

SUMMARY
Interferon-gamma (IFNG) has pleiotropic effects on cancer immune checkpoint blockade (ICB), including roles in ICB resistance. We analyzed gene expression in ICB-sensitive versus ICB-resistant tumor cells and identified a strong association between interferon-mediated resistance and expression of Ripk1, a regulator of tumor necrosis factor (TNF) superfamily receptors. Genetic interaction screening revealed that in cancer cells, RIPK1 diverted TNF signaling through NF-kB and away from its role in cell death. This promoted an immunosuppressive chemokine program by cancer cells, enhanced cancer cell survival, and decreased infiltration of T and NK cells expressing TNF superfamily ligands. Deletion of RIPK1 in cancer cells compromised chemokine secretion, decreased ARG1+ suppressive myeloid cells linked to ICB failure in mice and humans, and improved ICB response driven by CASP8-killing and dependent on T and NK cells. RIPK1-mediated resistance required its ubiquitin scaffolding but not kinase function. Thus, cancer cells co-opt RIPK1 to promote cell-intrinsic and cell-extrinsic resistance to immunotherapy.
eTOC BLURB
Interferon signaling in cancer cells can promote resistance to immune checkpoint blockade. Cucolo et al. describe how RIPK1 participates in IFN-driven resistance by regulating the consequences of signaling through TNF family of receptors. Deletion of RIPK1 and interference with its scaffolding function disables cancer cell survival signals, favors T and NK cell killing, and decreases chemokines that regulate suppressive macrophages.
Graphical Abstract
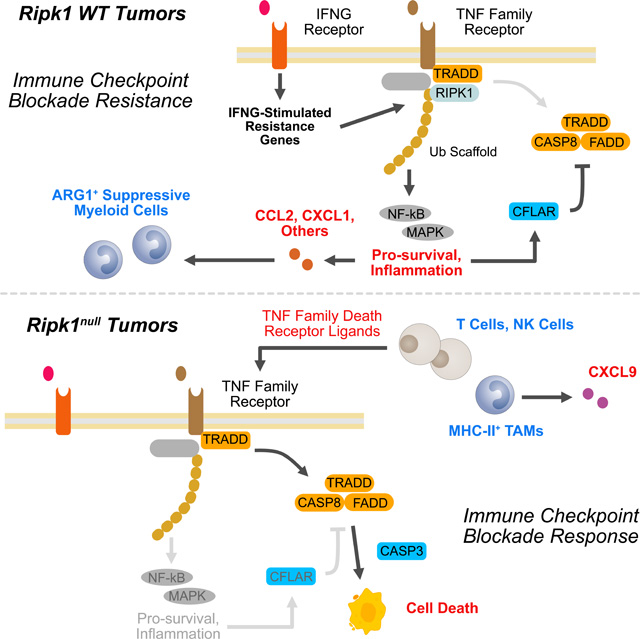
INTRODUCTION
Immune checkpoint blockade (ICB) therapy can result in impressive clinical responses of multiple cancer types, however resistance to this promising therapy is common (Hodi et al., 2010; Patel and Minn, 2018). Mechanisms of resistance can be broadly divided into cancer cell-intrinsic and extrinsic. Intrinsic mechanisms include those that interfere with tumor cell killing, such as loss of MHC-I or a paucity of high-affinity neoantigens (Sharma et al., 2017), and alteration of cancer cell signaling pathways, such as WNT, that can interfere with immune infiltration (Spranger et al., 2015). Other intrinsic mechanisms include the expression of immune inhibitory molecules by cancer cells such as PDL1, which antagonizes the function of multiple immune cell types (Freeman et al., 2000). Cancer cell extrinsic mechanisms often involve features of the tumor immune landscape. This includes poor infiltration of cytotoxic T cells and NK cells, and the accumulation of suppressive myeloid cells, which often belong to ill-defined subsets (Veglia et al., 2018).
Although intrinsic and extrinsic mechanisms of ICB resistance can be distinct, the underlying pathways that control each may overlap. This makes the identification of common pathways an important goal. One pathway that can influence ICB resistance is the interferon (IFN) pathway. Typically considered a pathway that promotes ICB response (Zaretsky et al., 2016), IFN can also orchestrate ICB resistance as a consequence of chronic stimulation of cancer cells (Benci et al., 2016). In such situations, blocking cancer cell signaling through either type one IFN (IFN-I) or IFN-gamma (IFNG) restores ICB responses. This is associated with cancer cell extrinsic effects such as enhanced immune infiltration and increased IFN production and signaling by immune cells. Cancer cell intrinsic changes that contribute to restored anti-tumor immunity include diminished expression of PDL1 and multiple other ligands for immune inhibitory receptors. Moreover, inhibiting cancer cell IFN signaling can also enhance perforin-independent killing through death receptors belonging to the tumor necrosis factor (TNF) family (Benci et al., 2019). Mechanistically, which IFN-stimulated genes (ISGs) control resistance and how these cancer cell ISGs might influence extrinsic effects on the immune microenvironment and/or intrinsic effects through pathways such as TNF signaling remain unclear.
Activation of the TNF receptor (TNFR1) and other members of the TNF receptor superfamily (TNFRSF) can result in two opposing downstream events that are tightly regulated through checkpoints (Annibaldi and Meier, 2018; Micheau and Tschopp, 2003). The first is the formation of complex I that directs NF-kB and MAPK pathway activation. This results in cell survival signals and expression of inflammatory cytokines and chemokines that help shape the immune landscape. The second is the formation of complex II that promotes caspase 8 (CASP8)-dependent apoptosis or RIPK3/MLKL-dependent necroptosis, which can improve cancer immunotherapy (Snyder et al., 2019; Wegner et al., 2017). One critical protein that regulates the outcome of TNFR1 signaling is the receptor interacting protein kinase I (RIPK1) (Weinlich and Green, 2014). RIPK1 is an ISG that serves as a node in homeostatic signaling where it can direct cell survival, inflammation, and cell death downstream not only of TNFR1 but other receptors as well. Mechanistically, RIPK1 has distinct kinase and scaffolding functions that regulate complex I and complex II signaling. The kinase activity of RIPK1 is essential for its cell death function, but is dispensable for its pro-survival NF-kB/MAPK signaling function (Berger et al., 2014; Polykratis et al., 2014). As a scaffold, RIPK1 is important for complex I stabilization and NF-kB/MAPK activation and provides an important checkpoint in regulating cell survival (Ea et al., 2006; Li et al., 2006). Thus, RIPK1 is a critical determinant of cell fate downstream of TNF family receptors.
In this study, we investigated immunotherapy resistance pathways that are regulated by inhibitory IFNG signaling in cancer cells. From among a collection of ISGs implicated in ICB resistance, we examined how RIPK1 coordinates both cancer cell-intrinsic and extrinsic mechanisms impacting the anti-tumor immune response. Using a combination of CRISPR genetic interaction screening and single-cell sequencing approaches we dissected how RIPK1 simultaneously reshapes the tumor immune cell landscape and regulates sensitivity to immune-mediated killing. Finally, we identified critical signaling domains in RIPK1 required for these resistance mechanisms.
RESULTS
Cancer cell RIPK1 is an ISG that regulates resistance to immune checkpoint blockade
We previously discovered that relapse after ICB in both the murine B16-F10 melanoma and TSA breast cancer models is associated with increased expression of a subset of ISG (Benci et al., 2016). In these resistant murine tumors, blocking IFN signaling by genetic deletion of the IFN-I receptor (IFNAR) and/or IFNG receptor (IFNGR) on cancer cells restores response, suggesting the existence of key ISGs that help promote resistance. In order to identify such ISGs, we utilized ICB-resistant Res 499 melanoma cells, which were derived from ICB-sensitive B16 tumors (Twyman-Saint Victor et al., 2015), and examined the in vivo expression of all genes shown to mediate resistance to anti-PD1-based therapy previously discovered using an in vivo CRISPR screen of highly expressed genes in the B16 model (Manguso et al., 2017). Indeed, expression of these resistant genes was enriched in Res 499 cells compared to B16 (Figure 1A, top left). Conversely, when IFN signaling was prevented in Res 499 cancer cells by CRISPR-mediated co-deletion of the genes encoding for IFNAR and IFNGR, the expression of these resistance genes was markedly diminished (Figure 1A, bottom left). To ascertain whether IFNGR or IFNAR exert the greater influence, we compared the IFNAR/IFNGR co-deletion to IFNAR or IFNGR single deletion (Figure 1A, right; Figure S1A). This analysis demonstrated that IFNGR deficiency has a larger impact on resistance genes, as measured by the normalized enrichment score (NES), compared to IFNAR. Moreover, inspection of the genes most strongly impacted by IFNGR deficiency revealed Ripk1 to be amongst the top (Figure 1A, top-right heatmap). Consistent with the ability of IFNG to regulate Ripk1, IFNG treatment in vitro confirmed induction of RIPK1 protein (Figures 1B and S1B). Thus, a multitude of ICB resistance genes are controlled by IFN signaling with Ripk1 being a resistance gene predominantly regulated by IFNG.
Figure 1. RIPK1 is an interferon stimulated gene that promotes resistance to cancer immune checkpoint blockade.

A. Gene set enrichment analysis (GSEA) of anti-PD1-based therapy resistance genes in untreated Res 499 melanoma cells vs. B16 parental cells (top left), Res 499 vs. Res 499 IFNGR + IFNAR deficient (bottom left), Res 499 IFNGR + IFNAR deficient vs. Res 499 IFNAR deficient (top right), or Res 499 IFNGR + IFNAR deficient vs Res 499 IFNGR deficient (bottom right). Cancer cells were sorted from in vivo tumors. Heatmaps for resistance genes most impacted by IFNGR or IFNAR deficiency are shown (leading edge genes) with blue indicating decreased expression.
B. Protein expression of RIPK1 in TSA breast cancer cells treated with 100 ng/ml recombinant murine IFNG at indicated timepoints.
C-D. Survival analysis of mice bearing Res 499 tumors (n=5–10, 1 independent experiment) (C) or B16 tumors (n=15–20, 3 independent experiments) (D) with gRNA control (WT) or RIPK1
CRISPR-mediated deletion (Ripk1null) and treated with or without anti-CTLA4 (aC4) +/− anti-PDL1 (aP1).
E. Tumor growth curves of mice with control or Ripk1null TSA breast cancer tumors treated with or without anti-CTLA4 (aCTLA4) or anti-PD1 (aPD1) (n=5–10, 1 independent experiment). P-values indicate interaction between treatment and genotype (i.e., effect of treatment is influenced by genotype).
F. Survival analysis of mice with WT or Ripk1null TSA tumors treated with or without anti-CTLA4 (n=15–30, 3 independent experiments).
G-H. RIPK1 protein expression (G) and tumor growth curves (H) of empty vector expressing WT or Ripk1null TSA cells, or Ripk1null cells with ectopic WT Ripk1. Mice were treated with or without anti-CTLA4 (n=5–10, representative of 3 independent experiments).
I. Association between RIPK1 copy number alterations (CNA) and mRNA expression (left) or progression-free survival (right) from pan-cancer TCGA patients (n=10,713).
P-values for survival were determined by log-rank test. Mixed effect model was used for tumor growth analysis. For comparison between two groups, a two-sided Wilcoxon test was used for non-parametric data, and for multiple groups a Kruskal-Wallis test is used.
To confirm that RIPK1 sustains acquired ICB resistance and to verify that RIPK1 can promote resistance even in treatment naïve tumors (Manguso et al., 2017), we deleted Ripk1 by CRISPR (Ripk1null) in both Res 499 and parental B16 tumors (Figure S1C). Wildtype control Res 499 tumors exhibited modest response to combination ICB with anti-CTLA4 plus anti-PDL1 but Ripk1null counterparts were potently re-sensitized (Figure 1C). Similarly, treatment-naïve B16 tumors, which responded well to anti-CTLA4 monotherapy, also exhibited improved response upon RIPK1 deletion (Figure 1D). These findings were also observed in the treatment naïve TSA breast cancer model whereby efficacy of both anti-CTLA4 and anti-PD1 monotherapies were improved with RIPK1 deletion (Figures 1E–F and S1D). RIPK1 deletion had no effect on cancer cell growth in vitro (Figure S1E–F). Conversely, increasing tumor RIPK1 above endogenous levels by ectopically expressing it in Ripk1null cancer cells worsened ICB efficacy compared to WT tumors (Figure 1G–H), suggesting that a gain in RIPK1 expression was detrimental. Indeed, in a pan-cancer analysis of The Cancer Genome Atlas (TCGA) whereby RIPK1 mRNA expression was correlated with DNA copy number alterations, copy number gains or amplification of RIPK1 was commonly observed and was associated with worse progression-free survival (Figure 1I). Thus, RIPK1 is an ISG that influences ICB efficacy in both treatment naïve and relapsed tumors and demonstrates altered expression across many human cancers.
Cancer cell RIPK1 promotes TNF-mediated cell survival and NF-kB signaling rather than cell death
Because RIPK1 is a critical node controlling inflammation, cell death, and cell survival signals through TNF and other receptors, its primary function in cancer is likely context dependent and influenced by interactions with other signaling partners. Therefore, to elucidate the nature of RIPK1-driven signaling and how disrupting RIPK1 enhances ICB response, we utilized an in vivo genetic interaction screening approach. For this, we designed a Cas12a-based CRISPR screen using a CRISPR RNA (crRNA) library to approximately 200 RIPK1-signaling associated proteins. For efficient combinatorial screening in B16 tumors, the Cas12a endonuclease, which allows for multigene editing using a single crRNA array (Figures 2A and S2A), was used for dual targeting of either Ripk1 or a negative control (targeting the Rosa26 locus) along with a library gene (Gier et al., 2020; Zetsche et al., 2017). We opted to perform the in vivo screen using untreated B16 tumors given that the CRISPR system enhances immunogenicity. In RIPK1 wildtype (WT) tumors, the NF-kB signaling kinase Map3k7 (Tak1) was a top hit (Figure 2B, red dots in left scatter plot) with most crRNAs exhibiting significant depletion (Figure 2B, right plots; Figure S2B–C). TAK1 is known to promote pro-survival and inflammatory responses through complex I signaling, while antagonizing cell death through complex II (Dondelinger et al., 2015). Therefore, depletion of crRNAs against Map3k7 suggested that complex I signaling was active in WT tumors. In contrast, in Ripk1null tumors the crRNAs to complex II genes Fadd, Casp8, and Tradd were enriched (Figure 2B, orange dots in left scatter plot; Figure S2D–E), suggesting that in the absence of RIPK1, complex II signaling dominated to actively promote apoptotic cell death. Genes that were depleted comparably in both genotypes or exhibited greater depletion in Ripk1null tumors included inhibitors of cell death (Cflar) and complex I associated linear ubiquitin assembly complex (LUBAC) proteins (Rnf31 and Rbck1). Networks of polyubiquitin chains conjugated to RIPK1 and other complex I proteins control the stability of the complex and the activation of NF-kB/MAPK signaling (Haas et al., 2009), suggesting that cell survival and ubiquitin scaffolding functions that regulate complex I were important in both genotypes but particularly so for Ripk1null tumors to counteract active cell death signaling. In total, this genetic interaction screen suggests that in WT tumors, cell survival and inflammatory signaling through complex I is favored, while in the absence of RIPK1, cells are poised to undergo cell death through complex II (Figure 2C).
