

Abstract
Anaerobic protists frequently harbour methanogenic archaea, which apparently contribute to the hosts’ fermentative metabolism by consuming excess H2. However, the ecological properties of endosymbiotic methanogens remain elusive in many cases. Here we investigated the ecology and genome of the endosymbiotic methanogen of the Cononympha protists in the hindgut of the termite Coptotermes formosanus. Microscopic and 16S rRNA amplicon sequencing analyses revealed that a single species, designated here “Candidatus Methanobrevibacter cononymphae”, is associated with both Cononympha leidyi and Cononympha koidzumii and that its infection rate in Cononympha cells varied from 0.0% to 99.8% among termite colonies. Fine-scale network analysis indicated that multiple 16S rRNA sequence variants coexisted within a single host cell and that identical variants were present in both Cononympha species and also on the gut wall. Thus, “Ca. Methanobrevibacter cononymphae” is a facultative endosymbiont, transmitted vertically with frequent exchanges with the gut environment. Indeed, transmission electron microscopy showed escape or uptake of methanogens from/by a Cononympha cell. The genome of “Ca. Methanobrevibacter cononymphae” showed features consistent with its facultative lifestyle: i.e., the genome size (2.7 Mbp) comparable to those of free-living relatives; the pseudogenization of the formate dehydrogenase gene fdhA, unnecessary within the non-formate-producing host cell; the dependence on abundant acetate in the host cell as an essential carbon source; and the presence of a catalase gene, required for colonization on the microoxic gut wall. Our study revealed a versatile endosymbiosis between the methanogen and protists, which may be a strategy responding to changing conditions in the termite gut.
Keywords: symbiosis, endosymbiont, gut microbiome, insect, protozoa, methanogen, Methanobrevibacter
Graphical Abstract
Graphical Abstract.
Introduction
Methanogenic archaea, or methanogens, play a key role in the final step of the decomposition of organic matter in various anaerobic environments [1]. In the guts of termites, for example, which are keystone animals in the terrestrial carbon cycle, methanogens accounted for 0%–10% of the prokaryotic gut community and possibly produce 1%–3% of the global methane [2, 3]. Methanogens are present in the guts of diverse termite lineages [4, 5] and are considered to contribute to the gut ecosystem as H2-sinks by producing methane from H2 and CO2 [6, 7]. Methanogens are found on the hindgut epithelium [4, 8, 9] or within the cells of certain gut protist species [10]. The protists in the termite gut belong to the phylum Parabasalia or the order Oxymonadida in the phylum Preaxostyla and play major roles in the digestion of lignocellulose [11]. These gut protists generally harbour endo- or ectosymbiotic prokaryotes, which likely contribute to the host nutrition by supplying nitrogenous compounds [11] and/or removing H2 [12, 13]. In addition to the mutualistic symbionts, commensal or parasitic endosymbiotic bacteria are occasionally present [14].
Endosymbiotic methanogens are observed, for example, in the parabasalid protist Trichomitopsis termopsidis in the gut of the termite Zootermopsis angusticolis [15, 16] and the oxymonad protist Dinenympha parva in the gut of the termite Reticulitermes speratus [17, 18]. It has been hypothesized that endosymbiotic methanogens promote wood decomposition of the protist hosts by consuming excess H2 [10], but their detailed physiology and ecology remain unclear. Indeed, although there are numerous examples of endosymbiosis between anaerobic protists and methanogens in various environments, the ecological properties of endosymbiotic methanogens, including whether they are obligate or facultative endosymbionts, are elusive in many cases [19, 20].
The Formosan subterranean termite Coptotermes formosanus is the most destructive pest of wooden constructions in southern China and Japan and has invaded Hawaii and the southern part of the USA [21]. The termite harbours a relatively simple protistan gut community consisting of five unculturable parabasalid species: Pseudotrichonympha grassii, Holomastigotoides hartmanii, Holomastigotoides minor, Cononympha [Spirotrichonympha] leidyi, and Cononympha koidzumii [22–24]. Among them, endosymbiotic methanogens are found only in Cononympha cells [25, 26], which are mainly localized in the posterior part of the hindgut [27, 28]. Cononympha protists numerically dominate the protistan gut community, occupying 2200–10 900 out of 3800–12 900 total protist cells per gut [28]. Transcriptomic analysis of Cononympha cells suggested that Cononympha hydrolyzed cellulose, hemicellulose, pectin, and chitin, contributing to both lignocellulose digestion and nitrogen recycling [24].
In the present study, we examined the ecology and metabolic capacity of the uncultured endosymbiotic methanogen of Cononympha species in the gut of C. formosanus using a combination of microscopy, fine-scale phylogenetic analysis, and genome sequence analysis. Here, we show evidence of facultative endosymbiosis with frequent horizontal acquisitions of the methanogen and its genome features adapted to the lifestyle. Our study sheds light on another aspect of the multilayered symbiotic system in the termite gut.
Materials and Methods
Termite collection and methanogen infection rate
C. formosanus (family Rhinotermitidae) were collected from four prefectures in Japan (Table S1). The termite colonies collected in 2019 or later were kept with their nest logs in plastic containers in a laboratory until use. Colonies collected in 2018 or earlier were kept in laboratories for years fed with red pine chips. The entire guts of three worker termites per colony were removed using sterile forceps, and the gut contents were suspended in sterile solution U [29]. The ratio of methanogen-containing Cononympha cells to total Cononympha cells in individual guts was examined on the basis of F420 autofluorescence under an Olympus BX51 epifluorescence microscope. The procedure of CH4 measurement emitted by termites and semi-quantitative PCR amplification of the gene for methyl coenzyme M reductase (McrA) are described in Supplemental Methods.
Collection of single Cononympha cells and sequencing of 18S rRNA genes
Single Cononympha cells were collected using a Leica AM6000 micromanipulation system and subjected to whole genome amplification (WGA) using the illustra GenomiPhi V2 kit (GE Healthcare) as described previously [30]. To identify the species of Cononympha, the 18S rRNA gene was amplified by PCR using Cononympha-specific primers designed in this study (Table S2). The PCR conditions are described in Supplemental Methods. The PCR products were purified, cloned, and sequenced using the Sanger method as described previously [30].
16S rRNA amplicon sequencing analysis
The V3–V4 region (ca. 400 bp) of the 16S rRNA gene was amplified by PCR with prokaryote-universal primers, Pro341F, and Pro805R (Table S2), using Phusion High-Fidelity DNA Polymerase (New England Biolabs), as described previously [31]. Purification, library preparation, and paired-end sequencing (300 bp × 2) on the MiSeq platform with the MiSeq Reagent Kit v3 (Illumina) were conducted as described previously [31]. The paired-end reads were trimmed, quality filtered, and sorted into amplicon sequence variants (ASVs) using DADA2 v1.6 [32]. The ASVs were classified using SINA v1.2.11 [33] with the SILVA v132 database [34]. ASVs classified as Eukarya or undetermined and ASVs of which frequency was <0.1% of the total reads were discarded from subsequent analyses.
Phylogenetic analysis of 16S rRNA genes
Near full-length 16S rRNA genes of methanogens were amplified by PCR using Phusion Hi-Fidelity DNA Polymerase. Primers M23F and M1382R broadly targeting methanogens were used for gut wall samples, while primer CfC-M23F adjusted to the sequence of the endosymbiotic methanogen was used as the forward primer for endosymbiotic methanogens (Table S2). The PCR conditions and procedures of sequencing and alignment are described in Supplemental Methods. A maximum-likelihood tree was constructed using IQ-TREE v1.6.12 [35] with the TVM + F + I + G4 nucleotide substitution model selected by ModelFinder implemented in IQ-TREE. Network construction of sequence variants (SVs) was conducted using the PopART program depending on Qt v4.8.5 with TCS algorithms [36].
