

Abstract
Recent advances in the field of immuno-oncology have brought transformative changes in the management of cancer patients. The immune profile of tumours has been found to have key value in predicting disease prognosis and treatment response in various cancers. Multiplex immunohistochemistry and immunofluorescence have emerged as potent tools for the simultaneous detection of multiple protein biomarkers in a single tissue section, thereby expanding opportunities for molecular and immune profiling while preserving tissue samples. By establishing the phenotype of individual tumour cells when distributed within a mixed cell population, the identification of clinically relevant biomarkers with high-throughput multiplex immunophenotyping of tumour samples has great potential to guide appropriate treatment choices. Moreover, the emergence of novel multi-marker imaging approaches can now provide unprecedented insights into the tumour microenvironment, including the potential interplay between various cell types. However, there are significant challenges to widespread integration of these technologies in daily research and clinical practice. This review addresses the challenges and potential solutions within a structured framework of action from a regulatory and clinical trial perspective. New developments within the field of immunophenotyping using multiplexed tissue imaging platforms and associated digital pathology are also described, with a specific focus on translational implications across different subtypes of cancer.
Keywords: multiplex immunofluorescence, multiplex immunohistochemistry, tumour infiltrating lymphocytes, multiplex imaging, digital image analysis, tumour immune profiling, cancer prognosis, therapy response, clinical integration
Fundamentals of immune infiltration in cancer
In earlier concepts of cancer, the specific role of the immune system in the pathogenesis of malignancy was not well recognised. Over the last 15 years, however, the crucial role that the host immune system plays in tumour evolution has been brought to the forefront of cancer research; indeed, the immune landscape of tumours has emerged as a key hallmark of cancer [1], During oncogenesis, alteration of the tumour microenvironment (TME) and tumour neoantigens trigger signals that facilitate immune responses in an attempt to eliminate preneoplastic cells [2], Immune cells of varying density can be found in most types of malignancy due to the immunogenic response triggered by cancer cells [3]. The accumulation of different immune cells can have both tumour-promoting and tumour-suppressive functions [4].
In the first phase of tumourigenesis, cytotoxic immune cells such as NK and cytotoxic CD8+ T cells identify and kill only highly immunogenic cancer cells. Consequently, less immunogenic cells escape the reach of both the adaptive and innate immune systems, fostering the progression to malignancy [5]. Thus, heterogeneous populations of immune infiltrates can drive tumour progression via a complex network of crosstalk between themselves and the other components of the TME. The qualitative characterisation of this interplay between immune infiltrates and cancer cells is called the immune contexture [6], which encompasses the density of each immune infiltrate and their spatial architecture across the tumour. Profiling immune contexture is one of the most significant ways to obtain insights into immune responses and has the potential to provide information with predictive and prognostic value [6]. Numerous studies over the years have provided strong evidence linking the presence of tumour-infiltrating lymphocytes (TILs) with clinical outcomes in various tumour types, such as melanoma, breast, colorectal, and non-small cell lung cancer (NSCLC) [7–11]. Hence, there is considerable interest in the scientific community with respect to approaches that can appropriately profile the immune contexture in cancer tissue samples.
Routine H&E staining in diagnostic laboratories allows for histopathological assessment of the general degree of immune infiltration, but possesses significant limitations. Significantly, subtyping of functionally distinct immune cell populations is not possible with H&E staining. Even though the presence of a specific immune population is important, more nuanced characteristics, including the quantity, functional state, spatial distribution, and interplay of immune subpopulations in the TME, collectively influence tumour progression [12]. Therefore, mapping multiple layers of biological characteristics is required to acquire information of maximum clinical value. To achieve this, many state-of-the-art multiplexed imaging technologies have been developed in recent years [13]. These platforms offer the ability to quantitatively assess multiple biomarkers to elucidate the biological characteristics, density, and spatial distribution of different classes of immune cells with objective quantitative data. The ability to perform multiparametric assessment of immune cells allows one to explore and extract a plethora of novel spatial features of translational and functional significance.
Advantages of multiplex immunostaining
Traditionally, H&E and conventional immunohistochemistry (IHC) have been considered the gold standard for evaluating histopathological biomarkers of clinical relevance. However, given that only a very limited number of markers can be assessed at the same time, such approaches can be extremely limiting. In the era of ever-increasing findings in immune-oncology research, it is becoming more and more crucial to obtain information on multiple biomarkers to make more accurate clinical decisions [14]. It has already been shown in a meta-analysis that multiplex immunostaining-based assays outperform other commonly used assay modalities in predicting response to anti-PD-l/PD-Ll therapy, including PD-L1 IHC, tumour mutational burden, and gene-expression profiling [15].
Multiplex IHC and immunofluorescence (mIHC/IF) allow simultaneous analysis of multiple markers on the same tissue section. Depending on different mIHC/IF technologies, the number of assessable markers generally varies between relatively discrete panels (two to eight markers) (Figure 1) to high-plex panels (up to 100) [13]. Simultaneous assessment of multiple markers provides mIHC/IF with key advantages over H&E or conventional IHC. First, with conventional IHC, it is often necessary to stain the same number of serial tissue sections as the number of markers that need to be assessed, risking tissue depletion, particularly on core needle biopsies that are the mainstay of primary diagnosis in many settings (Figure 2). Indeed, with these traditional approaches it can be very challenging to investigate an extensive panel of immune markers after exhausting most of the usable sections for routine diagnostic markers. Second, it is only possible to visually cross-compare up to two or three consecutive sections if one wants to study the colocalisation or spatial relationship between different markers. For example, an average lymphocyte is 10 μm in diameter, and with sections of standardised thickness (3–5 μm), it is possible to have a maximum of three consecutive sections of the same cell [13]. Alternatively, there is an option to use virtual multiplexing, where multiple single-stained serial images are digitally stacked on top of one another to provide a multiplex effect [16]. However, these images are not always reliable for cell phenotyping, especially where there are co-expressed markers, due to the similar limitation in that specific cells are not consistently present across multiple tissue sections. Also, these workflows are impractical for most high-throughput clinical study/trial requirements [17].
Figure 1.
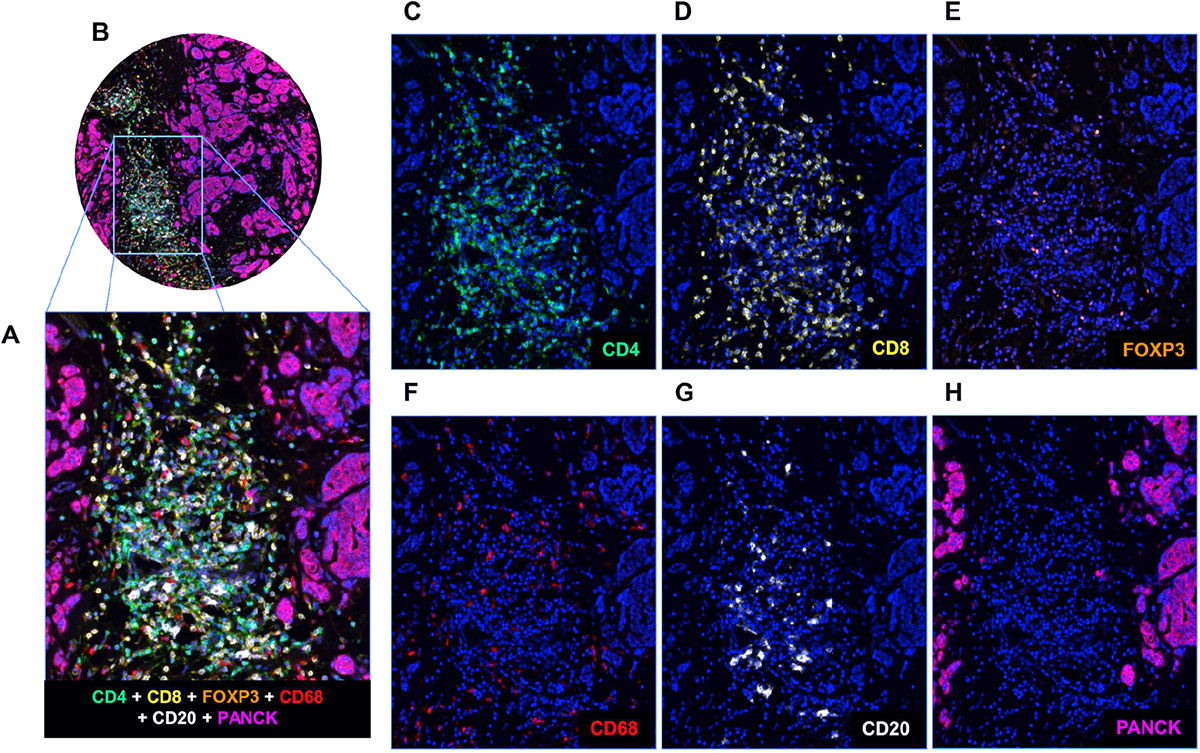
mIF staining of a panel of five immune markers plus one epithelial marker. (A and B) A breast cancer TMA core showing composite staining of each of the six markers in the mIF panel (CD4, CD8, F0XP3, CD68, CD68, and PanCK) together with DAPI. (C–H) Individual images of CD4 (green), CD8 (yellow), F0XP3 (orange), CD68 (red), CD20 (white), and PanCK (purple) with DAPI counterstain.
Figure 2.
