

ABSTRACT
Bacterial ribonuclease E (RNase E) is vital for posttranscriptional regulation by degrading and processing RNA. The RraA protein inhibits RNase E activity through protein-protein interactions, exerting a global regulatory effect on gene expression. However, the specific role of RraA remains unclear. In this study, we investigated rraA expression in Vibrio alginolyticus ZJ-T and identified three promoters responsible for its expression, resulting in transcripts with varying 5′-UTR lengths. During the stationary phase, rraA was significantly posttranscriptionally inhibited. Deletion of rraA had no impact on bacterial growth in rich medium Luria-Bertani broth with salt (LBS) but resulted in decreased biofilm formation and increased resistance to polymyxin B. Transcriptome analysis revealed 350 differentially expressed genes (DEGs) between the wild type and the rraA mutant, while proteome analysis identified 267 differentially expressed proteins (DEPs). Integrative analysis identified 55 genes common to both DEGs and DEPs, suggesting that RraA primarily affects gene expression at the posttranscriptional level. KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis demonstrated that RraA facilitates the conversion of fatty acids, propionic acid, and branched-chain amino acids to acetyl-CoA while enhancing amino acid and peptide uptake. Notably, RraA positively regulates the expression of virulence-associated genes, including those involved in biofilm formation and the type VI secretion system. This study expands the understanding of the regulatory network of RraA through transcriptome analysis, emphasizing the importance of proteomic analysis in investigating posttranscriptional regulation.
IMPORTANCE
RraA is an inhibitor protein of ribonuclease E that interacts with and suppresses its endonucleolytic activity, thereby playing a widespread regulatory role in the degradation and maturation of diverse mRNAs and noncoding small RNAs. However, the physiological functions and associated regulon of RraA in Vibrio alginolyticus have not been fully elucidated. Here, we report that RraA impacts virulence-associated physiological processes, namely, antibiotic resistance and biofilm formation, in V. alginolyticus. By conducting an integrative analysis of both the transcriptome and proteome, we revealed the involvement of RraA in carbon metabolism, amino acid catabolism, and transport, as well as in the type VI secretion system. Collectively, these findings elucidate the regulatory influence of RraA on multiple pathways associated with metabolism and pathogenesis in V. alginolyticus.
KEYWORDS: Vibrio alginolyticus, RraA, polymyxin B, biofilm formation, transcriptome and proteome, metabolism
INTRODUCTION
RNA stability plays a very important role in the posttranscriptional modulation of gene expression and is affected by many pathways in bacteria, including those involving endoribonucleases, exoribonucleases, and RNA pyrophosphate hydrolases (RppH values). Ribonuclease E (RNase E) is thought to be a major player in mRNA decay and processing, in addition to being involved in the maturation of rRNA and tRNA (1). The cellular levels and activity of RNase E in bacteria are tightly controlled (2). Ribonuclease activity regulator A (RraA), a conserved 17.4 kDa protein, is a protein inhibitor that interacts with RNase E and inhibits its cleavage (3).
The interaction between RraA and RNase E leads to an overall change in RNA abundance as well as functionally altered gene expression. Gene chip analysis has shown that the mutation of RraA results in dramatic changes in the global abundance of mRNA in Escherichia coli, affecting more than 15% of cellular transcripts (4). The overexpression of RraA in E. coli increases the relative abundance of 2,000 mRNAs, which are involved primarily in envelope biosynthesis and aerobic metabolism (3). RraA can also affect multiple components of RNA degradation vesicles in a dynamic, energy-dependent manner (5).
Numerous orthologs of E. coli RraA, including VvRraA1 and VvRraA2 in Vibrio vulnificus, RraA in Pseudomonas aeruginosa, and ScRraA1 and ScRraA2 in Streptomyces coelicolor, have been found (6–12). In S. coelicolor, compared with the wild-type strain, the rraAS1 gene deletion results in a greater growth rate and increased production of actinorhodin and undecylprodigiosin (11). In V. vulnificus, VvRraA1 modulated the pathogenicity of V. vulnificus in mice (12). Transcriptomic analysis indicated that VvRraA1 modulates the abundance of a large number of mRNA transcripts (12). Taken together, these findings suggest that RraA is a global regulator that controls a wide range of cellular and physiological processes in a species-specific manner. However, the ortholog of RraA in V. alginolyticus, a widespread pathogen of marine fish and humans, has not yet been investigated.
Here, we characterized the ortholog of RraA in V. alginolyticus, constructed a deletion strain, a complementary strain, and an overexpression strain of rraA, and subsequently evaluated their growth ability, motility, antibiotic resistance, and biofilm formation ability. In addition, the RraA regulon of V. alginolyticus during the early exponential growth phase in an LBS medium was investigated using integrated high-throughput RNA-seq and proteomics analysis.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. V. alginolyticus was routinely cultured in Luria–Bertani (LB) medium supplemented with 2.5% NaCl at 30°C. E. coli strains were cultured in LB media supplemented with appropriate antibiotics at 37°C. Thiosulfate citrate bile salts sucrose (TCBS) medium (HuanKai, Guangzhou, China) containing 5 µg/mL chloramphenicol (Cm) and 0.2% D-glucose was used for selecting transconjugants. To identify transgenic bacteria that underwent plasmid excision and allelic exchange, the ccdB gene was induced using TCBS medium supplemented with 0.2% arabinose plus 5 µg/mL Cm or not, after which bacteria that had lost the inserted plasmid were selected. When necessary, the E. coli media were supplemented with antibiotics at the following final concentrations: ampicillin, 100 µg/mL; Cm, 20 µg/mL; and, if needed, diaminopyrimidine at a final concentration of 0.3 mM in the growth media.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source |
|---|---|---|
| Vibrio alginolyticus strains | ||
| ZJ-T | Apr, translucent/smooth variant of wild strain ZJ51; isolated from diseased Epinephelus coioides off the South China coast | (13) |
| ZJ-T-ΔrraA | Apr; ZJ-T carrying a deletion of rraA | This study |
| ZJ-T-ΔrraA+ | Cmr; ZJ-T-ΔrraA carrying a rraA complementation plasmid pMMB207-rraA | This study |
| ZJ-T/over rraA-pSCT32 | Cmr; ZJ-T carrying a RraA expression plasmid over rraA-pSCT32 | This study |