Fluorescence in situ hybridization and transmission electron microscopy
To discriminate between Con. leidyi and Con. koidzumii cells, oligonucleotide probes specific to each 18S rRNA sequence were designed using ARB [37] (Table S2). Fluorescence in situ hybridization (FISH) was performed as described previously [38] with hybridization at 55°C for 2 h. Observations were conducted under the Olympus BX51 epifluorescence microscope. Transmission electron microscopy (TEM) was performed as described previously [39] using an H-7500 transmission electron microscope (Hitachi).
Genome sequencing
Single Cononympha cells were collected as described above, washed several times in droplets of solution U, and transferred to solution U containing 0.1% Tween 20 (Nacalai Tesque). Prokaryotic cells that leaked out from the ruptured host cell were collected with a glass capillary attached to the Leica AM6000 micromanipulation system and subjected to WGA as described above. Nine WGA samples of single Cononympha cells were prepared. These were subjected to paired-end sequencing on the MiSeq platform. After assembling and binning as below, one sample was selected for deeper paired-end sequencing and mate-pair sequencing on MiSeq, and long-read sequencing on the MinION platform (Oxford Nanopore Technologies), based on the estimated genome completeness of the target methanogen. The procedures for library preparation and sequencing are described in Supplemental Methods.
Genome assembly
The MiSeq paired-end reads were trimmed, quality filtered, and assembled into contigs using SPAdes v3.9 [40]. The contigs were binned using a combination of MyCC [41] and Contig Annotation Tool (CAT) [42]. The results were evaluated for the selection of the best sample. The MinION reads, after being quality trimmed, were assembled together with the quality-trimmed MiSeq paired-end and mate-pair reads into contigs using SPAdes v3.15.2 with the “hybrid-assembly” and “meta” mode [40]. The assembled contigs ≥500 bp were binned using a combination of MyCC, CAT, RAST (http://rast.nmpdr.org/rast.cgi), and BLASTn searches. The bin affiliated with Methanobrevibacter was further subjected to scaffolding using LINKS v2.0.1 [43] and gap-closing using TGS-GapCloser [44]. The quality-trimmed MiSeq and MinION reads were mapped onto the gap-closed bin using BBMap v38.96 and Minimap2 v2.24-r1122 [45]. The reads mapped onto the bin were then reassembled using SPAdes v3.15.2 with the “hybrid-assembly” and “sc” mode. Reassembled contigs ≥1000 bp were binned as described above. Details of quality filtering are described in Supplemental Methods.
Gene annotation
Finding and functional annotation of genes were performed using a combination of DFAST [46] and BLASTp searches of the NCBI non-redundant (nr) protein database. CRISPR-Cas systems were identified using CRISPRCasFinder [47]. Assignment of COG (clusters of orthologous genes) categories [48] was conducted by RPS-BLAST v2.6.0+ searches of the NCBI Conserved Domain Database v3.16, and the results were manually curated. Pseudogenes were manually identified as described previously [49]. Metabolic pathways were inferred using the KEGG automatic annotation server (KAAS) [50] and KEGG Mapper [51].
Phylogenomics
To construct a phylogenomic tree, we retrieved genome sequences of Methanobacteriaceae as references from GTDB r202 [52] and metagenome-assembled genomes (MAGs) assigned to Methanobacteriaceae recently obtained from termite guts [5, 53]. Gene prediction, extraction, and alignment are described in Supplemental Methods. A maximum-likelihood tree was constructed using IQ-TREE v1.6.12 with the LG + F + R5 amino acid substitution model selected by ModelFinder. The robustness of the tree topology was evaluated using 1000 ultrafast bootstrap resamplings. Average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using ANI/AAI-Matrix [54].
Comparative genome analysis
The relative abundance of genes assigned to COG functional categories was calculated for genome sequences with >80% estimated completeness (Table S3). Principal component analysis was performed after a central log-ratio transformation using a script (https://github.com/dkato2021/COGplot.git). To compare genome contents with an identical criterion, the identification of intact genes or pseudogenes was automatically performed using Pseudofinder v1.1.0 [55] and the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [48].
Results
Discovery of Cononympha cells lacking endosymbiotic methanogens
During our preliminary observations of protists in the guts of C. formosanus by epifluorescence microscopy, we unexpectedly found that, in certain C. formosanus colonies, no Cononympha cells emitted F420 autofluorescence as a characteristic signal of the presence of endosymbiotic methanogens, unlike in other C. formosanus colonies (Fig. 1A–D) or previous reports [25, 26, 28]. To confirm the absence of methanogens in Cononympha cells in such termite colonies, we performed amplicon sequencing analysis of the 16S rRNA V3–V4 region for prokaryotic microbiota associated with individual Cononympha cells. A total of 30 single Cononympha cells collected from two C. formosanus colonies (O2020a and O2020b in Table S1), in which almost all Cononympha cells were without F420 autofluorescence, were examined. For comparison, we also conducted the same experiment using 28 single Cononympha cells from two colonies (K2019b and K2019c in Table S1), in which almost all Cononympha cells exhibited F420 autofluorescence. No 16S rRNA sequences of methanogens were obtained from the former 30 Cononympha cells, whereas sequences of Methanobrevibacter were recovered from all of the latter 28 Cononympha cells (Fig. S1).
Figure 1.

Detection of endosymbiotic methanogens of Cononympha protists in the hindgut of Coptotermes formosanus. (A, C) Phase-contrast images of Cononympha cells. (B, D) Epifluorescent images. Endosymbiotic methanogens are visible with their greenish F420 autofluorescence in panel B, whereas no signal was detected in panel D. Amorphous yellow is the autofluorescence of wood particles. Bars indicate 10 μm. (E) Ratio of methanogen-containing Cononympha cells to total Cononympha cells in individual termite guts across 33 termite colonies. Values indicate methane emission rates (nmol−1 g termite−1 h−1) averaged between biological replicates. Asterisks attached to termite colony IDs indicate laboratory-reared termite colonies.
Infection rate of methanogens in Cononympha cells and methane emission rate
We examined the infection rate of methanogens in Cononympha cells for 33 colonies of C. formosanus collected from four prefectures in Japan (Table S1), based on the detection of F420 autofluorescence. The infection rate greatly varied from 0% to 99.8% depending on the termite colonies (Fig. 1E; Tables S1 and S4). This difference was irrespective of natural or laboratory-reared termite colonies and prefectures of the sampling sites (Fig. 1E; Tables S1 and S4).
To test whether the presence or absence of the endosymbiotic methanogens of Cononympha is linked to the methane emission rate from the termite hosts, we chose three C. formosanus colonies of the high and low endosymbiotic methanogen-infection rate types, respectively (Fig. 1E; Table S1; Supplementary Methods). The results showed that termites from the colonies of the “high infection rate” type emitted 4.5–7.8 times more methane than those from the “low infection rate” type colonies (Fig. 1E). Semi-quantitative PCR targeting the mcrA gene in the entire gut microbiota generated results consistent with the methane emission rates (Fig. S2; Supplementary Methods). No mcrA amplification was detected in any of the four tested C. formosanus colonies of the “low infection rate” type under the PCR condition, where mcrA amplicons were detected in all six tested colonies of the “high infection rate” type (Fig. S2; Table S1).
Because the hindgut wall of C. formosanus can be another habitat of methanogens [56], we examined F420 autofluorescence signals on gut wall fragments from termite colonies of both “high infection rate” and “low infection rate” types. We observed dense colonization of rod-shaped methanogens on fragments of the hindgut wall from two out of seven colonies of the “high infection rate” type, whereas almost no signals were detected in termites from four colonies of the “low infection rate” type (Fig. S3; Table S1).
