

Summary
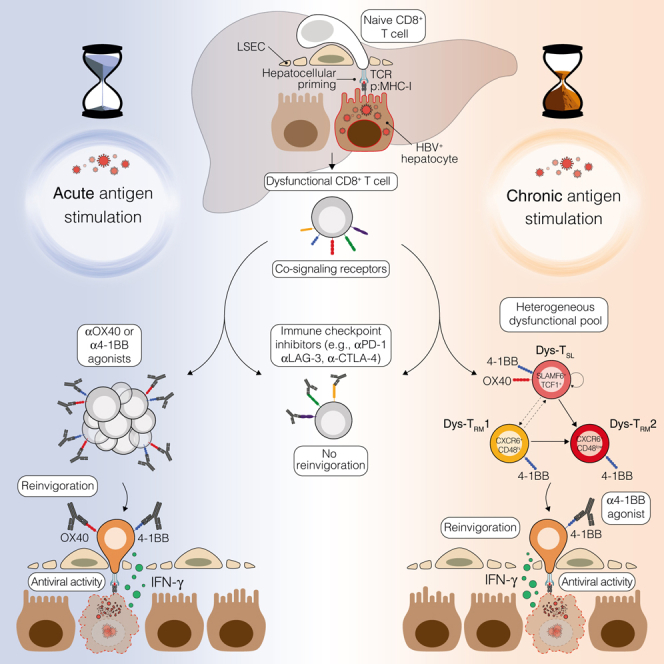
Reversing CD8+ T cell dysfunction is crucial in treating chronic hepatitis B virus (HBV) infection, yet specific molecular targets remain unclear. Our study analyzed co-signaling receptors during hepatocellular priming and traced the trajectory and fate of dysfunctional HBV-specific CD8+ T cells. Early on, these cells upregulate PD-1, CTLA-4, LAG-3, OX40, 4-1BB, and ICOS. While blocking co-inhibitory receptors had minimal effect, activating 4-1BB and OX40 converted them into antiviral effectors. Prolonged stimulation led to a self-renewing, long-lived, heterogeneous population with a unique transcriptional profile. This includes dysfunctional progenitor/stem-like (TSL) cells and two distinct dysfunctional tissue-resident memory (TRM) populations. While 4-1BB expression is ubiquitously maintained, OX40 expression is limited to TSL. In chronic settings, only 4-1BB stimulation conferred antiviral activity. In HBeAg+ chronic patients, 4-1BB activation showed the highest potential to rejuvenate dysfunctional CD8+ T cells. Targeting all dysfunctional T cells, rather than only stem-like precursors, holds promise for treating chronic HBV infection.
Keywords: hepatitis B virus, CD8+ T cells, T cell dysfunction, liver, 4-1BB, OX40, immunotherapy, chronic viral infection
Graphical abstract
Highlights
-
•
Hepatocellular priming induces key co-signaling receptors in dysfunctional CD8+ T cells
-
•
While checkpoint inhibition fails, 4-1BB and OX40 activation restores T cell function
-
•
Prolonged Ag stimulation yields a self-renewing, long-lived, heterogeneous T cell pool
-
•
In chronic settings, only 4-1BB remains effective among further co-receptor modulation
In this study, we traced the trajectory and fate of dysfunctional HBV-specific CD8+ T cells and analyzed the modulation of co-signaling receptors following hepatocellular priming. We identified 4-1BB agonism as the most promising strategy to convert these cells into antiviral effectors for treating chronic HBV infections.
Introduction
Hepatitis B virus (HBV) is a hepatotropic, non-cytopathic DNA virus known for its ability to cause persistent infections, often culminating in cirrhosis and hepatocellular carcinoma.1 HBV has a unique ability to evade detection by the innate immune system, resulting in adaptive cellular and humoral responses taking center stage in mediating the resolution of acute infections.1,2 In particular, CD8+ T cells have a crucial role in viral clearance through their production of antiviral cytokines and elimination of infected hepatocytes.1,2 Nonetheless, the persistence of HBV in most neonatal/perinatal infections and a small subset of adult-onset infections underscores a failure in this adaptive immune response.1
One of the prevailing enigmas in HBV research revolves around the timing and location of T cell priming during infection. Traditional immunological views suggest naive lymphocytes first encounter antigens (Ags) in secondary lymphoid organs. However, recent experiments in mouse models of neonatal HBV infection provide the intriguing alternative that the liver itself may be the main priming site of naive HBV-specific CD8+ T cells.3,4 This unusual setting, facilitated by the unique anatomy of liver sinusoids,5 could have profound implications for the ensuing immune response.
Notably, hepatocellular priming of CD8+ T cells triggers local activation and proliferation, but it does not direct them into effector cells.3,4 Instead, it initiates a unique differentiation program that culminates in a dysregulated T cell phenotype.4 Although these cells appear to respond to interleukin (IL)-2,4,6,7 the potential for various co-inhibitory receptor blockades or agonistic co-stimulation to reverse this dysfunction remains largely unexplored. The significance of this research deepens considering the underwhelming outcomes of the initial clinical trials involving PD-1 blockade.8
Recent studies have identified diverse T cell subsets (e.g., naive-like, cytotoxic, stem cell-like, and exhausted) in chronic infections and cancer.9,10,11,12,13,14,15,16,17,18 These subsets are evolutionarily linked and exhibit varied responsiveness to therapeutic co-signaling receptor modulation.9,10,11,12,13,14,15,16,17,18 The extent to which such heterogeneity arises from hepatocellular priming and its consequent impact on the responsiveness of each subset to immunotherapeutic interventions, however, remains uncertain.
Neonatal/perinatal HBV infections typically proceed through an initial stage of high HBV replication and low liver inflammation, historically labeled the “immunotolerant phase.” Recent studies challenge this notion, uncovering active HBV-specific T cell responses and hepatocarcinogenetic events even during this phase,19,20,21 renamed “high replication, low inflammatory phase” (HRLI). This underscored the need for earlier initiation of therapy. Since antiviral drugs are minimally effective during the early stages of infection,22 immune modulation emerges as a potential complementary strategy to enhance their antiviral impact. Regrettably, our limited understanding of immune responses in young patients restricts the development of novel immunotherapies.
This study seeks to address these gaps, exploring the unique characteristics of T cell dysfunction in HBV infection due to hepatocellular priming and evaluating therapeutic interventions to reverse it.
Results
Immune co-signaling landscape of HBV-specific dysfunctional CD8+ T cells uncovers targets for therapeutic intervention
To unravel the immune co-signaling landscape in HBV-specific dysfunctional CD8+ T cells, we analyzed the expression of co-inhibitory and co-stimulatory receptors (Figure 1A) induced upon HBV Ag recognition in the liver. Our exploration began by analyzing our previously published bulk RNA sequencing (RNA-seq) dataset,4 which offered a high sequencing depth. In this dataset, HBV-transgenic (Tg) mice expressing the HBV core protein in hepatocytes (lineage major urinary protein [MUP]-core23) were adoptively transferred with naive T cell receptor (TCR) Tg Cor93 T cells (Cor93 TN) recognizing an H-2Kb-restricted epitope within the HBV core protein.3 Intrahepatic Cor93 T cells were subsequently isolated at defined time points for bulk RNA-seq analysis (Figure 1B). We observed a selective upregulation of several co-signaling receptors compared with naive cells (Figure 1C). Prominent among the upregulated co-inhibitory receptors were Pdcd1, Ctla4, and Lag3, encoding for PD-1, CTLA-4, and LAG-3, respectively. Conversely, among the early upregulated co-stimulatory receptors were members of the tumor necrosis factor (TNF) receptor superfamily Tnfrsf4 and Tnfrsf9 (encoding for OX40 and 4-1BB, respectively), as well as Icos (ICOS) (Figure 1C).
Figure 1.

