

Abstract
Carabranolides present characteristic NMR resonances for the cyclopropane moiety, which distinctly differ from those of other compounds and were used for an NMR-guided isolation in this study. As a result, 11 undescribed carabranolides (1–11), along with five known ones (12–16), were isolated from the fruits of Carpesium abrotanoides L. Compounds 1–11 are new esters of carabrol at C-4 with different carboxylic acids. Their structures were elucidated by HRESIMS and NMR spectroscopic data analysis. The biological evaluation showed that compounds 2–4, 15, and 16 exhibited significant inhibitory activity against LPS-induced NO release with an IC50 value of 5.6–9.1 μM and dose-dependently decreased iNOS protein expression in RAW264.7 cells.
The genus Carpesium (Compositae family) comprises about 21 species, which are widely distributed in Asia and Europe, particularly in the mountainous areas of Southwest China.1 In China, 17 species and three varieties are known, and six species (Carpesium abrotanoides L., C. cernuum L., C. divaricatum Sieb. et Zucc., C. lipskyi Winkl., C. macrocephalum Franch. et Sav., and C. nepalense Less) have been used as traditional Chinese medicines to treat cold, fever, sore throat, tonsillitis, wound bleeding, swollen poison, herpes zoster, and snake bites.2,3 Sesquiterpene lactones and sesquiterpene dimers were reported as characteristic constituents of this genus with diverse biological activities including cytotoxic,4 anti-inflammatory,5 antibacterial,6 and insecticidal7 activities.
C. abrotanoides, a perennial
plant, has been extensively used as a medicinal plant for treating
sore throat, tonsillitis, bronchitis, trauma, bleeding, and snake
and insect bites in China, Korea, and Japan.7−10 Fruits of this species serve
as a significant anthelmintic in traditional Chinese medicine for
ascariasis, pinworm disease, and tapeworm infections.11 Although over 130 compounds have been identified from C. abrotanoides,12−14 the bioactive compounds from
its fruits have rarely been investigated.7,13 In
a recent study, seven new cytotoxic sesquiterpene lactone dimers with
a carabrol unit were isolated from the fruits of C. abrotanoides.13 Our preliminary NMR data analysis
revealed that a number of unknown carabrol derivatives were present
in the extract of the fruits of C. abrotanoides.
To identify these unknown carabranolides and their bioactivities,
an NMR-guided isolation was performed on the fruits of C.
abrotanoides, which led to the isolation of 11 new carabranolides
(1–11), along with five known derivatives
(12–16). The anti-inflammatory activity
of these sesquiterpenes was also evaluated, revealing that compounds 2–4, 15, and 16 significantly inhibited LPS-induced NO release with an IC50 value of 5.6–9.1 μM and dose-dependently decreased
iNOS protein expression in RAW264.7 cells.
Results and Discussion
Carabranolides present two NMR signals of characteristic methines at δH ∼0.44 and ∼0.34 (H-1 and H-5) as well as a quaternary carbon at δC ∼17.1 (C-10) in their 1H and 13C NMR spectra for the cyclopropane moiety.4,6,15 Chemical shifts of H-4 and C-4 in these compounds shifted downfield from δH ∼3.8 to ∼4.9 and from δC ∼68 to ∼71, respectively, due to the esterification of the hydroxy group at C-4 of carabrol.16 Therefore, an NMR-guided isolation method for carabranolide derivatives was established in this study. Signals for H-1, H-4, H-5, C-4, and C-10 were observed in 1H and 13C NMR spectra (Figure 1) of the ethanolic extract of C. abrotanoides fruits. A series of fractionation steps were then performed and monitored by NMR analyses. Eleven undescribed carabranolides (1–11) were isolated, along with the known compounds carabrol-4-O-linoleate (12) and carabrol-4-O-palmitate (13),17 carabrol (14),18 carabrone (15),15 and (4S)-acetyloxyl-11(13)-carabren-8β,12-olide (16),19 identified by comparison of experimental and reported spectroscopic data.
Figure 1.

1H (A) and 13C (B) NMR spectra of the PE fraction of the EtOH extract, the target fraction, a target compound, and carabrol (14) from the fruits of C. abrotanoides.
Compound 1 was obtained as a yellow oil. Its molecular formula was determined as C27H40O7 by (+)-HRESIMS analysis at m/z 477.2833 [M + H]+ (calcd for 477.2847). The IR spectrum of 1 showed the presence of carbonyl (1759 cm–1) and hydroxyl (3487 cm–1) bands. The 1H NMR spectrum of 1 (Table 2) exhibited 1H signals for two methyl groups at δH 1.06 (s, H-14) and 1.21 (d, J = 6.2 Hz, H-15), one exocyclic olefinic methylene at δH 5.56 (d, J = 2.5 Hz, H-13a) and 6.24 (d, J = 2.5 Hz, H-13b), and two methines of a characteristic cyclopropane moiety at δH 0.34 (ddd, J = 9.1, 7.0, 4.1 Hz, H-5) and 0.44 (td, J = 7.2, 4.1 Hz, H-1). The 13C NMR and DEPT135 spectra showed signals for 27 carbons (Table 1), including two methyl groups (δC 18.3 and 20.0, C-14 and C-15, respectively), 13 methylenes (one exocyclic methylene at δC 122.7, C-13), seven methines (four oxygenated methines at δC 69.0, 70.6, 75.8, and 84.7; C-10′, C-4, C-8, and C-9′), and five quaternary carbons (three carbonyl carbons at δC 170.6, 173.5, and 175.4, C-12, C-1′, and C-12′). 1H and 13C NMR spectroscopic data of 1 suggested the presence of a carabranolide unit and a furanoctanoic acid group in 1.
Table 2. 1H NMR (600 MHz) Data of 1–5 (CDCl3).
| no. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 0.44, td (7.2, 4.1) | 0.45, ttd (7.0, 3.9, 2.1) | 0.44, td (7.2, 4.1) | 0.36, td (7.1, 4.1) | 0.44, td (7.3, 4.1) |
| 2 | 1.31, m | 1.31, m | 1.31, m | 1.18, m | 1.30, m |
| 1.61, m | 1.64, m | 1.62, m | 1.29, m | 1.60, m | |
| 3 | 1.57, m | 1.57, m | 1.58, m | 1.56, m | 1.57, m |
| 1.69, m | 1.69, m | 1.71, m | 1.63, m | 1.69, m | |
| 4 | 4.91, q (6.3) | 4.92, q (6.3) | 4.96, qd (6.3, 6.3) | 4.90, ddd (7.4, 6.3, 5.3) | 4.92, q (6.3) |
| 5 | 0.34, ddd (9.1, 7.0, 4.1) | 0.34, ddd (9.0, 7.0, 4.1) | 0.35, ddd (9.2, 7.0, 4.1) | 0.26, ddd (8.8, 7.1, 4.1) | 0.34, ddd (8.7, 7.1, 4.1) |
| 6 | 0.90, m | 0.92, m | 0.94, ddd (14.5, 12.2, 9.2) | 0.87, m | 0.91, m |
| 2.35, m | 2.37, m | 2.36, dd (14.5, 7.0) | 2.33, m | 2.35, m | |
| 7 | 3.16, m | 3.17, m | 3.16, m | 3.14, m | 3.15, m |
| 8 | 4.79, ddd (12.3, 9.2, 6.1) | 4.79, ddd (11.5, 9.1, 6.1) | 4.78, ddd (11.5, 8.8, 6.1) | 4.76, ddd (11.5, 8.8, 6.2) | 4.78, ddd (11.5, 8.8, 6.1) |
| 9 | 0.96, m | 0.96, m | 0.97, dd (13.9, 11.5) | 0.90, m | 0.97, m |
| 2.33, m | 2.32, m | 2.32, dd (13.9, 6.1) | 2.29, m | 2.33, m | |
| 13 | 5.56, d (2.5) | 5.56, d (2.6) | 5.55, d (2.5) | 5.55, d (2.5) | 5.55, d (2.4) |
| 6.24, d (2.5) | 6.24, d (2.6) | 6.24, d (2.5) | 6.24, d (2.5) | 6.24, d (2.4) | |
| 14 | 1.06, s | 1.06, s | 1.07, s | 1.00, s | 1.07, s |
| 15 | 1.21, d (6.2) | 1.21, d (6.2) | 1.24, d (6.2) | 1.20, d (6.3) | 1.21, d (6.2) |
| 2′ | 2.27, t (7.4) | 2.27, t (7.4) | 2.42, dd (15.5, 9.7) | 3.52, d (13.7) | 2.26, m |
| 2.58, dd (15.5, 4.5) | |||||
| 3′ | 1.62, m | 1.62, m | 2.81, dq (9.7, 4.8) | 1.63, m | |
| 4′ | 1.32, m | 1.32, m | 1.63, m | ||
| 5′ | 1.32, m | 1.32, m | 5.88, br s | 6.65, d (2.7) | 1.57, m |
| 6′ | 1.32, m | 1.32, m | 4.13, q (6.5) | ||
| 7′ | 1.62, m | 1.62, m | 2.35, dd (10.2, 7.2) | 6.61, dd (8.2, 2.7) | 5.79, dd (15.5, 6.5) |
| 2.46, dd (10.2, 4.9) | |||||
| 8′ | 1.71, m | 1.57, m | 1.89, dtt (13.5, 7.0, 5.1) | 7.04, d (8.2) | 5.68, dd (15.5, 6.2) |
| 1.86, m | 1.63, m | 2.12, ddt (13.5, 10.0, 4.9) | |||
| 9′ | 4.35, ddd (8.8, 4.5, 3.8) | 4.33, ddt (8.3, 6.7, 3.1) | 1.97, s | 2.26, s | 3.93, t (6.2) |
| 10′ | 4.49, t (4.5) | 4.28, dt (6.7, 2.9) | 3.46, ddd (9.1, 6.2, 2.8) | ||
| 11′ | 2.56, dd (17.7, 1.0) | 2.52, dd (17.9, 4.1) | 1.41, m | ||
| 2.81, dd (17.7, 4.5) | 2.83, dd (17.9,6.7) | 1.50, m | |||
| 12′ | 1.63, m | ||||
| 13′ | 1.31, m | ||||
| 14′ | 1.31, m | ||||
| 15′ | 1.31, m | ||||
| 16′ | 1.29, m | ||||
| 17′ | 1.30, m | ||||
| 18′ | 0.89, t (6.9) |
Table 1. 13C NMR (150 MHz) Data of 1–11 (CDCl3).