Figure 2. RIPK1 genetic deletion in cancer cells alters the balance between TNFRSF complex I and complex II signaling as revealed by in vivo CRISPR-Cas12a screen.

A. Experimental workflow for double-gene deletion AsCas12a CRISPR screening.
B. Scatter plot of median Log2 fold-change of crRNAs associated with the indicated target gene after Cas12a-mediated co-deletion of Rosa26 control (WT) or Ripk1 (Ripk1null). Fold-change is calculated between in vitro and in vivo timepoints. Targets preferentially depleted in WT (red), Ripk1null (beige), or both (blue), or targets preferentially enriched in Ripk1null (orange) are highlighted and have a P-value < 0.05 (see Methods). Also shown are Log2 fold-change for individual crRNAs (red bars) for significant hits overlaid on the distribution for all crRNAs.
C. Select targets identified in (B) projected onto a schematic of the TNF signaling pathway in Ripk1 WT (top) and Ripk1null (bottom) cancer cells. Highlighted gene targets (non-opaque) are depleted in WT or enriched in Ripk1null tumors and illustrate inferred signaling bias for each genotype.
D-E. Expression and quantitation of NF-kB and MAPK pathway proteins (n=2–3) (D) and NF-kB transcriptional reporter activity (representative of 3 independent experiments) (E) in WT or Ripk1null B16 cancer cells after treatment with 100 ng/ml murine TNF.
F. CASP3 cleavage after TNF stimulation of TSA WT or Ripk1null cells for the indicated times under serum-free conditions.
G. In vitro dose response of TNF-mediated killing with 1 ug/ml cycloheximide for 24 hours for WT or Ripk1null B16 and TSA cells measured by normalized viability (representative of 2–3 independent experiments).
P-values for time course was determined by repeated measures ANOVA. For dose response and reporter assay, a non-linear model was fitted and significance determined by comparison to a reduced model using ANOVA.
To validate that the balance between complex I and II signaling in cancer cells is altered when RIPK1 is deleted, we examined NF-kB/MAPK and cell death signaling events after TNF stimulation in RIPK1 WT and gene-deleted conditions. These analyses revealed that deletion of RIPK1 in B16, Res 499, and TSA cells diminished phosphorylation of various NF-kB and MAPK family members including the p65 subunit of NF-kB, p38, and JNK (Figures 2D and S2F) indicative of blunted canonical NF-kB and MAPK signaling. This was accompanied by a decrease in NF-kB transcriptional activity (Figures 2E and S2G). Under conditions of RIPK1 deletion, TNF stimulation conversely increased cleavage of Caspase 3 (Figure 2F). Accordingly, TNF treatment in combination with cycloheximide, which blocks the NF-kB transcriptional checkpoint on cell death (Kreuz et al., 2001), increased in vitro killing in both the mouse cell lines (Figure 2G) and in human cancer cell lines as well (Figure S3A–B). Thus, these results corroborate that RIPK1 contributes to optimal NF-kB activation and promotes cell survival in response to TNF family ligands.
Shifting TNFRSF complex I and II signaling by RIPK1 alters the tumor immune landscape and interactions between cancer and myeloid cells
To better understand the in vivo immune consequences of altered TNFRSF signaling in Ripk1null tumors, we employed single-cell RNA sequencing (scRNA-seq) on untreated WT and Ripk1null TSA breast cancer tumors. From the CD45-positive immune cells, we identified 18 distinct immune cell subsets (Figure 3A). On a gross level, Ripk1null tumors displayed an increase in the frequency of CD8 T cells and NK cells compared to their WT counterparts (Figure 3A–B). However, the largest numerical change after RIPK1 deletion was a decrease in myeloid cells, which constitute the largest proportion of cells in the tumor microenvironment. These changes in lymphocyte and myeloid composition were confirmed by flow cytometry (Figures 3C and S4A). Thus, deleting RIPK1 in cancer cells increases intra-tumoral lymphocyte abundance and decreases myeloid cell infiltrate.
Figure 3. Deletion of RIPK1 in cancer cells results in changes in the tumor immune microenvironment and predicted chemokine-receptor interactions with myeloid cells.

A. UMAP of CD45+ immune cells from scRNA-seq of untreated TSA WT or Ripk1null tumors (n=2 for each). Each cluster is color-coded (left) and the density of cells in each cluster is shown (right).
B. Frequency of immune cells from (A) grouped by major immune subtypes.
C. Flow cytometric analysis of CD103+ DCs and F4/80+ macrophages (top), and CD8+ T cells and NKp46+ NK cells (bottom) (2 independent experiments).
D. Patterns of interactions between ligand-expressing cells and receptor-expressing cells (L-R patterns) using cancer and myeloid populations (left UMAP) from WT and Ripk1null tumors. Top row visualizes ligand expression patterns (red) predicted to interact with receptor expression patterns (beige) that are visualized in the bottom row. Thickness of connecting lines denotes cell-cell interaction (CCI) score for the L-R patterns, and blue lines indicate L-R patterns having the top 3 CCI scores in WT or Ripk1null tumors. L-R patterns whereby the ligand-expressing cells include cancer cells are outlined by a red box (e.g., L-R pattern 2–1 and 3–1 in WT tumors).
E. Expression of Ccl2 (top) and Ccr2 (bottom) in WT and Ripk1null tumors. Ccl2 and Ccr2 is the top ligand-receptor interaction pair from L-R pattern 3–1 from WT tumors shown in (D).
F. Cytokine protein levels secreted ex vivo from TSA WT or Ripk1null tumor explants (representative of 2 independent experiments).
G. TNF dose response for secretion of the indicated cytokines by WT or Ripk1null TSA cells transduced with empty vector, or Ripk1null cells ectopically expressing WT Ripk1 after in vitro stimulation for 48 hours.
For comparison between two groups, a two-sided T-test or Wilcoxon test was used for parametric or non-parametric data, respectively. For dose response, a non-linear model was fitted and significance determined by comparison to a reduced model using ANOVA.
A decrease in myeloid cells in tumors with RIPK1 deleted in cancer cells suggests a defect in myeloid recruitment due to the loss of complex I and associated inflammatory signals by cancer cells. To analyze this possibility, we extracted the macrophage/monocyte clusters and the CD45-negative cancer cell clusters from the scRNA-seq data and performed a ligand-receptor (L-R) interaction analysis using scTensor (Tsuyuzaki et al., 2019), which uses tensor decomposition to infer cell-cell interactions. Top patterns of predicted interactions between ligand-expressing cells with receptor-expressing cells (L-R patterns) from WT and Ripk1null tumors were assessed by a cell-cell interaction score (Figure 3D). Additionally, the identity of the ligands and receptors associated with these cell-cell interaction patterns (L-R pair) were also determined. This analysis revealed that two out of three top L-R patterns from WT tumors involved interactions between ligand-expressing cancer cells and receptor-expressing myeloid cells. This is visualized by the ligand-expressing cell patterns that involve cancer cells, which are outlined by a red box, and connected by blue lines to receptor-expressing cells (Figure 3D, top and bottom rows in WT L-R patterns). In contrast, the top three L-R patterns in Ripk1null tumors were myeloid-to-myeloid cell interactions, suggesting that RIPK1 deletion weakened interactions between cancer cell clusters and myeloid cell clusters. The top L-R pair from WT tumors was the chemokine ligand Ccl2 and its receptor Ccr2, which is a chemokine signaling axis important for monocyte recruitment to the tumor microenvironment (Jahchan et al., 2019). Ccl2 was expressed by a subset of cancer cells that was diminished in abundance after RIPK1 deletion, while Ccr2 was expressed by a subset of myeloid clusters that appeared relatively unaltered by RIPK1 status (Figure 3E). Among other top L-R pairs predicted between cancer and myeloid clusters were the chemokine Ccl5 and the receptors Ccr1 and Ccr5 (Figure S4B), which are also involved in myeloid cell recruitment (Jahchan et al., 2019). Thus, deletion of RIPK1 is predicted to interfere with chemokine-receptor interactions between cancer and myeloid cells.
To validate predictions from the L-R analysis that RIPK1 deletion decreases chemokine production, explants from untreated WT or Ripk1null TSA breast cancer tumors were examined ex vivo. This revealed that RIPK1 deletion markedly decreased production not only of CCL2, but also of inflammatory mediators involved in myeloid differentiation (G-CSF and GM-CSF), other chemokines involved in myeloid cell recruitment (CXCL1), and immunosuppressive cytokines (IL-10) (Figure 3F). Conversely, chemokines important in recruiting T and NK cells, such as CXCL9 (Jahchan et al., 2019), were increased in Ripk1null tumor explants (Figure 3F). Regulation of at least some of these inflammatory mediators by RIPK1 was cancer cell autonomous, as direct in vitro stimulation of cancer cells with TNF resulted in a RIPK1-dependent dose response in CXCL1 and CCL2 production, and ectopic RIPK1 expression restored the compromised production observed in Ripk1null cells (Figure 3G). RIPK1 deletion in human cancer cell lines also reduced TNF-mediated secretion of inflammatory cytokines involved in myeloid cell recruitment (Figure S3C–F). In total, these findings demonstrate that deleting RIPK1 and blunting complex I signaling decreases multiple inflammatory mediators that may impact the intra-tumoral myeloid compartment.
Loss of RIPK1 interferes with development of ARG1+ myeloid cells associated with ICB resistance
Not unexpectedly, simply restoring CCL2 or CCL2 in combination with CXCL1 in Ripk1null tumors failed to fully restore ICB resistance or changes in the myeloid cell compartment (Figure 4A; Figure S4C–F and data not shown). This suggests that RIPK1 signaling in cancer cells regulates multiple factors that determines the composition of tumor-associated myeloid cells. Therefore, we sought to characterize how interfering with RIPK1 signaling in cancer cells impacts the intra-tumoral myeloid compartment. For this, we re-examined the eight major clusters of myeloid cells from the scRNA-seq analysis (Figure 4B). Compared to WT tumors, Ripk1null tumors had a higher proportion of myeloid cells belonging to the Mac_1 cluster and a dramatic reduction in the Mac_4 population (Figure 4C). Cells in cluster Mac_1 are macrophages with high expression of MHC-II, as well as high expression of Cxcl9 (Figure 4D–E), which is consistent with our ex vivo cytokine studies (Figure 3F). In contrast, cells in the Mac_4 cluster are characterized by genes linked to an immunosuppressive phenotype including the immune checkpoint protein Lgals3, matrix remodeling protein Mmp12, the hypoxia-associated gene Hilpda, the anti-inflammatory heme enzyme Hmox1, and the hallmark suppressive myeloid marker Arg1 (Bronte et al., 2003), which appears particularly specific to Mac_4 (Figure 4D–E). Indeed, flow cytometry corroborated that ARG1+ macrophages decreased in tumors with RIPK1 deletion (Figures 4F and S5A). In mice with wildtype tumors, depletion of these ARG1+ macrophages with an anti-CSF1R antibody (Figure 5A) resulted in improved response to anti-PD1 similar to the improvement observed with RIPK1 deletion (Figures 5B and S5B). As further support that this Arg1+ Mac_4 population is likely immunosuppressive, there is a highly specific enrichment of genes expressed by ARG1+ TREM2+ myeloid subsets (Figure S5C), which are populations recently shown to be immunosuppressive in murine tumors (Katzenelenbogen et al., 2020; Molgora et al., 2020). Overall, these data suggest that tumor RIPK1 signaling influences a suppressive myeloid population that blunts tumor response to ICB.
Figure 4. RIPK1 regulates the intra-tumoral accumulation of ARG1+ suppressive macrophages.

A. Survival of mice bearing TSA WT tumors, Ripk1null tumors, or Ripk1null tumors with ectopic expression of Ccl2 alone (left; n=10–20, 3 independent experiments) or Ccl2 and Cxcl1 (right; n=5–20, 2 independent experiments), treated with anti-CTLA4.
B. UMAP of myeloid clusters from scRNA-seq of untreated TSA WT or Ripk1null tumors (n=2 for each). Each cluster is color-coded (left) and the density of cells in each cluster is shown (right).
C. Frequency of cells in the myeloid clusters shown in (B).
D. Expression of top 10 differentially expressed genes from each myeloid cluster. Select genes including genes from Mac_1 and Mac_4 clusters are highlighted in red and blue, respectively.
E-F. Expression of Cxcl9 and Arg1 in myeloid cells (E) and flow cytometric analysis of ARG1+ F4/80+ macrophages (F) from WT or Ripk1null tumors.
P-values for survival were determined by log-rank test. For comparison between two groups, a two-sided T-test or Wilcoxon test was used for parametric or non-parametric data, respectively.
Figure 5. ARG1+ suppressive macrophages predict clinical response to ICB and their loss through myeloid cell depletion phenocopies RIPK1 deletion.

A. Flow cytometric analysis of anti-CSF1R (aCSF1R) mediated depletion of F4/80+ macrophages (left) and ARG1+F4/80+ macrophages (right).
B. Survival of mice bearing WT or Ripk1null TSA tumors treated with anti-CSF1R, anti-PD1 (aPD1), or both (n=5–10, 1 independent experiment).
C. Schema for analyzing enrichment of genes from mouse myeloid clusters (see Figure 4B) in myeloid cells from human melanomas. Shown are UMAPs of myeloid clusters from human melanoma (bottom left), enrichment for differentially expressed genes from mouse myeloid clusters in each human myeloid cluster (top right), and the median expression of genes from mouse Mac_4 cluster overlaid on the human myeloid cluster UMAP (bottom right).
D. Density plot of myeloid clusters from human melanoma from patients treated with anti-PD1 +/− anti-CTLA4. Plots are stratified by ICB response (columns) and pre- and post-ICB biopsies (rows). For presentation purposes, densities for each condition are overlaid on UMAP from (C) (cyan dots).
E. Multivariable random forest model for probability of response for melanoma patients treated with anti-PD1 +/− anti-CTLA4. Shown are the variable importance scores, which represents the increase in classification error rate when the variable is perturbed, for each myeloid and T/NK clusters. The classification error rate for the model is 21%.
For comparison between two groups, a two-sided T-test or Wilcoxon test was used for parametric or non-parametric data, respectively.