Immune co-signaling landscape of HBV-specific dysfunctional CD8+ T cells uncovers targets for therapeutic intervention
(A) Schematic representation of co-signaling molecules.
(B) Experimental setup. Naive Cor93 T cells (Cor93 TN) were transferred into HBV-transgenic (HBV-Tg, lineage MUP-core) recipients. At the indicated time points, Cor93 T cells were sorted from the livers and processed for bulk RNA-seq. From Bénéchet et al.4
(C) Bubble plot of the upregulated co-inhibitory (left) and co-stimulatory (right) genes expressed by Cor93 T cells upon hepatocellular priming. Data are displayed according to the time point of analysis and represent normalized expression using bubble size and a color scale ranging from low (gray) to high (purple).
(D) Experimental setup.
(E) Uniform manifold approximation and projection (UMAP) representation of sorted cells (top left). Each dot corresponds to a single cell, and cells belonging to the same time point are encircled by dotted lines. Feature plots showing the expression of the most upregulated co-signaling receptor genes modulated after hepatocellular priming.
(F) Experimental setup.
(G and H) t-Distributed stochastic neighbor embedding (t-SNE) analysis clustered according to the time point (G) and showing the expression of the indicated co-signaling receptors (H) in Cor93 T cells.
(I) Mean fluorescence intensity (MFI) fold change (FC) of the indicated markers at the indicated time points compared with day 0. n = 3 per time point; two-way ANOVA test with Geisser-Greenhouse correction. Statistical significance compared with day 0 is shown.
See also Figure S1.
To add depth to our findings, single-cell RNA-seq (scRNA-seq) was performed under the same conditions (Figure 1D). This analysis confirmed the induction of Pdcd1, Ctla4, and Lag3 after hepatocellular priming, with Cd160, Tigit, Havcr2, and Cd244a (encoding for CD160, TIGIT, TIM-3, and CD244, respectively) minimally upregulated subsequently (Figure 1E). Also with this analysis, Tnfrsf4, Tnfrsf9, and Icos remained the most induced co-stimulatory genes (Figure 1E), with Tnfrsf9 expression sustained up until day 14. At this late stage, Icos and Tnfrsf4 were expressed in a small fraction of cells.
We then performed flow cytometry, validating the dynamic expression of the most upregulated co-signaling genes at the protein level. To this end, we transferred Cor93 TN cells into HBV replication-competent Tg mice (lineage 1.3.32) that express all HBV Ags and replicate HBV exclusively in the liver at high levels without any evidence of cytopathology24 (Figure 1F). This model is immunological equivalent to MUP-core recipients,4 but it additionally offers the evaluation of CD8+ T cell antiviral activity. t-distributed stochastic neighbor embedding (t-SNE) analyses validated the upregulation of PD-1, LAG-3, CTLA-4, ICOS, 4-1BB, and OX40 in Cor93 T cells upon intrahepatic recognition of HBV, with PD-1 and 4-1BB expression remaining elevated until day 14 (Figures 1G–1I). Similar results were observed using different TCR Tg CD8+ T cells (Env28 T cells, recognizing an H-2Ld-restricted epitope of the HBV envelope protein,3 Figures S1A–S1C) and in a distinct lineage of HBV-Tg mice (Figures S1D–S1F), confirming consistency in our observations.
Figure S1.

Analysis of co-signaling receptors induced in CD8+ T cells upon hepatocellular priming, related to Figure 1
(A) Experimental setup. 106 naive Env28 CD8+ T cells (Env28 TN) were adoptively transferred into HBV replication-competent transgenic mice (lineage 1.3.32; background C57BL/6 × BALB/c H-2bxd hybrids) mice. At the indicated time points, intrahepatic leukocytes were isolated and processed for multicolor flow cytometry. n = 3–4 per group.
(B) t-Distributed stochastic neighbor embedding (t-SNE) analysis of Env28 T cells (gates as live/CD90.1+/CD8+ cells) showing expression of selected co-signaling receptors at the indicated time point upon T cell transfer.
(C) Mean fluorescence intensity (MFI) of the indicated co-signaling receptors at the indicated time points after T cell transfer expressed as fold change (FC) relative to day 0.
(D) Experimental setup. 106 naive Cor93 T cells (Cor93 TN) were adoptively transferred into HBV-Tg mice (lineage MUP-core). At the indicated time points, intrahepatic leukocytes were isolated and processed for multicolor flow cytometry. n = 3 per group.
(E) t-SNE analysis of Cor93 T cells (gated as live/CD45.1+/CD8+ cells) showing expression of selected co-signaling receptors at the indicated time point upon T cell transfer.
(F) MFI of the indicated co-signaling receptors at the indicated time points after T cell transfer expressed as fold change (FC) relative to day 0. For (C) and (F), two-way ANOVA test with Geisser-Greenhouse correction. Statistical significance compared with day 0 is shown.
This investigation provided an atlas of the co-signaling receptors induced in dysfunctional HBV-specific CD8+ T cells upon hepatocellular priming and revealed potential targets for therapeutic intervention.
Intrahepatically primed, dysfunctional CD8+ T cells are unresponsive to co-inhibitory receptor blockade but can be revived by OX40 and 4-1BB agonism
After characterizing the co-signaling landscape in intrahepatically primed, dysfunctional CD8+ T cells, we investigated their sensitivity to co-signaling receptor modulation. To this end, we transferred Cor93 TN cells into HBV replication-competent Tg mice, followed by the administration of blocking monoclonal antibodies (mAbs) against PD-1, LAG-3, or CTLA-4 at saturating concentrations 24 h later (Figure 2A). Remarkably, neither of these treatments induced a substantial increase in the number of intrahepatic leukocytes (IHLs) and Cor93 T cells when compared with PBS-treated, Cor93-injected controls (Figures 2B and 2C). Similarly, immune checkpoint inhibitors did not alter the ability of Cor93 T cells to produce interferon (IFN)-γ upon cognate peptide stimulation or kill Ag-expressing hepatocytes, as shown by unchanged serum alanine transaminase (sALT) levels (Figures 2D–2F). These observations held true in a distinct lineage of HBV-Tg mice (Figures S2A–S2F). Consistent with the absence of a functional differentiation program, these treatments did not have any impact on intrahepatic HBV replication, as determined by southern blot analysis (Figure 2G). Accordingly, there was no difference observed in the cytoplasmic staining of HBcAg, an indirect measure of HBV replication24 (Figure 2H). Also, we found no histological variations or cleaved caspase-3 in hepatocytes (Figure 2H).
Figure 2.

Intrahepatically primed, dysfunctional CD8+ T cells are unresponsive to co-inhibitory receptor blockade but can be revived by OX40 and 4-1BB agonism
(A) Experimental setup.
(B) Numbers of IHL isolated from the indicated mice.
(C) Numbers of Cor93 T cells in the livers of the indicated mice.
(D) Representative plots of IFN-γ expression among Cor93 T cells in the liver of the indicated mice.
(E) Number of IFN-γ-producing Cor93 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 4–5; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(F) Serum alanine transaminases (sALTs) of the indicated groups of mice at the indicated time points.
(G) HBV DNA quantification by southern blot analysis of liver lysates from the indicated mice. Bands corresponding to the size of the integrated transgene (Tg), relaxed circular (RC), double-stranded (DS) linear, and single-stranded (SS) HBV DNAs are indicated.
(H) Representative micrographs of liver sections from the indicated groups of mice. Upper panels show hematoxylin-eosin (H&E) staining, middle panels show staining for HBcAg, and lower panels show staining for cleaved caspase 3 (ΔCas3). Scale bars represent 100 μm.
(I) Experimental setup.
(J) Numbers of IHL isolated from the indicated mice.
(K) Numbers of Cor93 T cells isolated from the liver of the indicated mice.
(L) Representative plots of IFN-γ expression among Cor93 T cells in the liver of the indicated mice.
(M) Number of IFN-γ-producing Cor93 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 4–5; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(N) sALT of the indicated group of mice at the indicated time points. n = 4–5; two-way ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(O) HBV DNA quantification by southern blot analysis of liver lysates from the indicated mice.
(P) Representative micrographs of liver sections from the indicated groups of mice with the indicated stainings. Scale bars represent 100 μm.
See also Figures S2 and S3.
Figure S2.

