

Abstract
Copy number variants (CNVs) have been implicated in many human diseases, including psychiatric disorders. Whole genome sequencing offers advantages in CNV calling compared to previous array-based methods. Here we present a robust and transparent CNV calling pipeline, PECAN (PEdigree Copy number vAriaNt calling), for short-read, whole genome sequencing data, comprised of a novel combination of four calling methods and structural variant genotyping. This method is scalable and can incorporate pedigree information to retain lower-confidence CNVs that would otherwise be discarded. We have robustly benchmarked PECAN using gold-standard CNV calls for two well-established evaluation samples, NA12878 and HG002, showing that PECAN performs with high precision and recall on both datasets, outperforming another pedigree-based CNV calling pipeline. As part of this work, we provide a list of high-confidence gold standard CNVs for the NA12878 reference sample, curated from multiple studies. We applied PECAN to a collection of pedigrees multiply affected with schizophrenia and identified a rare deletion that perfectly co-segregates with schizophrenia in one of the pedigrees. The CNV overlaps the gene PITRM1, which has been implicated in a complex phenotype including ataxia, developmental delay, and schizophrenia-like episodes in affected adults.
Subject terms: Bioinformatics, Genomics, Psychiatric disorders, Psychosis, Schizophrenia, Next-generation sequencing
Introduction
Copy number variants (CNVs) are a sub-type of structural variants (SVs) within the genome, usually described as deletions and duplications1. They are typically defined as the difference in the dosage of genomic segments greater than 50 base pairs when compared to a reference genome. As it is estimated that they make up 4.9–9.5% of the human genome2 much work has been done to evaluate their role in disease3–6. Historically, hybridization-based techniques (e.g. array CGH and SNP microarrays) have been used to detect and genotype CNVs7. These methods are highly dependent on the design of the hybridization probes, which tend to be sparse and unevenly distributed across the genome. This makes it difficult to accurately resolve CNV breakpoints, as well as limiting the size of detectable CNVs, and can lead to biases due to uneven genome coverage.
Whole genome sequencing (WGS) technologies can improve calling accuracy8 by identifying discrepancies in either read alignments or read depth to identify putative CNV regions9. Paired-end read (PR) tools detect CNVs by examining where the paired-end reads are significantly different from the expected insert size for a collection of reads, and split read (SR) tools examine where one read in a pair is properly mapped, but its mate does not map or only partially maps10. Read depth (RD) tools examine the number of reads within a region, under the assumption that this is correlated with the copy number of that segment of DNA10. Significant increases or decreases in read depth can indicate the presence of a duplication or deletion event respectively.
To date, no single computational method can detect all CNVs11,12. Combining the output of multiple CNV calling methods can increase the number of true CNVs detected, although this often comes at the expense of increasing the false discovery rate. Consequently, most investigators use a consensus calling strategy, assuming that CNVs called by multiple tools are more likely to be true positives13,14. However, the performance of many consensus methods has either not been formally examined, or has been tested using gold-standard CNVs generated using genotype arrays, which are not directly comparable to WGS data (missing smaller CNVs and lacking accurate breakpoints). A comprehensive evaluation of individual SV calling methods and pairs of methods has shown that different methods are better at detecting different classes of CNVs, with some combinations of pairs of methods performing better than others11. However, the performance of more complex consensus methods has received less attention. A downside of consensus approaches is that, by definition, CNV calls only identified by one tool (solo calls) are automatically excluded. In studies of individuals who are closely related, we might expect the breakpoints for the same CNV to be more comparable than in an unrelated cohort, offering within-family validation. Incorporating pedigree support for solo calls into a consensus may be important in reclaiming CNV calls that would otherwise be lost.
Here, we describe a novel consensus pipeline, PECAN (Pedigree Copy number vAriaNt calling), for CNV calling from short-read whole genome sequencing (WGS) data. This pipeline uses a unique combination of four different calling methods (CNVpytor15, ERDS16, LUMPY17, and Manta18) and structural variant genotyping (SV219). We show that incorporating relatedness information can increase the performance of CNV calling on pedigree-based data. Our method is flexible, transparent, parallelisable and makes use of the latest software versions to improve runtime, allowing scalability for large, unrelated cohorts in addition to pedigree data. We evaluate the performance of PECAN on two sets of curated ‘gold standard’ CNV calls: (1) NA12878 from the CEPH1463 trio20 and (2) HG002 from the Ashkenazim trio21. We compare the performance of our method on these datasets to a previously published CNV calling method for pedigree data22. Finally, we apply our method to WGS data from a collection of pedigrees enriched for schizophrenia23 to identify candidate causal CNVs.