Given the profound decrease in Mac_4 myeloid cells in our murine tumor model upon RIPK1 deletion in cancer cells, we sought to understand the potential clinical significance Mac_4-like myeloid cells might have on immunotherapy efficacy in human patients. For this, we examined a scRNA-seq dataset of human melanoma patients treated with immune checkpoint blockade (Sade-Feldman et al., 2018). Examination of all myeloid cells from 48 tumor biopsies taken from human melanoma patients revealed that differentially expressed genes from the murine Mac_4 cluster are highly and specifically enriched in myeloid cluster 4 of these human tumors (Figures 5C and S5D). Importantly, this human Mac_4-like cluster (hMac_4) is nearly exclusively found in pre-treatment and/or post-treatment biopsies of patients who failed anti-PD1 +/− anti-CTLA4 therapy (Figure 5D). Indeed, when all myeloid subsets along with T and NK cell populations in the human melanomas were used to predict resistance to ICB, the hMac_4 cluster was the second top predictor for ICB failure, as assessed by variable importance score from a random forest model yielding a 21% error rate (Figure 5E). In total, these results suggest that RIPK1 expression in cancer cells contributes to cell extrinsic mechanisms of ICB resistance by promoting the recruitment and development of immunosuppressive macrophages. These macrophages exhibit multiple immunosuppressive features and are associated with ICB resistance in both mice and humans.
Deletion of RIPK1 drives T and NK cell killing and sensitizes to cell death by TNFRSF death receptors
Concomitant with a decrease in suppressive myeloid cells, deletion of RIPK1 both promoted accumulation of macrophages expressing lymphoid recruiting chemokines (Figure 4D–E) and resulted in greater T and NK cell infiltration (Figure 3A–C). Thus, we next examined how T and NK cells might contribute to improved ICB response in Ripk1null tumors. All major T cell subsets and NK cells were increased by RIPK1 deletion in cancer cells, including effector-like CD8 T cells expressing Granzyme B (GZMB) (Figure S6A–B). To confirm the importance of CD8 T cells and NK cells, we depleted each population in mice prior to B16 or TSA tumor implantation (Figure S6C). This revealed that depletion of either cell-type markedly attenuated the efficacy of anti-CTLA4 and rendered the effect of RIPK1 deletion largely inconsequential (Figures 6A–B and S6D). Furthermore, response to CTLA4 blockade partially persisted when Ripk1null B16 tumors were implanted in Perforin germline deleted (Prf1−/−) mice (Figures 6C and S6E). Immune cells from Prf1−/− mice can still utilize TNF signaling to kill target cells (Kearney et al., 2017). This suggests that disabling RIPK1 in cancer cells might enhance TNFRSF-mediated killing by CD8 T cells and NK cells in vivo, consistent with the ability of RIPK1 deletion to enhance TNF killing in vitro (Figure 2G).
Figure 6. RIPK1 deletion sensitizes tumors to cell death by perforin-dependent and CASP8-dependent mechanisms.

A-B. Survival of mice bearing B16 WT or Ripk1null tumors and depleted of either CD8 T cells (n=5–10, representative of 2 independent experiments) (A) or NK cells (n=5–15, 2 independent experiments) (B) and treated with or without anti-CTLA4 (aC4). Depletion was performed using an anti-CD8 (aCD8) or anti-NK1.1 (aNK) antibody.
C. Survival of WT or Prf1−/− mice bearing Ripk1null B16 tumors and treated with or without anti-CTLA4 (n= 15, 2 independent experiments).
D. UMAP of lymphocytes and cancer cells from scRNA-seq of untreated TSA WT or Ripk1null tumors (n=2 for each) (right). Expression of Ripk1 in cancer cells and in the indicated T and NK cell subsets is shown in the heatmap. Black box represents p<0.05 for comparison between WT and Ripk1null groups.
E-F. Expression of a cell death metagene or Tradd (E) or of the indicated TNF superfamily receptor or ligand (F) in cancer or immune cells. Median expression in each cell population is shown in the heatmap (left), while per cell expression is overlaid on the UMAP from (D) but faceted by Ripk1 genotype and cell type (right). The cell death metagene is the average scaled expression of genes in the Hallmark apoptosis gene set. For the heatmap, cell types of interest for each comparison are highlighted in bold, and black boxes represent values with p<0.05 for comparison between WT and Ripk1null groups. Scale shows relative expression.
G-H. Expression of the indicated proteins (G) and in vitro TNF dose response for normalized viability after treatment with TNF plus 1 ug/ml of cycloheximide for 24 hours (H) for WT B16 cells and B16 cells genetically deleted for Ripk1 (Ripk1null), Casp8 (Casp8null), and Ripk1 and Casp8 (Ripk1null/Casp8null). Data for WT and single Ripk1 deletion groups shown in (H) are from Fig. 2G and presented here to facilitate comparison.
I. Survival of mice bearing B16 tumors with the indicated genotypes treated with or without antiCTLA4 (n=15–25, 3 independent experiments).
P values for survival analysis were determined by log-rank test. For dose response, a non-linear model was fitted and significance determined by comparison to a reduced model using ANOVA.
To investigate if RIPK1 deletion might impact TNFRSF interactions between cancer cells and T/NK cells in vivo, we analyzed the expression of apoptosis genes and genes encoding TNFRSF members and their cognate ligands by scRNA-sequencing. As expected, RIPK1 deletion in cancer cells resulted in decreased Ripk1 expression only in cancer cells (Figure 6D). This is mirrored by an enrichment of apoptosis-related genes also in cancer cells (Figure 6E, top). Among these upregulated apoptosis-related genes is Tradd (Figure 6E, bottom), a TNFR1 scaffolding protein that can associate with the complex II protein FADD. Conversely, expression of apoptosis inhibitors regulated by complex I signaling, such as Cflar and Tnfaip3 (A20), decreased in cancer cells from Ripk1null tumors (Figure S6F). After RIPK1 deletion in cancer cells, Tnfr1 expression also increased (Figure 6F, top), and although there was modest or no appreciable increase in gene expression for the TRAIL receptor (TRAILR2) or FASL receptor (FAS) (Figure S6G), an increase in genes encoding their cognate ligands TRAIL and FASL was observed in subsets of T and NK cells (Figure 6F, middle and bottom). This elevated expression in Trail and Fasl after RIPK1 deletion was associated with greater expression of Ifng and IFNG-related ISGs in T and NK cell subsets (Figure S7A–B), consistent with previous reports that TNFRSF ligands can be regulated by IFNG (Maciejewski et al., 1995; Sedger et al., 1999). Thus, these results suggest that interactions between TNF superfamily receptors and ligands as well as cell death signaling may be facilitated after RIPK1 deletion.
To assess whether engagement of receptors for TNF, TRAIL, and/or FASL might contribute to the ability of RIPK1 deletion to improve ICB efficacy, we deleted the effector Caspase, CASP8, in WT and Ripk1null B16 and TSA cancer cells (Figures 6G and S7C). CASP8 acts downstream of all three death receptor pathways and crRNAs against Casp8 were enriched specifically in Ripk1null tumors in our in vivo CRISPR genetic interaction screen (Figure 2B). This was confirmed in a validation in vivo CRISPR screen using only cell death genes. Here, crRNAs for Casp8 and the downstream executioner Caspase Casp3, but not other Caspases or regulators of intrinsic apoptosis, were enriched in Ripk1null tumors (Figure S7D–E), indicating the specific engagement of the extrinsic cell death pathway after RIPK1 deletion. Accordingly, deletion of CASP8 reversed the ability of RIPK1 deletion to sensitize to TNF killing in vitro (Figures 6H and S7F) and abrogated the improved response to anti-CTLA4 in vivo (Figure 5I). Notably, crRNAs to the receptors for TNF, TRAIL, and FASL were not enriched in Ripk1null tumors in our in vivo CRISPR screen (Figure S2D), consistent with multiple TNFRSF death receptors providing functional redundancy upstream of CASP8. Collectively, these data suggest that RIPK1 deletion may facilitate engagement of multiple TNFRSF interactions between cancer cells and T/NK cells. Because elimination of RIPK1 poises cancer cells to undergo TNFRSF-mediated cell death through complex II and CASP8, this results in greater ICB efficacy.
Scaffolding rather than kinase function of RIPK1 controls immunosuppressive chemokine and cell survival signals important for ICB resistance
RIPK1 has distinct kinase-dependent and scaffolding functions that can trigger cell death or inflammation and cell survival. Since current inhibitors of RIPK1 have focused on targeting the kinase domain (Degterev et al., 2008; Wang et al., 2018b), we tested the importance of the kinase and the scaffolding function in RIPK1-mediated ICB resistance in our tumor models. For this, we ectopically introduced a kinase dead RIPK1 mutant (K45A and D138N) (Berger et al., 2014; Polykratis et al., 2014), a scaffolding dead mutant (K376R) (Tang et al., 2019; Zhang et al., 2019), or RIPK1 with both types of mutations in Ripk1null cancer cells (Figure 7A). We then analyzed chemokine production after in vitro stimulation with TNF to assess the roles of these functions in RIPK1 dependent inflammatory signals by cancer cells. This revealed that the kinase function of RIPK1 was dispensable for restoring CCL2 and CXCL1 production, while the K376R scaffolding domain mutant was compromised in chemokine generation, particularly for CCL2 (Figure 7B). Similarly, the kinase domain mutants also fully reestablished resistance to TNF-mediated cytotoxicity in vitro, while the K376R scaffolding mutant was approximately 5-fold more sensitive to TNF-induced cell death compared to WT RIPK1 (Figure 7C–D). We next examined the impact of the scaffolding and kinase mutations on the anti-PD1 response in vivo. This revealed that when introduced into Ripk1null cells the K45A kinase mutant resembled WT RIPK1 in reestablishing ICB resistance as measured by tumor response after anti-PD1 (Figure 7E). In contrast, Ripk1null tumors expressing the K376R scaffolding domain mutant remained sensitive (Figure 7E; p=0.005 vs WT; p=N.S. vs Ripk1null), as indicated by a greater than 50% response rate and decreased tumor growth after anti-PD1 therapy (Figure 7F). Together, these data suggest that the scaffolding domain of RIPK1 promotes complex I signaling to orchestrate immunosuppressive cytokine secretion and pro-survival signaling. Thus, interfering with the scaffolding rather than kinase function of RIPK1 in cancer cells can improve ICB efficacy.
Figure 7. RIPK1 scaffolding but not kinase domain is important for intrinsic and extrinsic resistance mechanisms to immune checkpoint blockade.

A. Schematic of the primary protein and domain structure of RIPK1 and position of various mutants (left). K45A and D138N are kinase dead, while K376R is a scaffolding dead mutant. Expression of RIPK1 protein in WT or Ripk1null TSA cells expressing an empty vector, WT Ripk1, or the indicated Ripk1 mutant (right).
B. In vitro chemokine production at 48 hours after 10 ng/ml of TNF for WT TSA cells, Ripk1null TSA cells, or Ripk1null cells expressing the indicated Ripk1 mutants.
C-D. In vitro dose response for TNF-mediated killing with 1 ug/ml of cycloheximide (C) for the indicated cell lines. The effective dose 50 (ED50) for 24-hour TNF-mediated killing relative to WT cells are also shown with 95% confidence intervals (D).
E-F. Response rates (E) and tumor growth (F) of mice bearing the indicated TSA tumor and treated with or without anti-PD1 (n=10–19, 2 independent experiments). The p-values beneath the pie charts in (E) compare response rates with and without anti-PD1.
For comparison between two groups, a two-sided T-test or Wilcoxon test was used for parametric or non-parametric data, respectively. Mixed effect model was used for tumor growth analysis.
DISCUSSION
Understanding how cancer signaling pathways can govern acquired or primary resistance to immune checkpoint blockade is an important step in improving immunotherapy. Here, we show that RIPK1 exhibits genomic alterations that increase its expression across many human cancers and are associated with poor clinical outcome. In cancer cells, RIPK1 is predominantly an IFNG-driven ISG that contributes to ICB resistance through both cancer cell-intrinsic and extrinsic mechanisms. Cancer cell RIPK1 can orchestrate production of NF-kB/MAPK-dependent inflammatory cytokines downstream of the TNFRSF pathway that promote the accumulation of immunosuppressive ARG1+ myeloid cells, which are strongly associated with ICB failure in human melanoma patients. This complex I signaling arm also supports cell survival signals that impede immune-mediated killing. When RIPK1 is deleted or when its scaffolding function is crippled, TNFRSF signaling is shifted away from complex I and toward complex II. The net result from this rewired signaling circuit is greater sensitization to TNFRSF-mediated cell death due to decreased expression of pro-survival signals, such as Cflar, and increased expression of cell death genes. At the same time, chemokines that support suppressive myeloid cells, such as CCL2, decrease due to diminished complex I output. Because immunostimulatory macrophages that produce chemokines such as CXCL9 are supported instead, this results in greater recruitment of T and NK cells. Subtypes of these T and NK cells exhibit greater expression of TNFRSF ligands like Trail and Fasl. Together with greater Tnfr1 expression on cancer cells, multiple TNF family receptor-ligand interactions likely contribute to PRF1-independent immune-mediated killing of cancer cells that are poised to die through CASP8.
Our work underscores important discoveries made from in vivo and in vitro CRISPR screens demonstrating that TNFRSF pathway members contribute to tumor resistance to lymphocyte killing and to immunotherapy (Kearney et al., 2018; Manguso et al., 2017; Vredevoogd et al., 2019). Sensitizing cells to alternative cytotoxic killing mechanisms is important because low T cell to tumor ratios suggest that indirect killing mechanisms play a role in cytotoxic lymphocyte control of tumors (Beck et al., 2019; Hoekstra et al., 2020; Huang et al., 2017). Further, these ancillary killing mechanisms are likely important to reduce chronic antigen stimulation that could worsen T cell exhaustion. Such favorable effects afforded by complementary tumor killing mechanisms also apply to chimeric antigen receptor (CAR) T cells (Singh et al., 2020; Dufva et al., 2020). Moreover, the ability of innate lymphoid cells to utilize TNFRSF-mediated killing mechanisms may be particularly important when tumors have a poor neoantigen repertoire (Benci et al., 2019). Thus, TNFRSF-driven killing mechanisms can be an important complement to PRF1-dependent mechanisms and impact ICB efficacy.
While TNF family members have long been studied as an anti-tumor therapy, it is now appreciated that at baseline these cytokines are unlikely strongly cytotoxic but instead exert pleiotropic effects. These pleiotropic effects include inhibiting tumor growth through direct killing, or promoting tumor growth by enhancing cell survival and inhibiting anti-tumor immune responses (Montfort et al., 2019). Tumor resistance to TNFRSF killing is likely dominated by complex I signaling that activates NF-kB/MAPK pathways, resulting in cell survival and immunosuppressive chemokine generation. This is highlighted by top hits in our CRISPR screen that regulate complex I signaling, like TAK1 and LUBAC proteins. Thus, a potential therapeutic strategy is to bias the output of TNFRSF signaling toward complex II to promote cell death, or to block complex I while retaining complex II-driven cell death function. RIPK1 is an appealing target in this regard, because RIPK1 is a critical node in homeostatic signaling that has been shown to orchestrate signals from a variety of death receptor family inputs. We show that deletion of RIPK1 in cancer cells or antagonizing the scaffolding domain can achieve a favorable balance that primes cells to undergo cell death but also prevents development of immunosuppressive myeloid populations. This dual function is important because although dying cancer cells release DAMPs that stimulate innate immune responses (Aaes et al., 2016; Galluzzi et al., 2017), engulfment of dying cells can also promote development of suppressive myeloid cells (Cunha et al., 2018; Feng et al., 2019). However, inhibiting RIPK1 may be ineffective or counterproductive in situations where RIPK1 is needed to drive immunogenic cell death (Yatim et al., 2015; Snyder et al., 2019; Vredevoogd et al., 2019). The availability of multiple TNFRSF pathways due to ligand expression by T cells and NK cells, engagement of parallel pattern recognition receptor pathways that can contribute to cell death (Dillon et al., 2014), or altered levels of cell death inhibitory proteins may all mitigate reliance on RIPK1 for effective cell killing and allow its deletion to improve cancer immunotherapy. Thus, although the dichotomous nature of TNFRSF signaling needs to be considered when designing approaches to improve immunotherapy, targeting RIPK1 may be one promising strategy.