Fine-scale phylogenetic composition and host specificity of endosymbiotic methanogens
In the above 16S rRNA amplicon sequencing analysis (Fig. S1), multiple ASVs belonging to Methanobrevibacter were obtained from each Cononympha cell (Fig. S4). Nine closely related ASVs were obtained in total, showing only 1–4 mismatches out of 386 bp. Among them, the ASV-3 sequence was identical to the corresponding region of clone SlMeN10 (AB360373), which was derived from Cononympha cells in a C. formosanus gut [26]. Since the genome of the endosymbiotic methanogen contains only a single rRNA operon as described below, these multiple ASVs were not derived from intragenomic variation. These results indicated that multiple strains of a single Methanobrevibacter species frequently coexisted within a single Cononympha cell.
Next, we examined whether both Cononympha species, i.e., Con. leidyi and Con. koidzumii, can harbour methanogens. We obtained the 18S rRNA gene sequences of single Cononympha cells used in the above 16S rRNA amplicon analysis (Fig. S1) and found that both Cononympha species housed methanogens in colonies of the “high infection rate” type (Fig. S5; Table S5). Conversely, in colonies of the “low infection rate” type, both Cononympha species lacked methanogens (Fig. S5; Table S5). We observed that both Cononympha species, visually discriminated by FISH specifically targeting 18S rRNA of each species, harboured Methanobrevibacter-like rods (Fig. S6).
We further investigated whether the lineages of the endosymbiotic methanogens are distinct, even though closely related, between the two host Cononympha species or not. In addition, we examined the phylogenetic relationship between the endosymbionts and the methanogens on the gut wall. To examine these with higher resolution, we obtained near full-length 16S rRNA gene sequences of methanogens by PCR amplification from 14 single Con. leidyi cells, three single Con. koidzumii cells, and seven fragments of the hindgut wall (Fig. 2; Tables S1 and S5). Three out of four 16S rRNA gene SVs of methanogens associated with Con. koidzumii cells were also obtained from Con. leidyi cells. In addition, we obtained 16S rRNA gene amplicons of methanogens from three of the seven gut wall fragments and sequenced 24 clones in total, all of which showed 99%–100% sequence similarities to SVs of the endosymbionts (Fig. 2). These SVs from the gut wall or Cononympha cells formed clusters that were delineated mainly by the host C. formosanus colonies (Fig. 2); thus, the sequence variations of these methanogens were strongly related to the host termite colony and not to the Cononympha species.
Figure 2.
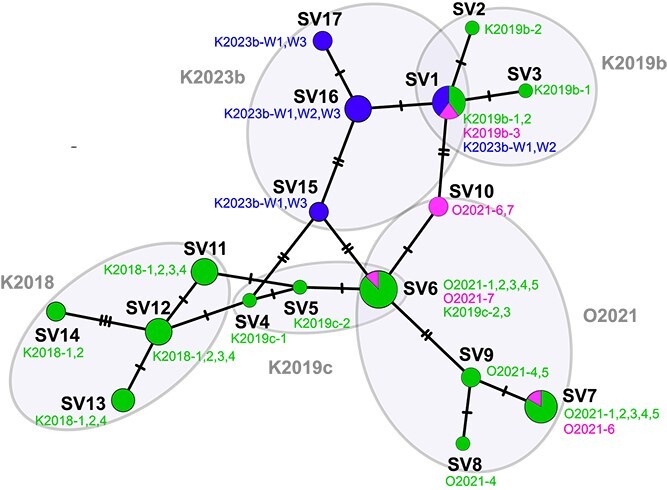
Network analysis of 16S rRNA gene sequence variants (SVs) of methanogens obtained from Cononympha leidyi (green), Cononympha koidzumii (magenta), and gut wall (blue). Unambiguously aligned 1312 nucleotide sites were used. The circle size of each SV is equivalent to the number of samples of single Cononympha cells or gut wall fragments, from which the corresponding SVs were detected. Slashes on branches indicate the number of nucleotide substitutions. The SVs were clustered by termite colonies, and their colony IDs and Cononympha cell IDs are shown (see Tables S1 and S5).
These data together indicated that the endosymbiosis between the Cononympha protists and the methanogens is facultative and that the two Cononympha species can horizontally acquire the same Methanobrevibacter species. Furthermore, the spatial distribution of the methanogen most likely extends to the gut wall of the termite host. The shape and size of the methanogens on the gut wall (Fig. S3A) and within Cononympha cells were similar. Both methanogens were straight rods, and the size was 1.1–1.8 μm by 0.4–0.5 μm (n = 50) in the former while it was 1.3–1.8 μm by 0.4–0.5 μm (n = 50) in the latter.
TEM of endosymbiotic methanogens
The facultative nature of this endosymbiosis was further corroborated by TEM. An electron micrograph showed that electron-dense, rod-shaped prokaryotic cells, morphologically indistinguishable, were present not only within a Cononympha cell but also on its surface (Figs 3 and S7A–D). They appeared to be in the process of phago- or exocytosis by the Cononympha cell (Figs 3 and S7A). These prokaryotic cells morphologically resembled cells of cultured Methanobrevibacter species [8, 9, 57]. The intracellular ones were surrounded by a host membrane (Figs 3 and S7A–D), and some were localized proximal to putative hydrogenosomes (Figs 3B and S7C). Intracellular Methanobrevibacter-like cells apparently during fission were observed (Fig. S7D); they likely proliferate within the host Cononympha cell. We hereafter designated this endosymbiotic methanogen as “Candidatus Methanobrevibacter cononymphae” (abbreviated as Mbv. cononymphae), and the species description is presented below.
Figure 3.
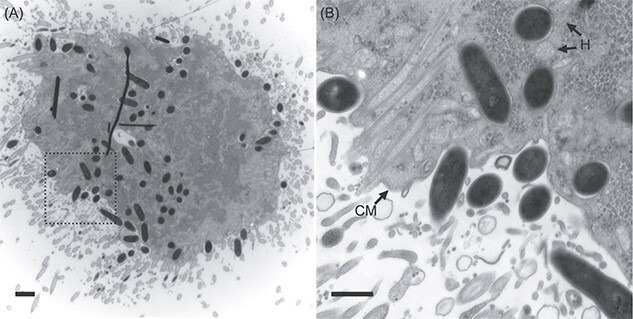
Transmission electron microscopy of a Cononympha cell. (A) Section of a Cononympha cell. Several black lines are artefacts. (B) Magnified view of the boxed region in panel (A). Electron-dense Methanobrevibacter-like cells were observed within the host cytoplasm and also on the host cell surface. H: Examples of putative hydrogenosomes; CM: Cytoplasmic membrane of Cononympha. Bars = 1 μm (A); 500 nm (B).
Phylogenetic position of Mbv. cononymphae based on 16S rRNA gene
Based on the near full-length 16S rRNA gene, Mbv. cononymphae was placed within a clade exclusively comprising uncultured clones derived from termite guts (Fig. S8). Within this clade, Mbv. cononymphae was closely related to clones obtained from the protists Microjoenia sp. and D. parva in the gut of R. speratus [17]. This clade further formed a monophyletic cluster with termite-gut-derived sequences, including cultured species inhabiting the gut epithelium of the protist-dependent termite Reticulitermes flavipes, i.e., Methanobrevibacter curvatus [8] and Methanobrevibacter filiformis [9]. Endosymbiotic Methanobrevibacter housed by an anaerobic ciliate, Nyctotherus ovalis, in the hindgut of the cockroach Blaptica dubia [58] and that housed by an aquatic anaerobic ciliate, Trimyema compressum [20], were more distantly related (Fig. S8).