Validation of co-signaling receptor modulation in independent mouse models, related to Figure 2
(A) Experimental setup. 106 naive Cor93 T (Cor93 TN) cells were adoptively transferred into HBV-Tg mice (lineage MUP-core). 24 h later, indicated groups of mice were injected intraperitoneally with PBS or 100 μg of monoclonal antibodies (mAbs) blocking PD-1, LAG-3, or CTLA-4. Livers were collected and analyzed at day 5.
(B) Total numbers of intrahepatic leukocytes (IHLs) isolated from the indicated mice.
(C) Total numbers of Cor93 T cells in the livers of the indicated mice.
(D) Representative density plots of IFN-γ expression among Cor93 T cells in the liver of the indicated mice. Numbers represent the percentage of cells within the indicated gates.
(E) Total number of IFN-γ-producing Cor93 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 3–4; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(F) Amount of serum alanine transaminases (sALTs) in the serum of the indicated groups of mice at the indicated time points.
(G) Experimental setup. HBV replication-competent transgenic mice (lineage 1.3.32) were injected intraperitoneally with PBS or with 100 μg of agonist mAbs activating OX40 or 4-1BB. Livers were collected and analyzed at day 4.
(H) Total numbers of IHL isolated from the indicated mice.
(I) Representative micrographs of liver sections from the indicated groups of mice. The upper panels show hematoxylin-eosin (H&E) staining, the middle panels show immunohistochemical staining for cleaved caspase 3 (ΔCas3, brown), and the lower panels show immunohistochemical staining for HBcAg (brown). Scale bar represents 100 μm.
(J) HBV DNA quantification by southern blot analysis of liver lysates from the indicated mice. Bands corresponding to the expected size of the integrated transgene (Tg), relaxed circular (RC), double-stranded (DS) linear, and single-stranded (SS) HBV DNAs are indicated.
(K) Amount of sALT in the serum of the indicated groups of mice at the indicated time points.
(L) Experimental setup. 106 Cor93 TN cells were adoptively transferred into HBV-Tg mice (lineage MUP-core). 24 h later, selected groups of mice were injected intraperitoneally with PBS or 100 μg of mAbs activating ICOS, OX40, or 4-1BB. Livers were collected and analyzed at day 5.
(M) Total numbers of IHL isolated from the indicated mice.
(N) Numbers of Cor93 T cells isolated from the liver of the indicated mice.
(O) Representative density plots of IFN-γ expression among Cor93 T cells in the liver of the indicated mice.
(P) Total number of IFN-γ-producing Cor93 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 3–4; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction for multiple comparisons. Each group was compared with control.
(Q) Amount of sALT in the serum of the indicated group of mice at the indicated time points. n = 3–4; two-way ANOVA test with Dunnett correction for multiple comparisons. Each group was compared with control group (simple effect within row).
(R) Experimental setup. 106 naive Env28 CD8+ TCR transgenic cells (Env28 TN) were adoptively transferred into HBV replication-competent transgenic mice (lineage 1.3.32; background C57BL/6 × BALB/c H-2bxd hybrids). 24 h later, selected groups of mice were injected intraperitoneally with PBS or 100 μg of mAbs activating OX40 or 4-1BB. Livers were collected and analyzed at day 5.
(S) Numbers of IHL isolated from the indicated mice.
(T) Numbers of Env28 T cells isolated from the liver of the indicated mice.
(U) Representative density plots of IFN-γ expression among Env28 T cells in the liver of the indicated mice.
(V) Percentages of IFN-γ-producing Env28 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 3–4; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(W) Amount of sALT in the serum of the indicated groups of mice at the indicated time points. n = 3–4; two-way ANOVA test with Dunnett correction for multiple comparisons. Each group was compared with PBS-injected controls (simple effect within row).
(X) Representative micrographs of liver sections from the indicated groups of mice. The upper panels show staining for HBcAg, and the lower panels show staining for cleaved caspase 3 (ΔCas3). Scale bar represents 100 μm.
(Y and Z) Representative histograms (Y) and percentages (Z) of in vitro differentiated Cor93 T effector (Cor93 TE) or Env28 T effector (Env28 TE) cells producing IFN-γ upon cognate in vitro peptide stimulation at the indicated concentrations. Results are representative of two independent experiments giving similar results.
We further evaluated the effect of agonist mAbs activating ICOS, OX40, or 4-1BB on dysfunctional CD8+ T cells under the same conditions (Figure 2I). Agonism of OX40 and 4-1BB, but not ICOS, triggered an increase in IHL and led to an almost 100-fold expansion of intrahepatic Cor93 CD8+ T cells with enhanced IFN-γ-producing and cytotoxic abilities (Figures 2J–2N). Consequentially, OX40 and 4-1BB agonism resulted in suppression of intrahepatic HBV replication, induced a complete loss of cytoplasmic HBcAg staining, and triggered the formation of necro-inflammatory clusters scattered throughout the liver parenchyma (Figures 2O and 2P). These findings were completely dependent on the transferred HBV-specific CD8+ T cells (Figures S2G–S2K), and the results were reproduced in an independent lineage of HBV-Tg mice (Figures S2L–S2Q). By extending our investigations to distinct TCR Tg CD8+ T cells (Env28 T cells), we found that αOX40 mAb-treatment moderately promoted T cell expansion and effector differentiation, while 4-1BB had a superior effect (Figures S2R–S2X). The underlying reason for this observation could be related to the lower TCR affinity of Env28 T cells compared with Cor93 T cells (Figures S2Y and S2Z).
Intravital imaging further revealed that OX40 and 4-1BB agonism induced dense clusters of largely immotile cells scattered throughout the liver parenchyma, contrasting the loose periportal clusters of relatively motile cells in dysfunctional Cor93 T cells (Figure S3; Videos S1 and S2).
Figure S3.

Spatiotemporal dynamics of HBV-specific CD8+ T cells upon 4-1BB and OX40 activation, related to Figure 2
(A) Experimental setup. 106 fluorescent naive Cor93 T (Cor93 TN) cells were adoptively transferred into HBV-Tg mice (lineage MUP-core). 24 h later, selected group of mice were injected intraperitoneally with 100 μg of monoclonal agonist antibody against OX40. 2 days later, mice were injected with Qtracker quantum dots (Qdots) to visualize blood vessels, and the dynamics of Cor93 T cells were assessed by multiphoton intravital microscopy (MP-IVM).
(B–D) Quantitative analyses of Cor93 T cell behavior in the mouse liver. Single-cell speed (B), meandering index (C), and arrest coefficient (D, calculated for a threshold of 3 μm/min). Mann-Whitney test.
(E) Representative time-lapse images (left) showing Cor93 T cells (red) and sinusoids (white) in PBS-injected controls or αOX40-treated mice. Scale bar represents 50 μm. Time-color coding is used to superimpose frames, providing a visualization of cell movement over time, with the outlines of the cells clearly delineated (right).
(F) Representative confocal micrograph of the liver of the indicated mice 3 days after T cell transfer. Sinusoids are stained with anti-CD38 Abs (white), while Cor93 T cells are depicted in red. Scale bars represent 100 μm.
(G) 106 fluorescent Cor93 TN were adoptively transferred into HBV-Tg mice (lineage MUP-core). 24 h later, selected group of mice were injected intraperitoneally with 100 μg of monoclonal agonist antibodies against 4-1BB. 2 days later, mice were injected with Qtracker quantum dots (Qdots) to visualize blood vessels, and the dynamics of Cor93 T cells were assessed by MP-IVM.
(H–J) Quantitative analyses of Cor93 T cell behavior in the mouse liver. Single-cell speed (H), meandering index (I), and arrest coefficient (J, calculated for a threshold of 3 μm/min). Two-tailed Mann-Whitney test.
(K) Representative time-lapse images (left) showing Cor93 T cells (red) and sinusoids (white) in PBS-injected controls or α4-1BB-treated mice. Scale bar represents 50 μm. Time-color coding is used to superimpose frames, providing a visualization of cell movement over time, with the outlines of the cells clearly delineated.
(L) Representative confocal microscopy of the liver of the indicated mice 3 days after T cell transfer. Sinusoids are stained with anti-CD38 Abs (white), while Cor93 T cells are red. Scale bars represent 100 μm.
In conclusion, while immune checkpoint inhibitors exhibit minimal effects on intrahepatically primed dysfunctional CD8+ T cells, OX40 and 4-1BB agonism can convert these cells into potent antiviral effectors.
Intrahepatic priming generates a stable and heterogeneous pool of dysfunctional memory-like CD8+ T cells
In the experiments described thus far, mice received treatment 1 day following the transfer of HBV-specific CD8+ T cells. We sought to investigate whether manipulating co-signaling receptors could counteract the deeper dysfunction resulting from hepatic priming and extended Ag stimulation, conditions mirroring chronic HBV (CHB) infection in humans. Recent studies in chronic infections and cancer have discerned unique T cell subsets (e.g., naive-like, cytotoxic, stem cell-like, and exhausted), which have evolutionary links and differ in their responsiveness to therapeutic co-signaling receptor modulation.9,10,11,12,13,14,15,16,17,18 Yet, it is unclear whether such phenotypic heterogeneity arises from hepatocellular priming and extended Ag stimulation and, eventually, how each subset responds to co-signaling receptor manipulation.
To address this, we examined Cor93 T cells 28 days after adoptive transfer into HBV-Tg mice. These hepatocellularly (HC)-primed dysfunctional cells were compared with control Cor93 T cells injected into wild-type (WT) mice transduced with a replication-defective recombinant lymphocytic choriomeningitis-based vector (rLCMV-core) that targets the same HBV core protein to Kupffer cells (KCs) and dendritic cells, which are not naturally infected by HBV (Figure 3A). Notably, in rLCMV-core-injected mice, as opposed to HBV-Tg mice, Cor93 T cells are primed by KC4 and evolve into genuine effector cells that eliminate Ags from the liver (Figure S4A). These cells, referred to as KC-primed, mature into functional IFN-γ-producing memory cells and stand in contrast to HC-primed cells, which lack the ability to kill Ag-expressing hepatocytes and produce antiviral cytokines (Figures S4B and S4C).
Figure 3.