A major mechanism of resistance to immunotherapy is the intra-tumoral accumulation of immune suppressive myeloid cells; however, details regarding myeloid subtypes and what regulates properties important for ICB resistance remains poorly understood. We identify a subset of ARG1+ myeloid cells present in RIPK1 WT tumors but markedly diminished in Ripk1null tumors. A similar myeloid population can also be identified in human melanomas that strongly predicts clinical resistance to anti-PD1 and/or anti-CTLA4 therapy. Other recent studies have found that a subset of myeloid cells that express ARG1 and TREM2 correlate with poor patient survival (Molgora et al., 2020) and transcriptionally overlap with the ARG1+ population identified in our study. Functional studies of ARG1+TREM2+ expressing macrophages in murine sarcoma models show that they promote tumor growth and can directly suppress T cell proliferation and activity in vitro (Katzenelenbogen et al., 2020; Molgora et al., 2020). Genetic deletion or therapeutic blockade of TREM2 resulted in decreased growth of MCA sarcoma tumors in vivo and increased cytotoxic T cell infiltration and activation. The underlying molecular mechanisms and origins of these ARG1+TREM2+ myeloid cells remain to be uncovered. We show that deletion of RIPK in cancer cells significantly reduces accumulation of ARG1+TREM2+ myeloid cells in our tumor models, resulting in replacement by macrophages that express high levels of MHC-II and Cxcl9, a chemokine important for cytotoxic lymphocyte recruitment and indicative of an anti-tumor phenotype. Thus, given the emerging biological and potential clinical importance of suppressive myeloid cells marked by ARG1 and/or TREM2 expression, targeting RIPK1 in cancer cells may offer a tractable therapeutic strategy to antagonize this myeloid population.
Previous work highlighting RIPK1 as a therapeutic target for pancreatic ductal adenocarcinoma, found that inhibiting RIPK1 kinase activity in tumor associated macrophages reprograms these cells towards an anti-tumor phenotype and helps to promote tumor clearance (Seifert et al., 2016; Wang et al., 2018b). Our work suggests that in cancer cells, a similar strategy that targets the RIPK1 kinase domain might be suboptimal or ineffective. We show that disrupting the kinase domain of RIPK1 in cancer cells has minimal effects on preventing suppressive chemokine production and does not enhance TNF-mediated cytotoxicity. In contrast, interfering with the scaffolding domain prevents CCL2 production and increases cancer cell sensitivity to TNFRSF-mediated cell death compared to WT RIPK1, resulting in improved ICB efficacy. Thus, although targeting RIPK1 offers a way to modulate the pleiotropic effects of TNFRSF signaling, important factors need to be considered including which function of RIPK1 to disable and in which cell type. For example, pharmacologically improving ICB efficacy may require targeting different domains of RIPK1 in cancer cells versus immune cells. Or, new classes of inhibitors, such as small molecules that bind to the kinase domain but then target the protein for degradation, could be used to simultaneously disrupt RIPK1 kinase-dependent and kinase-independent functions (Donovan et al., 2020). Notably, given that RIPK1 deletion in mice is embryonic lethal (Dillon et al., 2014), while loss of the kinase function is not (Berger et al., 2014; Polykratis et al., 2014), the systemic toxicity of targeting the scaffolding function of RIPK1 and/or kinase function needs to be carefully evaluated. Unraveling these complexities are warranted given the central role of RIPK1 in controlling cell intrinsic and extrinsic determinants of immunotherapy response that are downstream of TNFRSF signaling and ICB resistance pathways controlled by IFN.
LIMITATIONS OF THE STUDY
In this study, we show that cancer cell RIPK1 impacts ICB response through both cell-extrinsic and cell-intrinsic mechanisms. However, a more complete understanding of how RIPK1 regulates these two mechanisms is limited by the involvement of multiple chemokines (or other factors) downstream of TNFRSF signaling that impacts myeloid cell composition in the tumor microenvironment and by the multiple upstream TNFRSF receptors and ligands that converge on RIPK1 to direct cell killing. The cellular context that enables effective RIPK1-independent cell killing after deletion of RIPK1 also has not been comprehensively addressed in this work, or the situations in which cell-intrinsic versus cell-extrinsic mechanisms dominate. Additionally, our study relies on genetic deletion of RIPK1 to assess its potential as a therapeutic target. Development of therapeutics that can specifically interfere with the scaffolding function, the kinase function, or both will enable a better assessment of the therapeutic window impacting efficacy versus toxicity.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact, Andy Minn (andyminn@upenn.edu).
Materials availability
Plasmids generated in this study will be provided by the lead contact under a material transfer agreement.
Data and code availability
Single-cell RNA-seq data have been deposited at the GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources table.
This manuscript analyzes existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse RIPK1 Monoclonal Antibody | BD Biosciences | Cat#610458; RRID:AB_397831 |
| Anti-Rat Caspase-8 Monoclonal Antibody, Clone 1G12 | Enzo Life Sciences | Cat#ALX-804-447-C100; RRID:AB_2050952 |
| Anti-Rabbit GAPDH Monoclonal Antibody, Clone 14C10 | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| Anti-Rabbit SAPK/JNK Polyclonal Antibody | Cell Signaling Technology | Cat#9252; RRID:AB_2250373 |
| Anti-Mouse Phospho-SAPK/JNK (Thr183/Tyr185) Monoclonal Antibody | Cell Signaling Technology | Cat#9255; RRID:AB_2307321 |
| Anti-Rabbit NF-κB p65 Monoclonal Antibody, clone D14E12 | Cell Signaling Technology | Cat#8242; RRID:AB_10859369 |
| Anti-Rabbit Phospho-NF-κB p65 (Ser536) Polyclonal Antibody | Cell Signaling Technology | Cat#3031; RRID:AB_330559 |
| Anti-Rabbit p38 MAPK Polyclonal Antibody | Cell Signaling Technology | Cat#9212; RRID:AB_330713 |
| Anti-Rabbit Phospho-p38 MAPK (Thr180/Tyr182) Monoclonal Antibody, clone 3D7 | Cell Signaling Technology | Cat#9215; RRID:AB_331762 |
| Anti-Rabbit IkBa Polyclonal Antibody | Cell Signaling Technology | Cat#9242; RRID:AB_331623 |
| Anti-Rabbit Caspase-3 Polyclonal Antibody | Cell Signaling Technology | Cat#9662; RRID:AB_331439 |
| Anti-Rabbit Cleaved Caspase-3 (Asp175) Polyclonal Antibody | Cell Signaling Technology | Cat#9661; RRID:AB_2341188 |
| InVivoPlus Anti-Mouse PD-1 (CD279) Antibody, clone RMP1-14 | Bio X Cell | Cat#BE0146; RRID:AB_10949053 |
| InVivoPlus Anti-Mouse PD-L1 (B7-H1) Antibody, clone 10F.9G2 | Bio X Cell | Cat#BE0101; RRID:AB_10949073 |
| InVivoPlus Anti-Mouse CTLA-4 (CD152) Antibody, clone 9H10 | Bio X Cell | Cat#BE0131; RRID:AB_10950184 |
| InVivoMAb Anti-Mouse CD8α, clone 2.43 | Bio X Cell | Cat#BE0061; RRID:AB_1125541 |
| InVivoPlus Anti-Mouse NK1.1 Antibody, clone PK136 | Bio X Cell | Cat#BE0036; RRID:AB_1107737 |
| InVivoPlus Anti-Mouse CSF1R (CD115) Antibody, clone AFS98 | Bio X Cell | Cat#BE0213; RRID:AB_2687699 |
| InVivoPlus Rat IgG2a Isotype Control Antibody, clone 2A3 | Bio X Cell | Cat#BE0089; RRID:AB_1107769 |
| Anti-Mouse CD45.2, clone 104 | BioLegend | Cat#109822; RRID:AB_493731 |
| Anti-Mouse I-A/I-E, clone M5/114.15.2 | BioLegend | Cat#107631; RRID:AB_10900075 |
| Anti-Mouse F4/80, clone BM8 | BioLegend | Cat#123114; RRID:AB_893478 |
| Anti-Mouse CD11b, clone M1/70 | BioLegend | Cat#101243; RRID:AB_2561373 |
| Anti-Mouse CD11c, clone N418 | BioLegend | Cat#117347; RRID:AB_2563654 |
| Anti-Mouse CD103, clone 2E7 | BioLegend | Cat#121416; RRID:AB_2128621 |
| Anti-Mouse PD-L1, clone B7-H1 | BioLegend | Cat#124319; RRID:AB_2563619 |
| Anti-Mouse Ly6C, clone HK1.4 | BioLegend | Cat#128015; RRID:AB_1732087 |
| Anti-Mouse Ly6G, clone 1A8 | BioLegend | Cat#127617; RRID:AB_1877262 |
| Anti-Mouse PD-1, clone Rmp1-30 | BioLegend | Cat#109110; RRID:AB_572017 |
| Anti-Mouse CD3, clone 145-2C11 | BioLegend | Cat#100322; RRID:AB_389322 |
| Anti-Mouse B220, clone RA3-6B2 | BioLegend | Cat#103247; RRID:AB_2561394 |
| Anti-Mouse NKP46, clone 29A1.4 | BioLegend | Cat#137619; RRID:AB_2562452 |
| Anti-Mouse CD8, clone 53-6.7 | eBioscience | Cat#48-0081-82; RRID:AB_1272198 |
| Anti-Mouse CD4, clone rm4-5 | eBioscience | Cat#45-0042-82; RRID:AB_1107001 |
| NK1.1, clone PK136 | eBioscience | Cat#47-5941-82; RRID:AB_2735070 |
| F4/80, clone BM8 | eBioscience | Cat#11-4801-81; RRID:AB_2735037 |
| CD11c, clone HL3 | BD Biosciences | Cat#553801; RRID:AB_395060 |
| TCRb, clone H57-597 | BD Biosciences | Cat#742483; RRID:AB_2740816 |
| Anti-mouse CD11b, clone M1/70 | Thermo Fisher Scientific | Cat#12-0112-82; RRID:AB_2734869 |
| Arg1, clone A1exF5 | Thermo Fisher Scientific | Cat#56-3697-80; RRID:AB_2734832 |
| Granzyme B, clone GB11 | Thermo Fisher Scientific | Cat#GRB17; RRID:AB_2536540 |
| TruStain FcX (anti-mouse CD16/32) Antibody | Biolegend | Cat#101319 |
| IRDye 800CW Goat Anti-Rabbit IgG (H + L) | Licor | Cat#926-32211 |
| IRDye 800CW Goat anti-Mouse IgG (H + L) | Licor | Cat#925-32210 |
| IRDye 800CW Goat Anti-Rat IgG (H + L) | Licor | Cat#926-32219 |
| Bacterial and Virus Strains | ||
| MegaX DH10B electrocompetent cells | Thermo Fisher Scientific | Cat#C640003 |
| One Shot Stbl3 Chemically Competent E. coli | Thermo Fisher Scientific | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse TNF-α | Biolegend | Cat#575204 |
| Recombinant Murine IFN-γ | Pepro Tech | Cat#315-05 |
| Protease Inhibitor Cocktail | Sigma | Cat#P8340 |
| Polyethylenimine | Polysciences | Cat#02371-100 |
| Polybrene | Sigma | Cat#H9268 |
| Matrigel Basement Membrane Matrix, Phenol Red-Free, LDEV-Free, | Corning | Cat#356237 |
| Collagenase Type IV | Gibco | Cat#17104019 |
| Collagenase/Dispase | Sigma | Cat#11097113001 |
| DNAse I | Sigma | Cat#10-104-159-001 |
| ACK lysis buffer | Quality Bio | Cat#118156721 |
| Actinomycin D | Sigma | Cat#A1410 |
| Cycloheximide | Cell Signaling Technology | Cat#2112S |
| Lipofectamine 3000 Transfection Reagent | Invitrogen | Cat#L3000008 |
| NuPAGE LDS Sample Buffer (4X) | Thermo Fisher Scientific | Cat#NP0007 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat#11668027 |
| X-TremeGene™ 9 Transfection Reagent | Roche | Cat#6365779001 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10x Genomics | Cat#PN-120258 |
| Nano-Glo Luciferase Assay System | Promega | Cat#N1110 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7570 |
| GeneArt Site-Directed Mutagenesis Kit | Invitrogen | Cat#A13282 |
| BD Cytometric Bead Array Mouse Flex Set KC | BD Biosciences | Cat#558340 |
| BD Cytometric Bead Array Mouse Flex Set MCP-1 | BD Biosciences | Cat#558342 |
| BD Cytometric Bead Array Mouse Flex Set MIP-1a | BD Biosciences | Cat#558449 |
| BD Cytometric Bead Array Mouse Flex Set IL-10 | BD Biosciences | Cat#562263 |
| BD Cytometric Bead Array Mouse Flex Set GM-CSF | BD Biosciences | Cat#558347 |
| BD Cytometric Bead Array Mouse Flex Set G-CSF | BD Biosciences | Cat#560152 |
| BD Cytometric Bead Array Mouse Flex Set MIG | BD Biosciences | Cat#558341 |
| BD Cytometric Bead Array Human Flex Set RANTES (CCL5) | BD Biosciences | Cat#558324 |
| BD Cytometric Bead Array Human Flex Set IL-8 (CXCL8) | BD Biosciences | Cat#558277 |
| BD Cytometric Bead Array Human Flex Set MCP-1 (CCL2) | BD Biosciences | Cat#558287 |
| LIVE/dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34957 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| NextSeq 500/550 v2.5 Sequencing Kit | Illumina | Cat#20024906 |
| Deposited Data | ||
| Arg1+Trem2+ Myeloid Cell Single-cell RNA Sequencing Data | Katzenelenbogen et al., 2020 | GEO: GSE150877 |
| TCGA Pan-Cancer Data | cBioPortal | |
| Human Melanoma Single-cell RNA Sequencing Data | Sade-Feldman et al., 2018 | GEO: GSE120575 |
| TSA Breast Cancer Wild-type or RIPK1 Deletion Tumors Single-Cell RNA Sequencing Data for CD45+ and CD45- Cell Types | This Manuscript | GEO: GSE196178 |
| Experimental Models: Cell Lines | ||
| B16-F10 Murine Melanoma | ATCC | Cat#CRL-6475; RRID:CVCL_0159 |
| TS/A Murine Mammary Carcinoma (TSA) | Laboratory of Dr. Sandra Demaria | PMID: 19706802 |
| Res 499 (B16 derived) | Laboratory of Dr. Andy Minn | PMID:25754329 |
| MDA-MB-231 Breast Cancer Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0062 |
| Huh-7 Hepatocellular Carcinoma Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0336 |
| A549 Non-Small Cell Lung Cancer Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0023 |
| A375 Melanoma Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0132 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mus musculus | The Jackson Laboratory | Cat#000664; RRID:IMSR_JAX000664 |
| C57BL/6-Prf1tm1Sdz/J (Prf1 KO) Mus musculus | The Jackson Laboratory | Cat#002407; RRID:IMSR_JAX:002407 |