| ZJ-T/pSCT32-gfp-rraA-TL | Cmr; ZJ-T carrying a rraA translational fusion plasmid pSCT32-gfp-rraA-TL | This study |
| E. coli strain | ||
| GEB883 | WT; E. coli K12 ΔdapA::ermpir RP4-2 ΔrecA gyrA462, zei298::Tn10; donor strain for conjugation | (14) |
| Plasmids | ||
| pSW7848 | Cmr; suicide vector with an R6K origin, requiring the Pir protein for its replication, and the ccdB toxin gene | (15) |
| pSW7848-ΔrraA | Cmr; pSW848 containing the flanking region fragments in the mutant allele of rraA | This study |
| pMMB207 | Cmr; RSF1010 derivative, IncQ lacIq Ptac oriT | (16) |
| pSCT32 | Cmr; expression plasmid with a pBR322 and a f1 origin at the same time and a trc promoter | (17) |
| pMMB207-rraA | Cmr; pMMB207 containing the wild-type allele of rraA | This study |
| pSCT32-rraA | Cmr; pSCT32 containing the wild-type allele of rraA | This study |
| pSCT32-gfp | Cmr; pSCT32 containing promoterless reporter gene gfp encoding a green fluorescent protein | This study |
| pSCT32-gfp-rraA-TL | Cmr; rraA (including all three promoters and start codon, from −493 to +3 nt) are translationally fused to pSCT32-gfp | This study |
Sequence analysis
The species used in this analysis and their accession IDs are as follows: EcRraA (AAB01208.1AAB01208.1 E. coli strain_K12), StRraA (WFG57654.1 Salmonella enterica subsp. enterica serovar Typhimurium), VvRraA 1(ACK76427.1 V. vulnificus MO6-24/O), VvRraA 2(ACK76428.1 V. vulnificus MO6-24/O), VaRraA (WP_005397954.1 Vibrio alginolyticus), VcRraA (1VI4_A Chain A Vibrio cholerae)PaRraA (QPV51173.1 P. aeruginosa), MtRraA (ANZ84631.1 Mycobacterium tuberculosis), TtRraA (WVY30247.1 Thermus thermophilus), ScRraA1 (WMT37493.1 S. coelicolor), and ScRraA2 (5 × 15_A Chain A S. coelicolor). Sequence analysis was performed by constructing a phylogenetic tree based on amino acid differences using the maximum likelihood method with 300 bootstrap replicates. This analysis was carried out using MEGA X software (downloaded from http://www.megasoftware.net/, accessed on 6 June 2022). The Percent Identity Matrix among different strains, created by Clustal2.1, can be found at the following website: https://www.ebi.ac.uk/jdispatcher/msa/mafft.
Construction of the mutant, complementary, and overexpression strains
The mutant strains were constructed according to Grimm et al. (2003) (18). Briefly, the upstream and downstream regions of rraA were obtained through PCR using the primers RraA-UP-F/R and RraA-DOWN-F/R (Table S2). Simultaneously, the vector fragment pSW7848 was amplified through PCR employing the primers pSW7848-F/R (Table S2). The recombinant suicide plasmid pSW7848-ΔrraA was obtained through isothermal assembly and subsequently transformed into GEB883 cells (Table 1). The complementary strain was constructed with a pMMB207-based plasmid by insertion of the rraA open reading frame (ORF) under the Ptac promoter. The rraA overexpression strain was constructed with the high-copy-number plasmid pSCT32, which harbors the rraA gene under the Ptrc promoter. The details of transformation, recombination, and selection of target strains were performed as previously described by Liu et al. (19, 20).
Whole-transcriptome sequencing by RNA-seq
Three single colonies of each strain were cultured overnight. The cells were then diluted 1,000-fold in LBS medium and grown to the early logarithmic phase (OD600nm approximately ≈ 0.6). Subsequently, the cells were collected from 100 mL of culture. RNA was extracted using the high-purity TransZol Up Plus RNA Kit (TransGen Biotech, Beijing, China) following the extraction protocol. The purified RNA was subsequently sent to Genedenovo Biotech (Guangzhou, China) for the construction of single-end RNA-Seq libraries, which were subsequently sequenced using the Illumina NovaSeq 6000 platform with paired-end 150-base reads. The detailed data processing was carried out as previously described by Liu et al. (19).
The differentially expressed genes (DEGs) were identified based on a fold change ≥2 and a false discovery rate-adjusted P value (q value) <0.05. The DEGs were then subjected to enrichment analysis of GO (Gene Ontology) annotations and KEGG pathways using a threshold of q values < 0.05.
5'-RACE System for rapid amplification of cDNA ends
RNA extraction of wild-type ZJ-T was performed as previously described in the section “Whole-transcriptome sequencing by RNA-seq.” The primers used are listed in Table S2 and were designed with the NCBI Primer Design Tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Table S2). An Evo M-MLV reverse transcription kit (Accurate Biotechnology Co., Ltd., Hunan, China) was used for cDNA synthesis, and a 5′-RACE kit (Sangon Biotech, Shanghai, China) was used for 5′-UTR amplification, PCR product purification, and tailing.
Quantitative reverse transcription PCR analysis
RNA extraction and quantitative real-time fluorescent quantitative PCR (qRT-PCR) were performed as previously described (19). The ΔΔCt method was used to calculate relative expression levels, with recA serving as the reference gene (21). The experiment was repeated at least twice. The primers used for qRT-PCR are listed in Table S2, and statistical significance was assessed using Student’s t test (ns P > 0.05, *P < 0.05, and **P < 0.01).
Growth measurement
Growth measurements in rich medium LBS from ZJ-T, ZJ-T-ΔrraA, ZJ-T-ΔrraA+, and ZJ-T/over rraA-pSCT32 were carried out as previously described by Liu et al. (19, 20). The experiments were repeated three times.
Motility assay
Overnight cell cultures of ZJ-T, ZJ-T-ΔrraA, ZJ-T-ΔrraA+, and ZJ-T/over rraA-pSCT32 were adjusted to OD600nm = 1.0 in LBS medium. Five microlitres of each culture were spotted on 0.3% agar plates and 1.5% agar plates. The plates were inverted in a 30°C incubator for 24 hours. The diameter was subsequently measured to evaluate the motility of the strains. Statistical significance was assessed using one-way analysis of variance (ANOVA) with the least significant difference (LSD) method (*P < 0.05 and **P < 0.01).
Antibiotic sensitivity test
The antibiotic sensitivity test was carried out by using an antimicrobial drug sensitization tablet kit (Hangzhou Microbial Reagent Co., Ltd., Hangzhou, China). Briefly, single clones of V. alginolyticus ZJ-T, ZJ-T-ΔrraA, ZJ-T-ΔrraA+, and ZJ-T/over rraA-pSCT32 were inoculated into LBS liquid media and incubated overnight at 30°C with shaking at 200 rpm. Then, 200 µL of the overnight bacterial solution was mixed with 10 mL of fresh LBS liquid medium, poured onto LBS plates, and dried for 5 minutes. Antibiotic paper sheets containing different concentrations of antibiotics were added to the plates using sterile forceps, and the plates were incubated in an incubator at 30°C for 24 hours. The diameters of the inhibition zones were subsequently measured and recorded. The experiments were repeated at least three times.
