

SUMMARY
Phosphate (Pi) serves countless metabolic pathways and is involved in macromolecule synthesis, energy storage, cellular signaling, and bone maintenance. Herein, we describe the coordination of Pi uptake and efflux pathways to maintain mammalian cell Pi homeostasis. We discover that XPR1, the presumed Pi efflux transporter, separately supervises rates of Pi uptake. This direct, regulatory interplay arises from XPR1 being a binding partner for the Pi uptake transporter PiT1, involving a predicted transmembrane helix/extramembrane loop in XPR1, and its hitherto unknown localization in a subset of intracellular LAMP1-positive puncta (named “XLPVs”). A pharmacological mimic of Pi homeostatic challenge is sensed by the inositol pyrophosphate IP8, which functionalizes XPR1 to respond in a temporally hierarchal manner, initially adjusting the rate of Pi efflux, followed subsequently by independent modulation of PiT1 turnover to reset the rate of Pi uptake. These observations generate a unifying model of mammalian cellular Pi homeostasis, expanding opportunities for therapeutic intervention.
In brief
Li et al. demonstrate that organellar XPR1 regulates inorganic phosphate fluxes in mammalian cells. Control of phosphate influx by XPR1 is mediated through its interaction with the phosphate transporter PiT1 that prevents PiT1 degradation. The signaling molecule IP8 functionalizes XPR1-dependent regulation of PiT1 expression to supervise cellular phosphate homeostasis.
Graphical abstract
INTRODUCTION
The participation of cell signaling cascades in the dynamic maintenance of metabolite and nutrient homeostasis comprises one of the most fundamental biological processes; an improved molecular-level understanding of the underlying mechanisms is likely to offer therapeutic opportunities.1 However, an area of long-standing uncertainty has surrounded the mechanisms of short-term regulation of the cellular levels of one prominent nutrient: inorganic phosphate (Pi).2–4 The very survival of an organism depends upon this careful control of cellular Pi uptake, efflux, and its intracellular distribution; Pi is an essential component of bone maintenance, countless metabolic pathways, and macromolecule synthesis, as well as energy storage and cellular signaling processes, while also being one of the most pervasive metabolic inhibitors.5–7 Among prominent pathological examples of defects in Pi homeostasis are ectopic calcification and inflammation.8,9
In recent years, it has been determined that certain aspects of eukaryotic Pi homeostasis are modulated by inositol pyrophosphates (PP-IPs), particularly IP8 (Figure 1A).2,4,6,7,10–12 Cytosolic levels of IP8 rise and fall in response to corresponding fluctuations in cytosolic Pi, that is, IP8 is a Pi sensor.12–14 In mammals, a major downstream effector of IP8 is xenotropic and polytropic retrovirus receptor 1 (XPR1), a ubiquitous protein of approximately 80 kDa (Figure 1A) that is widely considered to mediate cellular Pi efflux. This plasma membrane transport activity was first proposed by Battini’s group15 and is consistent with cell surface imprinting of XPR1 with a fluorescent tag2,15 or immunogold16 and a number of additional genetic knockdown/knockout (KO) and rescue experiments (for access to this literature, see Hernando et al.,4 Jennings,6 Michigami et al.,7 and Wang et al.17). Nevertheless, it should be noted that Pi transport activity by XPR1 has not yet been directly validated (i.e., by reconstitution into liposomes), so it remains formally possible that this protein is instead a regulator of another, as yet unsuspected Pi transporter.18 On the other hand, an XPR1 ortholog from rice, OsPHO1:2, which is also present in plasma membranes, has been incorporated into liposomes and shown to transport Pi.19 The regulation of XPR1-dependent Pi transport activity is thought to be mediated by IP8 binding to lysine surface cluster within the N-terminal SYG1/Pho81/XPR1 (SPX) domain of XPR1.2,10 Although this SPX domain can also bind IP7 in vitro, only IP8 can promote Pi efflux.10
Figure 1. Subcellular localization of XPR1.

(A) Domain graphic of XPR1, as defined by InterPro (#Q9UBH6), highlighting the SPX domain (colored orange; the functional IP8 ligand10 is also depicted in its chair conformations; carbon atoms are numbered according to standard nomenclature). The graphic also shows the EXS domains (cyan) and the IDR (intrinsically disordered region; green). A region not functionally defined by InterPro is colored gray. Also noted are three lysosomal localization motifs (vertical red lines): one so-called “tyrosine-based” motif (φ = bulky, hydrophobic residue) and two “dileucine-based” motifs.20 Our leucine/isoleucine mutants 1 and 2 are noted (LM1; LM2; red font specifies mutated residues).
(B and C) Time courses of [33P]-Pi uptake and [33P]-Pi efflux, respectively, in the indicated strains of U2OS cells, color coded as shown in (B): WT (blue), XPR1 KO (pink), XPR1 KO + WT XPR1 (brown), and XPR1 KO + the LM1/2 XPR1 mutant (red). Data are means ± standard errors from three separate experiments. ***p < 0.001 (two-way ANOVA, with Tukey’s multiple-comparison test).
(D) Western blot analysis of PiT1 and PiT2 proteins in cell lysates (“input”) and an anti-Halo immunoprecipitate (“IP”) of Halo-tagged endogenous XPR1 from HCT116 cells. Blots were developed separately using anti-PiT1, anti-PiT2, or anti-Halo antibodies. The right panel is the quantification of IP PiT1 normalized to Input PiT1; data are means ± standard errors from three separate experiments. **p = 0.0076 (paired t test).
(E) Membrane fractions were prepared from U2OS cells and analyzed by western blots for XPR1 and the plasma membrane marker ATP1A1. The right-hand panel displays IM/PM ratios for each protein; data are means ± standard errors from three separate experiments. *p = 0.018 (paired t test). TM, total cell membrane fraction; PM, plasma membrane enriched fraction; IM, intracellular organelle membrane fraction.
(F) Western blot analysis of XPR1 and various organelle markers in cell lysate (“input”) or anti-HA immunoprecipitated (“IP”), TMEM192-[HA]3-tagged vesicles purified from WT, PPIP5K KO, or XPR1 KO HCT116 cells.
(G) Confocal analysis of endogenous LAMP1 in U2OS cells that were transiently transfected with either C-terminally mCherry-tagged WT XPR1 or one of the three dileucine mutants LM1, LM2, or LM1/2 (see A). Statistical analysis of three independent experiments is shown in Figure S3B.
(H) Confocal analysis of endogenous ERp46 in U2OS cells transiently transfected with C-terminally mCherry-tagged LM1/2 mutant of XPR1. Statistical analysis of three independent experiments is shown in Figure S3C. All scale bars depict 10 μm.
However, little is known about how this recently described regulatory paradigm might dynamically coordinate with cellular Pi uptake. This situation is a fundamental challenge to the goal of understanding the molecular basis for cellular Pi homeostasis.2,4 PiT1 (SLC20A1) and its isoform, PiT2 (SLC20A2), are widely expressed proteins that accumulate Pi from the extracellular milieu.4,6,7 A recent study reported that XPR1-mediated Pi efflux is enhanced by either overexpression of PiT2 or elevation of extracellular Pi.2 In this interplay between XPR1 and PiT2, the latter was given a relatively passive role—an increased capacity for Pi uptake rather than a downstream effector of Pi homeostasis—with the active regulatory component being PP-IP-mediated control over XPR1 activity [Pi].2 In such circumstances, in which both influx and efflux pathways are increased, it remains unclear how this provides the required adjustment—i.e., a net reduction in cytosolic [Pi]. Thus, in the current study, we have widened our understanding of the roles of IP8/XPR1 signaling in maintaining Pi homeostasis.
RESULTS
Organellar XPR1 regulates cellular Pi fluxes
We began this study by seeking possible roles for the IP8/XPR1 signaling axis in regulating mammalian-cell Pi uptake in U2OS and Saos-2 cells. For ease of comparison with earlier studies performed by Battini and colleagues,9,15 we initially monitored cellular accumulation of [33P]-Pi that was added to Pi-free culture media. This methodology usefully exploits the phenomenon that Pi uptake follows Michaelis-Menten kinetics,21 so by analogy to enzymatic reactions, our use of limiting substrate (i.e., Pi) reduces the initial uptake rate (v) to Vmax[S]/Km. Thus, we can qualitatively investigate experimental effects upon transport, irrespective of whether it is Km or Vmax that is modified. Nevertheless, in additional experiments described further, we have also recorded Pi fluxes in the presence of physiologically relevant Pi concentrations of 1 mM.
We found that the XPR1 KO reduced the rate of [33P]-Pi uptake by approximately 50% (Figures 1B and S1A). Since linear rates of [33P]-Pi uptake were recorded in 120 min time courses, only 30 min time points for uptake are shown hereafter. The XPR1 KO also reduced the rate of cellular [33P]-Pi efflux by 60%–70% (Figures 1C and S1B), consistent with previous studies.2,10,15,22
To understand how XPR1 may regulate cellular Pi uptake, we performed a subtractive proteomic screen to identify binding partners. To optimize physiological relevance, we performed this experiment with endogenous XPR1. For these experiments, we used CRISPR to insert the coding sequence for a Halo-tag23 immediately upstream of the stop codon for the XPR1 gene in HCT116 cells (Figures S2A and S2B). Putative XPR1-binding proteins were pulled down with Halo Nanotrap and analyzed by mass spectrometry (Table S1). We were intrigued that PiT1 (SLC20A1) emerged from this screen (Figures S2C and S2D). We confirmed PiT1 as a binding partner through its co-immunoprecipitation with Halo-tagged XPR1; PiT2 did not bind to XPR1 (Figure 1D).