| BALB/cAnNCrl Mus musculus | Charles River Laboratories | Cat#028; RRID:IMSR_CRL:28 |
| Oligonucleotides | ||
| CRISPR/Cas9 Guide RNA targeting murine Ripk1: Ripk1 gRNA #1: GGGTCTTTAGCACGTGCATC Ripk1 gRNA #2: CAGTCGAGTGGTGAAGCTAC | This Manuscript | N/A |
| CRISPR/Cas9 Guide RNA targeting murine Casp8: Casp8 gRNA #1: TGAGATCCCCAAATGTAAGC Casp8 gRNA #2: GTCATCTTCCAGCTTACATT | This Manuscript | N/A |
| CRISPR/Cas9 Guide non-targeting control for mouse experiments: Genomic control gRNA #1: GCGCTTCCGCGGCCCGTTCAA | This Manuscript | N/A |
| CRISPR/Cas9 Guide RNA targeting human RIPK1: hRIPK1 gRNA #1: CTTCCTCTATGATGACGCCC | This Manuscript | N/A |
| CRISPR/Cas9 Guide RNA targeting human RIPK1: hRIPK1 gRNA #2: TGGAAAAGGCGTGATACACA | This Manuscript | N/A |
| CRISPR/Cas9 Guide non-targeting control for human studies: non-targeting control gRNA #1: GAAGATGGGCGGGAGTCTTC | This Manuscript | N/A |
| See Table S1 Related to Figures 2 and S2 for CRISPR/Cas12 Guide RNAs used in CRISPR screens | This Manuscript | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 Plasmid | a gift from Feng Zhang | Addgene Cat#62988; RRID:Addgene_62988 |
| LentiV_Cas9_puro Plasmid | a gift from Christopher Vakoc | Addgene Cat#108100; RRID:Addgene_108100 |
| LRG2.1 Lentiviral expression plasmid of sgRNA with GFP | a gift from Christopher Vakoc | Addgene Cat#108098; RRID:Addgene_108098 |
| psPAX2 lentiviral packaging plasmid | a gift from Didier Trono | Addgene Cat#12260; RRID:Addgene_12260 |
| pMig-Flag-Myc-IRES-GFP retroviral plasmid | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1 retroviral plasmid expressing WT RIPK1 | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1(D138N) retroviral plasmid expressing kinase mutant RIPK1 | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1(K45A) retroviral plasmid expressing kinase mutant RIPK1 | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1(K376R) retroviral plasmid expressing scaffolding mutant RIPK1 | This Manuscript | N/A |
| pUMVC Retrovirus Packaging Plasmid | a gift from Bob Weinberg | Addgene Cat#8449; RRID:Addgene_8449 |
| pCMV-VSV-G Envelope Protein Plasmid | a gift from Bob Weinberg | Addgene Cat#8454; RRID:Addgene_8454 |
| pCDH-EF1-FHC Lentiviral Backbone Plasmid | a gift from Richard Wood | Addgene Cat#64874; RRID:Addgene_64874 |
| pCDH-EF1-FHC-CCL2 Lentiviral Plasmid Expressing CCL2 | This Manuscript | N/A |
| pCDH-EF1-FHC-CCL2-CXCL1 Lentiviral Plasmid Expressing CCL2 and CXCL1 | This Manuscript | N/A |
| pNL3.2.NF-κB-RE Plasmid with NF-κB luciferase reporter | Promega | Cat#N1111 |
| EFS-GFP-P2A-Neo-U6-crRNA (pRG212) AsCas12a crRNA expression vector | a gift from Junwei Shi | Addgene Cat#149722; RRID:Addgene_149722 |
| AsCas12a-6xNLS-E174R/S542R (pRG232) Lentiviral expression plasmid of AsCas12a-6xNLS-E174R/S542R | a gift from Junwei Shi | Addgene Cat#149723; RRID:Addgene_149723 |
| Software and Algorithms | ||
| CRISPRseek v3.11 | https://bioconductor.org/packages/CRISPRseek/ | DOI: 10.18129/B9.bioc.CRISPRseek |
| makeblastdb v2.6.0 | https://rdrr.io/github/mhahsler/rBLAST/man/makeblastdb.html | |
| CutAdapt v1.16 | https://cutadapt.readthedocs.io/en/v3.5/ | DOI: 10.14806/ej.17.1.200 |
| gCrisprTools v1.12.0 | https://bioconductor.org/packages/gCrisprTools/ | DOI: 10.18129/B9.bioc.gCrisprTools |
| CellRanger | 10X Genomics | https://www.10xgenomics.com/ |
| Seurat v3.1.5 | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| SAVERCAT | Huang et al., 2020 | |
| GSVA v1.32.0 | Hänzelmann et al., 2013 | DOI: 10.18129/B9.bioc.GSVA |
| scTensor v.2.0.0 | https://bioconductor.org/packages/scTensor/ | DOI: 10.18129/B9.bioc.scTensor |
| lmerTest v3.1-3 | Kuznetsova et al., 2017 | DOI: 10.18637/jss.v082.i13 |
| R Language and Environment | CRAN | https://cran.r-project.org/ |
| survival | Therneau T (2021). A Package for Survival Analysis in R | https://CRAN.R-project.org/package=survival |
| drc dose response analysis | Ritz et al., 2015 | https://CRAN.R-project.org/package=drc |
| randomForestSRC v2.9.3 | Ishwaran et al., 2011 | https://github.com/kogalur/randomForestSRC/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
B16-F10 murine melanoma cells (male derived), TSA murine mammary carcinoma cells (female derived), and resistant sublines were derived and cultured as previously described (Twyman-Saint Victor et al., 2015). In brief, B16-F10 cells and its derivatives were cultured in DMEM and TSA cells were cultured in RPMI media supplemented with 10% FBS, all cells were cultured at 37C. Human cell lines stably expressing Cas9 were a gift from Dr. Junwei Shi including MDA-MB-231 breast cancer cells (female derived), Huh-7 hepatocellular carcinoma cells (male derived), A549 non-small cell lung cancer cells (male derived), and A375 melanoma cells (female derived). Human cell lines were cultured at 37C in DMEM supplemented with 10% FBS, 100 U/mL streptomycin, 100 ug/mL penicillin, 2 mM L-glutamine. All cell lines were determined to be free of Mycoplasma (Lonza).
Mice
All animal experiments were performed according to protocols approved by the Institute of Animal Care and Use Committee of the University of Pennsylvania. Five- to seven-week-old female C57BL/6 (stock #000664) and Prf1 germline deletion (stock #002407) were obtained from The Jackson Laboratory (Bar Harbor, ME). Five- to seven-week-old female BALB/c (stock #028) were obtained from Charles River Laboratories. Mice were allowed to acclimate up to 1 week before experiment initiation and were maintained under pathogen-free conditions. Mice were randomly assigned to experimental groups.
METHOD DETAILS
CRISPR gene targeting
For murine cell lines, Ripk1 and Casp8 gene targeting by CRISPR/Cas9 was accomplished by co-transfection of a Cas9 plasmid (Addgene #62988) (Ran et al., 2013), the guide sequence (selected using the Zhang Lab CRISPR design tool http://crispr.mit.edu) cloned into the gBlock plasmid, and a plasmid with the puromycin selection marker. Gene blocks used contain a 20 bp target size (N), U6 promoter, gRNA scaffold, and termination signal. Tumor cells were transfected with respective targeting vectors using Lipofectamine 2000 reagent and 24 hours later selected with puromycin. Single cell clones were screened by western blot for respective protein expression. Confirmed gene-deleted clones were pooled. For controls, non-targeting gRNAs (gift from John Doench) were used and clones without gene deletion were also pooled. The common gene block sequence used is previously published (Benci et al., 2019). For human cell lines, cells were stably transduced with Cas9 using the lentiviral EFS-Cas9-P2A-Puro expression vector (Addgene #108100) (Tarumoto et al., 2018). sgRNAs targeting human RIPK1 (hRIPK1) or a non-targeting negative control sgRNA were cloned into a lentiviral expression vector (LRG2.1; Addgene #108098), which is a bicistronic vector expressing both an optimized sgRNA backbone and a GFP reporter marker (Grevet et al., 2018). Lentivirus was produced by transfecting HEK293T cells with helper plasmids VSVG and psPAX2 (Addgene #12260) using X-TremeGene™ 9 (Roche) in a mass ratio of 6:2:4 for plasmid DNA:VSVG:psPAX2. Media was replaced ~6–8 hours post transfection, and viral supernatant was collected several times within 24–72 hours of transfection. Supernatant was passed through a 0.45 μm PVDF filter before use (Millipore). Lentivirus was added to human target cell lines with 8 μg/mL Polybrene (Sigma) for two hours at 37C. Cells were sorted at least 48 hours post-infection for eGFP+ to ensure expression of the sgRNA constructs. Gene deletions were confirmed by western blotting. The sequences for each guide used include:
Ripk1 gRNA #1: GGGTCTTTAGCACGTGCATC
Ripk1 gRNA #2: CAGTCGAGTGGTGAAGCTAC
Casp8 gRNA #1: TGAGATCCCCAAATGTAAGC
Casp8 gRNA #2: GTCATCTTCCAGCTTACATT
Genomic control gRNA #1: GCGCTTCCGCGGCCCGTTCAA
hRIPK1 gRNA #1: CTTCCTCTATGATGACGCCC
hRIPK1 gRNA #2: TGGAAAAGGCGTGATACACA
non-targeting control gRNA #1: GAAGATGGGCGGGAGTCTTC
Viral transduction for gene over-expression
For RIPK1 re-expression or expression of mutant RIPK1, pMig-Flag-Myc-IRES-GFP control and mRIPK1(WT), mRIPK1(D138N), mRIPK1(K45A) mutant vectors were a gift from Dr. Douglas Green. Scaffolding mutant mRIPK1(K376R) was generated using GeneArt™ Site-Directed Mutagenesis kit (Invitrogen) and verified by DNA sequencing. These vectors were transfected into HEK293T cells using lipofectamine 2000 and packaged into retrovirus for transduction using VSVG and pUMVC helper plasmids. Retrovirus were collected 48 hours after transfection. Tumor cells were transduced with virus with polybrene for 72 hours and underwent fluorescence-activated cell sorting (FACS) for eGFP+ cells. For cytokine over-expression, the pCDH-EF1-FHC vector was a gift from Richard Wood (Addgene #64874). Cxcl1 and Ccl2 genes were cloned into this vector for constitutive over-expression. This or control vector was transfected into HEK293T cells using lipofectamine 2000 and packaged into lentivirus for transduction using VSVG and psPAX2 helper plasmids. Tumor cells were transduced with virus with polybrene for 72 hours and underwent puromycin selection for 1 week and then were utilized for in vitro gene expression studies and tumor implantation experiments. Over-expression efficiency was assessed by gene-specific quantitative PCR analysis and cytokine bead array.
In vivo mouse studies
For B16-F10 melanoma, 5 × 104 cells were mixed with an equal volume of Matrigel Membrane Matrix (Corning) and subcutaneously injected on the right flank of C57BL/6 mice. For TSA mammary carcinoma, 1× 105 cells were mixed with an equal volume of Matrigel and subcutaneously injected on the right flank of BALB/c mice. Antibody treatments were carried out on days 5, 8, and 11 post-implantation for B16-F10, and on days 7, 10, and 13 for TSA tumors unless otherwise specified. CD8+ T cell and NK cell depletion was carried out on days −2, 0, 4, 8, 12 and 16, myeloid cell depletion was carried out using anti-CSF1R beginning on the day of tumor implantation and dosed 3x/week until completion of the experiment. Depletion was confirmed by flow cytometric analysis of peripheral blood or tumors. All antibodies were administered intraperitoneally at 200 ug/dose, while anti-CSF1R was administered at 400 ug/dose. Antibodies were from Bio X Cell and include: PD1 (RMP1–14), PD-L1 (10F.9G2), CTLA4 (9H10), CD8 (2.43), NK1.1 (PK136), CSF1R (AFS98), Rat IgG2a Isotype control (2A3).
Western blotting
Where indicated, cells were treated with 10 ng/mL or 100 ng/mL mTNF (Biolegend), or 100 ng/mL mIFNg (PeproTech). For CASP3 western, TSA cells were treated with 10 ng/mL mTNF in serum free conditions for indicated timepoints. Cells were lysed in lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris HCl (pH 7.5), 1 mM Na3VO4, 5 mM NaF, 1 mM PMSF, 5 mM Na pyrophosphate, and Protease Inhibitor Cocktail (Sigma)). Insoluble material was removed by centrifugation at 15,000 rpm and lysates were quantitated using a BCA Protein Assay (Thermo Fisher) and protein concentration was normalized before the addition of LDS sample buffer (Nupage). Lysates were subject to SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad). The membrane was probed with primary antibodies, IRDye secondary antibodies (Licor) and visualized with the Licor Odyssey. The following antibodies were used for protein detection: RIPK1 (BD Biosciences 610458), Caspase-8 (Enzo Life Sciences ALX-804–447-C100), GAPDH (Cell Signaling Technology 2118), SAPK/JNK (Cell Signaling Technology 9252S), phospho-SAPK/JNK (Cell Signaling Technology 9255S), NF-kB p65 (Cell Signaling Technology 8242S), phospho-NF-kB p65 (Cell Signaling Technology 3031S), p38 MAPK (Cell Signaling Technology 9212S), phosphor-p38 MAPK (Cell Signaling Technology 9215S), IkBa (Cell Signaling Technology 9242S), Caspase-3 (Cell Signaling Technology 9662S), Cleaved Caspase-3 (Cell Signaling Technology 9661S). Protein expression was quantified using Licor software and target band intensity was normalized to control GAPDH expression, followed by normalization to the intensity of the target band at the 0-minute timepoint.
Cytokine Measurements
For murine samples, eleven days post-implantation tumors were harvested, weighed, and 200 ug were minced and placed in serum-free RPMI media for 1 hour at 37C. The media was then collected, spun to remove any contaminating cells, and analyzed for cytokine expression using the BD Cytometric Bead Array Mouse Flex Sets for KC, MCP-1, MIP-1a, IL-10, GM-CSF, G-CSF, MIG (BD Biosciences) according to manufacturer’s instructions. Cytokine levels were normalized by tumor weight for each sample. For in vitro cytokine expression, cells were cultured for 48 hours in serum free media or with mTNF (Biolegend) where indicated, and media was collected and analyzed as described. Wild type or RIPK1null human cancer cell lines were evaluated for in vitro cytokine expression using BD Cytometric Bead Array Human Flex Sets for CXCL8, CCL2, and CCL5 (BD Biosciences).
