

Abstract
Background
Aldehyde dehydrogenase 2 (ALDH2) is critical for alcohol metabolism by converting acetaldehyde to acetic acid. In East Asian descendants, an inactive genetic variant in ALDH2, rs671, triggers an alcohol flushing response due to acetaldehyde accumulation. As alcohol flushing is not exclusive to those of East Asian descent, we questioned whether additional ALDH2 genetic variants can drive facial flushing and inefficient acetaldehyde metabolism using human testing and biochemical assays.
Methods
After IRB approval, human subjects were given an alcohol challenge (0.25 g/kg) while quantifying acetaldehyde levels and the physiological response (heart rate and skin temperature) to alcohol. Further, by employing biochemical techniques including human purified ALDH2 proteins and transiently transfected NIH 3T3 cells, we characterized two newly identified ALDH2 variants for ALDH2 enzymatic activity, ALDH2 dimer/tetramer formation, and reactive oxygen species production after alcohol treatment.
Results
Humans heterozygous for rs747096195 (R101G) or rs190764869 (R114W) had facial flushing and a 2-fold increase in acetaldehyde levels, while rs671 (E504K) had facial flushing and a 6-fold increase in acetaldehyde levels relative to wild type ALDH2 carriers. In vitro studies with recombinant R101G and R114W ALDH2 enzyme showed a reduced efficiency in acetaldehyde metabolism that is unique when compared to E504K or wild-type ALDH2. The effect is caused by a lack of functional dimer/tetramer formation for R101G and decreased Vmax for both R101G and R114W. Transiently transfected NIH-3T3 cells with R101G and R114W also had a reduced enzymatic activity by ~ 50% relative to transfected wild-type ALDH2 and when subjected to alcohol, the R101G and R114W variants had a 2-3-fold increase in reactive oxygen species formation with respect to wild type ALDH2.
Conclusions
We identified two additional ALDH2 variants in humans causing facial flushing and acetaldehyde accumulation after alcohol consumption. As alcohol use is associated with a several-fold higher risk for esophageal cancer for the E504K variant, the methodology developed here to characterize ALDH2 genetic variant response to alcohol can lead the way precision medicine strategies to further understand the interplay of alcohol consumption, ALDH2 genetics, and cancer.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-024-05507-x.
Keywords: Genetic variant, Acetaldehyde, Alcohol, Aldehyde dehydrogenase 2 (ALDH2), Alcohol challenge, Enzyme kinetics
Background
Alcohol is consumed by at least 2 billion people world-wide annually [1, 2]. When alcohol is metabolized, acetaldehyde accumulation can lead to an overproduction of cellular reactive oxygen species (ROS); adding to the basal ROS production occurring when alcohol is metabolized to acetaldehyde and acetic acid [3, 4] (Fig. 1). Therefore, acetaldehyde accumulation is one of the important factors driving the pathophysiology of alcohol-induced cancer [5, 6]. Broadly, this biological process is not only important for alcohol-mediated carcinogenesis, but is also linked to the mechanism of drug-induced cellular toxicity [7–9].
Fig. 1.
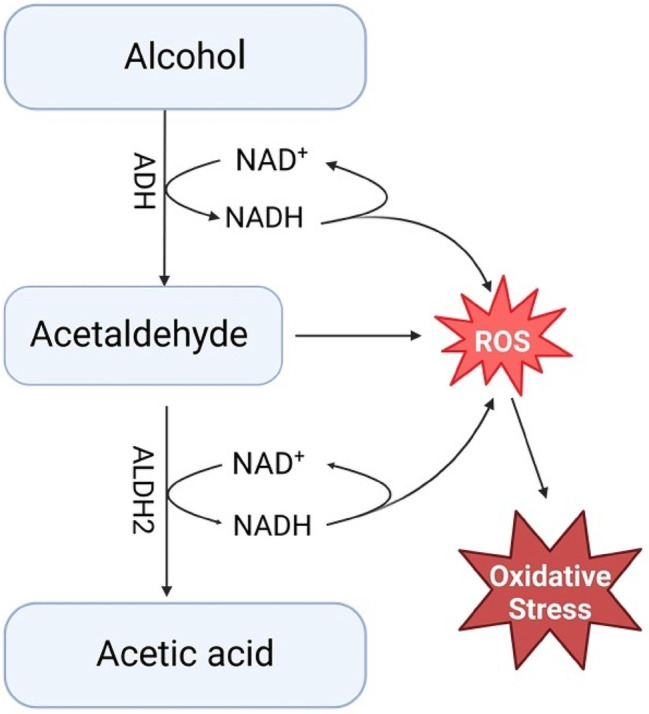
Alcohol metabolism and reactive oxygen species generation. Reactive oxygen species (ROS) are generated when ADH converts alcohol to acetaldehyde and when ALDH2 converts acetaldehyde to acetic acid. For both ADH and ALDH2, NADH is converted back to NAD+ and an electron is released reacting with oxygen to form ROS. Electrons from these steps can further react with dioxygen species (O2) to generate the superoxide (O2·−) and hydroxyl (OH.) radicals. Accumulation of acetaldehyde, which occurs with inactive ALDH2 variants, results in damage to the electron transport complexes leading to overproduction of ROS
In East Asian countries, ~ 40% of the population carry a genetic variant in the aldehyde dehydrogenase 2 (ALDH2) enzyme that metabolizes acetaldehyde to acetic acid [10, 11]. The ALDH2 genetic variant (ALDH2*2, rs671), causes acetaldehyde accumulation along with facial flushing and tachycardia after alcohol consumption [12]. The ALDH2*2 variant leads to an accumulation of acetaldehyde after alcohol consumption which contributes to an overproduction of ROS leading to carcinogenesis [13, 14]. Even moderate alcohol consumption for ALDH2*2 carriers increases the odds ratio for developing aerodigestive track cancer (2.61, 1.19–5.75) and esophageal cancer (3.12, 1.95–5.01) due to acetaldehyde accumulation from inefficient acetaldehyde metabolism [15, 16]. East Asia also has the highest world-wide incidence of esophageal cancer; likely due to the high frequency of the ALDH2*2 variant [17]. As nearly 540 million people of either sex carry the ALDH2*2 genetic variant, the frequency of ALDH2*2 carriers within the human population shines an initial light into understanding how alcohol use, an inactive ALDH2 genetic variant, and subsequent acetaldehyde-induced ROS production can lead to an increased risk for aerodigestive tract and esophageal cancers [12, 18–22].
Further, the genetic database gnomAD indicates there are ~ 570 missense ALDH2 variants occurring across all ethnicities which may cause a loss of function, gain of function, or no difference in ALDH2 enzyme activity. Whether other variants in ALDH2, besides rs671 (E504K), cause facial flushing and acetaldehyde accumulation after alcohol consumption remain uncharacterized in humans. To accelerate this process of discovery, a non-invasive method to quantify acetaldehyde levels along with the physiological response to alcohol in humans is needed. Here, we developed in humans a non-invasive technique to test how ALDH2 variants alter acetaldehyde metabolism and the physiological response to alcohol. We then confirmed these findings using biochemical assays in vitro and in cell culture. This is important to understand considering the cancer risk associated with facial flushing occurring for the rs671 variant [23, 24].
Methods
For this manuscript, we performed a comprehensive literature review using PubMed, Google Scholar, and Science Direct covering years 1998–2024. The literature review served as the foundation for the manuscript including assisting with the formulation of the research question.
