

Abstract
Disorders of sex development (DSD) can be classified as 46,XX DSD, 46,XY DSD, and sex chromosome DSD. Several underlying causes including associated genes have been reported. Steroidogenic factor-1 is encoded by the NR5A1 gene, a crucial regulator of steroidogenesis in the growth of the adrenal and gonadal tissues. It has been discovered to be responsible for 10 to 20% of 46, XY DSD cases. Here, we described a 2-month-old infant who had ambiguous genitalia and 46, XY. Using whole exome sequencing followed by polymerase chain reaction–Sanger sequencing, a novel heterozygous nonsense c.1249C > T (p.Gln417Ter) variant in the NR5A1 gene was identified. It is present in his mother but absent in his father and maternal aunt and uncle. At the age of 7 months, the patient received a monthly intramuscular injection of low-dose testosterone for 3 months in a row. His penile length and diameter increased from 1.8 to 3 cm and from 0.8 to 1.3 cm, respectively. The patient also had normal adrenal reserve function by adrenocorticotropic hormone stimulation test. This study identified a novel causative p.Q417X (c.1249C > T) variant in NR5A1 causing 46,XY DSD in a Thai boy which is inherited from his unaffected mother.
Keywords: disorders of sex development, NR5A1, novel, mutation
Introduction
Disorders of sex development (DSD) or differences of sexual development is a group of condition in which development of chromosomal, gonadal, or anatomical sex is atypical. 1
This condition is an umbrella term that covers a variety of anatomical phenotypes, hormonal phenotypes, gonadal phenotypes, and chromosome complements. 2 Perceptions, approach, and care of individuals with DSD have been updated by Lee et al. 3 DSD can be classified as 46, XX DSD; 46, XY DSD; and sex chromosome DSD. The relationship between genetic factors and DSD has been extensively studied and updated. 2 4 5 However, in ∼50% of 46, XY DSD children, the precise cause could be determined. 6 7 8
Steroidogenic factor-1 (SF-1) is a crucial regulator of steroidogenesis in the growth of the adrenal and gonadal tissues and is encoded by the NR5A1 gene. In males, NR5A1 also controls the expression of the steroidogenic enzymes StAR, CYP11A1, and CYP17A1 necessary for testosterone biosynthesis and the expression of insulin-like polypeptide 3 (INSL3) required for testicular descent. 9 10 Individuals with 46, XY complete gonadal dysgenesis caused by heterozygous mutations in the NR5A1 gene have also been reported to have adrenal insufficiency. 11 12 46,XY DSD patients with NR5A1 mutations may have either normal or low serum anti-Müllerian hormone (AMH) levels. Consequently, Müllerian structures may be seen in individuals with low serum AMH levels. 13 14 The genotype–phenotype correlations in reported cases of NR5A1 mutations have not yet been established and need to be clarified. Here, we describe a case of underdeveloped genitalia in 46, XY with a novel NR5A1 mutation in a Thai family.
Case Presentation
A 2-month-old infant was referred to King Chulalongkorn Memorial Hospital because of ambiguous genitalia. He was born at term from unrelated healthy parents with a birth weight of 4,565 g. He had both palpable gonads in labioscrotal folds with the presence of a urogenital sinus at the perineum upon physical examination. His phallus length and width were 1.5 and 0.5 cm, respectively. The external masculinization score was 3. 15
A chromosome analysis revealed 46, XY. At the age of 2 months, hormonal testing was done, and the results showed that the serum AMH level was 12 ng/mL (40–174) and the basal serum testosterone level was 1.85 ng/mL (0.14–3.63). The intravenous 125 g adrenocorticotropic hormone stimulation test demonstrated a normal increase in serum cortisol levels from 1.76 to 32.4 µg/dL. The uterus was not demonstrated by pelvic ultrasonography. To rule out androgen insensitivity syndrome and 5α-reductase type 2 deficiency, respectively, AR and SRD5A2 mutation analysis was performed. A low-dose testosterone intramuscular injection (Testoviron Depot 15 mg) was administered every month for 3 months starting at the age of 7 months. His penile length and diameter increased from 1.8 to 3 cm and from 0.8 to 1.3 cm, respectively. This suggests a positive response to the testosterone administered. At present, his parents decided to rear him as a boy. This child and his parents have received care from a multidisciplinary team that includes pediatric endocrinologists, geneticists, pediatric surgeons, and child psychiatrists.
