

Abstract
Dysregulated TGF-β signaling contributes to fibrotic liver disease and hepatocellular cancer (HCC), both of which are associated with fatty liver disease. SIRT6 limits fibrosis by inhibiting TGF-β signaling through SMAD2 and SMAD3 and limits lipogenesis by inhibiting SREBP1 and SREBP2 activity. Here, we showed that, compared to wild-type mice, high-fat diet-induced fatty liver is worse in TGF-β signaling-deficient mice (SPTBN1+/−) and the mutant mice had reduced SIRT6 abundance in the liver. Therefore, we hypothesized that altered reciprocal regulation between TGF-β signaling and SIRT6 contributes to these liver pathologies. We found that deficiency in SMAD3 or SPTBN1 reduced SIRT6 mRNA and protein abundance and impaired TGF-β induction of SIRT6 transcripts, and that SMAD3 bound to the SIRT6 promoter, suggesting that a SMAD3-SPTBN1 pathway mediated the induction of SIRT6 in response to TGF-β. Overexpression of SIRT6 in HCC cells reduced expression of TGF-β-induced genes, consistent with the suppressive role of SIRT6 on TGF-β signaling. Manipulation of SIRT6 abundance in HCC cells altered SREBP activity and overexpression of SIRT6 reduced the amount of acetylated SPTBN1 and the abundance of both SMAD3 and SPTBN1. Furthermore, induction of SREBP target genes in response to SIRT6 overexpression was impaired in SPTBN1 heterozygous cells. Thus, we identified a regulatory loop between SIRT6 and SPTBN1 that represents a potential mechanism for susceptibility to fatty liver in the presence of dysfunctional TGF-β signaling.
Keywords: TGF-β signaling, SIRT6, fatty liver, NASH
1. Introduction
The liver is a critical regulator of lipid metabolism. Regulation of lipid metabolism requires a delicate balance of signals and the ability to properly respond to changes in diet and environmental conditions. Non-alcoholic fatty liver disease (NAFLD) is a common component of metabolic syndrome and is associated with an increased risk of fibrotic liver disease and hepatocellular carcinoma (HCC). NAFLD and its attendant risk of HCC are on the rise throughout the world.1, 2
Transforming growth factor β (TGF-β) is involved in hepatic responses to stress, including the excess lipid accumulation associated with fatty liver.3 TGF-β stimulation of hepatic stellate cells (HSCs) in mice mediates the progression of fibrosis in NAFLD.4, 5 Mice with global deletion of SMAD3 are resistant to high-fat diet (HFD)- induced insulin resistance, obesity, and liver steatosis.6 Hepatocyte-specific deletion of TGFBR2 suppresses liver steatosis in mice fed with a choline-deficient amino acid diet.7
SIRT1 and SIRT6 are enzymes of the sirtuin family that are associated with liver metabolism. Activation of SIRT1 by resveratrol attenuates fatty liver in mice; SIRT1 heterozygous mice display hepatic steatosis that is exacerbated by a HFD.8, 9 Liver-specific deletion of SIRT1 in mice results in hyperglycemia, oxidative damage, and insulin resistance through impairment of a pathway involving mammalian target of rapamycin complex 2 (MTORC2) and AKT.10 SIRT6 inhibits cholesterol biosynthesis and fatty acid metabolism in the liver11, 12, and liver-specific deletion of SIRT6 in mice leads to increased triglyceride synthesis and fatty liver.13 Conversely, mice overexpressing SIRT6 exhibit lower levels of visceral fat, plasma low-density lipoprotein (LDL)-bound cholesterol, and triglycerides when challenged with a HFD.14, 15
SMAD2 and SMAD3 are substrates for SIRT6-mediated deacetylation.16 Hepatic steatosis progresses to fibrosis through a pathway involving HSC activation by SMAD2 and SMAD3 signaling in response to TGF-β.17 Mouse studies show that SIRT6 functions through the TGF-β–SMAD2/3 pathway to limit liver fibrosis.18, 19 The abundance of SIRT6 decreases in activated HSCs in human and mouse fibrotic livers.19 In hepatocytes, SMAD3 forms a complex with the adaptor protein SPTBN1 in response to TGF-β signaling to maintain genomic stability and prevent the development of HCC.20 Mouse studies also implicate SPTBN1 is involved in diet-induced NAFLD and NASH.21 Thus, we hypothesized that SIRT6 is a direct target of TGF-β signaling through SMAD3 and SPTBN1 and that impairment of this regulation contributes to steatosis.
Using mouse embryo fibroblasts (MEFs) and HCC cell lines, we found that TGF-β-stimulated SIRT6 expression through a mechanism that depended on SMAD3 and SPTBN1. Similar to the effect of SIRT6 on SMAD3,16 we found that overexpression of SIRT6 reduced SPTBN1 acetylation. Decreased SIRT6 abundance in mice deficient for SPTBN1 was associated with NAFLD phenotypes. Our results indicate that impaired regulation of SIRT6 downstream of the TGF-β pathway involving SMAD3 and SPTBN1 can contribute to the development of fatty liver disease through effects on hepatocytes.
2. Materials and methods
2.1. Bioinformatic analysis of SIRT6 promoter and spatial distribution of SIRT6 in liver tissue
The CTCFBSDB database 2.022 was used to identify consensus sequences for CTCF binding, potential SMAD3 binding sites were analyzed using the online tool JASPAR (http://jaspar.genereg.net) with an 80% score threshold. The sequences 1,000 basepairs (bp) upstream of the transcription start site (TSS) were inspected for human SIRT6.
Human and mouse spatial distribution of SIRT6 transcript abundance was evaluated in the dataset GSE146409 using the online tools developed by Hassan Massalha23 (https://itzkovitzwebapps.weizmann.ac.il/webapps/home/).
2.2. ChIP-seq and ChIP-qPCR
Chromatin immunoprecipitation (ChIP) was performed with SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling, 9003) according to the manufacturer’s instructions. Briefly, after overnight serum starvation, HepG2 cells were treated with 200 pM TGF-β for 2 h and were fixed for 10 min at 25°C with 1% formaldehyde. After incubation, glycine was added to a final concentration of 0.125 M to quench formaldehyde. Subsequently, cells were lysed, and chromatin was harvested and fragmented using enzymatic digestion followed by sonication. The chromatin was then subjected to immunoprecipitation with anti-SMAD3 (Cell Signaling, 9523) or anti-CTCF (Active Motif, 61311) antibody at 4°C overnight, and was incubated with protein G magnetic beads at 4°C for 2 h. A mock immunoprecipitation with a neutral, unrelated IgG (Cell Signaling, 2729) antiserum was carried out in parallel. The immune complexes were washed and eluted in 150 μl elution buffer. Eluted DNA and input DNA were incubated at 65°C to reverse the crosslinking. After digestion with proteinase K, DNA was purified with spin columns. The library was prepared with KAPA library preparation kit (Kapa Biosystems) and sequenced using a HiSeq 2000 platform (Illumina) at the Integrated Genomics Operation of MSKCC.
The relative abundance of precipitated DNA fragments was analyzed by qPCR using Power SYBR Green PCR Master Mix (Qiagen) and the enrichments were normalized to IgG group. The following primers were used for ChIP-qPCR:
hSIRT6 SBE1: GTAACTCTGCGTGGCATTCA (forward), GCCGCCTACGTGAGAGTTC (reverse)
hSIRT6 SBE2: CCATTGACCTTGAGCAGGAC (forward), TTGGGTTCAAGCGATTCTC (reverse)
ChIP-seq reads were first trimmed using Trimmomatic (v0.33). Reads were trimmed if the first or last 3 nucleotides had phred quality score of < 15 or at the point where a sliding window of 4 nucleotides averaged a phred quality score < 15. Illumina adapters were also removed. The reads were then aligned using bowtie2 (v 2.2.6). Multimapping reads were removed after alignment. Peak calling was done using MACS2 (v2.1.0) on pooled replicates and individual samples using a p-value cutoff of 0.01. The peaks were then filtered further using IDR to make sure the peaks are consistent among replicates. The promoter was annotated as region within 2,000 bp from TSS, intron was annotated as region within the gene body but not in the promoter region, and peaks at regions outside but close to gene body were annotated as intergenic.
