

Abstract
Background and objectives
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest malignancies. An epigenetic-based synthetic lethal strategy provides a novel opportunity for PDAC treatment. Finding more DNA damage repair (DDR)-related or cell fate-related molecules with aberrant epigenetic changes is becoming very important. Family with sequence similarity 110C (FAM110C) is a cell fate-related gene and its function in cancer remains unclear.
Methods
Seven cell lines, 34 cases of intraductal papillary mucinous neoplasm (IPMN), 15 cases of mucinous cystic neoplasm (MCN) and 284 cases of PDAC samples were employed. Methylation-specific PCR, western blot, CRISPR knockout, immunoprecipitation and a xenograft mouse model were used in this study.
Results
FAM110C is methylated in 41.18% (14/34) of IPMN, 46.67% (7/15) of MCN and 72.89% (207/284) of PDAC, with a progression trend from IPMN/MCN to pancreatic cancer (P = 0.0001, P = 0.0389). FAM110C methylation is significantly associated with poor overall survival (OS) (P = 0.0065) and is an independent prognostic marker for poor OS (P = 0.0159). FAM110C inhibits PDAC cells growth both in vitro and in vivo, serving as a novel tumor suppressor. FAM110C activates ATM and NHEJ signaling pathways by interacting with HMGB1. Loss of FAM110C expression sensitizes PDAC cells to VE-822 (an ATR inhibitor) and MK-8776 (a CHK1 inhibitor).
Conclusion
FAM110C methylation is a potential diagnostic and prognostic marker in PDAC, and its epigenetic silencing sensitizes PDAC cells to ATR/CHK1 inhibitors.
Key words: FAM110C, DNA methylation, pancreatic cancer, DNA damage repair, synthetic lethality
Introduction
Pancreatic ductal adenocarcinoma cancer (PDAC) is the most malignant tumor and is expected to be the second leading cause of cancer-related death by 2030.[1,2] There are three major PDAC precursor lesions, including intraductal papillary mucinous neoplasm (IPMN), pancreatic intraepithelial neoplasia (PanIN) and mucinous cystic neoplasm (MCN).[3,4] With the extensive application of next generation sequencing, precision medicine has been growing very fast in oncology. However, unlike other types of cancer, the development of tailored therapeutics for PDAC has been largely unsatisfactory. Oncogenetic mutations of KRAS account for 95% of PDAC patients, yet targeting their proteins is challenging due to the high affinity for GTP and/or ADP.[5,6] Despite the promising development of a KRASG12C inhibitor in preclinical models,[7] it is important to note that this specific mutation accounts for only approximately 1% of all KRAS mutations.[8] Due to the high levels of intratumoral and intertumoral heterogeneity as well as the presence of untargetable mutations in TP53, SMAD4, and CDKN2A/B genes, precision medicine in pancreatic cancer is still unapplicable.[5,9,10] BRCA1/2 mutations and other DNA damage repair (DDR) gene mutations provide new opportunities for PADC therapy by applying a synthetic lethal strategy.[11] Nevertheless, most of these genes are mutated by less than 5%, including BRCA1/2, PALB2, ATM and MLH1,[6] necessitating the search for additional actionable targets.
With exhaustive genomic resources, aberrant epigenetic changes may provide more avenues for cancer therapy. The classical epi-drugs primarily focus on targeting epigenetic regulators, including readers, writers, and erasers.[12] The ideal cancer therapeutic strategy is to target aberrantly changed cancer cells precisely, without damaging normal cells. Synthetic lethality can be employed by leveraging abnormal epigenetic changes to selectively eliminate cancer cells. For this purpose, the aberrant epigenetic changes of key components in DDR or cell fate-related pathways need to be identified.
Family with sequence similarity 110 (FAM110) family has been identified, consisting of FAM110A, B, C and D.[13,14] FAM110 proteins were found to be located to centrosomes and accumulated at the microtubule organization center in interphase and at spindle poles during mitosis.[14] The data for the FAM 110 family in cancer is very limited.[15,16] FAM 110C protein was reported to inhibit cell proliferation by inducing G1/S arrest.[14,17] Additionally, FAM 110C was found to interact with the microtubule cytoskeleton and suppress cell migration by inhibiting AKT signaling.[18] Nevertheless, the potential involvement of FAM110C in DDR and its role in pancreatic cancer remain to be elucidated.
Materials and methods
Cells and different types of pancreatic tissues
Seven PDAC cell lines were involved in this study, including MIAPaCa-2, Panc3.11, Panc5.04, Panc10.05, SW1990, JF-305 and PATU-8988T. All cell lines were authenticated by STR profiling and routinely tested for mycoplasma contamination.
Different types of pancreatic tissue samples, including 284 cases of PDAC, 34 cases of IPMN and 15 cases of MCN, were obtained from the Chinese PLA General Hospital. The tumors were staged according to the 8th edition of the AJCC Cancer Staging Manual. None of the patients received chemotherapy before surgery. Sample collection followed the guidelines approved by the institutional review board (IRB number: 20090701–015).
Cell treatment, RNA extraction, PCR
Cells were treated with 2 μM 5-aza-2’-deoxycytidine (5-aza, Sigma-Aldrich, # A3656, USA) for 96 h. Total RNA was extracted using TRIzol reagent (Invitrogen, #15596026, USA). Five micrograms of qualified RNA was used for cDNA synthesis following the manufacturer’s instruction (Thermo Scientific, #K1691, USA). The RT-PCR primer sequences for FAM110C and GAPDH (internal control) are listed in Supplementary Table 1.
Preparation of DNA and sodium bisulfite treatment
A phenol-chloroform extraction assay was used to prepare DNA. Bisulfite treatment, methylation-specific PCR (MSP), and bisulfite sequencing (BSSQ) followed previous protocols.[19,20] Normal lymphocyte DNA (NL) was used as a control for unmethylation, and in vitro methylated DNA (IVD) was used as a methylation control. The MSP and BSSQ primers for FAM110C are presented in Supplementary Table 1.
Immunohistochemistry
Immunohistochemistry (IHC) was carried out following a previous description.[20] The antibodies are listed in Supplementary Table 2.
Construction of cell lines stably expressing FAM110C
The coding sequence of the human FAM110C (NM_001077710.3) expression vector was constructed using the pCDH-CMV-MCS-puro plasmid (Genewiz, AA27980-1/M548037, USA). FAM110C expressing or empty vectors with plasmids (pLP1, pLP2, and VSVG) were packaged using HEK293T cells with LipofectamineTM 3000 Reagent (Invitrogen, #L3000008, USA). Lentivirus-transfected PDAC cells were screened with puromycin (MCE, #HY-15695, USA) at concentrations of 1.5 μg/ mL (MIAPaCa-2) and 0.5 μg/mL (JF-305) for 3 days. Thereafter, monoclonal cells were selected by limited dilution in 96-well plates and validated by western blot.
Construction of FAM110C knockout cell lines
The single guide RNA (sgRNA) sequences utilized in this study can be found in Supplementary Table 1. The sgRNAs targeting the first and second exons of FAM 110C were designed by the MIT CRISPR design tool (http:// crispr.mit.edu). The LentiCRISPR v2-gDNA plasmid was used to construct FAM110C knockout Panc10.05 cells. Monoclonal cells were selected with puromycin (2 μg/mL) following the above method.
MTT and colony formation
PDAC cells were seeded into 96-well plates at a density of 2×103 (MIAPaCa-2), 1.5×103 (JF-305), and 1.5×103 (Panc10.05) cells per well. An MTT assay was performed at 0, 24, 48, 72, and 96 h to determine cell viability (KeyGEN Biotech, # KGT5251, China). The results are shown as plotting curves, with the mean value ± standard deviation.
Table 1.