| no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 34.7, CH | 34.7, CH | 34.6, CH | 34.7, CH | 34.8, CH | 34.8, CH | 34.8, CH | 34.8, CH | 34.7, CH | 34.8, CH | 34.8, CH |
| 2 | 25.0, CH2 | 25.0, CH2 | 25.0, CH2 | 24.9, CH2 | 25.0, CH2 | 25.0, CH2 | 25.0, CH2 | 25.0, CH2 | 25.0, CH2 | 25.0, CH2 | 25.0, CH2 |
| 3 | 35.9, CH2 | 35.9, CH2 | 35.9, CH2 | 35.9, CH2 | 36.0, CH2 | 36.0, CH2 | 36.0, CH2 | 36.0, CH2 | 36.0, CH2 | 36.0, CH2 | 36.0, CH2 |
| 4 | 70.6, CH | 70.6, CH | 71.5, CH | 71.2, CH | 70.5, CH | 70.5, CH | 70.4, CH | 70.5, CH | 71.2, CH | 70.5, CH | 70.4, CH |
| 5 | 22.9, CH | 22.9, CH | 22.9, CH | 22.9, CH | 22.9, CH2 | 22.9, CH | 22.9, CH | 22.9, CH | 22.9, CH | 22.9, CH | 22.9, CH2 |
| 6 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 | 30.8, CH2 |
| 7 | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH | 37.8, CH |
| 8 | 75.8, CH | 75.8, CH | 75.6, CH | 75.7, CH | 75.7, CH | 75.7, CH | 75.7, CH | 75.7, CH | 75.6, CH | 75.7, CH | 75.7, CH |
| 9 | 37.4, CH2 | 37.4, CH2 | 37.3, CH2 | 37.3, CH2 | 37.4, CH2 | 37.4, CH2 | 37.4, CH2 | 37.4, CH2 | 37.4, CH2 | 37.4, CH2 | 37.4, CH2 |
| 10 | 17.1, C | 17.1, C | 17.1, C | 17.0, C | 17.1, C | 17.1, C | 17.1, C | 17.1, C | 17.1, C | 17.1, C | 17.1, C |
| 11 | 139.0, C | 139.0, C | 139.0, C | 139.1, C | 139.0, C | 139.0, C | 139.0, C | 139.0, C | 139.0, C | 139.0, C | 139.0, C |
| 12 | 170.6, C | 170.7, C | 170.5, C | 170.7, C | 170.6, C | 170.6, C | 170.6, C | 170.6, C | 170.5, C | 170.6, C | 170.5, C |
| 13 | 122.7, CH2 | 122.7, CH2 | 122.6, CH2 | 122.6, CH2 | 122.6, CH2 | 122.6, CH2 | 122.6, CH2 | 122.6, CH2 | 122.6, CH2 | 122.6, CH2 | 122.5, CH2 |
| 14 | 18.3, CH3 | 18.3, CH3 | 18.3, CH3 | 18.2, CH3 | 18.3, CH3 | 18.3, CH3 | 18.3, CH3 | 18.3, CH3 | 18.3, CH3 | 18.2, CH3 | 18.2, CH3 |
| 15 | 20.0, CH3 | 20.0, CH3 | 20.0, CH3 | 20.0, CH3 | 20.0, CH3 | 20.0, CH3 | 20.0, CH3 | 20.0, CH3 | 19.9, CH3 | 20.0, CH3 | 20.0, CH3 |
| 1′ | 173.5, C | 173.5, C | 171.5, C | 171.4, C | 173.5, C | 173.5, C | 173.5, C | 173.5, C | 172.7, C | 173.5, C | 173.5, C |
| 2′ | 34.6, CH2 | 34.6, CH2 | 36.5, CH2 | 38.8, CH2 | 34.7, CH2 | 34.7, CH2 | 34.7, CH2 | 34.7, CH2 | 41.6, CH2 | 34.7, CH2 | 34.7, CH2 |
| 3′ | 24.9, CH2 | 24.9a, CH2 | 36.4, CH | 125.3, C | 25.1a, CH2 | 25.0a, CH2 | 25.1, CH2 | 25.0, CH2 | 68.1, CH | 25.0, CH2 | 25.1, CH2 |
| 4′ | 29.2b, CH2 | 28.9b, CH2 | 163.1, C | 138.4, C | 25.1a, CH2 | 25.1a, CH2 | 29.1b, CH2 | 29.2b, CH2 | 36.6, CH2 | 28.9b, CH2 | 29.5b, CH2 |
| 5′ | 29.0b, CH2 | 29.0b, CH2 | 127.7, CH | 117.2, CH | 37.2, CH2 | 37.4, CH2 | 29.2b, CH2 | 29.1b, CH2 | 25.5, CH2 | 28.8b, CH2 | 29.3b, CH2 |
| 6′ | 28.9b, CH2 | 28.9b, CH2 | 198.8, C | 154.6, C | 72.3, CH | 72.1, CH | 29.4b, CH2 | 29.2b, CH2 | 29.6b, CH2 | 28.9b, CH2 | 29.2b, CH2 |
| 7′ | 25.4, CH2 | 25.1a, CH2 | 34.1, CH2 | 112.8, CH | 136.5, CH | 136.5, CH | 29.5b, CH2 | 25.7, CH2 | 29.6b, CH2 | 24.7, CH2 | 29.b, CH2 |
| 8′ | 28.3, CH2 | 33.1, CH2 | 27.4, CH2 | 131.3, CH | 129.9, CH | 129.5, CH | 29.6b, CH2 | 32.7, CH2 | 29.7b, CH2 | 34.1, CH2 | 27.2a, CH2 |
| 9′ | 84.7, CH | 87.5, CH | 22.7, CH3 | 19.7, CH3 | 75.5, CH | 75.6, CH | 37.5, CH2 | 63.0, CH2 | 29.7b, CH2 | 179.0, C | 130.0, CH |
| 10′ | 69.0, CH | 71.8, CH | 74.5, CH | 74.6, CH | 72.0, CH | 29.7b, CH2 | 129.7, CH | ||||
| 11′ | 39.4, CH2 | 37.7, CH2 | 32.9, CH2 | 32.9, CH2 | 37.4, CH2 | 29.7b, CH2 | 27.1a, CH2 | ||||
| 12′ | 175.4, C | 174.8, C | 25.0a, CH2 | 25.0a, CH2 | 25.7a, CH2 | 29.6b, CH2 | 29.7b, CH2 | ||||
| 13′ | 29.0b, CH2 | 29.0b, CH2 | 25.6a, CH2 | 29.4b, CH2 | 29.1b, CH2 | ||||||
| 14′ | 29.2b, CH2 | 29.0b, CH2 | 25.6a, CH2 | 29.7b, CH2 | 29.1b, CH2 | ||||||
| 15′ | 29.4b, CH2 | 29.4b, CH2 | 32.7, CH2 | 31.9, CH2 | 29.1b, CH2 | ||||||
| 16′ | 31.7, CH2 | 31.7, CH2 | 63.0, CH3 | 22.7, CH2 | 31.9, CH2 | ||||||
| 17′ | 22.6, CH2 | 22.6, CH2 | 14.1, CH3 | 22.7, CH2 | |||||||
| 18′ | 14.0, CH3 | 14.0, CH3 | 14.1, CH3 |
Assignments may be interchanged within the same column.
The spin system H-15–H-4–H-3–H-2–H-1–H-5–H-6–H-7–H-8–H-9 was observed in the COSY spectrum. HMBC correlations from H-13 to C-7, C-11, and C-12 and from H-14 to C-1, C-5, C-9, and C-10 (Figure 2) established the carabrol unit of 1 as the same in carabrol (14).18 The remaining 1H and 13C NMR data of 1 were almost identical to those of lonfuranacid A, a natural furanoctanoic acid,20 indicating 1 has a lonfuranacid A unit, which was supported by the 1H–1H correlations of H-2′/H-3′/H-4′/H-5′/H-6′/H-7′/H-8′/H-9′/H-10′/H-11′ and HMBC correlations from H-2′ to C-1′, H-10′ to C-8′ and C-12′, and H-9′ to C-12′ (Figure 2). By comparing with the NMR data of 14, the chemical shift of H-4 in 1 shifted from δH 3.82 (14) to 4.92 (1), suggesting the hydroxy group at C-4 of carabrol was esterified by the lonfuranacid A moiety in 1. This hypothesis was confirmed by analysis of HMBC data, which showed a correlation between H-4 (δH 4.91) and C-1′ (δC 173.5). NOE correlations observed for H-4/H-1/H-9β, H-5/H-14/H-9α/H-7, and H-14/H-8 (Figure S1) further confirmed that 1 has the same relative configuration as carabrol (14).15 The similar NMR data between the subunit of 1 and lonfuranacid A, along with the coupling constant between H-9′ and 10′ (J9′,10′ = 4.5 Hz), supported the relative configurations of C-9′ and C-10′ as being the same as in lonfuranacid A. Thus, the structure of 1 was determined as lonfuranacid A-4-O-carabrol and named carabrolate A.