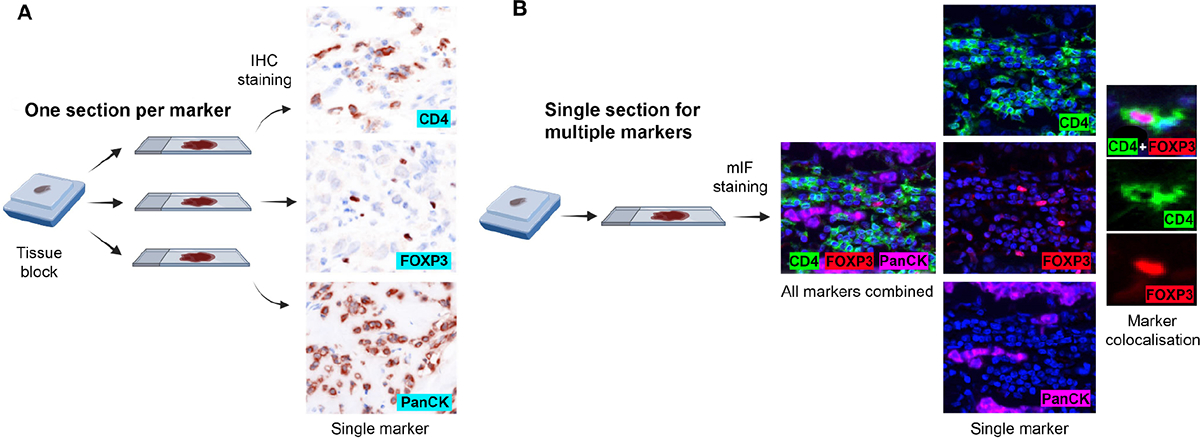
Comparison of experimental design between conventional chromogenic IHC and mIF. (A) In conventional chromogenic IHC, each marker requires a separate slide. Although multiplexing with chromogenic IHC is possible, it is severely restricted in terms of the number of markers that can be simultaneously stained. (B) With mIF, one section is enough to examine multiple markers. The figure was partly created using BioRender.com.
As far as biological information is concerned, multiplex imaging brings a wealth of additional information to the table, which is impossible to extract from H&E or conventional IHC (Figure 3). The majority of methodologies described for TIL scoring in histopathological samples use H&E staining [18,19]. However, H&E staining only reveals the degree of total lymphocyte infiltration. TIL populations may contain both antitumour and tumour-promoting immune cells; therefore, without further subtyping, it is not possible to capture an accurate representation of the immune contexture. Multiplex imaging techniques offer a huge advantage here, as it is possible to simultaneously phenotype multiple immune populations. Admittedly, it is possible to quantify the infiltration of specific populations with single-plex IHC staining of serial sections, but in most cases it is not enough to characterise their functional status in samples. For example, T cell populations may be labelled with CD3. However, T cells can co-exist in dozens of different states having significantly different biological functions in the TME, states that can only be characterised by further profiling the lineage markers in the cells. For instance, CD3+CD8+ co-expressing T cells represent cancer-killing cytotoxic T cells and CD3+CD4+FOXP3+ co-expressors represent tumour-promoting T regulatory cells (Tregs).
Figure 3.
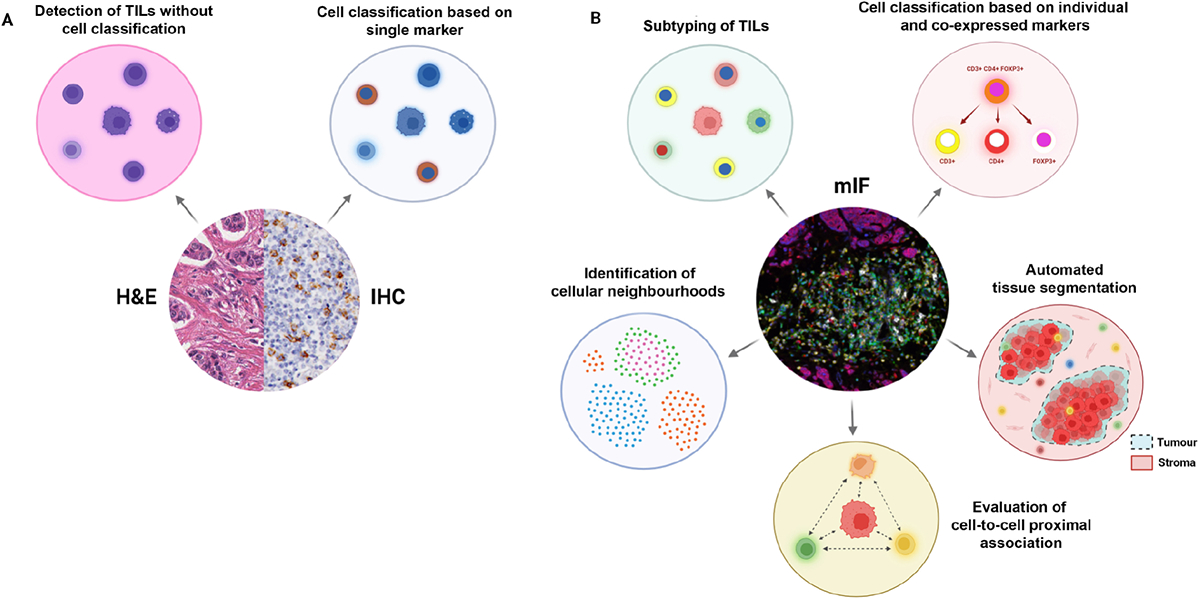
TIL profiling with H&E versus conventional chromogenic IHC versus mIF. (A) H&E staining enables the measurement of total TILs in tissue, whereas, with conventional IHC, a specific immune population can be profiled based on a single protein marker. (B) mIF staining allows the total TIL population to be subtyped based on multiple markers. It is also possible to further characterise cells based on marker colocalisation. With an epithelial or tumour differentiation marker, it is possible to automate tumour-stroma segmentation with image analysis software. Furthermore, multiplex images can be used to map spatial distributions of different cell phenotypes and examine their proximal associations, and further identify distinct cellular neighbourhoods. Created with BioRender.com.
Furthermore, multiplex staining provides a much deeper insight into the spatial characteristics of the targeted cell populations and allows any proximal association between cells expressing specific markers to be deciphered. Assessment of immune cells in a multiplex setting enhances spatial mapping of immune cells in relation to tumour cells as malignant cells can be labelled concurrently [20]. Combined with digital image analysis (DIA), this process allows faster and higher-throughput quantification of stromal, intratumoural, and peritumoural TILs compared with manual evaluation.
DIA and artificial intelligence
In a multiplex-based immune profiling workflow, the first step after staining involves digitising the stained section into a high-resolution image using a whole-slide imaging scanner. Multiplex images carry a massive amount of complex biological information, and to extract and manage this wealth of data, histopathological DIA software is used.
Prior to analysis, raw scan images usually undergo a number of preprocessing steps to eliminate autofluorescence and unmix overlapping fluorophore signals. The image analysis workflow for immune profiling in tumour tissue primarily involves three steps (Figure 4). In the first, tissue segmentation is performed, and involves the automated identification and partitioning of separate tissue compartments, such as tumour and stroma [21]. Tissue segmentation can also be carried out by manually annotating regions of interest (ROIs), especially in studies with fewer samples. However, in multiplex studies, an epithelial marker, like cytokeratin, is generally used to assist the software in segmenting tumour and stromal content in the tissue [22]. This streamlines and increases the efficiency of quantifying intratumoural and stromal TILs in a large number of tissue samples. The second step is cell segmentation, where each cell in the tissue is detected and segmented by the software, typically based on a nuclear counterstain such as DAPI. However, studies have shown that using signals from multiple markers, including membrane markers, significantly improves cell segmentation compared with single markers or counterstaining [23,24]. Following cell segmentation, each cell is phenotypically classified according to single or multiple markers. Cell or object classifying tools mainly fall into two categories. Threshold-based classifiers determine if a cell is positive or negative for a marker depending on the signal intensity. Artificial intelligence (Al)-based classifiers are algorithms that are trained by a user on a variety of staining and morphological features to detect specific objects/cells of interest. Post-training, they automatically perform object segmentation on the samples. Al-based classifiers allow the incorporation of the domain-specific knowledge of experienced pathologists in developing the image analysis algorithm. Generally, pathologists or specialists train these kinds of classifier by manually annotating objects, tissue, or different morphological and biological structures. Pathologists feed the algorithm such data until the classifier learns to distinguish objects with acceptable proficiency. This offers a much more rapid, unbiased, and potentially accurate assessment of features of interest.
Figure 4.
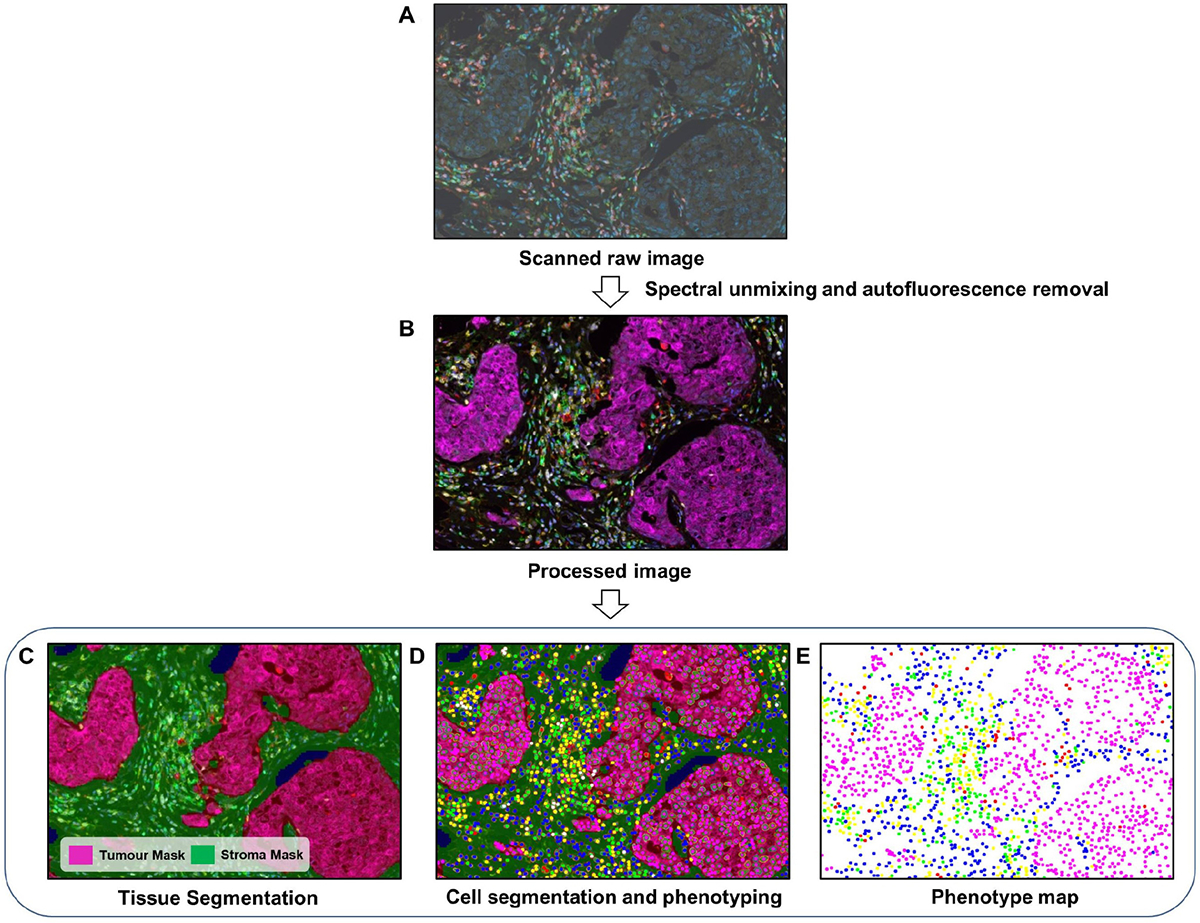
DIA workflow for mIF. (A) First, raw images are generated by scanning stained tissue slides. (B) Spectral unmixing and autofluorescence removal are performed with the software to extract the true signals from each marker. (C) A tissue segmentation algorithm is run to segment tumour and stromal areas. (D) Cell segmentation followed by cell phenotyping is performed to classify all cells based on the marker panel used. (E) Finally, spatial mapping of all cell classes is carried out for further proximity analysis. The images were generated using inForm® image analysis software, Akoya Bioscience.