If it were an interaction of XPR1 with PiT1 that regulates Pi uptake, we might expect this to occur in the plasma membrane. However, we identified three potential intracellular-targeting sequences near the C terminus of XPR1 (Figure 1A).20,24 Confocal analysis revealed that endogenous Halo-tagged XPR1 is largely distributed throughout the cell interior in a punctate manner (Figure S1C). Since this XPR1 signal coincided with a small proportion of the lysosomal-associated membrane protein 1 (LAMP1) continuum, we conclude it to be an organellar compartment, comprising XPR1/LAMP1-positive vesicles (XLPVs); plasma membrane XPR1 was below our detection limits. Additionally, in cell fractionation experiments, we found endogenous XPR1 to be 2.5-fold enriched in the cell’s intracellular membranes, rather than the ATP1A1-enriched plasma membrane fraction (Figures 1E and S1D).
Furthermore, cellular fractionation of HCT116 cells transfected with the lysosomal TMEM192-[HA]3 tag25 revealed it to co-purify with endogenous XPR1 and LAMP1, in preference to markers for Golgi, endoplasmic reticulum (ER), or mitochondria (Figure 1F). Overall, we conclude that the cell surface pool of XPR1 that others have detected2 may comprise only a minor proportion of total cellular XPR1.
Confocal analysis of U2OS cells after transient expression of either XPR1-mCherry or XPR1-YFP yielded the same conclusions that we derived by analyzing endogenous XPR1 expression (see above): there is no detectable plasma membrane signal, and XLPVs comprise a small proportion of the overall LAMP1-positive continuum (Figures 1G and S3A). We prepared XPR1 constructs in which [Ile/Leu] within two “dileucine motifs” were mutated to Ala (LM1 and LM2; Figure 1A). XLPVs were eliminated upon expression of the double mutant, LM1/2 (Figures 1G and S3B), which is predominantly mislocalized to the ER, as evidenced by LM1/2 colocalization with ER-resident protein 46 (Figures 1G, 1H, and S3C). Significantly, stable overexpression of wild-type (WT) XPR1-mCherry rescued [33P]-Pi uptake by XPR1 KO cells, whereas the LM1/2 mutant was only marginally effective (Figures 1B and S1A). Thus, we conclude that the organellar pool of XPR1 participates in regulating Pi uptake. Interestingly, the LM1/2 mutant of XPR1 also weakly promoted Pi efflux from XPR1 KO cells (Figures 1C and S1B). Mutation of the tyrosine-based lysosomal targeting motif eliminated XLPVs (Figures 1A and S3D); we could not test the functionality of this mutant in Pi homeostasis because this construct could not be stably expressed in our XPR1 KO cells.
Our demonstration of an intracellular, vesicular pool of mammalian XPR1 invites comparisons with the recent demonstration of a vesicular pool of the Drosophila ortholog, PXo.26 Furthermore, it has been proposed that vesicle exocytosis contributes to Pi export by PXo and XPR1 orthologs in some plants.26,27 We therefore performed a 15 min [33P]-Pi efflux experiment after which we used size-exclusion columns to isolate exosomes from the culture medium. No radiolabel co-purified with the extracellular, vesicular fraction (Figure S3E). It should also be noted that the PXo pool is not LAMP1 positive,26 suggesting it is not a functional ortholog of mammalian XPLVs.
The significance of the SPX domain for XPR1-mediated regulation of Pi uptake
The SPX domain of XPR1 is hypothesized to mediate IP8-dependent control over Pi efflux2,10; IP7 is not effective.10 The SPX domain crystal structure (residues 1–213) has previously been described at a resolution of 2.43 Å in complex with a sulfate ion, but not a PP-IP ligand.11 We prepared an SPX domain construct from residues 1–207, except we replaced an internal disordered region11 (see STAR Methods) with a GSGSG linker. We obtained SPX and SPX/IP8 crystal complexes with 1.4 and 1.85 Å resolution, respectively (Figures 2A, S4A, and S4B), which are both virtually superimposable (root-mean-square deviation = Å) upon the previously published SPX/SO42− complex, except for the α−6 helix (Figure S4A), which was previously noted to occupy alternate configurations.11
Figure 2. Characterization of the SPX domain of XPR1.

(A) Electrostatic surface presentation to show a stick model of IP8 binding to a surface lysine cluster on one molecule of the SPX domain. Blue indicates positive charge and red indicates negative charge, at physiological pH. An omit Fo-Fc different map for IP8 is shown as green mesh, contoured at σ = 3.
(B) Confocal analysis of U2OS cells transiently transfected with C-terminal, mCherry-tagged (mCh) full-length XPR1 or ΔSPX-XPR1 and stained for LAMP1. Punctate signals of co-localized XPR1 and LAMP1 are named “XPR1 and LAMP1-positive vesicles” (XLPVs; see text). Statistical analysis of three independent experiments is shown in Figure S4C. Scale bars depict 10 μm.
(C) Bar graphs depicting relative rates of [33P]-Pi influx in the following strains of U2OS cells: wild-type (blue) and XPR1-KO cells stably transfected with control vector (pink), wild-type XPR1-mCherry (brown), ΔSPX-XPR1-mCherry (red), or the SPX domain alone (yellow). Data are means ± standard errors from three separate experiments. **p < 0.01 (one-way ANOVA, with Tukey’s multiple-comparison test).
(D) Rates of [33P]-Pi efflux from the cell strains described in (C), using the same color coding. Data are means ± standard errors from three separate experiments. ***p < 0.001 (two-way ANOVA, with Tukey’s multiple-comparison test).
The asymmetric unit contains two protein molecules, which have multiple interactions with one molecule of IP8 (Figure S4B). These data break ground through their atomic-level rationalization of the SPX domain being an IP8 receptor, in particular by illustrating the importance of the 1- and 5-diphosphate groups, which make 10 polar contacts with the protein, including residues K2, K26, K158, K161, K162, and K165 (Figures 2A and S4B), that comprisea lysine surface cluster previously identified as functionally important to PP-IP-dependent Pi homeostasis.2,11 However, a specific role for the SPX domain in Pi uptake has not previously been reported. To address this point, we expressed ΔSPX-XPR1 in XPR1 KO cells; this ΔSPX construct localizes in XLPVs to the same extent as WT XPR1 (Figures 2B and S4C) but failed to rescue cellular Pi uptake or Pi efflux (Figures 2C and 2D). These data are consistent with a key role for SPX/IP8 in Pi uptake, but not XLPV biogenesis.
The IP8/XPR1 signaling axis regulates PiT1 protein expression
We next pursued the significance of our observation (see above) that PiT1 is a binding partner for XPR1. The KO of PiT1 in U2OS cells reduced [33P]-Pi uptake by 80% (Figures 3A and 3B), and uptake was rescued by exogenous expression of HA-tagged PiT1 (Figures 3A and 3B). These data illustrate the importance of this transporter to Pi homeostasis in this cell type. Unexpectedly, PiT1 KO also attenuates Pi efflux (Figure 3C). Interestingly, cellular Pi levels were increased in XPR1 KO, but not PiT1 KO cells (Figure 3D).
Figure 3. The kinase activity of PPIP5K regulates PiT1 expression and function in Pi uptake and release.

(A) Western blot analysis of PiT1 expression in three strains of U2OS cells: WT, PiT1 KO, and PiT1 KO in which PiT1-HA is stably expressed.
(B and C) [33P]-Pi uptake and [33P]-Pi efflux, respectively, in the cell strains indicated in (A).
(D) Total cellular Pi in the following strains of U2OS cells: WT, XPR1 KO, XPR1 KO + XPR1, PiT1 KO, and PiT1 KO + PiT1. Data are means and standard errors from three replicates. ***p < 0.001. One-way ANOVA, with Tukey’s multiple-comparison test.
(E) Western blot analysis of PiT1 and PiT2 expression in the following strains of U2OS cells: WT, XPR1 KO, XPR1 KO + XPR1, XPR1 KO + the XPR1 LM1/2 mutant, and PiT1 KO. Statistical analysis of three independent experiments is shown in Figure S5A and S5B.
(F) Western blot analysis of the siRNA validation of the PiT2 antibody in HCT116 cells.
(G) PiT1 expression levels in WT, PPIP5K KO, and PPIP5K KO Saos-2 cells stably transfected with either PPIP5K1 or one of the indicated mutants, following treatment of cells for 6 h with either 10 μM UNC7467 (see Figure S6A) or DMSO vehicle. Statistical analysis of three independent experiments is shown in Figure S6B.
(H) Confocal microscopy analysis of WT of PPIP5K KO U2OS cells or WT cells treated for 6 h with 10 μM UNC7467 and transiently transfected with C-terminally mCherry-tagged (mCh) XPR1; immunostaining of LAMP1 is also shown. All scale bars depict 10 μm.
(I) Total [33P]-Pi uptake after 30 min of labeling of the cell lines described in (G). All Pi transport assays (B, C, and I) were performed three times, and data shown are means and standard errors from three replicates. ***p < 0.001, ns = not significant. One-way ANOVA, with Tukey’s multiple-comparison test.
We found PiT1 expression is dramatically reduced in XPR1 KO cells; expression of PiT2 was unaffected (Figures 3E, 3F, S5A, and S5B). The ΔSPX-XPR1 construct is unable to rescue PiT1 expression (Figures S5A and S5C). Furthermore, PiT1 expression in XPR1 KO cells was restored by stable expression of WT XPR1, but only marginal rescue was elicited by stable expression of the LM1/LM2 mutant (Figure 3E), a cell model in which XLPVs are not detected (see above), indicating that it is this organellar pool of XPR1 that regulates PiT1 protein expression.