The association betweenClinical factors and FAM110C methylation status inpancreaticcancer patients
| Methylation status | ||||
|---|---|---|---|---|
| Clinical parameter |
|
|||
| Unmethylated | Methylated | |||
| NO. 284 | n = 77 (27.18%) | n = 207 (72.89%) | P value | |
| Gender | ||||
| Male | 185 | 45 | 140 | 0.1485 |
| Female | 99 | 32 | 67 | |
| Age (y) | ||||
| ≤50 | 45 | 12 | 33 | 0.9415 |
| >50 | 239 | 65 | 174 | |
| Differentiation | ||||
| Well/Moderately | 137 | 40 | 97 | 0.4456 |
| Poorly | 147 | 37 | 110 | |
| TNM stage | ||||
| I/II | 252 | 71 | 181 | 0.2586 |
| III/IV | 32 | 6 | 26 | |
| Lymph node metastasis | ||||
| Negative | 184 | 50 | 134 | 0.9749 |
| Positive | 100 | 27 | 73 | |
| Tumor size (cm) | ||||
| ≤ 4 cm | 217 | 67 | 150 | 0.0103* |
| >4 cm | 67 | 10 | 57 | |
| Tumor location | ||||
| Proximal | 178 | 46 | 132 | 0.5327 |
| Distal | 106 | 31 | 75 | |
| Smoking | ||||
| Yes | 113 | 27 | 86 | |
| No | 171 | 50 | 121 | 0.3212 |
| Alcohol consumption | ||||
| Yes | 130 | 29 | 101 | 0.0942 |
| No | 154 | 48 | 106 | |
P values are obtained from χ2 test, *P < 0.05.
For colony formation, cells were seeded at a density of 300 (MIAPaCa-2), 300 (JF-305) and 500 (Panc10.05) cells per well in 6-well plates. The results were evaluated after a growth of 2 weeks.
Cell Cycle and Apoptosis Analysis
FAM 110C silenced and re-expressed MIAPaCa-2 and JF-305 cells, as well as before and after FAM110C knockout Panc10.05 cells were synchronized to the G0/G1 phase by serum withdrawal for 12 h, followed by re-entry into the cell cycle by the addition of serum (10% FBS) for 36 h. Cells were fixed and stained with propidium iodide using a cell cycle detection kit (KeyGEN Biotech, #KGA512, China) in accordance with the instructions. For cell-cycle analysis, FACS Caliber (BD Biosciences, USA) and Modifit software (Verity Software House, USA) were employed. The apoptosis assay was performed following the instructions of the Annexin V-FITC/PI Apoptosis Detection Kit (KeyGEN Biotech, #KGA108, China).
Transwell Assay
For cell migration evaluation, 6×104 MIAPaCa-2, 2×104 JF-305, and 4×104 Panc10.05 cells were applied to the upper chamber (Corning, #3422, USA) for 30 h. In the invasion assay, 8×104 MIAPaCa-2, 5×104 JF-305, and 6×104 Panc10.05 cells were placed in the upper chamber coated with matrigel (BD Biosciences, #354234, USA) for 36 h.
Table 2.
Univariate and multivariate Coxregression analysis of prognosticfactors in PDAC patients(n = 186)
| Clinical parameter | Univariate analysis |
Multivariate analysis |
||
|---|---|---|---|---|
| HR (95%CI) | P value | HR (95%CI) | P value | |
| Gender (male vs. female) | 0.973 (0.649-1.460) | 0.896 | ||
| Age (y) (≤50 vs. >50) | 0.720 (0.394-1.316) | 0.285 | ||
| Differentiation (high or middle vs. low differentiation) | 0.696 (0.471-1.028) | 0.069 | 0.727 (0.492, 1.075) | 0.110 |
| TNM stage (I/II vs. III/IV) | 0.712 (0.417-1.216) | 0.213 | ||
| Lymph node metastasis (negative vs. positive) | 0.714 (0.485-1.052) | 0.089 | 0.794 (0.536, 1.177) | 0.250 |
| Tumor size (cm) (<4 vs. 4) ≥ | 0.822 (0.555-1.218) | 0.329 | ||
| Tumor location (distal vs. proximal) | 0.668 (0.443-1.008) | 0.054 | 0.727 (0.479, 1.103) | 0.134 |
| FAM110C (unmethylation vs. methylation) | 0.512 (0.313-0.837) | 0.008** | 0.544 (0.332, 0.893) | 0.016* |
| Smoking (no vs. yes) | 1.049 (0.707-1.556) | 0.811 | ||
| Alcohol consumption (no vs. yes) | 0.915 (0.621-1.349) | 0.654 | ||
HR: Hazard ratio; *P < 0.05; **P<0.01.
The procedures followed previous protocols.[21]
Western blot and immunoprecipitation
Antibodies for western blot and immunoprecipitation (IP) are listed in Supplementary Table 2. A Rabbit IgG (Beyotime, #A7016, China) was employed as a negative control for IP. The procedures followed the manufacturer’s instructions (Thermo Scientific, #87788, USA; YEASEN, #36403ES08, China). Visible specific bands were resected for mass spectrometry analysis after SDS-PAGE electrophoresis and silver staining.
SiRNA interference technique
SiRNAs against HMGB1 (siRNA#1, siRNA#2, and siRNA#3) and a negative control duplex (Scramble) were synthesized by JTSBIOCo. , Ltd (Wuhan, China) and transfected into the cells using RNAiMax (Invitrogen, #13778150, USA). The sequences of HMGB1-targeting siRNAs and the negative control duplex are listed in Supplementary Table 3. SiRNA#2 was determined to be the most efficient siRNA, and was applied for subsequent experiments.
Xenograft mouse model
The animal experiment procedures were approved by the Animal Ethics Committee of the Chinese PLA General Hospital. BALB/c nude mice (4 weeks old) were purchased from SPF (Beijing, China) Biotechnology Co., Ltd. and housed under standard pathogen-free conditions. The nude mice were randomly divided into two groups, each consisting of five mice. FAM110C unexpressed and stably expressed MIAPaCa-2 cells (6×106) were used to build xenograft mice model. Tumor volume (V) was detected every 4 days following inoculation and calculated by the formula V = (length × width2) / 2.
Half-inhibitory concentration analysis
Cells (MIAPaCa-2, JF-305, and Panc10.05) with or without FAM110C expression were seeded at a density of 3000 cells/well into 96-well plates. Cell viability was evaluated by MTT assay 48 h after treatment with gradient dilutions of ATR or CHK1 inhibitors (KeyGEN Biotech, #KGT5251, China). The absorbance was measured at a wavelength of 490 nm by a microplate reader (Thermo Multiskan MK3, USA). The value of the half-inhibitory concentration (IC50) was calculated using GraphPad Prism software (GraphPad Software Inc., USA).
Statistical analysis
SPSS 22.0 software (IBM, USA) and GraphPad Prism 7.0 software (GraphPad Software Inc., USA) were employed in this study. The chi-square test was used to analyze the associations between FAM110C methylation status and clinicopathological factors. Kaplan-Meier survival curves were plotted with log-rank tests to compare overall survival (OS). Univariate and multivariate Cox regression analyses were used to evaluate prognostic factors for OS. Quantitative data was analyzed by the Student’s two-tailed t test. P < 0.05 was considered statistically significant.
Results
The expression of FAM110C is regulated by promoter region methylation in human PDAC
K450 microarray methylation and mRNA expression data from 178 cases of available PDAC were extracted from the UCSC Xena Browser (http://xena.ucsc.edu/). The expression of FAM110C was reduced in PDAC compared to noncancerous pancreatic tissue samples (P = 0.0000, Supplementary Figure 1A). The mRNA levels of FAM110C were inversely associated with methylation status in the CpG sites around transcription start site (TSS) (Supplementary Figure 1B), suggesting the possibility for methylation regulation of FAM110C gene expression.