In immuno-oncology research, AI classifiers are now widely employed for tissue segmentation and lymphocyte detection within the context of H&E-based images [25–27]. In multiple key studies, AI-based approaches have shown a better or similar level of accuracy compared with expert pathologists [28,29]. AI algorithms are increasingly being integrated into multiplex image analysis workflows to improve structure and object identification and phenotypic segregation [30,31]. Due to tissue heterogeneity, phenotyping of simple objects becomes troublesome if the classifier only works with a few basic parameters, such as signal intensity or cell diameter. AI-based semi-automated or automated image analysis approaches are very useful in those scenarios. As the Al-based classifiers can be trained over a wide variety of tissue samples and utilise large datasets, the classifiers are able to gather a lot more information to identify specific morphological features. Zarubin et al [31] trained an AI algorithm on 219 manually annotated regions from different tissues (kidney, ureter, lung, ureter, lymph node, tonsil) and showed near human accuracy in segmenting various cell types (tumour, immune) in multiplexed images. For chromogenic mIHC, it is challenging to digitally evaluate more than four biomarkers in the same section due to a lack of readily available analysis tools. To solve this, Fassler and colleagues [32] developed two complementary deep learning-based tools (Color AE and U-Net) utilising pathologist annotations and constructed an ensemble method that reliably classified six distinctly chromogenically labelled immune populations.
After cell segmentation and phenotyping, the final analysis involves profiling the spatial architecture of the markers. With cell segmentation, DIA software can create a topographical map of every cell in the tissue using the individual cell coordinates [33]. Based on those data, any spatial feature of each cell phenotype can be analysed, and potential clinically significant associations can be assessed further. Currently, there are multiple high-end commercial and open-source DIA software packages that offer both AI- and non-AI-based analysis tools (listed in [34]).
Pitfalls of multiplex immunostaining
Multiplexed staining assays are unique sample-sparing tools that offer a superior histopathological interpretation of disease heterogeneity compared with conventional staining techniques. Nevertheless, they come with certain methodological pitfalls that should be considered before designing any experiment. In general, multiplex immunostaining workflows are more technically challenging and time-consuming. Compared with classical IHC, it involves a greater number of experimental variables and thus requires a more comprehensive and lengthy optimisation process.
Any IHC technique requires extensive validation of antibody specificity and optimisation of their staining conditions. In order to develop a mIHC/IF panel, the staining conditions for each antibody need to be further optimised in a multiplexed setting [35]. Comparison with standard single-plex IHC/IF represents the gold standard during this optimisation. Staining markers in the correct sequence can be critical, as changes in tissue antigenicity during multiple staining rounds can affect antibody performance. Simultaneous staining of multiple targets on the same tissue sample introduces additional complexities. For example, staining multiple targets with fluorophores having close spectral emission poses the risk of spectral overlap. This may lead to fluorophore signals leaking into the wrong channel and resulting in false-positive detections. Thus, variables such as reagent concentrations, antibody–dye pairing, and the antibody staining sequence in the panel must be thoroughly optimised to prevent spectral overlapping and accurate detection of targets. Parra et al [36] provided a detailed discussion on the procedural requirements and a step-by-step guideline for proper optimisation of a mIF staining panel. Some vendors now provide spectral unmixing tools in their DIA packages that can eliminate overlapping signals during post-processing of raw images [35]. Using these tools, users can build spectral libraries by profiling the exact emission spectrum of individual fluorophores. These libraries are then used to deconvolute overlapping signals. IF-based techniques are also inherently susceptible to issues related to autofluorescence, which can interfere with the detection of targets labelled with fluorophores that emit at wave-lengths near the autofluorescence emission spectrum. These issues can be reasonably resolved by following an optimal protocol for handling pre-analytical conditions, like tissue fixation and sample preparation, then removal of autofluorescence signal in the scanned images through DIA unmixing tools [35]. Additionally, multiple antibodies targeting closely spaced epitopes may create steric hindrances and impede antibody binding [37]. Adequate stripping of antibodies after each staining cycle is necessary to prevent this.
Together, the use of multiple antibodies, high-end equipment, and DIA packages makes multiplex staining considerably costlier than traditional IHC, particularly for high-throughput studies. Consequently, for most immune profiling studies with large sample sizes, tissue microarrays (TMA) are used instead of full-face sections. However, in some cases, tissue cores in a microarray may not adequately represent the heterogeneity of marker expression in the sample [38,39]. Some immune populations are often dispersed irregularly across tumour sections; thus, small-sized tissue cores are often not sufficient to capture the accurate spatial composition of the populations [40]. For similar reasons, TMAs are also unlikely to be ideal for profiling rare immune populations and spatial phenotypes. Several validation studies suggest that increasing the number of cores and carefully selecting cores that are more representative of the marker distribution leads to an improvement in the concordance between TMA cores and full-face sections, particularly for markers that are preferentially distributed [41,42].
Statistical considerations for multiplex immune profiling
mIHC/IF assays generate high-resolution, single-cell spatial data, usually spanning thousands of observations per sample across multiple ROIs. Each observation contains information on cellular phenotype, protein expression, and spatial location. Due to the complex and hierarchical nature of these data, statistical methods must be carefully vetted to minimise bias and maximise knowledge learned. One inherent challenge of immune profiling with mIHC/IF is intratumoural heterogeneity, whereby immune cell densities and/or protein levels can vary dramatically across ROIs within a single tumour sample [43–45]. This variation can be either random or related to anatomic features, such as the invasive tumour margin [46,47]. Without a standardised approach for analysing mIHC/IF data, heterogeneity increases the risk of bias and/or inter-observer discordance [43]. A customary statistical method to overcome heterogeneity is to sample tumours across multiple high-powered fields/ROIs, and to report outcomes as the mean of these ROIs [43]. This approach is simple and effective, and is the basis for clinically validated prognostic instruments, such as the breast cancer H&E stromal TIL score (which relies on whole-slide visual averaging) [19,43] and the colon cancer Immunoscore (which relies on averaging cell counts across four distinct spatial/cellular compartments) [48,49].
The process of averaging estimates across numerous high-powered fields requires significant manual and computational labour; therefore, it is of practical interest to ascertain how many ROIs must be sampled to overcome the effects of intratumoural heterogeneity. Recently, mIF data from a breast cancer preoperative immunotherapy clinical trial were used to characterise the impact of ROI sample size on statistical power [50]. Using bootstrapping simulations to emulate 1,000 trials under various ROI sample sizes, it was shown that undersampling of ROIs resulted in unstable estimates of treatment effect, whereas sampling of 15 or more ROIs per specimen resulted in consistent estimation of treatment effect. In addition to ROI sample size, the spatial location of sampled ROIs may also influence the estimation of mIHC/IF results. For example, acknowledging the natural inclination of immune cells to cluster at the invasive margin, oversampling of ROIs at the invasive margin relative to other areas would have the effect of inflating the cell density estimate of a given tumour. Statistical methods could be used to adjust estimates of the mean for anatomic confounders such as the invasive margin. Linear regression modelling could be employed, with the inclusion of spatial covariates into the model to account for proximity to the invasive margin, thus generating estimates of mean immune cell density that are independent of sampling effects of the invasive margin. In relation to the Immunoscore used in colon cancer, a similar but less statistically formalised method is employed, whereby CD3 and CD8 counts are estimated across each of two tumour compartments (invasive edge and tumour), and then averaged [49].
Finally, mIHC/IF output images bear an uncanny resemblance to topographical maps, with tumour epithelial nests and other anatomic structures mimicking geographical features (such as polygonal boundaries of continents), and immune cell locations representing spatial point locations of features on the map (such as locations of cities). As such, another promising direction is to use abundant spatial statistical techniques and software packages (e.g. Spatstat) borrowed from the fields of geography and ecology [51]. Using these programs, one can convert mIHC/IF output data into vector-format datasets that overlay immune cell location (spatial points) with anatomic feature location (spatial polygons). Software can then be used to estimate cell densities at the local level (across partitioned regions of an ROI), and to formally interrogate the relationship between cell location/density, the surrounding microanatomy, and neighbouring cells. It is of paramount importance to tackle such analyses using a multidisciplinary approach, whereby immunologists, pathologists, and statisticians work together to ensure proper hypothesis generation, histological annotation, and statistical modelling/testing. For detailed information on analysing and reporting spatial immune profiling data, please see the companion manuscript from The International Immuno-Oncology Biomarker Working Group [52].
Clinical and translational implications of tissue-based immune profiling in cancer
The remarkable success of immunotherapy, particularly the checkpoint inhibitor therapeutic strategies targeting CTLA-4 and PD-1/PD-L1, has revolutionised treatment for several types of malignancy, as well as our broader understanding of the clinical significance of immune contexture in cancer. Over the last couple of decades, numerous studies have confirmed the ability of tumour immune composition to significantly influence clinical outcomes in various cancer types [53]. This highlights the potential importance of immune profiling as a metric to be reported and used in clinical settings for tumour characterisation and for guiding clinical decisions.