2.3. Cell culture and transfection
Human liver cancer cell lines HepG2 and Huh7 and MEFs from SPTBN1+/−, SPTBN1−/−, and SMAD3−/− mice were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich, D5671) and supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, F2442) and 1% penicillin/streptomycin (Thermo Fisher Scientific, 15140122). For transfection, cells were transfected with empty vector (EV) or FLAG-SIRT6 (Addgene, 13817) plasmids or FLAG-tagged wild-type (WT) or mutant SMAD3 plasmids using Lipofectamine 3000 (Invitrogen, 11668019) according to the manufacturer’s instructions. Flag-tagged SMAD3 and the K333R and K378R mutant SMAD3 plasmids were obtained from Dr. X. Charlie Dong of Indiana University School of Medicine, Indianapolis, Indiana.
For siRNA experiments, 20 nM of siSMAD3 or control siRNA (Dharmacon, US) were transfected by Lipofectamine RNAiMAX reagent (Thermo Fisher, 13778150) for 48–72 h according to the manufacturer’s instructions.
2.4. Quantitative transcript analysis
Total RNA was extracted from cells using Trizol, separated with chloroform, and precipitated with isopropanol. cDNA was synthesized from 2 μg of total RNA using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, 4368814). Quantitative reverse-transcription PCR (qRT-PCR) was performed with iTaq™ Universal SYBR Green Supermix (Bio-Rad Laboratories, 172–5124) and specific primers for qRT-PCR (Table 1). The reaction was run in triplicate, and the transcription of each gene was normalized to the mean values of transcripts for ACTB (β-actin) or 18S rRNA. Transcript abundance was determined using the ΔΔCT method.
Table 1.
Primers for qRT-PCR. Human genes are preceded by h, mouse genes by m.
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| mSIRT6 | ATGTCGGTGAATTATGCAGCA | GCTGGAGGACTGCCACATTA |
| hSIRT6 | CCCACGGAGTCTGGACCAT | CTCTGCCAGTTTGTCCCTG |
| hSPTBN1 | ATCTAACGCACACTACAACCTG | TCAAGCACCTTTCCAATTCGT |
| hSMAD3 | CCCCAGAGCAATATTCCAGA | GACATCGGATTCGGGGATAG |
| hSERPINE1 | AGTGGACTTTTCAGAGGTGGA | GCCGTTGAAGTAGAGGGCATT |
| hHMGA2 | CGAAAGGTGCTGGGCAGCTCCGG | CCATTTCCTAGGTCTGCCTCTTG |
| hMMP9 | TGTACCGCTATGGTTACACTCG | GGCAGGGACAGTTGCTTCT |
| hSREBP1 | GTTGGCCCTACCCCTCC | CTTCAGCGAGGCGGCTT |
| hTERT | GCCGATTGTGAACATGGACTACG | GCTCGTAGTTGAGCACGCTGAA |
| mFASN | CTGCGTGGCTATGATTATGG | AGGTTGCTGTCGTCTGTAGT |
| mSCD1 | GCAAGCTCTACACCTGCCTCTT | CGTGCCTTGTAAGTTCTGTGGC |
| h18S | CTACCACATCCAAGGAAGCA | TTTTTCGTCACTACCTCCCCG |
| m18S | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| hACTB | CACCATTGGCAATGAGCGGTTC | AGGTCTTTGCGGATGTCCACGT |
| mACTB | CATTGCTGACAGGATGCAGAAGG | TGCTGGAAGGTGGACAGTGAGG |
2.5. Immunoblotting, immunoprecipitation, and immunohistochemical analyses
Cells were harvested, lysed with lysis buffer [(50 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 1% NP-40, 1 mM EDTA), Complete Protease Inhibitor Cocktail (Sigma, 5892953001), 1 mM PMSF, 1 mM NaF, and 1 mM sodium orthovanadate] and sonicated. Primary antibodies against SMAD3 (9523) and acetylated-lysine (9441) were from Cell Signaling Technologies. Antibodies against LDLR (sc-18823), β-ACTIN (sc-69879), VINCULIN (sc-73614), and GAPDH (sc-47724) were from Santa Cruz, and anti-FLAG (F3165) was from Sigma. The antibody against SPTBN1 was custom from Bio-Synthesis company (Texas, US).20 The antibody against SIRT6 is from NOVUS (NB100–2522).
For immunohistochemical analyses, mouse liver tissue samples were fixed in 10% formalin solution and stored in 70% ethanol before they were processed for embedding and sectioning at the Research Pathology Core Laboratory of George Washington University. Liver sections were stained with H&E. For staining of SIRT6, sections were deparaffinized and hydrated in 1 mmol/L EDTA buffer for antigen retrieval at 100°C for 5 minutes, and the liver sections then were incubated with normal horse serum for 1 hour. Next, the liver sections were incubated with the SIRT6 antibody at 4°C overnight. Diaminobenzidine was used as a chromogen for SIRT6 antibody detection, and hematoxylin was used as the counterstain. Semi-quantitative analysis of SIRT6 staining intensity was performed as previously described.24
2.6. Dual luciferase activity assay
Briefly, MEFs were seeded at a density of 1 × 104 cells per well in 96-well plates, and the cells were transfected with FLAG-SIRT6 using Lipofectamine 3000 (Thermo-Fisher, L3000015). The next day, the cells were transfected with Renilla (a gift from Dr. Ray-chang Wu) and with a luciferase reporter containing the LDLR promoter region (pLDLR-luc, Addgene, 14940) or the SCD1 promoter (pGL3-SCD1, a gift from Dr. Giovanni Sorrentino and Dr. Giannino Del Sal) and using Lipofectamine 3000. After 48 hours of transfection, the cells were treated with TNF-α (20 ng/ml) and cycloheximide (CHX, 10 μg/ml) for 3 h. The cells were then lysed with luciferase cell culture lysis reagent (100 μl). Luciferase and Renilla activity were analyzed in the lysates (20 μl) using a dual luciferase assay kit according to the manufacturer’s instructions (Promega, E1980). Luciferase activity was normalized to Renilla activity (AU) for each sample, and fold changes were calculated.
2.7. Mouse models and histology
All animal procedures performed in this work were approved by the Institutional Animal Care and Use Committee of George Washington University School of Medicine and Health Sciences in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. SPTBN1+/− mutant mice were generated as previously described.25, 26 Genotypes were determined by PCR. For HFD-induced liver steatosis, 10- to 12-week-old male mice were fed a normal chow diet or HFD (ENVIGO, TD.06414) for 12 to 16 weeks. Body weight and serum total cholesterol and triglyceride concentrations were measured. Routine tissue processing and histologic techniques were used for liver tissue sections stained with Hoechst and eosin (H&E).
To measure food intake, water intake, and urine output, mice fed a normal chow diet or HFD were housed individually in an MMC100 metabolic cage (Hatteras Instruments) for 24 hours. Food intake was measured by weight differences and water intake was measured by volume differences before and after 24 hours in the metabolic cage. The amount of urine output in 24 hours was measured by volume.
2.8. Statistical analysis
Differences between groups were evaluated using Student t-tests. Results are presented as mean ± standard deviation. For all statistical analyses, p < 0.05 was considered statistically significant.