Then, the mRNA level of FAM 110C was examined by RT-PCR. It was not detected in MIAPaCa-2 and JF-305 cells, exhibited reduced expression in SW1990 and PATU-8988T cells, and exhibited higher expression levels in Panc3.11, Panc5.04 and Panc10.05 cells (Figure 1A). FAM110C was completely methylated in MIAPaCa-2 and JF-305 cells, partially methylated in SW1990 and PATU-8988T cells and unmethylated in Panc3.11, Panc5.04 and Panc10.05 cells (Figure 1B). The regulation of FAM 110C expression by methylation was further validated by 5-aza treatment (Figure 1A). MSP efficiency and methylation density were validated by BSSQ in MIAPaCa-2, JF-305 and Panc10.05 cells (Figure 1C).
Figure 1.
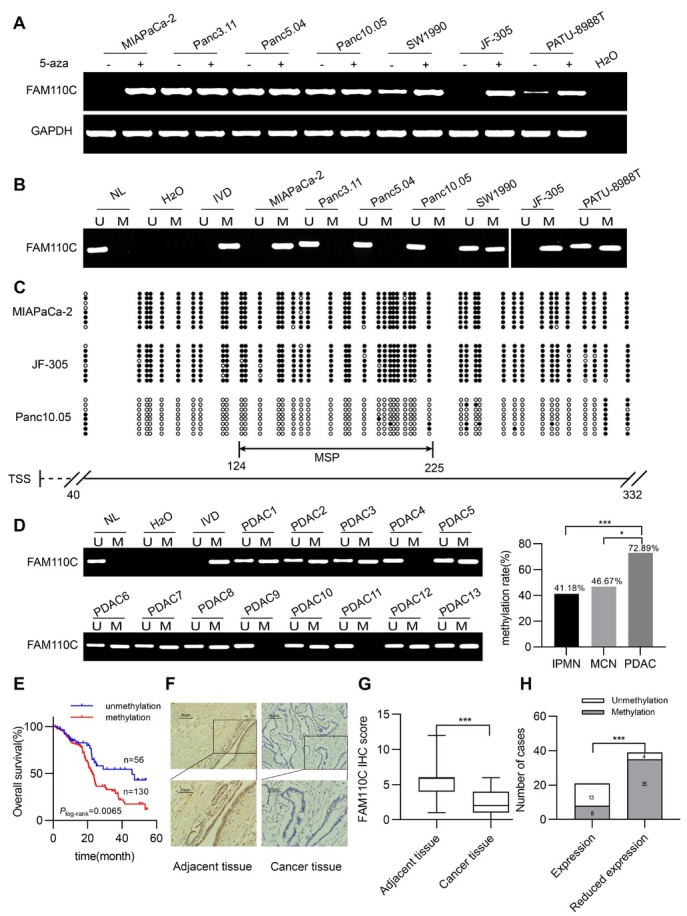
The expression and methylation status of FAM110C in pancreatic cancer cells and tissue samples. (A) MIAPaCa-2, Panc3.11, Panc5.04, Panc10.05, SW1990, JF-305, PATU-8988T are pancreatic cancer cells. H2O: negative control; GAPDH: internal control; 5-aza: 5-aza-2’-deoxycytidine;(-): absence of 5-aza;(+): presence of 5-aza. (B) MSP results present methylation status of FAM110C in pancreatic cancer cells. U: unmethylated alleles; M: methylated alleles; IVD: in vitro methylated DNA (methylation control); NL: normal lymphocytes DNA (unmethylation control); H2O: double distilled water. (C) BSSQ results of FAM110C in MIAPaCa-2, JF-305 and Panc10.05 cells. Double-headed arrow indicates the product size of MSP was 102 bp (from 124bp to 225bp) and bisulfite sequencing was conducted in a 293 bp region of the CpG island (from 40bp to 332bp) around the FAM110C transcription start site. Filled circles: methylated CpG sites; open circles: unmethylated CpG sites; TSS: transcription start site. (D) Representative MSP results of FAM110C in PDAC samples (*P < 0.05, ***P < 0.001). (E) The overall survival time was significantly shorter in methylated patients than in unmethylated individuals (P = 0.0065, Log-rank test). (F) Representative IHC results show FAM110C staining in pancreatic cancer tissue and adjacent tissue samples (top: 200×; bottom: 400×). (G) Box plots for FAM110C IHC score, horizontal lines represent the median score, vertical bars represent the range of score. ***P < 0.001. (H) Bar diagram indicates an inverse relationship between FAM110C expression levels and DNA methylation status. ***P < 0.001.
Methylation of FAM 110C was detected in 41.18% (14/34) of IPMN, 46.67% (7/15) of MCN and 72.89% (207/284) of PDAC. The ratio of methylation is increased with the progression of carcinogenesis (P = 0.0001, P = 0.0389, Figure 1D). FAM110C methylation was significantly associated with tumor size (P = 0.0103), while no association was observed between FAM 110C methylation and gender, age, smoking, alcohol consumption, tumor differentiation, TNM staging, lymph node metastasis or tumor location (Table 1). For 186 cases of patients with available survival data, log rank testing was performed. FAM110C methylation was significantly associated with poor OS (P = 0.0065, Figure 1E). Then, univariate and multivariate Cox regression analyses were used to analyze the prognostic markers for survival. FAM 110C methylation is an independent prognostic marker for poor OS (P = 0.0159, Table 2).
The IHC assay was used for FAM110C expression detection in 60 available PDAC and matched adjacent tissue samples. The levels of FAM110C were higher in adjacent tissue than in PDAC tissue samples (P = 0.0000, Figure 1F& G). The staining was located in both the nucleus and cytoplasm (Figure 1F). Among the tumor samples, a reduced level of FAM 110C was associated with promoter region methylation (P = 0.0000, Figure 1H), indicating the epigenetic regulation of FAM 110C expression.
FAM110C suppresses PDAC cell proliferation
The OD values obtained by MTT assay were used for evaluating cell viability. The OD values were 1.045 ± 0.086 vs. 0.665 ± 0.056 and 0.900 ± 0.044 vs. 0.640 ± 0.033 before and after the re-expression of FAM110C in MIAPaCa-2 and JF-305 cells, respectively (Figure 2A). The OD value was decreased significantly by FAM110C (P = 0.0000, P = 0.0000). In Panc10.05 cells, the OD values were 0.380 ± 0.026 vs. 0.529 ± 0.019 before and after deletion of FAM 110C (P = 0.0000, Figure 2A). The above results indicate the inhibitory role of FAM 110C in cell proliferation.
Figure 2.