A seminal study on colorectal cancer showed that the composition of TIL infiltrates is a better predictor of survival than routine TNM (tumour, node, metastasis) classification [10]. This finding first challenged the concept of only looking at neoplastic cell characteristics for assessing the risk of progression. Galon et al [54] developed the Immunoscore, which assigns a score of 0–4 based on the density of CD3+ and CD8+ populations in both the tumour centre and invasive margins [55]. Immunoscore correlates with disease-free and overall survival in colorectal cancer and other tumour types, including melanoma, breast, kidney, and lung cancers [56,57].
The efficacy of different forms of immunotherapy is critically affected by the immune composition of tumours before therapy [58] and the absence of T cells and tumour-specific T cell responses are key contributors to poor clinical responses [59]. These findings led to classifying immune composition in the TME into distinct phenotypes that correlate with patient responses to immunotherapy and prognosis. Several years of IHC-based spatiotemporal studies across different tumour types have established distinct immune phenotypes: hot tumours (immune-inflamed) and cold tumours (immune-excluded and immune-desert) [60]. The immune-inflamed phenotype is characterised by substantial infiltration of TILs in the tumour centre. It is associated with the presence of CD4+ and CD8+ T cells in the tumour parenchyma that reflects a preceding antitumour response mediated by an immune-permissive microenvironment. The immune-excluded phenotype is characterised by pronounced infiltration of immune cells localised at the tumour interface with surrounding tissue, instead of within the tumour centre. This class of tumour is hypothesised to be poorly immunogenic. The final phenotype, immune-desert tumours, is characterised by the absence of pre-existing T cells in either tumour parenchyma or stroma. These features indicate the absence of pre-existing antitumour immunity. Tumours with immune-desert phenotypes often exhibit poor response to immunotherapy [61].
Currently, with the ongoing discovery of these complex phenotypes of clinical significance, spatiotemporal multi-marker assessment of immune contexture is becoming a necessity in immune-oncology research. Hence, tissue-based multiplexing has evolved into an optimal investigating tool for identifying predictive and prognostic immune biomarkers [55]. A comprehensive list of studies focusing on the clinical relevance of immune profiling through mIHC/IF techniques is presented in Table 1.
Table 1.
Application of multiplex staining in clinical immune profiling studies.
| Tumour type | Marker panel | Summary | Reference |
|---|---|---|---|
| Prognostic studies | |||
| Breast cancer (HER2+ and TNBC) | CD4, CD8, CD20, FOXP3, CD68, PanCK | Higher B cell infiltration was associated with better overall survival | [62] |
| Breast cancer | CD8, CD103, CD69, PanCK, DAPI | Higher tissue-resident memory T cell infiltration was associated with better recurrence-free survival | [63] |
| Breast cancer (TNBC) | CD4, CD8, FOXP3, CD20, CD33, PD-1 | Higher PD-1+ CD8+ T cells, PD-1+CD4+ T cells were associated with better prognosis | [64] |
| Breast cancer (TNBC) | CD4, CD8, FOXP3, PD-1, PD-L1 | CD4/PD-L1, CD8/PD-1, and CD8/PD-L1 double-positive TILs were significantly associated with recurrence | [65] |
| Pancreatic ductal adenocarcinomas | CD8, KRT7 | High density of CD8+ T cells in tumour centre was associated with improved survival | [66] |
| Endometrial cancer | CD8, CD4, FOXP3 | High Treg counts and Treg/CD8+ ratios were significantly associated with worse distant metastasis-free survival | [67] |
| Lung cancer (NSCLC) | Panel 1: CD4, CD38, CD68, FOXP3, CD20 Panel 2: CD8, PD-L1, CD163, CD68, CD133 | Patients were classified into three subtypes based on their unique immune composition (immune activated, immune defected and immune exempted), where immune-activated patients showed the longest disease-free survival | [68] |
| Lung cancer (NSCLC) | CD3, CD8, CD20 | Higher CD3+ and CD8+ infiltration was associated with better outcome | [69] |
| Gastric cancer | CD68, CD163, CD206, IRF8, PD-L1 | Higher CD163+ (CD206−) tumour-associated macrophage density with high CD68 expression was associated with better patient survival | [70] |
| Gastric cancer | CD4, CD8, FOXP3, PD-1, PD-L1, TIM3 | Higher levels of CD8, PD-1, and PD-L1 following NAC were associated with better overall survival | [71] |
| Predictive studies | |||
| Melanoma | CD8, FOXP3, SOX10 PD-1, PD-L1 | CD8+ cells within 20 μm of a melanoma cell were predictive of PD-1-based immunotherapy. | [72] |
| Melanoma | CD4, CD8, CD20, CD3, GZMB, Ki67 | Pretreatment lymphocytic infiltration (CD3, CD8) was indicative of anti-PD-1 response | [73] |
| Lung cancer (NSCLC) | CD3, CD8, CD4, PD-1, CD57, FOXP3, CD25, Granzyme B | CD8+ T cells lacking PD-1 inhibitory receptor positively impacted nivolumab-treated patient survival | [74] |
| Lung cancer (NSCLC) | Panel 1 - AE1/AE3, PD-L1, CD3, CD4, CD8, and CD68 Panel 2 - AE1/AE3, PD-1, granzyme B, FOXP3, CD45RO, CD57. |
Higher intratumoural T helper cell and macrophage levels were associated with chemotherapy outcome | [75] |
| Breast cancer | CD3, CD20, CD8 | CD3, CD8, and CD20 infiltration were predictive of NAC response | [76] |
| Breast cancer (HER2+) | CD8, CD4, CD20, CD68, FoxP3, CK | CD4+, CD8+, CD20+ s-TILs, CD20+ s-TILs were independently associated with higher pCR in patients treated with neoadjuvant anti-HER2 therapy | [77] |
| Breast cancer | PD-L1, PanCK | PD-L1 was predictive of NAC response | [78] |
| Gastric cancer | CD8, PD-1, PD-L1, TIM-3, LAG-3, CD4, CD20, FoxP3, CTLA-4, CD68, CD163, CD66b, HLA-DR, STING | Multi-marker protein signature predicts anti-PD-1/PD-L1 therapy response | [79] |
| Lung cancer (NSCLC) | PD-L1, CD68, CD8 | High level of PD-1 in macrophages was linked with higher CD8+ cell infiltration and better survival outcome in PD-1 pathway blockade therapy | [80] |
pCR, pathological complete response; s-TIL, stromal tumour-infiltrating lymphocyte; TNBC, triple-negative breast cancer.
Cancer prognosis
The major fraction of immune infiltrates in cancer is comprised of T cells. T cell infiltration has been associated with survival outcomes in multiple different tumour types, including melanoma, breast, lung, colon, liver, and bladder [11,54,81–83]. CD8+ cytotoxic T cells are widely regarded as the central players in antitumour immunity, and a higher degree of CD8 infiltration is mostly associated with favourable clinical outcomes [84–87]. CD4+FOXP3+ Treg cells are critical subsets of helper T cells that suppress the antitumour immune response [88]. Recently, relatively small multiplex panels (three to four markers) are increasingly used to study T cell composition and their prognostic relevance in various cancers. Yamagami et al [67] assessed the composition of CD4+, CD8+, and Treg cell populations in endometrial cancer with mIF and found that patients with high Treg counts and Treg/CD8 ratios experience significantly worse survival. Several studies in different cancer types have shown that T cell aggregation in intratumoural regions was linked to better prognosis [10,66,89]. Spatial analysis of eight distinct immune subpopulations in pancreatic ductal adenocarcinoma with mIF found intratumoural T cell infiltration to be independently correlated with favourable patient survival [90]. In a similar study of NSCLC, cytotoxic T cell (CD3+, CD8+) infiltration was mapped using a multiplex panel and a higher level of intratumoural CD8+ cell infiltration independently correlated with better survival [69]. Another recent study of NSCLC showed increased CD8+ T cell density in the invasive margin to be positively associated with recurrence-free survival [91].
mIHC/IF assays are also widely used by researchers to study proximal associations between cancer and immune markers, as well as among different immune cell subtypes [92]. The proximity of malignant cells to specific immune subsets can suggest an effective antitumour or tumour-promoting environment that may be important for determining prognosis. Nearchou et al [93] simultaneously assessed the distribution of T cell infiltrates in intratumour locations and at invasive margins within the context of tumour budding in colorectal cancer. The study not only confirmed T cell infiltrates and tumour budding to be independent prognostic factors, but also found that the spatial relationship of lymphocyte infiltrates and tumour budding offers additional prognostic value. Combining all the features together into a prognostic index generated improved prognostic stratification for patients compared with any of the features individually. Another study explored the spatial relationship of 17 distinct leukocyte lineages with a 29-plex mIF platform. They found that the proximity of Treg and myeloid cells to tumour cells had a strong correlation with earlier cancer recurrence [94]. Similar results by multiplexing were also found in lung cancer and head and neck squamous cell carcinoma, where the proximity of Treg cells to carcinoma cells was linked to poor prognosis [91,95].
Together with T cell subsets, recent research has also highlighted the prognostic utility of other immune populations, such as B cells, macrophages, and dendritic cells. A recent study used multiplex staining for immune profiling of colon cancer and developed a highly prognostic signature comparable with the Immunoscore by combining the prognostic features of CD8+ T cells and CD68+/CD163+ macrophages. The signature further demonstrated significant prognostic efficacy in four other cancer types – oesophageal adenocarcinoma, bladder cancer, lung adenocarcinoma, and melanoma [96]. Multiplex immunostaining techniques also allow identifying complex phenotypes, such as tertiary lymphoid structures [97,98]. Tertiary lymphoid structures are ectopic lymphoid structures of cellular aggregates, generally comprised of a germinal core with proliferating B cells and follicular dendritic cells, surrounded by a CD3+ T cell zone [99]. The prevalence of tertiary lymphoid structures in tumours is generally suggestive of strong tumour immunity and is mostly correlated with a better prognosis [100].