Figure 2.

Key 1H–1H COSY and HMBC correlations of 1–11.
Compound 2 exhibited the same molecular formula as 1 by (+)-HRESIMS analysis at m/z 477.2838. The IR and NMR spectra of 2 were very similar to those of 1, indicating that 2 was also a carabrol derivative. A comparison of the 13C NMR data (Table 1) of 2 with those of 1 revealed that these compounds were nearly identical, except that the resonances of C-8′, C-9′, and C-10′ of 2 shifted downfield in a range of 2–5 ppm compared with those of 1, which could be due to the configurational change at C-9′ in the furanoctanoic acid unit of 2. The J9′,10′ coupling constant of 6.7 Hz supported that H-9′ and H-10′ were opposite, the same as in lonfuranacid B.20 Therefore, the structure of 2 was determined as lonfuranacid B-4-O-carabrol, and it was named carabrolate B.
Compound 3 was purified as a yellow oil. Its molecular formula (C24H32O5) was inferred by (+)-the HRESIMS analysis at m/z 423.2135 [M + Na]+ (calcd for 423.2142). The 1H and 13C NMR data (Tables 1 and 2) of 3 identified it as a carboxylic acid ester of carabrol at C-4. The 1H–1H correlations of H-2′/H-3′/H-8′/H-7′ and HMBC correlations from H-2′ to C-1′, H-3′ to C-1′, C-5′, and C-7′, H-9′ to C-3′ and C-5′, H-7′ and H-8′ to C-6′, and H-5′ to C-6′ and C-7′ (Figure 2) established the acid unit of 3 as 2-methyl-4-oxo-2-cyclohexene-1-acetic acid, an oxidation product of limonene.21 The key correlation between H-4 (δH 4.96) and C-1′ (δC 171.5) confirmed that the acid unit is attached to C-4. Thus, the structure of 3 was determined as shown and named carabrolate C.
Compound 4 was isolated as a yellow oil. HRESIMS analysis of 4 gave a [M + H]+ peak at m/z 399.216 (calcd for 399.2166), indicating the molecular formula C24H30O5, with 10 degrees of unsaturation. The presence of a carabrol group in 4 was supported by an analysis of 1H and 13C NMR data (Tables 1 and 2). In addition to resonances for carabrol, an ABX aromatic system δH 6.61 (dd, J = 8.2, 2.7 Hz, H-7′), 6.65 (d, J = 2.7 Hz, H-5′), and 7.04 (d, J = 8.2 Hz, H-8′) and six aromatic carbons (δC 112.8, 117.2, 125.3, 131.3, 138.4, and 154.6) were observed in the 1H and 13C NMR spectra of 4. The HMBC correlations from H-2′ to C-1′, C-4′, and C-8′, H-9′ to C-3′ and C-5′, and H-4 to C-1′ (Figure 2) were further indicative of a 2-methyl-4-hydroxyphenylacetic acid22 unit in 4, attached to C-4 of the carabraol group. Compared to those of 3, chemical shifts of H-1–6, H-9, H-14, and H-15 of 4 were upfield in a range of 0.04–0.33 ppm, which could be caused by the shielding effect, inductive effect, and spatial proximity of the 2-methyl-4-hydroxyphenylacetic acid moiety in 4. Thus, the structure of 4 was determined as shown and named carabrolate D.
Carabrolates E (5) and F (6) presented the same molecular formula C33H54O7 based on analysis of the HRESIMS and NMR data. The 1H and 13C NMR data (Tables 1–3) of 5 and 6 were similar, revealing that these compounds are carabraol-4-O-fatty acid esters. In the 1H NMR spectrum of 5, resonances assigned to an oxylipin group were observed, including a triplet methyl group at δH 0.89 (t, J = 6.9 Hz, H-18′), 11 methylenes at δH 1.29–2.26, two olefinic protons at δH 5.79 (dd, J = 15.5, 6.5 Hz, H-7′) and 5.68 (dd, J = 15.5, 6.2 Hz, H-8′), and three oxygenated methines at δH 4.13 (q, J = 6.5 Hz, H-6′), 3.93 (t, J = 6.2 Hz, H-9′), and 3.46 (ddd, J = 9.1, 6.2, 2.8 Hz, H-10′). The oxylipin group in 5 exhibited almost identical NMR data to those of a natural 6,9,10-trihydroxyoctadec-7-enoic acid,23 which were confirmed by 2D NMR analysis (Figure 2). The coupling constant of H-9′, 10′ (J9′,10′ = 6.2 Hz) and identical NMR data to those of 9R,10R-trihydroxyoctadec-7-enoic acid23 deduced the 9′β-OH, and 10′β-OH in 5. Thus, the structure of 5 was determined as trihydroxyoctadec-7-enate. 1D and 2D NMR data analysis revealed that 5 and 6 have the same planar structure. The major differences between them were the chemical shifts of C-5′ to C-8′ and the NOE correlation of H-6′/H-8′ in 5 but not in 6, which suggested that the hydroxy group at the C-6′ in 6 was opposite to that of 5.24 To establish the absolute configurations of the C-6′, C-9′, and C-10′ in 5 and 6, vicinal diols in 5 and 6 were first protected by ketals to give 5a and 6a (Figure 3) according to the previously described procedure.25 The secondary alcohol at C-6′ in 5a and 6a was then derivatized with (S)- and (R)-α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) chlorides,26,27 yielding their S- and R-MTPA esters (5b/5c, 6b/6c) (Figure 3). By analysis of ΔδS–R values of the 1H NMR chemical shift between 5b and 5c and between 6b and 6c, respectively, the absolute configurations of the C-6′ in 5 and 6 were designated as S and R, respectively (Figure 3). Based on the relative configuration of C-9′ and C-10′, the absolute configurations of oxylipin moieties in 5 and 6 were determined as 6′S,9′R,10′R and 6′R,9′R,10′R, respectively.
Table 3. 1H NMR (600 MHz) Data of 6–11 (CDCl3).
| no. | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|
| 1 | 0.44, td (7.3, 4.1) | 0.44, td (7.2, 4.1) | 0.44, td (7.2, 4.1) | 0.44, td (7.2,4.1) | 0.44, td (7.2, 4.1) | 0.44, td (7.2, 4.1) |
| 2 | 1.32, m | 1.31, m | 1.32, m, | 1.29, m | 1.31, m | 1.30, m |
| 1.63, m | 1.61, m | 1.60, m | 1.62, m | 1.63, m | 1.62, m | |
| 3 | 1.56, m | 1.57, m | 1.56, m | 1.59, m | 1.56, m | 1.55, m |
| 1.68, m | 1.69, m | 1.68, m | 1.72, m | 1.68, m | 1.67, m | |
| 4 | 4.92, q (6.3) | 4.91, q (6.3) | 4.91, q (6.3) | 4.96, dt (5.9 | 4.91, h (6.3) | 4.91, dq (12.1, 6.3) |
| 5 | 0.34, ddd (8.7, 7.0, 4.1) | 0.34, ddd (8.8, 7.1, 4.1) | 0.34, ddd (8.8, 7.1, 4.1) | 0.34, ddd (8.8, 7.0, 4.1 | 0.34, ddd (8.9, 7.1, 4.1) | 0.34, ddd (8.7, 7.0, 4.1) |
| 6 | 0.91, m | 0.91, m | 0.93, m | 0.91, m | 0.92, m | 0.93, m |
| 2.35, m | 2.36, m | 2.35, m | 2.36, m | 2.35, m | 2.36, dd (14.3,7.1) | |
| 7 | 3.15, m | 3.15, m | 3.25, m 3.15 | 3.15, m | 3.16, m | 3.15, m |
| 8 | 4.78, ddd (11.5, 8.8, 6.1) | 4.78, ddd (11.5, 8.8, 6.1) | 4.78, ddd (11.5, 8.8, 6.1) | 4.78, ddd (11.4, 8.8, 6.1) | 4.78, ddd (11.5, 8.8, 6.1) | 4.78, ddd (11.5, 8.8, 6.1) |
| 9 | 0.96, m | 0.96, m | 0.95, m | 0.96, m | 0.95, m | 0.96, dd (13.8, 11.5) |
| 2.32, m | 2.32, m | 2.32, m | 2.32, m | 2.32, m | 2.32, dd (13.8,6.2) | |
| 13 | 5.55, d (2.5) | 5.55, d (2.5) | 5.55, d (2.5) | 5.55, d (2.5) | 5.55, d (2.50) | 5.55, d (2.5) |
| 6.24, d (2.5) | 6.24, d (2.5) | 6.24, d (2.5) | 6.24, d (2.5) | 6.24, d (2.50) | 6.24, d (2.5) | |
| 14 | 1.06, s | 1.06, s | 1.06, s | 1.07, s | 1.06, s | 1.06, s |
| 15 | 1.21, d (6.2) | 1.21, d (6.2) | 1.21, d (6.2) | 1.23, d (6.3) | 1.21, d (6.2) | 1.21, d (6.3) |
| 2′ | 2.25, m | 2.26, t (7.6) | 2.26, t (7.6) | 2.38, dd (16.3,9.1) | 2.26, t (7.5) | 2.26, t (7.5) |
| 2.47, dd (16.3,3.1) | ||||||
| 3′ | 1.62, m | 1.61, m | 1.61, m | 3.99, tt (7.8, 4.4) | 1.31, m | 1.61, m |
| 4′ | 1.62, m | 1.30, m | 1.31 m | 1.43, m | 1.31, m | 1.30, m |
| 1.52, m | ||||||
| 5′ | 1.54, m | 1.30, m | 1.31 m | 1.29, m | 1.31, m | 1.30, m |
| 6′ | 4.16, q (6.2) | 1.30, m | 1.31 m | 1.25, m | 1.31, m | 1.30, m |
| 7′ | 5.83, dd (15.5, 6.0, 1.1) | 1.30, m | 1.31 m | 1.25, m | 1.61, m | 1.30, m |
| 8′ | 5.71, dd (15.5, 6.4, 1.2) | 1.30, m | 1.57, m | 1.25, m | 2.32, t (7.5) | 2.01, q (6.7) |
| 9′ | 3.94, t (6.4) | 1.43, m | 3.63, t (6.6) | 1.25, m | 5.34, m | |
| 10′ | 3.46, ddd (9.3, 6.4, 3.2) | 3.58, dq (8.4, 5.1) | 1.25, m | 5.35, m | ||
| 11′ | 1.41, m 1.50, m | 1.43, m | 1.25, m | 2.02, q (6.7) | ||
| 12′ | 1.63, m | 1.32, m | 1.25, m | 1.30, m | ||
| 13′ | 1.31, m | 1.32, m | 1.25, m | 1.30, m | ||
| 14′ | 1.31, m | 1.32, m | 1.25, m | 1.30, m | ||
| 15′ | 1.31, m | 1.57, m | 1.25, m | 1.30, m | ||
| 16′ | 1.29, m | 3.65, t (6.6) | 1.25, m | 1.26, m | ||
| 17′ | 1.30, m | 0.89, t (7.0) | 1.29, m | |||
| 18′ | 0.89, t (6.8) | 0.88, t (7.0) |
Figure 3.