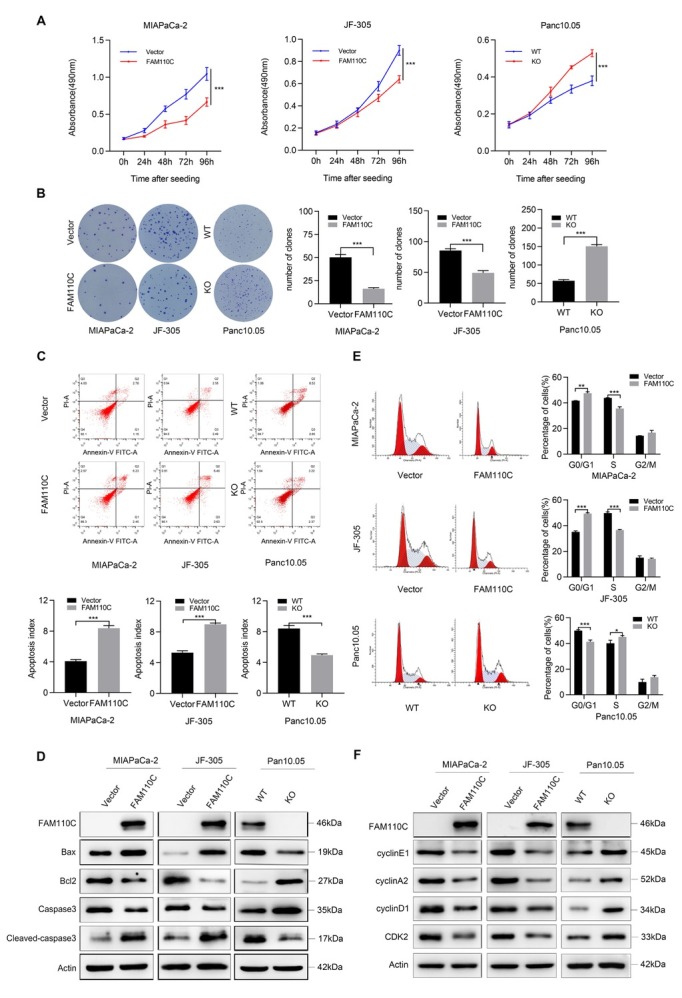
The roles of FAM110C play in cell proliferation, apoptosis and cell cycle. (A) OD value for FAM110C expressed and unexpressed MIAPaCa-2, JF-305 and Panc10.05 cells. (B) Representative colony formation results. (C) Apoptosis results of FAM110C expressed and unexpressed MIAPaCa-2, JF-305 and Panc10.05 cells. The bar diagram represents the percentage of apoptosis. (D) Western blot shows the effects of FAM110C on the expression levels of bax, bcl-2, caspase-3 and cleaved caspase-3. (E) Representative results of cell phase distribution. The bar diagram represents the percentage. (F) Western blot suggests the effects of FAM110C on expression levels of cyclin E1, cyclin A2, cyclinD1 and CDK2. Vector: control vector; FAM110C: FAM110C expressing vector; WT: wild type control; KO: FAM110C knockout. *P < 0.05; **P < 0.01; ***P < 0.001.
The clone numbers were 50.3 ± 3.06 vs. 16.3 ± 1.16 and 85.7 ± 2.89 vs. 49.3 ± 3.51 in FAM110C unexpressed and re-expressed MIAPaCa-2 and JF-305 cells, respectively (P = 0.0001, P = 0.0002, Figure 2B). In FAM110C highly expressed and knockout Panc 10.05 cells, the clone numbers were 82.0 ± 5.10 vs. 128.7 ± 2.62 (P = 0.0000, Figure 2B). These results demonstrated that FAM110C inhibited PDAC cell colony formation.
FAM110C induces cell apoptosis
The ratio of apoptotic cells was 4.09% ± 0.23% vs. 8.4% ± 0.35% and 5.31% ± 0.25% vs. 8.99% ± 0.17% in FAM110C unexpressed and re-expressed MIAPaCa-2 and JF-305 cells, respectively (P = 0.0001, P = 0.0000, Figure 2C). The ratio of apoptotic cells was 8.41% ± 0.42% vs. 4.96% ± 0.14% in Panc10.05 cells with high FAM110C expression and knockout (P = 0.0002, Figure 2C). These results suggested that FAM110C induces PDAC cell apoptosis. Restoration of FAM110C expression decreased caspase-3 and bcl-2 levels and increased cleaved caspase-3 and bax levels in MIAPaCa-2 and JF-305 cells, while knocking out FAM110C in Panc10.05 cells increased caspase-3 and bcl-2 levels and decreased cleaved caspase-3 and bax levels (Figure 2D), further suggesting the function of FAM110C in cell apoptosis.
FAM110C induces G1/S phase arrest
In FAM110C silenced and re-expressed MIAPaCa-2 cells, the cell phase distributions were as follows: 41.76% ± 0.10% vs. 47.49% ± 1.17% for G0/G1 (P= 0.0011), 43.89% ± 0.19% vs. 35.66% ± 1.35% for S (P = 0.0005), and 14.35% ± 0.14% vs. 16.84% ± 1.76% for G2/M. For FAM110C unexpressed and re-expressed JF-305 cells, the distribution of cell phases was as follows: G0/G1 phase: 35.14% ± 0.81% vs. 49.34% ± 0.69% (P = 0.0000), S phase: 49.73% ± 0.94% vs. 36.59% ± 0.50% (P = 0.0000) and G2/M phase: 15.13% ± 1.29% vs. 14.07% ± 0.62%. FAM110C was deleted by the CRISPR CAS9 technique in highly expressed Panc10.05 cells, and cell phases were distributed as follows: G0/G1 phase: 50.03% ± 0.67% vs. 41.25% ± 1.42% ( P = 0.0006), S phase: 40.09% ± 2.35% vs. 45.07% ± 1.07% (P = 0.0289), and G2/M phase: 9.88% ± 2.27% vs. 13.69% ± 1.45% before and after FAM110C knockout. These results indicate that G1/S arrest is induced by FAM110C (Figure 2E). Decreased cyclin D1, cyclin A2, cyclin E1 and CDK2 levels were observed by restoration of FAM110C expression in MIAPaCa-2 and JF-305 cells, while increased cyclin D1, cyclin A2, cyclin E1 and CDK2 levels were observed in Panc10.05 cells by knocking out FAM110C (Figure 2F), further validating that FAM110C induces G1/S arrest.
FAM110C suppresses cell migration and invasion
In FAM110C unexpressed and re-expressed MIAPaCa-2 and JF-305 cells, the number of migratory cells was 294.44 ± 1 3.91 vs. 147.11 ± 9.740 and 306.33 ± 18.33 vs. 115.89 ± 9.39, respectively (P = 0.0000, P = 0.0000, Figure 3A). The number of migratory cells before and after knockout of FAM110C in Panc10.05 cells was as follows: 108.67 ± 8.49 vs. 235.33 ± 10.74 (P = 0.0000, Figure 3A), further suggesting the inhibitory role of FAM110C in cell migration. For invasion analysis, in FAM110C-silenced and FAM110C-overexpressing MIAPaCa-2 and JF-305 cells, the invasive cells were 267.22 ± 16.08 vs. 105.33 ± 8.31 and 168.22 ± 7.97 vs. 57.56 ± 10.55, respectively (P = 0.0000, P = 0.0000, Figure 3A). The number of invasive cells was 94.67 ± 5.50 vs. 204.33 ± 12.70 in FAM110C highly expressed and knocking out Panc10.05 cells (P = 0.0000, Figure 3A). The above data demonstrates that FAM110C suppresses cell invasion. To further validate the influence of FAM110C on cell invasion and migration, MMP2, MMP7 and MMP9 were detected by western blot. Decreased MMP2, MMP7 and MMP9 were found in FAM110C re-expressed MIAPaCa-2 and JF-305 cells, and they were increased in FAM110C knockout Panc10.05 cells (Figure 3B), validating the results at the molecular level. The levels of MMP2, MMP7 and MMP9 were examined by IHC in FAM110C unexpressed and re-expressed MIAPaCa-2 cell xenografts. The levels of MMP2, MMP7 and MMP9 were decreased by re-expressing FAM110C, demonstrating the effect of FAM110C on invasion and migration in vivo (Figure 3F).
Figure 3.