3. Results
3.1. TGF-β signaling stimulates SIRT6 transcription through direct binding of SMAD3 and CTCF to the SIRT6 promoter
Previously, we found that SMAD3 and SPTBN1 functioned with the transcriptional regulator CCCTC-binding factor (CTCF) at promoters of genes (TERT, MYC) relevant to HCC.20 To evaluate if SIRT6 was regulated by a similar pathway, we used bioinformatics to identify potential SMAD3 or CTCF binding sites in the human SIRT6 promoter. We identified two consensus SMAD-binding elements (SBEs) (SBE1 and SBE2) and two consensus CTCF binding sites in the SIRT6 promoter (Figure 1A). CHIP-seq analysis of HepG2 cells indicated that SMAD3 and CTCF bind to the promoter region of SIRT6 and that binding increased in response to TGF-β (Figure 1B). With ChIP-qPCR assays, we detected SMAD3 bound to the SIRT6 promoter at both SBE1 and SBE2 in HepG2 cells exposed to TGF-β (Figure 1C). To determine if TGF-β signaling stimulated or repressed SIRT6 expression, we measured TGF-β-stimulated changes in transcript abundance in MEFs from WT, SPTBN1+/−, or SMAD−/− mice (Figure 1D). Using SERPINE1 as a positive control for a TGF-β-SMAD3 target gene, we found that TGF-β-stimulated expression of SERPINE1 and SIRT6 depended on both SMAD3 and SPTBN1. Consistent with the transcript results, SIRT6 protein abundance was decreased in SPTBN1+/− or SPTBN1−/− MEFs or in HepG2 cells in which SMAD3 was knocked down (Figure 1E, Figure S1A, S1B). These results indicated that TGF-β signaling positively regulates SIRT6 transcription in a pathway dependent on SMAD3 and SPTBN1.
FIGURE 1. SIRT6 is a target of TGF-β signaling.
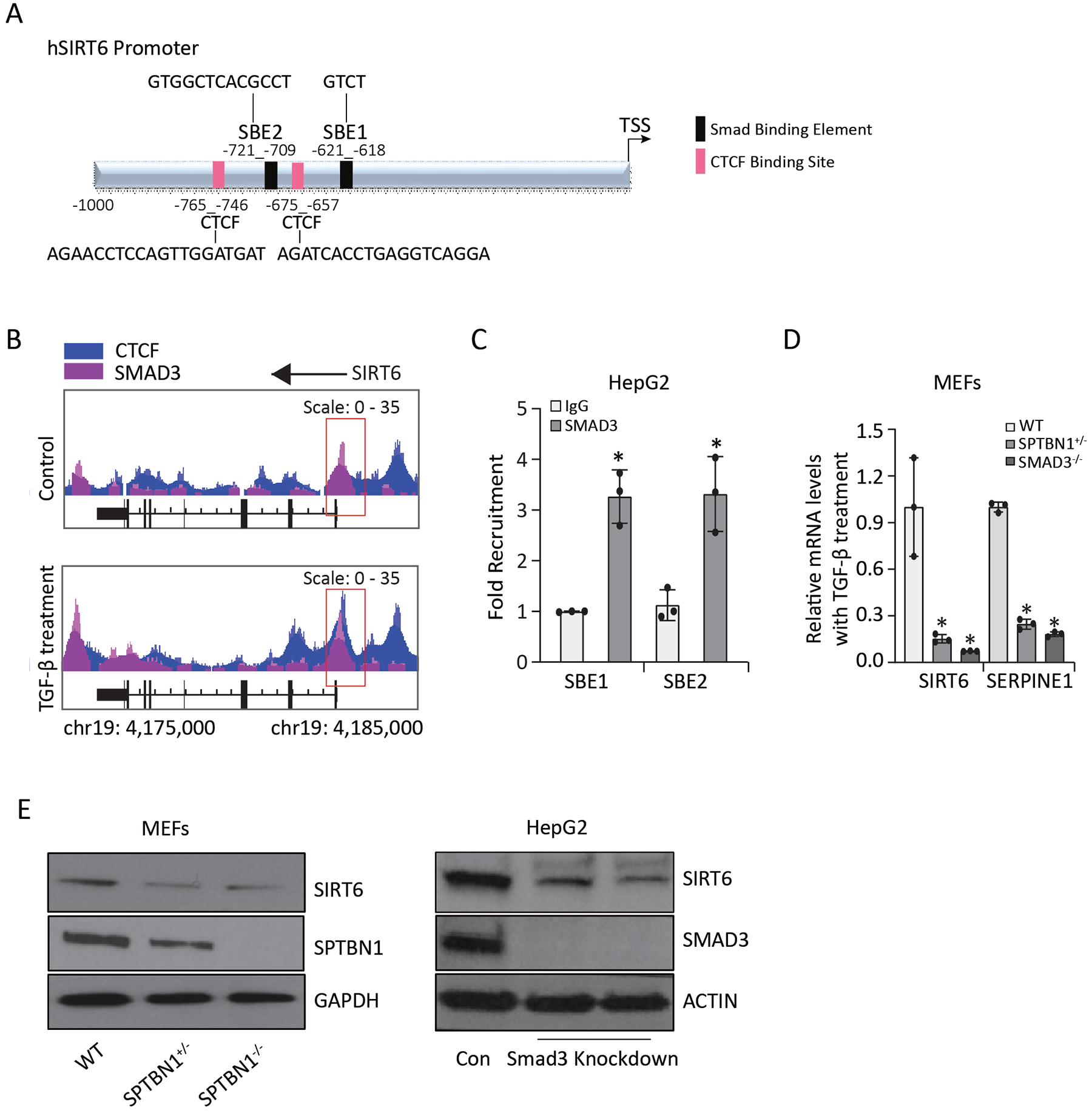
(A) Schematic of the human SIRT6 promoter with potential SBEs and CTCF binding sites. (B) Representative SMAD3 and CTCF binding peaks at the SIRT6 promoter and enhancer regions from one of three ChIP-Seq experiments in HepG2 cells with or without TGF-β treatment. The region of chromosome 19 (chr19) is indicated. (C) SMAD3 binding to SBE1 and SBE2 from ChIP-qPCR experiments is presented as the fold recruitment. Data are presented as the mean ± SEM of three independent experiments. *, p < 0.05; compared with IgG group. (D) Relative mRNA levels of SIRT6 and SERPINE1 were determined in MEFs exposed to 200 pM TGF-β for 24 h. *, p < 0.05; compared to WT. (E) SIRT6 was detected by Western blotting in MEFs with different genotyping (left) and HepG2 stable cell lines with or without SMAD3 knockdown (right). GAPDH or ACTIN served as the loading control.
Acetylation of SMAD3 occurs at Lys333 and Lys378, and SIRT6 reduces acetylation of these residues in HSCs.18 Therefore, we evaluated if expression of K333R or K378R, mutations that mimic deacetylated lysine, affected SIRT6 abundance when expressed in Huh7 cells (Figure S1C). We compared cells expressing Flag-tagged SMAD3 to those expressing the mutated forms of Flag-tagged SMAD3 with or without TGF-β treatment. SIRT6 abundance was similar in all conditions, suggesting that deacetylation of these residues was not necessary for SMAD3 to promote SIRT6 expression in these cells (Figure S1C).
3.2. SIRT6 reduces SPTBN1 acetylation
SPTBN1 forms a complex with SMAD3 to regulate TGF-β target genes,27 and SIRT6 reduces liver fibrosis by deacetylating and suppressing SMAD2 and SMAD3 activity in HSCs.18, 19 Here, we explored whether SIRT6 also affects SPTBN1. First, we established that SIRT6 inhibited TGF-β-induced gene expression in hepatocytes using HepG2 cells. We found that overexpression of Flag-SIRT6 reduced TGF-β–stimulated transcription of the genes MMP9, SERPINE1, SREBP1, and HMGA2, but had no effect on the expression of TERT (Figure 2A). SIRT6 interacts with SMAD3 to mediate deacetylation. Therefore, we immunoprecipitated Flag-SIRT6 from HepG2 cells with or without exposure to TGF-β and tested for the presence of SMAD3 and SPTBN1. Both SMAD3 and SPTBN1 coimmunoprecipitated with Flag-SIRT6 and TGF-β exposure increased this interaction (Figure 2B, Figure S2A).