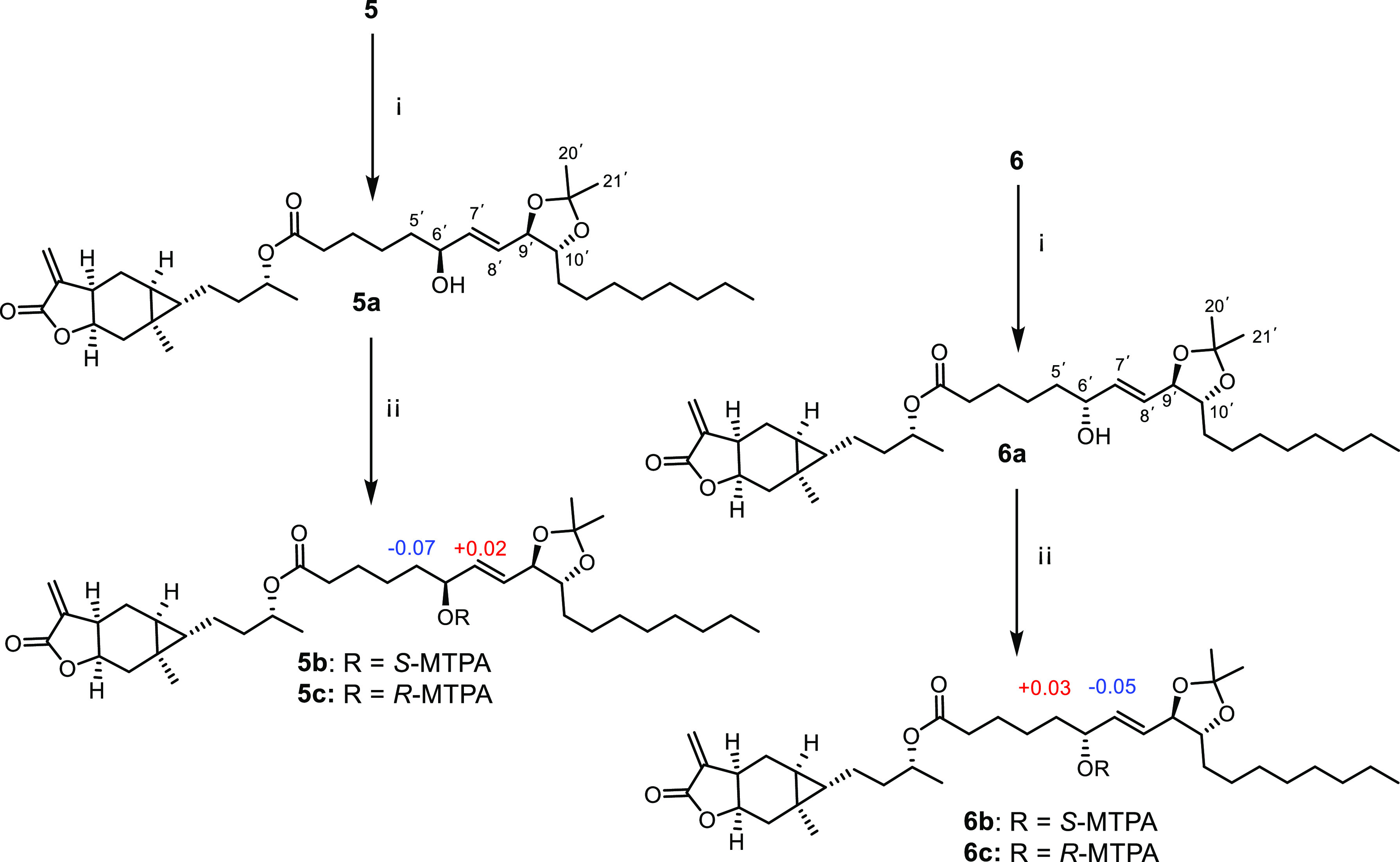
Ketalization and Mosher esterification for compounds 5 and 6. ΔδS–R values for Mosher esters (5b/5c and 6b/6c) are shown. Reagents and conditions: (i) Acetone, p-toluene sulfonic acid (PDSA), 37 °C, 2 h; (ii) DMAP, (S)- or (R)-MTPA chloride, room temperature, 2 h.
Compounds 7–9 were obtained as a yellow oil, with the molecular formulas C31H52O6, C24H38O5, and C32H54O5, determined by analysis of the corresponding HRESIMS ions [M + Na]+ at m/z 543.3656, 429.2608, and 541.3863, respectively. Compounds 7–9 are also esters of carabrol composed of fatty acids, as indicated by the analysis of NMR data (Tables 1 and 3), except for varying degrees of oxidation. The fatty acid in 7 was established as 10,16-dihydroxy hexadecanoic acid by the observation of an oxygenated methylene at δH 3.65 (t, J = 6.6 Hz, H-16′), an oxygenated methine δH 3.58 (dq, J = 8.4, 5.1 Hz, H-10′), and 13 methylenes at δH 1.30–2.26 (m), as well as 16 carbon signals including 14 methylenes (an oxygenated methylene at δC 63.0, C-16′), an oxygenated methine at δC 72.0, C-10, and a quaternary carbon at δC 173.5 (C-1′) in the 1H and 13C NMR spectra of 7, which were consistent with the data of 10,16-dihydroxy hexadecanoic acid.28 The position of the hydroxy group at C-10′ in 7 was further confirmed by characteristic fragment ions at m/z 271.2225, 187.1491, and 169.1209 in its HRESIMS/MS spectrum (Figure 4). By comparison with known spectroscopic data, the fatty acid groups of 8 and 9 were verified to be 9-hydroxynonanoic acid29 and 3-hydroxyheptadecanoic acid, respectively.30 The HMBC correlations from H-9′ to C-7′/C-8′ in 8 and from H-3′ to C-1′/C-2′/4′ in 8 also supported the existence of 9′-OH in 8 and 3′-OH in 9 (Figure 2). The secondary alcohol at C-3′ in 9 was derivatized with (S)- and (R)-MTPA chloride, yielding its S- and R-MTPA esters (9a and 9b) (Figure 5). The absolute configuration of C-3′ in 9 was determined as S based on the ΔδS–R values of its Mosher esters (9a and 9b). Moreover, for compounds 7–9, fatty acid groups attaching to C-4 of carabrol were established by their key HMBC correlations between H-4 (δH 4.91–4.96) and C-1′ (δC 172.7–173.5) (Figure 2). Hence, the structures of 7–9 were determined as shown and named carabrolates G (7), H (8), and I (9).
Figure 4.

Some diagnostic HRESIMS/MS (+) fragment ions of 7 and 11.
Figure 5.

ΔδS–R values for Mosher esters 9a and 9b.