Furthermore, PiT1 expression was strongly reduced in Saos-2 PPIP5K KO cell lines10 (Figure 3G) that cannot synthesize IP8 (see Figure S6A). We have previously demonstrated that IP7 levels are elevated when IP8 synthesis is completely blocked by the PPIP5K KO.28 Thus, we conclude that IP7 does not compensate for the reduction in PiT1 stability when IP8 is absent. PiT1 expression in PPIP5K KO cells was restored upon stable expression of PPIP5K1 in the KO cell line. PiT1 levels were not rescued by PPIP5K1D332A, the kinase-dead mutant, but PiT1 expression was restored by PPIP5K1R399A, the phosphatase-dead mutant that overproduces IP8 (Figures 3G and S6B).
We also treated cells for 6 h with a recently developed IP6K inhibitor, UNC7467 (Figure S6A), which deprives PPIP5Ks of the IP7 precursor for IP8 synthesis29 (and see below). This pharmacological approach reduced PiT1 expression in WT cells and also prevented PPIP5K1 and PPIP5K1R399A from rescuing PiT1 expression in PPIP5K KO cells (Figures 3G and S6B). These data are consistent with IP8 being the PP-IP with the dominant role in regulating PiT1 stability. However, we cannot exclude an ancillary role for IP7 in regulating PiT1 expression when IP8 is also present.
XLPVs remained evident in both UNC7467-treated cells and PPIP5K KO cells (Figure 3H). Interestingly, in both cases, an XPR1 signal was additionally observed at the cell periphery (Figures 3H and S6F). These data indicate that the IP8/SPX domain interaction might restrain XPR1 delivery to the plasma membrane. Nevertheless, [33P]-Pi uptake was compromised when IP8 synthesis was reduced by either the PPIP5K KO or treatment with UNC7467 (Figure 3I). Rescue of Pi uptake in PPIP5K KO cells by PPIP5K1 or PPIP5K1R399A was also antagonized by cell treatment with UNC7467 (Figure 3I). Separately, rescue by XPR1 of [33P]-Pi uptake in XPR1 KO cells was also blocked by UNC7467 (Figure S6C). It is striking that, in each of this series of experiments, the impacts of UNC7467 upon [33P]-Pi efflux quantitatively imitated the drug’s effects upon [33P]-Pi uptake (Figures 3I, S6D, and S6E). We conclude that both XPR1 and IP8 are required for maximal PiT1 protein expression and its functions in both [33P]-Pi uptake and [33P]-Pi efflux.
Evidence for a unifying model to coordinate Pi uptake and Pi efflux
Deletion of the SPX domain has no impact on XPR1/PiT1 binding (Figures 4A and S7A). However, the XPR1 ERD1/XPR1/SYG1 (EXS) domain (Figure 1A) participates in protein/protein interactions.30 We created a series of deletion mutants that were each expressed in HCT116 cells and screened for co-immunoprecipitation of PiT1. Three were identified that failed to pull down PiT1: Δ590–600, Δ620–627, and Δ603–627 (Figures 4A and S7A). An AlphaFold model predicts these residues include a transmembrane helix and a cytosol-facing loop (Figure 4B). The XPR1-LM1/2 mutant also shows weaker co-immunoprecipitation of PiT1, consistent with the importance of XLPVs to the XPR1/PiT1 interaction (Figures 4A and S7A).
Figure 4. Coordinated impact of XPR1-PiT1 interactions that contribute to a unifying model for regulation of cellular Pi uptake and release.

(A) Western blot analysis of PiT1 and mCherry (mCh)-tagged WT XPR1 and the indicated mutants, in both HCT116 cell lysates (“input”) and anti-mCherry immunoprecipitates (“IP”). Statistical analysis of three independent experiments is shown in Figure S7A.
(B) A molecular graphics representation (PyMol) of the AlphaFold2 model of human XPR1 (AF-Q9UBH6-F1-model_v4). Predicted α-helices are depicted as tubes that are colored lime green for the SPX domain and purple for the proposed transmembrane domain (TM), except for residues 590–619, which are highlighted in violet. C-terminal to that helix are residues 620–627 (depicted as spheres) that are predicted to form a loop (carbon is violet, oxygen is red, nitrogen is blue, and sulfur is yellow). The remainder of the C terminus (cyan) is truncated at residue 650, as depicted by the broken line, due to low confidence in structural predictions for IDRs.
(C) Confocal microscopy analysis of LAMP1, XPR1-mCherry (mCh), and PiT1-HA distribution in U2OS cells; the scale bar depicts 10 μm. The outlined (red and blue boxes) areas are magnified. Zones of PiT1 colocalization with XLPV are highlighted with white arrows. These areas are analyzed by recording fluorescence intensity of each signal across the lines drawn in each high-magnification image (orange, gray, blue, or purple). Line profile analysis on the right panel illustrates where fluorescence intensity peaks coincide. From the analysis of 20 images by ComDet Plugin of ImageJ, we have calculated that 5.46% ± 0.84% of the total PiT1 fluorescence colocalized with XLPVs. The yellow scale bar depicts 10 μm, and the white scale bars depict 2 μm.
(D) U2OS cells (WT [blue], PPIP5K KO [red], XPR1 KO [pink], or PiT1 [green]) were treated overnight with either 50 nM bafilomycin or DMSO vehicle, and then, western blot analysis of PiT1 and XPR1 was performed.
(E) Assays of [33P]-Pi uptake were performed on the cell strains described in (D), except that fresh bafilomycin was added for the Pi uptake protocol. Data are means ± standard errors from three separate experiments. ***p < 0.001, **p < 0.01, ns = not significant; one-way ANOVA, with Tukey’s multiple-comparison test.
(F) Densitometric analysis of western blots analyzing expression of PiT1 protein, normalized to actin controls, in WT, PPIP5K KO, and XPR1 KO U2OS cells, treated with either cycloheximide (CHX) or vehicle control for the indicated times. Data are means ± standard errors from three separate experiments.
***p < 0.001, two-way ANOVA, with Tukey’s multiple-comparison test.
Analysis by confocal microscopy revealed that a small portion of PiT1 colocalized with XLPVs both near the cell periphery and in the perinuclear zone (Figure 4C), which is where lysosomal function in degradation activities is prevalent.31 Indeed, others have reported that cellular PiT1 levels in HEK293 cells increase upon inhibition of lysosomal proteolysis by bafilomycin.3 We obtained similar data and further showed that bafilomycin stimulated Pi uptake and attenuated the degree to which PiT1 levels are reduced in both PPIP5K KO cells and XPR1 KO cells (Figures 4D, 4E, S8A). We also observed lesser but still statistically significant inhibition by bafilomycin of Pi efflux (Figures S8B, S8C, S8D, and S8E).
These data led us to posit that the XPR1/IP8 signaling axis might tonically stabilize PiT1 protein levels. We pursued this idea by using cycloheximide to inhibit protein synthesis in cultured cells. The levels of PiT1 protein declined about 15% within an hour and then stabilized (Figures 4F and S7B). In contrast, in XPR1- and PPIP5K-KO cells, PiT1 protein was more unstable and its levels declined 50% within 1 h (Figures 4F and S7B). In control experiments, we did not observe consistent reduction of PiT1 mRNA levels in the PPIP5K KO and XPR1 KO cells (Figure S7C). These data confirm a gatekeeper role for the XPR1/IP8 signaling axis that has overarching and rapid control over PiT1 protein expression and its function.
Pharmacological inhibition of IP6K represents a tool for more rapid intervention in PP-IP synthesis than is possible with genetic intervention. We therefore investigated if UNC7467 would provide temporal resolution of the separate impacts of IP8 upon Pi uptake and efflux. For these experiments, cells were preincubated for 20 h in culture medium containing 1 mM [33P]-Pi so as to radiolabel to steady state. (Figure S9A). The rates of [33P]-Pi efflux and uptake were very similar, consistent with near-equilibrium of the two activities (Figures 5A and S9B). The first dataset that we obtained indicated that the inhibition of Pi efflux by UNC7467 was at a maximal level within 2 h of drug treatment; inhibition of Pi uptake by UNC7467 appears to develop more slowly (Figure S9B). This conclusion was reinforced by a second set of experiments performed within a shorter time frame: UNC7467 inhibited Pi efflux by 60% within 0.5 h, whereas Pi uptake only began to be inhibited (by 15%) after 1 h (Figure 5A). Consistently, PiT1 protein levels began to decrease only after 1 h, suggesting that the inhibition of Pi uptake by UNC7467 is via regulating PiT1 protein stability (Figures 5B and 5C). In control experiments, we found that UNC7467 dramatically reduced IP8 and 5-IP7 levels within 0.5 or 1 h of treatment (Figures S9C and S9D), and it does not affect PiT1 binding to XPR1 (Figures S9E and S9F).
Figure 5. Temporal inhibition of inositol pyrophosphates regulates Pi efflux and influx separately.

(A) Steady-state rates of [33P]-Pi uptake and [33P]-Pi efflux in WT U2OS cells treated for either 30 min or 1 h with 10 μM UNC7467. All Pi transport and densitometric assays were performed three times, and data shown are means and standard errors from three replicates. Ns = not significant; *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA, with Tukey’s multiple-comparison test.
(B) Western blots analyzing PiT1 and PiT2 protein levels in U2OS cells treated with 10 μM UNC7467 for the indicated times.
(C) Densitometric analysis of PiT1 and PiT2 protein levels in (B), normalized to actin controls; data are means ± standard errors from three separate experiments. *p < 0.05; **p < 0.01, one-way ANOVA, with Tukey’s multiple-comparison test.
These data indicate Pi efflux is more rapidly susceptible to a fall in IP8 levels than is Pi uptake. This temporal resolution of regulatory processes that separately modulate Pi uptake and efflux comprises the basis for a unifying model of mammalian cellular [Pi] homeostasis (see Figure 6 and the discussion section).