FIGURE 2. SIRT6 reduces SPTBN1 acetylation.
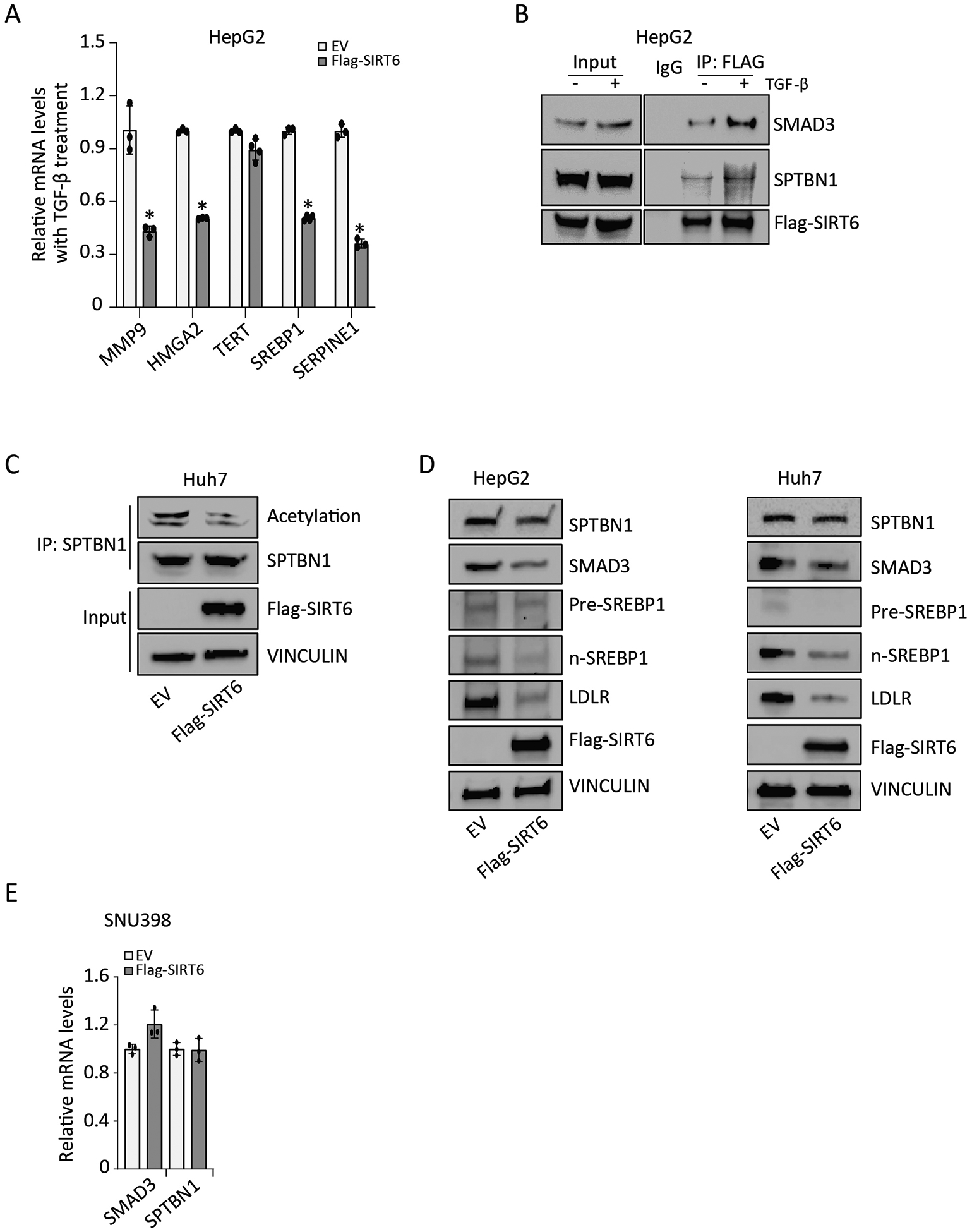
(A) Relative mRNA levels of the indicated TGF-β target genes in HepG2 cells transfected with empty vector (EV) or Flag-SIRT6 and exposed to 200 pM TGF-β for 24 h. Data are presented as mean ± SEM relative to the amount in EV-transfected cells exposed to TGF-β treatment (n = 3 – 4). *, p < 0.05; compared with EV group with TGF-β treatment. (B) Co-immunoprecipitation of SMAD3, SPTBN1, and Flag-SIRT6 was performed in HepG2 cells with or without exposure to 200 pM TGF-β for 3 h, using an irrelevant IgG antibody or anti-FLAG antibody. IP, immunoprecipitation. Representative data from 1 of 3 independent experiments are shown. (C) The effect of SIRT6 overexpression on SPTBN1 acetylation in Huh7 cells was determined by immunoprecipitating SPTBN1 and Western blotting for the presence of acetylation with SPTBN1. Representative data from 1 of 3 independent experiments are shown. (D) The effect of SIRT6 overexpression on SPTBN1, SMAD3, Pre-SREBP1, n-SREBP1, and LDLR abundance in HepG2 cells and Huh7 cells was determined by Western blotting. VINCULIN served as the loading control. EV, empty vector. Representative data from 1 of 3 independent experiments are shown. (E) Relative mRNA levels of SMAD3 and SPTBN1 were determined in SNU398 cells transfected with empty vector (EV) or Flag-SIRT6. Data are presented as mean ± SEM relative to the amount in EV-transfected cells (n = 3). No significant differences were detected with t-test, compared to EV group.
Acetylated SPTBN1 is detected in proteomic studies.28–30 Thus, we tested if SPTBN1 was acetylated in Huh7 cells. We detected acetylated SPTBN1 in these cells and found that overexpression of Flag-SIRT6 reduced the amount of acetylated SPTBN1 (Figure 2C, Figure S2B). SIRT6 overexpression in HepG2 and Huh7 cells appeared to slightly reduce SMAD3 and SPTBN1 abundance (Figure 2D, Figure S2C) and SIRT6 knockdown in HepG2 cells appeared to slightly increase SPTBN1 abundance (Figure S2D). SIRT6 overexpression did not affect transcript abundance of SPTBN1 and SMAD3 in SNU398 cells (Figure 2E). The coimmunoprecipitation and overexpression data indicated that TGF-β promotes an interaction between SIRT6 and SPTBN1 and reduces SPTBN1 acetylation. These data suggested that SIRT6 regulates SPTBN1 through deacetylation to contribute to SIRT6-mediated inhibition of TGF-β signaling.
We also examined the effect of SIRT6 overexpression and knockdown on pre-SREBP1, mature SREBP1 (n-SREBP1), and LDLR, which is encoded by an SREBP1 target gene (Figure 2D, Figure S2C, Figure S2D). Although the results were variable, we found that overexpression of SIRT6 appeared to reduce the abundance of n-SREBP1 and LDLR in both HepG2 and Huh7 cells.