General genome features of Mbv. cononymphae
We obtained a draft genome sequence of Mbv. cononymphae from a single Cononympha cell. The genome consisted of 245 contigs with 100% and 98.6% completeness estimated using CheckM [59] and CheckM2 [60], respectively (Table 1). Although only a single rRNA operon was identified in this genome as in many other Methanobrevibacter species (Table 1), we recovered four near full-length 16S rRNA gene SVs showing 3–5 base mismatches by PCR amplification from the same DNA sample. Thus, this genome sequence was derived from multiple, at least four, genomovars and here designated as the composite genome “CfCl-M3”.
Table 1.
General genome features of “Ca. Methanobrevibacter cononymphae” and related species.
| species | “Ca. Mbv. cononymphae” CfCl-M3 |
Mbv. curvatus
(GCA_001639295) |
Mbv. filiformis
(GCA_001639265) |
Mbv. cuticularis
(GCA_001639285) |
NOE (GCA_003315655) |
|---|---|---|---|---|---|
| Habitat | Intracellular/gut wall | Gut wall | Gut wall | Gut wall | Intracellular |
| Total length (Mb) | 2.71 (2.67d) | 2.41 | 2.61 | 2.61 | 1.91 |
| Contig | 244 | 232 | 295 | 169 | 70 |
| Completenessa (%) | 98.6 | 98.2 | 97.1 | 99.7 | 99.3 |
| G + C (%) | 27.2 | 25.7 | 27.0 | 26.8 | 25.3 |
| CDSb | 2319 (1834) | 1883 | 1985 | 1869 | 2175 |
| Coding density (%)b | 65.8 (59.7) | 68.5 | 68.5 | 66.2 | 68.3 |
| rRNA | 3 | 3 | 3 | 3 | 3 |
| tRNA | 28 | 31 | 30 | 32 | 33 |
| Pseudogenec | 29/174 (148) | 11/86 | 9/83 | 15/117 | 95/209 (187) |
Estimated using CheckM2 v.1.0.1.
Identified using Prodigal v2.6.3. Manually curated values are shown in parentheses.
Identified using Pgap/Pseudofinder with those within 1000 bp region from both contig ends being excluded. The number was manually curated for CfCl-M3 in this study and previously for NOE [57] and shown in parenthesis.
Total length with duplicated CDSs being excluded.
The total contig length was 2 709 899 bp, and 1834 protein-coding sequences (CDSs) were predicted, excluding 90 CDSs truncated by contig ends and 203 CDSs <100 amino acid sequences that were automatically predicted but with no significant hits in BLASTp searches of the NCBI nr protein database. At least, two CRISPR-Cas systems were identified with a total of 282 spacer sequences (Tables S6–S8). Most spacers showed no sequence similarity to those in CRISPRCasdb [61]. The number of pseudogenes was 148 after manual inspection (Table 1), and 14 were assigned to category [V] (defence mechanism) of COGs, including DNA restriction-modification systems and cas genes (Fig. S9; Table S6). Finding of duplicated regions and the frequency of single nucleotide polymorphisms in the CfCl-M3 composite genome are described in Supplemental Methods, Supplemental Results, and Table S9.
Phylogenomics and comparative genome analysis
A maximum-likelihood tree based on concatenated amino acid sequences of conserved single-copy genes (Fig. 4; Table S3) was basically congruent with the 16S rRNA gene tree (Fig. S8). Mbv. cononymphae belonged to a monophyletic cluster consisting exclusively of MAGs and two Methanobrevibacter isolates obtained from the guts of protist-dependent termites. These genomes shared at least 75% ANI and 60% AAI (Fig. S10). This clade was sister to a clade comprising genome sequences from protist-dependent/independent termite guts or other environments (Fig. 4).
Figure 4.

Phylogenetic position and characteristic genetic features of “Ca. Methanobrevibacter cononymphae”. Maximum likelihood tree was constructed based on 42 concatenated single-copy genes using the LG + F + R5 amino acid substitution model. Methanobacterium veterum MK4 and Methanobacterium bryantii M.o.H. were used as outgroups (Table S3). Ultrafast bootstrap values of 85–95% and ≥ 95% are indicated with open and closed circles, respectively. Genomes derived from protist-dependent and protist-independent termite species are highlighted in filled and unfilled diamonds, respectively. The genome completeness estimated using CheckM is shown by pie charts. The presence or absence of genes for formate dehydrogenase FdhAB, formate transporter FdhC, acetyl-CoA decarbonylase/synthase Cdh, cobalamin biosynthesis Cbi, and oxygen tolerance (catalase KatE, F420H2 oxidase FprA, and desulfoferredoxin Dfx) are linked to the phylogenetic tree. Existing and absent genes are indicated by filled (blue) and blank (grey) squares, respectively. Pseudogenes are indicated with “φ”. The fdhA gene of MAG_NG87 is at a contig end and truncated; therefore, it is impossible to specify whether it is intact or pseudogenized.
Genome sequences derived from termite guts in these two clades (Fig. 4) tended to possess more genes assigned to the COG categories [L] (replication, recombination, and repair) and [V] (defense mechanisms) (Fig. S11). Among the genes, those related to DNA methylation and drug resistance were especially abundant in termite-gut derived genomes (Fig. S12). No clear differences in the number and ratio of genes assigned to respective COG categories were observed among the five Methanobrevibacter species in Table 1, except for a much smaller number and ratio of genes assigned to [V] (defense mechanism) in Methanobrevibacter sp. NOE (Fig. S13), which is likely an obligate endosymbiont [57].
The number of pseudogenes of Mbv. cononymphae was approximately 1.5–2.1 times larger than those of the gut-wall dwelling Mbv. curvatus, Mbv. filiformis, and Methanobrevibacter cuticularis, but smaller (ca. 80%) than that of Methanobrevibacter sp. NOE (Table 1). The distribution pattern of pseudogenes among COG categories indicated that pseudogenes were characteristically accumulated in Mbv. cononymphae in categories [L] (replication, recombination, and repair), [V] (defence mechanism), and [X] (mobilome; prophages, transposons) (Fig. S14). No unique genes were identified in Mbv. cononymphae among COG-assigned 911 CDSs when compared to other known Methanobrevibacter species (Fig. S15; Table S10). Details are described in Supplemental Results.
Predicted metabolism of Mbv. cononymphae
All genes for enzymes required for energy conservation through methanogenesis were present in Mbv. cononymphae (Fig. 5). Similar to other Methanobrevibacter species, it likely produces methane from H2 and CO2. On the other hand, the formate dehydrogenase subunit alpha gene fdhA is pseudogenized, and the gene for formate transporter (fdhC) was missing (Fig. S16). Intact fdhA and fdhC genes were not found in sequence reads outside the assembled genome or in the other eight samples that were not selected for deeper sequencing analysis. In addition, genes for the acetyl-CoA decarbonylase/synthase (ACDS) multienzyme complex were absent; thus, Mbv. cononymphae apparently requires acetate to produce acetyl-CoA. The cobalamin biosynthetic pathway was absent, and instead, genes for the cobalamin/siderophore transporter FepBCD were identified (Table S6). These characteristics, i.e., loss or absence of genes for Fdh, ACDS, and cobalamin biosynthesis, were shared by most MAGs derived from the guts of protist-dependent termite species, but not shared by the gut wall-dwelling species Mbv. curvatus, Mbv. filiformis, and Mbv. cuticularis [8, 9] (Fig. 4).
Figure 5.