Laboratory Investigations
Three milliliters of peripheral blood were drawn from the patient, his parents, and any other available relatives after they gave their informed consent. Using the Puregene blood kit (Qiagen, Hilden, Germany), DNA was extracted from peripheral blood leukocytes. Trio-exome sequencing was performed by Macrogen, Inc. (Seoul, Korea) as previously described. 16 Polymerase chain reaction (PCR)–Sanger sequencing of the 478-bp PCR product with primers (5′-ATGCCCATGTCTTTGATGGT-3′ and 5′-CTCGGTGGGCATCAGAAA-3′) specific to exon 7 of the NR5A1 gene (NM_004959.4) was subsequently performed to confirm the presence of the identified c.1249C > T variant in the patient, his parents, maternal uncle, and aunt.
A novel heterozygous nonsense c.1249C > T (p.Gln417Ter) variant (ENST00000373588.9: NM_004959.5:c.1249C > T [ https://varsome.com/gene/hg38/nr5a1 ]) in exon 7 of NR5A1 was identified in the patient by whole exome sequencing analysis. The NR5A1:c.1249C > T variant is located in the GRCh37/hg19 chr9:127245174 G > A genomic position ( https://gnomad.broadinstitute.org/gene/ENSG00000136931?dataset=gnomad_r2_1 ). The c.1249C > T (p.Gln417Ter) variant showed (ref/alt; 54/46) in the patient and (ref/alt; 68/27) in his mother. In addition, no consanguinity between both parents was calculated by PI_HAT.
PCR–Sanger sequencing confirmed the presence of this mutation in the patient and mother. However, it was absent in the father, the maternal uncle, and the maternal aunt ( Fig. 1 ).
Fig. 1.
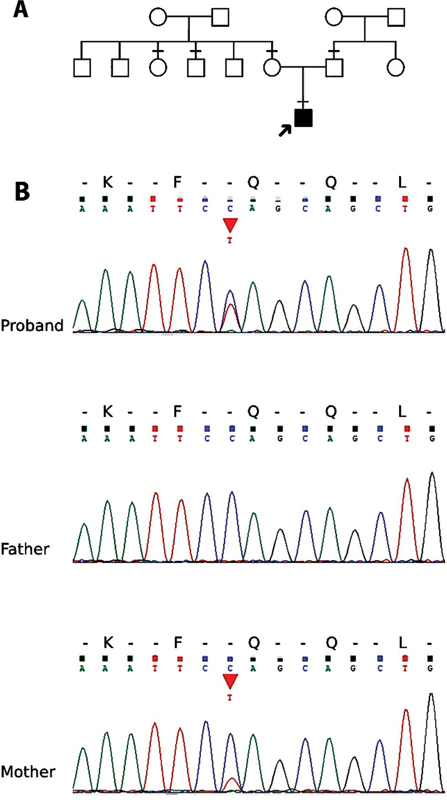
The pedigree and sequencing chromatograms. ( A ) The pedigree of the patient and his family. The horizontal line mark (-) in the pedigree represented the available blood samples in this study. ( B ) The chromatograms showing the c.1249C > T (p.Gln417Ter) variant in the proband and his mother but not in his father.
Discussion
In this study, we report on a child with ambiguous genitalia and preserved adrenal function. Genetic analysis revealed a heterozygous c.1249C > T (p.Gln417Ter) variant inherited from his mother. This causative variant has never been previously described.