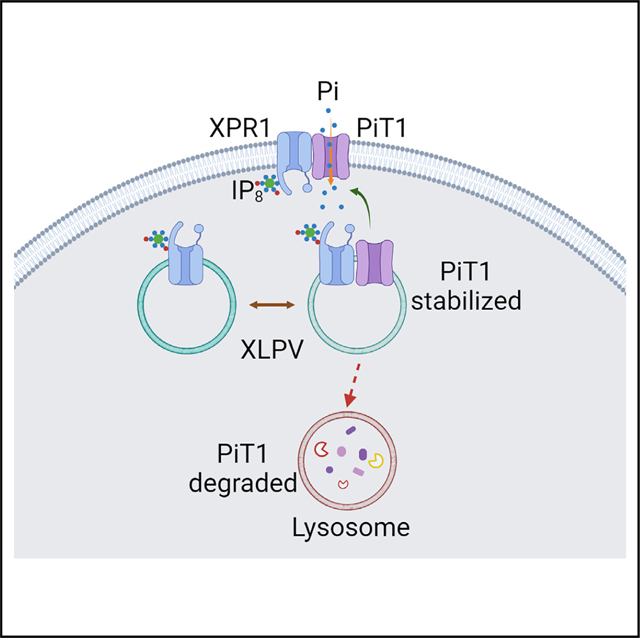
Figure 6. A model to account for the regulation by XPR1 of PiT1-mediated Pi uptake.

The model depicts two pools of XPLVs, one of which also contains PiT1. Since XLPVs are part of the lysosome/endosome continuum, some recycling of PiT1 between early endosomes and plasma membranes is expected.3 In our model, we additionally hypothesize that XPR1 is recycled between XLPVs and the plasma membrane, although the latter pool of XPR1 was below the limits of detection by confocal microscopy. We propose that the XLPV pool of PiT1 is stabilized through its interaction with IP8-bound XPR1. A decrease in the intracellular level of IP8 (as occurs during Pi scarcity10 or following cell treatment with UNC7467) initially compromises XPR1 functionality in Pi efflux (this is not represented in this figure). Subsequently, the decrease in IP8 commits PiT1 to a degradative pathway, reducing the availability of PiT1 that can be trafficked to the plasma membrane, lowering the rate of Pi influx and rebalancing Pi homeostasis.
DISCUSSION
The paradigm that emerges from the current study provides molecular- and cellular-level insights into the coordination of cellular Pi uptake and efflux at an interface of cellular signaling with metabolic homeostasis. As such, our model also extends the signaling context for our earlier demonstration that levels of IP8 comprise a sensor of fluctuations in cytosolic [Pi]13; this discovery, originally made with mammalian cells,13 has subsequently been recapitulated in experiments with fungi and plants.12,32 We now show how PPIP5K-mediated synthesis of IP8, which in turn acts through its cognate receptor XPR1 (Figure 2A), regulates the cellular levels of expression of a major Pi uptake protein, PiT1 (Figures 4D, 4E, 4F, 5B, and 5C). We describe a physical interaction between endogenous XPR1 and PiT1 (Figures 1D and 6) that is dependent upon a predicted helix within the EXS domain that includes a loop that faces into the cytosol (Figures 4A and 4B) and hitherto unrecognized amino acid motifs within the EXS and intrinsically disordered region domains (Figure 1A) that are required for the assembly of XPR1 into intracellular, LAMP1-positive puncta (i.e., XPLVs; Figures 1E, 1F, 1G, and 1H). This intracellular pool of XPR1 is necessary for PiT1-dependent Pi uptake (Figure 1B), by preventing PiT1 from entering a degradative pathway (Figure 6). Our discovery that rapid protein turnover is a regulatory mechanism for controlling Pi uptake may have wider pathological implications: anomalies in protein degradation have been linked to developmental disorders, cancer, and neurodegenerative disease; aging is also associated with a loss of proteostasis.33
These conclusions introduce PiT1 as a downstream effector of Pi homeostasis, a role that recently escaped PiT2.2 It is of further interest that XPR1 does not regulate either the expression of the PiT2 (Figure 3E) or its presence at the plasma membrane.2 Thus, it may be of additional biological significance that PiT2 does not share the “moonlighting” activities of intracellular pools of PiT1 that are independent of its ability to transport Pi, including regulation of cell growth and differentiation, apoptosis, ER stress, immune responses, and glucose metabolism.34–37
LAMP1-positive lysosomes are known to have secretory functions and/or act as signaling hubs that regulate nutrient and bioenergetic homeostasis; these particular vesicles tend to be directed toward the plasma membrane.31,38,39 The latter location has previously raised the possibility of recycling of PiT1 to and from the plasma membrane.3 XPR1 may undergo similar trafficking, but we were unable to pursue this idea by confocal microscopy, which lacked sufficient sensitivity to detect the plasma membrane pool of XPR1,2 although we did note accumulation of XPR1 at this location, in cells in which PiT1 levels were depleted upon our targeting IP8 synthesis (Figures 3G and 3H).
Our description of temporal separation of cellular control over Pi uptake and Pi release (Figures 5A–5C) represents a major contribution to the long-standing goal of understanding how these processes are coordinated. We have demonstrated that within minutes of the inhibition of IP8 synthesis by UNC746729 (i.e., a pharmacological mimic of the impact of cytosolic Pi deprivation13), which in turn divests XPR1 from its functionalizing ligand,10 there is a rapid reduction in the rate of cellular Pi efflux, without detectable changes in Pi uptake (Figure 5A), so as to quickly preserve cytosolic [Pi]. Shortly thereafter, the separate, XPR1-mediated regulation of Pi uptake comes into effect, through control over PiT1 protein levels (Figures 3I, 5A–5C, S9B, and 6). The closely controlled and sequential nature of these events indicates the importance to the cell of avoiding imbalance in the rates of Pi uptake and efflux over extended periods of time.
Limitations of the study
A major challenge to developing a molecular-level understanding of the regulation of Pi efflux from mammalian cells
Although it is now widely appreciated that mammalian XPR1 mediates PP-IP-dependent regulation of Pi efflux from mammalian cells, it remains unclear if XPR1 is a genuine Pi-transport protein or a regulator of another Pi transporter.18 This conundrum has limited our ability to further our molecular-level understanding of the regulation of Pi efflux from mammalian cells. Future application of methodologies to generate adequate levels of XPR1 in a mammalian expression system, then purify the protein to homogeneity, and incorporate it into phospholipid vesicles for Pi transport assays19 could serve as an informative experimental platform to screen for potential ancillary factors that might be required to recapitulate the actions of fully functional XPR1 in vitro.
Technical limitations to the study of Pi fluxes in intact cells
A technical challenge for our study has been to quantify the impact upon cellular IP8 synthesis following cell treatment with UNC7467, an IP6K inhibitor. IP8 is present in cells at low concentrations that challenge the sensitivity limits of current mass spectrometry techniques. Moreover, during the review of this manuscript, S.B.S. retired and our laboratory was closed. Consequently, we only had the opportunity to provide two biological replicates for the data described in Figures S9C and S9D.
Most of the experiments we performed in this study monitored cellular accumulation of [33P]-Pi that was added to Pi-free culture media. This strategy was taken for ease of comparison with earlier studies performed by Battini and colleagues.9,15 This methodology usefully exploits the phenomenon that Pi uptake follows Michaelis-Menten kinetics,21 so by analogy to enzymatic reactions, our use of limiting concentrations of Pi reduces the initial uptake rate (v) to Vmax[S]/Km. Thus, we performed qualitative experimental studies of Pi transport, irrespective of whether it is Km or Vmax that is modified. Nevertheless, a more physiologically relevant and informative analysis of cellular Pi fluxes can be obtained by incubating cells with the biologically relevant extracellular concentration of Pi, i.e., 1 mM. Indeed, we deployed this condition for some experiments (Figures 5A and S9B), which gave valuable, quantitative insight into steady-state values for Pi influx and efflux and their regulation by IP8.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chunfang Gu (guc2@nih.gov).
Materials availability
All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and code availability
Structure coordinates of XPR1 SPX domain have been deposited in the Protein DataBank, PDB: 8TYV, and PDB: 8TYU. This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
HCT116 cells (a colon cancer cell line) were cultured in DMEM/F12. WT, XPR1 KO, PiT1 KO U2OS cells were cultured in DMEM as well as WT, PPIP5K KO, XPR1 KO Saos2 cells. XPR1 KO U2OS or Saos2 cells stably expressing XPR1 or XPR1 mutants were cultured in DMEM with 100 μg/ml geneticin. All culture medium contains 10% fetal bovine serum (FBS) (Gemini Bio-product) and 100 U/mL Penicillin-Streptomycin (ThermoFisher Scientific) and cells were culture at 37°C with 5% CO2.
METHOD DETAILS
Cells and cell culture
The origin of our HCT116 cells and the culture conditions are as previously described.28 XPR1 in HCT116 cells was genomically-tagged with Halo right before the stop codon following a protocol described previously.23 Briefly, pX335-U6-Chimeric_BB-CBh-hSpCas9n(D10A) carrying sgRNA 5′-TTTTCCGAGCTCTTCCGGAT-3′ or 5′-TTCGGCTGGAAAGGTACTAG-3′ were transfected in HCT116 cells together with a plasmid DNA containing Halo-tag sequence which is flanked by ~800bp homology arms of insertion site. Knocked in clones were screened by PCR amplification of targeted region using a pair of primers (5′-AGAGATGGAAACGGG CACTC-3′, 5′-GAAATTCAGCCGGTGGTGTG-3′) followed by sequencing confirmation. To validate the PiT2 antibody, 3 × 105 HCT116 cells were seeded on a 6 well plate. Cells were transfected with ON-TARGETplus Human SLC20A2 siRNA or non-targeting Control siRNAs (Horizon) for 40h. Cells were then lysed in RIPA buffer including Halt protease and phosphatase inhibitor (ThermoFisher Scientific) and PiT2 levels were detected by western blot using anti-SLC20A2 antibodies (ProteinTech, 12820–1-AP).