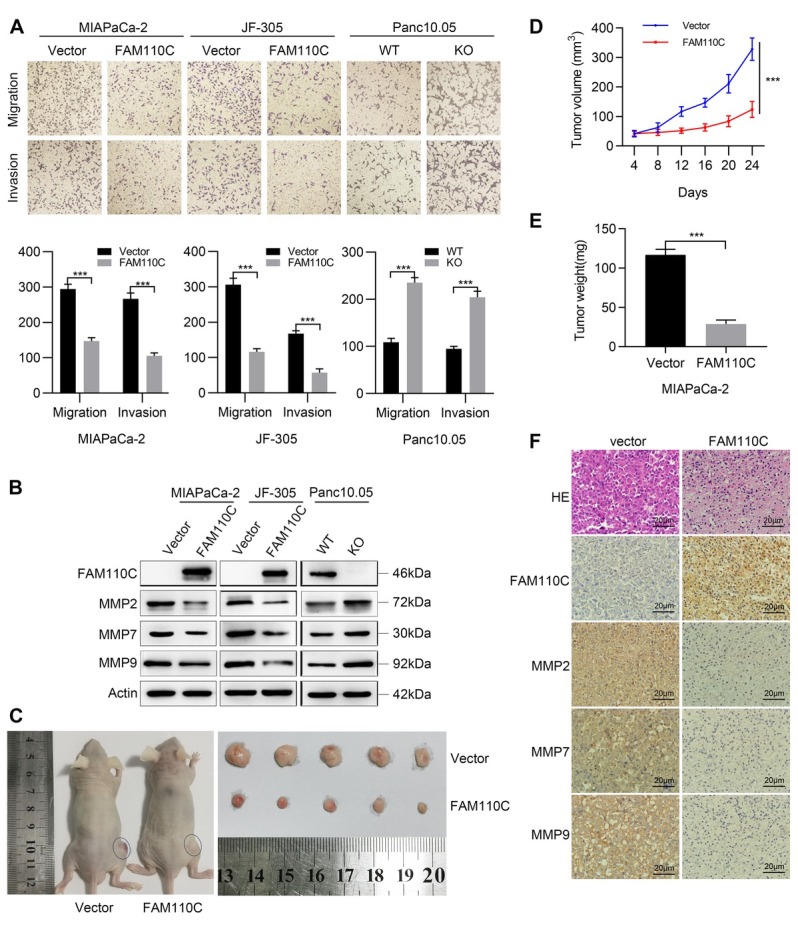
Effect of FAM110C on cell migration, invasion, and tumor xenograft model. (A) The migration and invasion results of FAM110C expressed and unexpressed MIAPaCa-2, JF-305 and Panc10.05 cells. The average number of migration and invasion cells was presented by the bar diagram. Vector: control vector; FAM110C: FAM110C expressing vector; WT: wild type control; KO: FAM110C knockout. ***P < 0.001. (B) Western blot shows the effects of FAM110C on the expression levels of MMP2, MMP7 and MMP9. (C) FAM110C unexpressed and re-expressed MIAPaCa-2 cells xenograft in nude mice. (D) Growth curves and (E) Tumor weight of FAM110C unexpressed and re-expressed MIAPaCa-2 cell xenografts (n = 5). ***P < 0.001. (F) The expression of MMP2, MMP7 and MMP9 in FAM110C unexpressed and re-expressed MIAPaCa-2 cell xenografts.
The growth of PDAC cell xenografts was suppressed by FAM110C
In FAM110C defect and force-expressed MIAPaCa-2 cell xenografts, the tumor volume was 328.65 ± 38.38 vs. 123.91 ± 27.19 mm3 (P = 0.0000). Smaller tumor volumes were observed in xenografts re-expressing FAM110C (Figure 3C& D). The tumor weight was 116.68 ± 7.24 vs. 28.98 ± 4.97 mg in MIAPaCa-2 cell xenografts without and with forced expression of FAM110C. The tumor weight was significantly reduced by FAM110C (P = 0.0000, Figure 3E).
FAM110C is involved in DNA damage repair by interacting with HMGB1
To gain a deeper understanding of the roles of FAM110C in PDAC, an immunoprecipitation technique was employed. Figure 4A illustrates that when comparing PDAC cells with and without FAM110C expression, two extra bands were observed in FAM110C re-expressing MIAPaCa-2 and JF-305 cells. To identify the proteins present in these bands, mass spectrometry was utilized. As listed in Supplementary Table 4 and 5, the proteins that were pulled down by FAM110C were found to be similar in the two experiments. After excluding keratin, actin, and other cytoskeletal proteins, the majority of the proteins were related to apoptosis, DDR and stress-related signaling pathways. High mobility group box 1 (HMGB1) was observed to be present in both protein complexes derived from FAM110C re-expressed MIAPaCa-2 and JF-305 cell lysates. As shown in Supplementary Table 4 and 5, HMGB1 exhibited a higher score level, in addition to apoptosis-related proteins. HMGB1 was reported to be involved in DDR and stress-related signaling pathways.[22, 23, 24, 25, 26] The interaction of FAM110C and HMGB1 was then validated by western blot and reciprocal co-IP assays (Figure 4B). Next, we focused on investigating the role of FAM110C through interacting with HMGB1 in PDAC cells under low dose cisplatin treatment, by comparing epigenetic silencing or deletion of FAM110C cells with FAM110C expressing cells.
Figure 4.

The role of FAM110C in DDR. (A) Co-IP assay and silver staining. Red arrow: specific band, which was subjected to mass spectrometry. IgG: negative control. FAM110C (-): without FAM110C expression FAM110C (+): FAM110C expression. (B) Validation of interaction between FAM110C and HMGB1. (C) cisplatin (-): without cisplatin treatment; cisplatin (+): cisplatin treatment. (D) Testing the efficiency of siRNAs for knocking down HMGB1. Scramble: siRNA negative control; siRNA#1, siRNA#2 and siRNA#3: siRNAs for HMGB1. (E) The effects of HMGB1 knockdown on ATM/CHK2 and NHEJ pathways. (F) Evaluation of IC50 for VE-822 and MK-8776 in PDAC cells.
The levels of phosphorylated ATR (p-ATR) and phosphorylated CHK1 (p-CHK1) were found to be elevated in FAM110C unexpresing MIAPaCa-2 and JF-305 cells compared to FAM110C re-expressing cells. Additionally, increased levels of p-ATR and p-CHK1 were observed after knockout of FAM110C in Panc 10.05 cells, indicating the inhibitory effect of FAM110C on the ATR/CHK1 pathway (Figure 4C). On the other hand, the levels of phosphorylated ATM (P-ATM) and phosphorylated CHK2 (p-CHK2) were increased after restoration of FAM110C expression in MIAPaCa-2 and JF-305 cells. Conversely, decreased levels of p-ATM and p-CHK2 were observed after knockout of FAM110C in Panc10.05 cells, demonstrating that FAM110C activated ATM signaling (Figure 4C). The effect of FAM110C on the non-homologous end joining (NHEJ) pathway was also assessed. In FAM110C expressing MIAPaCa-2, JF-305, and Panc10.05 cells, the levels of phosphorylated DNAPKcs (p-DNAPKcs) and XRCC4 were found to be higher than those in cells that did not express FAM110C, suggesting that FAM110C activated NHEJ signaling (Figure 4C).
To further validate the involvement of FAM110C in DDR through its interaction with HMGB1, siRNA was employed. The efficiency of siRNAs was tested, and siRNA#2 was found to be the most effective (Figure 4D). In FAM110C highly expressed MIAPaCa-2, JF-305 and Panc10.05 cells, the levels of p-ATM, p-CHK2, p-DNAPKcs and XRCC4 were reduced by knocking down HMGB1, further demonstrating that the effect of FAM110C on DDR is mediated through its interaction with HMGB1 (Figure 4E).