Biofilm formation assay
The overnight culture of the strains (ZJ-T, ZJ-T-ΔrraA, ZJ-T-ΔrraA+, and ZJ-T/over rraA-pSCT32) was adjusted to an OD600nm = 1.0. Fifty microlitres of the diluted cultures were inoculated into 5 mL of LBS liquid medium and incubated in glass tubes at 30°C. The cultures were removed, and the tubes were collected 3 hours, 12 hours, 24 hours, and 48 hours postinoculation. The tubes were washed three times with distilled water, after which the matrix attached to the glass tubes was stained with 200 µL of 2% crystal violet stain in ethanol for 5 minutes. The excess stain was gently rinsed away with water, and 200 µL of pure ethanol was added to dissolve the remaining crystal violet. The solutions were pipetted into a 96-well plate, and a spectrophotometer was used to measure the absorbance at 550 nm.
Translational fusion and analysis
To identify the promoter of rraA, a translational fusion based on the green fluorescent protein (GFP) reporter was constructed according to previous methods (19). In brief, fragments of different lengths from the upstream region of the rraA start codon, which includes the longest length and the start codon (from −493 to +3), were cloned and inserted ahead of the ORF of gfp in the pSCT32 vector (Table S2). The resulting plasmid was subsequently transformed into E. coli GEB883 receptor cells, which were confirmed via PCR analysis and sequencing.
Western blotting was used to quantify the target protein fusions. For the western blotting procedure, samples were collected at OD600nm readings of 0.5, 3.0, and 5.0, all of which were cultured in LBS containing 5 µg/mL Cm. Cells from three independent biological replicates were collected, centrifuged, and resuspended in 100 µL of 2 × SDS loading buffer per OD600nm unit (Sangon Biotech, Shanghai, China). Subsequently, the samples were incubated at 100°C for 10 minutes. SDS-PAGE, western blotting, and visualization were performed according to previous methods (19). The data presented represent the most representative results from three technical replicates.
RESULTS
Phylogenetic analysis and characterization of the rraA transcript of V. alginolyticus
The rraA gene is located on chromosome 1 of V. alginolyticus ZJ-T. A hypothetical protein (HP), separated by a 64 bp intergenic region, is located upstream of rraA (Fig. 1A). However, in E. coli, rraA is located downstream of the menA gene, which encodes a 1,4-dihydroxy-2-naphthoic acid octaprenyltransferase that catalyzes the prenylation of menaquinone (4, 22). The amino acid sequences of RraA were analyzed using the multiple sequence comparison tool MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/), which generated a similarity matrix (percent identity matrix). The similarity between VaRraA of V. alginolyticus and VvRraA2 of V. vulnificus reached a maximum of 86.90%. Moreover, the similarities between VvRraA2 of V. vulnificus and VcRraA of V. cholerae were found to be 41.885% and 39.52%, respectively (Fig. 1B; Fig. S1). Some of the conserved polar amino acids, considered interaction sites with RNase E, may be located on the surface of the molecule. Moreover, the two cysteines, Cys9 and Cys41, are identical in orthologs of all bacteria and are considered key amino acids for the formation of RraA homohexamers (23).
Fig 1.

Bioinformatic analysis of the rraA sequences of V. alginolyticus and their expression in the LBS. (A) Schematic diagram of the rraA coding position. The arrows indicate the transcriptional and coding directions. (B) Alignment of amino acid sequences of V. alginolyticus RraA and its orthologs in different bacteria. (C) Analysis of the sequence structure and promoter of rraA. (D) 5′-RACE experiments of the rraA gene. (E) Relative expression of rraA at different stages (Student’s t test, P values: *, <0.05; **, <0.01). The levels of rraA were normalized to the expression of the housekeeping gene recA. Error bars represent SDs. (F) Western blot of the rraA translational fusion at three growth stages at OD600nm values of 0.5, 3.0, and 5.0. Anti-RNA polymerase β as an internal reference.
The transcription of rraA was reported to originate from its promoter, and PrraA is located in the intergenic region in E. coli (4). Nevertheless, transcriptome analysis in this study revealed that rraA has a 493 nt-long 5′-UTR (untranslated region) that contains part of the upstream hp coding region, indicating that two or more promoters may be involved in its transcription in V. alginolyticus ZJ-T (Fig. 1C). Furthermore, 5′-RACE analysis revealed at least three transcripts with start points corresponding to −493 nt, −305 nt, and −38 nt (the A of the start codon is +1; Fig. 1D), consistent with the results of BPROM prediction. The −38 nt sequence may correspond to the conserved promoter of PrraA in E. coli.
A continuous increase in the rraA transcript abundance was observed with cell growth, as determined by quantitative RT-PCR. When the cell culture entered the stationary phase, there was a 12-fold increase compared to that in the early log phase. This observation is consistent with previous findings in E. coli (4) (Fig. 1E). However, there was a discrepancy between the mRNA level of rraA and its corresponding protein at different growth stages. While the RraA protein level gradually increased from the early to late log phase, it subsequently decreased during the early stationary phase (Fig. 1F). This finding suggested that the expression of rraA in V. alginolyticus may be significantly regulated posttranscriptionally, possibly at the translational efficiency level, during the stationary phase.
Positive regulatory effect of RraA on polymyxin B resistance and biofilm formation in V. alginolyticus ZJ-T
To investigate the role of rraA in V. alginolyticus ZJ-T, we constructed an in-frame deletion of rraA in ZJ-T, named ZJ-T-ΔrraA, and its complementary strain ZJ-T-ΔrraA+, which carried the low-copy-number plasmid pMMB207 harboring the entire coding region of rraA transcribed from the Ptet promoter. Additionally, we constructed an overexpression strain, ZJ-T/over rraA-pSCT32, which carried the high-copy-number plasmid pSCT32 with the same construction as above. qRT-PCR revealed that, compared with that in the WT, the expression of rraA in the complemented strain was completely rescued, while the overexpression strain exhibited an increase in rraA expression of approximately 17–18-fold (Fig. 2A).
Fig 2.

The physiological role of rraA in V. alginolyticus. (A) Relative expression of rraA in the wild-type, rraA mutant, complementary, and overexpression strains. (B) Growth curves of the wild-type, rraA mutant, complementary, and overexpression strains grown in LB +2.5% NaCl-rich medium. (C) The swimming and swarming abilities of ZJ-T, ZJ-T-ΔrraA, ZJ-T-ΔrraA+, and ZJ-T/over rraA-pSCT32 on 0.3% and 1.5% LBS agar plates. (D) Antibiotic resistance to polymyxin B in wild-type ZJ-T, ZJ-T-ΔrraA, ZJ-T-ΔrraA+, and ZJ-T/over rraA-pSCT32. (E) Relative mRNA expression levels of the outer membrane proteins, lipid A biosynthesis genes, and two-component regulator genes. The levels of genes were normalized to the internal control recA level. Error bars indicate SD (Student’s t test, P-value: ns, >0.05 and **, <0.01). (F) Biofilm synthesis of V. alginolyticus wild-type ZJ-T, the rraA mutant, the complemented strain, and the overexpression strain at different time points.