Intrahepatic priming generates a stable and heterogeneous pool of dysfunctional memory-like CD8+ T cells
(A) Experimental setup. 106 Cor93 TN cells were adoptively transferred into WT mice (C57BL/6) previously transduced with rLCMV-core 4 h before or into HBV-Tg mice (MUP-core). Cor93 T cells from the first group (rLCMV-core) will be referred to as “KC-primed” (blue), while those from the HBV-Tg group will be labeled “HC-primed” (red). Livers were collected at day 28, and Cor93 T cells were analyzed or FACS-sorted and processed for scRNA-seq.
(B) Number of Cor93 T cells in the liver of the indicated mice 28 days after transfer. n = 17, pooled from 4 independent experiments; two-tailed Mann-Whitney test.
(C) Heatmap of normalized and scaled expression values of 106 marker genes characterizing the 2 groups. Genes representative of each group are indicated.
(D) Heatmap showing the frequency of Cor93 T cells expressing the indicated markers by flow cytometry. n = 6–7, pooled from 2 experiments.
(E) UMAP projection of 18,489 sorted and sequenced Cor93 T cells. Each dot corresponds to a single cell, colored according to experimental groups (upper) or unbiasedly clustered using Louvain algorithm (lower). Dys-TSL, dysfunctional stem-like T cells; Dys-TRM1 and Dys-TRM2, dysfunctional tissue-resident memory T cells subset 1 and subset 2, respectively; TRM, tissue-resident memory T cells; TCM, central-memory T cells.
(F) Feature plot representation of the normalized expression level of selected genes.
(G) Bubble plot representation of the scaled expression level of a manually curated list of genes in clusters identified in (E). Dot size correlates with cell marker expression percentage per cluster, while color scale indicates average gene expression per cluster.
(H) Violin plots showing the normalized expression of Cd48 in the HC-primed clusters.
(I) Representative gating strategy (left) and frequencies (right) of the three subpopulations among total Cor93 T cells determined by flow cytometry. n = 8; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction.
(J) t-SNE representation of Cor93 T cell subpopulations determined by flow cytometry.
(K) t-SNE representation of the expression of indicated markers on Cor93 T cell clusters.
(L) Frequency of expression of the indicated markers in the three subpopulations determined by flow cytometry. n = 8, pooled from 2 independent experiments.
See also Figure S4.
Figure S4.

HC priming generates a long-lived and heterogeneous pool of dysfunctional memory-like CD8+ T cells, related to Figure 3
(A) Representative immunohistochemical micrographs of liver sections from the indicated groups of mice at day 28, showing HBcAg expression (brown). Scale bar represents 200 μm.
(B) Amount of sALT in the serum of the indicated group of mice at the indicated time points. n = 9–12.
(C) Percentage of IFN-γ-producing Cor93 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 8–10, pooled from 3 independent experiments; Mann-Whitney test.
(D) Multiphoton intravital microscopy was performed between days 28 and 35. Representative time-lapse images showing Cor93 T cells (green) and membranes of host cells (red) in the indicated groups (scale bars represent 10 μm, time mm:ss). Cell motion during time is visualized by white dotted tracks.
(E–G) Quantitative analyses of Cor93 T cell behavior in the mouse liver 28–35 days after T cell transfer. n = 11–14 mice pooled from 3 independent experiments. Single-cell speed (E), arrest coefficient (F, calculated for a threshold of 3 μm/min), and meandering index (G). Mann-Whitney test.
(H) Fraction of Ki67+ Cor93 T cells isolated from the liver of the indicated mice 28 days after adoptive transfer. n = 6–7 per group, pooled from 2 experiments; unpaired two-tailed t test.
(I) Experimental setup. 2 × 104 CD8+ Cor93 TN cells were adoptively transferred into HBV-Tg (MUP-core, HC-primed) or WT (C57BL/6) mice that had been transduced with rLCMV-HBc 4 h before (KC-primed). Livers were collected and analyzed on day 28.
(J) Total numbers of Cor93 T cells per liver in the indicated groups.
(K) Geometric mean fluorescent intensity (gMFI) of IFN-γ in Cor93 T cells isolated from the liver in the indicated groups at the indicated time point upon ex vivo stimulation with or without cognate peptide. n = 3–4; two-way ANOVA.
(L) Experimental setup. 106 naive CD8+ Cor93 TN cells were adoptively transferred into HBV-Tg (MUP-core, HC-primed) or WT (C57BL/6) mice that had been transduced with rLCMV-HBc 4 h before (KC-primed). Livers were analyzed after 3 months.
(M) Total numbers of Cor93 T cells per liver in the indicated groups.
(N) gMFI of IFN-γ in Cor93 T cells isolated from the liver in the indicated groups at the indicated time point upon stimulation with or without cognate peptide. n = 3–4; two-way ANOVA.
(O) Experimental setup. 106 CD8+ Cor93 TN cells were adoptively transferred into HBV-Tg (MUP-core, HC-primed) or WT (C57BL/6) mice that had been transduced with rLCMV-HBc 4 h before (KC-primed). Livers were analyzed after 6 months.
(P) Total numbers of Cor93 T cells per liver in the indicated groups.
(Q) gMFI of IFN-γ in Cor93 T cells isolated from the liver in the indicated groups at the indicated time point upon stimulation with or without cognate peptide. n = 4–5; two-way ANOVA.
(R) Experimental setup. 106 CD8+ Cor93 TN cells were adoptively transferred into HBV replication-competent transgenic (1.3.32, HC-primed) or WT (C57BL/6) mice that had been transduced with rLCMV-HBc 4 h before (KC-primed). Livers were analyzed after 28 days.
(S) Numbers of Cor93 T cells per liver in the indicated groups.
(T) gMFI of IFN-γ in Cor93 T cells isolated from the liver in the indicated groups at the indicated time point upon 4 h stimulation with or without cognate peptide. n = 5–6; two-way ANOVA.
(U) Representative immunohistochemical micrographs of liver sections from the indicated groups of mice, showing HBcAg expression (brown). Scale bar represents 100 μm.
(V) Heatmap showing the percentages of Cor93 T cells expressing the indicated markers in the indicated groups as determined by flow cytometry. Each column represents an individual mouse.
We next compared the motility behavior of dysfunctional and memory cells, as this parameter is linked to Ag recognition and effector functions.4 To this end, intravital microscopy was performed at day 28 using the same setup. Most cells in both conditions were observed crawling intravascularly along the sinusoids (Figure S4D; Video S3). Intravascular KC-primed Cor93 T cells exhibited higher speeds compared with HC-primed Cor93 T cells (Figure S4E), while HC-primed Cor93 T cells displayed a higher arrest coefficient (Figure S4F). Directionality, as indicated by the corrected meandering index, remained unchanged (Figure S4G). The reduced motility of HC-primed relative to KC-primed T cells is consistent with persistent Ag recognition.
To explore the potential heterogeneity within dysfunctional T cells, we performed scRNA-seq of HC- and KC-primed Cor93 T cells sorted from the livers of mice injected 28 days earlier (Figure 3A). HC-primed cells demonstrated remarkable longevity, as we recovered only slightly fewer cells from the liver of HC-primed mice compared with KC-primed (Figure 3B). Interestingly, dysfunctional Cor93 cells were proliferating more at day 28 compared to the memory control cells (Figure S4H), possibly because of persistent Ag presentation, indicating that more cells might be dying upon HC priming. We then determined the highest-ranking differentially expressed genes (DEGs) between KC-primed and HC-primed cells. The two different priming modalities elicited strikingly different gene expression patterns (Figure 3C; Table S1), with KC-primed cells showing high expression of genes linked to a central-memory phenotype (e.g., Lef1, Ccr7, and Tcf7). By contrast, HC-primed cells were characterized by the expression of the transcription factor Tox (critical for T cell exhaustion13), the chemokine receptor Cxcr6, typically expressed by liver-resident memory (TRM) cells,25 co-stimulatory (Tnfrsf9) and co-inhibitory (e.g., Lag3 and Pdcd1) receptors (Figure 3C; Table S1). We confirmed the presence of prominent marker genes at the protein level, demonstrating that HC-primed cells showed expression of the liver TRM markers CD69 and CXCR6, co-inhibitory receptors PD-1 and LAG-3, as well as TOX (Figure 3D). IFN-γ, however, was not expressed.
Similar results were obtained decreasing the number of transferred Cor93 T cells (Figures S4I–S4K), prolonging the experiment up to 3 (Figures S4L–S4N) and 6 months (Figures S4O–S4Q), and using HBV replication-competent Tg mice (Figures S4R–S4T). HBV-specific T cells persisted in similar numbers in all these settings and maintained their dysfunctional phenotype as they lacked IFN-γ production (Figures S4K, S4N, S4Q, and S4T). Consistently, these dysfunctional cells did not exert any antiviral activity, as shown by the persistence of cytoplasmic HBcAg in HBV replication-competent Tg mice (Figure S4U). Similarly, we detected the core HC-primed signature described above also in HBV replication-competent mice (Figure S4V). These results indicate that hepatocellular priming triggers a durable, memory-like T cell population with a dysfunctional phenotype.
To examine the potential heterogeneity in this population, we created a uniform manifold approximation and projection (UMAP) representation of the scRNA-seq dataset. This revealed that HC- and KC-primed T cells clustered distinctly (Figure 3E, upper panel). We conducted unbiased clustering and manually annotated the different clusters based on the characteristics of DEGs (Figure 3E lower panel). Whereas KC-primed cells could be classified into conventional TRM and central-memory cells (TCM), HC-primed T cells were annotated into four different clusters (Figure 3E, lower panel; Table S2). Since two of them showed a dysfunctional TRM-like gene signature, characterized by the expression of genes such as Cxcr6, as well as Pdcd1, Tox, Lag3, Klrd1, and Klrc1, they were termed Dys-TRM1 and Dys-TRM2. Moreover, we identified a cluster of cells with a stem-like phenotype (Dys-TSL), characterized by the expression of Tcf7, Slamf6, and Xcl1, and one cluster of Mki67high proliferating cells (Figures 3F and 3G; Table S2). Based on differential gene expression analysis, we identified Slamf6 and Cd48 as potential marker genes to distinguish the three dysfunctional subsets (Figures 3G and 3H). We validated these markers at the protein level and, using the gating strategy delineated in Figure 3I, we obtained a similar proportion of dysfunctional subpopulations, with the majority of cells being Dys-TRM2 (Figure 3I, right panel). We also confirmed the expression of key markers on the three subpopulations at the protein level (Figures 3J–3L). Consistent with earlier publications,26 TCF1 (encoded by Tcf7) showed a strong correlation with SLAMF6 (Figures 3J–3L), allowing them to be used interchangeably as Dys-TSL markers.
In conclusion, our findings indicate that hepatocellular priming induces a long-lasting, heterogeneous, dysfunctional CD8+ T cell population.
Developmental relationship between the three subpopulations induced by hepatocellular priming
To unravel the developmental relationships among the different subpopulations induced by HC priming, we carried out RNA velocity analysis. This method generates high-dimensional vectors that predict the future state of individual cells27 (Figure 4A). From this analysis, we identified that Dys-TRM1 and Dys-TSL, along with proliferating cells, primarily contributed to the major Dys-TRM2 subset (Figure 4A). This was corroborated by a higher percentage of Ki67+ cells in Dys-TRM1 and Dys-TSL compared with Dys-TRM2 (Figure 4B). We then devised an adoptive transfer experiment to examine the developmental potential of Dys-TSL, Dys-TRM1, and Dys-TRM2. We fluorescence-activated cell sorting (FACS)-sorted these 3 subsets 28 days after HC priming and transferred them into rLCMV-core-transduced WT mice (Figure 4C). While all subpopulations showed limited engraftment, Cor93 T cells derived from mice injected with Dys-TSL and Dys-TRM1 exhibited notably higher Ki67 staining compared with those from mice injected with Dys-TRM2 (Figure 4D). Next, we assessed the relative plasticity of each transferred subset using TCF1 and CD48 markers (Figure 4E). All transferred populations demonstrated a strong predilection for generating Dys-TRM2 (Figure 4F). Interestingly, Dys-TRM2 cells were found to only give rise to their own subset. By contrast, Dys-TSL and, to a lesser extent, Dys-TRM1 cells showed the ability to differentiate into all subsets (Figure 4F).
Figure 4.