3.3. SPTBN1 heterozygous mice develop a liver phenotype resembling NAFLD that is associated with reduced SIRT6 abundance
To explore the connection between TGF-β signaling and SIRT6 in fatty liver disease, SPTBN1+/− mice were fed a HFD for 12 – 16 weeks to induce a phenotype resembling human NAFLD. WT mice and SPTBN1+/− mice had similar food intake, water intake, and urine output on a normal diet; these characteristics were comparable between the two groups of mice on a HFD (Figure S3). Compared to the HFD WT mice, HFD SPTBN1+/− mice exhibited a 20% increase in serum triglycerides concentration (p < 0.05) without a change in body weight or serum cholesterol concentration (Figure 3A). Both WT mice and SPTBN1+/− mice had changes in the liver in response to the HFD (Figure 3B). However, the changes were more severe in the livers of the SPTBN1+/− mice: Liver architecture was more abnormal, lipid droplets were larger, and there was evidence of inflammation (Figure 3B).
FIGURE 3. SPTBN1+/− mice have reduced SIRT6 abundance and develop HFD-induced liver steatosis.
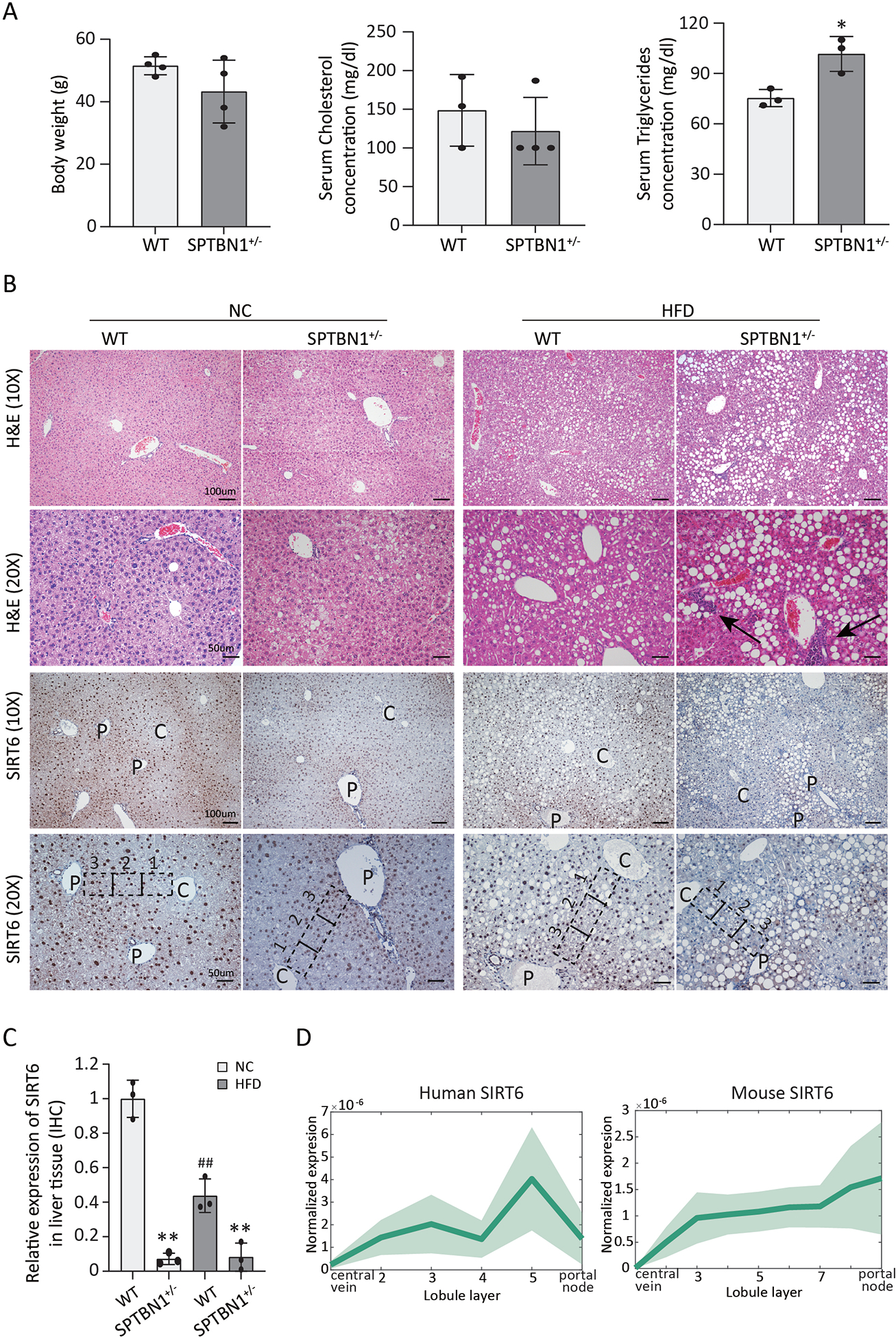
WT or SPTBN1+/− mice (10 – 12 weeks old) were fed a normal chow (NC) diet or a HFD for 12 – 16 weeks. (A) Body weight and serum cholesterol and serum triglyceride concentrations are shown for mice after receiving a HFD for 12 – 16 weeks. Data are presented as mean ± SEM of 3 – 4 mice per group. *, p < 0.05 compared to WT. (B) Liver histology and SIRT6 immunohistochemistry (IHC) results are shown for mice after 12–16 weeks on the indicated diets. Hematoxylin & eosin (H&E) and IHC images are shown at two magnifications: 10×, scale bar = 100 μm; 20×, scale bar = 50 μm. C, central vein; P, portal tracts; arrows indicate inflammation. Representative zones between central vein and portal tracts are marked by the black dashed line:1, indicates area close to central vein; 2, indicates area in the middle; 3, indicates area close to portal tracts. (C) SIRT6 abundance was quantified from SIRT6 IHC in liver tissue from mice of the indicated genotypes (n = 3). **, p < 0.01; compared with WT group on the same diet; ##, p < 0.01, compared with WT group on NC diet. (D) Profiles of SIRT6 transcripts in the regions of the liver from the central vein to the portal node in human (left) and mouse (right). Each profile is normalized to the maximal expression across zones. The line indicates the mean, and the shaded region indicates the SEM.
These severe liver phenotypes in the HFD SPTBN1+/− mice resemble the non-alcoholic steatohepatitis (NASH) phenotype of mice with liver-specific SIRT6 knockout.13 We performed immunohistochemistry to analyze the distribution and abundance of SIRT6 in liver tissue of WT mice and SPTBN1+/− mice fed normal chow or a HFD. SIRT6 displayed a distinct spatial distribution pattern with relatively higher abundance in the portal node region and lower abundance in the region close to central vein (Figure 3B). This pattern was present in livers from both WT mice and SPTBN1+/− mice. Semi-quantitative analysis of the intensity of SIRT6 staining throughout the liver revealed that WT mice fed either diet had higher amounts of SIRT6 in the liver tissue than did SPTBN1+/− mice on the matching diet (Figure 3C). Furthermore, HFD reduced the amount of SIRT6 in livers of WT mice, consistent with previous report that SIRT6 expression was decreased in HFD-induced liver steatosis.31 We found that the amount of SIRT6 was already low in the livers of the SPTBN1+/− mice and HFD did not further reduce it, consistent with impaired TGF-β-mediated induction of SIRT6. The spatial differences in the distribution of SIRT6 corresponded to differences in the pattern of SIRT6 transcripts in the published datasets23, 32 for human and mice liver tissues (Figure 3D). These results indicated that SPTBN1+/− mice developed a liver phenotype resembling NASH that is associated with reduced SIRT6 abundance.