Loss of FAM110C expression sensitizes pancreatic cancer cells to VE-822 and MK-8776
As FAM110C is involved in NHEJ and ATM signaling, and ATR/CHK1 signaling is the compensation pathway, we explored the sensitivity of PDAC cells to VE-822 (an ATR inhibitor) and MK-8776 (a CHK1 inhibitor), with or without FAM110C expression. The IC50 of VE-822 was 0.281 ± 0.074 μM vs. 2.011 ± 0.226 μM and 0.441 ± 0.071 μM vs. 2.096 ± 0.184 μM in FAM110C unexpressed and re-expressed MIAPaCa-2 and JF-305 cells under treatment with cisplatin, respectively. The IC50 of VE- 822 was reduced significantly in FAM110C-silenced cells (P = 0.0000, P = 0.0000, Figure 4F). The IC50 of VE-822 was 2.268 ± 0.469 μM vs. 0.337 ± 0.128 μM in Panc10.05 cells before and after knockout of FAM110C. The IC50 was significantly reduced after knockout of FAM110C (P = 0.0000, Figure 4F). These results indicate that loss of FAM110C expression sensitized PDAC cells to the ATR inhibitor.
The IC50 of MK-8776 was 26.330 ± 8.128 μM vs. 93.682 ± 11.243 μM and 13.442 ± 1.632 μM vs. 63.373 ± 12.309 μM in FAM110C unexpressed and re-expressed MIAPaCa-2 and JF-305 cells under treatment with cisplatin, respectively (P = 0.0000, P = 0.0000, Figure 4F). The ICof MK-8776 was 76.623 ± 9.618 μM vs. 34.748 ± 4.387 μM in Panc10.05 cells before and after knockout of FAM110C under cisplatin treatment (P = 0.0000, Figure 4F). These results demonstrated that loss of FAM110C expression sensitized pancreatic cancer cells to the CHK1 inhibitor.
Discussion
Abnormal epigenetic alterations have been reported in various cancers, and DNA methylation is regarded as a potential cancer detection, prediction, prognosis and chemo-radio therapeutic marker.[27, 28, 29] The nature of epigenetic changes is reversible, making it an attractive therapeutic target. Gene expression is regulated by epigenetic machinery, including writers (responsible for adding modifications to DNA or histones, such as DNA methyltransferase), readers (recognition of modifications and recruitment of effector proteins, such as methyl-binding domain proteins) and erasers (enzymes to remove chemical modifications, such as histone demethylase).[12,30] Many drugs have been developed to target epigenetic machinery.[28,31] Two demethylating agents, decitabine and azacytidine, were approved by the FDA in hematological malignancies and myelodysplastic syndromes.[31,32] Significant toxicity was found with high-dose treatment in solid tumors, without improving overall survival.[30,33] Accumulating evidence demonstrates that the efficacy of mono epi-drug therapy is very limited, suggesting that specific restoration of epigenetically silenced gene expression is challenged markedly by epi-drugs. Clinical trials involving the combination of chemotherapy, radiotherapy, or immunotherapy with epi-drug therapy are currently underway.[34, 35, 36] An epigenetic-based therapeutic strategy employing synthetic lethality may precisely target cancer cells with aberrant epigenetic changes, without hurting normal cells.[11,33] The concept of synthetic lethality stems from the study of fruit flies, a genetic model. A lethal outcome is observed when both specific genes are mutated, whereas individual mutations of either gene alone do not affect viability.[37] This principle was applied to cancer therapy with PARP inhibitors in BRAC1/2 mutated cells.[38,39] The rationale was extended to “BRCAness” for other DDR gene mutants.[40,41] Beyond “BRCAness”, epigenetic silencing of DDR-related or cell fate-related genes is also suitable for synthetic lethal therapeutic strategies.[21,42] It is desirable to look for novel DDR-related or cell fate-related genes, that are regulated by epigenetics and have aberrant epigenetic changes in tumors to broaden the scope of therapeutic targets.
FAM110A and FAM110B were reported to play important roles in pancreatic, lung, and prostate cancers.[16,43,44] However, the role of FAM110C in cancer remains unclear. It is important to understand the epigenetic regulation and mechanism of FAM110C in pancreatic cancer to develop novel treatment strategies. Our results demonstrated that FAM110C was frequently methylated in PDAC, with an accumulating tendency during carcinogenesis. FAM110C methylation was significantly associated with tumor size. These results indicate that methylation of FAM110C may serve as a potential early PDAC detection marker. The log rank test was conducted on 186 cases of patients with available survival data, revealing a significant association between FAM110C methylation and poor OS. Subsequently, both univariate and multivariate Cox regression analyses were employed, confirming that FAM110C methylation is an independent prognostic marker for poor OS.
FAM110C suppressed PDAC cell proliferation, migration and invasion, and induced apoptosis and G1/S arrest. FAM110C suppressed PDAC cell xenograft growth in mice, implying its potential as a novel tumor suppressor in PDAC. To gain further insights into the mechanism of FAM110C in PDAC, an IP assay and mass spectrometry analysis were applied. The interaction of FAM110C and HMGB1 was discovered, and confirmed through western blot and reverse immunoprecipitation. HMGB1 is reported to be involved in the PI3K/AKT, NF-kB and JAK/STAT signaling pathways to regulate DNA replication and gene transcription.[25,26] HMGB1 was also revealed to be involved in different DDR signaling pathways, including mismatch repair (MMR), base excision repair (BER), nucleotide excision (NER), and NHEJ.[25,45, 46, 47, 48] Both the PI3K/AKT and NF-κB signaling pathways were reported to promote DDR and are involved in inflammation.[11,49, 50, 51, 52, 53] Under the inflammatory environment, HMGB1 may promote carcinogenesis, especially during hepatocarcinogenesis.[26,54] Therefore, HMGB1 may play conflicting roles in various cancers under different environments, potentially exerting both antitumor and protumor effects. HMGB1 was recently found to be a damage-associated molecule in dying cancer cells that enhances immunogenic cell death.[55] Under DNA-damaging agent treatment in various malignant tumors, HMGB1 has been reported to primarily regulate the DNA damage response checkpoint and cell survival.[22,56,57] Notably, chemotherapy agents predominantly induce DNA double strand breaks, which are repaired through the classical pathways of homologous recombination repair (HR) and NHEJ. HR is composed of the ATR/CHK1 and ATM/ CHK2 signaling pathways.[58] Subsequently, an investigation was conducted to examine the role of FAM110C in DDR. These findings indicate that FAM110C activates the NHEJ and ATM/CHK2 signaling pathways while inhibiting the ATR/CHK1 pathway in PDAC cells.
Epigenetic-based synthetic lethality emerges as a novel strategy for cancer therapy. Consequently, an exploration was undertaken to assess the synthetic lethal effects between ATR/CHK1 inhibitors and the loss of FAM110C expression. ATR and CHK1 inhibitors have shown promise in treating PARP inhibitor-resistant PDAC, and ongoing clinical trials are evaluating the combination of ATR/CHK1 inhibitors with other therapeutics.[59, 60, 61] Our findings indicated that the epigenetic silencing or deletion of FAM110C sensitized PDAC cells to ATR and CHK1 inhibitors when exposed to low doses of cisplatin (Figure 5). It is noteworthy that epigenetic abnormalities are more frequently observed in DDR and cell fate signaling pathways in various types of cancer.[10] Therefore, conducting further investigations on FAM110C may lead to the development of more effective therapeutic strategies for PDAC.
Figure 5.
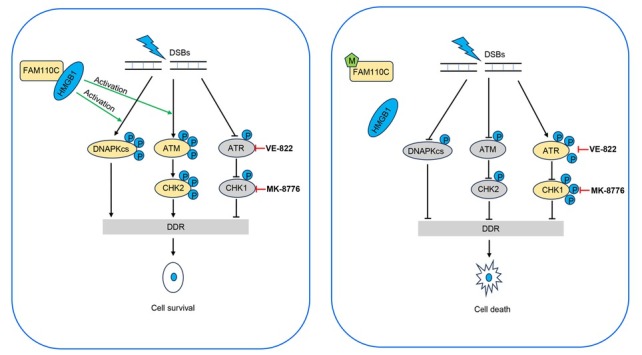
The schematic illustration of synthetic lethality between epigenetic silencing of FAM110C and ATR/CHK1 inhibitors. A working model for synthetic lethality of FAM110C methylation and ATR/CHK1 inhibitor in PDAC cells. DSBs: double-strand breaks; M: DNA methylation; P: phosphorylation; DDR: DNA damage repair.
