

Abstract
BACKGROUND
Sudden unexpected death in epilepsy (SUDEP) is a fatal complication experienced by otherwise healthy epilepsy patients. Dravet syndrome (DS) is an inherited epileptic disorder resulting from loss of function of the voltage-gated sodium channel, NaV 1.1, and is associated with particularly high SUDEP risk. Evidence is mounting that NaVs abundant in the brain also occur in the heart, suggesting that the very molecular mechanisms underlying epilepsy could also precipitate cardiac arrhythmias and sudden death. Despite marked reduction of NaV 1.1 functional expression in DS, pathogenic late sodium current (INa,L) is paradoxically increased in DS hearts. However, the mechanisms by which DS directly impacts the heart to promote sudden death remain unclear.
OBJECTIVES
In this study, the authors sought to provide evidence implicating remodeling of Na+ -and Ca2+ -handling machinery, including Nav 1.6 and Na+/Ca2+exchanger (NCX) within transverse (T)-tubules in DS-associated arrhythmias.
METHODS
The authors undertook scanning ion conductance microscopy (SICM)-guided patch clamp, super-resolution microscopy, confocal Ca2+ imaging, and in vivo electrocardiography studies in Scn1a haploinsufficient murine model of DS.
RESULTS
DS promotes INa,L in T-tubular nanodomains, but not in other subcellular regions. Consistent with increased NaV activity in these regions, super-resolution microscopy revealed increased NaV 1.6 density near Ca2+release channels, the ryanodine receptors (RyR2) and NCX in DS relative to WT hearts. The resulting INa,L in these regions promoted aberrant Ca2+ release, leading to ventricular arrhythmias in vivo. Cardiac-specific deletion of NaV 1.6 protects adult DS mice from increased T-tubular late NaV activity and the resulting arrhythmias, as well as sudden death.
CONCLUSIONS
These data demonstrate that NaV 1.6 undergoes remodeling within T-tubules of adult DS hearts serving as a substrate for Ca2+ -mediated cardiac arrhythmias and may be a druggable target for the prevention of SUDEP in adult DS subjects.
Keywords: Dravet syndrome, NaV1.6, sodium channels, sudden cardiac death, sudden unexpected death in epilepsy
Sudden unexpected death in epilepsy (SUDEP) is a fatal complication, occurring in patients with epilepsy who were otherwise healthy. Patients with inherited epileptic disorders, such as Dravet syndrome (DS), are at a particularly high risk of sudden death.1 At the subcellular level, DS is characterized by the loss of function or expression of a specific voltage-gated sodium channel (NaV), NaV1.1, encoded by the SCN1A gene.2 In SUDEP cases where an autopsy is performed, no clear cause of death is found.3 SUDEP shares this feature with cases of sudden cardiac death—unexpected death resulting from cardiac arrhythmias. Prior work on the cardiovascular contribution to SUDEP has largely focused on autonomic modulation of cardiac function.4,5 However, therapies based on this notion have demonstrated limited efficacy in preventing SUDEP.6 NaVs expressed at high levels in the brain (commonly labeled “neuronal-type” NaVs) are also expressed in cardiomyocytes.7–10 Further, DS-causing mutations in SCN1A have been shown to alter cardiomyocyte function in both preclinical animal models and induced pluripotent stem cells derived from patients with DS.11,12 Taken together, these studies suggest that the molecular mechanisms underlying the epileptic condition in the brain may also directly affect the heart, independent of autonomic control, predisposing the epileptic heart to arrhythmias and sudden cardiac death.
Although DS is characterized by a marked reduction of functional NaV1.1, previous studies have demonstrated a paradoxical increase in the late component of the voltage-gated sodium current in these hearts (INa,L).11 At present, however, the precise mechanisms by which NaV1.1 loss of function leads to these paradoxical changes in INa,L and how these, in turn, contribute to arrhythmia remain unknown.11,13 Further, although many investigations of SUDEP in DS have focused on death in the pediatric population, which may reflect the availability of preclinical models with a specific focus on early mortality rather than the clinical scenario in many DS patients. There is a precipitous loss of life among DS patients not only during early childhood but also during late adolescence and early adulthood.14–16 The second wave of mortality associated with DS in early adulthood remains understudied.
In this study, we directly address this gap in knowledge using an adult (>100 days of age) murine model of DS to study how Scn1a haploinsufficiency contributes to cardiac arrhythmias. Mechanistically, we demonstrate that DS results in an increase in functional NaV1.6 expression within transverse tubule (T-tubule) nanodomains of cardiomyocytes, which promotes ectopic Ca2+ release at the cellular level and arrhythmias in vivo. We further demonstrate that a cardiac-specific reduction in Scn8a (encoding NaV1.6) is sufficient to reduce arrhythmia burden and improve survival in DS mice. Collectively, these data strongly suggest that maladaptive remodeling of NaV1.6 within the heart results in sudden death, which contributes to mortality in adult DS. These results highlight cardiac NaV1.6 as a potential therapeutic target for preventing sudden death in DS.
METHODS
A detailed description of the methods is provided in the Supplemental Appendix.
STATISTICS.
Experiments and analyses were conducted in an unblinded fashion. We performed all statistical analyses using GraphPad Prism 9.4.1. To test for data normality, we performed the Shapiro-Wilk test. To compare 2 independent data sets, we performed the unpaired 2-tailed Student’s t-test or the Mann-Whitney U test where appropriate, depending on the normality of the data. Differences in distributions were tested with the 2-sample Kolmogorov-Smirnov test. A P value of <0.05 was considered significant. To compare more than 2 data sets, we performed an ordinary 1-way analysis of variance or Kruskal-Wallis test depending on the normality of the data. To correct for multiple comparisons, we performed a post hoc analysis with the original method of Benjamini and Hochberg. A q value of <0.05 was considered significant. All data in this report are presented as the mean ± SEM or as box-and-whisker plots. For survival analysis, curves were compared using the log-rank (Mantel-Cox) test and corrected for multiple comparisons post hoc via Tukey’s method.
STUDY APPROVAL.
All animal procedures were approved by Institutional Animal Care and Use Committee at The Ohio State University and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (publication no. 85–23, revised 2011).
RESULTS
CARDIAC-SPECIFIC ABLATION OF SCN8A REDUCES ALL-CAUSE MORTALITY AND ARRHYTHMIAS IN DS.