Prior to recruitment and testing of humans, IRB approval was obtained from Stanford University (IRB 46095). This study is a basic experimental study involving humans with the manipulation or task used (consuming alcohol) expressly used for measurement and is not an intervention. Written informed consent was received from participants prior to inclusion in the study. The experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Participant recruitment
Participants were recruited from flyers posted on the Stanford University campus in succession. The initial flyer recruited people who flush after they consume alcohol for an alcohol study. The flyer was then modified to recruit people only of non-Asian descent.
Participants were initially screened for exclusion using a questionnaire. Participants were also instructed to complete a CAGE questionnaire to screen out participants with potential alcoholism. Those participants eligible then provided a saliva sample for DNA extraction and purification (Quick-DNA Miniprep Kit, Zymo Research). DNA was amplified using 12 primers designed to cover the ALDH2 exome for sequencing (Supplemental Table 1). After amplification, samples were sent for Sanger sequencing (Eton Bioscience).
Human alcohol challenge
Participants were selected for the alcohol challenge portion of the study based on genotyping results. At this stage, participants were also excluded if homozygous for rs671 (secondary to the expected severity of the alcohol response). Regarding patient selection, the human volunteers for this study were healthy volunteers without a past medical history or taking any medications. None of the patients had a history of cardiovascular disease or diabetes mellitus. East Asian participants heterozygous for rs671 (E504K) were called back for participation in the alcohol challenge. In addition, people without sequenced variants were also selected to closely match the age, sex and weight of the participants heterozygous for rs671 (E504K). Additional participants were selected for the alcohol challenge study based upon genotyping results identifying people who were carriers of the ALDH2 variants including rs747096195 (R101G) and rs190764869 (R114W). Details regarding patient demographics are reported (Supplemental Table 2).
Participants were instructed prior to the alcohol challenge not to eat or drink for 2 h prior to testing. Testing was performed between 12pm and 4pm on a weekday. Upon arrival, patients were consented and weighed. To assess physiologic measurements, a five lead EKG was placed and EKG measurements were recorded (ADInstruments). Skin temperature was measured under the left cheek bone using liquid crystal technology (Crystaline II Temperature Indicators, Sharn Anesthesia). Baseline hemodynamic data including heart rate and skin temperature were collected. Prior to testing, a hand-held alcohol breath test meter confirmed no alcohol was consumed before testing (BACtrack Element Pro). Breath samples were collected by a 3 L Tedlar bag (Zefon International) that were sealed after collection. Breath samples were analyzed within 4 h of sample collection.
After establishing a baseline, participants were given a 415 mL (14oz) drink consisting of 0.25 g/kg Ketel One vodka, water, and Minute Maid lemonade. We chose vodka for the alcohol challenge to minimize the effect of congeners on our study [25]. The lemonade was used as a mixer. For reference in the United States, a standard drink is 14 g of alcohol which is equal to one 12 oz glass of 5% beer or one 5oz glass of 12% wine [26]. As an example, a 70 kg person in this study drank 17.5 g of alcohol. Participants consumed the first 7oz of the 14oz drink during the first 5 min of the study and the second 7oz during the second 5 min. After baseline measurements taken 3 min before alcohol consumption, physiologic measurements of heart rate, skin temperature, and breath metabolites were taken at 5, 10, 15, 30, 60, 90, 120, and 150 min during and after alcohol consumption (Fig. 2A). After completion of the study, a breath alcohol test and a standard field sobriety test were administered. The study lasted a total duration of 3 h. When collecting the data, we considered heart rate and facial skin temperature as the physiological measurements to analyze in relation to the acetaldehyde levels measured. Participants completing the alcohol challenge were given $125 compensation.
Fig. 2.

Flowchart and experimental protocol for alcohol challenge. (A) Alcohol challenge protocol. After 10 min baseline, subjects were given an alcohol challenge (0.25 g/kg); consumed over 10 min. Subjects were then monitored while measuring physiologic parameters (heart rate and skin temperature). (B) Recruitment of 16 subjects of East Asian descent for an alcohol challenge after genotyping. 8 subjects (4 male and 4 female) were selected for alcohol challenge with an ALDH2*1*2 genotype. Additionally, 8 subjects with an ALDH2*1/*1 genotype were age and sex matched to those with an ALDH2*1/*2 genotype. (C) Recruitment of subjects self-identified as non-East Asian descent. We identified 2 subjects that were carriers for uncharacterized genetic variants in ALDH2 that were subjected to an alcohol challenge
Breath metabolite analysis
The breath metabolite acetaldehyde was measured from a Tedlar bag using selective ion flow mass spectrometry (Syft Voice Ultra, New Zealand). Soft ionization was carried out using H3O+ and NO+ with the mass and reaction ratios described for measuring acetaldehyde (Supplemental Table 3). Helium was used a carrier gas.
In Silico ALDH2 Modeling
Protein data bank (PDB) entry 1O02 was imported into PyMOL (Schrödinger, Inc.) for molecular visualization of wild-type ALDH2 and to mutate R101 and R114 by using the protein mutagenesis function.
ALDH2 site-directed variant construction
Protein Expression and Purification: Based upon genotyping results, site-directed mutagenesis of human ALDH2 was performed by PCR using a QuikChange site-directed mutagenesis kit (Agilent Technologies) and a kanamycin resistance template plasmid, pET-28a-c (+) containing full length cDNA of wild type ALDH2 gene. Oligonucleotides were used to generate ALDH2 variants (R101G, R114W and E504K) (Supplementary Table 4). The mutated plasmids were sequenced at Sequetech Corporation (Mountain View, CA) to check for the presence of the desired mutations and absence of unwanted base changes within the plasmids. Co-expression of wild-type ALDH2 and its site directed variants (R101G, R114W and E504K) was achieved using the pEDuet-1 vector plasmid (Millipore Sigma, Milwaukee, WI). Briefly, we cloned one copy of wild-type ALDH2 into the His-tag cloning site of the pETDuet-1 vector plasmid by EcoRI and HindIII and another copy of wild-type ALDH2 into the S-tag using NdeI and XhoI. For co-expression of wild-type ALDH2 with the variants, we cloned one copy of wild type ALDH2 into the His-tag cloning site as described above and the ALDH2 variants into the S-tag cloning site of pETDuet-1 plasmid using NdeI and XhoI. Co-expression of the recombinant proteins was carried out at 30 °C in the presence of GroEL chaperone using E. coli BL21 (DE3) host cells. Cells were induced with 0.5 M IPTG and grown for 16 h at 30 °C. Cells were recovered by centrifugation at 5000 rpm and recombinant proteins were lysed with B-PER complete bacterial protein extraction reagent (Thermo fisher Scientific, South San Francisco, CA). Protein purification was achieved with a His-trap nickel affinity column (Millipore Sigma, Milwaukee, WI). Following purification, the amount of protein was determined using the Pierce BCA Protein Assay Kit (Thermo fisher Scientific, South San Francisco, CA).
Expression of wild-type ALDH2 and its variant enzymes in NIH-3T3 cells
Transient transfection of wild type ALDH2 and ALDH2 missense variants was performed in the NIH-3T3 cell line (BPS Bioscience #79,469) cultured in Growth 5 A medium. Once the 3T3 cells had reached 80% confluence, they were transfected with either the mammalian expression pCMV3-ALDH2-C-FLAG plasmid harboring wild-type ALDH2 or the ALDH2 variants. Additional cells were transfected with an empty pCMV3-C-FLAG vector. Transfection complexes were prepared using a 1:5 ratio (1 µg of plasmid/5µL of Lipofectamine 2000) in Opti-MEM-I-Reduced Serum Medium and incubated for 2 h. After incubation, the reduced serum medium was replaced with Growth 5 A medium and transfection was allowed to proceed for 72 h. Protein expression analysis was conducted 72 h post-transfection using an enzymatic activity assay as described below.