The molecular formula C24H36O6 of 10 was established by analysis of (+)-HRESIMS at m/z 421.2577 [M + H]+ (calcd for 421.2585). The 1H and 13C NMR spectroscopic data (Tables 1 and 3) of 10 showed the presence of the carabrol moiety. Moreover, the resonances of an azelaic acid group in 10 were revealed by the seven methylenes (δH 1.31–2.32 and δC 24.7–34.7) and two carbonyl carbons (δC 173.5, C-1′ and 179.0, C-9′) in the 1H and 13C NMR spectra. Furthermore, these data were identical to those of the natural azelaic acid.31 The HMBC cross-peak between H-4 (δH 4.91) and C-1′ (δC 173.5) proved the connection of the carabranolide moiety and azelaic acid (Figure 2). Thus, the structure of 10 was determined as shown and named carabrolate J.
Compound 11, a yellow oil, presented the molecular formula C33H54O4 based on analysis of (+)-HRESIMS at m/z 537.391 [M + H]+ (calcd for 537.3914). Its 1H and 13C NMR spectroscopic data (Tables 1 and 3) were extremely close to those of carabrol-4-O-linoleate (12)17 in CDCl3, except for the absence of signals for a double-bond group at C-12′ and C-13′. Compound 11 was comprised of a carabrol unit and oleic acid group through HRMS and NMR data analysis.32 The position of the double bond at C-9′ and C-10′ in 11 was verified by fragment ions at m/z 155.0859 and 143.0819 through HRESIMS/MS analysis (Figure 4). Moreover, the oleic acid group attaching to C-4 of carabrol was defined by the key HMBC cross-peak (Figure 2) between H-4/C-1′. Therefore, compound 11 was established as carabrolate K.
Given the anti-inflammatory activity of carabrol,33 the activity of carabrol derivatives (1–16) on LPS-induced NO production in RAW264.7 cells was investigated. As shown in Table 4, compounds 1–8, 9, and 14–16 exhibited notable inhibitory effects on LPS-induced NO release in RAW264.7 cells, with IC50 values ranging from 5.8 to 15.1 μM. All tested compounds had no obvious effect on cell viability at a concentration of 20 μM. Structurally, the principal distinction among compounds 1–16 is the substituents at the C-4 position. The unsubstituted carabrol (14) exhibited considerable inhibitory activity against NO production with an IC50 value of 10.96 μM. When the 4-OH of carabrol was oxidized (15), acetylated (16), or esterified with 2-methyl-4-hydroxyphenylacetic acid (4), the IC50 values of 4 (5.82 μM), 15 (6.77 μM), and 16 (5.64 μM) decreased almost one-fold compared with carabrol. In contrast, the esterification of carabrol with the long-chain fatty acids diminished the bioactivity, while cyclic or unsaturated and oxidized fatty acid esters of carabrol (2, 3, 5–8, 10) exhibited a similar activity to that of carabrol (Table 4).
Table 4. IC50 Values of 1–16 for NO Inhibitory Activity.
| no. | IC50 (μM) | no. | IC50 (μM) |
|---|---|---|---|
| 1 | 15.06 ± 1.01 | 9 | >20 |
| 2 | 8.67 ± 0.74 | 10 | 9.35 ± 0.78 |
| 3 | 9.15 ± 0.96 | 11 | >20 |
| 4 | 5.82 ± 0.43 | 12 | >20 |
| 5 | 11.80 ± 0.82 | 13 | >20 |
| 6 | 11.56 ± 0.67 | 14 | 10.96 ± 0.43 |
| 7 | 9.87 ± 0.75 | 15 | 6.77 ± 0.57 |
| 8 | 9.75 ± 0.54 | 16 | 5.64 ± 0.32 |
| Dex | 8.52 ± 0.61 |
Inducible nitric oxide synthase (iNOS) is a key enzyme for LPS-induced NO release, and the effects of selected compounds (2–4, 15, and 16) on LPS-stimulated iNOS protein expression were then evaluated in RAW264.7 cells. As a result, compounds 2–4, 15, and 16 dose-dependently inhibited LPS-induced iNOS expression at 5, 10, and 20 μM (Figure 6). These results suggested that carabrol derivatives are anti-inflammatory agents.
Figure 6.

Anti-inflammatory activity of tested compounds on LPS-induced RAW264.7 cells. (A) Compounds 2–4, 15, and 16 inhibited LPS-induced NO production in a concentration-dependent manner. (B) Compounds 2–4, 15, and 16 dose-dependently decreased LPS-induced iNOS expression in RAW264.7 cells. Dexamethasone (DEX) was used as a positive control. (Data are presented as mean ± SD, ###p < 0.001, compared with the blank control group; *p < 0.05, **p < 0.01, and ***p < 0.001, compared with the LPS group.)
Experimental Section
General Experimental Procedures
Optical rotations were defined by an Autopol I polarimeter (Rudolph, Flanders, USA). UV and ECD spectra were recorded with a J-1500 circular dichroism spectrometer (JASCO, Japan). IR data were measured with an IR Affinity-1S spectrometer (Shimadzu, Japan). NMR experiments were recorded with a Bruker Ascend-600 spectrometer (Bruker, Germany) with TMS used as an internal standard. An Agilent-6230 Q-TOF mass spectrometer with an Agilent 1260 UHPLC system was used to obtain HRESIMS data (Agilent, USA). HRESIMS/MS analyses for 7 and 11 were performed on an Agilent-6230 Q-TOF with the energy variable from 10 to 40 eV. Medium-pressure liquid chromatography (MPLC) was performed on a Buchi C-620 system (Buchi, Switzerland) equipped with a Siliabond C18 column (ODS gel, 5 μm, 49 × 460 mm) with a flow rate of 100 mL/min. Semipreparative HPLC was performed on an Agilent 1260 HPLC system (Agilent, USA) coupled with a DAD detector by using an XBrdige C18 column (5 μm, 10 × 250 mm, Waters, USA) at a flow rate of 3 mL/min. Silica gel (200–300 mesh, Qingdao Haiyang Company, People’s Republic of China) was used for column chromatography (CC). The analysis by thin-layer chromatography (TLC) was carried out by silica gel GF254 (Merck, Darmstadt, Germany), and potassium bismuth iodide was used as a visualizing reagent.
Analytical grade petroleum ether (PE), ethyl acetate (EtOAc), and methanol (MeOH) and HPLC grade MeOH and acetonitrile (MeCN) were purchased from Anaqua Global International Inc. (Cleveland, OH, USA). CDCl3 containing TMS (1%) was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). Deionized water was obtained using a Millipore Milli-Q-plus system (Millipore, Bedford, MA, USA).
The primary antibody against iNOS (CST no. 13120S) was purchased from Cell Signaling Technology (Beverly, MA, USA). The primary antibody against GAPDH (cat. no. 60004-1-Ig) was obtained from Proteintech Group (Chicago, IL, USA). Secondary antibodies, goat anti-mouse (cat. no. 926-32210) and goat anti-rabbit (cat. no. 926-32211) IgG, were bought from LI-COR 800CW (Lincoln, NE, USA). The anti-inflammatory agent dexamethasone (lot no. 923H058) was purchased from Solarbio (China). LPS was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Plant Material
The fruits of C. abrotanoides were purchased in May 2022 from the Bozhou medicinal materials market in Anhui Province, People’s Republic of China, and identified by Dr. Guo-yuan Zhu from Macau University of Science and Technology. The voucher specimen (CA-2022-05) is already stored in the State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology.
Extraction and Isolation
The dried fruits of C. abrotanoides (15 kg) were powdered and extracted with 80% EtOH at room temperature. The extract was concentrated under reduced pressure to afford 1.6 L of the concentrated extract, which was then partitioned successively with PE and EtOAc. The 1H and 13C NMR spectra of PE and EtOAc extracts demonstrated the characteristic signals indicative of carabranolide derivatives, which were used to guide the further isolation of targeted compounds.
The PE extract (241g) was separated by silica gel CC (PE–EtOAc–MeOH, 1:0:0 to 0:1:1), and the eluates were analyzed by TLC and combined to give 6 fractions. NMR analyses of these fractions showed that fraction P.3 contained carabranolide derivatives. Fraction P.3 (26 g) was then subjected to preparative MPLC (MeCN–H2O, 40:60 to 100:0) to give 9 subfractions (P.3.1–9). Fraction P.3.6 (2.4 g) was further fractionated by MPLC (MeCN–H2O, 85:15) to yield 4 subfractions (P.3.6.1–4). Fraction P.3.6.2 was purified by semipreparative HPLC (MeCN–H2O, 90:10) to afford 12 (tR 18 min, 12 mg). Afterward, compounds 11 (tR = 15 min, 16 mg) and 13 (tR = 32 min, 28 mg) were purified by HPLC (MeOH–H2O, 90:10) from fraction P.3.6.3.