Predictive tool for therapy response
PD-L1 is the most well-studied and well-accepted predictive biomarker in the clinical setting for immune therapies. IHC-based PD-L1 assays have already been approved as companion diagnostic testing for selecting patients to receive checkpoint blockade therapies in several different cancer types [101,102]. However, PD-L1 alone has not been sufficient for optimal patient stratification [103] and several other components of the TME appear to affect the likelihood of therapy response. Strong evidence from multiple studies suggests that TIL infiltration with a T cell-inflamed phenotype is associated with an anti-PD-Ll therapy response [104]. In mismatch repair-proficient colorectal cancer CD8+PD-1+ T cell infiltration was found to be the only biomarker predicting response to neoadjuvant immunotherapy [105]. De Vries et al [106] demonstrated evidence of γδ T cells contributing to an immune checkpoint blockade response in HLA class I-negative mismatch repair-deficient colon cancer patients. Multiplex immunostaining assays can serve as an ideal diagnostic tool to assess these kinds of complex immune subset in clinical settings.
When CD8+ T cell distribution was assessed in melanoma TME, high CD8+ cell density at the invasive margin was found to be associated with anti-PD-Ll therapy response [107]. In another recent mIF-based study with melanoma samples, spatial interactions between T cell populations and malignant cells were investigated, and higher CD8+ cell density within close proximity to melanoma cells was found to be associated with a better response to anti-PD-1 therapy [72]. Interestingly, in Merkel cell carcinoma, the proximal association between PD-1 and PD-L1 was found to be predictive of an anti-PD-1 response [108].
In addition to predicting therapy response in patients using tissues sampled prior to therapy, immune profiling of early on-treatment patient biopsies has also been found to be very effective in predicting long-term therapy response. A multiplexed immune profiling study in a cohort of melanoma patients treated with combined CTLA-4 and PD-1 blockade therapy showed increased accumulation of CD8+ cells in the tumour centre that was significantly correlated with the therapeutic response [109]. All of these studies support the clinical value of TIL profiling, and PD-L1 as a companion diagnostic test, with better predictive accuracy. As IHC-based techniques are already routinely used for clinical assays, tissue-based multiplexed platforms will be ideal tools for combining TIL scores with PD-L1 data. Similar strategies should also be adopted for designing immunotherapy clinical trials. This is crucial, as patient selection for clinical trials is still based on conventional toxicity and efficacy patterns observed with chemotherapy and targeted agents [110]. Very few clinical trials are adopting standard immune biomarkers for patient selection, which is a major impediment to proper characterisation of the biological response.
Apart from modulating the tumour response for immunotherapy, the immune composition of the TME has also been found to heavily influence the clinical outcome of other kinds of cancer treatment [111–113]. It was originally thought that chemotherapy only had an immunosuppressive effect. However, recent studies have shown that certain types of chemotherapy can facilitate an antitumoural immune response by inducing tumour-associated neoantigen expression [114]. Parra et al [75] investigated the change in immune composition after neoadjuvant chemotherapy (NAC) in tumour tissues from 112 NSCLC patients using two different six-plex IF panels to quantify 12 tumour-associated immune phenotypes. Higher levels of PD-L1 with T cell and tumour-associated macrophage cell infiltration were observed in samples from patient treated with NAC compared with those who had not received NAC, indicating that NAC induces a discrete immune response. Moreover, patients receiving NAC who had higher levels of helper T cells and tumour-associated macrophages showed better survival. In a similar study of HER2-positive breast cancer patients, an assessment of CD8+ cells together with PD-L1 was found to be valuable in predicting the response to anti-HER2 neoadjuvant therapy [115].
Implementing mIHC/IF technologies in daily clinical practice
Although multiplex imaging technologies have critical utility in clinical research, very few of these technologies have been adequately validated for clinical application. To date, no multiplex staining assays have been approved by the Food and Drug Administration for use in clinics as in vitro diagnostics [116]. With the growing need to better understand the TME for clinical decision-making, incorporating these technologies into clinical pathology is becoming increasingly necessary. There are several challenges to overcome for mIHC/IF techniques to become widely adopted in clinics.
To begin with, there are extensive infrastructure requirements to facilitate mIHC/IF and digital pathology in a clinical institution. In general, mIHC/IF images are composite files of separate images generated for each marker, creating a large file size. With a moderate-sized panel (five to seven markers), the whole-slide image size will probably fall between 0.5 and 4 gigabytes [117]. Consequently, a hospital will generate hundreds of terabytes of imaging data each year, and it is essential to have access to infrastructure that can store, process, and facilitate the sharing of this amount of data. Additionally, Al-based quantitative and spatially resolved image analysis of these images requires expensive high-end work-stations with powerful graphics processing units [118]. It also requires additional human resources with considerable statistical and bioinformatic expertise for downstream statistical analysis and robust data interpretation.
In addition to infrastructure requirements, ensuring consistency of these new mIHC/IF techniques by standardisation is also a critical impediment to their integration into clinics. Most mIHC/IF techniques are specific in their methodology, having unique staining and imaging platforms, different analysis packages, and image formats [13]. These factors inherently introduce variability, which raises the question whether data from independent laboratories can be compared.
In order to meet these challenges, The Cancer Immune Monitoring and Analysis Center (CIMAC) conducted a multistep harmonisation study to compare assay performance among independent laboratories and to determine whether it is feasible to generate comparable data regardless of the platform and site [119]. They have compared the staining of a five-marker immune panel (PD-L1, PD-1, CD3, CD8, and PanCK) on head and neck tumour samples among three different institutions using two different multiplexed imaging platforms – a mIF-based tyramide signal amplification system and chromogenic mIHC. Both platforms were evaluated for sensitivity, specificity, and reproducibility, followed by the harmonisation of three aspects – staining, image acquisition, and image analysis procedures across the two platforms. Post-harmonisation of the data, for most markers, the correlation coefficient exceeded 0.85; combining all markers, it was over 0.7, with a median coefficient of variation below 0.1, indicative of excellent precision between measurements. These findings demonstrated that despite differences in protocols, platforms, reagents, and image analysis applications, independent multiplex immunostaining platforms could produce harmonised data without imposing rigid standardisation.
One of the significant factors that affected the harmonisation effort was pre-analytical variables, such as sample procurement and processing. It is generally recognised that pre-analytical variables pose challenges to IHC standardisation [120,121]. Thus, CIMAC has formulated an ‘umbrella’ protocol for standardising pre-analytical conditions, which can be adopted to minimise analytical variability across laboratories [122]. They are also putting considerable effort into analytically validating assay platforms based on their ability to perform the most robust and unbiased analyses, allowing them to prioritise specific assays for clinical trials.
A six-institution intrasite collaboration in 2019, termed the MITRE study, developed a standardised end-to-end workflow for a six-plex mIF assay (PD-L1, PD-1, CD8, CD68, FOXP3, and CK) suitable for multisite trials [123]. Assay optimisation led to sensitive and reproducible results between and within all sites. Further similar efforts must be made to address standardisation issues with mIHC/IF. The Society for Immunotherapy of Cancer (SITC) has taken a crucial step in that direction by forming a 21-member task force of pathologists and research leads from academia and pharmaceutical companies to develop best practice guidelines for optimising and validating multiplex immunostaining assays [37]. Overall, for validating these multiplex imaging assays, a harmonised, systematic approach should be designed and adopted for clinical use. Throughout the process, there needs to be more collaboration between the clinical and scientific communities. To meet high infrastructure requirements, solutions like cloud-based analysis pipelines can be adopted as an alternative to developing storage and analysis infrastructure in hospitals. This will enable easy access to the heavy computational requirements and facilitate collaboration among researchers and pathologists by providing shared access to data and algorithms. In addition, there should be free public databases for multiplexed images, with widely accepted minimum information standards [124]. This will encourage meta-analyses and help to develop more reliable algorithms. Ultimately, the future adoption of multiplex imaging technologies in clinics will require more harmonisation, standardisation, and validation studies addressing all the factors contributing to variability. This will not only ensure improved accuracy and reproducibility of immune profiling, but also facilitate a faster and more streamlined process of test development.