3.4. Overexpression of SIRT6 inhibits lipogenesis in the context of heterozygous loss of SPTBN1
Because the SPTBN1+/− mice had reduced SIRT6 abundance and developed severe liver steatosis phenotypes that resemble NASH and NASH is associated with stress and inflammatory signaling,33, 34 we hypothesized that the reduction in SIRT6 contributes to the increased lipogenesis in the context of SPTBN1 deficiency. To mimic the stress conditions associated with NASH, we exposed WT and SPTBN1+/− MEFs to the inflammatory cytokine TNF-α and cycloheximide (CHX) and evaluated the effect of SIRT6 overexpression on SREBP activity. We used both reporter gene assays (Figure 4A) and analysis of endogenous transcripts of SREBP target genes (Figure 4B). Compared with vehicle-treated cells, stressed MEFs of both genotypes had significantly increased LDLR and SCD1 reporter gene expression (Figure 4A).
FIGURE 4. SIRT6 blunts stress-induced expression of SREBP target genes in SPTBN1+/− MEFs.
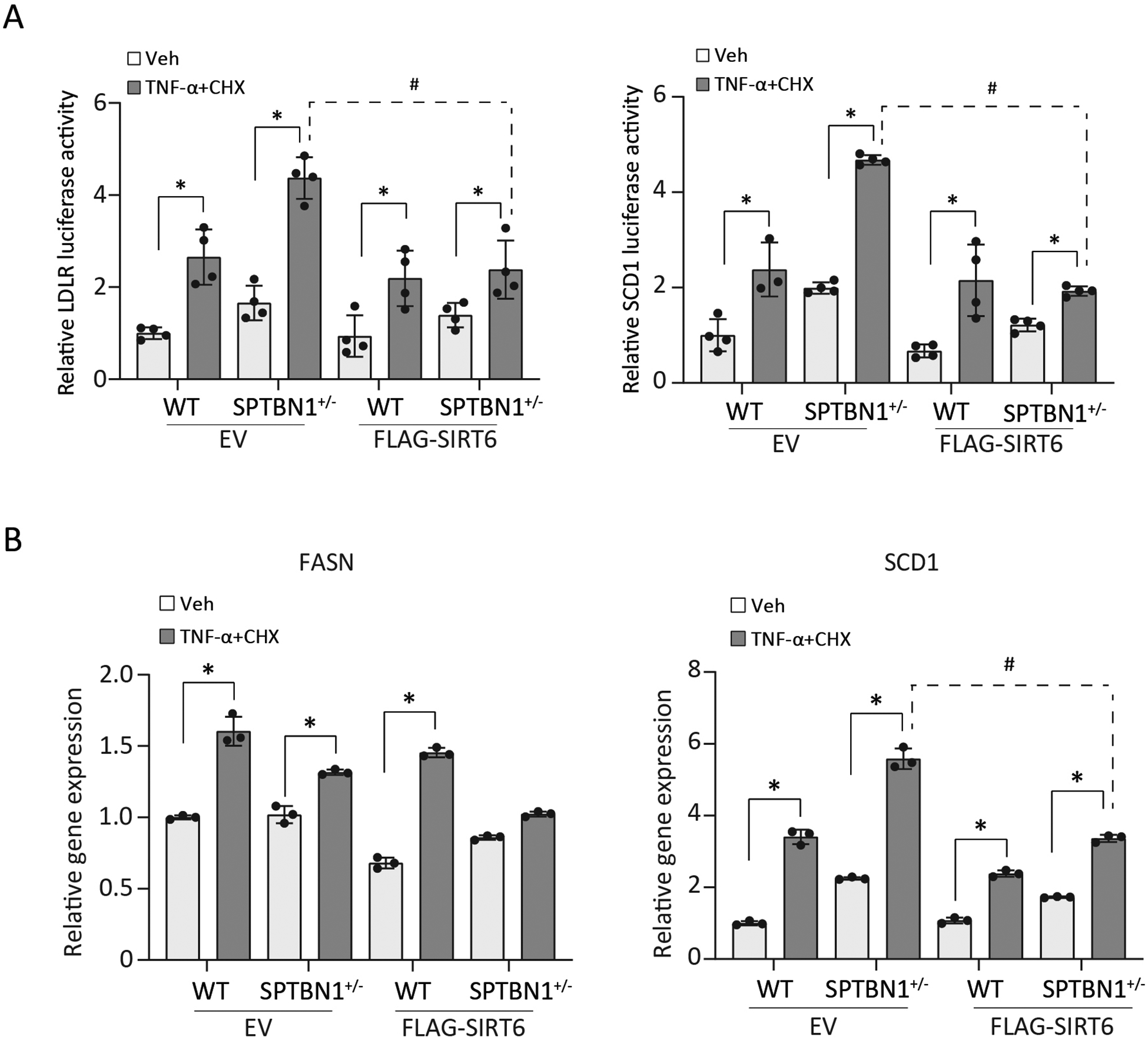
MEFs were transfected with empty vector (EV) or FLAG-SIRT6 and exposed to vehicle (Veh) or stressed with TNF-α (20 ng/ml) and cycloheximide (CHX, 10 μg/ml) for 3 h. (A) In addition to the EV or FLAG-SIRT6, MEFs were transfected with plasmids encoding Renilla and luciferase controlled by the indicated promoter. Luciferase activity from the LDLR promoter or the SCD1 promoter was normalized using the activity of Renilla luciferase and the mean ± SEM relative to the vehicle-treated WT were plotted (n = 3 – 4 independent experiments). (B) Transcript abundance of the indicated SREBP target genes is presented relative to that in WT MEFs transfected with empty vector and exposed to vehicle. Data are presented as the mean ± SEM (n = 3). In A and B, *, p < 0.05, compared with vehicle-treated cells; #, p < 0.05, stressed SPTBN1+/− EV versus FLAG-SIRT6.
Overexpression of SIRT6 blunted the stress-induced increase in reporter gene expression in MEFs of both genotypes. Consistently, for FASN and SCD1, stress induced the expression of both genes in WT and SPTBN1+/− cells (Figure 4B). Overexpression of Flag-SIRT6 blunted the stress-induced increase in FASN and SCD1 transcripts especially in the SPTBN1+/− MEFs. Thus, these data indicated that impaired regulation of SIRT6 in response to stress in cells with SPTBN1 deficiency contributes to excess expression of SREBP target genes.
4. DISCUSSION
Fibrotic liver disease can result from progression of NAFLD. Both TGF-β and SIRT6 are important regulators of fibrosis in multiple tissues, including the liver and kidney. Whereas inhibition of TGF-β signaling by SIRT6 is established in HSCs in the liver,13, 16, 35 regulation of SIRT6 by TGF-β in hepatocytes has not been reported. Furthermore, the involvement of the SMAD3 adaptor SPTBN1 in this process has not been investigated previously. Our results showed that TGF-β signaling positively regulates SIRT6 expression in hepatocytes through a mechanism that depends on SMAD3 and SPTBN1. The finding that TGF-β signaling increase SIRT6 expression is consistent with a study of renal epithelial cells,36 which showed that TGF-β increased SIRT6 at the transcript and protein level, SIRT6 limited TGF-β–induced fibrotic changes in mouse kidney cells, and increased SIRT6 abundance occurred in experimentally induced fibrotic renal disease in mice.36 Our data indicated that a similar reciprocal regulatory mechanism is involved in the regulation of hepatocyte lipid metabolism.