Developmental relationship between the three subpopulations induced by hepatocellular priming
(A) RNA velocity plotted in the UMAP space for HC-primed Cor93 T cells at day 28. Arrows indicate the location of the estimated future cell state. Each dot corresponds to a single cell, colored according to the unbiased clusters identified in Figure 3E.
(B) Frequencies of Ki67+ Dys-TSL, Dys-TRM1, and Dys-TRM2 cell populations 28 days after transfer. n = 8; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction.
(C) Experimental setup. Cor93 T cells were isolated from the livers at day 28 after HC priming, and Dys-TSL, Dys-TRM1, and Dys-TRM2 were FACS-sorted according to the expression of CD48 and SLAMF6. Sorted populations were transferred into rLCMV-core-transduced WT recipients analyzed 5 days later.
(D) Ki67 expression of Cor93 T cells recovered in the liver of the indicated mice 5 days after transfer of FACS-sorted Dys-TSL, Dys-TRM1, or Dys-TRM2. n = 3; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction.
(E and F) Representative plots (E) and frequencies (F) of Dys-TSL, Dys-TRM1, and Dys-TRM2 output subpopulations recovered in the livers of mice injected with the indicated input cell populations. n = 3 per group; two-way ANOVA.
(G) Experimental setup. Cor93 T cells were isolated from the livers at day 28 after HC priming, and Dys-TSL and Dys-TRM were CD45.1 MACS-enriched and then FACS-sorted according to the expression of SLAMF6. Sorted subpopulations were transferred into HBV-Tg mice (lineage 1.3.32), and the recipients were analyzed 5 days later.
(H) Representative plots indicating the purity of the indicated cells after sorting.
(I and J) Representative plots (I) and frequencies (J) of Dys-TSL and Dys-TRM output subpopulations recovered in the livers of mice injected with the indicated input cell populations. n = 3 per group; two-way ANOVA.
(K) Percentage of Ki67+ Cor93 T cells recovered from the liver of the indicated mice 5 days after transfer. n = 3 per group; two-tailed Mann-Whitney test.
(L) Graphical representation depicting the current model of Dys-T developmental hierarchy.
To provide deeper insights into the potential fates of these T cell subsets within the context of chronic hepatic Ag exposure, we conducted additional transfer experiments into HBV-Tg mice using Dys-TSL and Dys-TRM subsets sorted from HC-primed mice (Figure 4G). Dys-TSL cells exhibited the capacity to differentiate into both Dys-TSL and Dys-TRM subsets, highlighting their potential for plasticity and proliferation (Figures 4H–4K). By contrast, transferred Dys-TRM cells maintained their TRM phenotype, reinforcing their state of terminal differentiation and limited proliferative ability (Figures 4H–4K).
Collectively, our findings outline the hierarchical relationship among the subpopulations induced by HC priming (Figure 4L).
Intrahepatically primed CD8+ T cells show a unique dysfunctional profile that does not completely overlap with exhaustion
Next, we aimed to compare the gene expression profile in the three T cell subsets produced by HC priming with that of classical T cell exhaustion. To do so, we utilized a previously published scRNA-seq dataset26 of splenic CD8+ T cells from mice at day 28 after chronic LCMV infection. Specifically, we compared the gene expression program of the T cell subsets induced in chronic LCMV infection (i.e., effector-like, exhausted [TEX], and precursor of exhausted [TPEX]) with the three T cell subpopulations resulting from HC priming. Although Dys-TRM shared a core signature with TEX, including prototypical genes such as Tox and Pdcd1 (Figure 5A, red squares), they expressed a unique set of genes, including Tnfrsf9, Prf1, Klrc1, and Klrd1 (Figure 5A, orange squares). Likewise, Dys-TSL exhibited a shared set of core genes with TPEX, such as Slamf6 and Tcf7 (Figure 5A, green squares), but uniquely expressed genes like Tnfrsf4 (Figure 5A, blue squares). Unlike chronic LCMV infection, HC priming did not give rise to effector-like cells (Figure 5A, right column). This is also reflected by the weak enrichment of the effector-like signature in the Dys-TRM subsets (Figure 5B). This comparison revealed that the dysfunctionality caused by HC priming is different from the T cell exhaustion observed during chronic LCMV infection, indicated by the poor enrichment of the TEX signature on the Dys-TRM population (Figure 5B). By contrast, a strong enrichment of the TPEX signature was observed on the Dys-TSL population (Figure 5B), indicating that these cells are transcriptionally similar.
Figure 5.