Acknowledgements
GB was funded by the Gilead Breast Cancer Research Grant 2023. JSR-F was funded in part by the Breast Cancer Research Foundation, a Susan G Komen Leadership Grant, and NIH/NCI P50 CA247749 01 grant. SV was supported by Interne Fondsen KU Leuven/Internal Funds KU Leuven. BA received the Swedish Society for Medical Research (Svenska Sällskapet för Medicinsk Forskning) Postdoctoral Grant and the Swedish Breast Cancer Association (Bröstcancerförbundet) Research Grant 2021. GC was supported by the Peer Reviewed Cancer Research Program (Award W81XWH-21-1-0160) from the US Department of Defense and the Mayo Clinic Breast Cancer SPORE Grant P50 CA116201 from the NIH. CF-M was funded by the Horizon 2020 European Union research and innovation programme under the Marie Sklodowska Curie grant agreement no. 860627 (CLARIFY Project). SBF: NHMRC GNT1193630. SG was partially supported by NIH Grant CA224319, DK124165, CA263705, and CA196521. AG was supported by Breast Cancer Now (and their legacy charity Breakthrough Breast Cancer) and Cancer Research UK (CRUK/07/012, KCL-BCN-Q3). TRK: Japan Society for the Promotion of Science (JSPS) KAKENHI (21K06909). UK was funded by the Horizon 2020 European Union Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement no. 860627 (CLARIFY Project). JKL was in part supported by National Institutes of Health (NIH) (R37CA225655). AM was supported by the National Cancer Institute under award numbers R01CA268287A1, U01CA269181, R01CA26820701A1, R01CA249992-01A1, R01CA202752-01A1, R01CA208236-01A1, R01CA216579-01A1, R01CA220581-01A1, R01CA257612-01A1, 1U01CA239055-01, 1U01CA248226-01, 1U54CA254566-01, National Heart, Lung and Blood Institute 1R01HL15127701A1, R01HL15807101A1, National Institute of Biomedical Imaging and Bioengineering 1R43EB028736-01, VA Merit Review Award IBX004121A from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program (W81XWH-19-1-0668), the Prostate Cancer Research Program (W81XWH-20-1-0851), the Lung Cancer Research Program (W81XWH-18-1-0440, W81XWH-20-1-0595), the Peer Reviewed Cancer Research Program (W81XWH-18-1-0404, W81XWH-21-1-0345, W81XWH-21-1-0160), the Kidney Precision Medicine Project (KPMP) Glue Grant and sponsored research agreements from Bristol Myers Squibb, Boehringer-Ingelheim, Eli Lilly and AstraZeneca. SKM thanks Kay Pogue-Geile, Director of Molecular Profiling at NSABP for her constant support and encouragement, Roberto Salgado, for getting me initiated into the wonderful subject of immuno-oncology and its possibilities. FuAM received funding from EPSRC EP/W02909X/1 and PathLAKE consortium. FP-L received research grants from Fondation ARC, La Ligue contre le Cancer. RDP received the Melbourne Research Scholarship and a scholarship from the Peter MacCallum Cancer Centre. JS: UH3CA225021, U24CA215109. ST was supported by Interne Fondsen KU Leuven/Internal Funds KU Leuven. JT was supported by institutional grants of the KWF Kankerbestrijding and the Dutch Ministry of Health, Welfare and Sport. EAT received the Breast Cancer Research Foundation Grant 22–161. GEV was supported by Breast Cancer Now (and their legacy charity Breakthrough Breast Cancer) and Cancer Research UK (CRUK/07/012, KCL-BCN-Q3). TW was supported by the French Government under management of Agence Nationale de la Recherche as part of the ‘Investissements d’avenir’ program, reference ANR-19-P3IA-0001 (PRAIRIE 3IA Institute) and by Q-Life (ANR-17-CONV-0005). HYW was funded in part by the NIH/NCI P50 CA247749 01 grant. YY received funding from Cancer Research UK Career Establishment Award (CRUK C45982/A21808). PS received funding support from the National Health and Medical Research Council, Australia. SL was supported by the National Breast Cancer Foundation of Australia (NBCF) (APP ID: EC-17-001), the Breast Cancer Research Foundation, New York (BCRF (APP ID: BCRF-21-102), and a National Health and Medical Council of Australia (NHMRC) Investigator Grant (APP ID: 1162318). RS was supported by the Breast Cancer Research Foundation (BCRF, grant no. 17–194). WMG was supported by the North–South Research Programme administered by the Higher Education Authority on behalf of the Department of Further and Higher Education, Research, Innovation and Science and the Shared Island Fund (AICRIstart: A Foundation Stone for the All-Island Cancer Research Institute (AICRI): Building Critical Mass in Precision Cancer Medicine https://www.aicri.org/aicristart), Irish Cancer Society (Collaborative Cancer Research Centre BREAST-PREDICT; CCRC13GAL; https://www.breastpredict.com), the Science Foundation Ireland Investigator Programme (OPTi-PREDICT; 15/IA/3104), the Science Foundation Ireland Strategic Partnership Programme (Precision Oncology Ireland; 18/SPP/3522; https://www.precisiononcology.ie). Open access funding provided by IReL.
Conflict of interest statement:
DBP: Speaker’s Bureau: Genentech, Novartis, Clinical Care Options, Oncocyte; research support: WindMIL, Brooklyn Immunotherapeutics, Merck, Bristol Myers Squibb, IMV; consulting: Merck, Biotheranostics, Puma, Gilead, Lilly, Sanofi, NGM Bio, Sanford Burnham Prebys, AstraZeneca. GB: Speaker’s fee from MSD and Novartis, advisory boards for Roche and MSD, consultant for MSD, Novartis, and Roche, travel and conference support from Roche, MSD, and Gilead. JSR-F is an Associate Editor of The Journal of Pathology and has received personal/consultancy fees from Goldman Sachs, Bain Capital, REPARE Therapeutics, Saga Diagnostics, and PaigeAl, membership of the scientific advisory boards of VolitionRx, REPARE Therapeutics, and Paige. Al, membership of the Board of Directors of Grupo Oncoclinicas, and ad hoc membership of the scientific advisory boards of AstraZeneca, Merck Daiichi Sankyo, Roche Tissue Diagnostics, and Personalis, outside the scope of this study. AIH: Research fund from Visiopharm A/S. ZK: Paid advisory role for Eli Lilly and AstraZeneca Canada. JT: Employee of Visiopharm A/S. KRMB: Scientific Advisory Board for CDI Labs, research funding form Carevive. FC: Chair of the Scientific and Medical Advisory Board of TRIBVN Healthcare, France, and advisory board fees from TRIBVN Healthcare, France in the last 5 years. Shareholder of Aiosyn BV, the Netherlands. LADC: Participation in the Tempus Algorithm Advisors program. AC: Contracted researcher for Oncoinvent AS and Novocure and a consultant for Sotio a.s. and Epics Therapeutics SA. ME: Egyptian missions sector. SBF: Expert advisory panel for AXDEV Group. JMG: Employee and stockholder of Roche/Genentech. SG: Research funding from Regeneron Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Genentech, EMD Serono, Pfizer, and Takeda, unrelated to the current work; named co-inventor on an issued patent for multiplex immunohistochemistry to characterise tumours and treatment responses. The technology is filed through Icahn School of Medicine at Mount Sinai (ISMMS) and is currently unlicensed. NH: Patent on a technology to measure immune infiltration in cancer to predict treatment outcome (W02012038068A2). MGH: Consultant for PaigeAl, VolastraTx, and advisor for PathPresenter. JH: Speaker’s honoraria or advisory board remunerations from Roche, Novartis, AstraZeneca, Eli Lilly, and MSD. Co-founder and shareholder of Stratipath AB. KK Employee and stockholder of Roche. GA: Employee of Merck & Co Inc. AIK: Honorarium from Roche, MSD, and Pfizer, member of the Advisory Board of Pfizer. A-VL Institutional grants from AstraZeneca and personal grants from AstraZeneca (travel and honorarium from advisory board), MSD (honorarium from advisory board), and Daiichi Sankyo (travel). XL: Eli Lilly Company, Advisor, Cancer Expert Now, Advisor, Champions Oncology, Research fund. AM: Equity holder in Picture Health, Elucid Bioimaging and Inspirata Inc, advisory board of Picture Health, Aiforia Inc and SimBioSys, consultant for SimBioSys, sponsored research agreements with AstraZeneca, Boehringer-lngelheim, Eli-Lilly, and Bristol Myers Squibb, technology licensed to Picture Health and Elucid Bioimaging involvement in three different R01 grants with Inspirata Inc DKM: Consulting: Astrazeneca, Lilly USA LLC Hologic. Sponsored Research: Merck Agendia. SM: Scientific Committee Study member. Roche, data and safety monitoring member of clinical trials: Sensorion, Biophytis, Servier, IQVIA, Yuhan, Kedrion. FuAAM: Research studentship funding from GSK DAM: Speaker fees from AstraZeneca, Eli Lilly, and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, M/M Software, Bristol Myers Squibb, and Eli Lilly, educational support from Takeda and Amgen. FP-L: Personal financial interests: AbbVie, Agendia, Amgen, Astellas, AstraZeneca, Bayer, BMS, Daiichi-Sankyo, Eisai, Exact Science, GSK lllumina, Incyte, Janssen, Lilly, MERCK lifa, Merck-MSD, Myriad, Novartis, Pfizer, Pierre-Fabre, Roche, Sanofi, Seagen, Takeda, Veracyte, Servier. Institutional financial interests: AstraZeneca, Bayer, BMS, MSD, Myriad, Roche, Veracyte. Congress invitations: AbbVie, Amgen, AstraZeneca, Bayer, BMS, Gilead, MSD, Novartis, Roche, Lilly, Pfizer. NMR: Co-Founder, Director and CSO of Histofy Ltd, UK AS: Advisory Board/Speaker’s Bureau: Aignostics, AstraZeneca, Bayer, BMS, Eli Lilly, lllumina, Incyte, Janssen, MSD, Novartis, Pfizer, Roche, Seagen, Takeda, and Thermo Fisher; grants from Bayer, Bristol Meyers Squibb, Chugai, and Incyte. TT: Employee of Tempus Labs. JT: Shareholder of EllogonAI BV. TT: Speaker’s fee from Pfizer. JvdL: Member of the advisory boards of Philips, the Netherlands and ContextVision, Sweden, research funding from Philips, the Netherlands, ContextVision, Sweden, and Sectra, Sweden in the last 5 years. Chief scientific officer and shareholder of Aiosyn BV, the Netherlands. TW: Collaboration with TRIBUN Health on automatic grading of biopsies for head and neck cancer, patent on the prediction of homologous recombination deficiency in breast cancer. YW: Employee of CellCarta. HYW: Advisory faculty of AstraZeneca. YY: Speaker/consultant for Roche and Merck PS: Consultant (uncompensated) to Roche-Genentech. SL Research funding to institution from Novartis, Bristol Meyers Squibb, Merck Puma Biotechnology, Eli Lilly, Nektar Therapeutics, AstraZeneca, Roche-Genentech, and Seattle Genetics. Consultant (not compensated) to Seattle Genetics, Novartis, Bristol Meyers Squibb, Merck, AstraZeneca, Eli Lilly, Pfizer, and Roche-Genentech. Consultant (paid to her institution) to Aduro Biotech, Novartis, GlaxoSmithKline, Roche-Genentech, AstraZeneca, Silverback Therapeutics, GI Therapeutics, PUMA Biotechnologies, Pfizer, Gilead Therapeutics, Seattle Genetics, Daiichi-Sankyo, Amunix, Tallac therapeutics, Eli Lilly, and Bristol Meyers Squibb. RS: Non-financial support from Merck and Bristol Myers Squibb, research support from Merck Puma Biotechnology and Roche; personal fees from Roche, Bristol Myers Squibb and Exact Sciences for advisory boards. WMG: Co-founder, shareholder and part-time Chief Scientific Officer of OncoAssure Limited, shareholder in Deciphex and member of the Scientific Advisory Board of Carrick Therapeutics.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39: 1–10. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 2018; 32: 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci 2011; 7:651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ribatti D The concept of immune surveillance against tumors. The first theories. Oncotarget 2017; 8: 7175–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman WH, Zitvogel L, Sautès–Fridman C, et al. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017; 14: 717–734. [DOI] [PubMed] [Google Scholar]