Methods
PECAN: CNV calling and quality control
Taking a consensus of callers both within and across calling classes has previously been shown to improve CNV detection compared to using the tools individually11. PECAN combines two read depth (RD) tools and two paired-read/split-read (PR/SR) tools. The RD tools selected were CNVpytor15 (an updated version of CNVnator24) and ERDS16, as CNVnator and ERDS have been shown to outperform several other RD-based callers for WGS data25. The PR/SR tools selected were LUMPY17 and Manta18. These two tools have been shown to perform well individually and as a pair26. Since both RD tools are known to have reduced performance for CNVs of length less than 1 kb25, such calls were removed from the output of the RD callers.
To reduce the runtime of the CNV calling process, several author-recommended modifications and updates to the original four software tools were considered. We selected CNVpytor over the previous version CNVnator24. For ERDS, we used the “TCAG” code suggested on the GitHub repository (https://github.com/igm-team/ERDS). LUMPY was implemented as part of smoove, which also lowers the false positive rate (https://github.com/brentp/smoove). Finally, we modified the configuration file for Manta to disable remote read retrieval for insertions, as suggested by the authors on the GitHub repository. As insertions are not part of our analysis, this is not expected to impact the performance of Manta. Apart from the above, the four tools were run using the default settings recommended by the authors.
SV219 calculates empirical genotype quality (GQ) scores based on the read data as well as single nucleotide variant (SNV) and indel calls in the CNV region. These scores were calculated for the output calls of each of the four callers. To control false positive CNV calls while maximising retention of true positive CNVs, we removed those with GQ = 0.
We observed that the raw output of the four calling tools sometimes generated CNV calls that were tens or hundreds of mega base pairs (Mbps) long. This was more prevalent in the PR/SR callers. Since these CNVs most likely represent false positive calls, we removed CNVs greater than a pre-specified length, to further improve the runtime of PECAN. As the largest CNV in the gnomAD database is 28.5 Mbps in length27, we removed CNV calls that were longer than 30 Mbps. This aided the SV2 genotyping, as reads underlying a CNV region are evaluated when assigning genotype quality scores.
Combining CNV calls
During the preliminary examination of the four calling methods, we noted that several overlapping CNV regions were called, which likely represent the same CNV, and was more of an issue with the two PR/SR callers. To resolve this issue, we implemented a collapsing strategy to identify sets of equivalent CNVs (see Supplementary Fig. S1), comparable to that described in Trost et al.25. Briefly, the overlapping regions were collapsed to a single set as follows: (i) if two CNVs of the same type (either deletion or duplication) overlap reciprocally by at least 25%, then they are added to the same set. (ii) If only one of the two CNVs is already in a set, then the other is added to that set. If both CNVs are already in sets, then the two sets are combined. (iii) Once all sets have been created, each set is collapsed down to one region by taking the union of all CNVs within the set.
A consensus CNV call across all tools is generated by merging calls of the same type (deletion or duplication) that overlap reciprocally by 50%, first considering calls within calling method types (CNVpytor vs ERDS, and LUMPY vs Manta), and then across the resulting calling method types (PR/SR vs RD). This merging strategy allows for differences in the ability of each of the four calling methods to define the breakpoints of a CNV, allowing the same CNV, albeit with variable breakpoints, to be identified across methods. Lastly, as recommended by Trost et al.25, CNV calls for which over 75% of their length comprise of repeat/low-complexity regions (RLCR) were removed. RLCRs were defined as: (i) assembly gaps, (UCSC “gap”' table); (ii) segmental duplications (UCSC “genomicSuperDups” table); and (iii) the pseudo-autosomal regions of the sex chromosomes. A workflow diagram for PECAN is shown in Fig. 1.
Figure 1.

Workflow of PECAN per individual. Input for PECAN consists of a BAM file and a VCF file of SNVs and indels. RD callers are shown in green (ERDS and CNVpytor), and PR/SR callers are shown in red (LUMPY and Manta). The SV2 genotyper is shown in purple. RLCR: repeat/low-complexity region.
Incorporating family information
All calls within a pedigree were combined, taking the union of calls of the same type (deletion or duplication) with 50% reciprocal overlap. Following Khan et al., solo calls that were not detected by at least two callers in any of the individual’s direct relatives were removed22. This ensured that the final list of CNVs for any individual in the pedigree either had support from at least two calling methods or was also present with confidence in a relative. The kinship2 package from R was used to determine whether two samples were related or not based on the pedigree structure28.