In vitro proliferation and cell death assays
Tumor cells were seeded at 3,000 cells per well in triplicate in a 96 well plate. The next day, cells were treated with 1 ug/mL of cycloheximide (Cell Signaling Technologies) and the indicated concentration of mTNF. 24 hours later media and dead cells were removed, and replaced with fresh RPMI media and cell viability was measured by CellTiter-Glo (Promega). Data were normalized to DMSO control and/or TNF dose 0. Dose response curves were analyzed using drc R package (v 3.0–1) (Ritz et al., 2015). For proliferation assay, cells were seeded at 2,000 cells per well in a 96 well plate, at indicated timepoints cell viability was measured by CellTiter-Glo (Promega). Data were normalized to background control followed by Day 0 control measurement.
NF-kB reporter assay
B16 and Res 499 cells were seeded at 15,000 cells per well, and TSA cells were seeded at 20,000 cells per well in triplicate in a 96 well plate. The next day, cells were transfected with the NanoLuc® Reporter Vector with NF-κB Response Element (pNL3.2.NF-κB-RE; Promega) using Lipofectamine 3000 transfection reagent (Invitrogen) at a ratio of 1:3, DNA:Lipofectamine. 24 hours later media was removed and replaced with fresh DMEM or RPMI media without FBS and treated with 10 ng/mL mTNF (Biolegend) over the indicated time-course. Reporter expression was read out using the Nano-Glo® Luciferase Assay System (Promega) according to the manufacturer’s protocol. Data were normalized to TNF 0 timepoint for reporter fold change calculations. Time-course response curves were analyzed using drc R package (v 3.0–1).
Flow cytometry and cell sorting
Two weeks post-implantation, tumors were harvested, digested with collagenase IV (EMSCO/Fisher) for B16 tumors or collagenase/dispase (Sigma) for TSA tumors, supplemented with DNAse I (Sigma), for 30 minutes at 37C. The suspension was filtered, treated with ACK lysis buffer (Quality Bio) for 1 minute on ice. Staining was carried out first in LIVE/dead Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) and TruStain FcX (Biolegend) for 15 minutes at 4C, followed by surface antibody staining for 30 minutes at 4C. Intracellular staining was carried out using Foxp3 Fix/Perm kit (Thermo Fisher Scientific). Cell sorting was carried out on LSR II or FACS Aria II flow cytometers (BD) when necessary. Fluorescently labeled anti-mouse antibodies against CD45 (104), I-A/I-E (M5/114.15.2), F4/80 (BM8), CD11b (M1/70), CD11c (N418), CD103 (2E7), PD-L1 (B7-H1), Ly6C (HK1.4), Ly6G (1A8), PD-1 (Rmp1–30), CD3 (145–2C11), B220 (RA3–6B2), and NKP46 (29A1.4) were obtained from BioLegend; CD8 (53–6.7), CD4 (rm4–5), NK1.1 (PK136), and F4/80 (BM8) from eBioscience; CD11c (HL3) and TCRb (H57–597) from BD Biosciences; CD11b (M1/70), Arg1 (A1exF5), and Granzyme B (GB11) from Thermo Fisher Scientific.
Double-gene deletion CRISPR library assembly
For double-gene deletion CRISPR screening, a dual-crRNA library was cloned into a AsCas12a crRNA expression vector pRG212 (EFS-GFP-P2A-Neo-U6-crRNA, Addgene #149722). To construct a dual-crRNA library, the crRNAs targeting gene of interest were assembled in the position#1 of the dual-crRNA array, while the negative control Rosa26 crRNA or two independent Ripk1 targeting crRNAs (g2.2 and g4.1) were assembled in the position#2 of the dual-crRNA array. The AsCas12a library gene list was generated using STRING database (v11) for RIPK1, IFNGR, and IFNAR protein-protein association networks consisting of approximately 200 genes. For each gene, approximately five 23-nucleotide crRNA sequences were designed to target functional protein domains using the photospacer adjacent motif (PAM) 5’-TTTV. crRNAs were designed using the CRISPRseek R package (v3.11) (Zhu et al., 2021) modified to identify Cas12a 5’ PAMs targeting protein domains selected using the Pfam database and mapped back to the UCSC mm10 Mouse reference genome. crRNA oligonucleotides were synthesized individually with overhangs for BsmbI recognition sites. The oligos were phosphorylated, annealed, pooled in equal molar ratio, and then ligated into the BsmBI-digested pRG212 backbone vector using Golden Gate cloning. The ligation product was electroporated into MegaX DH10B electrocompetent cells (Invitrogen) as previously described (Gier et al., 2020). The cloned library was cultured overnight at 30C, and DNA was extracted using PureLink HiPure Plasmid Maxiprep Kit (Invitrogen). Diversity of crRNA abundance was confirmed by sanger sequencing of randomly selected bacterial colonies. To confirm cloning of all dual-crRNAs into the pRG212 vector, deep sequencing was performed using the Illumina NextSeq. The pooled library lentivirus was produced by transfecting HEK293T cells with helper plasmids VSVG and psPAX2 (Addgene #12260) using polyethylenimine (Polysciences, PEI 25000) in a mass ratio of 4:2:3 for plasmid DNA:VSVG:psPAX2. Media was replaced ~6–8 hours post transfection, and viral supernatant was collected several times within 24–72 hours of transfection. Supernatant was passed through a 0.45 μm PVDF filter before use (Millipore). Lentivirus was added to target cell lines with 8 μg/mL Polybrene (Sigma #H9268) and centrifuged at 1,750 r.p.m. for 25 minutes at room temperature. Media was changed 15 hours post-infection. The lentivirus was titrated to achieve an MOI of ~0.3–0.5 to ensure single copy viral integration.
Double-gene deletion CRISPR screening
B16-F10 melanoma cells were stably transduced with an optimized opAsCas12a lentivirus vector (pRG232, Addgene #149723). B16 opAsCas12a cells were then stably transduced with the dual-crRNA array library at an infection rate of 30%, with either crRNA targeting the Rosa26 locus (a negative control crRNA targeting the editing safe harbor Rosa26 locus) or Ripk1 in the second position of the array, and the 200 gene library, consisting of 5 crRNAs per gene (Table S1), in the first. Cells were maintained at exponential growth. Cells were harvested at day 3 post-infection and sorted for GFP+ cells that expressed the crRNAs. Sorted cells recovered for 72–96h prior to injection in vivo. On the day of injection, cells were harvested as a reference for the fully represented pooled CRISPR RNA libraries. For tumor injections, 5 × 105 cells were mixed with an equal volume of Matrigel Membrane Matrix (Corning) and subcutaneously injected on the right and left flank of C57BL/6 mice (n=6 per library). Remaining cells were then cultured for the duration of the in vivo growth and harvested on day 17 post-implantation as a control for in vitro crRNA dropout. On day 17 post-implantation, tumors were harvested and digested with collagenase IV (EMSCO/Fisher) for 30 minutes at 37C. Tumor cells were sorted for GFP+ crRNA expression using the FACS Aria II (BD) and libraries were pooled by biological replicate. Harvested cells were washed in PBS, pelleted, and stored at −80C until genomic DNA extraction. For the in vitro screen targeting ~84 cell death related genes (Table S3), cells were treated with 3 uM Actinomycin D (Sigma) or DMSO control for 48 hours, harvested according to normal tissue culture protocols, and cell pellets were washed with PBS and stored at −80C until genomic DNA extraction.
Detection of library crRNAs was performed by deep sequencing as described previously (Gier et al., 2020). Briefly, genomic DNA was isolated from the stated initial and final timepoints for both in vitro and in vivo samples with the Quick-DNA Miniprep Plus kit (Zymo Research) following the manufacturer’s protocol. CRISPR RNA was PCR amplified and annealed to stacking barcodes for each sample, to ensure 1000x crRNA library representation. Illumina sequencing adapters were added during pre-capture PCR. CRISPR RNA cassette quantification was conducted through deep sequencing, the dual-crRNA libraries were pooled in equal molar ratios, and single end sequenced on the Illumina NextSeq with the NextSeq Mid-Output v2.5 150-cycle kit (Illumina).
Single Cell RNA sequencing sample preparation
Two weeks post-implantation, TSA tumors were harvested, digested with collagenase IV (EMSCO/Fisher) for 30 minutes at 37C. Tumor cells and infiltrating immune cells were sorted by live/dead negative and CD45+/− populations. Two samples per condition (WT or Ripk1null tumors) were used. Single cell emulsions were obtained using the 10x Chromium Controller, and libraries were prepared using the Chromium Single Cell 3’ Library & Gel Bead Kit v2 (10x Genomics) following the manufacturer’s protocol. Libraries were sequenced on an Illumina NextSeq using a NextSeq 500/550 v2.5 High Output Kit (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of tumor growth and survival
Mice were randomly assigned to treatment groups and tumor volume was determined using perpendicular tumor diameters measured using calipers. Volume was calculated using the formula L × W2 × 0.52, where L is the longest dimension and W is the perpendicular dimension. Differences in survival were determined for each group by the Kaplan–Meier method and the overall P value was calculated by the log-rank test using the survival R package (Therneau and Grambsch, 2000). An event was defined as when tumor burden reached a protocol-specified size of 1.5 cm in maximum dimension or developed skin ulceration above a protocol-specified size of 4 mm to minimize morbidity. A mixed effect linear model using the lmerTest (v3.1–3) R package (Kuznetsova et al., 2017) was used to determine differences in tumor growth curves. To determine the impact of treatment or genotype, multivariable linear regression was used to determine the significance of main and interaction effects.
Double-gene deletion CRISPR screening data analysis
The data analysis was performed similarly as described previously (Gier et al., 2020). Briefly, all library dual-crRNA cassette sequences were assembled into a BLAST reference index using makeblastdb (v2.6.0). Adapter sequences were trimmed using CutAdapt (v1.16) for each sample and then sequences were mapped to the BLAST index using blastn. BLAST queried sequence mapping was restricted with three rules: (1) having only one high-scoring dual-crRNA, (2) reporting only one aligned target (-max_hsps 1 -max_target_seqs 1), and (3) allowing no more than two mismatching nucleotides (including mismatches, indels and unaligned tails). crRNA cassette read counts were normalized to total read counts of the reference time point library.
For data analysis gCrisprTools (v1.12.0) was used to calculate Log2 fold change of crRNA expression between conditions. Briefly, cassette abundance counts and sample identifiers were aggregated, low abundance cassettes were removed (read.floor=10), and cassette counts were normalized using the “scale” method. A design matrix was generated by aggregating biological replicates (n=1 Rosa26 control, n=2 Ripk1) and classifying samples by in vivo and in vitro, and in vitro samples by early (initial) and late (final) collection timepoints. Counts were then converted to log2-counts per million based on the design matrix using voom and fitted to a linear model using lm.fit. Differentially expressed crRNAs were calculated using the linear modeling framework implemented in the limma package to generate contrasts of interest, ct.generateResults was used to generate target level P-values and Q-values for the depletion and enrichment of each crRNA and associated target gene, and Rho statistics for ranking targets. The following contrasts were used: Ripk1 contrast: ((final crRNA in vivo abundance – initial crRNA in vitro abundance) – (final crRNA in vitro abundance – initial crRNA in vitro abundance)); Rosa contrast: ((final crRNA in vivo abundance – initial crRNA in vitro abundance) – (final crRNA in vitro abundance – initial crRNA in vitro abundance)); Ripk1.diff contrast: (Ripk1 contrast – Rosa contrast). crRNAs with a P-value >0.05 for each contrast were considered significant. For target gene level plots, median log2 fold change estimate was used. For individual target genes candidates log2 fold change for each crRNA was visualized.
Single cell RNA-seq processing
The CellRanger pipeline (10x Genomics) was used to process scRNA-seq data. cellranger mkfastq was used to demultiplex raw BCL files into FASTQ files. cellranger count was used to align reads to the mm10 Mouse genome (cellranger-mm10–3.0.0), count UMI barcodes, and generate gene-barcode expression matrices. Poor quality libraries were filtered by removing cells with fewer than 500 detected genes and cells with over 10% of mitochondrial reads mapping to chrM. Non-expressed genes (expressed in <1% of cells) were also removed. Downstream analyses were performed with Seurat (v3.1.5) for sctransform normalization, clustering, and dimensionality reduction. Biological replicates for WT or Ripk1null tumors were aggregated, and sparse matrices were converted to dense matrices. CD45+ cells from WT and Ripk1null conditions were harmonized following the Seurat v3 integration workflow and cell type annotation was performed on the integrated reference using known markers for immune cells and by gene set enrichment, as described below. Gene expression data were denoised with SAVERCAT (Huang et al., 2020) using default options. SAVERCAT adopts a well-established Poisson-Gamma mixture to model the gene expression data (Kim et al., 2015; Wang et al., 2018) built based on the model of SAVER (Huang et al., 2018). By coupling the conditional variational auto-encoder with Bayesian shrinkage framework, SAVERCAT corrects for batch effects alongside denoising gene expression data. It takes a post-QC gene expression matrix and batch information as input and outputs the denoised expression value estimates based on the posterior mean. This estimate emulates the data if there were no batch effects and if deeper sequencing were performed on the sample. Denoised estimates were used for visualization while Bayesian posterior sampled values were used for gene set enrichment and metagene calculations.
Human melanoma single-cell RNA sequencing data and TCGA data
Single-cell RNA sequencing data from 48 melanoma samples (Sade-Feldman et al., 2018) from patients treated with and without ICB therapy were downloaded from the GEO (GSE120575). The TPM data were then denoised using SAVER (Huang et al., 2018). Additional normalization with sctransform, clustering, and dimensionality reduction were performed with Seurat (v3.1.5.). Immune cell clusters were annotated using marker gene expression and by enrichment of human immune gene sets using GSVA, as described below. T cell, innate lymphoid cell, and myeloid cell clusters were retained for further analysis. For TCGA patient data analysis, RIPK1 DNA copy number and mRNA expression data from the TCGA pan-cancer data set were downloaded from cBioPortal (https://www.cbioportal.org/).
Immune cell cluster assignment
To assign shared nearest neighbor (SNN) clusters to an immune cell type, gene sets for various mouse immune cells were used to determine gene set enrichment scores. For general immune cell types, dendritic cell subsets, and myeloid cell subsets, previously published gene sets were used (Zilionis et al., 2019), excluding genes with a log2 fold-change less than 0.5 compared to genes in other like gene sets. Contaminating stromal cell clusters were identified using gene sets for cancer associated fibroblasts, stromal cells, and endothelial cells previously described (Jerby-Arnon et al., 2018) and removed. GSVA (v1.32.0) and default parameters were used to calculate gene set enrichment on the aggregated gene expression values for all cells in each cluster. Each cluster was then assigned to the immune cell type or subset with the greatest enrichment score. Differential gene expression analysis for myeloid cells was calculated using Seurat v3 FindAllMarkers default settings (Table S2). Top 10 differentially expressed genes for each cluster were visualized.