Consistent with previous findings in mice with DS, we observed a ~20% mortality in our cohort within the first 60 days.17 Notably, a second wave of sudden death was apparent after 100 days (Figure 1A), consistent with mortality trends observed in patients with DS when adjusted for species-dependent age differences.14–16,18 Importantly, this mortality phenotype was independent of the mouse strain used in this study (Supplemental Figure 1). Previous studies have demonstrated that reducing NaV1.6 (encoded by Scn8a) can ameliorate seizure burden and prevent sudden death in DS.19–22 In line with these findings, we observed that Scn8a haploinsufficient DS mice (DSxNaV1.6Het) are protected from sudden death (Figure 1A, Supplemental Figure 1). To test for a role of cardiac NaV1.6 in DS arrhythmia development, we crossed DS mice with a previously validated cardiac-specific NaV1.6-knockout mouse (DSxcNav1.6KO).23,24 This strategy reduced mortality relative to DS (Figure 1A, Supplemental Figure 1). In contrast to global reduction in NaV1.6 expression (DSxNaV1.6Het), cardiac-specific NaV1.6 deletion (DSxcNaV1.6KO) did not confer protection from sudden death within the first 60 days but substantially mitigated sudden death in the adult population. Our findings thus suggest mechanistic differences in mortality in early childhood vs adulthood in DS, implicating a primary cardiac defect in excitability and consequent sudden death as a driver of mortality in the latter subpopulation. Consistent with previous findings,11 adult DS mice displayed QT-interval prolongation relative to the wild type (WT), an established marker of arrhythmia susceptibility (Figures 1B and 1C).25 Conversely, adult DSxNaV1.6Het and DSxcNaV1.6KO evidenced QT intervals comparable to those observed in WT mice, suggesting that changes in functional expression of cardiac NaV1.6 contribute to the long-QT-interval phenotype observed in DS (Figure 1C). Our observation of QT-interval prolongation, coupled with reports of SUDEP during exercise14,15 and sympathetic dominance in the DS population26 prompted us to further investigate the arrhythmia susceptibility of adult DS mice with an in vivo catecholamine challenge. Our studies show that adult DS mice have a significantly higher catecholamine-induced arrhythmia burden than WT mice. Importantly—and consistent with the hypothesis that changes in functional expression of cardiac NaV1.6 in DS contribute to cardiac arrhythmias—arrhythmia burden during catecholamine challenge was significantly reduced in both adult DSxNaV1.6Het and DSxcNaV1.6KO mice (Figures 1D and 1E). Taken together, our in vivo findings suggest that NaV1.6 is a critical component of arrhythmogenesis that contributes to sudden death in DS mice.
FIGURE 1. DS Is Associated With an Increase in Mortality and Predisposition to Arrhythmia Development, Which Are Mitigated by Reduction in NaV1.6.

(A) Kaplan-Meier survival curves for WT (n = 163), DS (n = 238), DSxNaV1.6Het (n = 68), and DSxcNaV1.6KO (n = 92) mice. The data from all strains tested for WT and DS were pooled. *P < 0.001 relative to WT, &P < 0.001 relative to DS. (B) Representative ECG from WT and DS mice. (C) QT-interval prolongation in DS, mitigated by NaV1.6 reduction (WT: n = 11; DS: n = 18; DSxNaV1.6Het: n = 17; DSxcNaV1.6KO: n = 29 mice). (D) Representative ECGs following catecholamine challenge with caffeine and epinephrine illustrate SR followed by VT. (E) Summary data showing significant increase in arrhythmia burden in DS mice (WT: n = 38; DS: n = 59; DSxNaV1.6Het: n = 17; DSxcNaV1.6KO: n = 40). Arrhythmia scores: 0 = no abnormalities; 1 = premature ventricular complexes observed; 2 = bigeminy; 3 = VT; 4 = ventricular fibrillation. *q < 0.05, ****q < 0.0001 relative to DS. DS = Dravet syndrome; ECG = electrocardiogram; NaV = voltage-gated sodium channel; cNaV1.6KO = cardiac-specific NaV1.6 deletion; NaV1.6Het = Scn8a haploinsufficient; SR = sinus rhythm; VT = ventricular tachycardia; WT = wild type.
NaV1.6 CONTRIBUTES TO LATE NaV ACTIVITY WITHIN CARDIAC T-TUBULAR NANODOMAINS.
Recent reports have linked DS with a paradoxical increase in INa,L,11,12 which may contribute to the increased arrhythmia burden, as observed in our DS mice (Figures 1D and 1E). To further investigate this, we measured INa,L in DS hearts. Consistent with previous reports, we observed a significant increase in whole-cell INa,L in cardiomyocytes isolated from DS mice (Figure 2A, Supplemental Figure 2A). In contrast, no changes were observed in peak INa (Figure 2B), INa activation, or INa inactivation kinetics (Supplemental Figures 2B and 2C).
FIGURE 2. Whole-Cell Patch Clamp Recordings Reveal Increased Late INa in DS Cardiomyocytes, Which Are Mitigated by Reduction in NaV1.6.

(A) Representative late INa recordings from cardiomyocytes of each experimental group; increased late INa in DS cardiomyocytes is mitigated by NaV1.6 reduction (WT: n = 10 cells, 4 mice; DS: n = 15 cells, 8 mice; DSxNaV1.6Het: n = 13 cells, 6 mice; DSxcNaV1.6KO: n = 9 cells, 5 mice). (B) Peak INa (WT: n = 5 cells, 3 mice; DS: n = 8 cells, 7 mice; DSxNaV1.6Het: n = 9 cells, 5 mice; DSxcNaV1.6KO: n = 12 cells, 4 mice). *q < 0.05, **q < 0.01 relative to DS. INa,L = voltage-gated sodium current; other abbreviations as in Figure 1.