WGS data for reference samples
To evaluate PECAN, we took advantage of two publicly available and commonly used reference trios: a subset of the CEPH 1463 pedigree (proband: NA12878; father: NA12891; mother: NA12892); and the Ashkenazim trio (proband: HG002; father: HG003; mother: HG004) from the Genome in a Bottle (GIAB) consortium21. FASTQ files for the CEPH 1463 trio were downloaded from the Illumina Platinum Genomes project20. BAM files for the Ashkenazim trio were obtained from the GIAB FTP site, and were reverted to FASTQ files as described previously23. Reads for all six samples were aligned to the GRCh38 reference genome using the bwa-mem algorithm29, and each sample had an average depth of coverage of approximately 50 ×. Standard read processing following the GATK ‘Best Practices’ was applied, which involved marking duplicate reads, local read re-alignment around indels, and base quality score recalibration30. SNVs and indels were called from the BAM files for each of the six samples.
Curated gold standard CNV calls
Despite the extensive study of sample NA12878, it is unlikely that all true CNVs in their genome have been identified, since no single technology can detect all CNVs and there is a highly variable degree of concordance between CNV calling algorithms across different sequencing technologies (short-read sequencing, long-read sequencing, optical genome mapping, etc.)31. With this in mind, we created a high confidence set of calls by examining five studies which published lists of ‘gold standard’ NA12878 CNV calls to build our own gold standard dataset (Supplementary Table 1). To maximise both size and veracity of our gold standard CNV calls (i.e., maximise the number of true positives), we chose to include CNVs present in two or more of the individual studies. CNVs present in only one study are not considered to be gold standard calls as they are more likely to be false positive calls. As a quality control measure, we removed duplicates taken from the same source dataset (for example, the 1000 Genomes Project NA12878 CNVs present in both the DGV and Kosugi et al. datasets) and CNVs with contradictory classes across datasets (e.g., annotated as a deletion in one dataset and a duplication in a second). As many of the individual datasets only include deletions and the number of duplications were extremely limited (the total number of duplications was less than 5% of the total number of deletions, see Supplementary Fig. S2), we only included deletions in our gold standard data. For the HG002 reference sample, we used the Tier 1 v0.6 calls from the Genome in a Bottle consortium as our gold standard CNV calls21. From the VCF file, we retained simple deletions and contractions that passed quality control filters. As a supplementary benchmark, we also extracted duplications for HG002 that passed quality control filters and were annotated as not overlapping with a tandem repeat locus (TRall = "FALSE"). These two gold standard CNV datasets constitute the True Positives (TP) in our benchmarking analysis. Since our schizophrenia pedigrees had been previously investigated using data aligned to GRCh38, we lifted the gold standard datasets to GRCh37 with liftOver32. Any CNV present in the output data that are not TPs are considered False Positives (FP). Similarly, any CNVs from the gold standard datasets that are not present in the output are considered False Negatives (FN).
Benchmarking
Benchmarking was performed using the query CNV sets generated by: (i) PECAN; (ii) the four individual callers that are used in PECAN; and (iii) the pedigree CNV calling method by Khan et al.22. To evaluate the performance of the CNV calling methodologies, we calculated the precision and recall of the output CNVs relative to the high-confidence gold standard CNV calls for NA12878 and HG002. Here, we define the recall as the proportion of the gold standard CNV calls identified in the query CNV call set (i.e., TP / (TP + FP)), and we define the precision as the proportion of the query CNV call set that are found in the gold standard CNV calls (i.e., TP / (TP + FN)).
WGS data from Utah pedigrees multiply affected with schizophrenia
As an application of PECAN, we called CNVs from WGS data on 35 samples across six Utah pedigrees multiply affected with schizophrenia. Details of the cohort, phenotypes, sample selection, and sequencing have been described previously23. Sequencing reads were aligned to the GRCh38 reference genome, and SNVs and indels were called following the GATK ‘Best Practices’30. CNVs were manually annotated if they were private to one of the pedigrees, i.e., there was no variant of the same type with a 50% reciprocal overlap present in another pedigree. Secondly, CNVs were annotated based on their co-segregation pattern using the FilterVcf module from picard with custom JavaScript code. We annotated CNVs with a full co-segregation pattern (carried by all schizophrenia-affected samples in-family and absent from both unaffected and marry-in samples) or a reduced co-segregation pattern (carried by all but one schizophrenia-affected samples in-family and absent from both unaffected and marry-in samples).