The NR5A1 gene, located on chromosome 9q33, has one untranslated exon (exon 1) with six coding exons (exons 2–7). SF-1, 461 amino acid residues, has two zinc finger DNA-binding domains, a ligand-binding domain (LBD), and two functional activation domains. 17 18 19 In 46, XY individuals, SF-1 induces AMH expression in Sertoli cells, which leads to the regression of Müllerian structures and induces the production of steroidogenic enzymes in Leydig cells, causing the virilization of genitalia and testes descending. 20 21 22 23 However, it promotes follicle development and maturation in 46, XX individuals. 24 25 According to some studies, mutations in NR5A1 account for approximately 10 to 20% of 46, XY DSD cases. 26 27 Previous studies had shown phenotypic variations without any genotype–phenotype correlations. 28 29 30 A study of 30 Chinese children with NR5A1 mutations also demonstrated a wide range of external genitalia and identified p.R87C and p.R313C as the most common mutations in this cohort. In addition, exon 4 is the most frequently affected exon in 40% of the patients. 27 They found de novo mutations in 80% of their patients and inherited mutations from either their mothers or fathers in the remaining 20%. According to a study by Tantawy et al, Sertoli cells in people with NR5A1 mutations are more severely impaired than Leydig cells are, and their function declines over time. 26 Therefore, in 46, XY DSD and 46, XX DSD patients, respectively, oligozoospermia or azoospermia and premature ovarian failure may be seen. 26 31 This may be the cause of the low luteinizing hormone (LH)/follicle-stimulating hormone (FSH) ratio seen in people with NR5A1 mutations. Unfortunately, we did not measure LH or FSH to support this hypothesis in our patient. A recent analysis of 188 NR5A1 mutations from 238 cases reported in the literature showed a wide range of different phenotypes without mutation hotspots. The phenotype variation caused by the same mutation can be used to infer the influence of additional genetic modifiers. In 17% of 46,XY cases, the uterus was found. In 25% of 46,XX, the absence of Müllerian derivative was reported. Adrenal insufficiency is undoubtedly uncommon. 32 Previous studies showed that even within the same family, the NR5A1 p.R92W variant causes different levels of masculinization in 46,XX DSD. 33 34 They postulated that the key molecules in the female development pathway would be antagonized by the NR5A1 p.R92W variant, resulting in a decreased inhibition of the male development pathway. 33 35
In this study, the heterozygous c.1249C > T (p.Gln417Ter) variant was identified in the patient and his mother. This variant is located next to the c.1250delA (p.Gln417Argfs*13) that was previously described by Song et al. 27 Both were in the LBD of SF-1. The c.1249C > T (p.Gln417Ter) variant in NR5A1 is predicted to cause 45 amino acid residues missing from the mature protein starting from residue 417. MutationTaster2 predicted it to be deleterious. The VarSite predicted this nonsense variant to be fatal. ESEFinder and MMSplice showed no potential splice sites for 25 nucleotides flanking this nucleotide change.
The mother of our patient is 44 years old, has regular periods, and has no clinical evidence of ovarian failure. A previous study revealed that two from five mothers (40%) with heterozygous NR5A1 mutations experienced premature ovarian failure, but the rest (60%) had fertility preserved. 27 However, time of follow-up is required.
Adrenal insufficiency is uncommon condition found in individuals with NR5A1 mutations. 36 According to a previous study, 10 of the 175 people whose adrenal function was assessed had some degree of adrenal dysfunction. 32 In our patient, a normal adrenal reserve function and normal testosterone level were demonstrated. When testosterone levels were measured in 46, XY DSD with NR5A1 variants, the results were inconsistent. 37 38 Our patient's low AMH level raises the possibility that Sertoli cell function is compromised. However, the 46, XY DSD with the NR5A1 variant cannot be ruled out by normal testosterone and AMH levels. 37
46, XY boys with NR5A1 mutations can have spontaneous puberty and preserve their fertility. 31 39 Our patient responded to exogenous testosterone, suggesting the possibility to achieve the male external genitalia when growing up.
Conclusion
We report a novel p.Q417X (c.1249C > T) mutation in NR5A1 causing 46, XY DSD with normal adrenal function in a Thai boy that is inherited from his unaffected mother.
Acknowledgment
We would like to thank the patient and the family members for participating in this study.
Funding Statement
Funding This work was supported by the Rachadapiseksompotch Fund (RA 59/008), Faculty of Medicine, Chulalongkorn University.
Footnotes
Conflict of Interest None declared.