Upon chronic antigenic stimulation, only 4-1BB but not OX40 stimulation results in significant T cell reinvigoration
(A) Heatmap displaying the expression of a selection of genes. Data on the left derived from the scRNA-seq dataset conducted in our study, while the data on the right originates from the dataset presented in Miller et al.26 Specific cell subsets are labeled above the heatmap. Highlighted squares indicate comparisons between subsets identified in both studies: genes common to both the Dys-TRM and TEX subsets (red), genes exclusive to the Dys-TRM subsets (orange), genes shared between the Dys-TSL and TPEX subsets (green), and genes specific to Dys-TSL (blue). Color scale indicates the normalized/scaled expression.
(B) UMAP projection (top left) and feature plots of signatures extracted from the re-analysis of the scRNA-seq dataset of Miller et al.,26 showing the enrichment of the TPEX (top right), TEX (bottom left), and effector-like (bottom right) signatures on the UMAP from Figure 3E. Color scale indicates the signature score.
(C and D) Violin plots showing the normalized expression profile of Tnfrsf4 (C) and Tnfrsf9 (D) on the subsets described in (A).
(E) Experimental setup.
(F) Numbers of IHL isolated from the liver of the indicated mice.
(G) Numbers of Cor93 T cells isolated from the liver of the indicated mice. n = 4–5; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(H and I) Representative FACS plots (H) and percentages (I) of Dys-TSL, Dys-TRM1, and Dys-TRM2 Cor93 T cell subpopulations in the groups described in (E).
(J and K) Representative plots (J) and fractions (K) of IFN-γ-producing Dys-TSL, Dys-TRM1, and Dys-TRM2 Cor93 T cell subpopulations in the groups described in (E).
(L and M) Representative plots (L) and percentages (M) of CCL5+ Dys-TSL, Dys-TRM1, and Dys-TRM2 Cor93 T cell subpopulations in the groups described in (E).
(N and O) Representative plots (N) and percentages (O) of Granzyme A (GzmA)+ Dys-TSL, Dys-TRM1, and Dys-TRM2 Cor93 T cell subpopulations in the groups described in (E). n = 4–5; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(P) sALT of the indicated groups of mice at the indicated time points. n = 4–5; two-way ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls (simple effect within row).
(Q) Representative immunohistochemical micrographs of liver sections from the indicated groups of mice stained for cleaved caspase 3 (ΔCas3). Scale bar represents 100 μm.
(R) Experimental setup.
(S) Representative plots of IFN-γ expression among Cor93 T cells in the liver of the indicated mice. Numbers represent the percentage of cells within the indicated gates.
(T) Frequencies of IFN-γ-producing Cor93 T cells in the livers of the indicated mice upon ex vivo cognate peptide stimulation. n = 3–4; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
(U) Representative micrographs of liver sections from the indicated groups of mice showing immunohistochemical staining for HBcAg. Scale bars represent 100 μm.
Upon chronic antigenic stimulation, only 4-1BB but not OX40 stimulation results in significant T cell reinvigoration
The abovementioned scRNA-seq analysis revealed that Tnfrsf4 (encoding for OX40) is expressed exclusively by the Dys-TSL population, while Tnfrsf9 (encoding for 4-1BB) is expressed by all HC-primed dysfunctional cells (Figures 5C and 5D). To evaluate the effects of OX40 and 4-1BB activation on dysfunctional T cells under chronic Ag stimulation, we transferred Cor93 T cells into HBV-Tg recipients and injected them with agonist antibodies activating OX40 or 4-1BB 28 days later (Figure 5E). Interestingly, we found that only 4-1BB activation, not OX40, led to an increase in the total IHL (Figure 5F) and in the number of Cor93 T cells (Figure 5G). Looking at the distribution among dysfunctional subpopulations, we noted that 4-1BB activation boosted the expansion of the SLAMF6+CD48low Dys-TSL population, at the expense of SLAMF6−CD48low Dys-TRM2, while SLAMF6−CD48high Dys-TRM1 cells remained unaffected by the treatments (Figure 5I). When assessing CD8+ T cell effector functions, only Dys-TSL produced IFN-γ in response to OX40 activation (Figures 5J and 5K), along with the secretion of CCL5 (Figures 5L and 5M) and Granzyme A (Figures 5N and 5O), as assessed by decreased staining upon cognate Ag stimulation. This aligns with the notion that OX40 expression is limited to this subset. By contrast, 4-1BB activation endowed all distinct TRM populations with the ability to produce and secrete effector molecules (Figures 5J–5O). Consistent with this, 4-1BB—but not OX40—activation promoted CD8+ T cell ability to lyse Ag-expressing hepatocytes, as evidenced by increased sALT levels (Figure 5P) and cleaved caspase-3 staining (Figure 5Q). Moreover, when tested in HBV replication-competent mice, 4-1BB activation—but not OX40 activation or PD-1 inhibition—induced cytoplasmic HBcAg clearance, indicating inhibition of HBV replication (Figures 5R–5U).
Collectively, our findings suggest that, in CD8+ T cells with established dysfunction, 4-1BB is a promising therapeutic target to restore the unique dysfunctional state of HBV-specific T cells in chronic settings.
Mechanistic insights into 4-1BB-mediated reinvigoration of dysfunctional T cells
To unravel the mechanisms behind the functional impairments of HC-primed T cells and the restorative effects of 4-1BB agonism, we analyzed the Ag-sensing capabilities of dysfunctional T cells. We found diminished expression of TCR and CD3 in HC-primed T cells compared with KC-primed counterparts, indicating persistent intrahepatic Ag engagement (Figures S5A–S5C). This notion was reinforced by evidence of unimpaired CD3ζ phosphorylation following peptide stimulation, suggesting intact proximal TCR signaling (Figure S5D). Intravital microscopy already underscored this by showing reduced motility among HC-primed T cells—a hallmark of active Ag sensing (Figures S4D–S4G). Remarkably, unlike in KC-primed conditions, the systemic administration of the cognate peptide did not alter the motility of HC-primed cells, highlighting their continuous engagement with Ag in vivo (Figures S5E and S5F).
Figure S5.

Antigen sensing, cytolytic function, and effector molecule expression in HC-primed CD8+ T cells before and after 4-1BB treatment, related to Figure 6
(A) Experimental setup. 106 Cor93 TN cells were adoptively transferred into WT mice (C57BL/6 background) previously transduced with rLCMV-core 4 h before (KC-primed) or into HBV-Tg mice (MUP-core). Livers were collected and analyzed at day 7.
(B) Representative histograms (left) and median fluorescence intensity (MFI, right) of Cor93–100:Dimer staining on Cor93 T cells (defined as live/CD45.1+/CD8+) isolated from the liver of the indicated mice.
(C) Representative histograms (left) and MFI (right) of CD3 staining on Cor93 T cells isolated from the liver or the spleen of the indicated mice.
(D) Representative histograms (left) and MFI quantification (right) of CD3ζ-pY142 detected by BD Phosflow protocol on Cor93 T cells isolated from the liver of the indicated mice at the indicated conditions. n = 3/4 per group; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction for multiple comparisons.
(E) Experimental setup. 106 fluorescent Cor93 TN cells were adoptively transferred into WT mice (C57BL/6 background) previously transduced with rLCMV-core 4 h before or into HBV-Tg mice (lineage MUP-core). Mice were injected or not with Cor93–100 peptide at day 28, and Cor93 T cell behavior was assessed by multiphoton intravital microscopy (MP-IVM) 4 h later.
(F) Single-cell speed of Cor93 T cells in the liver of the indicated mice in the indicated condition. Mann-Whitney test.
(G) Schematic representation of the experimental setup. HBV-Tg mice (1.3.32 lineage) were adoptively transferred with Cor93 TN and challenged 24 h later with or without 4-1BB agonist antibody. 5 days after T cell transfer, intrahepatic Cor93 T cells were isolated and plated with a mixture of C57BL/6 splenocytes pulsed or not with cognate Cor93–100 peptide. Unpulsed and pulsed target cell populations were labeled with low or high amount of cell trace violet (CTV), respectively, and mixed at 1:1 ratio prior to coculture. Cocultured cells were incubated for 4 or 16 h and analyzed by flow cytometry.
(H and I) Representative histograms (H) and quantification (I) depicting the specific killing of CTVhigh peptide-pulsed population in the indicated conditions. n = 3/4 per group; two-way ANOVA test with Geisser-Greenhouse correction.
(J) Schematic representation of the experimental setup. HBV replication-competent transgenic mice (lineage 1.3.32) were adoptively transferred with Cor93 TN and challenged 24 h later with or without 4-1BB agonist antibody. Livers were collected and analyzed 5 days after adoptive transfer.
(K–O) Representative histograms (left) and fractions (right) of FasL (K), TNF-α (L), IFN-γ (M), GzmB (N), and CD107a (O) in the indicated cell populations isolated from the liver of the indicated mice. n = 3/4 per group; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction. Each group was compared with PBS-injected controls.
Despite such engagement and signaling, HC-primed T cells demonstrated a pronounced inability to eliminate peptide-pulsed targets in vitro, a limitation not observed in T cells post α4-1BB treatment (Figures S5G–S5I). We posited that this cytolytic shortfall stemmed from insufficient expression of key cytotoxic molecules. Supporting this, HC-primed T cells lacked substantial levels of critical antiviral mediators like FasL, TNF-α, IFN-γ, and negligible GzmB expression (Figures S5J–S5N). By contrast, 4-1BB activation triggered the production of these effector molecules, alongside increased degranulation (assessed by CD107a staining, Figures S5J–S5O), indicating a bottleneck in effector molecule expression as a critical deficit.
Further explorations aimed to ascertain whether 4-1BB agonism fosters the expansion of a latent functional subset or catalyzes a broad functional conversion (Figure 6A). We observed no significant T cell proliferation 24 h after 4-1BB agonisms, as evidenced by stable cell counts and subpopulation frequencies in both treated and untreated mice (Figures 6B and 6C). Despite this, we detected enhanced IFN-γ production across multiple T cell subsets (Figures 6D and 6E), suggesting that 4-1BB promotes functional conversion at early time points, independent of proliferation. This was corroborated by in vitro studies revealing that 4-1BB agonism prompted functional conversion across diverse T cell subsets, as evidenced by augmented IFN-γ production among both proliferating (Ki67+) and quiescent cells (Figures 6F–6I).
Figure 6.