- 7.Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer 2017; 117:451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen B, Li H, Liu C, et al. Prognostic value of the common tumour-infiltrating lymphocyte subtypes for patients with non-small cell lung cancer: a meta-analysis. PLoS One 2020; 15: e0242173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu Q, Chen N, Ge C, et al. Prognostic value of tumor-infiltrating lymphocytes in melanoma: a systematic review and meta-analysis. Oncoimmunology 2019; 8: el593806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964. [DOI] [PubMed] [Google Scholar]
- 11.Schulze AB, Evers G, Görlich D, et al. Tumor infiltrating T cells influence prognosis in stage I–III non-small cell lung cancer. J Thorac Dis 2020; 12: 1824–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wells DK, Chuang Y, Knapp LM, et al. Spatial and functional heterogeneities shape collective behavior of tumor-immune networks. PLoS Comput Biol 2015; 11: e 1004181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan WCC, Nerurkar SN, Cai HY, et al. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun 2020; 40: 135–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ventola CL. Cancer immunotherapy, part 3: challenges and future trends. P T 2017; 42: 514–521. [PMC free article] [PubMed] [Google Scholar]
- 15.Lu S, Stein JE, Rimm DL, et al. Comparison of biomarker modalities for predicting response to PD-l/PD-Ll checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol 2019; 5: 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan RCK, Li JJX, Yeung W, et al. Virtual multiplex immunohistochemistry: application on cell block of effusion and aspiration cytology. Diagn Cytopathol 2020; 48: 417–423. [DOI] [PubMed] [Google Scholar]
- 17.Locke D, Hoyt CC. Companion diagnostic requirements for spatial biology using multiplex immunofluorescence and multispectral imaging. Front Mol Biosci 2023; 10: 1051491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendry S, Salgado R, Gevaert T, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international Immunooncology biomarkers working group: part 1: assessing the host immune response, TILs in invasive breast carcinoma and ductal carcinoma in situ, metastatic tumor deposits and areas for further research. Adv Anat Pathol 2017; 24: 235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salgado R, Denkert C, Demaria S, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an international TILs working group 2014. Ann Oncol 2015; 26: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halse H, Colebatch AJ, Petrone P, et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci Rep 2018; 8: 11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mori H, Bolen J, Schuetter L, et al. Characterizing the tumor immune microenvironment with tyramide-based multiplex immunofluorescence. J Mammary Gland Biol Neoplasia 2020; 25: 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SL, Cabanero M, Hyrcza M, et al. Computer-assisted image analysis of the tumor microenvironment on an oral tongue squamous cell carcinoma tissue microarray. Clin Transl Radiat Oncol 2019; 17: 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schüffler PJ, Schapiro D, Giesen C, et al. Automatic single cell segmentation on highly multiplexed tissue images. Cytometry A 2015; 87: 936–942. [DOI] [PubMed] [Google Scholar]
- 24.Hoyt CC. Multiplex immunofluorescence and multispectral imaging: forming the basis of a clinical test platform for Immuno-oncology. Front Mol Biosci 2021; 8: 674747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abousamra S, Gupta R, Hou L, et al. Deep learning-based mapping of tumor infiltrating lymphocytes in whole slide images of 23 types of cancer. Front Oncol 2022; 11: 806603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Z, Xu S, Shao W, et al. Deep-leaming-based characterization of tumor-infiltrating lymphocytes in breast cancers from histopathology images and multiomics data. JCO Clin Cancer Inform 2020; 4: 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulten W, Bándi P, Hoven J, et al. Epithelium segmentation using deep learning in H&E-stained prostate specimens with immunohistochemistry as reference standard. Sci Rep 2019; 9: 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagpal K, Foote D, Liu Y, et al. Development and validation of a deep learning algorithm for improving Gleason scoring of prostate cancer. NPJ Digit Med 2019; 2: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehteshami Bejnordi B, Veta M, van Diest PJ, et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 2017; 318: 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vance K, Alitinok A, Winfree S, et al. Machine learning analyses of highly-multiplexed immunofluorescence identifies distinct tumor and stromal cell populations in primary pancreatic tumors. Cancer Biomark 2022; 33: 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zarubin D, Belotskiy V, Xiang Z, et al. A clinical AI-driven multiplex immunofluorescence imaging pipeline to characterize tumor microenvironment heterogeneity. J Clin Oncol 2022; 40: 3020.35436146 [Google Scholar]
- 32.Fassler DJ, Abousamra S, Gupta R, et al. Deep learning-based image analysis methods for brightfield-acquired multiplex immunohistochemistry images. Diagn Pathol 2020; 15: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parra ER. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Front Mol Biosci 2021; 8: 668340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rojas F, Hernandez S, Lazcano R, et al. Multiplex immunofluorescence and the digital image analysis workflow for evaluation of the tumor immune environment in translational research. Front Oncol 2022; 12: 889886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viratham Pulsawatdi A, Craig SG, Bingham V, et al. A robust multiplex immunofluorescence and digital pathology workflow for the characterisation of the tumour immune microenvironment. Mol Oncol 2020; 14: 2384–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parra ER, Jiang M, Solis L, et al. Procedural requirements and recommendations for multiplex immunofluorescence tyramide signal amplification assays to support translational oncology studies. Cancers (Basel) 2020; 12: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taube JM, Akturk G, Angelo M, et al. The Society for Immunotherapy of cancer statement on best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) staining and validation. J Immunother Cancer 2020; 8: e000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khouja MH, Baekelandt M, Sarab A, et al. Limitations of tissue microarrays compared with whole tissue sections in survival analysis. Oncol Lett 2010; 1: 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obeid JM, Hu Y, Erdag G, et al. The heterogeneity of tumor-infiltrating CD8+ T cells in metastatic melanoma distorts their quantification: how to manage heterogeneity? Melanoma Res 2017; 27: 211–217. [DOI] [PubMed] [Google Scholar]
- 40.Nederlof I, De Bortoli D, Bareche Y, et al. Comprehensive evaluation of methods to assess overall and cell-specific immune infiltrates in breast cancer. Breast Cancer Res 2019; 21: 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoos A, Cordon-Cardo C. Tissue microarray profiling of cancer specimens and cell lines: opportunities and limitations. Lab Invest 2001; 81: 1331–1338. [DOI] [PubMed] [Google Scholar]
- 42.Camp RL, Charette LA, Rimm DL. Validation of tissue microarray technology in breast carcinoma. Lab Invest 2000; 80: 1943–1949. [DOI] [PubMed] [Google Scholar]
- 43.Kos Z, Roblin E, Kim RS, et al. Pitfalls in assessing stromal tumor infiltrating lymphocytes (sTILs) in breast cancer. NPJ Breast Cancer 2020; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mani NL, Schalper KA, Hatzis C, et al. Quantitative assessment of the spatial heterogeneity of tumor-infiltrating lymphocytes in breast cancer. Breast Cancer Res 2016; 18: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Althobiti M, Aleskandarany MA, Joseph C, et al. Heterogeneity of tumour-infiltrating lymphocytes in breast cancer and its prognostic significance. Histopathology 2018; 73: 887–896. [DOI] [PubMed] [Google Scholar]
- 46.König L, Mairinger FD, Hoffmann O, et al. Dissimilar patterns of tumor-infiltrating immune cells at the invasive tumor front and tumor center are associated with response to neoadjuvant chemotherapy in primary breast cancer. BMC Cancer 2019; 19: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steele KE, Tan TH, Korn R, et al. Measuring multiple parameters of CD8+ tumor-infiltrating lymphocytes in human cancers by image analysis. J Immunother Cancer 2018; 6: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galon J, Pages F, Marincola FM, et al. The immune score as a new possible approach for the classification of cancer. J Transl Med 2012; 10: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018; 391: 2128–2139. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez K, Kim I, Chun B, et al. Multiplex immunofluorescence to measure dynamic changes in tumor-infiltrating lymphocytes and PD-L1 in early-stage breast cancer. Breast Cancer Res 2021; 23: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.spatstat: Spatial Point Pattern Analysis, Model-Fitting, Simulation, Tests. Available from: https://CRAN.R-project.org/package=spatstat
- 52.Page DB, Broeckz G, Jahangir CA, et al. Spatial analyses of immune cell infiltration in cancer: current methods and future directions: a report of the International Immuno-oncology Biomarker Working Group on Breast Cancer. J Pathol 2023; 260: 514–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giraldo NA, Sanchez-Salas R, Peske JD, et al. The clinical role of the TME in solid cancer. Br J Cancer 2019; 120: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galon J, Mlecnik B, Marliot F, et al. Validation of the immunoscore (IM) as a prognostic marker in stage I/II/III colon cancer: results of a worldwide consortium-based analysis of 1,336 patients. J Clin Oncol 2016; 34: 3500. [Google Scholar]
- 55.Angell HK, Bruni D, Barrett JC, et al. The immunoscore: colon cancer and beyond. Clin Cancer Res 2020; 26: 332–339. [DOI] [PubMed] [Google Scholar]
- 56.Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 2014; 232: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lyons YA, Wu SY, Overwijk WW, et al. Immune cell profiling in cancer: molecular approaches to cell-specific identification. NPJ Precis Oncol 2017; 1: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bai R, Lv Z, Xu D, et al. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res 2020; 8: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 2020; 20: 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anandappa AJ, Wu CJ, Ott PA. Directing traffic: how to effectively drive T cells into tumors. Cancer Discov 2020; 10: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ochoa De Olza M, Navarro Rodrigo B, Zimmermann S, et al. Turning up the heat on non-immunoreactive tumours: opportunities for clinical development. Lancet Oncol 2020; 21: e419–e430. [DOI] [PubMed] [Google Scholar]
- 62.Garaud S, Buisseret L, Solinas C, et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight 2019; 4:el29641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Egelston CA, Avalos C, Tu TY, et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 2019; 4: el30000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He T-F, Yost SE, Frankel PH, et al. Multi-panel immunofluorescence analysis of tumor infiltrating lymphocytes in triple negative breast cancer: evolution of tumor immune profiles and patient prognosis. PLoS One 2020; 15: e0229955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayashi K, Nogawa D, Kobayashi M, et al. Quantitative high-throughput analysis of tumor infiltrating lymphocytes in breast cancer. Front Oncol 2022; 12: 901591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masugi Y, Abe T, Ueno A, et al. Characterization of spatial distribution of tumor-infiltrating CD8+ T cells refines their prognostic utility for pancreatic cancer survival. Mod Pathol 2019; 32: 1495–1507. [DOI] [PubMed] [Google Scholar]