Aldh2 enzymatic activity assay
NAD+ conversion to NADH in the presence of acetaldehyde was used to determine ALDH2 enzyme activity of purified recombinant protein (5 µg/mL) or total lysate (200 µg/mL) for cultured cells at pH = 9.0 and pH = 9.5, respectively as previously described [23]. For Alda-1 studies, wild type or ALDH2 variants were incubated with Alda-1 (20µM final concentration) for 2 min and assayed at pH = 9.0 as described elsewhere [27].
Reactive oxygen species (ROS) production
For cellular ROS production, NIH-3T3 cells were seeded to 70–80% confluence. The cells were then incubated with 2,7-dichlorofluorescein diacetate (DCFDA, 5 µM) in basal medium at 37 °C for 30 min in the dark. After 30 min, fluorescence was measured at 37 °C (485/535 nm for excitation/emission) to measure the basal level of ROS (Growth 5 A medium was used a vehicle). The cells were then treated with 50 mM ethanol and fluorescence was again measured. Fluorescence intensity was normalized to the basal level for each protein. The dose of 50mM ethanol was chosen for this study because it is used in prior experimental studies when using cell culture assays studying the effects of alcohol [23, 28, 29]. This level is considered a physiologic exposure to a cell as in humans, the circulating blood ethanol levels are within the 10–15 mM range after 0.25 g/kg-0.75 g/kg alcohol consumption [30].
ALDH2 cross-linking and western blot analysis
ALDH2 cross-linking was carried out by mixing bis(sulfosuccinimidyl)suberate (62.5 mM final concentration) with purified wild-type ALDH2 or ALDH2 variants (500 µg/mL) followed by adding SDS-loading buffer (5 µL, 120 mM Tris-HCl, pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 5% (w/v) bromophenol blue, 1% (w/v) DTT) in a total volume of 20µL at 4 °C [31]. MagicMark XP Western Protein Standard (ThermoFisher- Cat #: LC5603) was used to detect the molecular weight markers on western blot. The crosslinking mixture was analyzed by an 8% SDS-page gel (100 volts, 2h30min) at 4 °C and transferred to a nitrocellulose membrane (20 V, 16 h) at 4 °C. The next day membranes were washed with TBST buffer (4 times) and blocked with blotting-grade blocker (Biorad, Cat# 1,706,404) for 1 h. After 1 h, the membranes were probed using antibodies to ALDH2 (goat polyclonal, 1:1000, Abcam, Fremont, CA) or S-tag (rabbit monoclonal, 1:1000, Cell Signaling Technology, Danvers, MA) for 2 h and then washed 4 times with TBST buffer. Following washing, the membranes were incubated with anti-goat (1:2000 concentration, Abcam, Cat# ab97110) or anti-rabbit (1:2000 concentration, Cell Signaling Technology, Cat# 70,745) secondary antibodies for 1 h. After 1 h, the membranes were again washed with TBST buffer and proteins were visualized by SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, South San Francisco, CA). Images were acquired with Azure Biosystems C300 for analysis.
Statistical analysis
All data is presented as mean ± SEM. The number needed to recruit for this study is based upon prior studies examining differences in acetaldehyde metabolism in the blood for carriers of the r671 variant versus non-carriers [30, 32, 33]. Based upon the dose of alcohol (0.25 g/kg) producing a 5-fold difference in blood acetaldehyde levels [30], we predicted that at a minimum there was a need to recruit 4 people per group to identify statistical differences between people that are wild type ALDH2 relative to people that are heterozygous for rs671 (E504K). For analysis of differences in metabolites or physiological data, a two-way ANOVA followed by Bonferroni correction for multiplicity was used in order to compare each group to the control group or to compare measurements performed within a group over time. When comparing heart rate or skin temperature changes with acetaldehyde levels, a Pearson correlation test with a 2-tailed p-value was used. A two-tailed Student’s t-test was performed to compare the treated and untreated recombinant ALDH2 using Alda-1 and in ROS generation experiments. In addition, a one-way ANOVA was performed to determine the statistical difference of recombinant ALDH2 proteins or the ALDH2 transient transfection studies. Statistical analysis was performed using GraphPad Prism 6. *p < 0.01 was considered statistically significant between groups compared at the same time-point or end-point and ^p < 0.01 was considered statistically significant within groups with respect to vehicle or baseline.
Results
Human subjects studies
After genotyping human subjects, we recruited relevant participants to partake in an alcohol challenge. Initially, we recruited positive and negative controls for this study by recruiting those of Asian descent with and without the rs671 (E504K) genetic variant. For this portion of the study, 58 people contacted the laboratory regarding participation. Of those, 51 people participated in the initial phone screen with 29 people providing a saliva sample for genotyping, and 16 people were given an alcohol challenge (Fig. 2A); 8 were heterozygotes for rs671 and 8 were not (Fig. 2B). We then modified the protocol to recruit participants only of non-Asian descent. We had 69 people who underwent a phone screen to identify self-reporting flushing after ethanol exposure, 38 people provided saliva samples, and based on the genotyping results, we identified people heterozygous for rs747096195 (R101G) or rs190764869 (R114W).
To explore the phenotype after alcohol consumption based on the ALDH2 genotyping from human subjects, we subjected participants who were heterozygous for rs747096195 (R101G), rs190764869 (R114W), and rs671 (E504K) to an alcohol challenge and compared the response to those participants carrying wild type ALDH2. We considered the rs747096195 and rs190764869 variants together as these variants are within the same alpha helix for ALDH2 and each occur in a low frequency for humans. When comparing heterozygotes for rs671 (E504K, ALDH2*1/*2) with respect to those with a wild type ALDH2 genotype, breath acetaldehyde levels peaked 9-fold 5 min after completing alcohol consumption (Figs. 3A and 15-minute time point measurement, peak: 2.1 ± 0.4* versus 0.2 ± 0.3 ppm, n = 8/group). Acetaldehyde levels remained significantly elevated for 60 min after initial alcohol consumption (Fig. 3A, 1.2 ± 0.2* versus 0.13 ± 0.02 ppm, n = 8/group). Further, rs747096195 (R101G) and rs190764869 (R114W) accumulated acetaldehyde and took longer to metabolize acetaldehyde relative to those subjects that were wild type ALDH2 (Fig. 3A). This effect was not as prominent as compared to heterozygotes for rs671 (E504K). The total area under the acetaldehyde curve was 6-fold greater for the ALDH2*1/*2 genotype versus the ALDH2*1/*1 genotype (Figs. 3B and 143 ± 14* versus 22 ± 3 ppm-minutes, n = 8/group). Further, total acetaldehyde had a two-fold accumulation for rs747096195 and rs190764869 (Figs. 3B and 46 ± 7 ppm-minutes).
Fig. 3.