The EtOAc extract (678 g) was fractionated by silica gel CC (PE–EtOAc–MeOH, 1:0:0 to 0:1:1). A total of 50 fractions (1000 mL/each) were collected. After TLC analysis, the fractions were combined into 9 fractions. Carabrol derivatives were found in fractions E.6 and E.9 by NMR analysis. Fraction E.6 (98 g) was further separated into 10 fractions (E.6.1–10) by MPLC with a gradient mobile phase of MeCN–H2O (30:100 to 100:0). Fraction E.6.8 was separated by HPLC with MeCN and H2O (78:22) as eluent to yield 8 subfractions (E.6.8.1–8). Fraction E.6.8.2 was purified by preparative HPLC eluted with MeCN–H2O (80:20) to give 3 (tR 23 min, 6.2 mg). Compounds 8 (tR 28 min, 3.1 mg) and 10 (tR 31 min, 2.8 mg) were obtained from fraction E.6.8.8 via semipreparative HPLC eluted with a MeCN–H2O mobile phase (90:10). Fraction E.7 (50 g) was subjected to MPLC eluted with MeCN–H2O (30:100 → 100:0), giving 11 fractions (E.7.1–11). Fraction E.7.4 (2.5 g) was separated again into 9 subfractions (E.7.4.1–9) by MPLC with 70% MeCN. Compound 9 (tR 30 min, 2.2 mg) was obtained by HPLC eluted with 95% ACN from fraction E.7.4.4. Fraction E.7.4.5 was purified by semipreparative HPLC (MeOH–H2O, 60:40) to give 15 (tR 12 min, 11 mg) and 14 (tR 18 min, 3.5 mg). Fraction E.7.5 was separated by MPLC (MeCN–H2O, 70:30 → 100:0) to give 11 fractions (E.7.1–11). Fraction E.7.5.5 was applied to semipreparative HPLC (MeOH–H2O, 61:39) to yield 16 (tR 21 min, 3.6 mg). Moreover, compound 4 (tR 28 min, 1.1 mg) was obtained from Fr.E.7.5.6. via semipreparative HPLC eluted with 75% MeCN. Fraction E.7.7 (3.3 g) was separated by HPLC (MeCN–H2O, 60:40) to yield 12 fractions (E.7.7.1–12). Fraction E.7.7.12 was purified by semipreparative HPLC eluted with 82% MeOH to give 7 (tR 21 min, 3.2 mg). Fraction E.9 (95 g) was separated by MPLC (MeCN–H2O 70:100 to 100:0) to obtain 8 fractions (E.9.1–8). Fraction E.9.4 (4.5 g) was subjected to MPLC eluted with isocratic MeCN–H2O (80:20) to get 11 fractions (E.9.4.1–11). Subsequently, fraction E.9.4.9 (79 mg) was further purified by semipreparative HPLC (MeOH–H2O, 85:15) to give 1 (tR 31 min, 3.8 mg) and 2 (tR 39 min, 3.2 mg). Fraction E.9.4.11 was fractionated using HPLC eluted with 89% MeOH to get 6 fractions (E.9.4.11.1–6). Subsequently, compounds 5 (tR 31 min, 1.2 mg) and 6 (tR 39 min, 2.9 mg) were isolated via semipreparative HPLC eluted with 85% MeOH from fraction E.9.4.11.4.
Carabrolate A (1)
Yellow oil; [α]25D +12.6 (c 0.5, MeOH); IR (KBr) νmax 3487, 2940, 2862, 2353, 1759, 1543, 1450, 1258, and 1188 cm–1; UV (MeOH) λmax (log ε) 195 (3.80) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 477.2833 [M + H]+ (calcd for C27H40O7, 477.2847).
Carabrolate B (2)
Yellow oil; [α]25D +21.9 (c 0.5, MeOH); IR (KBr) νmax 3487, 2932, 2862, 2361, 2338, 1759, 1366, 1265, and 1180 cm–1; UV (MeOH) λmax (log ε) 195 (4.00) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 477.2838 [M + H]+ (calcd for C27H40O7, 477.2847).
Carabrolate C (3)
Yellow oil; [α]25D +48.2 (c 0.5, MeOH); IR (KBr) νmax 2940, 2870, 2361, 2338, 1728, 1667, 1265, 1180, and 1150 cm–1; UV (MeOH) λmax (log ε) 229 (4.05) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 423.2135 [M + Na]+ (calcd for C24H32O5, 423.2142).
Carabrolate D (4)
Yellow oil; [α]25D +25.4 (c 0.5, MeOH); IR (KBr) νmax 3726, 2940, 2870, 2361, 2338, 1736, 1667, 1505, 1458, 1265, 1227, 1157, and 995 cm–1; UV (MeOH) λmax (log ε) 197 (4.21) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 399.216 [M + H]+ (calcd for C24H30O5, 399.2166).
Carabrolate E (5)
Yellow oil; [α]25D +17.1 (c 0.5, MeOH); IR (KBr) νmax 3688, 3372, 2932, 2862, 1728, 1458, 1373, 1350, 1265, 1188, and 1150 cm–1; UV (MeOH) λmax (log ε) 195 (3.90) nm; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 580.4204 [M + NH4]+ (calcd for C33H54O7, 580.4208).
Carabrolate F (6)
Yellow oil; [α]25D +26.60 (c 0.5, MeOH); IR (KBr) νmax 3726, 3402, 2932, 2862, 2361, 1759, 1728, 1667, 1458, 1373, 1265, 1188, 1142, 1103, 1042, and 988 cm–1; UV (MeOH) λmax (log ε) 195 (3.92) nm; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS m/z 580.4202 [M + NH4]+ (calcd for C33H54O7, 580.4208).
Carabrolate G (7)
Yellow oil; [α]25D +23.5 (c 0.5, MeOH); IR (KBr) νmax 3626, 2931.80 2862.36 2360.87 2338, 1759, 1728, 1458, 1373, 1265, 1180, and 995 cm–1; UV (MeOH) λmax (log ε) 195 (4.14) nm; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS m/z 543.3656 [M + Na]+ (calcd for C31H52O4, 543.36525); (+)-HRESIMS/MS: 233.1531 [C15H21O2]+, 271.2225 [C16H31O3]+, 187.1491 [C10H19O3]+, and 169.1209 [C10H17O2]+.
Carabrolate H (8)
Yellow oil; [α]25D +22.0 (c 0.5, MeOH); IR (KBr) νmax 3726, 2932, 2862, 2361, 1728, 1373, 1188, and 995 cm–1; UV (MeOH) λmax (log ε) 195 (3.83) nm; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS m/z 429.2608 [M + Na]+ (calcd for C24H38O5, 429.2611).
Carabrolate I (9)
Yellow oil; [α]25D +23.5 (c 0.5, MeOH); IR (KBr) νmax 3726, 2924, 2855, 2361, 2338, 1759, 1728, 1265, 1180, 1150, and 995 cm–1; UV (MeOH) λmax (log ε) 211 (3.80) nm; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS m/z 541.3854 [M + Na]+ (calcd for C32H54O5, 541.3863).
Carabrolate J (10)
Yellow oil; [α]25D +20.0 (c 0.5, MeOH); IR (KBr) νmax 3726, 2932, 2862, 2361, 2338, 1759, 1728, 1458, 1265, 1180, 1150, 1096, 995, and 964 cm–1; UV (MeOH) λmax (log ε) 212 (4.17) nm; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS m/z 421.2577 [M + H]+ (calcd for C24H36O6, 421.2585).
Carabrolate K (11)
Yellow oil; [α]25D +12.8 (c 0.5, MeOH); IR (KBr) νmax 2361, 2338, 1728, 1659, 1188, and 995 cm–1; UV (MeOH) λmax (log ε) 195 (3.82) nm; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS m/z 537.3910 [M + Na]+ (calcd for C33H54O4, 537.3914). (+)-HRESIMS/MS: 233.1542 [C15H21O2]+, 155.0859 [C9H15O2]+, and 143.0819 [C8H14O2]+.
(S)-MTPA ester of 5a (5b):
1HNMR (C5D5N, 600 MHz) δH 0.42 (1H, m, H-1), 1.29 (2H, m, H-2), 1.59 (1H, m, H-3a), 1.74 (1H, m, H-3b), 5.11 (1H, m, H-4), 0.24 (1H, m, H-5), 0.82 (1H, m, H-6a), 2.22 (1H, d, J = 13.2 Hz, H-6b), 3.06 (1H, m, H-7), 4.77 (1H, m, H-8), 0.94 (1H, m, H-9a), 2.21 (1H, m, H-9b), 5.51 (1H, br s, H-13a), 6.30 (1H, br s, H-13b), 0.96 (3H, s, H-14), 0.84 (3H, d, J = 6.2 Hz, H-15), 2.37 (2H, m, H-2′), 1.57–1.78 (4H, overlapped, H-3′ and 4′), 1.24 (2H, m, H-5′), 5.27 (1H, m, H-6′), 5.64 (1H, overlapped, H-7′), 5.68 (1H, overlapped, H-8′), 4.26 (1H, t, J = 6.1 Hz, H-9′), 3.84 (1H, m, H-10′), 1.17–1.34 (14H, overlapped, H-11′–17′), 0.83 (3H, t, J = 6.3 Hz, H-18′), 1.22 (3H, s, H-20′), 1.23 (3H, s, H-21′).
(R)-MTPA ester of 5a (5c):
1HNMR (C5D5N, 600 MHz) δH 0.42 (1H, m, H-1), 1.30 (2H, m, H-2), 1.60 (1H, m, H-3a), 1.75 (1H, m, H-3b), 5.10 (1H, m, H-4), 0.24 (1H, m, H-5), 0.84 (1H, m, H-6a), 2.22 (1H, d, J = 13.2 Hz, H-6b), 3.05 (1H, m, H-7), 4.77 (1H, m, H-8), 0.92 (1H, m, H-9a), 2.19 (1H, m, H-9b), 5.51 (1H, d, J = 2.6 Hz, H-13a), 6.30 (1H, d, J = 2.6 Hz, H-13b), 0.97 (3H, s, H-14), 0.84 (3H, d, J = 6.5 Hz, H-15), 2.38 (2H, m, H-2′), 1.57–1.77 (4H, overlapped, H-3′ and 4′), 1.31 (2H, m, H-5′), 5.27 (1H, m, H-6′), 5.62 (1H, dd, J = 15.6, 6.1 Hz, H-7′), 5.64 (1H, dd, J = 15.6, 6.2 Hz, H-8′), 4.26 (1H, t, J = 6.2 Hz, H-9′), 3.85 (1H, m, H-10′), 1.19–1.33 (14H, overlapped, H-11′∼17′), 0.85 (3H, t, J = 6.7 Hz, H-18′), 1.19 (3H, s, H-20′), 1.20 (3H, s, H-21′).