References
- 1.. International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology . Lee P A, Houk C P, Ahmed S F, Hughes I A. Consensus statement on management of intersex disorders. Pediatrics. 2006;118(02):e488–e500. doi: 10.1542/peds.2006-0738. [DOI] [PubMed] [Google Scholar]
- 2.Parivesh A, Barseghyan H, Délot E, Vilain E. Translating genomics to the clinical diagnosis of disorders/differences of sex development. Curr Top Dev Biol. 2019;134:317–375. doi: 10.1016/bs.ctdb.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Global DSD Update Consortium . Lee P A, Nordenström A, Houk C P et al. Global disorders of sex development update since 2006: perceptions, approach, and care. Horm Res Paediatr. 2016;85(03):158–180. doi: 10.1159/000442975. [DOI] [PubMed] [Google Scholar]
- 4.The EU COST Action . Audi L, Ahmed S F, Krone N et al. GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 ‘DSDnet’. Eur J Endocrinol. 2018;179(04):R197–R206. doi: 10.1530/EJE-18-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Délot E C, Vilain E. Towards improved genetic diagnosis of human differences of sex development. Nat Rev Genet. 2021;22(09):588–602. doi: 10.1038/s41576-021-00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morel Y, Rey R, Teinturier C et al. Aetiological diagnosis of male sex ambiguity: a collaborative study. Eur J Pediatr. 2002;161(01):49–59. doi: 10.1007/s00431-001-0854-z. [DOI] [PubMed] [Google Scholar]
- 7.. LWPES Consensus Group ; ESPE Consensus Group . Hughes I A, Houk C, Ahmed S F, Lee P A. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91(07):554–563. doi: 10.1136/adc.2006.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ata A, Özen S, Onay H et al. A large cohort of disorders of sex development and their genetic characteristics: 6 novel mutations in known genes. Eur J Med Genet. 2021;64(03):104154. doi: 10.1016/j.ejmg.2021.104154. [DOI] [PubMed] [Google Scholar]
- 9.Parker K L, Schimmer B P. Steroidogenic factor 1: a key determinant of endocrine development and function. Endocr Rev. 1997;18(03):361–377. doi: 10.1210/edrv.18.3.0301. [DOI] [PubMed] [Google Scholar]
- 10.Schimmer B P, White P C. Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol. 2010;24(07):1322–1337. doi: 10.1210/me.2009-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Achermann J C, Ito M, Ito M, Hindmarsh P C, Jameson J L. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22(02):125–126. doi: 10.1038/9629. [DOI] [PubMed] [Google Scholar]
- 12.Brandt T, Blanchard L, Desai K et al. 46,XY disorder of sex development and developmental delay associated with a novel 9q33.3 microdeletion encompassing NR5A1. Eur J Med Genet. 2013;56(11):619–623. doi: 10.1016/j.ejmg.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suntharalingham J P, Buonocore F, Duncan A J, Achermann J C. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab. 2015;29(04):607–619. doi: 10.1016/j.beem.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baetens D, Stoop H, Peelman F et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet Med. 2017;19(04):367–376. doi: 10.1038/gim.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed S F, Khwaja O, Hughes I A. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 2000;85(01):120–124. doi: 10.1046/j.1464-410x.2000.00354.x. [DOI] [PubMed] [Google Scholar]
- 16.Chongsrisawat V, Damrongphol P, Ittiwut C, Ittiwut R, Suphapeetiporn K, Shotelersuk V. The phenotypic and mutational spectrum of Thai female patients with ornithine transcarbamylase deficiency. Gene. 2018;679:377–381. doi: 10.1016/j.gene.2018.09.026. [DOI] [PubMed] [Google Scholar]
- 17.Wong M, Ramayya M S, Chrousos G P, Driggers P H, Parker K L. Cloning and sequence analysis of the human gene encoding steroidogenic factor 1. J Mol Endocrinol. 1996;17(02):139–147. doi: 10.1677/jme.0.0170139. [DOI] [PubMed] [Google Scholar]
- 18.Little T H, Zhang Y, Matulis C K et al. Sequence-specific deoxyribonucleic acid (DNA) recognition by steroidogenic factor 1: a helix at the carboxy terminus of the DNA binding domain is necessary for complex stability. Mol Endocrinol. 2006;20(04):831–843. doi: 10.1210/me.2005-0384. [DOI] [PubMed] [Google Scholar]
- 19.Hoivik E A, Lewis A E, Aumo L, Bakke M.Molecular aspects of steroidogenic factor 1 (SF-1) Mol Cell Endocrinol 2010315(1-2):27–39. [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann S, Schwärzler A, Buth S, Engel W, Adham I M. Transcription of the Leydig insulin-like gene is mediated by steroidogenic factor-1. Mol Endocrinol. 1998;12(05):706–713. doi: 10.1210/mend.12.5.0107. [DOI] [PubMed] [Google Scholar]