Mechanism of action and plasticity of dysfunctional T cell subpopulations to 4-1BB agonism upon chronic antigenic stimulation
(A) Experimental setup.
(B) Numbers of Cor93 T cells isolated from the liver of the indicated mice.
(C) Representative FACS plots (left) and percentages (right) of Dys-TSL, Dys-TRM1, and Dys-TRM2 Cor93 T cell subpopulations in the indicated groups.
(D and E) Representative FACS plots (D) and fractions (E) of IFN-γ-producing Dys-TSL, Dys-TRM1, and Dys-TRM2 Cor93 T cell subpopulations in the indicated groups. n = 3–5; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction.
(F) Experimental setup. 106 Cor93 TN cells were adoptively transferred in HBV-Tg mice (MUP-core). 28 days later, Dys-TSL, Dys-TRM1, and Dys-TRM2 subsets were FACS-sorted. Cells were cultured in the presence of cognate Cor93–100 peptide with or without 4-1BB antibody and analyzed 16 h later.
(G–I) Fractions of proliferating Ki67+ and non-proliferating Ki67− IFN-γ-producing cells among Dys-TSL (G), Dys-TRM1 (H), and Dys-TRM2 (I). n = 3/group; one-way Brown-Forsythe and Welch ANOVA test with Dunnett correction.
(J) Experimental setup. Cor93 TN cells were transferred in HBV-Tg mice. 28 days later, livers were collected, and IHL were cultured in the presence of cognate Cor93–100 peptide with or without αOX40 or α4-1BB agonist antibody. Cells were analyzed 16 h later by flow cytometry.
(K and L) Representative histograms (left) and MFI quantification (right) of OX40 (K) and 4-1BB (L) basal expression levels in endogenous CD8+ T cells, Dys-TRM (TCF1− SLAMF6−), and Dys-TSL (TCF1+ SLAMF6+) Cor93 T cells in unstimulated conditions.
(M and N) Representative histograms (left) and frequencies (right) of IFN-γ (M) and Ki67 (N) expression in Dys-TRM and Dys-TSL Cor93 T cells in the indicated conditions. n = 4 per group; two-way ANOVA.
(O) Representative plots showing different dysfunctional T cell population in the indicated conditions with the same experimental design depicted in (F).
(P–R) Fractions of different CD8+ T cell subsets recovered after culture of FACS-sorted Dys-TSL (P), Dys-TRM1 (Q), and Dys-TRM2 (R) in the indicated conditions. n = 3 per group; two-way ANOVA test with Geisser-Greenhouse correction.
See also Figures S5 and S6.
Addressing the differential outcomes of 4-1BB and OX40 stimulation during chronic Ag exposure, we found OX40 expression limited to the Dys-TSL subset, contrasting with widespread 4-1BB presence across multiple dysfunctional subsets, including Dys-TSL and Dys-TRM (Figures 6J–6L). This broad expression enables 4-1BB to initiate a more comprehensive T cell reinvigoration compared with OX40 more targeted approach (Figures 6M and 6N). Moreover, 4-1BB activation not only boosted IFN-γ production and cell proliferation more effectively than OX40 but also increased genes crucial for survival, proliferation, and effector functions, as demonstrated by bulk RNA-seq (Figure S6; Table S3). Overall, the divergent expression patterns and the potent activation capacity of 4-1BB elucidate the distinct reinvigoration outcomes in the context of chronic Ag stimulation.
Figure S6.

Differential gene expression induced by 4-1BB vs. OX40 stimulation in dysfunctional T cells, related to Figure 6
(A) Experimental setup. 106 Cor93 TN cells were adoptively transferred into HBV replication-competent transgenic mice (lineage 1.3.32). 24 h later, indicated groups of mice were injected intraperitoneally with PBS or 100 μg of αOX40 or α4-1BB agonist. Livers were collected and analyzed at day 5, and bulk RNA-seq was performed on FACS-sorted Cor93 T cells.
(B) Principal-component analysis (PCA) of samples in the indicated conditions.
(C) Bar plots displaying a manually curated set of pathways enriched in Cor93 T cells upon in vivo αOX40 (red) and α4-1BB (orange) treatment compared with PBS HC-primed condition. Pathways were retrieved from the MSigDB database and belong to Gene Ontology, Reactome, and Hallmarks collections. NES, normalized enrichment score.
(D) Clustering of top significant (EnrichR combined score > 100, false discovery rate [FDR] < 0.05) Gene Ontology biological processes, BioPlanet, and MSigDB Hallmark pathways of processes upregulated in Cor93 T cells upon in vivo αOX40 (left) and α4-1BB (right) treatment compared with PBS HC-primed condition. The thermal scale represents the Jaccard similarity coefficient between every gene set pair (blue representing a similarity coefficient of 0 and red a similarity coefficient of 1).
(E) Heatmaps of manually curated list of genes linked to the indicated pathway comparing PBS, αOX40, and α4-1BB treatments. Values are Z scores, calculated from scaling by row the log2(TPM) values.
Finally, we delved into the dynamics among different CD8+ T cell subpopulations following HC- priming and post-4-1BB agonism. Sorted Dys-TRM1, Dys-TRM2, and Dys-TSL cells from HC-primed environments were cultured with the cognate peptide, with or without 4-1BB agonist, to observe their differentiation potential (Figure 6F). This exploration revealed that Dys-TSL cells serve as a foundational progenitor, capable of self-renewing and differentiating into both Dys-TRM1 and Dys-TRM2 (Figures 6O and 6P). Dys-TRM1 cells displayed some degree of plasticity, predominantly maintaining their phenotype while occasionally transitioning to Dys-TRM2 (Figures 6O and 6Q). By contrast, Dys-TRM2 cells, more terminally differentiated, exhibited minimal transitions, suggesting a committed effector state (Figures 6O and 6R). Notably, 4-1BB agonism appeared to rejuvenate these cells, promoting a less differentiated, more progenitor-like state across the dysfunctional T cell spectrum (Figures 6O–6R).
4-1BB agonism reinvigorates dysfunctional CD8+ T cells from patients with HBeAg+ CHB infection
Lastly, we explored whether and to what degree modulation of co-signaling receptors proved effective when assessed in samples from young patients with HBeAg+ CHB infection—the phase of HBV infection that is best represented by the aforementioned mouse models. To do so, we generated short-term T cell lines from the peripheral blood mononuclear cells (PBMCs) of eight patients (clinical parameters in Table S4) by stimulating them with HBV core or polymerase peptide pools in the presence or absence of recombinant human 4-1BB ligand (4-1BBL), OX40 ligand (OX40L), or blocking anti-human PD-1 mAb. On the 10th day, we evaluated cytokine production and CD107a expression in CD8+ T cells (Figures 7A and 7B). Only the treatment with 4-1BBL induced a significant increase in the production of both IFN-γ and TNF-α, as well as CD107a upregulation, by CD8+ T cells stimulated with either core or polymerase peptide pools (Figures 7C–7K). By contrast, OX40L induced only a modest increase in IFN-γ production by CD8+ T cells stimulated with polymerase peptide pools, and PD-1 blockade did not show any effect (Figures 7C–7K). Considering the fraction of patients with an increase in cytokine production or CD107a upregulation greater than 2-fold, 4-1BBL confirmed to be the most effective treatment for all the tested parameters (Figures 7E, 7H, and 7K). All individual patient data and dose-response curves are reported in Figure S7.
Figure 7.