Mouse studies indicate that both SIRT6 and TGF-β signaling play key roles in lipid metabolism in the liver.6, 7 Lipid accumulation in the liver is the hallmark of NAFLD and TGF-β signaling contributes to the progression to the fibrotic NASH condition, which can result in compromised liver function and is a risk factor for HCC.1, 2 In the liver, TGF-β signaling is complex, because multiple cells within the liver produce and respond to TGF-β.3 Our data here indicated that paracrine or autocrine TGF-β signaling in hepatocytes stimulates SIRT6 expression through a SMAD3- and SPTBN1-dependent transcriptional mechanism that involves binding of SMAD3 to the SIRT6 promoter. These results provide a mechanistic basis for our previous findings in fibroblasts that SMAD3−/− MEFs, SPTBN1+/− MEFs, and SMAD3−/−, SPTBN1+/− MEFs had reduced SIRT6 transcripts27 and for the TGF-β-mediated stimulation of SIRT6 expression in kidney epithelial cells and HSCs.18, 36
Finding that SIRT6 reduced SPTBN1 acetylation represents another potential mechanism for SIRT6 to limit TGF-β signaling. SIRT6 is already known to deacetylate SMAD219 and SMAD318 and reduce TGF-β responses. Additionally, SPTBN1 is involved in acetaminophen-induced liver damage37 and in diet-induced steatosis and NASH in mice.21 Liver-specific knockout of SPTBN1 protects mice from diet-induced NASH through a mechanism involving regulation of SREBP1 activity.21 How SIRT6-mediated deacetylation of SPTBN1 influences these other roles of SPTBN1 remains to be investigated. Our data suggested that a negative feedback loop between SIRT6 and SPTBN1 and that the positive regulation of SIRT6 by TGF-β signaling may both be important for limiting SREBP1-dependent gene expression and liver steatosis.
The physiological relevance of this regulatory loop was demonstrated by studying the effect of a HFD on SPTBN1 heterozygous mice. SREBP1 and SREBP2 are key transcription factors that promote de novo lipogenesis and control cellular lipid uptake,38 and both are negatively regulated by SIRT6.11, 39 SPTBN1+/− mice developed liver steatosis with decreased SIRT6 protein abundance, suggesting that impaired induction of SIRT6 contributes to the severe liver phenotype. SPTBN1+/− MEFs had less SIRT6 and exhibited increased activity of SREBP reporter genes in response to stress. Furthermore, overexpression of SIRT6 in SPTBN1+/− MEFs subjected to stress reduced the activity of SREBPs. Together, these findings indicated that the severe diet-induced liver steatosis in SPTBN1+/− mice involves impaired TGF-β-mediated induction of SIRT6, resulting in a physiologically relevant reduction in this inhibitor of de novo lipogenesis.
The role of TGF-β signaling in the liver is complex. TGF-β signaling is critical for the response to stress or recovery from liver injury.40 However, TGF-β signaling is implicated in liver diseases, including NAFLD, NASH, and HCC. This pathway has distinct roles in various stages of gastrointestinal disease and cancer.3 Reciprocal regulation by the products of TGF-β target genes provides a means to tightly control the activity of TGF-β signaling.41, 42 Similar to the deacetylating activity of SIRT6 on SMAD2 and SMAD3,18, 19 we observed that SIRT6 also reduced acetylated SPTBN1.
Like SIRT6, SIRT1 is also an important inhibitor of de novo lipogenesis43 and is implicated in fatty liver disease.10, 44, 45 Previously, we reported that SIRT1 abundance was reduced in SMAD3 deficient MEFs or in HCC cell lines in which SPTBN1 was knocked down.27 However, transcript abundance of SIRT1 was not changed, suggesting a posttranslational mechanism of regulation by TGF-β signaling of SIRT1.
Our results here indicate that impaired reciprocal regulation between SIRT6 and TGF-β signaling that involves SPTBN1 contributes to diet-induced NASH. These results, along with our previous findings of regulation of SIRT1 by a pathway involving SMAD3 and SPTBN1,27 suggest that TGF-β signaling regulates de novo lipogenesis in the liver through a transcriptional mechanism involving SIRT6 and a posttranslational mechanism involving SIRT1. Future investigation of these regulatory connections, including if impairment of the SIRT6-TGF-β pathway feedback loop occurs in human liver disease, may yield novel opportunities to intervene and prevent NAFLD from progressing to NASH.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Bao-Ngoc Nguyen for her critical review of the manuscript and technical support for Western blot and animal studies. We also thank Dr. Giovanni Sorrentino and Dr. Giannino Del Sal for the plasmid with the SCD1 promoter (pGL3-SCD1), Dr. Ray-chang Wu for the Renilla plasmid, and Dr. X. Charlie Dong for the Flag-tagged wild-type SMAD3 plasmids and the K333R and K378 mutants. This research work was supported by NIH grants R01AA023146 (L. Mishra), NIH R01 CA236591-01 (L. Mishra), NIH U01 CA230690-01 (L. Mishra), and Elaine H. Snyder Cancer Research Award (L. Mishra).
Funding:
This research work was supported by NIH grants R01AA023146 (L. Mishra), NIH R01 CA236591-01 (L. Mishra), NIH U01 CA230690-01 (L. Mishra), and Elaine H. Snyder Cancer Research Award (L. Mishra).
Abbreviations:
- TGF-β
transforming growth factor-beta
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- HCC
hepatocellular carcinoma
- HFD
high-fat diet
- NC
normal chow
- HSC
hepatic stellate cell
- TSS
transcriptional start site
- SBE
SMAD binding element
- MEF
mouse embryo fibroblast
- TNF-α
tumor necrosis factor-alpha
- CHX
cycloheximide
- SIRT
sirtuin
- CTCF
CCCTC-binding factor
- SREBP
Sterol regulatory element binding protein
Footnotes
CONFLICT OF INTEREST STATEMENT
These authors have no conflict of interest to declare.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
REFERENCES
- 1.Kumar R, Priyadarshi RN, and Anand U (2020) Non-alcoholic Fatty Liver Disease: Growing Burden, Adverse Outcomes and Associations. J Clin Transl Hepatol 8, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, and Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 [DOI] [PubMed] [Google Scholar]
- 3.Gough NR, Xiang X, and Mishra L (2021) TGF-β Signaling in Liver, Pancreas, and Gastrointestinal Diseases and Cancer. Gastroenterology 161, 434–452.e415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, and Brenner DA (2001) The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 34, 89–100 [DOI] [PubMed] [Google Scholar]
- 5.Kim J, Kang W, Kang SH, Park SH, Kim JY, Yang S, Ha SY, and Paik YH (2020) Proline-rich tyrosine kinase 2 mediates transforming growth factor-beta-induced hepatic stellate cell activation and liver fibrosis. Sci Rep 10, 21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan CK, Leuenberger N, Tan MJ, Yan YW, Chen Y, Kambadur R, Wahli W, and Tan NS (2011) Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 60, 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, and Seki E (2014) Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 59, 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou R, Yi L, Ye X, Zeng X, Liu K, Qin Y, Zhang Q, and Mi M (2018) Resveratrol Ameliorates Lipid Droplet Accumulation in Liver Through a SIRT1/ATF6-Dependent Mechanism. Cell Physiol Biochem 51, 2397–2420 [DOI] [PubMed] [Google Scholar]
- 9.Xu F, Gao Z, Zhang J, Rivera CA, Yin J, Weng J, and Ye J (2010) Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/− mice: a role of lipid mobilization and inflammation. Endocrinology 151, 2504–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, and Deng CX (2011) Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest 121, 4477–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elhanati S, Kanfi Y, Varvak A, Roichman A, Carmel-Gross I, Barth S, Gibor G, and Cohen HY (2013) Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep 4, 905–912 [DOI] [PubMed] [Google Scholar]