The heart expresses various NaV isoforms with distinct patterns of subcellular distribution. The predominant cardiac NaV isoform, NaV1.5, is heavily enriched at cardiac cell-cell junctions, known as intercalate discs (IDs), with some also localized to the lateral membrane and T-tubules (Figure 3A, Supplemental Figure 3).24,27,28 Meanwhile, NaV1.1 and NaV1.6 are predominantly located near Ca2+-handling proteins within T-tubules (Figure 3A).8,24,29,30 To gain insight into the molecular identity and distribution of the NaV responsible for the increased INa,L, we first directly assessed NaV activity using cell-attached patch clamp at the ID (Figure 3B). There were no significant differences in NaV activity at the ID between WT and DS cardiomyocytes (Figure 3C). One key limitation of the conventional cell-attached patch clamp approach is the inability to place the pipette at specific subcellular regions and thereby discriminate between subcellular features. Therefore, to examine NaV activity at the lateral membrane crest as well as at the T-tubules, we used the scanning ion-conductance microscopy (SICM)–guided “smart” patch clamp approach.24 Similar to the ID, NaV activity at the crests of the lateral membrane (Figure 3D) did not reveal significant differences in late NaV activity between DS and WT (Figure 3E). Conversely, smart patch recordings from T-tubular regions (Figure 4) revealed increased late NaV activity in DS cardiomyocytes relative to WT cardiomyocytes (Figure 4A), which was significantly reduced in both DSxNaV1.6Het and DSxcNaV1.6KO (Figure 4B). Together, these subcellular findings suggest that NaV1.6 within T-tubules is the main source of excess INa,L in DS cardiomyocytes.
FIGURE 3. No Changes in Late NaV Activity Within the Intercalated Disc or the Lateral Membrane Crests of DS Cardiomyocytes.
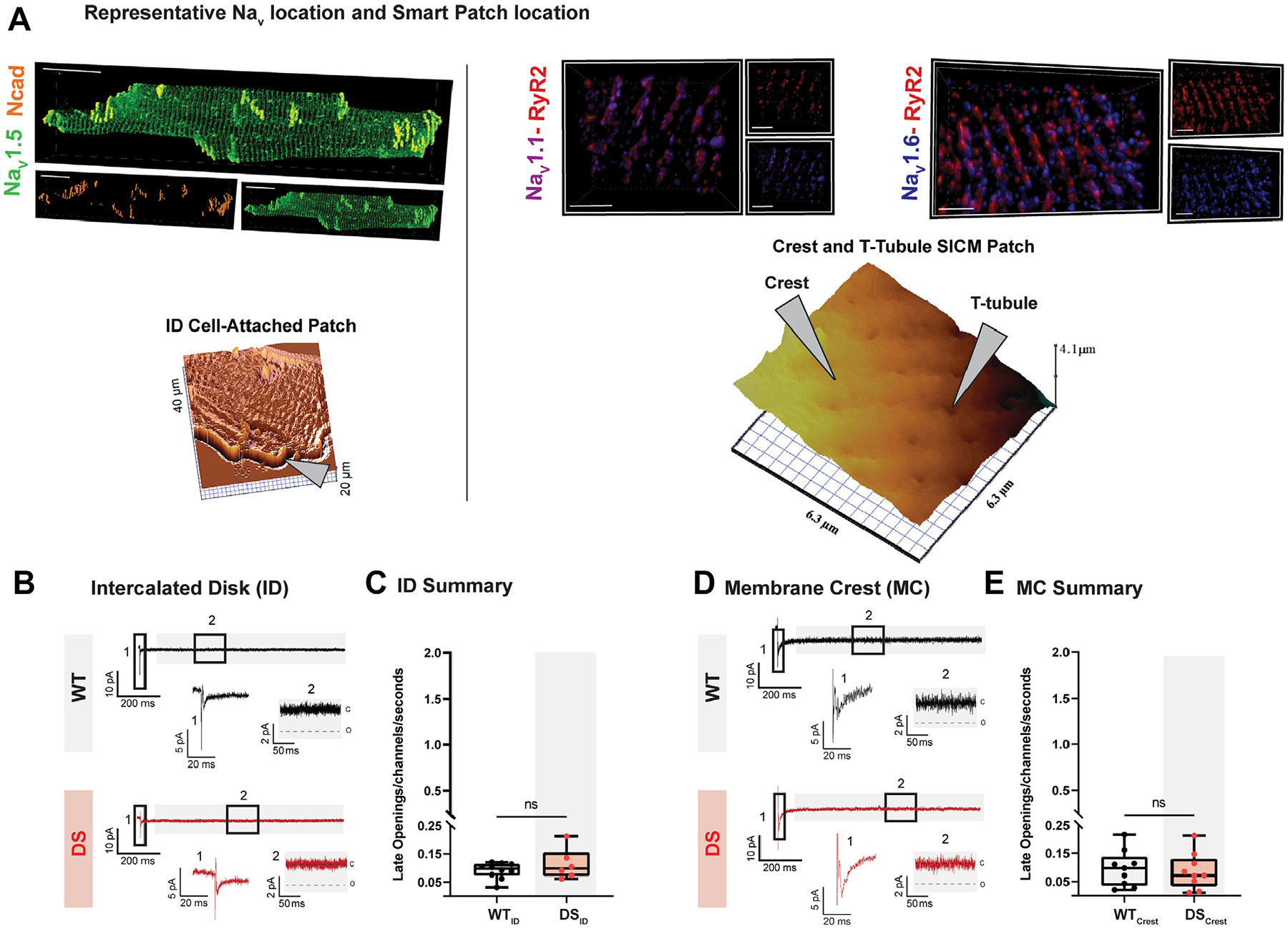
(A, top) Representative immunofluorescence confocal microscopy images of NaV (NaV1.5: green; NaV1.1: purple; NaV1.6: blue) distributions relative to ID (N-cadherin: orange) and T-tubular (RyR2: red) regions and (bottom) representative scanning ion conductance microscopy images of cardiomyocyte topography (specific regions indicated by markers; left ID, right crest and T-tubule). Scale bars: (left) 20 μm and (right) 2 μm. (B) Cell-attached patch INa recordings from the ID. (D) Scanning ion conductance microscopy-guided “smart” patch INa recordings from the lateral membrane crest. (C and E) Summary data of late activity at the ID (WT: n = 10 cells, 3 mice; DS: n = 6 cells, 3 mice) and later membrane crest (WT: n = 9 cells, 3 mice; DS: n = 9 cells, 3 mice). ID = intercalate disc; ns = not significant; RyR2 = ryanodine receptors; T-tubule = transverse tubule; other abbreviations as in Figures 1 and 2.
FIGURE 4. NaV1.6 Contributes to Increased Late NaV Activity Within the T-Tubules of DS Cardiomyocytes.