U2OS and Saos-2 cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) (Gemini Bio-product) and 100 U/mL Penicillin-Streptomycin (ThermoFisher Scientific) at 37°C with 5% CO2. Knockout of PPIP5Ks and XPR1 in U2OS cells and Saos-2 cells was performed using the methods described previously.10,28 PiT1 KO cells were generated using a guide messenger RNA that targets exon 2 of PiT1 (5′-GTCCTTTGGTGGACTACCTA-3′).
The pReceiver-M56(mCherry) and M07(HA) vectors carrying the ORF sequences of WT XPR1 and PiT1, respectively, were purchased from GeneCopoeia. All mutants of XPR1 plasmids were generated based on WT XPR1 plasmid. To generate stable cell lines, pReceiver vectors carrying the ORF sequences of specific genes were used to transfect recipient cells (U2OS or Saos-2), after which the cells were treated with geneticin to select against non-transfected cells. Specifically, 1×105 cells/well were seeded in 1mL of complete growth medium in 12-well tissue culture plates. After 48 h, the cells reached 60–80% confluence and were then transfected and 100 μg/ml geneticin was added. Replacement media with 100 μg/ml geneticin was added every 2–3 days for up to two weeks. The geneticin selected cells were single cell plated in 96 well plate for clone expansion. The expression of specific genes in single clones were verified by western blot.
Screening of endogenous XPR1 binding proteins
HCT116 cells and HCT116 cells with Halo-tagged XPR1 were cultured in duplicate in 150mm dishes and harvested around 80% confluency. Cells were lysed in buffer containing 50mM Tris pH7.5, 250mM NaCl, 1mM EDTA, 0.5% IGEPAL CA-630 (Sigma), 1× protease inhibitor cocktail (thermoFisher Scientific), 1× PhosSTOP (Sigma)). The lysate was sonicated for 3min (37 HZ, 30% power) and centrifuged at 13500g for 10 min at 4°C. The supernatant was diluted with one volume dilution buffer (50mM Tris pH7.5, 150mM NaCl, 1mM EDTA, 1× protease inhibitor cocktail, 1X PhosSTOP) and incubated with ChromoTek Halo-Trap Agarose beads (Proteintech) overnight at 4°C. The beads were washed four times with dilution buffer and eluted with 2× sample buffer (Biorad) containing 5% β-me (Sigma).
The eluted proteins were separated by SDS-PAGE; SDS-PAGE gel lanes were manually cut into 12 fractions each. These gel regions were then minced, and protein digests performed with a ProGest robotic digester (Genomic Solutions) where the gel pieces were destained by incubation in 25 mM ammonium bicarbonate/50% v/v acetonitrile (2 × 15 min). The gel pieces were dehydrated in acetonitrile, followed by drying under a nitrogen stream, and then incubated with 250 ng trypsin (Promega) for 8 h at 37°C. The digests were collected, and peptides were extracted from the gel three times with the extractions pooled for each sample. Samples were removed from the robot, lyophilized, and individually resuspended in 0.1% formic acid.
Samples were analyzed by LC/MS on a Q Exactive Plus mass spectrometer (ThermoFisher Scientific) interfaced with a nanoAcquity UPLC system (Waters Corporation) equipped with a 75 mm × 200 mm HSS T3 C18 column (1.8 mm particle, Waters Corporation) and a Symmetry C18 trapping column (180 mm × 20 mm) with 5 mm particle size; the flow rate was 450 nL/min. The trapping column was positioned in-line of the analytical column and upstream of a micro-tee union which was used both as a vent for trapping and as a liquid junction. Trapping was performed using the initial solvent composition. 5 mL of digested sample was injected onto the column. Peptides were eluted by using a linear gradient from 99% solvent A (0.1% formic acid in water (v/v)) and 1% solvent B (0.1% formic acid in acetonitrile (v/v)) to 40% solvent B over 100 min. For the mass spectrometry, a data-dependent acquisition method was employed with an exclusion time of 15 s and an exclusion of +1 charge states. The mass spectrometer was equipped with a NanoFlex source and was used in the positive ion mode. Instrument parameters were as follows: sheath gas, 0; auxiliary gas, 0; sweep gas, 0; spray voltage, 2.7 kV; capillary temperature, 275°C; S-lens, 60; scan range (m/z) of 200–2000; 2 m/z isolation window; resolution: 70,000; automated gain control (AGC), 2 × 10e5 ions; and a maximum IT of 200 ms. Mass calibration was performed before data acquisition using the Pierce LTQ Velos Positive Ion Calibration mixture (ThermoFisher Scientific). Peak lists were generated from the LC/MS data using Mascot Distiller (Matrix Science) and the resulting peak lists were searched using the Spectrum Mill software package (Agilent) against the entire Swissprot database. Searches were performed using trypsin specificity and allowed for one missed cleavage and variable methionine oxidation. Mass tolerances were 20 ppm for MS scans and 50 ppm for MSMS scans.
Subcellular fractionation
The Minute Plasma Membrane Protein Isolation Kit (Invent Biotechnologies, SM-005) was used to isolate both plasma membranes and intracellular membranes from approximately 3 × 107 cells cultured in 150mm dishes. RIPA buffer (ThermoFisher Scientific) supplemented with Halo protease and phosphatase inhibitor cocktail (ThermoFisher Scientific) was used to resuspend the membrane fraction, the plasma membrane fraction and the intracellular organelle membrane fraction (200μl, 150ul and 50ul respectively). Samples were treated with SDS-PAGE and Western analysis was performed using antibodies for XPR1 (proteintech, 14174–1-AP), PiT1 (Proteintech, 12423–1-AP) and ATP1A1 (Proteintech, 14418–1-AP).
Intracellular tagging with TMEM192-[HA]3 in HCT116 cells was performed as previously described.40 Next, approximately 10 million tagged or non-tagged cells were lysed in 1 mL of 136 mM KCl, 10 mM KH2PO4 (pH 7.25), and gently homogenized with 20 strokes of a 2 mL homogenizer. The homogenate was then centrifuged at 1000 x g for 2 min at 4°C and the supernatant was incubated with 100 μL anti-HA magnetic beads (ThermoFisher Scientific) on a gentle rotator shaker for 15 min. The beads were gently washed three times with KPBS on a DynaMag Spin Magnet, and the immunoprecipitates were eluted in 2× sample buffer (Biorad) containing 5% β-mecaptoethanol (Biorad).
Cellular phosphate flux assays
Phosphate transport was measured as previously described by multiple laboratories.10,15,22 Cells were seeded (U2OS, 3 × 105 cells; Saos-2, 6 × 105 cells) per well in 1.5 mL medium in 12-well plates; Pi transport assays were performed 48 h later at 37°C. For uptake assays, cells were incubated in Pi-free DMEM [Gibco catalog number: 11971–025] plus 10% FBS and approx. 0.5 μCi/mL [33P]-Pi (ARC; ARP 0153) for 30 min, unless otherwise stated. Assays were terminated by washing cells three times with 1 mL Pi-free DMEM, followed immediately by lysis in 1 mL PBS plus 1% Triton X-100, and the accumulated [33P]-Pi was determined with a liquid scintillation counter. DPM data were normalized to cell protein (determined by a BCA protein assay; Pierce) and statistical significance was determined by using either one- or two-way ANOVA as indicated, with Tukey’s multiple-comparison test. Data were then normalized to control values for graphical presentation.
Phosphate efflux assays were performed for 2 h (unless otherwise stated) exactly as described previously10 after 30 min of [33P]-Pi uptake as described above. Efflux is depicted as a percentage of the [33P]-Pi that was accumulated by each cell type prior to the efflux assay; statistical significance was determined by using two-way ANOVA, with a post-hoc Tukey test. All [33P]-Pi flux data are given as means ± standard errors from either 3 or 4 replicates.
For the steady-state Pi flux assays, cells cultured in normal medium containing 1mM Pi were labeled with approximately 7.5uCi/ml [33P]-Pi for 24 h. Parallel wells were cultured without [33P]-Pi labeling. Cells were then incubated 10 μM UNC7467 for the indicated time. Then the incubation media from labeled and unlabeled cells were swapped after cells washed with DMEM to measure Pi influx (unlabeled cells) and efflux (labeled cells) respectively. Both influx and efflux assays were terminated after 30 min. Protein concentration of the cell lysate was determined by BCA (Thermo-Fisher Scientific).
Confocal microscope imaging
The XPR1 coding sequence (NM_004736.4) was tagged with YFP (pcDNA6.2-C-YFP vector) or mCherry (pReceiver-M56 vector) at the C terminus. XPR1 mutants were constructed using Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to the vendor’s manual. WT or XPR1 mutants were overexpressed in U2OS cells for 24 h. Cells were fixed and stained with anti-LAMP1(dshb # 1D4B), anti-calreticulin (Abcam # ab22683), anti-GM130 (Cell Signaling Technology #12480), anti-ERp46 (Protein-tech, #19834–1-AP) or anti-TOMM20 (Cell Signaling Technology #42406) followed by an Alex Fluor conjugated secondary antibody (ThermoFisher Scientific). In certain experiments, cells were treated with 10μM UNC7467 for 6h before fixation and staining. PiT1 with HA tagged at the C terminus was overexpressed in U2OS cells and stained with anti-PiT1 antibody (Cell Signaling Technology #12765) following by staining with LAMP1 (dshb # 1D4B). To stain endogenous PiT1, we used anti-PiT1 antibody from Cell Signaling Technology (#12765). All images were obtained using Zeiss LSM 780 UV confocal with 633/1.4 PlanApo oil objective, and images were analyzed by ImageJ. Representative images from 1 of 3 replicates are provide throughout. Colocalization of WT XPR1 or XPR1 mutants with LAMP1 was quantified using JaCob plugin with threshold settings consist with all conditions; plasma membrane localization of XPR1 were analyzed by line profile of ImageJ; Colocalization of PiT1 with XLPV was analyzed by ComDet v.0.5.5 plugin (https://github.com/ekatrukha/ComDet; settings: particle size = 5 pixels, co-localization distance = 4 pixels) which detects particles and analyzes the co-localization based on the distance between the center of two spots in different channels. Threshold (in SD) for each channel was chosen around 100 to ensure appropriate particle selection.