Breath metabolite and physiologic characteristics after alcohol challenge. (A) Breath concentrations (in parts per million) of acetaldehyde. (B) The total area under the acetaldehyde curve. (C) Heart rate change after alcohol consumption (D) Change in heart rate versus change in breath acetaldehyde concentration. (E) Skin temperature change after alcohol consumption and (F) Change in skin temperature versus change in breath acetaldehyde concentration. Black lines are wild type subjects, dark blue lines are R101G heterozygote, light blue lines are R114W heterozygote and red lines are E504K heterozygote subjects. *p < 0.01 by one-way or two-way ANOVA versus wild type subjects at the same time-points, ^p < 0.01 by two-way ANOVA with respect to baseline values within groups, n = 8/group for wild-type or E504K (4 male and 4 female) and n = 2/group (1 female R101G and 1 female R114W).
Concomitantly, heart rate markedly increased in ALDH2*1/*2 participants relative to ALDH2*1/*1 participants peaking at 30 min (Figs. 3C and 104 ± 3* versus 73 ± 4 beats per minute, n = 8/group). No relative changes in heart rate occurred for those participants carrying rs747096195 (R101G) or rs190764869 (R114W) variants (Fig. 3C). The change in heart rate versus change in breath acetaldehyde concentration also correlated during peak acetaldehyde concentrations (Fig. 3D, r2 = 0.78). Additionally, skin temperature changes relative to baseline significantly increased for rs671 at 30 min followed by rs747096195 (R101G) and rs190764869 (R114W) at 30 min after the start of alcohol consumption (Fig. 3E). However, skin temperature changes had less of a correlation with acetaldehyde levels measured at 30 min (Fig. 3F, r2 = 0.31).
In vitro and cell culture studies
When examining the crystal structure of ALDH2, R101G and R114W are located in distinct regions of the α-B helix that are separate from E504K (Fig. 4A and B). A closer look at the ALDH2 tetramer suggests that R101 is important in the interaction between two opposite ALDH2 subunits of the tetramer, while R114 is important in the interaction of α-B and α-E helices within the same monomer (Fig. 4C and D, respectively). In addition, E504 located in the oligomerization domain is important for the formation of homodimers at the dimer interface (Fig. 4E). In wild-type ALDH2, R101 of one subunit forms a hydrogen bond with S517 of the opposite ALDH2 subunit via a conserved water molecule, while R114 forms an H-bond with E227 of the same subunit (Fig. 4C and D). The E504 residue form hydrogen bonds with R281 of the same subunit and R492 of the adjacent dimer partner [34]. When R101 is mutated to a glycine, a hydrogen bond via a conserved water molecule is lost between R101 and S517 (Fig. 4F). Upon R114W mutation, the two hydrogen bonds between R114 of α-B helix and E227 of the adjacent α-E helix within the same subunit are lost (Fig. 4G). The E504K variant disrupts the hydrogen bonding network between E504 and residues, R281 and R492, leading to a change in conformation of these residues at the dimer interface (Fig. 4H).
Fig. 4.

Location of R101G R114W and E504K variants in ALDH2 and impact of these variants. (A) Crystal structure of ALDH2, with 3 amino acids within the tetramer highlighted (R101 (light blue), R114 (dark blue) and E504 (red)). (B) Location of R101 (light blue), R114 (dark blue) and E504 (red) in the ALDH2 subunit. H-bond interactions between (C) R101 with S517 of another subunit via conserved water. (D) R114 with E227 of the same subunit. (E) E504K with R281 of one subunit and R492 of another ALDH2 subunit. (F) The R101G variant leads to loss of H-bonds with S517 of the opposite subunit. (G) R114W variant leads to loss of H-bonds with E227 within the same subunit and (H) E504K leads to loss of H-bonds with R281 within the same subunit and R492 of another subunit at the dimer interface
To mimic the heterozygous condition in humans, we co-expressed in bacteria wild type and the ALDH2 variants into the pETDuet-1 vector. The co-expressed variants R101G and R114W with wild type ALDH2 had a significant decrease in Vmax compared to wild-type ALDH2 (Fig. 5A and B, 1.1 ± 0.0* and 1.1 ± 0.1* versus 2.8 ± 0.0 mmol/min/mg of protein for wild type ALDH2, respectively, n = 5/group). In addition, E504K co-expressed with wild type ALDH2 had a significant decrease in Vmax relative to wild type ALDH2 (Fig. 5A and B, 0.8 ± 0.0* versus 2.8 ± 0.0 mmol/min/mg of protein for wild type, n = 5/group). Furthermore, the Km for R114W co-expressed with wild-type ALDH2 was similar to wild type ALDH2 and E504K (Fig. 5B, Km = 104 ± 13 versus 129 ± 13 and 111 ± 9 µM for wild type ALDH2 and E504K, respectively). However, R101G co-expressed with wild type ALDH2 had a ~ 0.3-0.4-fold decrease of Km compared to wild type ALDH2 (Figs. 5B and 75 ± 10* vs. 129 ± 13 µM).
Fig. 5.

R101G and R114W variants cause inefficient acetaldehyde metabolism. (A) The Michaelis–Menten plot showing rate of reaction (y-axis) against NAD+ concentration for proteins expressed with petDuet-1 plasmid. These in vitro results support inefficient acetaldehyde metabolism for WT/R101G and WT/R114W. (B) Vmax of WT/R101G and WT/R114W are significantly different from WT/E504K and wild type ALDH2. Km of WT/R101G is significantly different from WT/R114W, WT/E504K and wild type ALDH2. n = 5/group, *p < 0.01 versus wild type ALDH2 enzyme
ALDH2 exists as a tetramer comprising of four identical subunits [35]. The tetramer functions as a dimer of dimers, where each of the four monomeric subunits contains a dinucleotide (NAD+)-binding domain, oligomerization domain and a catalytic domain with three key cysteine residues and a glutamate, which are required for the catalysis at the active site [34]. To determine whether wild type ALDH2 or the variants can form dimers and tetramers in solution, we employed a cross linker, bis(sulfosuccinimidyl)suberate, to capture these two forms of ALDH2. Furthermore, we performed western blot analysis of the crosslinked proteins to determine the presence of S-tagged proteins expressed with the petDuet-1 plasmid. A western blot with the S-tag antibody showed that R101G formed few dimers and tetramers relative to wild type and WT/E504K (Fig. 6A and Supplementary Fig. 1A). Additionally, there was no significant difference between dimers and tetramers of WT/R114W compared to wild type or WT/E504K using the S-tag antibody (Fig. 6A). When using an ALDH2 antibody to detect proteins expressed in the His-tag and S-tag cloning sites of petDuet-1 plasmid, WT/R101G formed dimers and tetramers albeit at a lower level compared wild type or WT/E504K (Fig. 6B and Supplementary Fig. 1B). However, WT/R114W had similar levels of dimers and tetramers relative to the wild type ALDH2 or the WT/E504K variant.
Fig. 6.

Formation of ALDH2 dimers and tetramers. (A) Representative western blot of wild type ALDH2 and the ALDH2 variants using a S-tag antibody with quantification of dimer and tetramer formation detected by the S-tag antibody. The R101G variant did not form dimers and tetramers. (B) Representative western blot of wild type ALDH2 and the ALDH2 variants using an ALDH2 antibody with quantification of dimer and tetramer formation by the ALDH2 antibody. MW = molecular weight markers, n = 5 or 6/group, *p < 0.01 by one-way ANOVA relative to wild type, R114W and E504K
To determine whether Alda-1, a specific ALDH2 activator, increases the activity of the variants, we measured the activity of purified enzymes in the presence of Alda-1 (20µM). Alda-1 significantly enhanced the activities of wild-type ALDH2 and E504K relative to vehicle (Fig. 7, 2.0 ± 0.1^ and 0.7 ± 0.0^ versus 3.5 ± 0.1 and 1.5 ± 0.1 mmol/min/mg of protein for wild type and E504K). In addition, Alda-1 significantly enhanced the activities of R101G and R114W relative to vehicle (Fig. 7, 0.8 ± 0.1^ and 1.0 ± 0.1^ versus 1.5 ± 0.1 and 2.0 ± 0.1 mmol/min/mg of protein for R101G and R114W, respectively). Therefore, Alda-1 brought the activity of these less-active variants close to the activity of wild type ALDH2 enzyme.