In conclusion, FAM110C acts as a potential tumor suppressor in PDAC. FAM110C methylation is a potential diagnostic and prognostic marker for PDAC. Epigenetic silencing of FAM110C sensitized pancreatic cancer cells to ATR/CHK1 inhibitors.
Supplementary Material
Supplementary Material
Acknowledgements
None.
Funding Statement
This work was supported by grants from National Key Research and Development Program of China (2018YFA0208902, 2020YFC2002705); National Science Foundation of China (NSFC No. 82272632, 81672138).
Footnotes
Author Contributions
Fengna Liu: Investigation, Formal analysis, Methodology, and Writing - Original Draft; Aiai Gao: Investigation, Visualization, Editing; Meiying Zhang: Validation, Project administration, Visualization; Yazhuo Li: Formal analysis; Fan Zhang: Investigation; James G. Herman: Editing, Supervision; Mingzhou Guo: Conceptualization, Resources, Supervision, Funding acquisition, Writing - Review & Editing.
Informed Consent
Not applicable.
Ethical Approval
The study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Chinese PLA General Hospital (IRB number: 20090701-015). The animal experiments were performed according to procedures approved by the Animal Ethics Committee of the Chinese PLA General Hospital.
Conflict of Interest
The authors declare no competing interest.
Data Availability Statement
Data is available for readers on request.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7. doi: 10.3322/caac.21708. –. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matri-sian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913. doi: 10.1158/0008-5472.CAN-14-0155. –. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi J, Yokoyama Y, Kokuryo T, Ebata T, Nagino M. Cells of origin of pancreatic neoplasms. Surg Today. 2018;48:9. doi: 10.1007/s00595-017-1501-2. –. [DOI] [PubMed] [Google Scholar]
- 4.Riva G, Pea A, Pilati C, Fiadone G, Lawlor RT, Scarpa A. Histo-molecular oncogenesis of pancreatic cancer: From precancerous lesions to invasive ductal adenocarcinoma. World J Gastrointest Oncol. 2018;10:317. doi: 10.4251/wjgo.v10.i10.317. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krantz BA, O’Reilly EM. Biomarker-Based Therapy in Pancreatic Ductal Adenocarcinoma: An Emerging Reality? Clin Cancer Res. 2018;24:2241. doi: 10.1158/1078-0432.CCR-16-3169. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perkhofer L, Gout J, Roger E, Kude de Almeida F, Baptista Simões C, Wiesmüller L. DNA damage repair as a target in pancreatic cancer: state-of-the-art and future perspectives. Gut. 2021;70:606. doi: 10.1136/gutjnl-2019-319984. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172:578. doi: 10.1016/j.cell.2018.01.006. et al . –. [DOI] [PubMed] [Google Scholar]
- 8.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med. 2018;8:a031435. doi: 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dreyer SB, Chang DK, Bailey P, Biankin AV. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin Cancer Res. 2017;23:1638. doi: 10.1158/1078-0432.CCR-16-2411. –. [DOI] [PubMed] [Google Scholar]
- 10.Guo M, Peng Y, Gao A, Du C, Herman JG. Epigenetic heterogeneity in cancer. Biomark Res. 2019;7:23. doi: 10.1186/s40364-019-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y, Guo M. Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci. 2020;111:3111. doi: 10.1111/cas.14565. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan W, Herman JG, Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. 2016;8:119. doi: 10.2217/epi.15.84. –. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Sun Y, Zhang X, Shan X, Li J, Yao Y. Three Novel Genetic Variants in the FAM110D, CACNA1A, and NLRP12 Genes Are Associated With Susceptibility to Hypertension Among Dai People. Am J Hypertens. 2021;34:874. doi: 10.1093/ajh/hpab040. et al . –. [DOI] [PubMed] [Google Scholar]
- 14.Hauge H, Patzke S, Aasheim HC. Characterization of the FAM110 gene family. Genomics. 2007;90:14. doi: 10.1016/j.ygeno.2007.03.002. –. [DOI] [PubMed] [Google Scholar]
- 15.Xie M, Cai L, Li J, Zhao J, Guo Y, Hou Z. FAM110B Inhibits Non-Small Cell Lung Cancer Cell Proliferation and Invasion Through Inactivating Wnt/β-Catenin Signaling. Onco Targets Ther. 2020;13:4373. doi: 10.2147/OTT.S247491. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H, Li H, Zhao T, Khan AA, Pan R, Wang S. TSPAN1-elevated FAM110A promotes pancreatic cancer progression by transcriptionally regulating HIST1H2BK. J Cancer. 2022;13:906. doi: 10.7150/jca.66404. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li F, Jang H, Puttabyatappa M, Jo M, Curry TE Jr. Ovarian FAM110C (family with sequence similarity 110C): induction during the periovula-tory period and regulation of granulosa cell cycle kinetics in rats. Biol Reprod. 2012;86:185. doi: 10.1095/biolreprod.112.099259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauge H, Fjelland KE, Sioud M, Aasheim HC. Evidence for the involvement of FAM110C protein in cell spreading and migration. Cell Signal. 2009;21:1866. doi: 10.1016/j.cellsig.2009.08.001. –. [DOI] [PubMed] [Google Scholar]
- 19.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821. doi: 10.1073/pnas.93.18.9821. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia Y, Yang Y, Liu S, Herman JG, Lu F, Guo M. SOX17 antagonizes WNT/β-catenin signaling pathway in hepatocellular carcinoma. Epigenetics. 2010;5:743. doi: 10.4161/epi.5.8.13104. –. [DOI] [PubMed] [Google Scholar]
- 21.Du W, Gao A, Herman JG, Wang L, Zhang L, Jiao S. Methylation of NRN1 is a novel synthetic lethal marker of PI3K-Akt-mTOR and ATR inhibitors in esophageal cancer. Cancer Sci. 2021;112:2870. doi: 10.1111/cas.14917. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X, Cong J, Lin Z, Sun J, Yang B, Li A. Inhibition of HMGB1 Overcomes Resistance to Radiation and Chemotherapy in Nasopharyngeal Carcinoma. Onco Targets Ther. 2020;13:4189. doi: 10.2147/OTT.S239243. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stros M, Cherny D, Jovin TM. HMG1 protein stimulates DNA end joining by promoting association of DNA molecules via their ends. Eur J Biochem. 2000;267:4088. doi: 10.1046/j.1432-1327.2000.01450.x. –. [DOI] [PubMed] [Google Scholar]
- 24.Lange SS, Vasquez KM. HMGB 1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol Carcinog. 2009;48:571. doi: 10.1002/mc.20544. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chikhirzhina E, Starkova T, Beljajev A, Polyanichko A, Tomilin A. Functional Diversity of Non-Histone Chromosomal Protein HmgB1. Int J Mol Sci. 2020;21:7948. doi: 10.3390/ijms21217948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Idoudi S, Bedhiafi T, Pedersen S, Elahtem M, Alremawi I, Akhtar S. Role of HMGB1 and its associated signaling pathways in human malignancies. Cell Signal. 2023;112:110904. doi: 10.1016/j.cellsig.2023.110904. et al . [DOI] [PubMed] [Google Scholar]
- 27.Ma K, Cao B, Guo M. The detective, prognostic, and predictive value of DNA methylation in human esophageal squamous cell carcinoma. Clin Epigenetics. 2016;8:43. doi: 10.1186/s13148-016-0210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davalos V, Esteller M. Cancer epigenetics in clinical practice. CA Cancer J Clin. 2023;73:376. doi: 10.3322/caac.21765. –. [DOI] [PubMed] [Google Scholar]