(A) Representative scanning ion conductance microscopy-guided “smart” patch INa recordings from T-tubule openings. (B) Summary data of late activity at the T-tubules (WT: n = 9 cells, 5 mice; DS: n = 13 cells, 7 mice; DSxNaV1.6Het: n = 13 cells, 5 mice; DSxcNaV1.6KO: n = 7 cells, 4 mice). *q < 0.05, **q < 0.01, ***q < 0.001 relative to DS. Abbreviations as in Figures 1 to 3.
NaV1.6 IS REMODELED WITHIN T-TUBULAR CA2+-HANDLING NANODOMAINS IN DS.
Our findings from subcellular electrophysiology assays suggest that the paradoxical increase in INa,L in DS is a result of increased NaV1.6 activity within the T-tubules. Consistently, Western blots of DS hearts revealed an increase in total NaV1.6 protein relative to WT (Figures 5A and 5B, Supplemental Figure 4A) without concomitant alterations to the expression or localization of the predominant cardiac NaV isoform, NaV1.5 (Supplemental Figures 4B and 4C and 3, respectively). Although supportive of our hypothesis, Western blotting provides no information on the subcellular region that evidences change in NaV1.6 expression. Based on our SICM-guided smart patch experiments, we expect Ca2+-handling machinery-rich regions of T-tubules to harbor these changes. Therefore, to confirm the subcellular localization of changes in NaV1.6 expression, we performed proximity ligation assays. Our data demonstrate an increase in NaV1.6-ryanodine receptors (RyR2) and NaV1.6-sodium/calcium exchanger (NCX) puncta in DS mice relative to WT (Supplemental Figures 5A and 5B). To further assess NaV1.6 nanodomain spatial organization, we performed superresolution imaging, specifically stochastic optical reconstruction microscopy, which enables assessment of protein distribution in cardiomyocytes with ~20-nm lateral and <50-nm axial resolution.31 Analysis with a previously validated machine learning-based cluster analysis tool (STORM-RLA31) revealed that in DS hearts, NaV1.6 is in closer proximity to both RyR2 and NCX than in WT (Figure 6). These results suggest that Ca2+-handling nanodomains within T-tubules of DS hearts are further enriched with NaV1.6, which may provide a structural substrate for the aberrant Ca2+ release that underlies in vivo arrhythmias and sudden death.
FIGURE 5. Increased NaV1.6 Expression in DS Hearts.

(A) Representative Western blots of NaV1.6 and GAPDH (loading control) in WT and DS hearts. (B) Summary Western blot data (n = 6). *P < 0.05 relative to DS. GAPDH = glyceraldehyde 3-phosphate dehydrogenase; other abbreviations as in Figures 1 to 3.
FIGURE 6. STORM Reveals Enhanced NaV1.6 Clustering in Close Proximity to NCX and RyR2 in DS Hearts.

Representative stochastic optical reconstruction microscopy (STORM) images from (A and C) WT and (B and D) DS hearts immunolabeled for RyR2 (top, red), NaV1.6 (top and bottom, blue), and NCX (bottom, yellow). Protein distribution measured as percentage of molecules (E: NaV1.6 to RyR2; G: NaV1.6 to NCX) and percentage of clusters (F: NaV1.6 to RyR2; H: NaV1.6 to NCX) (n = 5 images/heart from 3 WT hearts and 3 DS hearts). Differences in distributions and medians were tested with the 2-sample Kolmogorov-Smirnov test and Wilcoxon rank-sum test test, respectively (P > 0.05: ns; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). (I) Probability distribution and (J) cumulative distribution plots of NaV1.6 clusters relative to RyR2 in WT and DS mice. (K) Probability distribution and (L) cumulative distribution plots of NaV1.6 clusters relative to NCX in WT and DS mice (n = 5 images/heart from 3 WT hearts and 3 DS hearts). Scale bars: (A, left) 7 μm, (A, right) 2 μm, (B, left) 7 μm, (B, right) 2 μm, (C, left) 5 μm, (C, right) 1 μm, (D, left) 6 μm, and (D, right) 1 μm. NCX = sodium/calcium exchanger; other abbreviations as in Figures 1 to 3.
Next, we performed Monte Carlo simulations to gain insight into the potential mechanism responsible for changes in NaV1.6 distribution vis-à-vis Ca2+-handling machinery within the T-tubules. Using a model that assumes channel-channel interactions via passive biophysics without recourse to active biochemical processes (see Methods), we were able to replicate the pattern of clustering between NCX and NaV1.6 (Figure 7) that we observed experimentally in WT hearts (Figure 6). Importantly, simulations of DS (partial NaV1.1 reduction) result in an increase of NaV1.6 near NCX (Figures 7B to 7D), and the distribution of NCX near NaV1.6 was unchanged, paralleling the experimental findings (Figure 6, Supplemental Figure 6). The computational results, therefore, led us to hypothesize that channel-channel functional interactions may be sufficient to explain the compensatory changes in NaV1.6 cluster distribution in DS without recourse to other compensatory mechanisms. To test this further, we assessed Scn8a transcript levels in WT and DS hearts. Consistent with the hypothesis derived from numerical simulations, we did not detect significant changes in Scn8a messenger RNA levels between WT and DS hearts (Supplemental Figure 4D). Furthermore, in line with previous observations,32 these results together suggest the hypothesis that channel-channel functional interaction between NaV1.6 and NaV1.1 may help shape the cluster size of NaV1.6, which in turn can effect nanodomain ionic composition.
FIGURE 7. Monte Carlo Clustering Simulations Predict That Channel-Channel Interactions Are Sufficient to Explain Enhanced NaV1.6 Clustering in DS.

(A) Representative map showing simulated NCX, NaV1.6, and NaV1.1 clustering. (B) Representative map of NCX, NaV1.6, and NaV1.1 clustering in DS. Note the smaller NaV1.1 cluster. (C) Distribution and cumulative distribution plots showing the distribution of NaV1.6 relative to NCX. Reduction in NaV1.1 in DS shifts NaV1.1 distribution to the left (red), without other changes in expression or NaV1.6-NCX interactions. (D) Distribution and cumulative distribution plots showing the distribution of NCX relative to NaV1.6. Reduction in NaV1.1 in DS does not change this distribution. The data shown in C and D are pooled from 5 simulations for each case. Abbreviations as in Figures 1 to 3 and 6.