Next, CNV allele frequencies from v4.127 were annotated with SVAFotate33, using the supplied allele frequency databases from GRCh38 and taking a 50% reciprocal overlap. Finally, CNVs were annotated using AnnotSV34, again taking a 50% reciprocal overlap. The MANE transcript was selected when multiple transcripts were present35. Of particular interest from AnnotSV was the implementation of the American College of Medical Genetics and Genomics (ACMG) CNV clinical significance ranking36. Prioritised CNVs were visualised by examining the raw sequencing reads using samplot37. CNVs were rejected if the sequencing read profiles were not consistent with the presence/absence of a CNV call across the pedigree samples.
Results
Calling CNVs from NA12878 and HG002 using PECAN
PECAN called 2312 deletions and 166 duplications for NA12878 (Supplementary Fig. S2), which increased to 2375 deletions and 182 duplications when family information was incorporated. For HG002, PECAN called 2352 deletions and 82 duplications (Supplementary Fig. S3), which increased to 2437 deletions and 100 duplications with family information. With optimisations for speed, PECAN took on average 10.2 h to run on an Intel Xeon Gold 6130 server with four CPU cores per individual (see Supplementary Table S2).
Curation of the gold standard datasets
Across the five NA12878 CNV datasets, 2505 deletions were present in at least two datasets (Supplementary Fig. S4). This set of CNVs was used as the high-confidence NA12878 CNVs and is available as part of this publication (see the supporting GitHub repository). Of these, 726 were greater than 1 kb in length, which is the recommended length for the RD callers25. Therefore, to assess performance we calculated metrics based on the full NA12878 CNV set, the subset greater than 1 kb and the subset less than 1 kb in length. For the HG002 sample, 5,141 deletions were retained in the gold standard CNV calls, of which 498 were greater than 1 kb in length. We extracted a total of 529 duplications for this sample, of which twelve were greater than 1 kb in length.
Performance on gold standard datasets
The overall performance of PECAN on the gold standard deletion calls (both with and without pedigree information) is shown in Fig. 2, with both achieving precision scores of approximately 80% on both NA12878 and HG002 datasets, higher than any of the individual callers (Supplementary Fig. S5). In contrast, the method presented by Khan et al. had less than 50% precision on both datasets. Similarly, the recall of PECAN on the > 1 kb deletions across both datasets exceeded 77%, and the recall on the NA12878 < 1 kb deletions was comparable at 72% (Supplementary Fig. S6). While the recall on the HG002 < 1 kb deletions was lower at 32%, this was still markedly better than that of Khan et al. on the same sample (3% recall). Overall, PECAN displayed substantial improvement in performance compared to that described by Khan et al., both with and without the inclusion of pedigree information.
Figure 2.

Deletion calling performance on the two reference samples NA12878 and HG002 split by CNV length. This plot shows the performance metrics (precision and recall) for: PECAN with the pedigree information (TRIO); PECAN without the pedigree information (INDIVIDUAL); and the CNV pipeline described by Khan et al.
When we included family information, the recall of PECAN increased by 1.6–2.9% across the reference samples. We also examined the precision of the solo calls reclaimed by family information, which accounted for approximately 8–14% of all solo calls. While the precision of all solo calls was modest (12.7–40.8%), it was noticeably increased in the reclaimed solo calls (47.5–65.9%) (Fig. 3). This shows that incorporating family information can help retain true positive CNV calls while keeping some control over the false discovery rate.
Figure 3.

Investigating the value of the reclaimed solo calls. Precision values for PECAN on (a) NA12878 and (b) HG002, broken down by the evidence level of the CNV. CNVs were called by multiple callers (Consensus) or by one caller (Solo CNV). Solo calls with support from pedigree members were reclaimed, and those with no pedigree support were lost.
PECAN performed modestly on the duplications (Supplementary Fig S6), achieving an overall recall of 2.5% and recall of 15.9%. Including pedigree information resulted in a slight improvement of the recall to 2.8%. In comparison, the pipeline of Khan et al. achieved similar recall of 2.3%, but with a reduced precision of 2.2%.
Application: pedigrees enriched for schizophrenia
We used PECAN to identify CNVs from WGS data for 35 samples across six pedigrees from Utah multiply affected with schizophrenia (Supplementary Table S3). Across the six pedigrees, PECAN identified 6524 deletions and 895 duplications. We prioritised family-private CNVs with a full or reduced co-segregation pattern that were rare in gnomAD (allele frequency < 1%) and predicted to be pathogenic or likely pathogenic (see Methods). Five deletions survived this filtering strategy, two with a full co-segregation pattern and three with a reduced co-segregation pattern. A breakdown of the deletion and duplication counts for the pedigree CNVs is given in Supplementary Table S4. Following manual inspection of the sequencing reads, two deletions were removed as the sequencing read profiles for both indicated that each of the CNVs was likely carried by an unaffected or marry-in sample. Details for the three remaining CNVs are given in Supplementary Table S5, and pedigree diagrams for the two families carrying the three deletions are shown in Fig. 4 and Supplementary Fig. S7.