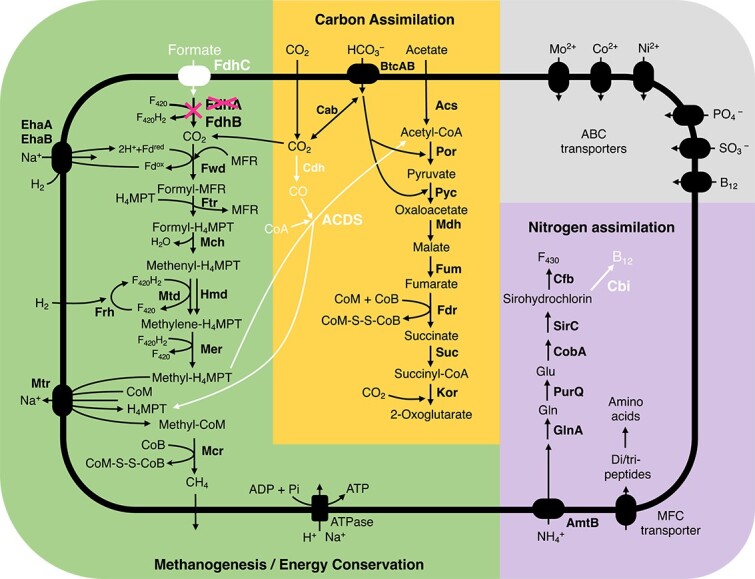
The predicted metabolic pathway of “Ca. Methanobrevibacter cononymphae.” Genes or pathways characteristically absent in Mbv. cononymphae are shown in white. The gene for formate dehydrogenase subunit alpha (fdhA) is pseudogenized.
Mbv. cononymphae possesses genes for ammonium transporter AmtB and glutamine synthetase GlnA. The genome also encoded a major facilitator superfamily transporter for di- and tripeptides, which may be other nitrogen sources (Fig. 5). The amino acid and cofactor synthesis capabilities were similar to those of other Methanobrevibacter species (Table S11). Genes for proteins involved in oxygen tolerance, including catalase KatE, were identified (Fig. 4), and details are described in Supplemental Results. No genes related to motility were found.
Discussion
This study unveiled the ecology and genomic features of the endosymbiotic methanogen Mbv. cononymphae harbored by two Cononympha species in the gut of C. formosanus. A series of evidence obtained by a combination of F420 autofluorescence detection, small subunit (SSU) rRNA gene sequence analysis, and TEM, clearly indicated that their association is facultative and that the methanogens are most likely transmitted in “mixed mode” [62], i.e., both vertically and horizontally among Cononympha cells across the two Cononympha species. In addition, our data suggest that Mbv. cononymphae colonizes the hindgut wall. This facultative lifestyle is consistent with its genomic features such as the genome size comparable to those of free-living relatives, the number of pseudogenes intermediate between free-living relatives and the putatively obligate endosymbiont NOE of the anaerobic ciliate N. ovalis [58] (Table 1), and the presence of a catalase gene (CfCl-M3_0439, Table S6).
Catalase activity was previously observed in Mbv. filiformis, Mbv. curvatus, and Mbv. cuticularis, all of which attach to the microoxic hindgut wall of R. flavipes [8, 9]. The catalase gene katE was commonly found in the clade containing Mbv. cononymphae and its sister clade; however, it is pseudogenized in Methanobrevibacter sp. NOE (Fig. 4). This is possibly because NOE, unlike Mbv. cononymphae, has become an obligate endosymbiont that rarely encounters a high concentration of oxygen.
Assuming that Cononympha cells produce H2, CO2, and acetate as the main fermentation products but do not produce formate, as observed in other cellulolytic parabasalids [63, 64] and predicted based on the transcriptome of Cononympha [24] (see Supplemental Methods), the loss of fdhC and pseudogenization of fdhA (Fig. S16) are likely attributable to endosymbiosis. In the hindgut of termites, the amount of formate is generally limited, but still appreciable amounts were detected [7, 65]. Indeed, the fdh operon is apparently intact in the genomes of the gut wall-dwelling species Mbv. filiformis, Mbv. curvatus, and Mbv. cuticularis (Fig. S16), even though these isolates exhibited only weak or no growth on formate [8, 9]. It is conceivable that the reduced opportunity and/or requirement to use formate has allowed the loss of functional fdh genes in Mbv. cononymphae. The lack of ACDS in Mbv. cononymphae may also be related to endosymbiosis. The expectedly high concentration of acetate within the host protist cell may have allowed the endosymbiont to depend solely on acetate to produce acetyl-CoA (Fig. 5). However, as the concentration of acetate is also high in the termite hindgut [7, 65], other factors may be involved in the absence of ACDS.
The absence of the functional biosynthetic pathway of cobalamin, which is essential in methanogenesis [66] (Fig. 5), was previously reported also in the endosymbiotic methanogens Methanobrevibacter sp. NOE and Methanocorpusculum sp. MCE [58]. Thus, its absence and dependence on a transporter may be a common trait in endosymbiotic methanogens. These characteristics, i.e., the absence of functional fdh, ACDS genes, cobalamin biosynthetic pathway, and the presence of the catalase gene were common in most MAGs from the guts of protist-dependent termites (Fig. 4). This implies that these methanogens possibly have a lifestyle similar to that of Mbv. cononymphae, i.e., facultative endosymbiosis, although their localizations are unidentified. Among them, MAG_NG87 from Coptotermes elisae [53] and MAG_GCA031280375 from Prorhinotermes canalifrons [5] showed high ANIs, 98.8% and 96.2%, respectively, with Mbv. cononymphae, and both termite genera harbour Cononympha species [67]; these MAGs are most likely of endosymbionts of Cononympha with similar lifestyles.
It is widely believed that hydrogenotrophic methanogens critically contribute to the fermentative metabolism of H2-producing anaerobic protist hosts as syntrophic mutualists [19]. Indeed, retarded growth of anaerobic ciliates that had lost their endosymbiotic methanogens was previously observed [68, 69]. In particular, removal of “Candidatus Methanoregula pelomyxae”, an endosymbiont of Pelomyxa schiedti, led to the death of the anaerobic amoeba host [70]. In the case of termite-gut protists, Messer and Lee (1989) [16] reported that Trichomitopsis termopsidis treated with 2-bromoethanesulfonate (BES) inhibiting methanogenesis was viable in the gut of Z. angusticollis. However, in an experiment using cultured Trichomitopsis termopsidis, the growth of the protist was severely retarded when methanogenesis was suppressed by BES treatment [64]. Its growth was recovered when fed with a more favorable nutrient source in addition to cellulose, i.e., autoclaved cells of a specific bacterial strain [64]. This reminded other reports that anaerobic ciliates during successive cultures under optimal conditions tended to lose their endosymbiotic methanogens, whereas ciliates under food-limiting conditions or at low temperatures kept housing methanogens [71, 72]. These previous results imply that the loss of Mbv. cononymphae may be related to the nutritional condition of the Cononympha host. It is unclear whether Mbv. cononymphae contributes to the nutrition of the Cononympha host, not only by consuming H2, but also by supplementing nitrogenous compounds as suggested in several cases of obligate endosymbioses between termite-gut protists and bacteria [12, 49, 73–75].
Although Mbv. cononymphae can also likely colonize the gut wall, the methane emission rate shown in Fig. 1E indicated that the absence of the endosymbiont was not compensated by methanogens outside Cononympha cells. Large differences in the methane emission rate among colonies were previously observed in other termite species, i.e., R. speratus [2], Neotermes sugioi [koshunensis] [56], and Cryptotermes secundus [2, 7]. Thus, the facultative association of methanogens not only with gut protists but also with termite hosts may be not rare phenomena. Although what factors determine the infection rate of methanogens in gut protists and their total abundance in the termite gut remain unclear, methanogens might play a role in adjusting the fermentation process of the protist hosts and the whole termite gut ecosystem in response to changing nutritional conditions with their versatile localization and abundance.