It has been reported that the overexpression of RraAV1 results in a moderately slower growth rate than that of the WT in V. vulnificus (7, 24). However, in the present study, changes in the rraA transcript abundance did not significantly affect the growth of LBS in rich media (Fig. 2B). Furthermore, no variations were observed in either swimming or swarming motility among the strains (Fig. 2C), which contrasts with the decreases in swimming and swarming motility observed in the RraAV1 mutant strain of V. vulnificus (12, 24).
Thirty antibiotics were used to test the antibiotic resistance of the cells. As shown in Table S1, the alteration of rraA resulted in no change in resistance to any of the tested antibiotics except polymyxin B (Fig. 2D). Interestingly, both the deletion and overexpression of rraA increased the resistance of the cells to polymyxin B (Fig. 2D). In order to further explore the susceptibility of V. alginolyticus to polymyxin B in the rraA-free background, we have studied expression levels of genes related to outer membrane proteins, lipid A biosynthesis genes, and two-component regulator genes (Fig. 2E). In strain ZJ-T-ΔrraA, the expression levels of ompR, ompA, lpxH, and oprF were all increased (Fig. 2E).
To evaluate the possible effect of rraA on biofilm formation, we quantified the biofilm formation of the four strains at different time points during growth. As shown in Fig. 2F, the biofilm formation ability of ZJ-T increased during the log phase and remained stable at its peak during the stationary phase. In comparison, the attached biomass of ZJ-T-ΔrraA continued to increase but was significantly less than that of ZJ-T during the log phase. Moreover, the concentration decreases as the cells enter the stationary phase. At 48 h postinoculation, biofilm formation in ZJ-T-ΔrraA decreased by approximately 50% (Fig. 2F). Moreover, the profiles of the complemented and overexpression strains were similar to those of the wild type. These results suggest that RraA positively regulates biofilm formation in V. alginolyticus.
RraA regulon revealed by integrative transcriptome and proteome analysis
To investigate the regulatory role of RraA, we conducted high-throughput RNA-seq and DIA label-free proteomic analyses of ZJ-T and ZJ-T-ΔrraA cells harvested at the early log phase (OD600 nm=0.5).
By comparing the ZJ-T group with the ZJ-T-ΔrraA group, we identified 350 DEGs in ZJ-T-ΔrraA, including 174 upregulated genes and 176 downregulated genes (Fig. 3A and B), with criteria set at FDR < 0.05 and |log2FC| > 1. All DEGs are listed in Table S3. Quantitative proteomic analysis revealed 267 differentially expressed proteins (DEPs), comprising 58 upregulated and 209 downregulated proteins, in ZJ-T-ΔrraA (FDR < 0.05 Fig. 4A and B). These genes and proteins were further subjected to KEGG pathway enrichment analysis (Fig. 3C and 4C ). All DEPs are listed in Table S4. To validate the transcriptome analysis, we selected five genes, luxO (BAU10_06930), csgD (BAU10_22065), etk (BAU10_21860), tssC1(BAU10_20240), and tssM1 (BAU1020265) and BAU10_21865, to measure their transcript abundances using qRT-PCR. As shown in Fig. 5, the expression profiles are generally in agreement with the -omics data.
Fig 3.

Overview of the RNA transcriptomic profiles of the wild-type and rraA mutant strains. (A) Volcano plot of differentially expressed genes (DEGs) in the transcriptome [FDR < 0.05, | log2(fold change) | ≥ 1]. Upregulated expression is displayed in red, downregulated expression is displayed in green (judged by FDR < 0.05 and differential expression greater than twofold), and black points show no differences. (B) Statistical column chart of DEGs. WT: wild-type strain V. alginolyticus ZJ-T; RraA: rraA mutant strain ZJ-T-ΔrraA. (C) Histogram of the enrichment of the top 20 KEGG pathways identified via transcriptomics after rraA inactivation.
Fig 4.

Overview of the proteomic profiles of the wild-type and rraA mutant strains. (A) Volcano plot of DEGs identified via proteomics (FDR < 0.05, |fold change| ≥ 1.5). The red dots represent upregulated differences; the blue dots represent downregulated differences; and the black dots represent no differences. (B) Statistical column chart of the DEPs. WT: wild-type strain V. alginolyticus ZJ-T; RraA: rraA mutant strain ZJ-T-ΔrraA. (C) Histogram of the enrichment of the top 20 KEGG pathways identified by proteomics when rraA was knocked out. (D) Venn diagram of DEGs and DEPs between the wild-type and rraA mutant strains. (E) Nine-quadrant plots of the transcriptome and proteome of the wild-type ZJ-T strain and the rraA mutant strain. The black dots indicate non-DEPs and genes, the red dots indicate consistent or opposite trends in gene and protein changes, the green dots indicate DEGs but non-DEPs, and the blue dots indicate non-DEGs but DEPs (if differential ploidy is reached and the p-value is not reached, the dots are shown in gray).
Fig 5.
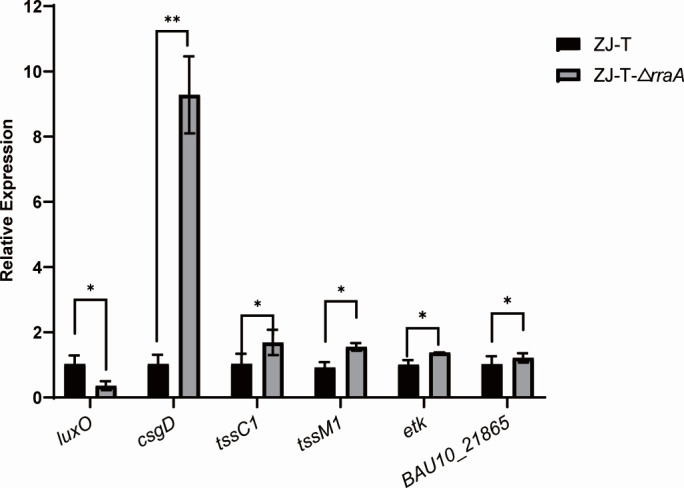
Relative expression of genes checked by qRT-PCR. The levels of genes were normalized to the internal control recA level. Error bars indicate SD (Student’s t test, P-value: ns, >0.05 and **, <0.01).
The combined transcriptomic and proteomic analysis revealed a total of 55 genes that were concurrently present among both the DEGs and DEPs (Fig. 4E). Additionally, 212 genes were identified exclusively as DEPs and nondifferentially expressed genes (NDEGs), while 183 genes were unique to nondifferentially expressed proteins (NDEPs) and DEGs. Furthermore, a substantial number of 2,739 genes were observed solely in the categories of NDEGs and NDEPs. Interestingly, 466 genes exhibited increased protein levels, and 192 genes exhibited decreased protein levels. The mutant had no variation in transcript abundance, suggesting that more than half of the differentially expressed genes are primarily regulated at the translational level (Fig. 4D).