Structure analysis of the SPX domain
The codon-optimized cDNA encoding residues 1–207 of human XPR1 was purchased from Genescript Inc and subcloned into the pDest-566 vector. Residues 94–129 were replaced with a GSGSG linker by Q5 site-directed mutagenesis kit (Biolabs). The resultant plasmid was transformed into E. coli BL21(DE3) and cultured into nutrient-rich 2xYT medium (7.5; 37°C) to an optical density of 0.7 at 600 nm. Isopropyl β-d-thiogalactopyranoside (0.1 mM) was added and cultures were continued at 15°C for 24 h. The cells were harvested and disrupted using a Constant Cell Disruption System (Constant System Ltd) at 20 KPsi. Recombinant protein was purified at 4°C with Ni-NTA agarose (Qiagen) and HiTrap Heparin HP column (Cytiva). After removing the maltose-binding protein tag with tobacco etch virus protease, the protein was further purified with another HiTrap Heparin HP column. Finally, the protein was applied to a Superdex 200 size exclusion column (Cytiva) and eluted with 150 mM NaCl, 20 mM Tris-HCl pH 7.2, concentrated to 15 mg/mL and stored at −80°C; purity (>95%) was validated by SDS-PAGE.
Crystallization was accomplished by the hanging drop vapor diffusion method using 1.5 μL of protein solution plus 1.5 μL of the crystallization medium (6% PEG 3350, 30% Ethelyn Glycol, 0.1 M sodium Citrate, 0.1 M MgCl2, pH 6.0). Subsequently, 2 mM IP8 was soaked in the crystallization buffer for 3 days. Diffraction data were collected using APS beamline 22-BM and processed with the HKL2000 algorithm41). The initial structure was determined by molecular replacement using apo human SPX domain (PDB: 5IJH) as the template. The structure was manually rebuilt with COOT42 and refined with Phoenix.9 The molecular graphics representations were prepared with the program PyMol (Schrö dinger, LLC). Atomic coordinates and structure factors have been deposited with the Protein DataBank with accession codes 8TYU and 8TYV (Table S2).
Quantitative PCR
Total RNA in U2OS or Saos2 cells were extracted by RNeasy mini kit (Qiagen) and reverse transcripted into cDNA using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). PiT1 mRNA levels were then determined by real-time quantitative PCR using iQ SYBR Green Supermix (Bio-Rad, 1708880) in a QuantStudio 5 Flex PCR System (Life Technologies) with the following paired primers: Forward = 5′TCCTTTGGTGGACTACCTATGG3’; Reverse = 5′CGGATGGTTTCGCTCACTTTG3’.
Quantification of inositol pyrophosphates
U2OS cells cultured in 150mm dishes (80–90% confluency) were treated with 10 μM UNC7467 for 0.5 or 1hr and lysed immediately with 2 mL of 1M perchloric acid. The supernatant was collected and incubated with 5mg TiO2 beads for 30 min at 4°C to capture inositol pyrophosphates.43 The beads were washed twice with 1 mL water and then inositol phosphates were eluted with 10% ammonium hydroxide by incubation at 4°C for 20 min. The elute were dried in a vacuum centrifuge and the pallets were resuspended using 10 μL of water. Capillary electrophoresis (CE) coupled with mass spectrometry (MS) through electrospray ionization (ESI) analysis44 was performed on An Agilent CE-QQQ system, which included an Agilent 7100 CE unit, an Agilent 6495C Triple Quadrupole, and an Agilent Jet Stream electrospray ionization source, all connected via an Agilent CE-ESI-MS interface. An Agilent 1200 LC pump fed the sheath liquid (a 50:50 combination of isopropanol and water) at a steady flow rate of 10 μL/min through a splitter. Separation occurred within a 100 cm fused silica capillary having an internal diameter of 50 μm and an external diameter of 365 μm.
The background electrolyte (BGE) consisted of 35 mM ammonium acetate that had been adjusted to pH 9.75 using ammonium hydroxide. Before each sample run, the capillary was flushed with BGE for 400 s. Samples were injected for 15 s at a pressure of 100 mbar (30 nL). The MS source parameters were adjusted to 150°C, 11 L/min flow rate, 8 psi nebulizer pressure, 175°C sheath gas temperature, −2000V capillary voltage, and 2000V nozzle voltage. Additionally, negative high-pressure radio frequency (RF) and negative low-pressure RF were kept at 70 and 40 V, respectively. Table S3 details the parameters for multiple reaction monitoring (MRM).
To prepare the internal standard (IS) stock solution, specific concentrations were used: 40 μM [13C6] IP6, 2 μM [13C6] 5-IP7, and 2 μM [13C6] 1,5-IP8. These IS compounds were added to the samples to assist with isomer assignment and quantification of IPs and PP-IPs. Each sample received 5 μL of the IS stock solution, which was thoroughly mixed with 5 μL of the sample. Quantification of IP 8, 5-IP7, and IP6 was done by injecting known amounts of the relevant heavy isotopic references into the samples. After spiking, the final concentrations in the samples were as follows 20 μM[ 13C6] IP6,1 μM [13C6] 5- IP7, and 1 μM [13C6] 1,5- IP8. Dorothea Fiedler contributed all [13C6] inositols.45
Other assays and reagents
Cycloheximide was obtained from Cell Signaling Technology (#2112). Bafilomycin A1 was supplied by MilliporeSigma (5084090001). For Western blot analyses, the sources of the antibodies are as follows: Halo, Promega, G9211; PiT1, Proteintech, 12423–1-AP; PiT2, Proteintech, 12820–1-AP (the specificity of this antibody has previously been verified by siRNA46 and we reverified specificity using identical procedures); β-Actin (C4), Santa Cruz Biotechnology, sc-47778; and XPR1, Proteintech, 14174–1-AP; mCherry, ThermoFisher Scientific, PA5–34974; LAMP1, Cell Signaling Technology, #9091; GM130, Cell Signaling Technology, #12480; calreticulin, Cell Signaling Technology, #12238; TOMM20, Cell Signaling Technology, 42406. Representative blots from 1 of 3 replicates are provide throughout.