NaV1.6 REMODELING IN DS PROMOTES PROARRHYTHMIC CA2+ RELEASE.
To examine the functional consequences of NaV1.6 remodeling within Ca2+-handling nanodomains on Na+/Ca2+ exchange and proarrhythmic Ca2+ release, we performed confocal line scan Ca2+ imaging. Confocal Ca2+ imaging revealed a significant increase in the frequency, amplitude, and duration of Ca2+ sparks in DS cardiomyocytes relative to WT (Figures 8A and 8B, Supplemental Figure 7D), without affecting other Ca2+ spark parameters or sarcoplasmic reticulum Ca2+ load (Supplemental Figure 7). Likewise, we observed a significant increase in spontaneous Ca2+ wave frequency (Figure 8C). Consistent with the involvement of NaV1.6 in proarrhythmic Ca2+ release, Ca2+ spark and Ca2+ wave frequency were reduced in DSxNaV1.6Het and DSxcNaV1.6KO hearts relative to DS. Together, our data suggest that structural remodeling of NaV1.6 within Ca2+-handling-rich nanodomains underlies the development of cardiac arrhythmias that contributes to sudden death in adult DS mice, presenting NaV1.6 as a viable therapeutic target.
FIGURE 8. NaV1.6 Remodeling Within T-Tubules in DS Contributes to Aberrant Ca2+ Release.

(A) Representative Ca2+ line scans of isolated cardiomyocytes (top) and corresponding Ca2+ transients (bottom). (B) Summary of frequency and distribution of Ca2+ sparks (WT: n = 121 cells, 7 mice; DS: n = 102 cells, 13 mice; DSxNaV1.6Het: n = 211 cells, 7 mice; DSxcNaV1.6KO: n = 126 cells, 11 mice) and (C) Ca2+ waves. WT: n = 123 cells, 7 mice; DS: n = 181 cells, 15 mice; DSxNaV1.6Het: n = 103 cells, 4 mice; DSxcNaV1.6KO: n = 152 cells, 11 mice. *q < 0.05 relative to DS. Abbreviations as in Figures 1 to 3.
DISCUSSION
Patients with inherited epileptic disorders, such as DS, are at a particularly high risk of sudden death.1 Several population-level studies investigating SUDEP in epileptic populations have indicated cardiorespiratory arrest as one of the leading causes of death.33,34 This has been supported by findings in murine models of DS.5,35,36 It is important to note that these studies have focused on postictal SUDEP in the young DS subpopulation. Similarly, previous studies investigating the cardiac component of SUDEP have focused on how an epileptic brain may dysregulate the heart via autonomic input during the peri-ictal period.4,5,11 To our knowledge, studies investigating the causes for non-seizure-related sudden death in adult DS models are lacking. Importantly, however, many epilepsy-causing mutations occur in proteins that are endogenously expressed not only in the brain but also in the heart. This includes several voltage-gated ion channels integral to ionic homeostasis, including multiple Nav isoforms.8,9,24,29,30 Based on this rationale, we postulated that sudden death associated with DS is, in part, a consequence of direct dysregulation of cardiac physiology by mutated proteins expressed in the heart. Recently, we and others have demonstrated that alterations in tetrodotoxin-sensitive NaVs (NaV1.1 and NaV1.6) directly promote cardiac arrhythmias in vivo by disrupting Na+/Ca2+ cycling at the subcellular level.7,24,29,37–40 Here, we show, for the first time to our knowledge, that Scn1a haploinsufficiency results in structural remodeling of NaV1.6 within cardiac T-tubules, a microdomain rich in Na+/Ca2+-handling proteins. Maladaptive remodeling of NaV1.6 promotes excessive NaV activity in close proximity to NCX, resulting in ectopic Ca2+ influx, which then triggers arrhythmogenic Ca2+ release from intracellular stores. Ultimately, NaV1.6 remodeling predisposes the animal to arrhythmia development and sudden death. We further demonstrate that cardiac-specific deletion of NaV1.6 is sufficient to significantly mitigate both arrhythmia and mortality in adult DS mice.
SUDDEN CARDIAC DEATH IN DS.
Clinically, sudden death in patients with DS first manifests in early childhood (<4 years old), followed by a second peak in mortality during the transition from late adolescence into early adulthood (~18 years old).14–16 With adjustment for age differences between the 2 species,18 the DS murine models used in this study phenocopy both the pediatric and early-adult sudden death trends in clinical populations (Figure 1A, Supplemental Figure 1). Historically, the abnormally high incidence of sudden death in DS pediatric populations has been attributed to postictal generalized electroencepha-logram suppression,41 respiratory arrest,35,42 acute cardiac bradycardia followed by asystole,5,11 or hyperthermia-induced ventricular tachycardia.13 Because sudden death in adult DS models has been understudied, little is known about the potential etiologies of sudden death in adult DS populations.14–16
Despite not having a clear understanding of how death occurs in adult DS, multiple studies have demonstrated that suppressing NaV1.6 can reduce mortality in murine models of DS.19–22 However, it remains unclear to what extent NaV1.6 inhibition can rescue cardiac dysfunction in DS models. To answer this question, we genetically ablated NaV1.6 from the hearts of our DS mouse model (DSxcNaV1.6KO). Early in life, the mortality in our DSxcNaV1.6KO model parallels DS mice. Unlike the DS mice, however, our DSxcNaV1.6KO mice do not experience a second wave of death in adulthood. Our data thus support the notion that DS-related deaths during adulthood may be, in part, a direct consequence of endogenous cardiac dysfunction, whereas those in infancy and childhood are likely caused by defects in the central or autonomic nervous system and their effect on downstream organ systems.35,42
In support of the hypothesis that cardiac death contributes to sudden death in DS, it is well established that DS results in altered cardiac physiology.5,11–13 At the level of an isolated cardiomyocyte, there is an increase in cardiomyocyte excitability and INa,L, both of which would predispose the heart to the development of arrhythmias.11 In the DS population, QT-interval prolongation has been documented, though it must be noted that this cohort did not evidence major ventricular arrhythmias during the peri-ictal period.43 Additionally, DS mice in our study were not observed via video monitoring; therefore, the precise mechanism of death remains unknown. It is therefore difficult to ascertain whether death in DS mice was preceded by seizure, post-ictal apnea and bradycardia, cardiac tachyarrhythmia, or another sentinel event. However, the significant increase in survival that accompanies cardiac-specific NaV1.6 ablation suggests that the heart contributes significantly to mortality in our adult DS models (Figure 1).