Positive regulation of fatty acid catabolism
The genes identified as DEGs or DEPs were enriched in various metabolic pathways (Fig. 3C and 4C). Notably, carbohydrate pathways such as those related to the degradation of fatty acids and propionic acid were significantly affected. The degradation of fatty acids is mediated by β-oxidation, which involves enzymes such as FadI (acetyl-CoA C-acyltransferase), FadD (ATP-dependent acyl-CoA synthase), FadE (acyl-CoA dehydrogenase), FadB (3-hydroxyacyl-CoA dehydrogenase), and FadA (3-ketoacyl-CoA thiolase) in bacteria (25–28). β-Oxidation can yield acetyl-CoA or both acetyl-CoA and propionyl-CoA, depending on the carbon number (even or odd) of the fatty acid chain. In this study, the protein abundances of FadE (BAU10_10485), FadB (BAU10_15270), FadA (BAU10_15265), and FadI (BAU10_10085) decreased by 1.67–3.17-fold at the protein level, although the abundance of fadE decreased in the mutant (Fig. 6A and B), suggesting that RraA positively regulates fatty acid degradation in V. alginolyticus.
Fig 6.

Heatmaps of DEGs and DEPs in the wild-type and rraA mutant strains. The pathways shown included carbohydrate metabolism, amino acid metabolism, ABC transporters, biofilm formation, and the type VI secretion system. Hierarchical clustering of samples and the relationships between genes and proteins were analyzed based on gene expression, measured as fragments per kilobase of exon model per million mapped fragments (FPKM), and protein expression data. Each column represents a sample, and each row represents a gene. The expression of genes in different samples is indicated by different colors. A redder color indicates higher expression, and a bluer color indicates lower expression.
Propionyl-CoA is generated through the methylcitrate cycle, which is the primary pathway for propionyl-CoA utilization in bacteria (25). In Vibrio species, this sequence of reactions is catalyzed by four core enzymes: methylcitrate synthase (PrpC), methylcitrate dehydratase (PrpD), aconitase (AcnD), and 2-methylaconitate cis-trans isomerase (PrpF). These enzymes are presumably encoded by the operon prpDB-acnD-prpFE, and they convert propionyl-CoA to pyruvate in a 1:1 molar ratio. Here, the levels of the proteins AcnD (BAU10_07690) and PrpC (BAU10_07695) were reduced by 2.53- and 6.01-fold, respectively, in the mutant (Table S4), indicating that impairment of RraA slows the methylcitrate cycle pathway, leading to a decrease in the pyruvate pool within the cell.
Notably, two subunits of pyruvate dehydrogenase, which is responsible for catalyzing the conversion of pyruvate to acetyl-CoA and is encoded by PdhAB (BAU10_20470 and BAU10_20475), were also significantly reduced by more than 12.17- and 22-fold, respectively, in the mutant (Table S4).
Positive role in amino acid transport and metabolism
Branched-chain amino acids (BCAAs) are initially converted to α-keto acids by the branched-chain amino acid transaminase, followed by decarboxylation by the branched-chain α-keto acid dehydrogenase complex, resulting in branched-chain acyl-CoA intermediates. These intermediates are further converted to propionyl-CoA or acetyl-CoA through β-oxidation catalyzed by enzymes such as methylmalonic acid-semialdehyde dehydrogenase (mmsA), hydroxyisobutyrate dehydrogenase (mmsB), isovaleryl coenzyme A dehydrogenase (ivd), and enoyl-CoA hydratase (paaF). In the present study, the BCAA degradation pathway was not significantly altered in the mutant. However, the impairment of rraA resulted in decreases of 13.31- and 18.44-fold in MmsA (BAU10_20645 and BAU10_18450, respectively), 10.54-fold in MmsB (BAU10_20625), and 9.26-fold in PaaF (BAU10_18460 and BAU10_20670, respectively; Table S4), suggesting that β-oxidation of branched-chain acyl-CoA is also positively regulated by RraA (Fig. 6A and B).
In addition, several key enzymes related to amino acid metabolism were affected. Alanine dehydrogenase (Ald BAU10_04925) and glutamate dehydrogenase (Gdh BAU10_07330), which are responsible for the catabolism of alanine, aspartate, and glutamate and the corresponding carbon skeletons, were downregulated by 1.70–3.32-fold at the protein level. Interestingly, the expression of the large subunit of glutamate synthase (GltB BAU10_01395), which is involved in converting glutamine and 2-oxoglutarate into glutamate, increased 2.45-fold in the mutant (Table S4).
Furthermore, core enzymes in the arginine synthesis pathway, including argininosuccinate lyase ArgH (BAU10_13655), acetylglutamate kinase ArgB (BAU10_13665), N-acetyl-gamma-glutamyl-phosphate reductase ArgC (BAU10_13670), bifunctional N-succinyl diaminopimelate-aminotransferase or acetylornithine transaminase protein ArgD (BAU10_13850), ornithine carbamoyltransferase ArgF (BAU10_13125), and argininosuccinate synthase ArgG (BAU10_13660), were positively regulated by RraA (Fig. 6A and B; Tables S3 and S4).
Additionally, several genes encoding ABC transporters were upregulated (BAU10_06495 ousX, up 2.05-fold; BAU10_06505 ousV, up 2.39-fold; BAU10_09575 oppF, up 2.16-fold; BAU10_20535 artP, up 4.18-fold); and several ABC transporters were downregulated (BAU10_05985 oppA, down 3.37-fold; BAU10_18030, down 6.38-fold; BAU10_19275 glnP down 2.21-fold; BAU10_19280 aapP down 2.33-fold; BAU10_20520 artM, down 2.24-fold). However, in their protein levels, a large amount of the amino acid ABC transporters showed decreased trends in the rraA mutant, including MppA (BAU10_15390, down 2.52-fold), ArgT (BAU10_04000, down 3.42-fold), AapJ (BAU10_07410, down 3.37-fold), OusX (BAU10_06495, down 3.81-fold), OppA (BAU10_05985, down 24.48-fold), AotP (BAU10_03995, down 2.23-fold), OusV (BAU10_06505, down 3.29-fold), ArtP (BAU10_20535, down 1.78-fold), BAU10_18335(down 17.77-fold), SdaC (BAU10_20390, down 4.55-fold), OppA(BAU10_09280, down 1.86-fold), OppF (BAU10_05965, down 12.57-fold), and GlnQ (BAU10_07585, down 1.54-fold), suggesting that RraA may play a role in positively regulating the uptake of amino acids and maintaining the balance between carbon and nitrogen sources within the cell.