Fig. 7.
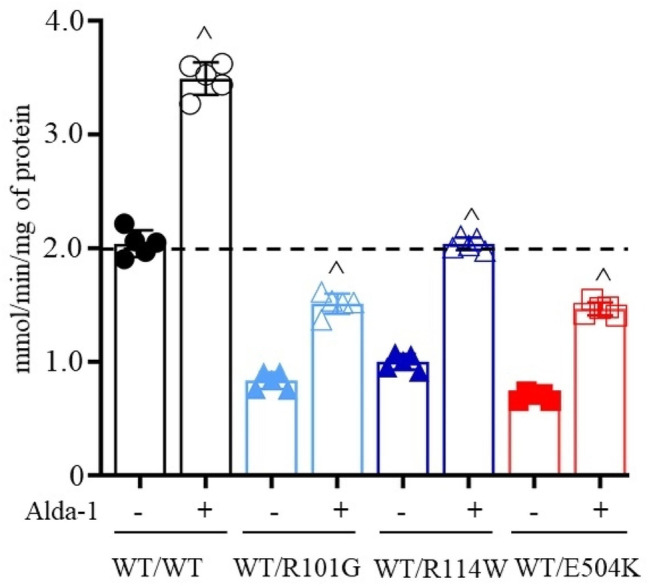
Alda-1 enhances the activity of wild type ALDH2 and the ALDH2 variants. The activity of wild type ALDH2 and ALDH2 variants expressed with petDuet-1 plasmid measured in the presence of Alda-1 (20µM) or vehicle. R101G and R114W activities were increased by ~ 82% and ~ 104% relative to vehicle. In addition, activity of wild type ALDH2 and E504K was increased by ~ 71% and ~ 110% relative to vehicle, n = 5/group, ^p < 0.01 with unpaired two-tailed Student’s t-test relative to untreated
To elucidate the cellular consequences of the reduced activity ALDH2 variants, we transiently transfected NIH-3T3 cells with wild-type ALDH2 and the three ALDH2 variants. Interestingly, the activity of R101G and R114W variants transiently transfected in NIH-3T3 cells was approximately 50% lower than that of wild-type, and was slightly higher than E504K (Fig. 8A, 0.2 ± 0.0* and 0.2 ± 0.0*, respectively versus 0.4 ± 0.1 mmol/min/mg of protein for wild type).
Fig. 8.

ALDH2 variants display a reduced enzymatic activity and increased cellular ROS. (A) ALDH2 activity in transiently transfected NIH-3T3 cells. n = 5/group, *p < 0.01 by one-way ANOVA versus wild type ALDH2. (B) ROS levels measured by DFCA in cells treated 50 mM ethanol and compared with untreated cell, n = 6 or 7/group, ^P < 0.01 with unpaired two-tailed Student’s t-test relative to untreated
Next, we measured cellular ROS in a NIH-3T3 cell line overexpressing wild-type ALDH2 and the three ALDH2 variants. Our results showed a marked increase in cellular ROS for the wild-type and the three variants when 3T3 cells were treated with 50 mM ethanol compared to cells treated with vehicle (Fig. 8B). Ethanol treatment led to a 2-fold increase in cellular ROS for wild-type transfected cells relative to vehicle (Figs. 8B and 3036 ± 301^ vs. 1489 ± 458, respectively, relative florescence units). All three variants demonstrated a higher than 2-fold increase in cellular ROS production upon exposure to ethanol relative to vehicle (Fig. 8B, R101G:5434 ± 913^ vs. 2712 ± 784, R114W:4377 ± 836^ vs. 1725 ± 337, and E504K:7188 ± 1426^ vs. 2431 ± 402, relative florescence units, respectively). The baseline ROS levels for R101G were also increased compared to WT transfected cells.
Discussion
By developing a non-invasive human assay to quantify the physiological response and acetaldehyde levels after alcohol consumption, we identified ALDH2 genetic variants rs747096195 (R101G) and rs190764869 (R114W) cause facial flushing and a 2-fold increase in acetaldehyde following alcohol consumption. Expectantly, carriers of rs671 (E504K) also triggered facial flushing and a 6-fold acetaldehyde accumulation compared to those without an ALDH2 variant. As acetaldehyde accumulation after alcohol consumption leads to higher risks of aerodigestive tract and esophageal cancer [12, 24, 36], this assay can more precisely provide a better understanding of alcohol-associated cancer risk in humans by quantifying genetic differences that are present in acetaldehyde metabolism.
Compared to prior research, using selective ion flow mass spectrometry (SIFT-MS) to detect breath levels of acetaldehyde after alcohol consumption have several advantages. As opposed to using gas chromatography to measure acetaldehyde [37, 38], SIFT-MS can be performed without additional sample processing, in real-time, and as a high-throughput assay. Further, SIFT-MS can detect acetaldehyde levels in the parts per billion range, providing sensitivity equivalent to laser spectroscopy [39, 40]. SIFT-MS also identified differences in acetaldehyde metabolism occurring for less common ALDH2 genetic variants, such as ALDH2 R101G and R114W. By using SIFT-MS to quantify acetaldehyde levels over a time-course after alcohol consumption, we also identified that heart rate changes correlate with breath acetaldehyde levels after an alcohol challenge. This builds upon prior human volunteer studies that describe an increased heart rate after an alcohol challenge [33, 41, 42]. Therefore, heart rate and acetaldehyde levels after alcohol consumption can provide biomarkers in humans to understand individual genetic differences in alcohol metabolism.
We identified that ALDH2 R101G and R114W variants limit acetaldehyde metabolism by two distinct means using complementary biochemical techniques with in vitro and cell culture studies. The R101G variant causes a disruption of the dimer-dimer interaction and the R114W variant causes a disruption of a H-bonding interaction within the same subunit. This is opposed to the limited enzymatic activity for the E504K variant which is attributed to the disruption of hydrogen bonding interactions at the dimer interface and coenzyme NAD+ binding site [34]. In the case of E504K variant, Alda-1, an allosteric ALDH2 agonist, increases acetaldehyde metabolism by providing structural stability to the enzyme, which rescues hydrogen-bond perturbations caused by the E504K variant [43]. Alda-1 binds near the exit of the substrate binding tunnel and can also significantly enhance ALDH2 activity for the R101G and R114W variants [43]. However, Alda-1 is not a universal activator for all ALDH2 variants as Alda-1 did not improve the activities of ALDH2*3 (I41V), ALDH2*6 (V304M) and ALDH2*7 (R339W) [23]. Rather than activating ALDH2 to increase acetaldehyde metabolism, targeting ALDH3A1 with a small molecule Alda-89 to enable ALDH3A1 to metabolize acetaldehyde can be an alternative approach to improve acetaldehyde metabolism for carriers of less active ALDH2 variants [44].