One note of particular interest is that population-level studies of DS-associated SUDEP reveal accidental death by drowning as a recurrent etiology.14,15 These reports hint that exercise and physical exertion coupled with increased sympathetic tone may precipitate sudden death in these patients. Consistent with this hypothesis, prior studies have demonstrated sympathetic dominance in adult DS patients, secondary to a reduction in parasympathetic tone.26,43,44 Further, sympathetic dominance has long been linked to increased susceptibility to lethal ventricular arrhythmias that underlie sudden cardiac death.45 To test whether adult DS mice are more susceptible to sympathetically induced arrhythmias, we subjected anesthetized mice to an in vivo catecholamine challenge and observed a significant increase in arrhythmia burden associated with DS. Further, consistent with our overall mortality data, we found that a cardiac-specific deletion of NaV1.6 significantly decreased the arrhythmia burden in these mice.
Other causes of death, such as respiratory dysfunction before bradycardia, have been proposed as possible contributors to SUDEP.35,42 These studies suggest that brainstem respiratory centers could be affected to compromise respiration under interictal conditions. However, evidence from mice with Scn1a haploinsufficiency in forebrain gamma-aminobutyric acid interneurons points to dysfunction in the autonomic nuclei in the brainstem that regulate cardiac and other essential functions as drivers of primary bradycardia promoting SUDEP in DS mice.5 These findings collectively suggest that multifactorial etiologies may contribute to SUDEP in DS. Importantly, our study adds remodeling of NaV1.6 channels in the heart as an additional key contributor to sudden death in DS.
DYSREGULATION OF CARDIAC NaV1.6 UNDERLIES PROARRHYTHMIC CA2+ RELEASE.
A majority of previous work investigating the cardiovascular contribution to SUDEP has focused on altered autonomic modulation of the heart. However, emerging evidence suggests that the very molecular mechanisms that underlie epileptic disorders could also directly affect the heart, leading to arrhythmias and sudden death.46 Until now, however, the mechanisms by which DS may promote sudden cardiac death remained unclear. To gain mechanistic insight into DS-associated cardiac dysfunction, we used an SICM-guided smart patch to study NaV activity in different nanodomains of the cardiomyocyte. Doing so, we determined that there was an increase in NaV activity within the T-tubules but not in other subcellular compartments, such as the lateral membrane crest or the intercalated disc.
Prior work by us and others has demonstrated that NaV clusters within the T-tubules are composed mainly of NaV1.1 and NaV1.6.8–10,23,37 Given that DS is associated with a functional reduction in NaV1.1, our data suggest that increased T-tubular NaV1.6, although apparently small but in such a location, exerts a potent functional effect, which gives rise to the paradoxically increased late NaV activity within these nanodomains. Excess late NaV activity, in turn, engages NCX and results in spontaneous Ca2+ waves, which underlie cardiac arrhythmias.
Notably, using superresolution microscopy and electrophysiology, coupled with computational modeling and protein and transcript analysis, we demonstrate that NaV1.6 increases in proximity to Ca2+-handling machinery (RyR2 and NCX), compensating for the loss of NaV1.1 in DS (Figure 9). This notion is consistent with previous experimental observations that have demonstrated an inhibitory effect of NaV1.1 on NaV1.6 functional expression.32 It is important to note that changes in T-tubular NaV1.6 could be a result of enhanced messenger RNA trafficking and local translation, reduced NaV1.6 degradation, or both and require further investigation. Importantly, the relevance of NaV1.6 in the arrhythmogenic process was further confirmed through germline NaV1.6 reduction and cardiac-specific NaV1.6 knockout, both of which prevented ectopic Ca2+ release and lowered the resulting arrhythmias and sudden death in DS.
FIGURE 9. Remodeling of NaVs Within the Ca2+-Handling Machinery–Rich Regions of Cardiac T-Tubules in DS Serves as a Substrate for Arrhythmogenic Ca2+ Release.

Reduction in NaV1.1 evidenced in DS facilitates the repopulation of cardiac sarcolemma (SL) sites typically occupied by NaV1.1 by NaV1.6. The resulting increase in subsarcolemmal Na+ through NaV1.6 promotes Na+/Ca2+ exchange and intracellular Ca2+ loading, thereby setting a stage for aberrant Ca2+ release from the sarcoplasmic reticulum (SR) via ryanodine receptors (RyR). Abbreviations as in Figures 1 to 3 and 6.
CONCLUSIONS
We provide here the first direct evidence, to our knowledge, that dysregulation of the heart significantly contributes to death in this adult murine model of DS. We demonstrate that Scn1a haploinsufficiency results in remodeling of NaV1.6 in cardiac T-tubules, which drives aberrant Ca2+ release, promotes arrhythmogenesis, and ultimately results in untimely death (Central Illustration). We further demonstrate that cardiac-specific ablation of Scn8a is sufficient to rescue the cardiac arrhythmia phenotype and consequent mortality observed in adult DS mice.
CENTRAL ILLUSTRATION. Targeting the Heart to Prevent Sudden Death in Dravet Syndrome.