Gene set enrichment and expression analysis
Enrichment of gene sets for single-cell RNA-sequencing data was determined using GSVA (v1.32.0) (Hänzelmann et al., 2013) and default parameters for all CD45+ immune cell subsets and CD45− cancer cell subsets. To compare enrichment scores of cells belonging to two groups, a two-sided Wilcoxon test was used. For analysis using all immune cells, gene expression values for all cells in each cluster were aggregated first for computational speed and a per cluster enrichment score was calculated. To estimate the significance of the enrichment scores, observed enrichment scores were compared to the distribution of scores from 1000 random gene sets. The Hallmark gene set for apoptosis was used as a gene set for cell death associated genes. For myeloid cell analyses of murine tumors, gene sets for myeloid subsets previously described (Katzenelenbogen et al., 2020) were used and include:
Cluster 1: H2-Aa, H2-Ab1, H2-Eb1, H2-Eb1, Mgl2, Cd209a
Cluster 2: Ly6c2, Plac8, S100a6, Ly6a, Ms4a4c, Irf7, Fcgr1, Rsad2, Ifit1, Ifit2, Ifit3, Isg15, Cxcl10
Cluster 3: Ace, Ear2, Chil3
Cluster 4: Mmp12, Hmox1, Gpnmb, Hilpda, Il7r, Clec4d, Trem2, Arg1
Cluster 5: Ctsl, Pf4, Spp1, Pdpn, Trem2, Arg1; c6: C1qa, C1qb, C1qc, Hexb, Cx3cr1, Ms4a7, Lgmn, Apoe
For myeloid cell analyses of human melanoma tumors, genes differentially expressed by murine myeloid clusters (Table S2) and previously published gene sets from human dendritic cell subsets were used (Zilionis et al., 2019). Similarly, for T and NK cell analysis of human melanoma tumors, gene differentially expressed by murine T and NK cells were used (Table S2). Enrichment of gene sets for Res 499 or B16 melanoma tumors was determined using genes discovered by in vivo CRISPR screening to promote resistance of B16 tumors to anti-PD1-based therapy as previously described (Manguso et al., 2017).
Analysis of human melanoma samples and predictors of clinical response
To assess the ability of myeloid cell, T cell, or innate lymphoid cell clusters in either pre-treatment or post-treatment biopsies to predict clinical response to ICB, we determined the relative frequencies of each of these immune cell clusters for 48 melanoma samples (Sade-Feldman et al., 2018). These immune cell cluster frequencies and the timing of the biopsy (pre-treatment vs. post-treatment) were then used to predict ICB response using random forest as implemented in the randomForestSRC (v2.9.3) R package (Ishwaran et al., 2011). Because the two class sizes were imbalanced, we used the imbalanced function with nodesize = 1 and other parameters set to default. Model error rate and variable importance scores were then determined. This procedure was repeated only on the pre-treatment biopsy samples, since these samples represent the immune cell composition of the tumor prior to ICB. This confirmed that similar to when all samples were used, the “hMac_4” cluster was the top predictor by variable importance score.
Cell-cell interaction analysis
The cancer and myeloid cell interaction analysis was carried out using scTensor (v.2.0.0). Using the scRNA-seq data from CD45+ and CD45− cells from murine TSA tumors, we first subset all monocyte and macrophage cells from the intra-tumoral CD45+ cells and combined these subsets with all CD45− cancer cells and performed PCA and cluster identification. We ran scTensor on the specified cell subsets for both WT and Ripk1null samples and identified cell interaction patterns and their interaction strength. We extracted and plotted the ligands and receptors that make up the cancer and myeloid cell interactions and calculated the averaged expression of ligands and receptors, respectively.
Statistical analysis
The significance of two-way comparisons was determined by two-sample, two-tailed t-test for parametric data and by a Wilcoxon test for non-parametric data. A Shapiro test was used to assist in determining whether response variables were normally distributed. To determine the impact of treatment or genotype, multivariable linear regression was used to determine the significance of main and interaction effects. For dose response, a non-linear model was fitted and significance was determined by comparison to a reduced model using ANOVA. Statistical details of the experiments including subject number (n) and independent experimental replicates can be found in the figure legends. Unless specified otherwise, error bars represent SEM. Significance was determined using a standard alpha of 0.05.
Supplementary Material
Table S2. Top 50 differentially expressed genes in mouse myeloid clusters and in T and NK cell clusters (Related to Figures 4–5 and S5).
Table S3. CRISPR screen crRNA library for cell death pathway (Related to Figure S7).
HIGHLIGHTS.
Cancer cell RIPK1 controls IFNG-driven immune checkpoint blockade (ICB) resistance
Tumors with RIPK1 resist immune cell killing and secrete suppressive cytokines
Deleting RIPK1 decreases ARG1+ macrophages, promotes TNF killing, and improves ICB
RIPK1-mediated ICB resistance requires scaffolding but not kinase function
ACKNOWLEDGMENTS
A.J.M. and L.C. are also supported by the Breast Cancer Research Foundation. A.J.M., J.S., Q.C., Y.Y, M.K., K.B., Y.S., I.E.B., and N.R.Z. are supported by The Mark Foundation for Cancer Research. A.J.M. and J.Q. are supported by the Parker Institute for Cancer Immunotherapy and by a program project grant from the NIH (1P01CA210944-01).
Footnotes
DECLARATION OF INTERESTS
A.J.M. has received research funding from Merck. He is a scientific advisor for Takeda, H3Biomedicine, Xilio, and Related Sciences. A.J.M. is an inventor on patents related to the IFN pathway and an inventor on a filed patent related to modified CAR T cells. A.J.M. is a scientific founder for Dispatch Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, et al. (2016). Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 15, 274–287. [DOI] [PubMed] [Google Scholar]
- Annibaldi A, and Meier P. (2018). Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 24, 49–65. [DOI] [PubMed] [Google Scholar]
- Beck RJ, Slagter M, and Beltman JB (2019). Contact-Dependent Killing by Cytotoxic T Lymphocytes Is Insufficient for EL4 Tumor Regression In Vivo. Cancer Res. 79, 3406–3416. [DOI] [PubMed] [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. (2019). Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 178, 933–948.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SB., Kasparcova V., Hoffman S., Swift B., Dare L., Schaeffer M., Capriotti C., Cook M., Finger J., Hughes-Earle A., et al. (2014). Cutting Edge: RIP1 Kinase Activity Is Dispensable for Normal Development but Is a Key Regulator of Inflammation in SHARPIN-Deficient Mice. J. Immunol. 192, 5476–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Serafini P, Mazzoni A, Segal DM, and Zanovello P. (2003). L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 24, 301–305. [DOI] [PubMed] [Google Scholar]
- Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, et al. (2018). LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 175, 429–441.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, Giansanti P, Heck AJR, Dejardin E, Vandenabeele P, et al. (2015). NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 60, 63–76. [DOI] [PubMed] [Google Scholar]
- Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, Liu X, et al. (2020). Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 183, 1714–1731.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, Pölönen P, Hohtari H, Saeed K, Hannunen T, et al. (2020). Immunobiology and Immunotherapy: Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 135, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea C-K, Deng L, Xia Z-P, Pineda G, and Chen ZJ (2006). Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol. Cell 22, 245–257. [DOI] [PubMed] [Google Scholar]
- Feng M, Jiang W, Kim BYS, Zhang CC, Fu Y-X, and Weissman IL (2019). Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ., Long AJ., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz LJ., Malenkovich N., Okazaki T., Byrne MC., et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, and Kroemer G. (2017). Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111. [DOI] [PubMed] [Google Scholar]
- Gier RA, Budinich KA, Evitt NH, Cao Z, Freilich ES, Chen Q, Qi J, Lan Y, Kohli RM, and Shi J. (2020). High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun. 11, 3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O, et al. (2018). Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, et al. (2009). Recruitment of the Linear Ubiquitin Chain Assembly Complex Stabilizes the TNF-R1 Signaling Complex and Is Required for TNF-Mediated Gene Induction. Mol. Cell 36, 831–844. [DOI] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J. (2013). GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra ME, Bornes L, Dijkgraaf FE, Philips D, Pardieck IN, Toebes M, Thommen DS, van Rheenen J, and Schumacher TNM (2020). Long-distance modulation of bystander tumor cells by CD8+ T-cell-secreted IFN-γ. Nat. Cancer 1, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, Murray JI, Raj A, Li M, and Zhang NR (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Zhang Z, and Zhang NR (2020). Dimension reduction and denoising of single-cell RNA sequencing data in the presence of observed confounding variables. BioRxiv 2020.08.03.234765. [Google Scholar]
- Ishwaran H, Kogalur UB, Chen X, and Minn AJ (2011). Random survival forests for high-dimensional data. Statistical Analysis and Data Mining 4, 115–132. [Google Scholar]
- Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, and Krummel MF (2019). Tuning the Tumor Myeloid Microenvironment to Fight Cancer. Front. Immunol. 10, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L., Shah P., Cuoco MS., Rodman C., Su M-J., Melms JC., Leeson R., Kanodia A., Mei S., Lin J-R., et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, Bornstein C, Moshe A, Keren-Shaul H, Cohen M, et al. (2020). Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 182, 872–885.e19. [DOI] [PubMed] [Google Scholar]
- Kearney CJ, Lalaoui N, Freeman AJ, Ramsbottom KM, Silke J, and Oliaro J. (2017). PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death and Differentiation 24, 1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, Pijpers L, Michie J, Brown KK, Knight DA, et al. (2018). Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 3, eaar3451. [DOI] [PubMed] [Google Scholar]
- Kim JK, Kolodziejczyk AA, Ilicic T, Illicic T, Teichmann SA, and Marioni JC (2015). Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat. Commun. 6, 8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuz S, Siegmund D, Scheurich P, and Wajant H. (2001). NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 21, 3964–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova A, Brockhoff PB, and Christensen RHB (2017). lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 82, 1–26. [Google Scholar]
- Li H, Kobayashi M, Blonska M, You Y, and Lin X. (2006). Ubiquitination of RIP Is Required for Tumor Necrosis Factor α-induced NF-κB Activation. J. Biol. Chem. 281, 13636–13643. [DOI] [PubMed] [Google Scholar]
- Maciejewski J, Selleri C, Anderson S, and Young NS (1995). Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 85, 3183–3190. [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, and Tschopp J. (2003). Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 114, 181–190. [DOI] [PubMed] [Google Scholar]
- Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, et al. (2020). TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182, 886–900.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, and Ségui B. (2019). The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SA, and Minn AJ (2018). Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 48, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykratis A., Hermance N., Zelic M., Roderick J., Kim C., Van T-M., Lee TH., Chan FKM., Pasparakis M., and Kelliher MA. (2014). Cutting Edge: RIPK1 Kinase Inactive Mice Are Viable and Protected from TNF-Induced Necroptosis In Vivo. J. Immunol. 193, 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D. (2015). Dose-Response Analysis Using R. PLOS ONE 10, e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedger LM, Shows DM, Blanton RA, Peschon JJ, Goodwin RG, Cosman D, and Wiley SR (1999). IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 163, 920–926. [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. (2016). The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A. (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, Lu XM, Pajarillo R, Agarwal S, Kuramitsu S, et al. (2020). Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 10, 552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D, and Oberst A. (2019). Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 4, eaaw2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Tang Y, Tu H, Zhang J, Zhao X, Wang Y, Qin J, and Lin X. (2019). K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat. Commun. 10, 4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarumoto Y, Lu B, Somerville TDD, Huang Y-H, Milazzo JP, Wu XS, Klingbeil O, El Demerdash O, Shi J, and Vakoc CR (2018). LKB1, Salt-Inducible Kinases, and MEF2C Are Linked Dependencies in Acute Myeloid Leukemia. Mol. Cell 69, 1017–1027.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Introduction. In Modeling Survival Data: Extending the Cox Model, Therneau TM, and Grambsch PM, eds. (New York, NY: Springer; ), pp. 1–6. [Google Scholar]
- Tsuyuzaki K, Ishii M, and Nikaido I. (2019). Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data. BioRxiv 566182. [Google Scholar]
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. (2015). Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Perego M, and Gabrilovich D. (2018). Myeloid-derived suppressor cells coming of age. Nat. Immunol. 19, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, Krijgsman O, Huang X, Kenski JCN, Lacroix R, et al. (2019). Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 178, 585–599.e15. [DOI] [PubMed] [Google Scholar]
- Wang J., Huang M., Torre E., Dueck H., Shaffer S., Murray J., Raj A., Li M., and Zhang NR. (2018a). Gene expression distribution deconvolution in single-cell RNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 115, E6437–E6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, Taunk PS, Wu N, Su W, Wu J, et al. (2018b). RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 34, 757–774.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner KW, Saleh D, and Degterev A. (2017). Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol. Sci. 38, 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, and Green DR (2014). The Two Faces of Receptor Interacting Protein Kinase-1. Mol. Cell 56, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Silva R.B. da, Sousa C.R. e, Green DR, Oberst A, and Albert ML (2015). RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, et al. (2017). Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang H, Xu C, Li X, Li M, Wu X, Pu W, Zhou B, Wang H, Li D, et al. (2019). Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nat. Commun. 10, 4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Holmes BR, Pagès H, Mao H, Lawrence M, Veksler-Lublinsky I, Ambros V, Aronin N, and Brodsky M. (2021). CRISPRseek: Design of target-specific guide RNAs in CRISPR-Cas9, genome-editing systems (Bioconductor version: Release (3.12)). [Google Scholar]
- Zilionis R., Engblom C., Pfirschke C., Savova V., Zemmour D., Saatcioglu HD., Krishnan I., Maroni G., Meyerovitz CV., Kerwin CM., et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Top 50 differentially expressed genes in mouse myeloid clusters and in T and NK cell clusters (Related to Figures 4–5 and S5).
Table S3. CRISPR screen crRNA library for cell death pathway (Related to Figure S7).
Data Availability Statement
Single-cell RNA-seq data have been deposited at the GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources table.