QUANTIFICATION AND STATISTICAL ANALYSIS
For the quantification of the colocalization of XPR1 with LAMP1, the acquired images from at least three independent experiments were analyzed by JaCob plugin of ImageJ. In order to analyze mCherry-tagged XPR1 localization at the plasma membrane, we recorded its relative fluorescence intensity across a 6 μm line drawn perpendicular to the cell perimeter were drawn across cell edges followed by Plot Profile by ImageJ.47 Quantification of XPR1 colocalization with PiT1 on XLPVs were analyzed using the ComDet plugin48 for ImageJ to calculate the percentage of PiT1 spots colocalized with XPR1 and LAMP1 double-positive spots (XLPVs). Western blots were quantified from at least three replicates. Band intensities were quantified using ImageJ. The ratio of the intensity of each band relative to total bands intensities of a single gel were calculated and then normalized to loading controls.49 For statistical analysis, paired two-tailed Student’s t-test was performed to compare two groups, while analysis of variance (ANOVA) test with Tukey’s multiple-comparison test was used for three or more groups. The specific statistical method used for each dataset is described in the corresponding figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| XPR1 | Proteintech | 14174–1-AP; RRID:AB_10642569 |
| PiT1 | Proteintech | 12423–1-AP; RRID:AB_2286212 |
| PiT1 | Cell Signaling Technology | 12765; RRID:AB_2798019 |
| PiT2 | Proteintech | 12820–1-AP; RRID:AB_2191004 |
| ATP1A1 | Proteintech | 14418–1-AP; RRID:AB_2227873 |
| Pierce™ Anti-HA Magnetic Beads | ThermoFisher Scientific | 88836; RRID:AB_2749815 |
| LAMP1 | DSHB | 1D4B; RRID:AB_2134500 |
| LAMP1 | Cell Signaling Technology | 9091; RRID:AB_2687579 |
| Calreticulin | Abcam | ab22683; RRID:AB_447253 |
| calreticulin | Cell Signaling Technology | 12238; RRID:AB_2688013 |
| GM130 | Cell Signaling Technology | 12480; RRID:AB_2797933 |
| ERP46 | Proteintech | 19834–1-AP; RRID:AB_10644285 |
| TOMM20 | Cell Signaling Technology | 42406; RRID:AB_2687663 |
| Halo | Promega | G9211 |
| mCherry | ThermoFisher Scientific | PA5–34974; RRID:AB_2552323 |
| β-Actin | Santa Cruz Biotechnology | sc-47778; RRID:AB_626632 |
| ChromoTek Halo-Trap Agarose beads | Proteintech | Ota; RRID:AB_2827595 |
| ChromoTek RFP-Trap Agarose | Proteintech | Rta; RRID:AB_2631362 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | C2987H |
| BL21 (DE3) Competent Cells | Agilent | 200131 |
|
| ||
| Chemicals and recombinant proteins | ||
|
| ||
| DMEM, high glucose | ThermoFisher Scientific | 11965092 |
| DMEM/F12 | ThermoFisher Scientific | 11320033 |
| Pi-free DMEM | ThermoFisher Scientific | 11971025 |
| Fetal bovine serum | Gemini Bio-product | 100–106 |
| Geneticin | ThermoFisher Scientific | 10131035 |
| IGEPAL CA-630 | Sigma | I8896–50ML |
| Halt™ Protease Inhibitor Cocktail | ThermoFisher Scientific | 78429 |
| PhosSTOP | Sigma | 4906837001 |
| 2x Laemmli Sample Buffer | Bio-Rad | 1610737EDU |
| 2-Mercaptoethanol | Sigma | M3148–100ML |
| RIPA buffer | ThermoFisher Scientific | 89901 |
| Dipotassium hydrogen phosphate [33P] | ARC | ARP 0153 |
| iQ SYBR Green Supermix | Bio-Rad | 1708880 |
| SuperScript™ III Reverse Transcriptase | ThermoFisher Scientific | 18080044 |
| Cycloheximide | Cell Signaling Technology | 2112 |
| Bafilomycin A1 | MilliporeSigma | 5084090001 |
| UNC7467 | This paper | N/A |
| Isopropyl-beta-D-thiogalactoside | Gold BioTechnology | I2481C25 |
| Ni-NTA Agarose | Qiagen | 30210 |
| IP8 | Jessen Henning lab | N/A |
| XPR SPX domain (1–207) recombinant protein | This paper | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat #23225 |
| The Minute Plasma Membrane Protein Isolation Kit | Invent Biotechnologies | SM-005 |
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs | E0554S |
| RNeasy mini kit | Qiagen | 74104 |
| SuperScript™ III Reverse Transcriptase | ThermoFisher Scientific | 18080044 |
| HiTrapTM Heparin HP column | Cytiva | 17040703 |
|
| ||
| Deposited data | ||
|
| ||
| Crystal structure of the SPX domain of XPR1 in complex with IP8 |
This paper | PDB ID: 8TYV |
| High-resolution crystal structure of the SPX domain of XPR1 at 1.4 Å |
This paper | PDB ID: 8TYU |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| U2OS | ATCC | HTB-96 |
| HCT116 | ATCC | CCL-247 |
| Saos2 | ATCC | HTB-85 |
| XPR1-Halo tagged HCT116 cells | This paper | N/A |
| XPR1 KO U2OS | This paper | N/A |
| PiT1 KO U2OS | This paper | N/A |
| XPR1 KO Saos2 | This paper | N/A |
| PPIP5K KO Saos2 | Li et al.10 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| PiT1 qPCR Forward primer: 5′-TCCTTTGGTGGACTACCTATGG-3′ |
PrimerBank | 327365348c1 |
| PiT1 qPCR Reverse primer: 5′-CGGATGGTTTCGCTCACTTTG-3′ |
PrimerBank | 327365348c1 |
| XPR1-Halo genomic amplification Forward primer: 5′-AGAGATGGAAACGGGCACTC-3′ | This paper | N/A |
| XPR1-Halo genomic amplification Reverse primer: 5′-GAAATTCAGCCGGTGGTGTG-3′ | This paper | N/A |
| XPR1-gRNA targeting sequence sernse: 5′-TTTTCCGAGCTCTTCCGGAT-3′ |
This paper | N/A |
| XPR1-gRNA targeting sequence anti sense: 5′-TTCGGCTGGAAAGGTACTAG-3′ |
This paper | N/A |
| PiT1-gRNA targeting sequence: 5′-GTCCTTTGGTGGACTACCTA-3′ |
This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pReceiver-M56-XPR1 | GeneCopoeia™ | EX-T0271-M56 |
| pReceiver-M56 vector | GeneCopoeia™ | EX-NEG-M56 |
| pReceiver-M07-PiT1 | GeneCopoeia™ | EX-G0139-M07 |
| pcDNA6.2-XPR1-C-YFP | This paper | N/A |
| pReceiver-M56-XPR1LM1 | This paper | N/A |
| pReceiver-M56-XPR1LM2 | This paper | N/A |
| pReceiver-M56-XPR1LM1/2 | This paper | N/A |
| pReceiver-M56-XPR1Y590A/I563A | This paper | N/A |
| pReceiver-M56-XPR1Δ603−627 | This paper | N/A |
| pReceiver-M56-XPR1Δ590−600 | This paper | N/A |
| pReceiver-M56-XPR1ΔSPX | This paper | N/A |
| pReceiver-M56-XPR1Δ620−627 | This paper | N/A |
| pCRISPR-CG02-PiT1 gRNA | GeneCopoeia™ | HCP309933 -CG02–3 |
| pX335-XPR1 sense gRNA | This paper | N/A |
| pX335-XPR1 anti sense gRNA | This paper | N/A |
| pDest-566-XPR11–207 | This paper | N/A |
| pDest-566-XPR11–193–GSGSG−130−207 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Software | https://imagej.nih.gov/ij/ |
| GraphPad Prism 9 | Software | https://www.graphpad.com/scientific-software/prism/ |
| SigmaPlot | Software | https://systatsoftware.com/sigmaplot/ |
| Mascot Distiller | Software | https://www.matrixscience.com/distiller.html |
| Spectrum Mill MS Proteomics Software | Software | https://proteomics.broadinstitute.org/millhome.htm |
| HKL2000 | Software | https://hkl-xray.com/hkl-2000 |
| COOT | Software | https://www.ccp4.ac.uk/download/#os=windows |
| Phoenix | Software | https://phenix-online.org/ |
| Pymol | Software | https://pymol.org/2/ |
Highlights.
Organellar XPR1 regulates cellular Pi fluxes
XPR1 physically interacts with PiT1 to temporally coordinate Pi uptake and Pi efflux
The IP8/XPR1 signaling axis determines PiT1 protein expression and hence Pi uptake
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (NIEHS), the University Cancer Research Fund (UNC), and the Deutsche Forschungsgemeinschaft (DFG) under Germany’s excellence strategy (CIBSS, EXC-2189, Project ID 390939984, to H.J.J.). We acknowledge support from the NIEHS Collaborative Crystallography Group and the Advanced Photon Sciences (APS) SE Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beam lines for assistance with crystallographic data collection. We are grateful to Jason Williams (Mass Spectrometry Research and Support Group at the NIEHS) for the mass spectrometry analysis.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114316.
REFERENCES
- 1.Wang Y-P, and Lei Q-Y (2018). Metabolite sensing and signaling in cell metabolism. Signal Transduct. Target. Ther 3, 30. 10.1038/s41392-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopez-Sanchez U, Tury S, Nicolas G, Wilson MS, Jurici S, Ayrignac X, Courgnaud V, Saiardi A, Sitbon M, and Battini JL (2020). Interplay between PFBC-associated SLC20A2 and XPR1 phosphate transporters requires inositol polyphosphates for control of cellular phosphate homeostasis. J. Biol. Chem 295, 9366–9378. 10.1074/jbc.RA119.011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zechner C, Henne WM, Sathe AA, Xing C, Hernandez G, Sun S, and Cheong MC (2022). Cellular abundance of sodium phosphate cotransporter SLC20A1/PiT1 and phosphate uptake are controlled post-transcriptionally by ESCRT. J. Biol. Chem 298, 101945. 10.1016/j.jbc.2022.101945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hernando N, Gagnon K, and Lederer E. (2021). Phosphate Transport in Epithelial and Nonepithelial Tissue. Physiol. Rev 101, 1–35. 10.1152/physrev.00008.2019. [DOI] [PubMed] [Google Scholar]
- 5.Alam MT, Olin-Sandoval V, Stincone A, Keller MA, Zelezniak A, Luisi BF, and Ralser M. (2017). The self-inhibitory nature of metabolic networks and its alleviation through compartmentalization. Nat. Commun 8, 16018. 10.1038/ncomms16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jennings ML (2023). Role of transporters in regulating mammalian intracellular inorganic phosphate. Front. Pharmacol 14, 1163442. 10.3389/fphar.2023.1163442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michigami T, Kawai M, Yamazaki M, and Ozono K. (2018). Phosphate as a Signaling Molecule and Its Sensing Mechanism. Physiol. Rev 98, 2317–2348. 10.1152/physrev.00022.2017. [DOI] [PubMed] [Google Scholar]
- 8.Voelkl J, Egli-Spichtig D, Alesutan I, and Wagner CA (2021). Inflammation: a putative link between phosphate metabolism and cardiovascular disease. Clin. Sci 135, 201–227. 10.1042/CS20190895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JRM, Sears RL, Ramos EM, Spiteri E, Sobrido MJ, et al. (2015). Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet 47, 579–581. 10.1038/ng.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Gu C, Hostachy S, Sahu S, Wittwer C, Jessen HJ, Fiedler D, Wang H, and Shears SB (2020). Control of XPR1-dependent cellular phosphate efflux by InsP8 is an exemplar for functionally-exclusive inositol pyrophosphate signaling. Proc. Natl. Acad. Sci. USA 117, 3568–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wild R, Gerasimaite R, Jung JY, Truffault V, Pavlovic I, Schmidt A, Saiardi A, Jessen HJ, Poirier Y, Hothorn M, and Mayer A. (2016). Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 352, 986–990. [DOI] [PubMed] [Google Scholar]