(S)-MTPA ester of 6a (6b):
1HNMR (C5D5N, 600 MHz) δH 0.42 (1H, td, J = 7.3, 3.8 Hz, H-1), 1.28 (2H, m, H-2), 1.59 (1H, m, H-3a), 1.74 (1H, m, H-3b), 5.11 (1H, m, H-4), 0.26 (1H, td, J = 7.9, 4.2 Hz, H-5), 0.81 (1H, m, H-6a), 2.20 (1H, d, J = 13.4 Hz, H-6b), 3.02 (1H, m, H-7), 4.78 (1H, ddd, J = 11.5, 8.7, 6.6 Hz, H-8), 0.96 (1H, m, H-9a), 2.18 (1H, dd, J = 13.1, 6.5 Hz, H-9b), 5.51 (1H, d, J = 2.5 Hz, H-13a), 6.30 (1H, d, J = 2.5 Hz, H-13b), 0.97 (3H, s, H-14), 0.84 (3H, d, J = 6.6 Hz, H-15), 2.38 (2H, m, H-2′), 1.57–1.74 (4H, overlapped, H-3′ and 4′), 1.73 (2H, m, H-5′), 5.73 (1H, q, J = 6.6 Hz, H-6′), 5.95 (1H, dd, J = 15.2, 6.6 Hz, H-7′), 5.98 (1H, dd, J = 15.2, 6.1 Hz, H-8′), 4.16 (1H, t, J = 6.1 Hz, H-9′), 3.80 (1H, m, H-10′), 1.19–1.38 (14H, overlapped, H-11′∼17′), 0.83 (3H, t, J = 7.8 Hz, H-18′), 1.24 (3H, s, H-20′), 1.26 (3H, s, H-21′).
(R)-MTPA ester of 6a (6c):
1HNMR (C5D5N, 600 MHz) δH 0.41 (1H, td, J = 7.1, 3.6 Hz, H-1), 1.29 (2H, m, H-2), 1.56 (1H, m, H-3a), 1.73 (1H, m, H-3b), 5.11 (1H, m, H-4), 0.26 (1H, td, J = 7.7, 4.1 Hz, H-5), 0.80 (1H, m, H-6a), 2.18 (1H, d, J = 13.2 Hz, H-6b), 3.03 (1H, m, H-7), 4.76 (1H, m, H-8), 0.92 (1H, m, H-9a), 2.16 (1H, d, J = 13.0 Hz, H-9b), 5.51 (1H, s, H-13a), 6.30 (1H, s, H-13b), 0.96 (3H, s, H-14), 0.84 (3H, d, J = 6.4 Hz, H-15), 2.37 (2H, m, H-2′), 1.58–1.75 (4H, overlapped, H-3′ and 4′), 1.70 (2H, m, H-5′), 5.73 (1H, q, J = 6.8 Hz, H-6′), 6.00 (1H, dd, J = 15.1, 6.3 Hz, H-7′), 5.98 (1H, dd, J = 15.1, 6.8 Hz, H-8′), 4.16 (1H, t, J = 6.8 Hz, H-9′), 3.79 (1H, m, H-10′), 1.18–1.38 (14H, overlapped, H-11′∼17′), 0.83 (3H, t, J = 7.3 Hz, H-18′), 1.24 (3H, s, H-20′), 1.26 (3H, s, H-21′).
(S)-MTPA ester of 9 (9a):
1HNMR (C5D5N, 600 MHz) δH 0.43 (1H, td, J = 7.1, 3.9 Hz, H-1), 1.30 (2H, m, H-2), 1.63 (1H, m, H-3a), 1.76 (1H, m, H-3b), 5.11 (1H, dt, J = 12.7, 6.4 Hz, H-4), 0.28 (1H, td, J = 8.0, 4.2 Hz, H-5), 0.85 (1H, m, H-6a), 2.21 (1H, d, J = 13.1 Hz, H-6b), 3.01 (1H, m, H-7), 4.78 (1H, ddd, J = 11.6, 8.8, 6.2 Hz, H-8), 0.95 (1H, m, H-9a), 2.17 (1H, dd, J = 10.8, 5.0 Hz, H-9b), 5.51 (1H, d, J = 1.3 Hz, H-13a), 6.32 (1H, d, J = 1.3 Hz, H-13b), 0.98 (3H, s, H-14), 0.87 (3H, d, J = 5.8 Hz, H-15), 2.93 (2H, dd, J = 10.5, 3.0 Hz, H-2′), 5.87 (1H, m, H-3′), 1.75 (2H, m, H-4′), 1.22–1.31 (24H, overlapped, H-5′∼16′), 0.88 (3H, t, J = 7.3 Hz, H-17′).
(R)-MTPA ester of 9 (9b):
1HNMR (C5D5N, 600 MHz) δH 0.42 (1H, td, J = 7.2, 4.0 Hz, H-1), 1.32 (2H, m, H-2), 1.58 (1H, m, H-3a), 1.74 (1H, m, H-3b), 5.08 (1H, dt, J = 12.7, 6.4 Hz, H-4), 0.26 (1H, td, J = 8.0, 4.2 Hz, H-5), 0.84 (1H, m, H-6a), 2.22 (1H, d, J = 13.9 Hz, H-6b), 3.01 (1H, m, H-7), 4.78 (1H, ddd, J = 12.9, 8.8, 6.7 Hz, H-8), 0.96 (1H, m, H-9a), 2.17 (1H, dd, J = 12.8, 6.8 Hz, H-9b), 5.51 (1H, d, J = 2.1 Hz, H-13a), 6.31 (1H, d, J = 2.1 Hz, H-13b), 0.98 (3H, s, H-14), 0.87 (3H, d, J = 5.7 Hz, H-15), 2.90 (2H, dd, J = 10.0, 2.0 Hz, H-2′), 5.90 (1H, m, H-3′), 1.82 (2H, m, H-4′), 1.21–1.30 (24H, overlapped, H-5′∼16′), 0.87 (3H, t, J = 7.1 Hz, H-17′).
Ketalization for Compounds 5 and 6
Treatment of 5 and 6 (1.0 mg) with PDSA (30 μg) in 400 μL of acetone afforded the acetone ketal at 37 °C for 2 h. The reaction was monitored by UHPLC-QTOF-MS analysis. Then the mixtures were partitioned with CHCl3 and H2O to give 5a and 6a.
Preparation of (S)- and (R)-MTPA Esters of 5a, 6a, and 9
Compounds 5a, 6a, and 9 (0.5 mg) were dissolved in 500 μL of deuterated pyridine. Then, 1.0 mg of 4-dimethylaminopyridine and 5 μL of (S)- or (R)-MTPA chlorides were added to the above solutions, which were allowed to react at room temperature for 2 h. The mixtures were subjected to an NMR spectrometer, and their 1H and 1H–1H COSY NMR spectra were recorded.
Cell Lines and Cell Culture
The RAW264.7 cell line was obtained from ATCC (USA) and cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) (Gibco, UK) in a humidified atmosphere with 5% CO2 at 37 °C.
Assay for NO Inhibitory Activity
NO inhibitory activity of selected compounds was evaluated by using an LPS-induced cell model. Briefly, RAW264.7 cells were inoculated into 96-well plates (1 × 105 cells/well) and were treated with the test compounds or dexamethasone (DEX, a positive control) at concentrations of 1.25, 2.5, 5, 10, and 20 μM in triplicate. After being induced with LPS (100 ng/mL) for 24 h, the cell culture supernatants were collected to measure levels of NO by using the Griess reagent (Beyotime Biotechnology, People’s Republic of China) according to the manufacturer’s instructions. Finally, the absorbance at 550 nm was measured with a microplate reader. Concurrently, the MTT assay was employed to assess the viability of RAW264.7 cells, thereby evaluating the cytotoxic effects of the compounds under investigation.34,35
Western Blot Analysis
In brief, RAW264.7 cells were seeded in six-well plates at a density of 5 × 105 cells/well and treated with various concentrations (10, 20, and 40 μM) of compounds 2–4, 15, and 16 or DEX (5 μM), followed by stimulation with LPS (0.5 μg/mL) for 24 h. Subsequently, cells were collected and lysed by using RIPA buffer. The extracted proteins were then separated by 8% SDS-PAGE and transferred onto PVDF membranes. These membranes were incubated overnight at 4 °C with primary antibodies of iNOS and GAPDH. After washing thrice with TBST, the membranes were exposed to a mouse monoclonal IgG conjugated secondary antibody for 1 h at room temperature. The protein bands were visualized by the LI-COR Odyssey imaging system (Lincoln, NE, USA).