RraA regulates genes involved in quorum sensing and the type VI secretion system
LuxQ-LuxU-LuxO is an important pathway for regulating biofilm formation in Vibrio parahaemolyticus (26).https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/deletion-mutantDeletion mutants of luxQ, luxU, and luxO formed weaker biofilms than did the WT strain. The data showed here that luxN (BAU10_09015) was downregulated by 2.26-fold at the mRNA level, while LuxO (BAU10_06930) was downregulated by 4.97-fold at the protein level, indicating that RraA may control biofilm formation by regulating the LuxQ-LuxU-LuxO pathway.
Bacterial secretion systems are macromolecular complexes that release virulence factors into the medium or translocate them into the target host cell (27). The type VI secretion system (T6SS) is widespread among pathogens and nonpathogens, and studies suggest that this system may be associated with interspecific competition and bacterial immunity to protect pathogens (28–30). Differentially expressed genes and proteins related to the T6SS were screened via transcriptomics and proteomics. Transcriptomic data revealed that T6SS-associated genes (tssA (BAU10_20250), tssB1 (BAU10_20245), tssC1 (BAU10_20240), tssC2 (BAU10_20235), tssG (BAU10_20215), BAU10_20275, tssM1 (BAU10_20265), tssH (BAU10_20210), and vasD (BAU10_20280) were significantly upregulated twofold to fourfold. In the rraA mutant strain, the expression of tssM1 and tssC1 was consistent with the transcriptome (Fig. 5). According to the proteomics data, only TagH was upregulated significantly, while the other DEGs exhibited no significant changes in protein levels. Mutations in the rraA gene activate the expression of most of the T6SS-related genes and several T6SS-related proteins, which suggests that rraA negatively and posttranscriptionally regulates the type VI secretion system.
DISCUSSION
RraA has widespread effects on mRNA abundance and plays important physiological roles in growth and biofilm formation in E. coli and V. vulnificus (6, 7, 10). In this study, we characterized the physiological role of RraA in V. alginolyticus ZJ-T and identified its regulon, which is summarized in Fig. 7.
Fig 7.

V. alginolyticus RraA regulon. Boxes indicate major pathways regulated by RraA. Solid arrows indicate those that have been confirmed by genetics. Genes or proteins in red are downregulated by rraA (upregulated in the rraA mutant), while genes or proteins in blue are upregulated by rraA (downregulated in the rraA mutant).
It was reported that the deletion of rraA resulted in reduced motility in V. vulnificus and Salmonella Typhimurium (12, 31). However, no significant impact on motility was observed for V. alginolyticus. Overexpression of RraA in E. coli was found to rescue cells from growth arrest, but this led to a decreased growth rate in V. vulnificus (12). These findings suggest that the function of RraA may have evolved to be species-specific.
Limited data exist on the regulation of rraA. An earlier study proposed that rraA in E. coli is transcribed from only one promoter, PrraA, located in the menA-rraA intergenic region, and the transcription of rraA increases during the stationary phase in a sigma(s)-dependent manner, as shown by primer extension and transcriptional fusion (4). Our study revealed the presence of at least three transcripts of different lengths, indicating the involvement of multiple promoters, including the conserved PrraA gene. Interestingly, in V. alginolyticus, the rraA mRNA level increased more than 12-fold during the stationary phase, but no detectable RraA protein was observed. This finding suggested the likelihood of posttranscriptional regulation, possibly through the inhibition of protein translation by an unknown factor. Similar expression patterns of the RraAV1 protein have been observed in V. vulnificus, indicating a potentially conserved mechanism.
This study utilized integrative transcriptome and proteome analysis to identify the regulon of rraA in V. alginolyticus. In comparison to the findings of a previous study conducted in V. vulnificus, the rraA regulon in V. alginolyticus exhibited significantly fewer transcriptional changes. This difference could be attributed to two factors. First, in a previous study, samples collected during the early stationary phase were used, while in our study, samples from the early log phase were used. Second, in a previous study, the criterion for differential gene expression was set at a 1.4-fold change, whereas we used a more stringent threshold of twofold.
Interestingly, only a few genes exhibited similar expression patterns for both DEGs and DEPs. This finding suggested that the regulation of rraA targets occurs primarily at the posttranscriptional level. For instance, in our study, the potential citrate catabolism operons citCDEFXG and citS-oadGAB, as well as the two-component system gene citAB, exhibited a significant decrease in transcript abundance (10–100-fold), but no difference in their protein products except for CitC was detected in the rraA mutant. One possible explanation is that RNase E is able to degrade both target mRNAs and small RNAs (sRNAs) that bind to target mRNAs and affect their translation. The impairment of rraA could lead to increased RNase E activity, resulting in decreased transcript abundance and the presence of unknown sRNAs that function as translational inhibitors. These opposing effects on protein synthesis could explain why the protein products of the affected genes showed no difference in the citrate metabolism operons. Notably, transcriptional, posttranscriptional, and translational regulation of citrate operons was reported in Lactococcus lactis, which further supports this hypothesis (32, 33).
Polymyxin B is a member of the colistin family of antibiotics that act by interacting with both the outer membrane and the inner membrane of bacteria (32). While resistance to this cationic lipopeptide is exceptionally rare (32), our findings indicate that disruption of the rraA gene leads to increased resistance to polymyxin B. Although the detailed mechanism remains unclear, previous reports have suggested a potential association between the reassembly of lipopolysaccharide and polymyxin B resistance in gram-negative bacteria (33, 34). In our investigation, we observed downregulation of the lpxH gene (BAU10_05120), which is known to be involved in lipid A biosynthesis and plays a crucial role in conferring polymyxin B resistance in gram-negative bacteria (33, 34). This finding suggested that V. alginolyticus RraA may facilitate the synthesis of lipid A, thereby conferring protection against polymyxin B-induced damage to cells.
Quorum sensing (QS) is a bacterial signaling pathway utilized to coordinate population behaviors, including biofilm formation, pathogenesis, and bioluminescence. In this study, we observed the downregulation of two QS proteins, LuxU (a phosphate transferase) and LuxO (a transcription factor activated by LuxU phosphorylation), at the protein level in the mutant. This finding suggested that RraA potentially has a positive impact on the QS signaling pathway by enhancing the translation of luxU and luxO. Previous reports have indicated that the deletion of luxO leads to reduced biofilm formation in V. parahaemolyticus and decreased expression of the cps biosynthesis gene cluster, which is responsible for producing major components of the biofilm matrix (26). Hence, it is reasonable to speculate that the decreased biofilm formation observed in the rraA mutant may be attributed to its positive influence on the LuxO and LuxU proteins.