Figure 4.
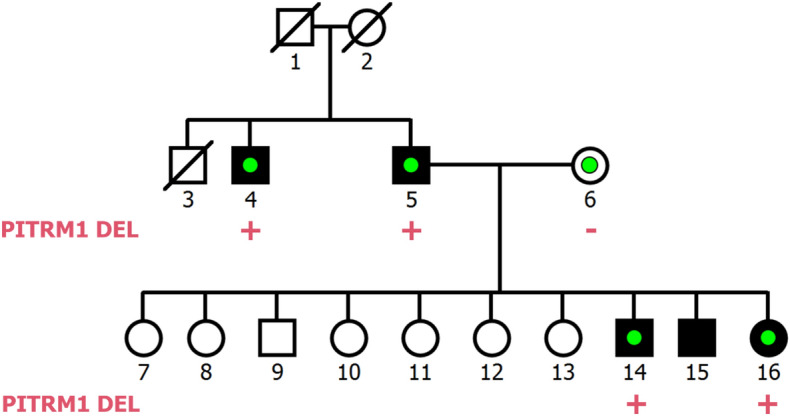
Pedigree image for family K1494. Fully shaded boxes denote samples with schizophrenia and the green dot indicates samples selected for WGS. The PITRM1 DEL carrier status is indicated under each sequenced sample (“+” for carrier or “−” for non-carrier). DEL deletion.
Discussion
We have developed PECAN, a novel consensus CNV calling pipeline for short-read WGS data, combining four CNV callers (two paired-end/split-read and two read-depth approaches) with SV genotyping. We have utilised both the latest software and recommended modifications/settings to enable PECAN to run more quickly than with older versions of the individual tools. We used empirical genotype quality scores to help control the number of false positive CNV calls from the four callers. While we selected the lowest possible genotype quality threshold for filtering, increasing this threshold results in small gains of precision at the expense of substantial losses in recall (see Supplementary Fig. S8). In addition, we have shown that incorporating pedigree information can provide support for lower-confidence CNV calls that would otherwise be discarded. An important factor when combining CNV calls generated by different tools is how to combine them to represent a set of unique, non-overlapping CNVs. This issue is under active development in the field of CNV calling, with individual research groups making decisions on how to achieve this38. In addition to the unique combination of steps described above, our method also benefits from a within tool and within individual CNV collapsing strategy to remove overlapping CNV calls representing the same CNV. While we have developed PECAN with human genomes in mind, it can also be applied to WGS data from other species where high-quality reference genomes are available.
Another important factor when performing any benchmarking analysis is the quality of the reference data. When curating our own gold standard CNV call set for NA12878, we chose to look for CNVs that had support across multiple NA12878 reference datasets and found little overlap existed across the selected studies (Supplementary Fig. S4). One explanation for this is that CNVs were called with different technologies across the five call sets and using different data types (SNP genotype arrays, aCGH, cytogenic techniques, short-/long-read sequencing, etc.), which might have led to the detection of different subsets of CNVs. This is a reminder that all that glitters is not gold standard and caution should be taken when selecting a gold standard dataset for benchmarking. To that end, we feel that the gold standard CNV calls we have used here (available with this publication) are of higher confidence than many other NA12878 datasets because they have support from multiple different, independent sources.
We have shown that our consensus method performs well, with high recall and precision on two independent gold standard CNV datasets. The recall is lower for sample HG002 (37%) than for sample NA12878 (76%), but this difference is driven by the CNVs of length less than or equal to 1kb (Supplementary Fig. S6). HG002 has a high recall on the subset of CNVs of length greater than 1kb (78%). One main difference between the gold standard CNV calls for the two samples is that long-read sequencing was more prevalent in the construction of the HG002 calls compared to the NA12878 calls. Long-read sequencing discovers nearly twice the number of structural variants compared to short-read sequencing39, so this may explain the modest recall achieved by PECAN on the shorter CNVs from HG002.
As part of our benchmarking, we have compared the performance of PECAN to a different CNV calling pipeline for pedigrees developed by Khan et al.22. To the best of the authors knowledge, this is the only other pedigree-based calling strategy available for this kind of data. We have shown that PECAN outperforms Khan et al.’s pipeline on both reference samples. One reason for this might be the selection of tools used, as some tools are known to perform better together than others11. While the inclusion of pedigree data did not dramatically improve the recall of our method on the gold standard data compared to not including the pedigree data, the reference families are trios rather than more complex multi-generational pedigree data. Considering the precision for the reclaimed solo calls was 1.6–3.7 times larger than that of all solo calls (Fig. 3), incorporating family information allows the retention of additional true positive CNV calls (thus improving recall), while keeping some control over the false discovery rate. Indeed, relaxing the pedigree inclusion criteria further in an attempt to improve recall results in noticeable increase in the false discovery rate (see Supplementary Fig. S6). We therefore think that inclusion of pedigree information when investigating large, complex pedigrees could achieve a greater improvement in recall than seen in the trios.