Description of “Candidatus Methanobrevibacter cononymphae” sp. nov
Methanobrevibacter cononymphae (co.no.nym’phae. N.L. fem. n. Cononymphae, of Cononympha, a genus of flagellated protist hosts). The archaea are straight rods with dimensions of 1.3–1.8 μm (mean ± SD, 1.58 ± 0.1; n = 50) by 0.4–0.5 μm (0.4 ± 0.0). The archaea are non-motile and facultatively colonize the cytoplasm of Cononympha leidyi and Cononympha koidzumii in the gut of Coptotermes formosanus. The archaea occasionally colonize the hindgut wall of C. formosanus. The assignment is based on the 16S rRNA gene (LC802674). The draft genome sequence CfCl-M3 showed 96.2% ANI to Methanovirga procula, which has been named under SeqCode on the basis of a MAG (GCA_031280375) obtained from the gut of Prorhinotermes canalifrons [5]. Since the genus Methanobrevibacter is highly divergent, Protasov et al (2023) have proposed division of Methanobrevibacter into at least nine genera, including Methanovirga, based on the genomic phylogeny and distance under SeqCode [5, 76].
Supplementary Material
Acknowledgements
We are grateful to Hirokazu Kuwahara for supporting the collection of protist cells and genome sequencing, to Takumi Murakami for handling sequence data, and to Katsuhiro Aminaka for helping with micromanipulation. We thank Yukihiro Kinjo, Osamu Kitade, Shuji Itakura, Satoko Noda, Wakako Omura, Masao Oya, Kazuki Takahashi, Gaku Tokuda, and Akinori Yamada for their cooperation in termite collection. We also thank Gillian Gile for her helpful advice on the morphological discrimination of Cononympha species, Satoshi Hattori for his advice on the metabolism of Methanobrevibacter, and Naoya Shinzato for unpublished information on endosymbiotic methanogens of Trimyema compressum. We thank Michiru Shimizu, Jun-ichi Inoue, and Masahiro Yuki for assisting with Sanger sequencing, which was performed in the RIKEN BSI. Sanger sequencing was also performed in the Biomaterial Analysis Center in the Tokyo Institute of Technology. TEM was assisted by Keiko Ikeda in the latter facility.
Contributor Information
Masayuki Kaneko, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan.
Tatsuki Omori, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan.
Katsura Igai, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan.
Takako Mabuchi, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan.
Miho Sakai-Tazawa, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan.
Arisa Nishihara, Japan Collection of Microorganisms, RIKEN BioResource Research Center, Tsukuba, Ibaraki 305-0074, Japan.
Kumiko Kihara, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan; Department of Biological and Chemical Systems Engineering, National Institute of Technology, Kumamoto College, Yatsushiro, Kumamoto 866-8501, Japan.
Tsuyoshi Yoshimura, Innovative Humano-habitability Laboratory, Research Institute for Sustainable Humanosphere, Kyoto University, Uji, Kyoto 611-0011, Japan.
Moriya Ohkuma, Japan Collection of Microorganisms, RIKEN BioResource Research Center, Tsukuba, Ibaraki 305-0074, Japan.
Yuichi Hongoh, Department of Life Science and Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan; Japan Collection of Microorganisms, RIKEN BioResource Research Center, Tsukuba, Ibaraki 305-0074, Japan.
Author contributions
Yuichi Hongoh and Moriya Ohkuma conceptualized the study and acquired fundings; Yuichi Hongoh, Kumiko Kihara, and Tsuyoshi Yoshimura administrated the project; Yuichi Hongoh Kumiko Kihara, Tsuyoshi Yoshimura, Masayuki Kaneko, Tatsuki Omori, and Miho Sakai-Tazawa collected termites; Tatsuki Omori, Masayuki Kaneko, Miho Sakai-Tazawa, and Katsura Igai performed experiments, Masayuki Kaneko, Tatsuki Omori, and Yuichi Hongoh analyzed and interpreted the data; Takako Mabuchi performed TEM; Arisa Nishihara, Masayuki Kaneko, and Katsura Igai measured methane; Masayuki Kaneko, Katsura Igai, and Yuichi Hongoh wrote the paper with input from the other authors.
Conflicts of interest
The authors declare no competing interests.
Funding
This study was financially supported by NEXT and KAKENHI grants from JSPS (Japan Society for the Promotion of Science) to Y.H. (GS009, 22 241 046, 16H04840, 20H02897, 20H05584, and 22 K19342) and to M.O. (17H01447 and 19H05689), and by JST (Japan Science and Technology Agency)-CREST (14532219) to Y.H. and by JST-GteX to M.O. (JPMJGX23B0).
Data availability
The 16S rRNA amplicon and metagenome sequence data generated in this study have been deposited in DDBJ under the accession numbers DRR534530–534599 (BioProject: PRJDB17265). Accession numbers for the CfCl-M3 genome are BAABUN010000001–BAABUN010000245. Representative near full-length SSU rRNA gene sequences will appear under the accession numbers LC802720–61 (18S rRNA) and LC802666–82 (16S rRNA).
References
- 1. Jabłoński S, Rodowicz P, Łukaszewicz M. Methanogenic archaea database containing physiological and biochemical characteristics. Int J Syst Evol Microbiol 2015;65:1360–8. 10.1099/ijs.0.000065 [DOI] [PubMed] [Google Scholar]
- 2. Sugimoto A, Inoue T, Tayasu Iet al. . Methane and hydrogen production in a termite-symbiont system. Ecol Res 1998;13:241–57. 10.1046/j.1440-1703.1998.00262.x [DOI] [Google Scholar]
- 3. Brauman A, Doré J, Eggleton Pet al. . Molecular phylogenetic profiling of prokaryotic communities in guts of termites with different feeding habits. FEMS Microbiol Ecol 2001;35:27–36. 10.1111/j.1574-6941.2001.tb00785.x [DOI] [PubMed] [Google Scholar]
- 4. Brune A. Methanogens in the digestive tract of termites. In: Hackstein JHP (ed), Microbiology Monographs: (Endo) symbiotic Methanogenic Archaea. Switzerland: Springer Nature, 2018, 81–101. 10.1007/978-3-319-98836-8_6 [DOI] [Google Scholar]
- 5. Protasov E, Nonoh JO, Kastle Silva JMet al. . Diversity and taxonomic revision of methanogens and other archaea in the intestinal tract of terrestrial arthropods. Front Microbiol 2023;14:e1281628. 10.3389/fmicb.2023.1281628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breznak JA, Switzer JM. Acetate synthesis from H2 plus CO2 by termite gut microbes. Appl Environ Microbiol 1986;52:623–30. 10.1128/aem.52.4.623-630.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pester M, Brune A. Hydrogen is the central free intermediate during lignocellulose degradation by termite gut symbionts. ISME J 2007;1:551–65. 10.1038/ismej.2007.62 [DOI] [PubMed] [Google Scholar]
- 8. Leadbetter JR, Breznak JA. Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Appl Environ Microbiol 1996;62:3620–31. 10.1128/aem.62.10.3620-3631.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leadbetter JR, Crosby LD, Breznak JA. Methanobrevibacter filiformis sp. nov., a filamentous methanogen from termite hindguts. Arch Microbiol 1998;169:287–92. 10.1007/s002030050574 [DOI] [PubMed] [Google Scholar]
- 10. Hongoh Y, Ohkuma M. Termite gut flagellates and their methanogenic and eubacterial symbionts. In: Hackstein JHP (ed), Microbiology Monographs: (Endo) symbiotic Methanogenic Archaea. Switzerland: Springer Nature, 2018, 55–79. 10.1007/978-3-319-98836-8_5 [DOI] [Google Scholar]
- 11. Brune A. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol 2014;12:168–80. 10.1038/nrmicro3182 [DOI] [PubMed] [Google Scholar]