The less active R101G and R114W ALDH2 variants also produce higher levels of ROS when transfected cells are exposed to alcohol. This is consistent with a prior study finding the less active ALDH2 variants I41V, P92T, T244M, V304M, and R338W generate higher levels of ROS after alcohol treatment [23]. Further, even without alcohol exposure, basal levels of ROS were higher for ALDH2*2 knock-in rodents in the tongue, lung, heart, kidney, and brain tissues with respect to wild type ALDH2 mice [45, 46]. Further, heterozygote ALDH2*2 iPSC endothelial cells have higher levels of ROS production compared to their genome-edited isogenic iPSC endothelial cell lines resulting in 1460 differentially expressed genes related to ROS, angiogenesis, and inflammatory pathways [47]. Taken together, exposure to alcohol in carriers of less active ALDH2 variants can lead to higher ROS levels that modulate cellular pathways creating an oncogenic environment [48].
Besides the link between alcohol consumption and cancer, the association of alcohol use and cardiovascular disease continues to be a focus of research as elevated levels of ROS can drive cellular pathways that lead to cardiovascular disease [49]. When ALDH2 knockout mice are exposed to alcohol for 6 weeks, higher levels of ROS within the ALDH2 knockout mice with respect to wild type ALDH2 mice were associated with reduced left ventricular ejection fraction and increased myocardial fibrosis [50]. A study with diabetic-induced ALDH2 knockout mice showed that ALDH2 deficiency increases diabetes-induced oxidative stress by increasing the lipid peroxidation product, 4-hydroxynonenal [51]. However, studies suggest in humans the ALDH2*2 genetic variant limits excessive alcohol drinking, and the impact of ALDH2 genetic variants on cardiovascular disease may be mitigated [52]. For example, when examining the association of alcohol consumption with carotid plaque burden in 22,384 adults from the China Kadoorie Biobank, individuals with the wild type ALDH2 genotype had a higher association with carotid plaque burden relative to carriers of the ALDH2*2 genetic variant [53]. These findings are also supported by a study examining coronary artery calcification in 1029 Japanese men who consume alcohol where carriers of the ALDH2*2 variant had a lower coronary artery calcification burden relative to those without the ALDH2*2 variant [54]. A 12-year observational study of 512,000 adults within China Kadoorie Biobank revealed an association between alcohol consumption with 61 different diseases but genetic evidence whether the ALDH2*2 variant modified this risk was limited in scope due to inadequate statistical power [55]. Together, these studies suggest that the interplay between genetics and alcohol consumption is complex and requires future experimental and translational research to understand the interplay between alcohol, ALDH2 genetic, and cardiovascular disease.
The health risks of alcohol use may also be influenced by congeners within alcoholic beverages that are produced during ethanol production by fermentation or distillation. The types of congeners formed include phenols, esters, and ketones in addition to alcohols, such as methanol and butanol, and acetaldehyde. Unregulated beverages high in congeners consumed in East and Southern Africa such as chang’aa, gongo, and kachasu, may contribute to why Eastern and Southern Africa has the second highest risk of esophageal cancer in the world [17, 56]. In addition, home-made and fruit-based spirits have higher acetaldehyde content relative to grain-based or industry produced spirits [57]. Further, congeners can also intensify hangover symptoms [58, 59] and spirits with less congeners such as vodka produce less hangover symptoms in study participants relative to whiskey [60]. Taken together, the impact of congeners on health risks such as cancer and cardiovascular disease require further investigation.
Our results need to be interpreted within the realm of potential limitations. We focused on ALDH2 variants instead of alcohol dehydrogenase (ADH) which converts alcohol to acetaldehyde. As the activity of ALDH2 correlates with ADH activity in the liver [61], the subtle differences within groups for our study could be due to genetic variability of ADH. As our study did not consider examining mitochondrial localization of ALDH2 or the ALDH2 variants, we measured total cellular ROS instead of mitochondrial ROS. Alcohol can also cause DNA damage via ROS and acetaldehyde which was not measured for the ALDH2 variants identified in this study. As the recruitment flyers were designed to identify people that have facial flushing after they consume alcohol, it is untested whether acetaldehyde accumulation may occur without facial flushing. Furthermore, variants other than rs671 are relatively rare in the general population, which prevented the collection of a large sample size for rs747096195 (R101G) and rs190764869 (R114W) that we identified. Our study focused on how ALDH2 variants impact the metabolism of acetaldehyde but did not consider how congeners in alcoholic beverages may influence aldehyde metabolism. Regardless, the non-invasive assay we developed can provide individualized assessments of acetaldehyde metabolism; leading to more personalized medicine strategies by identifying those that have impaired acetaldehyde metabolism after alcohol consumption versus those that do not.
Conclusions
Here we developed a methodology to non-invasively quantify acetaldehyde metabolism after alcohol consumption and identified two variants in ALDH2 besides rs671 that cause acetaldehyde accumulation after an alcohol challenge. Future work will involve leveraging this alcohol challenge assay to further phenotype human ALDH2 genetic variants in combination with supporting in vitro and cell culture studies to more precisely define the interplay between alcohol consumption, ALDH2 genetic variants, and cancer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Martina Steffen for her initial support as a clinical coordinator for this study and Kayla Anchondo-Johnson, RN for her assistance in collecting physiologic data.
Abbreviations
- ALDH2
Aldehyde Dehydrogenase 2
- DCFDA
2,7-Dichlorofluorescein Diacetate
- ROS
Reactive Oxygen Species
- IRB
Institutional Review Board
- SIFT-MS
Selective Ion-Flow Mass Spectrometry
Author contributions
MA, DMR, CHC and ERG participated in study design. FR, RCRH, XY, KNZ, JRW, LM, and ERG collected and analyzed the data for this manuscript. FR, ERG and JRW wrote the manuscript with input from RCRH, XY, MA, CHC and DMR. All the authors have read and agreed to the published version of the manuscript.
Funding
Funding for this project was supported by Stanford ChEM-H (ERG and DMR), and from the National Institutes of Health, NIAAA AA011147 (DMR), and NIGMS GM119522 (ERG).
Data availability
All data generated or analyzed during this study are included in this published article.
Declarations
Ethics approval and consent to participate
All human studies were approved by the Institutional Review Board (IRB) at Stanford University (IRB 46095). Written informed consent was received from participants prior to inclusion in the study. The experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Consent for publication
Not applicable.
Competing interests
Eric Gross holds a patent related to the ALDH2 activator Alda-1, and is a consultant for Chiima Therapeutics. Daria Mochly-Rosen and Che-Hong Chen hold patents related to Alda-1. Dr. Angst has funding for a research study from Alkahest Inc. Additional authors have no disclosures.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World Health Organization. Global status report on alcohol and health. Geneva; 2018.
- 2.Ritchie H, Roser M. Alcohol Consumption. OurWorldInData.org. 2018 [cited 2023 Jul 7].