Statistical Analysis
In the graph, data are presented as mean ± SD. The one-way ANOVA was used to assess the differences among the groups. Analysis of data was derived from the results of GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA). Values of p < 0.05 were considered to indicate statistical significance.
Acknowledgments
This work was funded by grants from the Macao Science and Technology Development Fund (0019/2022/AGJ and 006/2023/SKL).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.4c00338.
NMR, HRESIMS, IR, and UV spectra of compounds 1–11 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wu J.-W.; Tang C.; Ke C.-Q.; Yao S.; Liu H.-C.; Lin L.-G.; Ye Y. RSC Adv. 2017, 7 (8), 4639–4644. 10.1039/C6RA27626A. [DOI] [Google Scholar]
- Flora of China Editorial Committee, Chinese Academy of Sciences . Flora of China [M]; Science Press: Beijing, 1979; p 293. [Google Scholar]
- Zhang J.; Xu X.; Ye J.; Yang Y.; Gao S.; Li H.; Zhang W. Fitoterapia 2016, 110, 72–76. 10.1016/j.fitote.2016.02.013. [DOI] [PubMed] [Google Scholar]
- Jie-Wei W.; Chun-Ping T.; Sheng Y.; Chang-Qiang K.; Yang Y. Chin. J. Nat. Med. 2021, 19 (11), 868–873. [DOI] [PubMed] [Google Scholar]
- Yuan J.; Wen X.; Ke C.-Q.; Zhang T.; Lin L.; Yao S.; Goodpaster J. D.; Tang C.; Ye Y. Org. Chem. Front. 2020, 7 (11), 1374–1382. 10.1039/D0QO00093K. [DOI] [Google Scholar]
- Wang J.-F.; He W.-J.; Zhang X.-X.; Zhao B.-Q.; Liu Y.-H.; Zhou X.-J. Bioorg. Med. Chem. Lett. 2015, 25 (19), 4082–4084. 10.1016/j.bmcl.2015.08.034. [DOI] [PubMed] [Google Scholar]
- Wu H.-B.; Wu H.-B.; Wang W.-S.; Liu T.-T.; Qi M.-G.; Feng J.; Li X.-Y.; Liu Y. Ind. Crop. Prod. 2016, 92, 77–83. 10.1016/j.indcrop.2016.07.046. [DOI] [Google Scholar]
- Mayur B.; Sandesh S.; Shruti S.; Sung-Yum S. J. Med. Plants Res. 2010, 4 (15), 1547–1553. [Google Scholar]
- Wang F.; Yang K.; Ren F.-C.; Liu J.-K. Fitoterapia 2009, 80 (1), 21–24. 10.1016/j.fitote.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Qian Q.-G.; Gong L.-M.; Yang S.-H.; Zhao B.-Q.; Cai J.; Zhang Z.-J.; Wang Y.-H.; Zhou X.-J. Phytochem. Lett. 2020, 40, 5–9. 10.1016/j.phytol.2020.08.012. [DOI] [Google Scholar]
- Chinese Pharmacopoeia Commission . Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2020. [Google Scholar]
- Yang B.-J.; Wang J.; Zeng Z.-Q.; Yang X.; Huang A.-Y.; Hao X.-J.; Ding X.; Li S.-L. Nat. Prod. Res. 2022, 36 (12), 3207–3210. 10.1080/14786419.2021.1955881. [DOI] [PubMed] [Google Scholar]
- Wang Y.-F.; Fu Y.; Ji Y.-N.; Shi N.-N.; Sauriol F.; Lu X.-H.; Gu Y.-C.; Shi Q.-W.; Huo C.-H. Phytochemistry 2022, 203, 113389 10.1016/j.phytochem.2022.113389. [DOI] [PubMed] [Google Scholar]
- Ibrahim S. R. M.; Fadil S. A.; Fadil H. A.; Hareeri R. H.; Abdallah H. M.; Mohamed G. A. Plants 2022, 11 (12), 1598. 10.3390/plants11121598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. X. China. J. Chin. Mater. Med. 2016, 41 (11), 2105–2111. [DOI] [PubMed] [Google Scholar]
- Feng J.-T.; Wang H.; Ren S.-X.; He J.; Liu Y.; Zhang X. J. Agric. Food Chem. 2012, 60 (15), 3817–3823. 10.1021/jf205123d. [DOI] [PubMed] [Google Scholar]
- Hu Q.-L.; Wu P.-Q.; Liu Y.-H.; Qi F.-M.; Yu C.-X.; Zhao Y.; Yu Y.-F.; Fei D.-Q.; Zhang Z.-X. Phytochem. Lett. 2018, 27, 154–159. 10.1016/j.phytol.2018.07.010. [DOI] [Google Scholar]
- Liu C. Z.; Xu J.; Gui L. P.; Guo Y. Q. Studies on chemical constituents of Carpesii Fructus. Drug Eval. Res. 2010, 33 (3), 220–221. [Google Scholar]
- Li X.-W.; Weng L.; Gao X.; Zhao Y.; Pang F.; Liu J.-H.; Zhang H.-F.; Hu J.-F. Bioorg. Med. Chem. Lett. 2011, 21 (1), 366–372. 10.1016/j.bmcl.2010.10.138. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Cheng Z.; He Q.; Lin B.; Gao P.; Li L.; Liu Q.; Song S. Fitoterapia 2016, 110, 44–51. 10.1016/j.fitote.2016.02.011. [DOI] [PubMed] [Google Scholar]
- Kataoka S.; Hanada Y. Bull. Agric. Chem. Soc. Jpn. 1956, 20 (sup1), 223–242. 10.1080/03758397.1956.10857340. [DOI] [Google Scholar]
- Sebej P.; Lim B. H.; Park B. S.; Givens R. S.; Klan P. Org. Lett. 2011, 13 (4), 644–647. 10.1021/ol102887f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides A.; Napolitano A.; Bassarello C.; Carbone V.; Gazzerro P.; Malfitano A.; Saggese P.; Bifulco M.; Piacente S.; Pizza C. J. Nat. Prod. 2009, 72 (5), 813–817. 10.1021/np8006205. [DOI] [PubMed] [Google Scholar]
- Saikia B.; Devi T. J.; Barua N. C. Tetrahedron 2013, 69 (9), 2157–2166. 10.1016/j.tet.2012.12.076. [DOI] [Google Scholar]
- Venkatesha N. J.; Bhat Y. S.; Prakash B. S. J. RSC Adv. 2016, 6 (23), 18824–18833. 10.1039/C6RA01437B. [DOI] [Google Scholar]
- Huynh T.-H.; Kim H. K.; Lee J.; Ban Y. H.; Jang Y.-J.; Heo B. E.; Nguyen T. Q.; An J. S.; Kwon Y.; Nam S.-J.; Jang J.; Oh K.-B.; Shin M.-K.; Oh D.-C. J. J. Nat. Prod. 2024, 87 (3), 591–599. 10.1021/acs.jnatprod.3c01043. [DOI] [PubMed] [Google Scholar]
- Kwon H. C.; Kauffman C. A.; Jensen P. R.; Fenical W. J. Am. Chem. Soc. 2006, 128 (5), 1622–1632. 10.1021/ja0558948. [DOI] [PubMed] [Google Scholar]
- Ahmed A.; Crawford T.; Gould S.; Ha Y. S.; Hollrah M.; Noor-E-Ain F.; Dickman M. B.; Dussault P. H. Phytochemistry 2003, 63 (1), 47–52. 10.1016/S0031-9422(03)00003-7. [DOI] [PubMed] [Google Scholar]
- Chapuis C.; Robvieux F.; Cantatore C.; Saint-Léger C.; Maggi L. Helv. Chim. Acta 2012, 95 (3), 428–447. 10.1002/hlca.201100398. [DOI] [Google Scholar]
- Senthil Balan S.; Ganesh Kumar C.; Jayalakshmi S. Process. Biochem. 2019, 80, 171–180. 10.1016/j.procbio.2019.02.005. [DOI] [Google Scholar]
- Martins I. C. B.; Sardo M.; Santos S. M.; Fernandes A.; Antunes A.; André V.; Mafra L.; Duarte M. T. Cryst. Growth Des. 2016, 16 (1), 154–166. 10.1021/acs.cgd.5b01057. [DOI] [Google Scholar]
- Ding W.; Li Y.; Tian X.; Xiao Z.; Li R.; Zhang S.; Yin H. Molecules 2023, 28 (5), 2133. 10.3390/molecules28052133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J.; Lim H. J.; Lee D. Y.; Jung H.; Kim M.-R.; Moon D.-C.; Kim K. I.; Lee M.-S.; Ryu J.-H. Biochem. Bioph. Res. Co. 2010, 391 (3), 1400–1404. 10.1016/j.bbrc.2009.12.073. [DOI] [PubMed] [Google Scholar]
- Lyu H. Y.; Bao M. Y.; Io C. C.; Xiong H. M.; Chen F.-L.; Bai L. P.; Zhang W.; Jiang Z. H.; Zhu G. Y. Fitoterapia 2023, 169, 105604 10.1016/j.fitote.2023.105604. [DOI] [PubMed] [Google Scholar]
- Ren W.-J.; Io C.-C.; Jiang R.; Ng K.-F.; Liu J.-Z.; Bai L.-P.; Zhang W.; Jiang Z.-H.; Liu Y.-H.; Zhu G.-Y. J. Nat. Prod. 2023, 86 (5), 1230–1239. 10.1021/acs.jnatprod.2c01136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.