One limitation of PECAN is the apparent modest performance on the gold-standard duplication calls for HG002 (Supplementary Fig. S7). However, short-read WGS can identify over twice the number of duplications per genome than those observed in the gold standard set27,40, and long-read WGS can identify larger numbers again41. As such, the gold standard duplications considered here likely do not fully reflect all duplications in the evaluation sample’s genome. We therefore advise caution when interpreting these results, as the metrics may be not fully reflective on the actual performance of PECAN. In general, calling duplications from short-read WGS data is known to be challenging, and individual tools often suffer from limited true positive discovery in an effort to control false positives11.
As an application of PECAN, we called CNVs using WGS data from six pedigrees with multiply affected individuals with schizophrenia, in which rare SNVs had been previously investigated23. Functional prioritisation identified three rare, family-private, likely pathogenic deletions that co-segregate with schizophrenia (see Supplementary Table S4). Of note was a 3.2 kb deletion at 10p15.2 in pedigree K1494, carried by all four sequenced schizophrenia samples and absent from the unaffected marry-in sample (see Fig. 4 and Supplementary Table S5). This CNV overlaps an intron–exon junction of PITRM1 and was ranked by AnnotSV as ‘likely pathogenic’. The other two deletions had reduced co-segregation patterns, and so show less evidence of association with schizophrenia in these pedigrees (Supplementary Fig. S1).
PITRM1 (Pitrilysin Metallopeptidase 1) encodes an ATP-dependent metalloprotease that is known to degrade post-cleavage mitochondrial transit peptides42. This protein is known to be expressed across multiple human tissue types including brain tissues43, and has previously been shown to degrade the amyloid-β protein, suggesting a role in Alzheimer’s disease and neurodegeneration44. In the ClinVar database45, pathogenic SNVs in PITRM1 have been reported for autosomal recessive spinocerebellar ataxia. At least four independent affected families have been identified with deleterious PITRM1 SNVs and a core phenotype which includes developmental delay, ataxia, and seizures46. In the two families where the affected individuals have reached adulthood, they have developed schizophrenia-like symptoms and other psychiatric features47,48. While PITRM1 has not yet been implicated in schizophrenia from large-scale rare-variant49 or common variant studies50 our analysis indicates that this is a plausible candidate for involvement in schizophrenia and psychosis etiology51.
In conclusion, we have developed a novel consensus CNV calling pipeline, PECAN, which carefully balances CNV discovery over control of false positives. The method is flexible and can be applied to both related and unrelated cohorts. By making use of the latest versions of well-known, frequently used CNV calling tools, we have streamlined our pipeline to run more quickly than older versions of the individual tools, making it scalable for larger cohorts. We have shown that incorporating family-based information can help validate lower confidence calls that did not achieve a consensus, further improving our ability to identify potentially pathogenic CNVs from pedigree data. By performing robust benchmarking of our method, we have a good understanding of its performance and have shown that it outperforms another method for investigating CNVs in pedigrees22. We provide the NA12878 gold standard data as part of our publication to allow for fair and open comparison against other methods in the future. Lastly, by applying our method to a collection of pedigrees we have identified a deletion perfectly co-segregating with schizophrenia overlapping a gene that has previously been implicated in families with a complex phenotype with neurological and psychiatric symptoms, including psychosis, a core feature of schizophrenia.
Supplementary Information
Acknowledgements
The authors acknowledge the support of the Trinity Centre for High Performance Computing (ResearchIT).
Author contributions
CO and NMR contributed equally to the conceptualisation, investigation, and methodology of this project, with equal contribution of project supervision and support from EH and AC. CO generated the pipeline code, with support from NMR. NMR generated the NA12878 dataset, with support from CO. WB provided the schizophrenia pedigrees for sequencing and CO analysed these data. CO and NMR contributed equally to the original draft of the manuscript. CO, NMR, EH and AC all equally contributed to the editing of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Institutes of Health [5U01MH 109499-04 and 5R01MH124875 to A.C.]; and Science Foundation Ireland [16/SPP/3324 to A.C.].