- 12. Ohkuma M, Noda S, Hattori Set al. . Acetogenesis from H2 plus CO2 and nitrogen fixation by an endosymbiotic spirochete of a termite-gut cellulolytic protist. Proc Natl Acad Sci USA 2015;112:10224–30. 10.1073/pnas.1423979112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeuchi M, Kuwahara H, Murakami Tet al. . Parallel reductive genome evolution in Desulfovibrio ectosymbionts independently acquired by Trichonympha protists in the termite gut. ISME J 2020;14:2288–301. 10.1038/s41396-020-0688-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi K, Kuwahara H, Horikawa Yet al. . Emergence of putative energy parasites within Clostridia revealed by genome analysis of a novel endosymbiotic clade. ISME J 2023;17:1895–906. 10.1038/s41396-023-01502-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee MJ, Schreurs PJ, Messer ACet al. . Association of methanogenic bacteria with flagellated protozoa from a termite hindgut. Curr Microbiol 1987;15:337–41. 10.1007/BF01577591 [DOI] [Google Scholar]
- 16. Messer AC, Lee MJ. Effect of chemical treatments on methane emission by the hindgut microbiota in the termite Zootermopsis angusticollis. Microb Ecol 1989;18:275–84. 10.1007/BF02075814 [DOI] [PubMed] [Google Scholar]
- 17. Tokura M, Ohkuma M, Kudo T. Molecular phylogeny of methanogens associated with flagellated protists in the gut and with the gut epithelium of termites. FEMS Microbiol Ecol 2000;33:233–40. 10.1111/j.1574-6941.2000.tb00745.x [DOI] [PubMed] [Google Scholar]
- 18. Hara K, Shinzato N, Oshima Tet al. . Endosymbiotic Methanobrevibacter species living in symbiotic protists of the termite Reticulitermes speratus detected by fluorescent in situ hybridization. Microbes Environ 2004;19:120–7. 10.1264/jsme2.19.120 [DOI] [Google Scholar]
- 19. Fenchel T, Finlay BJ. Free-living protozoa with endosymbiotic methanogens. In: Hackstein JHP (ed.), Microbiology Monographs: (Endo) symbiotic Methanogenic Archaea, 2nd edn. Switzerland: Springer Nature, 2018, 1–11. [Google Scholar]
- 20. Shinzato N, Takeshita K, Kamagata Y. Methanogenic and bacterial endosymbionts of free-living anaerobic ciliates. In: Hackstein JHP (ed.), Microbiology Monographs: (Endo) symbiotic Methanogenic Archaea. Switzerland: Springer Nature, 2018, 37–53. 10.1007/978-3-319-98836-8_4 [DOI] [Google Scholar]
- 21. Evans TA, Forschler BT, Grace JK. Biology of invasive termites: a worldwide review. Annu Rev Entomol 2013;58:455–74. 10.1146/annurev-ento-120811-153554 [DOI] [PubMed] [Google Scholar]
- 22. Koidzumi M. Studies on the intestinal protozoa found in the termites of Japan. Parasitology 1921;13:235–309. 10.1017/S0031182000012506 [DOI] [Google Scholar]
- 23. Jasso-Selles DE, De Martini F, Velenovsky JF IVet al. . The complete protist symbiont communities of Coptotermes formosanus and Coptotermes gestroi: morphological and molecular characterization of five new species. J Euk Microbiol 2020;67:626–41. 10.1111/jeu.12815 [DOI] [PubMed] [Google Scholar]
- 24. Nishimura Y, Otagiri M, Yuki Met al. . Division of functional roles for termite gut protists revealed by single-cell transcriptomes. ISME J 2020;14:2449–60. 10.1038/s41396-020-0698-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsunoda K, Ohmura W, Yoshimura TKet al. . Methane emission by the termite, Coptotermes formosanus Shiraki (Isoptera: Rhinotermitidae). Wood Res 1993;79:34–40. [Google Scholar]
- 26. Inoue J, Noda S, Hongoh Yet al. . Identification of endosymbiotic methanogen and ectosymbiotic spirochetes of gut protists of the termite Coptotermes formosanus. Microbes Environ 2008;23:94–7. 10.1264/jsme2.23.94 [DOI] [PubMed] [Google Scholar]
- 27. Lai P, Tamashiro M, Fujii JK. Abundance and distribution of the three species of symbiotic protozoa in the hindgut of Coptotermes formosanus (Isoptera: Rhinotermitidae). Proc Hawaiian Entomol Soc 1983;24:271–6. [Google Scholar]
- 28. Yoshimura T. Contribution of the protozoan fauna to nutritional physiology of the lower termite, Coptotermes formosanus Shiraki (Isoptera: Rhinotermitidae). Wood Res 1995;82:68–129. [Google Scholar]
- 29. Trager W. The cultivation of a cellulose-digesting flagellate, Trichomonas termopsidis, and of certain other termite protozoa. Biol Bull 1934;66:182–90. 10.2307/1537331 [DOI] [Google Scholar]
- 30. Igai K, Kitade O, Fu Jet al. . Fine-scale genetic diversity and putative ecotypes of oxymonad protists coinhabiting the hindgut of Reticulitermes speratus. Mol Ecol 2022;31:1317–31. 10.1111/mec.16309 [DOI] [PubMed] [Google Scholar]
- 31. Murakami T, Segawa T, Dial Ret al. . Bacterial microbiota associated with the glacier ice worm is dominated by both worm-specific and glacier-derived facultative lineages. Microbes Environ 2017;32:32–9. 10.1264/jsme2.ME16158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Callahan BJ, McMurdie PJ, Rosen MJet al. . DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 2016;13:581–3. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012;28:1823–9. 10.1093/bioinformatics/bts252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quast C, Pruesse E, Yilmaz Pet al. . The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2013;41:D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nguyen L-T, Schmidt HA, von Haeseler Aet al. . IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015;32:268–74. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leigh JW, Bryant D. popart: full-feature software for haplotype network construction. Methods Ecol Evol 2015;6:1110–6. 10.1111/2041-210X.12410 [DOI] [Google Scholar]
- 37. Ludwig W, Strunk O, Westram Ret al. . ARB: a software environment for sequence data. Nucleic Acids Res 2004;32:1363–71. 10.1093/nar/gkh293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Utami YD, Kuwahara H, Murakami Tet al. . Phylogenetic diversity and single-cell genome analysis of ‘Melainabacteria’, a non-photosynthetic cyanobacterial group, in the termite gut. Microbes Environ 2018;33:50–7. 10.1264/jsme2.ME17137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sato T, Kuwahara H, Fujita Ket al. . Intranuclear verrucomicrobial symbionts and evidence of lateral gene transfer to the host protist in the termite gut. ISME J 2014;8:1008–19. 10.1038/ismej.2013.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bankevich A, Nurk S, Antipov Det al. . SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin H-H, Liao Y-C. Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci Rep 2016;6:e24175. 10.1038/srep24175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. von Meijenfeldt FAB, Arkhipova K, Cambuy DDet al. . Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol 2019;20:e217. 10.1186/s13059-019-1817-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Warren RL, Yang C, Vandervalk BPet al. . LINKS: scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience 2015;4:e35. 10.1186/s13742-015-0076-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu M, Guo L, Gu Set al. . TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 2020;9:1–11. 10.1093/gigascience/giaa094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34:3094–100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018;34:1037–9. 