- 29.Recillas-Targa F.. Cancer Epigenetics: An Overview. Arch Med Res. 2022;53:732. doi: 10.1016/j.arcmed.2022.11.003. –. [DOI] [PubMed] [Google Scholar]
- 30.Cossío FP. Esteller M, Berdasco M. Towards a more precise therapy in cancer: Exploring epigenetic complexity. Curr Opin Chem Biol. 2020;57:41. doi: 10.1016/j.cbpa.2020.04.008. –. [DOI] [PubMed] [Google Scholar]
- 31.Babar Q, Saeed A, Tabish TA, Pricl S, Townley H, Thorat N. Novel epigenetic therapeutic strategies and targets in cancer. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166552. doi: 10.1016/j.bbadis.2022.166552. [DOI] [PubMed] [Google Scholar]
- 32.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502. doi: 10.1038/cr.2011.24. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao A, Guo M. Epigenetic based synthetic lethal strategies in human cancers. Biomark Res. 2020;8:44. doi: 10.1186/s40364-020-00224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bates SE. Epigenetic Therapies for Cancer. N Engl J Med. 2020;383:650. doi: 10.1056/NEJMra1805035. –. [DOI] [PubMed] [Google Scholar]
- 35.Mondal P, Natesh J, Penta D, Meeran SM. Progress and promises of epigenetic drugs and epigenetic diets in cancer prevention and therapy: A clinical update. Semin Cancer Biol. 2022;83:503. doi: 10.1016/j.semcancer.2020.12.006. –. [DOI] [PubMed] [Google Scholar]
- 36.Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19:776. doi: 10.1038/s41573-020-0077-5. –. [DOI] [PubMed] [Google Scholar]
- 37.Setton J, Zinda M, Riaz N, Durocher D, Zimmermann M, Koehler M. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021;11:1626. doi: 10.1158/2159-8290.CD-20-1503. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917. doi: 10.1038/nature03445. et al . –. [DOI] [PubMed] [Google Scholar]
- 39.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913. doi: 10.1038/nature03443. et al . –. [DOI] [PubMed] [Google Scholar]
- 40.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110. doi: 10.1038/nrc.2015.21. –. [DOI] [PubMed] [Google Scholar]
- 41.Murai J, Pommier Y. BRCAness, Homologous Recombination Deficiencies, and Synthetic Lethality. Cancer Res. 2023;83:1173. doi: 10.1158/0008-5472.CAN-23-0628. –. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Yang W, Zhang M, He T, Zhou F, G Herman J. Methylation of TMEM176A, a key ERK signaling regulator, is a novel synthetic lethality marker of ATM inhibitors in human lung cancer. Epigenomics. 2021;13:1403. doi: 10.2217/epi-2021-0217. et al . –. [DOI] [PubMed] [Google Scholar]
- 43.Zhong H, Shi Q, Wen Q, Chen J, Li X, Ruan R. Pan-cancer analysis reveals potential of FAM110A as a prognostic and immunological bio-marker in human cancer. Front Immunol. 2023;14:1058627. doi: 10.3389/fimmu.2023.1058627. et al . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vainio P, Wolf M, Edgren H, He T, Kohonen P, Mpindi JP. Integrative genomic, transcriptomic, and RNAi analysis indicates a potential oncogenic role for FAM110B in castration-resistant prostate cancer. Prostate. 2012;72:789. doi: 10.1002/pros.21487. et al . –. [DOI] [PubMed] [Google Scholar]
- 45.Reeves R. High mobility group (HMG) proteins: Modulators of chromatin structure and DNA repair in mammalian cells. DNA Repair (Amst) 2015;36:122. doi: 10.1016/j.dnarep.2015.09.015. –. [DOI] [PubMed] [Google Scholar]
- 46.Yuan F, Gu L, Guo S, Wang C, Li GM. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J Biol Chem. 2004;279:20935. doi: 10.1074/jbc.M401931200. –. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Prasad R, Wilson SH. HMGB1: roles in base excision repair and related function. Biochim Biophys Acta. 2010;1799:119. doi: 10.1016/j.bbagrm.2009.11.008. - [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawchuk DJ, Mansilla-Soto J, Alarcon C, Singha NC, Langen H, Bianchi ME. Ku70/Ku80 and DNA-dependent protein kinase catalytic subunit modulate RAG-mediated cleavage: implications for the enforcement of the 12/23 rule. J Biol Chem. 2004;279:29821. doi: 10.1074/jbc.M403706200. et al . –. [DOI] [PubMed] [Google Scholar]
- 49.Feng GS. Conflicting roles of molecules in hepatocarcinogenesis: paradigm or paradox. Cancer Cell. 2012;21:150. doi: 10.1016/j.ccr.2012.01.001. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Li N, Zhang X, Wu H, Yu S. Hexavalent chromium caused DNA damage repair and apoptosis via the PI3K/AKT/FOXO1 pathway triggered by oxidative stress in the lung of rat. Ecotoxicol Environ Saf. 2023;267:115622. doi: 10.1016/j.ecoenv.2023.115622. [DOI] [PubMed] [Google Scholar]
- 51.Fang L, Choudhary S, Zhao Y, Edeh CB, Yang C, Boldogh I. ATM regulates NF-κB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res. 2014;42:8416. doi: 10.1093/nar/gku529. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramachandiran S, Adon A, Guo X, Wang Y, Wang H, Chen Z. Chromosome instability in diffuse large B cell lymphomas is suppressed by activation of the noncanonical NF-κB pathway. Int J Cancer. 2015;136:2341. doi: 10.1002/ijc.29301. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jenni R, Chikhaoui A, Nabouli I, Zaouak A, Khanchel F, Hammami-Ghorbel H. Differential Expression of ATM, NF-KB, PINK1 and Foxo3a in Radiation-Induced Basal Cell Carcinoma. Int J Mol Sci. 2023;24:7181. doi: 10.3390/ijms24087181. et al . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu T, Zhang W, Yang G, Li H, Chen Q, Song R. HMGB1 overex-pression as a prognostic factor for survival in cancer: a meta-analysis and systematic review. Oncotarget. 2016;7:50417. doi: 10.18632/oncotarget.10413. et al . –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013. doi: 10.1038/s41419-020-03221-2. et al . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao Y, Yi J, Tao L, Huang G, Chu X, Song H. Wnt signaling induces radioresistance through upregulating HMGB1 in esophageal squamous cell carcinoma. Cell Death Dis. 2018;9:433. doi: 10.1038/s41419-018-0466-4. et al . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ke S, Zhou F, Yang H, Wei Y, Gong J, Mei Z. Downregulation of high mobility group box 1 modulates telomere homeostasis and increases the radiosensitivity of human breast cancer cells. Int J Oncol. 2015;46:1051. doi: 10.3892/ijo.2014.2793. et al . –. [DOI] [PubMed] [Google Scholar]
- 58.Kantidze OL, Velichko AK, Luzhin AV, Petrova NV, Razin SV. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer. 2018;4:755. doi: 10.1016/j.trecan.2018.09.007. –. [DOI] [PubMed] [Google Scholar]
- 59.Gupta N, Huang TT, Horibata S, Lee JM. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor-resistant cancer. Pharmacol Res. 2022;178:106162. doi: 10.1016/j.phrs.2022.106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klomp JE, Lee YS, Goodwin CM, Papke B, Klomp JA, Waters AM. CHK1 protects oncogenic KRAS-expressing cells from DNA damage and is a target for pancreatic cancer treatment. Cell Rep. 2021;37:110060. doi: 10.1016/j.celrep.2021.110060. et al . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laquente B, Lopez-Martin J, Richards D, Illerhaus G, Chang DZ, Kim G. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer. 2017;17:137. doi: 10.1186/s12885-017-3131-x. et al . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material