CD31+ vasculature (bottom). Data are representative of three independent experiments with more than 1×103 TAMs quantified in each experiment. Scale bars: 50 μm, 20 μm, 50 μm, and 20 μm.
(B) Representative images of tumor sections from Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT mice with CD31 (red), Fibrinogen (Fg, white), Cleaved Caspase 3 (cyan), E-Cadherin (green), and DAPI (blue) (left). Isolated CD31 staining is indicated by magenta arrows and extravascular Fg+ events are indicated by orange arrows. Quantification of isolated CD31 staining (top right) and extravascular Fg+ events (bottom right) in randomly selected Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT tumor tissues per 1 mm2 (n = 5-14). Scale bars: 200 μm, 50 μm, 200 μm, and 50 μm.
(C) Representative images (left) and quantification (right) of pimonidazole-positive hypoxic area in randomly selected Tsc1fl/flRag1−/−PyMT and CD11cCreTsc1fl/flRag1−/−PyMT tumor tissues (n = 13 each). Scale bars: 50 μm and 50 μm.
Data are representative of three independent experiments. All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05, **: p < 0.01, ***: p < 0.001, and n.s. = not significant.
Figure S5. Enriched transcripts in TAM clusters as well as wild-type and TSC1-deficient TAMs
(A) UMAP projection of single cells in two-dimensions colored by normalized counts of macrophage lineage genes Adgre1 (left), Spi1 (middle), and Csf1r (right) encoding F4/80, PU1.1, and CD115 proteins, respectively.
(B) Violin plot of top 5 most significantly differential genes (ranked by adjusted p-value) for all 5 clusters. See also Tables S1 and S2.
(C and D) Top10 enriched KEGG pathways in wild-type (cluster 1 and 2) (C) and TSC1-deficient (cluster 4) (D) TAMs.
Figure S6. Loss of TSC1 does not affect chemokine receptor expression in TAMs, but enhances apoptosis under a hypoxic condition
(A) Representative flow cytometric analysis (left) and quantification (right) of MHCII expression in TAMs from Tsc1fl/flPyMT (n = 9) and CD11cCreTsc1fl/flPyMT (n = 8) mice.
(B) Representative flow cytometric analysis (left) and quantification (right) of CCR2 (top) and CX3CR1 (bottom) expression in TAMs and monocytes from Tsc1fl/flPyMT (n = 4) and CD11cCreTsc1fl/flPyMT (n = 4) mice.
(C) TAMs from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice were cultured under normoxic or hypoxic (1% oxygen) conditions overnight. Cleaved Caspase 3 signals were determined by immunoblotting. Quantification of cleaved Caspase 3 level is normalized to that of β-actin.
All data are shown as mean ± SEM, unpaired t-test, two-tailed, ****: p < 0.0001 and n.s. = not significant.
Figure S7. Tsc1-deficient TAMs outcompete endothelial progenitor cells in intratumoral regions but not peritumoral regions
(A) Representative peritumoral images of tumor sections from Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT mice with F4/80 (white), CD31 (red), protein C receptor (PROCR, green), cleaved Caspase 3 (CC3, cyan), and DAPI staining (left). Quantification of PROCR+ fraction among CD31+ endothelial cells by Manders’ colocalization analysis (top right) as well as CD31+CC3+ events differentiated by PROCR expression (bottom right) in Tsc1fl/flPyMT and CD11cCreTsc1fl/flPyMT tumor tissue sections (n = 10). Scale bars: 50 μm, 10 μm, 50 μm, and 10 μm.
(B) SCENITH (single cell energetic metabolism by profiling translation inhibition) assay to reveal puromycin incorporation-based protein biosynthesis among PROCR+ and PROCR− endothelial cells treated with phosphate-buffered saline (PBS) or oligomycin.
(C) PROCR+ and PROCR− endothelial cells were isolated from Tsc1fl/flPyMT tumors and cultured for 3 days. Fold change of cell number was determined.
All data are shown as mean ± SEM, unpaired t-test, two-tailed, *: p < 0.05 and n.s. = not significant.
Data Availability Statement
The datasets supporting the current study are available from the corresponding author on request.