Data availability
The source code for PECAN is available on GitHub at https://github.com/cathaloruaidh/PECAN_calling and can be freely accessed. The source code for the read alignment and short variant calling pipeline is available on GitHub at https://github.com/cathaloruaidh/WGS_Alignment_Calling and can be freely accessed. The WGS data for the CEPH1463 trio is available at the European Nucleotide Archive and can be accessed with the accession number PRJEB3381. The BAM files for the Ashkenazim trio are available from the Genome in a Bottle FTP site at https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/. All code and benchmarking data, including the curated list of high-confidence, gold standard CNV calls for NA12878 are available on GitHub at https://github.com/cathaloruaidh/PECAN_Paper_Data/ and can be freely accessed.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Cathal Ormond and Niamh M. Ryan.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-66021-0.
References
- 1.Shaikh, T. H. Copy number variation disorders. Curr. Genet. Med. Rep.5(4), 183–190 (2017). 10.1007/s40142-017-0129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zarrei, M., MacDonald, J. R., Merico, D. & Scherer, S. W. A copy number variation map of the human genome. Nat. Rev. Genet.16(3), 172–183 (2015). 10.1038/nrg3871 [DOI] [PubMed] [Google Scholar]
- 3.Girirajan, S., Campbell, C. D. & Eichler, E. E. Human copy number variation and complex genetic disease. Annu. Rev. Genet.45(45), 203–226 (2011). 10.1146/annurev-genet-102209-163544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stankiewicz, P. & Lupski, J. R. Structural variation in the human genome and its role in disease. Annu. Rev. Med.61, 437–455 (2010). 10.1146/annurev-med-100708-204735 [DOI] [PubMed] [Google Scholar]
- 5.Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J. O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet.14(2), 125–138 (2013). 10.1038/nrg3373 [DOI] [PubMed] [Google Scholar]
- 6.Shil, A. et al. Comparison of three bioinformatics tools in the detection of ASD candidate variants from whole exome sequencing data. Sci. Rep.13(1), 18853 (2023). 10.1038/s41598-023-46258-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet.12(5), 363–376 (2011). 10.1038/nrg2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou, B. et al. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J. Med. Genet.55(11), 735–743 (2018). 10.1136/jmedgenet-2018-105272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao, M., Wang, Q., Wang, Q., Jia, P. & Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform.14(Suppl 11), S1 (2013). 10.1186/1471-2105-14-S11-S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pirooznia, M., Goes, F. S. & Zandi, P. P. Whole-genome CNV analysis: Advances in computational approaches. Front. Genet.6, 138 (2015). 10.3389/fgene.2015.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kosugi, S. et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol.20(1), 117 (2019). 10.1186/s13059-019-1720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarwal, V. et al. A comprehensive benchmarking of WGS-based deletion structural variant callers. Brief Bioinform.23(4), 221 (2022). 10.1093/bib/bbac221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedrich, S., Barbulescu, R., Helleday, T. & Sonnhammer, E. L. L. MetaCNV: A consensus approach to infer accurate copy numbers from low coverage data. BMC Med. Genomics13(1), 76 (2020). 10.1186/s12920-020-00731-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarate, S. et al. Parliament2: Accurate structural variant calling at scale. Gigascience9(12), 145 (2020). 10.1093/gigascience/giaa145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suvakov, M., Panda, A., Diesh, C., Holmes, I. & Abyzov, A. CNVpytor: A tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing. Gigascience10(11), 074 (2021). 10.1093/gigascience/giab074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu, M. et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am. J. Hum. Genet.91(3), 408–421 (2012). 10.1016/j.ajhg.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol.15(6), R84 (2014). 10.1186/gb-2014-15-6-r84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics32(8), 1220–1222 (2016). 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- 19.Antaki, D., Brandler, W. M. & Sebat, J. SV2: Accurate structural variation genotyping and de novo mutation detection from whole genomes. Bioinformatics34(10), 1774–1777 (2018). 10.1093/bioinformatics/btx813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eberle, M. A. et al. A reference data set of 54 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res.27(1), 157–164 (2017). 10.1101/gr.210500.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zook, J. M. et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data3, 160025 (2016). 10.1038/sdata.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan, F. F. et al. Whole genome sequencing of 91 multiplex schizophrenia families reveals increased burden of rare, exonic copy number variation in schizophrenia probands and genetic heterogeneity. Schizophr. Res.197, 337–345 (2018). 10.1016/j.schres.2018.02.034 [DOI] [PubMed] [Google Scholar]