- 21.Parker K L, Rice D A, Lala D S et al. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. 2002;57:19–36. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]
- 22.Lin L, Achermann J C.Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development Sex Dev 20082(4-5):200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anamthathmakula P, Miryala C SJ, Moreci R S et al. Steroidogenic factor 1 (Nr5a1) is required for sertoli cell survival post sex determination. Sci Rep. 2019;9(01):4452. doi: 10.1038/s41598-019-41051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao H, Li Z, Cooney A J, Lan Z J. Orphan nuclear receptor function in the ovary. Front Biosci. 2007;12(09):3398–3405. doi: 10.2741/2321. [DOI] [PubMed] [Google Scholar]
- 25.Buaas F W, Gardiner J R, Clayton S, Val P, Swain A. In vivo evidence for the crucial role of SF1 in steroid-producing cells of the testis, ovary and adrenal gland. Development. 2012;139(24):4561–4570. doi: 10.1242/dev.087247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tantawy S, Lin L, Akkurt I et al. Testosterone production during puberty in two 46,XY patients with disorders of sex development and novel NR5A1 (SF-1) mutations. Eur J Endocrinol. 2012;167(01):125–130. doi: 10.1530/EJE-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Y, Fan L, Gong C. Phenotype and molecular characterizations of 30 children from China with NR5A1 mutations. Front Pharmacol. 2018;9:1224. doi: 10.3389/fphar.2018.01224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domenice S, Machado A Z, Ferreira F M et al. Wide spectrum of NR5A1-related phenotypes in 46,XY and 46,XX individuals. Birth Defects Res C Embryo Today. 2016;108(04):309–320. doi: 10.1002/bdrc.21145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faienza M F, Chiarito M, Baldinotti Fet al. NR5A1 gene variants: variable phenotypes, new variants, different outcomes Sex Dev 201913(5-6):258–263. [DOI] [PubMed] [Google Scholar]
- 30.Na X, Mao Y, Tang Y et al. Identification and functional analysis of fourteen NR5A1 variants in patients with the 46 XY disorders of sex development. Gene. 2020;760:145004. doi: 10.1016/j.gene.2020.145004. [DOI] [PubMed] [Google Scholar]
- 31.Warman D M, Costanzo M, Marino R et al. Three new SF-1 (NR5A1) gene mutations in two unrelated families with multiple affected members: within-family variability in 46,XY subjects and low ovarian reserve in fertile 46,XX subjects. Horm Res Paediatr. 2011;75(01):70–77. doi: 10.1159/000320029. [DOI] [PubMed] [Google Scholar]
- 32.Fabbri-Scallet H, de Sousa L M, Maciel-Guerra A T, Guerra-Júnior G, de Mello M P. Mutation update for the NR5A1 gene involved in DSD and infertility. Hum Mutat. 2020;41(01):58–68. doi: 10.1002/humu.23916. [DOI] [PubMed] [Google Scholar]
- 33.Members of UDN . Bashamboo A, Donohoue P A, Vilain E et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. 2016;25(16):3446–3453. doi: 10.1093/hmg/ddw186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knarston I M, Robevska G, van den Bergen J A et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum Mutat. 2019;40(02):207–216. doi: 10.1002/humu.23672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Igarashi M, Takasawa K, Hakoda A et al. Identical NR5A1 missense mutations in two unrelated 46,XX individuals with testicular tissues. Hum Mutat. 2017;38(01):39–42. doi: 10.1002/humu.23116. [DOI] [PubMed] [Google Scholar]
- 36.Achermann J C, Ozisik G, Ito M et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. 2002;87(04):1829–1833. doi: 10.1210/jcem.87.4.8376. [DOI] [PubMed] [Google Scholar]
- 37.Coutant R, Mallet D, Lahlou N et al. Heterozygous mutation of steroidogenic factor-1 in 46,XY subjects may mimic partial androgen insensitivity syndrome. J Clin Endocrinol Metab. 2007;92(08):2868–2873. doi: 10.1210/jc.2007-0024. [DOI] [PubMed] [Google Scholar]
- 38.Wu J Y, McGown I N, Lin L et al. A novel NR5A1 variant in an infant with elevated testosterone from an Australasian cohort of 46,XY patients with disorders of sex development. Clin Endocrinol (Oxf) 2013;78(04):545–550. doi: 10.1111/cen.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciaccio M, Costanzo M, Guercio G et al. Preserved fertility in a patient with a 46,XY disorder of sex development due to a new heterozygous mutation in the NR5A1/SF-1 gene: evidence of 46,XY and 46,XX gonadal dysgenesis phenotype variability in multiple members of an affected kindred. Horm Res Paediatr. 2012;78(02):119–126. doi: 10.1159/000338346. [DOI] [PubMed] [Google Scholar]