Fatty acids serve as important carbon sources within cells when easily utilized carbon sources such as glucose are available. However, these compounds are degraded to support cell survival when these carbon sources become depleted. Fatty acids also play critical roles in maintaining membrane integrity and affecting various cellular processes (35, 36). According to the results of the proteomics analysis conducted in our study, V. alginolyticus RraA potentially positively regulates the expression of genes involved in the degradation of long-chain organic acids derived from fats or branched-chain amino acids through processes such as β-oxidation, the methylcitrate cycle, and branched-chain catabolism. These pathways collectively convert long-chain carbohydrates into important intermediates such as acetyl-CoA, propionyl-CoA, and pyruvate, which are required for energy production and metabolic remodeling.
In a previous study, the small RNA molecule FarS (fatty acid-regulated sRNA) in V. cholerae, produced through RNase E-mediated maturation of the fadB 3′-UTR, was reported to inhibit the expression of FadE, which is the rate-limiting enzyme involved in the oxidation of acyl-CoA (34). Based on this information, it is reasonable to speculate that rraA may derepress the expression of FadE by negatively affecting the production of a FarS homolog in V. alginolyticus.
In summary, our study identified RraA as a global regulator in V. alginolyticus. For the first time, we characterized its regulon at both the transcriptomic and proteomic levels, revealing complex interactions and mechanisms. However, further investigations are needed to determine the regulatory role of RraA in response to environmental cues and its involvement in posttranscriptional regulatory networks.
ACKNOWLEDGMENTS
This study was funded by the National Natural Science Foundation of China (31872605).
We are grateful to Guangzhou Genedenovo Biotechnology Co., Ltd., for assisting in sequencing and bioinformatics analysis. We are thankful for the data archive support from the South China Sea Ocean Data Center (http://data.scsio.ac.cn, accessed on 16 July 2022) and the National Science and Technology Infrastructure of China (http://www.geodata.cn, accessed on 16th July 2022).
Chang Chen contributed reagents and materials and conceived and designed the experiments. Huizhen Chen, Qian Gao, Bing Liu, Ying Zhang, Songbiao Wang, and Youqi Chen performed the experiments. Huizhen Chen and Qian Gao analyzed the data. Bing Liu, Ying Zhang, Youqi Chen, and Jianxiang Fang performed the data visualization. Huizhen Chen and Bing Liu prepared the original draft, and Chang Chen reviewed and edited the manuscript. All the authors have read and agreed to the published version of the manuscript.
Contributor Information
Chang Chen, Email: chen.chang@scsio.ac.cn.
Nicolas E. Buchler, NC State University, Raleigh, North Carolina, USA
DATA AVAILABILITY
All RNA-sequencing data are deposited in GenBank (wild-type biosample: SAMN29675353, SRA: SRR20124473-SRR20124475; rraA mutant strain biosample: SAMN30175538, SRA: SRR22012560-SRR22012562; accessed on 10 December 2022). All proteome data are deposited in iProX (Integrated Proteome Resources) database (https://www.iprox.cn/page/home. html, accessed on 22 September 2023); the accession ID is IPX0007150000. The other data presented in this study are available in the article.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/msphere.00020-24.
Figure S1 and Tables S1 to S3.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Régnier P, Arraiano CM. 2000. Degradation of mRNA in bacteria: emergence of ubiquitous features. Bioessays 22:235–244. doi: [DOI] [PubMed] [Google Scholar]
- 2. Mudd EA, Higgins CF. 1993. Escherichia coli endoribonuclease RNase E: autoregulation of expression and site‐specific cleavage of mRNA. Mol Microbiol 9:557–568. doi: 10.1111/j.1365-2958.1993.tb01716.x [DOI] [PubMed] [Google Scholar]
- 3. Lee K, Zhan X, Gao J, Qiu J, Feng Y, Meganathan R, Cohen SN, Georgiou G. 2003. RraA: a protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 114:623–634. doi: 10.1016/j.cell.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 4. Zhao M, Zhou L, Kawarasaki Y, Georgiou G. 2006. Regulation of RraA, a protein inhibitor of RNase E-mediated RNA decay. J Bacteriol 188:3257–3263. doi: 10.1128/JB.188.9.3257-3263.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Górna MW, Pietras Z, Tsai YC, Callaghan AJ, Hernández H, Robinson CV, Luisi BF. 2010. The regulatory protein RraA modulates RNA-binding and helicase activities of the E. coli RNA degradosome. RNA 16:553–562. doi: 10.1261/rna.1858010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee K, Cohen SN. 2003. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol Microbiol 48:349–360. doi: 10.1046/j.1365-2958.2003.03435.x [DOI] [PubMed] [Google Scholar]
- 7. Lee M, Yeom JH, Sim SH, Ahn S, Lee K. 2009. Effects of Escherichia coli RraA orthologs of Vibrio vulnificus on the ribonucleolytic activity of RNase E in vivo. Curr Microbiol 58:349–353. doi: 10.1007/s00284-009-9360-8 [DOI] [PubMed] [Google Scholar]
- 8. Tang JA, Luo MA, Niu SQ, Zhou H, Cai XF, Zhang WL, Hu YA, Yin YB, Huang AL, Wang DQ. 2010. The crystal structure of hexamer RraA from Pseudomonas aeruginosa reveals six conserved protein-protein interaction sites. Protein J. 29:583–590. doi: 10.1007/s10930-010-9293-x [DOI] [PubMed] [Google Scholar]
- 9. Heo J, Kim D, Joo M, Lee B, Seo S, Lee J, Song S, Yeom JH, Ha NC, Lee K. 2016. RraAS2 requires both scaffold domains of RNase ES for high-affinity binding and inhibitory action on the ribonucleolytic activity. J Microbiol 54:660–666. doi: 10.1007/s12275-016-6417-9 [DOI] [PubMed] [Google Scholar]
- 10. Kim D, Kim YH, Jang JY, Yeom JH, Jun JW, Hyun S, Lee K. 2016. Functional analysis of Vibrio vulnificus orthologs of Escherichia coli RraA and RNase E. Curr Microbiol 72:716–722. doi: 10.1007/s00284-016-1007-y [DOI] [PubMed] [Google Scholar]
- 11. Seo S, Kim D, Song W, Heo J, Joo M, Lim Y, Yeom JH, Lee K. 2017. RraAS1 inhibits the ribonucleolytic activity of RNase ES by interacting with its catalytic domain in Streptomyces coelicolor. J Microbiol 55:37–43. doi: 10.1007/s12275-017-6518-0 [DOI] [PubMed] [Google Scholar]
- 12. Lee J, Shin E, Park J, Lee M, Lee K. 2021. Regulator of ribonuclease activity modulates the pathogenicity of Vibrio vulnificus. J Microbiol 59:1133–1141. doi: 10.1007/s12275-021-1518-5 [DOI] [PubMed] [Google Scholar]