- 23.Ormond, C. et al. Ultra-rare missense variants implicated in Utah pedigrees multiply affected with schizophrenia. Biol. Psychiatry7, 797–802 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res.21(6), 974–984 (2011). 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trost, B. et al. A comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am. J. Hum. Genet.102(1), 142–155 (2018). 10.1016/j.ajhg.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong, T., Hayes, V. M. & Chan, E. K. F. Detection of somatic structural variants from short-read next-generation sequencing data. Brief Bioinform.22(3), 056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins, R. L. et al. A structural variation reference for medical and population genetics. Nature581(7809), 444–451 (2020). 10.1038/s41586-020-2287-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinnwell, J. P., Therneau, T. M. & Schaid, D. J. The kinship2 R package for pedigree data. Hum. Hered.78(2), 91–93 (2014). 10.1159/000363105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM (Springer, 2013). [Google Scholar]
- 30.Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform.43, 11101–11133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaisson, M. J. P. et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun.10(1), 1784 (2019). 10.1038/s41467-018-08148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haeussler, M. et al. The UCSC genome browser database: 2019 update. Nucleic Acids Res.47(D1), D853-d858 (2019). 10.1093/nar/gky1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicholas, T. J., Cormier, M. J. & Quinlan, A. R. Annotation of structural variants with reported allele frequencies and related metrics from multiple datasets using SVAFotate. BMC Bioinform.23(1), 490 (2022). 10.1186/s12859-022-05008-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geoffroy, V. et al. AnnotSV and knotAnnotSV: A web server for human structural variations annotations, ranking and analysis. Nucleic Acids Res.49(W1), W21-w28 (2021). 10.1093/nar/gkab402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morales, J. et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature604(7905), 310–315 (2022). 10.1038/s41586-022-04558-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med.22(2), 245–257 (2020). 10.1038/s41436-019-0686-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belyeu, J. R. et al. Samplot: A platform for structural variant visual validation and automated filtering. Genome Biol.22(1), 161 (2021). 10.1186/s13059-021-02380-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirsche, M. et al. Jasmine and Iris: Population-scale structural variant comparison and analysis. Nat. Methods20(3), 408–417 (2023). 10.1038/s41592-022-01753-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao, X. et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet.108(5), 919–928 (2021). 10.1016/j.ajhg.2021.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werling, D. M. et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet.50(5), 727–736 (2018). 10.1038/s41588-018-0107-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavrichenko, K., Johansson, S. & Jonassen, I. Comprehensive characterization of copy number variation (CNV) called from array, long- and short-read data. BMC Genomics22(1), 826 (2021). 10.1186/s12864-021-08082-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res.44(D1), D733–D745 (2016). 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lonsdale, J. The genotype-tissue expression (GTEx) project. Nat. Genet.45(6), 580–585 (2013). 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King, J. V. et al. Molecular basis of substrate recognition and degradation by human presequence protease. Structure22(7), 996–1007 (2014). 10.1016/j.str.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landrum, M. J. et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res.46(D1), D1062-d1067 (2018). 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tolomeo, D. et al. Learning from massive testing of mitochondrial disorders: UPD explaining unorthodox transmission. J. Med. Genet.58(8), 543–546 (2021). 10.1136/jmedgenet-2020-107644 [DOI] [PubMed] [Google Scholar]
- 47.Brunetti, D. et al. Defective PITRM1 mitochondrial peptidase is associated with Aβ amyloidotic neurodegeneration. EMBO Mol. Med.8(3), 176–190 (2016). 10.15252/emmm.201505894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langer, Y. et al. Mitochondrial PITRM1 peptidase loss-of-function in childhood cerebellar atrophy. J. Med. Genet.55(9), 599–606 (2018). 10.1136/jmedgenet-2018-105330 [DOI] [PubMed] [Google Scholar]
- 49.Singh, T. et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature604(7906), 509–516 (2022). 10.1038/s41586-022-04556-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature604(7906), 502–508 (2022). 10.1038/s41586-022-04434-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coe, B. P. et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet.46(10), 1063–1071 (2014). 10.1038/ng.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code for PECAN is available on GitHub at https://github.com/cathaloruaidh/PECAN_calling and can be freely accessed. The source code for the read alignment and short variant calling pipeline is available on GitHub at https://github.com/cathaloruaidh/WGS_Alignment_Calling and can be freely accessed. The WGS data for the CEPH1463 trio is available at the European Nucleotide Archive and can be accessed with the accession number PRJEB3381. The BAM files for the Ashkenazim trio are available from the Genome in a Bottle FTP site at https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/. All code and benchmarking data, including the curated list of high-confidence, gold standard CNV calls for NA12878 are available on GitHub at https://github.com/cathaloruaidh/PECAN_Paper_Data/ and can be freely accessed.