- 12.Chabert V, Kim G, Qiu D, Mayer LM, Jessen HJ, and Mayer A. (2023). Inositol pyrophosphate dynamics reveals control of the yeast PHO starvation program through 1,5-IP8 and the SPX domain of Pho81. Elife 12, RP87956. 10.7554/elife.87956.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu C, Nguyen HN, Hofer A, Jessen HJ, Dai X, Wang H, and Shears SB (2017). The Significance of the Bifunctional Kinase/Phosphatase Activities of Diphosphoinositol Pentakisphosphate Kinases (PPIP5Ks) for Coupling Inositol Pyrophosphate Cell Signaling to Cellular Phosphate Homeostasis. J. Biol. Chem 292, 4544–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung JY, Ried MK, Hothorn M, and Poirier Y. (2018). Control of plant phosphate homeostasis by inositol pyrophosphates and the SPX domain. Curr. Opin. Biotechnol 49, 156–162. 10.1016/j.copbio.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Giovannini D, Touhami J, Charnet P, Sitbon M, and Battini JL (2013). Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 3, 1866–1873. [DOI] [PubMed] [Google Scholar]
- 16.Mailer RK, Allende M, Heestermans M, Schweizer M, Deppermann C, Frye M, Pula G, Odeberg J, Gelderblom M, Rose-John S, et al. (2021). Xenotropic and polytropic retrovirus receptor 1 regulates procoagulant platelet polyphosphate. Blood 137, 1392–1405. 10.1182/blood.2019004617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Kuo H-F, and Chiou T-J (2021). Intracellular phosphate sensing and regulation of phosphate transport systems in plants. Plant Physiol. 187, 2043–2055. 10.1093/plphys/kiab343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu C, Li X, Zong G, Wang H, and Shears SB (2024). IP8: A quantitatively minor inositol pyrophosphate signaling molecule that punches above its weight. Adv. Biol. Regul 91, 101002. 10.1016/j.jbior.2023.101002. [DOI] [PubMed] [Google Scholar]
- 19.Che J, Yamaji N, Miyaji T, Mitani-Ueno N, Kato Y, Shen RF, and Ma JF (2020). Node-Localized Transporters of Phosphorus Essential for Seed Development in Rice. Plant Cell Physiol. 61, 1387–1398. [DOI] [PubMed] [Google Scholar]
- 20.Staudt C, Puissant E, and Boonen M. (2016). Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci 18, 47. 10.3390/ijms18010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck L, Leroy C, Salaün C, Margall-Ducos G, Desdouets C, and Friedlander G. (2009). Identification of a novel function of PiT1 critical for cell proliferation and independent of its phosphate transport activity. J. Biol. Chem 284, 31363–31374. 10.1074/jbc.M109.053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson MS, Jessen HJ, and Saiardi A. (2019). The inositol hexakisphosphate kinases IP6K1 and −2 regulate human cellular phosphate homeostasis, including XPR1-mediated phosphate export. J. Biol. Chem 294, 11597–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koch B, Nijmeijer B, Kueblbeck M, Cai Y, Walther N, and Ellenberg J. (2018). Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing. Nat. Protoc 13, 1465–1487. 10.1038/nprot.2018.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braulke T, and Bonifacino JS (2009). Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793, 605–614. 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Schroder B, Wrocklage C, Hasilik A, and Saftig P. (2010). Molecular characterisation of ‘transmembrane protein 192’ (TMEM192), a novel protein of the lysosomal membrane. Biol. Chem 391, 695–704. 10.1515/BC.2010.062. [DOI] [PubMed] [Google Scholar]
- 26.Xu C, Xu J, Tang HW, Ericsson M, Weng JH, DiRusso J, Hu Y, Ma W, Asara JM, and Perrimon N. (2023). A phosphate-sensing organelle regulates phosphate and tissue homeostasis. Nature 617, 798–806. 10.1038/s41586-023-06039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arpat AB, Magliano P, Wege S, Rouached H, Stefanovic A, and Poirier Y. (2012). Functional expression of PHO1 to the Golgi and trans-Golgi network and its role in export of inorganic phosphate. Plant J. 71, 479–491. 10.1111/j.1365-313X.2012.05004.x. [DOI] [PubMed] [Google Scholar]
- 28.Gu C, Nguyen HN, Ganini D, Chen Z, Jessen HJ, Gu Z, Wang H, and Shears SB (2017). KO of 5-InsP7 kinase activity transforms the HCT116 colon cancer cell line into a hypermetabolic, growth-inhibited phenotype. Proc. Natl. Acad. Sci. USA 114, 11968–11973. 10.1073/pnas.1702370114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y, Mukherjee S, Huang D, Chakraborty M, Gu C, Zong G, Stashko MA, Pearce KH, Shears SB, Chakraborty A, et al. (2022). Development of Novel IP6K Inhibitors for the Treatment of Obesity and Obesity-Induced Metabolic Dysfunctions. J. Med. Chem 65, 6869–6887. 10.1021/acs.jmedchem.2c00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bondeson DP, Paolella BR, Asfaw A, Rothberg MV, Skipper TA, Langan C, Mesa G, Gonzalez A, Surface LE, Ito K, et al. (2022). Phosphate dysregulation via the XPR1-KIDINS220 protein complex is a therapeutic vulnerability in ovarian cancer. Nat. Cancer 3, 681–695. 10.1038/s43018-022-00360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cabukusta B, and Neefjes J. (2018). Mechanisms of lysosomal positioning and movement. Traffic 19, 761–769. 10.1111/tra.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Lau K, Puschmann R, Harmel RK, Zhang Y, Pries V, Gaugler P, Broger L, Dutta AK, Jessen HJ, et al. (2019). Two bifunctional inositol pyrophosphate kinases/phosphatases control plant phosphate homeostasis. Elife 8, e43582. 10.7554/eLife.43582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Correa Marrero M, and Barrio-Hernandez I. (2021). Toward Understanding the Biochemical Determinants of Protein Degradation Rates. ACS Omega 6, 5091–5100. 10.1021/acsomega.0c05318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beck-Cormier S, and Beck L. (2020). The Need of a Paradigm Shift to Better Understand PiT1 and PiT2 Biology: Response to “Why Is There No PiT1/SLC20A1 Pathogenic Variants Yet Linked to Primary Familial Brain Calcification? J. Bone Miner. Res 35, 825–826. 10.1002/jbmr.3969. [DOI] [PubMed] [Google Scholar]
- 35.Couasnay G, Bon N, Devignes CS, Sourice S, Bianchi A, Véziers J, Weiss P, Elefteriou F, Provot S, Guicheux J, et al. (2019). PiT1/Slc20a1 Is Required for Endoplasmic Reticulum Homeostasis, Chondrocyte Survival, and Skeletal Development. J. Bone Miner. Res 34, 387–398. 10.1002/jbmr.3609. [DOI] [PubMed] [Google Scholar]
- 36.Forand A, Koumakis E, Rousseau A, Sassier Y, Journe C, Merlin J-F, Leroy C, Boitez V, Codogno P, Friedlander G, and Cohen I. (2016). Disruption of the Phosphate Transporter Pit1 in Hepatocytes Improves Glucose Metabolism and Insulin Signaling by Modulating the USP7/IRS1 Interaction. Cell Rep. 16, 2736–2748. 10.1016/j.celrep.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Koumakis E, Millet-Botti J, Benna JE, Leroy C, Boitez V, Codogno P, Friedlander G, and Forand A. (2019). Novel function of PiT1/SLC20A1 in LPS-related inflammation and wound healing. Sci. Rep 9, 1808. 10.1038/s41598-018-37551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inpanathan S, and Botelho RJ (2019). The Lysosome Signaling Platform: Adapting With the Times. Front. Cell Dev. Biol 7, 113. 10.3389/fcell.2019.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barral DC, Staiano L, Guimas Almeida C, Cutler DF, Eden ER, Futter CE, Galione A, Marques ARA, Medina DL, Napolitano G, et al. (2022). Current methods to analyze lysosome morphology, positioning, motility and function. Traffic 23, 238–269. 10.1111/tra.12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z, and Minor W. (1997). Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 42.Emsley P, and Cowtan K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 43.Wilson MSC, Bulley SJ, Pisani F, Irvine RF, and Saiardi A. (2015). A novel method for the purification of inositol phosphates from biological samples reveals that no phytate is present in human plasma or urine. Open Biol. 5, 150014. 10.1098/rsob.150014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiu D, Wilson MS, Eisenbeis VB, Harmel RK, Riemer E, Haas TM, Wittwer C, Jork N, Gu C, Shears SB, et al. (2020). Analysis of inositol phosphate metabolism by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Commun 11, 6035. 10.1038/s41467-020-19928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harmel RK, Puschmann R, Nguyen Trung M, Saiardi A, Schmieder P, and Fiedler D. (2019). Harnessing (13)C-labeled myo-inositol to interrogate inositol phosphate messengers by NMR. Chem. Sci 10, 5267–5274. 10.1039/c9sc00151d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren X, Zhou Q, Bedar M, Foulad D, Huang KX, Dejam D, Dahan NJ, Kolliopoulos V, Harley BAC, and Lee JC (2023). Modulating Temporospatial Phosphate Equilibrium by Nanoparticulate Mineralized Collagen Materials Induces Osteogenesis via PiT-1 and PiT-2. Adv. Healthc. Mater 12, 2202750. 10.1002/adhm.202202750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Unen J, Botman D, Yin T, Wu YI, Hink MA, Gadella TWJ Jr., Postma M, and Goedhart J. (2018). The C-terminus of the oncoprotein TGAT is necessary for plasma membrane association and efficient RhoA-mediated signaling. BMC Cell Biol. 19, 6. 10.1186/s12860-018-0155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katrukha E. (2022). Ekatrukha/ComDet: ComDet 0.5.5 (Zenodo). [Google Scholar]
- 49.Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, and Kholodenko BN (2014). Evaluating strategies to normalise biological replicates of Western blot data. PLoS One 9, e87293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structure coordinates of XPR1 SPX domain have been deposited in the Protein DataBank, PDB: 8TYV, and PDB: 8TYU. This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.