This manuscript analyzes existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse RIPK1 Monoclonal Antibody | BD Biosciences | Cat#610458; RRID:AB_397831 |
| Anti-Rat Caspase-8 Monoclonal Antibody, Clone 1G12 | Enzo Life Sciences | Cat#ALX-804-447-C100; RRID:AB_2050952 |
| Anti-Rabbit GAPDH Monoclonal Antibody, Clone 14C10 | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| Anti-Rabbit SAPK/JNK Polyclonal Antibody | Cell Signaling Technology | Cat#9252; RRID:AB_2250373 |
| Anti-Mouse Phospho-SAPK/JNK (Thr183/Tyr185) Monoclonal Antibody | Cell Signaling Technology | Cat#9255; RRID:AB_2307321 |
| Anti-Rabbit NF-κB p65 Monoclonal Antibody, clone D14E12 | Cell Signaling Technology | Cat#8242; RRID:AB_10859369 |
| Anti-Rabbit Phospho-NF-κB p65 (Ser536) Polyclonal Antibody | Cell Signaling Technology | Cat#3031; RRID:AB_330559 |
| Anti-Rabbit p38 MAPK Polyclonal Antibody | Cell Signaling Technology | Cat#9212; RRID:AB_330713 |
| Anti-Rabbit Phospho-p38 MAPK (Thr180/Tyr182) Monoclonal Antibody, clone 3D7 | Cell Signaling Technology | Cat#9215; RRID:AB_331762 |
| Anti-Rabbit IkBa Polyclonal Antibody | Cell Signaling Technology | Cat#9242; RRID:AB_331623 |
| Anti-Rabbit Caspase-3 Polyclonal Antibody | Cell Signaling Technology | Cat#9662; RRID:AB_331439 |
| Anti-Rabbit Cleaved Caspase-3 (Asp175) Polyclonal Antibody | Cell Signaling Technology | Cat#9661; RRID:AB_2341188 |
| InVivoPlus Anti-Mouse PD-1 (CD279) Antibody, clone RMP1-14 | Bio X Cell | Cat#BE0146; RRID:AB_10949053 |
| InVivoPlus Anti-Mouse PD-L1 (B7-H1) Antibody, clone 10F.9G2 | Bio X Cell | Cat#BE0101; RRID:AB_10949073 |
| InVivoPlus Anti-Mouse CTLA-4 (CD152) Antibody, clone 9H10 | Bio X Cell | Cat#BE0131; RRID:AB_10950184 |
| InVivoMAb Anti-Mouse CD8α, clone 2.43 | Bio X Cell | Cat#BE0061; RRID:AB_1125541 |
| InVivoPlus Anti-Mouse NK1.1 Antibody, clone PK136 | Bio X Cell | Cat#BE0036; RRID:AB_1107737 |
| InVivoPlus Anti-Mouse CSF1R (CD115) Antibody, clone AFS98 | Bio X Cell | Cat#BE0213; RRID:AB_2687699 |
| InVivoPlus Rat IgG2a Isotype Control Antibody, clone 2A3 | Bio X Cell | Cat#BE0089; RRID:AB_1107769 |
| Anti-Mouse CD45.2, clone 104 | BioLegend | Cat#109822; RRID:AB_493731 |
| Anti-Mouse I-A/I-E, clone M5/114.15.2 | BioLegend | Cat#107631; RRID:AB_10900075 |
| Anti-Mouse F4/80, clone BM8 | BioLegend | Cat#123114; RRID:AB_893478 |
| Anti-Mouse CD11b, clone M1/70 | BioLegend | Cat#101243; RRID:AB_2561373 |
| Anti-Mouse CD11c, clone N418 | BioLegend | Cat#117347; RRID:AB_2563654 |
| Anti-Mouse CD103, clone 2E7 | BioLegend | Cat#121416; RRID:AB_2128621 |
| Anti-Mouse PD-L1, clone B7-H1 | BioLegend | Cat#124319; RRID:AB_2563619 |
| Anti-Mouse Ly6C, clone HK1.4 | BioLegend | Cat#128015; RRID:AB_1732087 |
| Anti-Mouse Ly6G, clone 1A8 | BioLegend | Cat#127617; RRID:AB_1877262 |
| Anti-Mouse PD-1, clone Rmp1-30 | BioLegend | Cat#109110; RRID:AB_572017 |
| Anti-Mouse CD3, clone 145-2C11 | BioLegend | Cat#100322; RRID:AB_389322 |
| Anti-Mouse B220, clone RA3-6B2 | BioLegend | Cat#103247; RRID:AB_2561394 |
| Anti-Mouse NKP46, clone 29A1.4 | BioLegend | Cat#137619; RRID:AB_2562452 |
| Anti-Mouse CD8, clone 53-6.7 | eBioscience | Cat#48-0081-82; RRID:AB_1272198 |
| Anti-Mouse CD4, clone rm4-5 | eBioscience | Cat#45-0042-82; RRID:AB_1107001 |
| NK1.1, clone PK136 | eBioscience | Cat#47-5941-82; RRID:AB_2735070 |
| F4/80, clone BM8 | eBioscience | Cat#11-4801-81; RRID:AB_2735037 |
| CD11c, clone HL3 | BD Biosciences | Cat#553801; RRID:AB_395060 |
| TCRb, clone H57-597 | BD Biosciences | Cat#742483; RRID:AB_2740816 |
| Anti-mouse CD11b, clone M1/70 | Thermo Fisher Scientific | Cat#12-0112-82; RRID:AB_2734869 |
| Arg1, clone A1exF5 | Thermo Fisher Scientific | Cat#56-3697-80; RRID:AB_2734832 |
| Granzyme B, clone GB11 | Thermo Fisher Scientific | Cat#GRB17; RRID:AB_2536540 |
| TruStain FcX (anti-mouse CD16/32) Antibody | Biolegend | Cat#101319 |
| IRDye 800CW Goat Anti-Rabbit IgG (H + L) | Licor | Cat#926-32211 |
| IRDye 800CW Goat anti-Mouse IgG (H + L) | Licor | Cat#925-32210 |
| IRDye 800CW Goat Anti-Rat IgG (H + L) | Licor | Cat#926-32219 |
| Bacterial and Virus Strains | ||
| MegaX DH10B electrocompetent cells | Thermo Fisher Scientific | Cat#C640003 |
| One Shot Stbl3 Chemically Competent E. coli | Thermo Fisher Scientific | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse TNF-α | Biolegend | Cat#575204 |
| Recombinant Murine IFN-γ | Pepro Tech | Cat#315-05 |
| Protease Inhibitor Cocktail | Sigma | Cat#P8340 |
| Polyethylenimine | Polysciences | Cat#02371-100 |
| Polybrene | Sigma | Cat#H9268 |
| Matrigel Basement Membrane Matrix, Phenol Red-Free, LDEV-Free, | Corning | Cat#356237 |
| Collagenase Type IV | Gibco | Cat#17104019 |
| Collagenase/Dispase | Sigma | Cat#11097113001 |
| DNAse I | Sigma | Cat#10-104-159-001 |
| ACK lysis buffer | Quality Bio | Cat#118156721 |
| Actinomycin D | Sigma | Cat#A1410 |
| Cycloheximide | Cell Signaling Technology | Cat#2112S |
| Lipofectamine 3000 Transfection Reagent | Invitrogen | Cat#L3000008 |
| NuPAGE LDS Sample Buffer (4X) | Thermo Fisher Scientific | Cat#NP0007 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat#11668027 |
| X-TremeGene™ 9 Transfection Reagent | Roche | Cat#6365779001 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10x Genomics | Cat#PN-120258 |
| Nano-Glo Luciferase Assay System | Promega | Cat#N1110 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7570 |
| GeneArt Site-Directed Mutagenesis Kit | Invitrogen | Cat#A13282 |
| BD Cytometric Bead Array Mouse Flex Set KC | BD Biosciences | Cat#558340 |
| BD Cytometric Bead Array Mouse Flex Set MCP-1 | BD Biosciences | Cat#558342 |
| BD Cytometric Bead Array Mouse Flex Set MIP-1a | BD Biosciences | Cat#558449 |
| BD Cytometric Bead Array Mouse Flex Set IL-10 | BD Biosciences | Cat#562263 |
| BD Cytometric Bead Array Mouse Flex Set GM-CSF | BD Biosciences | Cat#558347 |
| BD Cytometric Bead Array Mouse Flex Set G-CSF | BD Biosciences | Cat#560152 |
| BD Cytometric Bead Array Mouse Flex Set MIG | BD Biosciences | Cat#558341 |
| BD Cytometric Bead Array Human Flex Set RANTES (CCL5) | BD Biosciences | Cat#558324 |
| BD Cytometric Bead Array Human Flex Set IL-8 (CXCL8) | BD Biosciences | Cat#558277 |
| BD Cytometric Bead Array Human Flex Set MCP-1 (CCL2) | BD Biosciences | Cat#558287 |
| LIVE/dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34957 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| NextSeq 500/550 v2.5 Sequencing Kit | Illumina | Cat#20024906 |
| Deposited Data | ||
| Arg1+Trem2+ Myeloid Cell Single-cell RNA Sequencing Data | Katzenelenbogen et al., 2020 | GEO: GSE150877 |
| TCGA Pan-Cancer Data | cBioPortal | |
| Human Melanoma Single-cell RNA Sequencing Data | Sade-Feldman et al., 2018 | GEO: GSE120575 |
| TSA Breast Cancer Wild-type or RIPK1 Deletion Tumors Single-Cell RNA Sequencing Data for CD45+ and CD45- Cell Types | This Manuscript | GEO: GSE196178 |
| Experimental Models: Cell Lines | ||
| B16-F10 Murine Melanoma | ATCC | Cat#CRL-6475; RRID:CVCL_0159 |
| TS/A Murine Mammary Carcinoma (TSA) | Laboratory of Dr. Sandra Demaria | PMID: 19706802 |
| Res 499 (B16 derived) | Laboratory of Dr. Andy Minn | PMID:25754329 |
| MDA-MB-231 Breast Cancer Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0062 |
| Huh-7 Hepatocellular Carcinoma Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0336 |
| A549 Non-Small Cell Lung Cancer Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0023 |
| A375 Melanoma Cells Expressing Cas9 | Laboratory of Dr. Junwei Shi | N/A; RRID:CVCL_0132 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mus musculus | The Jackson Laboratory | Cat#000664; RRID:IMSR_JAX000664 |
| C57BL/6-Prf1tm1Sdz/J (Prf1 KO) Mus musculus | The Jackson Laboratory | Cat#002407; RRID:IMSR_JAX:002407 |
| BALB/cAnNCrl Mus musculus | Charles River Laboratories | Cat#028; RRID:IMSR_CRL:28 |
| Oligonucleotides | ||
| CRISPR/Cas9 Guide RNA targeting murine Ripk1: Ripk1 gRNA #1: GGGTCTTTAGCACGTGCATC Ripk1 gRNA #2: CAGTCGAGTGGTGAAGCTAC | This Manuscript | N/A |
| CRISPR/Cas9 Guide RNA targeting murine Casp8: Casp8 gRNA #1: TGAGATCCCCAAATGTAAGC Casp8 gRNA #2: GTCATCTTCCAGCTTACATT | This Manuscript | N/A |
| CRISPR/Cas9 Guide non-targeting control for mouse experiments: Genomic control gRNA #1: GCGCTTCCGCGGCCCGTTCAA | This Manuscript | N/A |
| CRISPR/Cas9 Guide RNA targeting human RIPK1: hRIPK1 gRNA #1: CTTCCTCTATGATGACGCCC | This Manuscript | N/A |
| CRISPR/Cas9 Guide RNA targeting human RIPK1: hRIPK1 gRNA #2: TGGAAAAGGCGTGATACACA | This Manuscript | N/A |
| CRISPR/Cas9 Guide non-targeting control for human studies: non-targeting control gRNA #1: GAAGATGGGCGGGAGTCTTC | This Manuscript | N/A |
| See Table S1 Related to Figures 2 and S2 for CRISPR/Cas12 Guide RNAs used in CRISPR screens | This Manuscript | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 Plasmid | a gift from Feng Zhang | Addgene Cat#62988; RRID:Addgene_62988 |
| LentiV_Cas9_puro Plasmid | a gift from Christopher Vakoc | Addgene Cat#108100; RRID:Addgene_108100 |
| LRG2.1 Lentiviral expression plasmid of sgRNA with GFP | a gift from Christopher Vakoc | Addgene Cat#108098; RRID:Addgene_108098 |
| psPAX2 lentiviral packaging plasmid | a gift from Didier Trono | Addgene Cat#12260; RRID:Addgene_12260 |
| pMig-Flag-Myc-IRES-GFP retroviral plasmid | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1 retroviral plasmid expressing WT RIPK1 | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1(D138N) retroviral plasmid expressing kinase mutant RIPK1 | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1(K45A) retroviral plasmid expressing kinase mutant RIPK1 | a gift from Douglas Green | N/A |
| pMig-Flag-Myc-IRES-GFP-mRIPK1(K376R) retroviral plasmid expressing scaffolding mutant RIPK1 | This Manuscript | N/A |
| pUMVC Retrovirus Packaging Plasmid | a gift from Bob Weinberg | Addgene Cat#8449; RRID:Addgene_8449 |
| pCMV-VSV-G Envelope Protein Plasmid | a gift from Bob Weinberg | Addgene Cat#8454; RRID:Addgene_8454 |
| pCDH-EF1-FHC Lentiviral Backbone Plasmid | a gift from Richard Wood | Addgene Cat#64874; RRID:Addgene_64874 |
| pCDH-EF1-FHC-CCL2 Lentiviral Plasmid Expressing CCL2 | This Manuscript | N/A |
| pCDH-EF1-FHC-CCL2-CXCL1 Lentiviral Plasmid Expressing CCL2 and CXCL1 | This Manuscript | N/A |
| pNL3.2.NF-κB-RE Plasmid with NF-κB luciferase reporter | Promega | Cat#N1111 |
| EFS-GFP-P2A-Neo-U6-crRNA (pRG212) AsCas12a crRNA expression vector | a gift from Junwei Shi | Addgene Cat#149722; RRID:Addgene_149722 |
| AsCas12a-6xNLS-E174R/S542R (pRG232) Lentiviral expression plasmid of AsCas12a-6xNLS-E174R/S542R | a gift from Junwei Shi | Addgene Cat#149723; RRID:Addgene_149723 |
| Software and Algorithms | ||
| CRISPRseek v3.11 | https://bioconductor.org/packages/CRISPRseek/ | DOI: 10.18129/B9.bioc.CRISPRseek |
| makeblastdb v2.6.0 | https://rdrr.io/github/mhahsler/rBLAST/man/makeblastdb.html | |
| CutAdapt v1.16 | https://cutadapt.readthedocs.io/en/v3.5/ | DOI: 10.14806/ej.17.1.200 |
| gCrisprTools v1.12.0 | https://bioconductor.org/packages/gCrisprTools/ | DOI: 10.18129/B9.bioc.gCrisprTools |
| CellRanger | 10X Genomics | https://www.10xgenomics.com/ |
| Seurat v3.1.5 | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| SAVERCAT | Huang et al., 2020 | |
| GSVA v1.32.0 | Hänzelmann et al., 2013 | DOI: 10.18129/B9.bioc.GSVA |
| scTensor v.2.0.0 | https://bioconductor.org/packages/scTensor/ | DOI: 10.18129/B9.bioc.scTensor |
| lmerTest v3.1-3 | Kuznetsova et al., 2017 | DOI: 10.18637/jss.v082.i13 |
| R Language and Environment | CRAN | https://cran.r-project.org/ |
| survival | Therneau T (2021). A Package for Survival Analysis in R | https://CRAN.R-project.org/package=survival |
| drc dose response analysis | Ritz et al., 2015 | https://CRAN.R-project.org/package=drc |
| randomForestSRC v2.9.3 | Ishwaran et al., 2011 | https://github.com/kogalur/randomForestSRC/ |