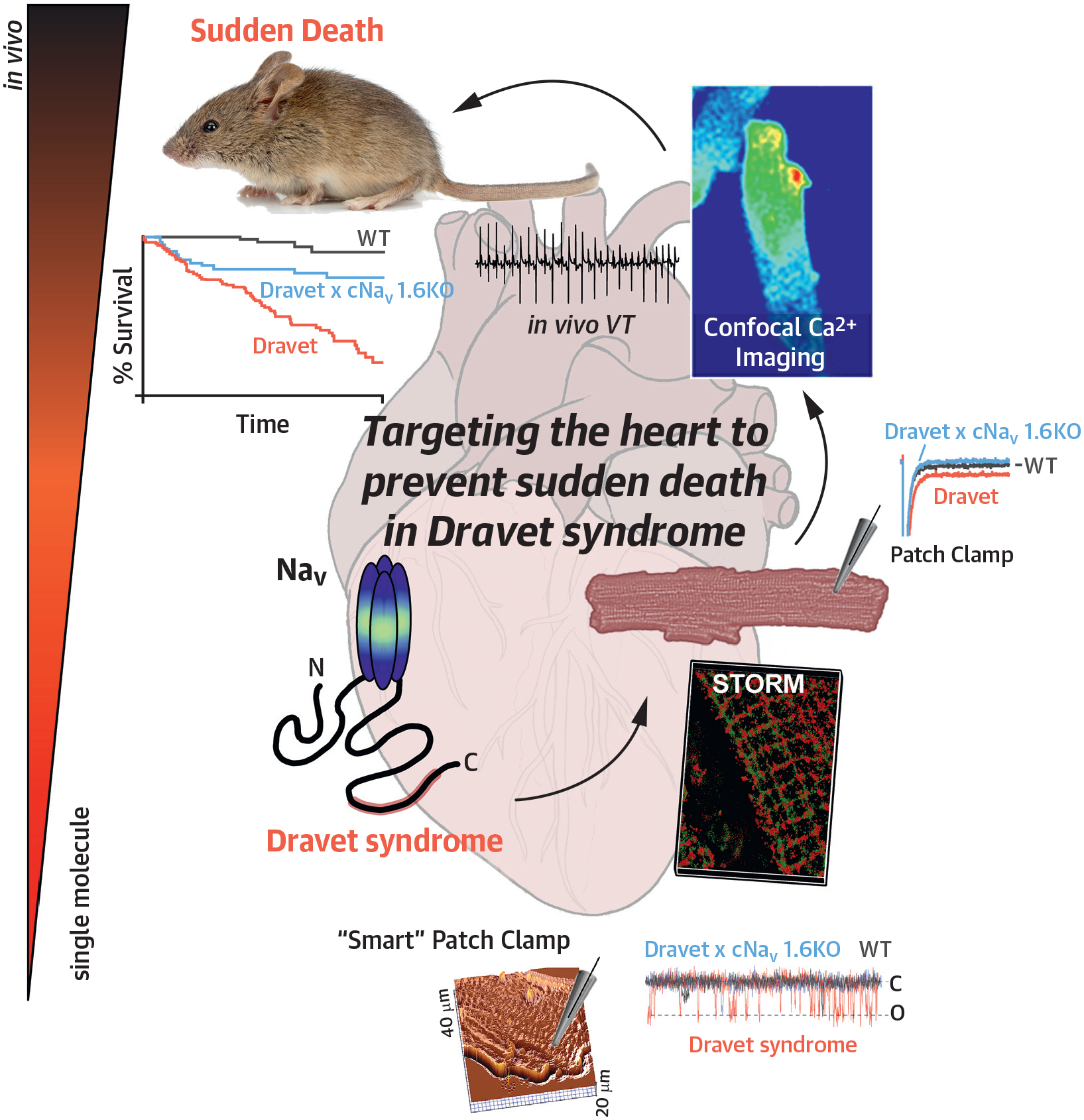
NAv = voltage-gated sodium channel; VT = ventricular tachycardia; WT = wild-type.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE:
Our results highlight a previously unappreciated role for the remodeling of sodium channels, including NaV1.6, in cardiac T-tubular nanodomains and their contribution to sudden death in DS.
TRANSLATIONAL OUTLOOK:
Given that NaV1.6 undergoes remodeling within T-tubules of DS hearts, serving as a substrate for Ca2+-mediated cardiac arrhythmias, NaV1.6 may be a druggable target for the prevention of sudden death in DS.
FUNDING SUPPORT AND AUTHOR DISCLOSURES
This work was supported by National Institutes of Health grants R01NS121234 and R01HL155378 (to Dr Radwański), R01HL148736 (to Dr Veeraraghavan), R01HL156652 (to Dr Hund), K99HL157684 (to Dr Nassal), and T32HL149637-02 and L40NS129034 (to Dr King); National Science Foundation Graduate Research Fellowship, NSF Fellow ID: 2019259354 (to Dr Struckman); and American Heart Association postdoctoral fellowships 915300 (to Dr Tarasov) and 908824 (to Dr Moise). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- cNav 1.6KO
cardiac-specific NaV1.6 deletion
- DS
Dravet syndrome
- ID
intercalate disc
- I Na,L
voltage-gated sodium current
- NaV
voltage-gated sodium channel
- Nav1.6Het
Scn8a haploinsufficient
- NCX
sodium/calcium exchanger
- RyR2
ryanodine receptors
- SICM
scanning ion-conductance microscopy
- SUDEP
sudden unexpected death in epilepsy
- T-tubule
transverse tubule
- WT
wild type
Footnotes
APPENDIX For an expanded Methods section as well as a supplemental table and figures, please see the online version of this paper.