10.1093/bioinformatics/btx713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Couvin D, Bernheim A, Toffano-Nioche Cet al. . CRISPRCasFinder, an update of CRISPRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 2018;46:W246–51. 10.1093/nar/gky425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tatusova T, DiCuccio M, Badretdin Aet al. . NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 2016;44:6614–24. 10.1093/nar/gkw569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hongoh Y, Sharma VK, Prakash Tet al. . Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc Nat Acad Sci USA 2008;105:5555–60. 10.1073/pnas.0801389105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moriya Y, Itoh M, Okuda Set al. . KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 2007;35:W182–5. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kanehisa M, Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci 2020;29:28–35. 10.1002/pro.3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Parks DH, Chuvochina M, Chaumeil PAet al. . A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol 2020;38:1079–86. 10.1038/s41587-020-0501-8 [DOI] [PubMed] [Google Scholar]
- 53. Arora J, Kinjo Y, Šobotník Jet al. . The functional evolution of termite gut microbiota. Microbiome 2022;10:e78. 10.1186/s40168-022-01258-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 2016;4:e1900v1.27123377 [Google Scholar]
- 55. Syberg-Olsen MJ, Garber AI, Keeling PJet al. . Pseudofinder: detection of pseudogenes in prokaryotic genomes. Mol Biol Evol 2022;39:msac153. 10.1093/molbev/msac153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shinzato N, Yoshino H, Yara K. Methane production by microbial symbionts in the lower and higher termites of the Ryukyu archipelago. In: Sato S, Ishida M, Ishikawa H (eds.), Endocytobiology V: Proceedings of the Fifth International Colloquium on Endocytobiology and Symbiosis, Kyoto, June 23–27, 1992. Kyoto: Tübingen University Press, 1993, 161–6. [Google Scholar]
- 57. Miller TL. Methanobrevibacter. In: Boone DR, Castenholz RW, Carrity GM (eds.), Bergey’s Manual of Systematics Bacteriology, 2nd edn. New York: Springer Verlag, 1989, 218–26. [Google Scholar]
- 58. Lind AE, Lewis WH, Spang Aet al. . Genomes of two archaeal endosymbionts show convergent adaptations to an intracellular lifestyle. ISME J 2018;12:2655–67. 10.1038/s41396-018-0207-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Parks DH, Imelfort M, Skennerton CTet al. . CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 2015;25:1043–55. 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chklovski A, Parks DH, Woodcroft BJet al. . CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 2023;20:1203–12. 10.1038/s41592-023-01940-w [DOI] [PubMed] [Google Scholar]
- 61. Pourcel C, Touchon M, Villeriot Net al. . CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res 2020;48:D535–44. 10.1093/nar/gkz915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ebert D. The epidemiology and evolution of symbionts with mixed-mode transmission. Annu Rev Ecol Evol Syst 2013;44:623–43. 10.1146/annurev-ecolsys-032513-100555 [DOI] [Google Scholar]
- 63. Yamin MA. Cellulose metabolism by the flagellate Trichonympha from a termite is independent of endosymbiotic bacteria. Science 1981;211:58–9. 10.1126/science.211.4477.58 [DOI] [PubMed] [Google Scholar]
- 64. Odelson DA, Breznak JA. Nutrition and growth characteristics of Trichomitopsis termopsidis, a cellulolytic protozoan from termites. Appl Environ Microbiol 1985;49:614–21. 10.1128/aem.49.3.614-621.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tholen A, Brune A. Impact of oxygen on metabolic fluxes and in situ rates of reductive acetogenesis in the hindgut of the wood-feeding termite Reticulitermes flavipes. Environ Microbiol 2000;2:436–49. 10.1046/j.1462-2920.2000.00127.x [DOI] [PubMed] [Google Scholar]
- 66. Weiss DS, Thauer RK. Methanogenesis and the unity of biochemistry. Cell 1993;72:819–22. 10.1016/0092-8674(93)90570-G [DOI] [PubMed] [Google Scholar]
- 67. Yamin MA. Flagellates of the orders Trichomonadida Kirby, Oxymonadida Grasse, and Hypermastigida Grassi & Foa reported from lower termites (Isoptera families Mastotermitidae, Kalotermitidae, Hodotermitidae, Termopsidae, Rhinotermitidae, and Serritermitidae) and from the wood-feeding roach Cryptocercus (Dictyoptera: Cryptocercidae). Sociobiology 1979;4:5–119. [Google Scholar]
- 68. Fenchel T, Finlay BJ. Endosymbiotic methanogenic bacteria in anaerobic ciliates - significance for the growth efficiency of the host. J Protozool 1991;38:18–22. 10.1111/j.1550-7408.1991.tb04788.x [DOI] [Google Scholar]
- 69. Yamada K, Kamagata Y, Nakamura K. The effect of endosymbiotic methanogens on the growth and metabolic profile of the anaerobic free-living ciliate Trimyema compressum. FEMS Microbiol Lett 1997;149:129–32. 10.1111/j.1574-6968.1997.tb10319.x [DOI] [Google Scholar]
- 70. Treitli SC, Hanousková P, Beneš Vet al. . Hydrogenotrophic methanogenesis is the key process in the obligately syntrophic consortium of the anaerobic ameba Pelomyxa schiedti. ISME J 2023;17:1884–94. 10.1038/s41396-023-01499-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wagener S, Pfennig N. Monoxenic culture of the anaerobic ciliate Trimyema compressum Lackey. Arch Microbiol 1987;149:4–11. 10.1007/BF00423128 [DOI] [Google Scholar]
- 72. Finlay BJ, Embley TM, Fenchel T. A new polymorphic methanogen, closely related to Methanocorpusculum parvum, living in stable symbiosis within the anaerobic ciliate Trimyema sp. J Gen Microbiol 1993;139:371–8. 10.1099/00221287-139-2-371 [DOI] [PubMed] [Google Scholar]
- 73. Hongoh Y, Sharma VK, Prakash Tet al. . Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 2008;322:1108–9. 10.1126/science.1165578 [DOI] [PubMed] [Google Scholar]
- 74. Ikeda-Ohtsubo W, Strassert JF, Köhler Tet al. . ‘Candidatus Adiutrix intracellularis’, an endosymbiont of termite gut flagellates, is the first representative of a deep-branching clade of Deltaproteobacteria and a putative homoacetogen. Environ Microbiol 2016;18:2548–64. 10.1111/1462-2920.13234 [DOI] [PubMed] [Google Scholar]
- 75. Strassert JFH, Mikaelyan A, Woyke Tet al. . Genome analysis of ‘Candidatus Ancillula trichonymphae’, first representative of a deep-branching clade of Bifidobacteriales, strengthens evidence for convergent evolution in flagellate endosymbionts. Environ Microbiol Rep 2016;8:865–73. 10.1111/1758-2229.12451 [DOI] [PubMed] [Google Scholar]
- 76. Hedlund BP, Chuvochina M, Hugenholtz Pet al. . SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat Microbiol 2022;7:1702–8. 10.1038/s41564-022-01214-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S rRNA amplicon and metagenome sequence data generated in this study have been deposited in DDBJ under the accession numbers DRR534530–534599 (BioProject: PRJDB17265). Accession numbers for the CfCl-M3 genome are BAABUN010000001–BAABUN010000245. Representative near full-length SSU rRNA gene sequences will appear under the accession numbers LC802720–61 (18S rRNA) and LC802666–82 (16S rRNA).