- 12.Naiman S, Huynh FK, Gil R, Glick Y, Shahar Y, Touitou N, Nahum L, Avivi MY, Roichman A, Kanfi Y, Gertler AA, Doniger T, Ilkayeva OR, Abramovich I, Yaron O, Lerrer B, Gottlieb E, Harris RA, Gerber D, Hirschey MD, and Cohen HY (2019) SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPARα. Cell Rep 29, 4127–4143.e4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, Gao B, and Deng CX (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, and Cohen HY (2010) SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9, 162–173 [DOI] [PubMed] [Google Scholar]
- 15.Anderson JG, Ramadori G, Ioris RM, Galiè M, Berglund ED, Coate KC, Fujikawa T, Pucciarelli S, Moreschini B, Amici A, Andreani C, and Coppari R (2015) Enhanced insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6. Mol Metab 4, 846–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maity S, Muhamed J, Sarikhani M, Kumar S, Ahamed F, Spurthi KM, Ravi V, Jain A, Khan D, Arathi BP, Desingu PA, and Sundaresan NR (2020) Sirtuin 6 deficiency transcriptionally up-regulates TGF-β signaling and induces fibrosis in mice. J Biol Chem 295, 415–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu F, Liu C, Zhou D, and Zhang L (2016) TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem 64, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong X, Huang M, Kim HG, Zhang Y, Chowdhury K, Cai W, Saxena R, Schwabe RF, Liangpunsakul S, and Dong XC (2020) SIRT6 Protects Against Liver Fibrosis by Deacetylation and Suppression of SMAD3 in Hepatic Stellate Cells. Cell Mol Gastroenterol Hepatol 10, 341–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Li Y, Liu Q, Huang Y, Li R, Wu T, Zhang Z, Zhou J, Huang H, Tang Q, Huang C, Zhao Y, Zhang G, Jiang W, Mo L, Zhang J, Xie W, and He J (2021) Sirt6 Alleviated Liver Fibrosis by Deacetylating Conserved Lysine 54 on Smad2 in Hepatic Stellate Cells. Hepatology 73, 1140–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Yao ZX, Chen JS, Gi YJ, Muñoz NM, Kundra S, Herlong HF, Jeong YS, Goltsov A, Ohshiro K, Mistry NA, Zhang J, Su X, Choufani S, Mitra A, Li S, Mishra B, White J, Rashid A, Wang AY, Javle M, Davila M, Michaely P, Weksberg R, Hofstetter WL, Finegold MJ, Shay JW, Machida K, Tsukamoto H, and Mishra L (2016) TGF-β/β2-spectrin/CTCF-regulated tumor suppression in human stem cell disorder Beckwith-Wiedemann syndrome. J Clin Invest 126, 527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao S, Yang X, Ohshiro K, Zaidi S, Wang Z, Shetty K, Xiang X, Hassan MI, Mohammad T, Latham PS, Nguyen BN, Wong L, Yu H, Al-Abed Y, Mishra B, Vacca M, Guenigault G, Allison MED, Vidal-Puig A, Benhammou JN, Alvarez M, Pajukanta P, Pisegna JR, and Mishra L (2021) β2-spectrin (SPTBN1) as a therapeutic target for diet-induced liver disease and preventing cancer development. Sci Transl Med 13, eabk2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziebarth JD, Bhattacharya A, and Cui Y (2013) CTCFBSDB 2.0: a database for CTCF-binding sites and genome organization. Nucleic Acids Res 41, D188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massalha H, Bahar Halpern K, Abu-Gazala S, Jana T, Massasa EE, Moor AE, Buchauer L, Rozenberg M, Pikarsky E, Amit I, Zamir G, and Itzkovitz S (2020) A single cell atlas of the human liver tumor microenvironment. Mol Syst Biol 16, e9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowe AR, and Yue W (2019) Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Shukla V, Farci P, Andricovich J, Jogunoori W, Kwong LN, Katz LH, Shetty K, Rashid A, Su X, White J, Li L, Wang AY, Blechacz B, Raju GS, Davila M, Nguyen BN, Stroehlein JR, Chen J, Kim SS, Levin H, Machida K, Tsukamoto H, Michaely P, Tzatsos A, Mishra B, Amdur R, and Mishra L (2017) Loss of the transforming growth factor-β effector β2-Spectrin promotes genomic instability. Hepatology 65, 678–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra A, Yan J, Xia X, Zhou S, Chen J, Mishra L, and Li S (2017) IL6-mediated inflammatory loop reprograms normal to epithelial-mesenchymal transition(+) metastatic cancer stem cells in preneoplastic liver of transforming growth factor beta-deficient β2-spectrin(+/−) mice. Hepatology 65, 1222–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Zaidi S, Rao S, Chen JS, Phan L, Farci P, Su X, Shetty K, White J, Zamboni F, Wu X, Rashid A, Pattabiraman N, Mazumder R, Horvath A, Wu RC, Li S, Xiao C, Deng CX, Wheeler DA, Mishra B, Akbani R, and Mishra L (2018) Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-β Pathway. Gastroenterology 154, 195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 29.Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, and Choudhary C (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 4, 842851. [DOI] [PubMed] [Google Scholar]
- 30.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, and Olsen JV (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ka SO, Bang IH, Bae EJ, and Park BH (2017) Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. Faseb j 31, 3999–4010 [DOI] [PubMed] [Google Scholar]
- 32.Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, Brandis A, Giladi A, Avihail AS, David E, Amit I, and Itzkovitz S (2017) Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, and George J (2004) Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 40, 46–54 [DOI] [PubMed] [Google Scholar]
- 34.Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, Teufel A, Itzel T, Longerich T, Maier O, Fischer R, Kontermann RE, Pfizenmaier K, Schulze-Osthoff K, and Bantel H (2020) TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis 11, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marquardt JU, Fischer K, Baus K, Kashyap A, Ma S, Krupp M, Linke M, Teufel A, Zechner U, Strand D, Thorgeirsson SS, Galle PR, and Strand S (2013) Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology 58, 1054–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai J, Liu Z, Huang X, Shu S, Hu X, Zheng M, Tang C, Liu Y, Chen G, Sun L, Liu H, Liu F, Cheng J, and Dong Z (2020) The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking β-catenin target gene expression. Kidney Int 97, 106–118 [DOI] [PubMed] [Google Scholar]
- 37.Baek HJ, Lee YM, Kim TH, Kim JY, Park EJ, Iwabuchi K, Mishra L, and Kim SS (2016) Caspase-3/7-mediated Cleavage of β2-spectrin is Required for Acetaminophen-induced Liver Damage. Int J Biol Sci 12, 172–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao W, and Espenshade PJ (2012) Expanding roles for SREBP in metabolism. Cell Metab 16, 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang AR, Ferrer CM, and Mostoslavsky R (2020) SIRT6, a Mammalian Deacylase with Multitasking Abilities. Physiol Rev 100, 145–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bird TG, Müller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, Lu WY, Jamieson T, Govaere O, Campbell AD, Ferreira-Gonzalez S, Cole M, Hay T, Simpson KJ, Clark W, Hedley A, Clarke M, Gentaz P, Nixon C, Bryce S, Kiourtis C, Sprangers J, Nibbs RJB, Van Rooijen N, Bartholin L, McGreal SR, Apte U, Barry ST, Iredale JP, Clarke AR, Serrano M, Roskams TA, Sansom OJ, and Forbes SJ (2018) TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, Dooley S, French SW, Mishra L, Petrovic L, Jeong JH, and Machida K (2013) Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest 123, 2832–2849 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Sun H, Peng Z, Tang H, Xie D, Jia Z, Zhong L, Zhao S, Ma Z, Gao Y, Zeng L, Luo R, and Xie K (2017) Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-β signaling in and promote progression of hepatocellular carcinoma. Oncogene 36, 2957–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, Wu SY, Chiang CM, Veenstra TD, and Kemper JK (2010) SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem 285, 33959–33970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng J, Liu C, Hu K, Greenberg A, Wu D, Ausman LM, McBurney MW, and Wang XD (2017) Ablation of systemic SIRT1 activity promotes nonalcoholic fatty liver disease by affecting liver-mesenteric adipose tissue fatty acid mobilization. Biochim Biophys Acta Mol Basis Dis 1863, 2783–2790 [DOI] [PubMed] [Google Scholar]
- 45.Juanola O, Martínez-López S, Francés R, and Gómez-Hurtado I (2021) Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int J Environ Res Public Health 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.