- 67.Yamagami W, Susumu N, Tanaka H, et al. Immunofluorescence-detected infiltration of CD4+FOXP3+ regulatory T cells is relevant to the prognosis of patients with endometrial cancer. Int J Gynecol Cancer 2011; 21: 1628–1634. [DOI] [PubMed] [Google Scholar]
- 68.Peng H, Wu X, Zhong R, et al. Profiling tumor immune microenvironment of non-small cell lung cancer using multiplex immunofluorescence. Front Immunol 2021; 12: 750046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schalper KA, Brown J, Carvajal-Hausdorf D, et al. Objective measurement and clinical significance of TILs in non-small cell lung cancer. J Natl Cancer Inst 2015; 107: dju435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang Y-K, Wang M, Sun Y, et al. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat Commun 2019; 10: 3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu Y, Ma X, Zhang Y, et al. Changes in expression of multiple checkpoint molecules and infiltration of tumor immune cells after neoadjuvant chemotherapy in gastric cancer. J Cancer 2019; 10: 2754–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gide TN, Silva IP, Quek C, et al. Close proximity of immune and tumor cells underlies response to anti-PD-1 based therapies in metastatic melanoma patients. Oncoimmunology 2019; 9: 1659093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong PF, Wei W, Smithy JW, et al. Multiplex quantitative analysis of tumor-infiltrating lymphocytes and immunotherapy outcome in metastatic melanoma. Clin Cancer Res 2019; 8: 2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mazzaschi G, Madeddu D, Falco A, et al. Low PD-1 expression in cytotoxic CD8+ tumor-infiltrating lymphocytes confers an immune-privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin Cancer Res 2018; 24: 407–419. [DOI] [PubMed] [Google Scholar]
- 75.Parra ER, Villalobos P, Behrens C, et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J Immunother Cancer 2018; 6: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown JR, Wimberly H, Lannin DR, et al. Multiplexed quantitative analysis of CD3, CD8, and CD20 predicts response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 2014; 20: 5995–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Angelis C, Nagi C, Hoyt CC, et al. Evaluation of the predictive role of tumor immune infiltrate in patients with HER2-positive breast cancer treated with neoadjuvant anti-HER2 therapy without chemotherapy. Clin Cancer Res 2020; 26: 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wimberly H, Brown JR, Schalper K, et al. PD-L1 expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res 2015; 3: 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Y, Jia K, Sun Y, et al. Predicting response to immunotherapy in gastric cancer via multi-dimensional analyses of the tumour immune microenvironment. Nat Commun 2022; 13: 4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y, Zugazagoitia J, Shabbir Ahmed F, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res 2020; 26: 970–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clemente CG, Mihm MC, Bufalino R, et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996; 77: 1303–1310. [DOI] [PubMed] [Google Scholar]
- 82.Liu F, Lang R, Zhao J, et al. CD8+ cytotoxic T cell and FOXP3+ regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res Treat 2011; 130: 645–655. [DOI] [PubMed] [Google Scholar]
- 83.Hülsen S, Lippolis E, Ferrazzi F, et al. High stroma T-cell infiltration is associated with better survival in stage pTl bladder cancer. Int J Mol Sci 2020; 21: 8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Menares E, Gálvez-Cacino F, Cáceres-Morgado P, et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat Commun 2019; 10: 4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kmiecik J, Poli A, Brons NHC, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. JNeuroimmunol 2013; 1–2: 71–83. [DOI] [PubMed] [Google Scholar]
- 86.Piersma SJ, Jordanova ES, van Poelgeest MIE, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res 2007; 67: 354–361. [DOI] [PubMed] [Google Scholar]
- 87.Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J 2018; 17: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saleh R, Elkord E. FoxP3+ T regulatory cells in cancer: prognostic biomarkers and therapeutic targets. Cancer Lett 2020; 490: 174–185. [DOI] [PubMed] [Google Scholar]
- 89.Tuminello S, Veluswamy R, Lieberman-Cribbin W, et al. Prognostic value of immune cells in the tumor microenvironment of early-stage lung cancer: a meta-analysis. Oncotarget 2019; 10: 7142–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carstens JL, Correa de Sampaio P, Yang D, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 2017; 8: 15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang L, Zhang W, Sun J, et al. Functional status and spatial interaction of T cell subsets driven by specific tumor microenvironment correlate with recurrence of non-small cell lung cancer. Front Immunol 2023; 13: 1022638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsujikawa T, Mitsuda J, Ogi H, et al. Prognostic significance of spatial immune profiles in human solid cancers. Cancer Sci 2020; 111: 3426–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nearchou LP, Lillard K, Gavriel CG, et al. Automated analysis of lymphocytic infiltration, tumor budding, and their spatial relationship improves prognostic accuracy in colorectal cancer. Cancer Immunol Res 2019; 7: 609–620. [DOI] [PubMed] [Google Scholar]
- 94.Banik G, Betts CB, Liudahk SM, et al. High-dimensional multiplexed immunohistochemical characterization of immune contexture in human cancers. Methods Enzymol 2020; 635: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barua S, Fang P, Sharma A, et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer 2018; 117: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mezheyeuski A, Backman M, Mattsson J, et al. An immune score reflecting pro- and anti-tumoural balance of tumour microenvironment has major prognostic impact and predicts immunotherapy response in solid cancers. EBioMedicine 2023; 88: 104452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Werner F, Wagner C, Simon M, et al. A standardized analysis of tertiary lymphoid structures in human melanoma: disease progression- and tumor site-associated changes with germinal center alteration. Front Immunol 2021; 12: 675146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lynch KT, Young SJ, Meneveau MO, et al. Heterogeneity in tertiary lymphoid structure B-cells correlates with patient survival in metastatic melanoma. J Immunother Cancer 2021; 9: e002273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dieu-Nosjean M-C, Goc J, Giraldo NA, et al. Tertiary lymphoid structures in cancer and beyond. Trends Immunol 2014; 35: 571–580. [DOI] [PubMed] [Google Scholar]
- 100.Attrill GH, Ferguson PM, Palendira U, et al. The tumour immune landscape and its implications in cutaneous melanoma. Pigment Cell Melanoma Res 2021; 34: 529–549. [DOI] [PubMed] [Google Scholar]
- 101.Hersom M, Jørgensen JT. Companion and complementary diagnostics-focus on PD-L1 expression assays for PD-1/PD-L1 checkpoint inhibitors in non-small cell lung cancer. Ther Drug Monit 2018; 40: 9–16. [DOI] [PubMed] [Google Scholar]
- 102.US Food and Drug Administration. List of cleared or approved companion diagnostic devices (in vitro and imaging tools) |FDA. [Accessed 15 March 2023]. Available from: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools [Google Scholar]
- 103.Li H, Van Der Merwe PA, Sivakumar S. Biomarkers of response to PD-1 pathway blockade. Br J Cancer 2022; 126: 1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Herbst RS, Soria J-C, Kowanetz M, et al. Predictive correlates of response to the anti-PD-Ll antibody MPDL3280A in cancer patients. Nature 2014; 515: 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chalabi M, Fanchi LF, Dijkstra KK, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 2020; 26: 566–576. [DOI] [PubMed] [Google Scholar]
- 106.de Vries NL, van de Haar J, Veninga V, et al. γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature 2023; 613: 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tumeh PC, Harvew CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giraldo NA, Nguyen P, Engle EL, et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J Immunothey Cancer 2018; 6: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen P-L, Roh W, Reuben A, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov 2016; 6: 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anagnostou V, Yarchoan M, Hansen AR, et al. Immuno-oncology trial endpoints: capturing clinically meaningful activity. Clin Cancer Res 2017; 23: 4959–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes As an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010; 28: 105–113. [DOI] [PubMed] [Google Scholar]
- 112.Chua W, Charles KA, Baracos VE, et al. Neutrophil/lymphocyte ratio predicts chemotherapy outcomes in patients with advanced colorectal cancer. Br J Cancer 2011; 104: 1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vitkin N, Nersesian S, Siemens DR, et al. The tumor immune contexture of prostate cancer. Front Immunol 2019; 10: 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jiang T, Shi T, Zhang H, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol 2019; 12: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hou Y, Nitta H, Wei L, et al. Evaluation of immune reaction and PD-L1 expression using multiplex immunohistochemistry in HER2-positive breast cancer: the association with response to anti-HER2 neoadjuvant therapy. Clin Breast Cancer 2018; 18: e237–e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harms PW, Frankel TL, Moutafi M, et al. Multiplex immunohistochemistry and immunofluorescence: a practical update for pathologists. Mod Pathol 2023; 36: 100197. [DOI] [PubMed] [Google Scholar]
- 117.Abels E, Pantanowitz L, Aeffner F, et al. Computational pathology definitions, best practices, and recommendations for regulatory guidance: a white paper from the digital pathology association. J Pathol 2019; 249: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tizhoosh HR, Pantanowitz L. Artificial intelligence and digital pathology: challenges and opportunities. J Pathol Inform 2018; 9: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akturk G, Parra ER, Gjini E, et al. Multiplex tissue imaging harmonization: a multicenter experience from CEVLAC-CIDC immuno-oncology biomarkers network. Clin Cancer Res 2021; 27: 5072–5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Agrawal L, Engel KB, Greytak SR, et al. Understanding preanalytical variables and their effects on clinical biomarkers of oncology and immunotherapy. Semin Cancer Biol 2018: 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Engel KB, Moore HM. Effects of preanalytical variables on the detection of proteins by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med 2011; 135: 537–543. [DOI] [PubMed] [Google Scholar]
- 122.Chen HX, Song M, Maecker HT, et al. Network for biomarker immunoprofiling for cancer immunotherapy: cancer immune monitoring and analysis centers and cancer immunologic data commons (CIMAC-CIDC). Clin Cancer Res 2021; 27: 5038–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Taube JM, Roman K, Engle EL, et al. Multi-institutional TSA-amplified multiplexed immunofluorescence reproducibility evaluation (MITRE) study. J Immunother Cancer 2021; 9: e002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bodenmiller B Multiplexed epitope-based tissue imaging for discovery and healthcare applications. Cell Syst 2016; 2: 225–238. [DOI] [PubMed] [Google Scholar]