- 3.Brocardo PS, Gil-Mohapel J, Christie BR. The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res Rev. 2011;67:209–25. 10.1016/j.brainresrev.2011.02.001 [DOI] [PubMed] [Google Scholar]
- 4.Joya X, Garcia-Algar O, Salat‐Batlle J, Pujades C, Vall O. Advances in the development of novel antioxidant therapies as an approach for fetal alcohol syndrome prevention. Birth Defects Res Part Clin Mol Teratol. 2015;103:163–77. 10.1002/bdra.23290 [DOI] [PubMed] [Google Scholar]
- 5.Johnson CH, Golla JP, Dioletis E, Singh S, Ishii M, Charkoftaki G, et al. Molecular mechanisms of Alcohol-Induced Colorectal Carcinogenesis. Cancers (Basel). 2021;13:4404. 10.3390/cancers13174404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Q, Chen H, Huo J-R. Alcohol consumption and corresponding factors: a novel perspective on the risk factors of esophageal cancer. Oncol Lett. 2016;11:3231–9. 10.3892/ol.2016.4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lakey PSJ, Berkemeier T, Tong H, Arangio AM, Lucas K, Pöschl U, et al. Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract. Sci Rep. 2016;6:32916. 10.1038/srep32916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA, et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019;10:851. 10.1038/s41419-019-2081-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohta S, Ohsawa I, Kamino K, Ando F, Shimokata H. Mitochondrial ALDH2 Deficiency as an oxidative stress. Mitochondrial pathog. Berlin, Heidelberg: Springer Berlin Heidelberg; 2004. pp. 36–44. [DOI] [PubMed] [Google Scholar]
- 10.Gross ER, Zambelli VO, Small BA, Ferreira JCB, Chen C-H, Mochly-Rosen D. A personalized medicine approach for Asian americans with the aldehyde dehydrogenase 2*2 variant. Annu Rev Pharmacol Toxicol. 2015;55:107–27. 10.1146/annurev-pharmtox-010814-124915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo H-R, Wu G-S, Pakstis AJ, Tong L, Oota H, Kidd KK, et al. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2⁎487Lys. Gene. 2009;435:96–103. 10.1016/j.gene.2008.12.021 [DOI] [PubMed] [Google Scholar]
- 12.Brooks PJ, Enoch M-A, Goldman D, Li T-K, Yokoyama A. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. 10.1371/journal.pmed.1000050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flashner S, Shimonosono M, Tomita Y, Matsuura N, Ohashi S, Muto M, et al. ALDH2 dysfunction and alcohol cooperate in cancer stem cell enrichment. Carcinogenesis. 2024;45:95–106. 10.1093/carcin/bgad085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo W, Gao Y, He Y, Sun J, Xu H, Feng D, et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol. 2019;71:1000–11. 10.1016/j.jhep.2019.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai S-T, Wong T-Y, Ou C-Y, Fang S-Y, Chen K-C, Hsiao J-R, et al. The interplay between alcohol consumption, oral hygiene, ALDH2 and ADH1B in the risk of head and neck cancer. Int J Cancer. 2014;135:2424–36. 10.1002/ijc.28885 [DOI] [PubMed] [Google Scholar]
- 16.Yang SJ, Yokoyama A, Yokoyama T, Huang YC, Wu SY, Shao Y, et al. Relationship between genetic polymorphisms of ALDH2 and ADH1B and esophageal cancer risk: a meta-analysis. World J Gastroenterol. 2010;16:4210. 10.3748/wjg.v16.i33.4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 18.Yokoyama T, Yokoyama A, Kato H, Tsujinaka T, Muto M, Omori T, et al. Alcohol flushing, alcohol and aldehyde dehydrogenase genotypes, and risk for esophageal squamous cell carcinoma in Japanese men. Cancer Epidemiol Biomarkers Prev. 2003;12:1227–33. [PubMed] [Google Scholar]
- 19.Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, et al. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007;8:292–3. 10.1016/S1470-2045(07)70099-2 [DOI] [PubMed] [Google Scholar]
- 20.Dong J, Thrift AP. Alcohol, smoking and risk of oesophago-gastric cancer. Best Pract Res Clin Gastroenterol. 2017;31:509–17. 10.1016/j.bpg.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 21.Heymann HM, Gardner AM, Gross ER. Aldehyde-Induced DNA and protein adducts as Biomarker Tools for Alcohol Use Disorder. Trends Mol Med. 2018;24:144–55. 10.1016/j.molmed.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McAllister SL, Sun K, Gross ER. Developing precision medicine for people of east Asian descent. J Biomed Sci. 2016;23:80. 10.1186/s12929-016-0299-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C-H, Ferreira JCB, Joshi AU, Stevens MC, Li S-J, Hsu JH-M, et al. Novel and prevalent non-east Asian ALDH2 variants; implications for global susceptibility to aldehydes’ toxicity. EBioMedicine. 2020;55:102753. 10.1016/j.ebiom.2020.102753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang JS, Hsiao J-R, Chen C-H. ALDH2 polymorphism and alcohol-related cancers in asians: a public health perspective. J Biomed Sci. 2017;24:19. 10.1186/s12929-017-0327-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodda LN, Beyer J, Gerostamoulos D, Drummer OH. Alcohol congener analysis and the source of alcohol: a review. Forensic Sci Med Pathol. 2013;9:194–207. 10.1007/s12024-013-9411-0 [DOI] [PubMed] [Google Scholar]
- 26.National Institutes of Health. What Is A Standard Drink? [cited 2023 Jul 6].
- 27.Chen C-H, Budas GR, Churchill EN, Disatnik M-H, Hurley TD, Mochly-Rosen D. Activation of Aldehyde Dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5. 10.1126/science.1158554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrera M, Molina P, Souza-Smith FM. Ethanol-induced lymphatic endothelial cell permeability via MAP-kinase regulation. Am J Physiol Physiol. 2021;321:C104–16. 10.1152/ajpcell.00039.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gómez-Quiroz L, Bucio L, Souza V, Escobar C, Farfán B, Hernández E, et al. Interleukin 8 response and oxidative stress in HepG2 cells treated with ethanol, acetaldehyde or lipopolysaccharide. Hepatol Res. 2003;26:134–41. 10.1016/S1386-6346(03)00010-X [DOI] [PubMed] [Google Scholar]
- 30.Kim S-W, Bae K-Y, Shin H-Y, Kim J-M, Shin I-S, Youn T, et al. The role of Acetaldehyde in human psychomotor function: a double-blind placebo-controlled crossover study. Biol Psychiatry. 2010;67:840–5. 10.1016/j.biopsych.2009.10.005 [DOI] [PubMed] [Google Scholar]
- 31.Datta S, Annapure US, Timson DJ. Characterization of Cd36_03230p, a putative vanillin dehydrogenase from Candida Dubliniensis. RSC Adv. 2016;6:99774–80. 10.1039/C6RA22209A [DOI] [Google Scholar]
- 32.Enomoto N, Takase S, Yasuhara M, Takada A. Acetaldehyde Metabolism in different Aldehyde Dehydrogenase-2 genotypes. Alcohol Clin Exp Res. 1991;15:141–4. 10.1111/j.1530-0277.1991.tb00532.x [DOI] [PubMed] [Google Scholar]
- 33.Chen Y-C, Yang L-F, Lai C-L, Yin S-J. Acetaldehyde enhances Alcohol Sensitivity and protects against Alcoholism: evidence from Alcohol metabolism in subjects with variant ALDH2*2 Gene Allele. Biomolecules. 2021;11. [DOI] [PMC free article] [PubMed]