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
REFERENCES
- 1.Skluzacek JV, Watts KP, Parsy O, Wical B, Camfield P. Dravet syndrome and parent associations: the IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia. 2011;52(suppl 2):95–101. [DOI] [PubMed] [Google Scholar]
- 2.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panelli RJ. SUDEP: a global perspective. Epilepsy Behav. 2020;103(pt B):106417. 10.1016/j.yebeh.2019.07.018 [DOI] [PubMed] [Google Scholar]
- 4.Delogu AB, Spinelli A, Battaglia D, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome: cardiac autonomic function in Dravet syndrome. Epilepsia. 2011;52:55–58. [DOI] [PubMed] [Google Scholar]
- 5.Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123(4):1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Granbichler CA, Nashef L, Selway R, Polkey CE. Mortality and SUDEP in epilepsy patients treated with vagus nerve stimulation. Epilepsia. 2015;56(2):291–296. [DOI] [PubMed] [Google Scholar]
- 7.Frasier CR, Wagnon JL, Bao YO, et al. Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy. Proc Natl Acad Sci U S A. 2016;113(45):12838–12843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maier SKG, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99(6):4073–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maier SKG, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 2004;109(11):1421–1427. [DOI] [PubMed] [Google Scholar]
- 10.Radwański PB, Brunello L, Veeraraghavan R, et al. Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc Res. 2015;106(1):143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One. 2013;8(10):e77843. 10.1371/journal.pone.0077843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frasier CR, Zhang H, Offord J, et al. Channelopathy as a SUDEP biomarker in Dravet syndrome patient-derived cardiac myocytes. Stem Cell Rep. 2018;11(3):626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasano T, Horigome Y, Takahashi Y, Ogiwara I, Furukawa T. Hyperthermia-induced ventricular tachycardia and sudden death in mouse model of Dravet syndrome. J Arrhythmia. 2011;27(suppl): BPB_4–BPB_4. 10.4020/jhrs.27.BPB_4 [DOI] [Google Scholar]
- 14.Sakauchi M, Oguni H, Kato I, et al. Mortality in Dravet syndrome: search for risk factors in Japanese patients. Epilepsia. 2011;52(suppl 2):50–54. [DOI] [PubMed] [Google Scholar]
- 15.Cooper MS, Mcintosh A, Crompton DE, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–47. [DOI] [PubMed] [Google Scholar]
- 16.Shmuely S, Sisodiya SM, Gunning WB, Sander JW, Thijs RD. Mortality in Dravet syndrome: a review. Epilepsy Behav. 2016;64(pt A): 69–74. [DOI] [PubMed] [Google Scholar]
- 17.Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2014;13(2):163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox JG, ed. The Mouse in Biomedical Research. 2nd ed. Elsevier; 2007. [Google Scholar]
- 19.Lenk GM, Jafar-Nejad P, Hill SF, et al. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann Neurol. 2020;87(3):339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson LL, Hawkins NA, Thompson CH, Kearney JA, George AL. Unexpected efficacy of a novel sodium channel modulator in Dravet syndrome. Sci Rep. 2017;7(1):1682. 10.1038/s41598-017-01851-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41(3):655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16(23):2892–2899. [DOI] [PubMed] [Google Scholar]
- 23.Struckman HL, Baine S, Thomas J, et al. Superresolution imaging using a novel high-fidelity antibody reveals close association of the neuronal sodium channel NaV1.6 with ryanodine receptors in cardiac muscle. Microsc Microanal. 2020;26(1):157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarasov M, Struckman HL, Olgar Y, et al. NaV1. 6 dysregulation within myocardial T-tubules by D96V calmodulin enhances proarrhythmic sodium and calcium mishandling. J Clin Invest. 2023;133(7):e152071. 10.1172/JCI152071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elming H, Brendorp B, Køber L, Sahebzadah N, Torp-Petersen C. QTc interval in the assessment of cardiac risk. Card Electrophysiol Rev. 2002;6(3): 289–294. [DOI] [PubMed] [Google Scholar]
- 26.Perulli M, Battista A, Sivo S, et al. Heart rate variability alterations in Dravet syndrome: the role of status epilepticus and a possible association with mortality risk. Seizure. 2022;94:129–135. [DOI] [PubMed] [Google Scholar]
- 27.Veeraraghavan R, Hoeker GS, Alvarez-Laviada A, et al. The adhesion function of the sodium channel beta subunit (β1) contributes to cardiac action potential propagation. eLife. 2018;7:e37610. 10.7554/eLife.37610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makara MA, Curran J, Little SC, et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. 2014;115(11):929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munger MA, Olğar Y, Koleske ML, et al. Tetrodotoxin-sensitive neuronal-type Na+ channels: a novel and druggable target for prevention of atrial fibrillation. J Am Heart Assoc. 2020;9(11): e015119. 10.1161/JAHA.119.015119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koleske M, Bonilla I, Thomas J, et al. Tetrodotoxin-sensitive Navs contribute to early and delayed afterdepolarizations in long QT arrhythmia models. J Gen Physiol. 2018;150(7): 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veeraraghavan R, Gourdie RG. Stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) for quantitative nanoscale assessment of spatial protein organization. Mol Biol Cell. 2016;27(22):3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia. 2012;53(1):87–100. [DOI] [PubMed] [Google Scholar]
- 33.Ryvlin P, Nashef L, Lhatoo SD, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12(10):966–977. [DOI] [PubMed] [Google Scholar]
- 34.Vilella L, Lacuey N, Hampson JP, et al. Post-convulsive central apnea as a biomarker for sudden unexpected death in epilepsy (SUDEP). Neurology. 2019;92(3):e171–e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuo FS, Cleary CM, LoTurco JJ, Chen X, Mulkey DK. Disordered breathing in a mouse model of Dravet syndrome. eLife. 2019;8. 10.7554/eLife.43387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalume F Sudden unexpected death in Dravet syndrome: respiratory and other physiological dys-functions. Respir Physiol Neurobiol. 2013;189(2): 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radwański PB, Ho HT, Veeraraghavan R, et al. Neuronal Na+ channels are integral components of pro-arrhythmic Na+/Ca2+ signaling nanodomain that promotes cardiac arrhythmias during β-adrenergic stimulation. J Am Coll Cardiol Basic Trans Science. 2016;1(4):251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biet M, Morin N, Lessard-Beaudoin M, et al. Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: a potential mechanism for sudden death in epilepsy. Circ Arrhythm Electrophysiol. 2015;8(4):912–930. [DOI] [PubMed] [Google Scholar]
- 39.Lin X, O’Malley H, Chen C, et al. Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol. 2015;593(6):1389–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN, Undrovinas A. Contribution of sodium channel neuronal isoform Nav1.1 to late sodium current in ventricular myocytes from failing hearts. J Physiol. 2015;593(6): 1409–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duble S, Thomas S. Sudden unexpected death in epilepsy. Indian J Med Res. 2017;145(6):738. 10.4103/ijmr.IJMR_548_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y, Bravo E, Thirnbeck CK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest. 2018;128(3):1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shmuely S, Surges R, Helling RM, et al. Cardiac arrhythmias in Dravet syndrome: an observational multicenter study. Ann Clin Transl Neurol. 2020;7(4):462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ergul Y, Ekici B, Tatli B, Nisli K, Ozmen M. QT and P wave dispersion and heart rate variability in patients with Dravet syndrome. Acta Neurol Belg. 2013;113(2):161–166. [DOI] [PubMed] [Google Scholar]
- 45.Vaseghi M, Shivkumar K. The role of the autonomic nervous system in sudden cardiac death. Prog Cardiovasc Dis. 2008;50(6): 404–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdel-Mannan O, Venkatesan TC, Sutcliffe AG. Paediatric sudden unexpected death in epilepsy (SUDEP): is it truly unexplained? Paediatr Child Health. 2022;32(10):382–387. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.