- 13. Chang C, Jin X, Chaoqun H. 2009. Phenotypic and genetic differences between opaque and translucent colonies of Vibrio alginolyticus. Biofouling 25:525–531. doi: 10.1080/08927010902964578 [DOI] [PubMed] [Google Scholar]
- 14. Nguyen AN, Disconzi E, Charrière GM, Destoumieux-Garzón D, Bouloc P, Le Roux F, Jacq A. 2018. csrB gene duplication drives the evolution of redundant regulatory pathways controlling expression of the major toxic secreted metalloproteases in Vibrio tasmaniensis LGP32. mSphere 3:e00582-18. doi: 10.1128/mSphere.00582-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Val ME, Skovgaard O, Ducos-Galand M, Bland MJ, Mazel D. 2012. Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet. 8:e1002472. doi: 10.1371/journal.pgen.1002472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morales VM, Bäckman A, Bagdasarian M. 1991. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97:39–47. doi: 10.1016/0378-1119(91)90007-x [DOI] [PubMed] [Google Scholar]
- 17. Zhang YJ, Chen GZ, Lin HY, Wang P, Kuang BZ, Liu J, Chen SY. 2017. Development of a regulatable expression system for the functional study of Vibrio vulnificus essential genes. Antonie van Leeuwenhoek 110:607–614. doi: 10.1007/s10482-016-0827-x [DOI] [PubMed] [Google Scholar]
- 18. Grimm C, Evers A, Brock M, Maerker C, Klebe G, Buckel W, Reuter K. 2003. Crystal structure of 2-methylisocitrate lyase (PrpB) from Escherichia coli and modelling of its ligand bound active centre. J Mol Biol 328:609–621. doi: 10.1016/s0022-2836(03)00358-9 [DOI] [PubMed] [Google Scholar]
- 19. Liu B, Fang JX, Chen HZ, Sun YH, Yang S, Gao Q, Zhang Y, Chen C. 2022. GcvB regulon revealed by transcriptomic and proteomic analysis in Vibrio alginolyticus. Int J Mol Sci 23:9399. doi: 10.3390/ijms23169399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu B, Gao Q, Zhang X, Chen HZ, Zhang Y, Sun YH, Yang S, Chen C. 2021. CsrA regulates swarming motility and carbohydrate and amino acid metabolism in Vibrio alginolyticus. Microorganisms 9:2383. doi: 10.3390/microorganisms9112383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 22. Shineberg B, Young IG. 1976. Biosynthesis of bacterial menaquinones-membrane-associated 1,4-dihydroxy-2-naphthoate octaprenyltransferase of Escherichia coli. Biochemistry 15:2754–2758. doi: 10.1021/bi00658a007 [DOI] [PubMed] [Google Scholar]
- 23. Alvarez AF, Rodríguez C, González-Chávez R, Georgellis D. 2021. The Escherichia coli two-component signal sensor BarA binds protonated acetate via a conserved hydrophobic-binding pocket. J Biol Chem 297:101383. doi: 10.1016/j.jbc.2021.101383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song S, Hong S, Jang J, Yeom JH, Park N, Lee J, Lim Y, Jeon JY, Choi HK, Lee M, Ha NC, Lee K. 2017. Functional implications of hexameric assembly of RraA proteins from Vibrio vulnificus. PLoS One 12:e0190064. doi: 10.1371/journal.pone.0190064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Upton AM, McKinney JD. 2007. Role of the methylcitrate cycle in propionate metabolism and detoxification in Mycobacterium smegmatis. Microbiology 153:3973–3982. doi: 10.1099/mic.0.2007/011726-0 [DOI] [PubMed] [Google Scholar]
- 26. Liu M, Zhu XY, Zhang C, Zhao Z. 2021. LuxQ-LuxU-LuxO pathway regulates biofilm formation by Vibrio parahaemolyticus. Microbiol Res 250:126791. doi: 10.1016/j.micres.2021.126791 [DOI] [PubMed] [Google Scholar]
- 27. Cascales E. 2008. The type VI secretion toolkit. EMBO Rep 9:735–741. doi: 10.1038/embor.2008.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brooks TM, Unterweger D, Bachmann V, Kostiuk B, Pukatzki S. 2013. Lytic activity of the Vibrio cholerae type VI secretion toxin VgrG-3 is inhibited by the antitoxin TsaB. J Biol Chem 288:7618–7625. doi: 10.1074/jbc.M112.436725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dong C, Zhang H, Gao ZQ, Wang WJ, She Z, Liu GF, Shen YQ, Su XD, Dong YH. 2013. Structural insights into the inhibition of type VI effector Tae3 by its immunity protein Tai3. Biochem J 454:59–68. doi: 10.1042/BJ20130193 [DOI] [PubMed] [Google Scholar]
- 30. Hood RD, Singh P, Hsu F, Güvener T, Carl MA, Trinidad RRS, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, Li M, Schwarz S, Wang WY, Merz AJ, Goodlett DR, Mougous JD. 2010. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7:25–37. doi: 10.1016/j.chom.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee M, Ryu M, Joo M, Seo YJ, Lee J, Kim HM, Shin E, Yeom JH, Kim YH, Bae J, Lee K. 2021. Endoribonuclease-mediated control of hns mRNA stability constitutes a key regulatory pathway for Salmonella Typhimurium pathogenicity Island 1 expression. PLoS Pathog 17:e1009263. doi: 10.1371/journal.ppat.1009263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Mastrigt O, Mager EE, Jamin C, Abee T, Smid EJ. 2018. Citrate, low pH and amino acid limitation induce citrate utilization in Lactococcus lactis biovar diacetylactis. Microb Biotechnol 11:369–380. doi: 10.1111/1751-7915.13031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drider D, Santos JM, Arraiano CM, López P. 1998. RNA processing is involved in the post-transcriptional control of the citQRP operon from Lactococcus lactis biovar diacetylactis. Mol Gen Genet 258:9–15. doi: 10.1007/s004380050701 [DOI] [PubMed] [Google Scholar]
- 34. Huber M, Fröhlich KS, Radmer J, Papenfort K. 2020. Switching fatty acid metabolism by an RNA-controlled feed forward loop. Proc Natl Acad Sci U S A 117:8044–8054. doi: 10.1073/pnas.1920753117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schönfeld P, Wojtczak L. 2016. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res 57:943–954. doi: 10.1194/jlr.R067629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chintalapati S, Kiran MD, Shivaji S. 2004. Role of membrane lipid fatty acids in cold adaptation. Cell Mol Biol (Noisy-le-grand) 50:631–642. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 and Tables S1 to S3.
Data Availability Statement
All RNA-sequencing data are deposited in GenBank (wild-type biosample: SAMN29675353, SRA: SRR20124473-SRR20124475; rraA mutant strain biosample: SAMN30175538, SRA: SRR22012560-SRR22012562; accessed on 10 December 2022). All proteome data are deposited in iProX (Integrated Proteome Resources) database (https://www.iprox.cn/page/home. html, accessed on 22 September 2023); the accession ID is IPX0007150000. The other data presented in this study are available in the article.