- 34.Larson HN, Weiner H, Hurley TD. Disruption of the Coenzyme binding site and Dimer Interface revealed in the Crystal structure of mitochondrial aldehyde dehydrogenase Asian variant. J Biol Chem. 2005;280:30550–6. 10.1074/jbc.M502345200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez-Zavala JS, Weiner H. Structural aspects of Aldehyde dehydrogenase that Influence dimer – tetramer formation. Biochemistry. 2002;41:8229–37. 10.1021/bi012081x [DOI] [PubMed] [Google Scholar]
- 36.Choi CK, Yang J, Kweon S-S, Cho S-H, Kim H-Y, Myung E, et al. Association between ALDH2 polymorphism and esophageal cancer risk in south koreans: a case-control study. BMC Cancer. 2021;21:254. 10.1186/s12885-021-07993-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Himemiya-Hakucho A, Tanaka T, Liu J, Fujimiya T. Effect of Alcohol Sensitivity in healthy young adults on Breath Pharmacokinetics of Acetaldehyde after Mouth washing with Alcohol. Alcohol Clin Exp Res. 2018;42:2100–6. 10.1111/acer.13878 [DOI] [PubMed] [Google Scholar]
- 38.Matsuura K, Kushio S, Imai H, Kudo H, Wakuda H, Otani N, et al. Association between psychomotor function and ALDH2 genotype after consuming barley shochu: a randomized crossover trial. Clin Transl Sci. 2023;16:686–93. 10.1111/cts.13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamat PC, Roller CB, Namjou K, Jeffers JD, Faramarzalian A, Salas R, et al. Measurement of acetaldehyde in exhaled breath using a laser absorption spectrometer. Appl Opt. 2007;46:3969. 10.1364/AO.46.003969 [DOI] [PubMed] [Google Scholar]
- 40.Jones AW. Drug-alcohol flush reaction and Breath Acetaldehyde Concentration: no interference with an Infrared Breath Alcohol Analyzer. J Anal Toxicol. 1986;10:98–101. 10.1093/jat/10.3.98 [DOI] [PubMed] [Google Scholar]
- 41.Iwase S, Matsukawa T, Ishihara S, Tanaka A, Tanabe K, Danbara A, et al. Effect of oral ethanol intake on muscle sympathetic nerve activity and cardiovascular functions in humans. J Auton Nerv Syst. 1995;54:206–14. 10.1016/0165-1838(95)00022-P [DOI] [PubMed] [Google Scholar]
- 42.Sagawa Y, Kondo H, Matsubuchi N, Takemura T, Kanayama H, Kaneko Y, et al. Alcohol has a dose-related effect on parasympathetic nerve activity during sleep. Alcohol Clin Exp Res. 2011;35:2093–100. 10.1111/j.1530-0277.2011.01558.x [DOI] [PubMed] [Google Scholar]
- 43.Perez-Miller S, Younus H, Vanam R, Chen C-H, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17:159–64. 10.1038/nsmb.1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C-H, Cruz LA, Mochly-Rosen D. Pharmacological recruitment of aldehyde dehydrogenase 3A1 (ALDH3A1) to assist ALDH2 in acetaldehyde and ethanol metabolism in vivo. Proc Natl Acad Sci. 2015;112:3074–9. 10.1073/pnas.1414657112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Chen C-H, Yang J, Mochly-Rosen D. Aldehyde dehydrogenase 2*2 knock-in mice show increased reactive oxygen species production in response to cisplatin treatment. J Biomed Sci. 2017;24:33. 10.1186/s12929-017-0338-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu X, Zeng X, Xiao F, Chen R, Sinharoy P, Gross ER. E-cigarette aerosol exacerbates cardiovascular oxidative stress in mice with an inactive aldehyde dehydrogenase 2 enzyme. Redox Biol. 2022;54:102369. 10.1016/j.redox.2022.102369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo H, Yu X, Liu Y, Paik DT, Justesen JM, Chandy M et al. SGLT2 inhibitor ameliorates endothelial dysfunction associated with the common ALDH2 alcohol flushing variant. Sci Transl Med. 2023;15. [DOI] [PMC free article] [PubMed]
- 48.Liou G-Y, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–96. 10.3109/10715761003667554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panth N, Paudel KR, Parajuli K. Reactive oxygen species: a key Hallmark of Cardiovascular Disease. Adv Med. 2016;2016:1–12. 10.1155/2016/9152732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen C, Wang C, Han S, Wang Z, Dong Z, Zhao X, et al. Aldehyde dehydrogenase 2 deficiency negates chronic low-to-moderate alcohol consumption-induced cardioprotecion possibly via ROS-dependent apoptosis and RIP1/RIP3/MLKL-mediated necroptosis. Biochim Biophys Acta - Mol Basis Dis. 2017;1863:1912–8. 10.1016/j.bbadis.2016.11.016 [DOI] [PubMed] [Google Scholar]
- 51.Wang C, Fan F, Cao Q, Shen C, Zhu H, Wang P, et al. Mitochondrial aldehyde dehydrogenase 2 deficiency aggravates energy metabolism disturbance and diastolic dysfunction in diabetic mice. J Mol Med. 2016;94:1229–40. 10.1007/s00109-016-1449-5 [DOI] [PubMed] [Google Scholar]
- 52.Oroszi G, Goldman D. Alcoholism: genes and mechanisms. Pharmacogenomics. 2004;5:1037–48. 10.1517/14622416.5.8.1037 [DOI] [PubMed] [Google Scholar]
- 53.Zhou T, Im PK, Hariri P, Du H, Guo Y, Lin K, et al. Associations of alcohol intake with subclinical carotid atherosclerosis in 22,000 Chinese adults. Atherosclerosis. 2023;377:34–42. 10.1016/j.atherosclerosis.2023.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hisamatsu T, Miura K, Tabara Y, Sawayama Y, Kadowaki T, Kadota A, et al. Alcohol consumption and subclinical and clinical coronary heart disease: a mendelian randomization analysis. Eur J Prev Cardiol. 2022;29:2006–14. 10.1093/eurjpc/zwac156 [DOI] [PubMed] [Google Scholar]
- 55.Im PK, Wright N, Yang L, Chan KH, Chen Y, Guo Y, et al. Alcohol consumption and risks of more than 200 diseases in Chinese men. Nat Med. 2023;29:1476–86. 10.1038/s41591-023-02383-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menya D, Kigen N, Oduor M, Maina SK, Some F, Chumba D, et al. Traditional and commercial alcohols and esophageal cancer risk in Kenya. Int J Cancer. 2019;144:459–69. 10.1002/ijc.31804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boffetta P, Kaihovaara P, Rudnai P, Znaor A, Lissowska J, Swiatkowska B, et al. Acetaldehyde level in spirits from central European countries. Eur J Cancer Prev. 2011;20:526–9. 10.1097/CEJ.0b013e328348fbe4 [DOI] [PubMed] [Google Scholar]
- 58.Zhu H, Xing Y, Akan OD, Yang T. Alcohol-Induced Headache with Neuroinflammation: recent progress. Fermentation. 2023;9:184. 10.3390/fermentation9020184 [DOI] [Google Scholar]
- 59.Swift R, Davidson D. Alcohol hangover: mechanisms and mediators. Alcohol Health Res World. 1998;22:54–60. [PMC free article] [PubMed] [Google Scholar]
- 60.Rohsenow DJ, Howland J, Arnedt JT, Almeida AB, Greece J, Minsky S, et al. Intoxication with Bourbon Versus Vodka: effects on Hangover, Sleep, and Next-Day Neurocognitive performance in young adults. Alcohol Clin Exp Res. 2010;34:509–18. 10.1111/j.1530-0277.2009.01116.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao N, Li J, Li M-R, Qi B, Wang Z, Wang G-J, et al. Higher activity of Alcohol dehydrogenase is correlated with hepatic fibrogenesis. J Pharmacol Exp Ther. 2018;367:473–82. 10.1124/jpet.118.249425 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.