4-1BB agonism reinvigorates dysfunctional CD8+ T cells from patients with HBeAg+ chronic HBV infection
(A) Experimental setup.
(B) Gating strategy (left) and representative plots (right) of cytokine production and CD107a upregulation on day 10.
(C and D) Paired percentages of IFN-γ-producing CD8+ T cells in short-term T cell lines generated by HBV core (C) or polymerase (Pol, D) peptide stimulation in the presence or absence of α-PD-1, OX40L, or 4-1BBL. One PBS control per patient is used for pairwise comparison against each of the other conditions. Figure S7 presents all individual patient data along with dose-response curves. For each stimulation, the dosage yielding the maximum enhancement for every cytokine was chosen. n = 8; two-way ANOVA.
(E) Pie charts represent the percentage of patients showing an increase in IFN-γ production by CD8+ T cells generated in short-term T cell lines by HBV core (left) or polymerase (right) peptide stimulation in the presence or absence of α-PD-1, OX40L, or 4-1BBL. Results are percentage of patients showing fold increase <2 (gray) or >2 (red).
(F and G) Paired percentages of TNF-α+ CD8+ cells in short-term T cell lines generated by HBV core (F) or polymerase (G) peptides as in (C) and (D).
(H) Pie charts represent the increase of TNF-α production by CD8+ T cells as in (E).
(I and J) Percentage of CD107a+ CD8+ cells in short-term T cell lines generated by HBV core (I) or polymerase (J) peptides as in (C) and (D).
(K) Pie charts represent CD107a upregulation in CD8+ T cells as in (E).
See also Figure S7.
Figure S7.

4-1BB agonism reinvigorates dysfunctional CD8+ T cells from patients with HBeAg+ chronic HBV infection, related to Figure 7
Percentage of IFN-γ, TNF-α, and CD107a-positive CD8+ cells in short-term T cell lines for each patient (n = 8) with HBeAg+ chronic HBV infection in the presence or absence of anti-PD-1 (yellow histograms; 1, 5, and 10 μg/mL), recombinant OX40L (red histograms; 30, 50, 100, 500, and 1,000 ng/mL), or recombinant 4-1BBL (orange histograms; 0.5, 1, 2, and 4 μg/mL). T cell lines were generated by HBV core (left) or polymerase (right) peptide pool stimulation.
In conclusion, our analysis suggests that, in HBeAg+ CHB patients, 4-1BB activation shows the highest potential to rejuvenate dysfunctional CD8+ T cells.
Discussion
In this study, we conducted an analysis of co-signaling receptors induced by hepatocellular priming and traced the trajectory and long-term fate of dysfunctional HBV-specific CD8+ T cells. We used mouse models that mirror the initial immune responses seen in young patients acquiring CHB infection neonatally or perinatally. Our findings suggest that immune checkpoint inhibitors offer limited effects on intrahepatically primed dysfunctional CD8+ T cells. By contrast, we observed that by targeting co-stimulatory members of the TNF receptor family, particularly 4-1BB, these cells could be transitioned into antiviral effectors. Consistent with this, in patients with HBeAg+ CHB infection, 4-1BB activation showed the highest potential to rejuvenate dysfunctional CD8+ T cells. These findings support the theory that CD8+ T cell priming by hepatocytes induces a unique dysfunctional program separate from exhaustion.4 They further imply that tailored strategies might be essential during various phases of CHB infection to effectively rejuvenate protective antiviral T cell responses.
In regions with high HBV endemicity, most CHB patients typically acquire their infection perinatally or during childhood. These patients, primarily young, HBeAg+, and highly viremic, exhibit a phase with high HBV replication but low liver inflammation. This stage is commonly known as the immunotolerant phase of CHB infection. They often respond poorly to direct-acting antiviral drugs. Despite its prevalence, our understanding of CD8+ T cell behavior during this phase is limited. Most detailed analyses have concentrated on the HBeAg-negative “immunologically active phase” (which can succeed the immunotolerant phase). In the HBeAg-negative phase, co-inhibitory receptors are upregulated and coincide with CD8+ T cell impairment. Yet, in vitro PD-1/PD-L1 blockade only marginally revives T cell activity.28 Furthermore, recent clinical trials on nucleoside analog-treated patients with suppressed viral loads reveal that only a very minor fraction benefit from checkpoint blockade.8 There is a glaring lack of data on checkpoint inhibition for the HBeAg+ stage.
Challenging conventional wisdom, recent studies cast doubt on the immunotolerant phase being a dormant stage and not requiring early treatment. Indeed, HBV-specific T cell responses in young immunotolerant individuals were found to be no more impaired than those in adult “immunologically active” patients.19 Furthermore, evidence of clonal hepatocyte expansion and HBV DNA integration during the immunotolerant phase suggests potential early-stage hepatocarcinogenetic events.20 These findings have sparked discussions around the need for earlier therapeutic intervention. Yet, the effectiveness of existing antiviral treatments during this phase of infection remains uncertain22 as numerous clinical trials have shown suboptimal impact on HBeAg and HBsAg loads after administering polyethylene glycol (PEG)-IFN-α and nucleoside analogtreatments, whether used individually or in combination.29,30,31 This underscores the potential necessity for supplementary immune modulatory approaches to enhance the performance of direct antiviral therapies. However, our limited understanding of immune responses in young HBeAg+ patients hinders the development of novel immunotherapies, emphasizing the need for thorough phenotypic and functional characterization of HBV-specific CD8+ T cells in the HRLI phase of CHB infection.
Whereas priming by hepatocytes in a non-inflamed liver, likely to occur in the first phases of CHB infection, triggers a unique dysfunctional gene program, Ag persistence and liver inflammation may gradually activate a gene signature progressively more similar to exhaustion.4,32,33 Yet a recent study found no clear evidence of classical exhausted T cells expressing all co-inhibitory receptors or showing hierarchical T cell exhaustion.34 Furthermore, the dysfunctionality of HBV-specific T cells did not correspond with the linear accumulation of co-inhibitory receptors, implying that these cells were not entirely functionally inert, further reinforcing the distinctiveness of their phenotype.34
Remarkably, extended Ag stimulation of intrahepatically primed dysfunctional T cells resulted in a self-sustaining, long-lived, heterogeneous population consisting of a dysfunctional progenitor or stem-like (TSL) population and two distinct dysfunctional tissue-resident memory (TRM) cell populations. All three subsets maintained 4-1BB expression, while OX40 expression was limited to TSL. Contrary to other chronic infections and cancer,26,35,36 our results showed that targeting all three subpopulations with 4-1BB activation and not only the stem-like T cells with OX40 activation endowed HBV-specific CD8+ T cells with robust antiviral activity. The mechanism behind this observation remains uncertain and may be linked to lower TSL numbers, their potential distinct localization, or the differential biological effects downstream of these two co-stimulatory receptors.37,38
The potential of 4-1BB agonism for initiating anti-tumor T cell responses is well recognized.39 Despite setbacks in early clinical trials with first-generation agonists due to toxicity and limited efficacy,40 promising preclinical results and a better understanding of off-target effects have led to the development of second-generation 4-1BB agonistic drugs.40 These drugs, noted for their improved safety and tolerability, are currently undergoing clinical evaluation.
In conclusion, our study outlines the behavior and potential therapeutic targeting of dysfunctional HBV-specific CD8+ T cells, providing insights that could guide the development of immunomodulators to end HBV infection and its complications.
Limitations of the study
This study underscores the potential of 4-1BB agonism in reinvigorating T cells but acknowledges some limitations. The use of monoclonal TCR Tg CD8+ T cells may not accurately reflect the complexity of natural, polyclonal responses to HBV Ag s, indicating a need for broader research on polyclonal cell sensitivity to 4-1BB agonism. Additionally, the experimental models lack covalently closed circular DNA (cccDNA), limiting insights into 4-1BB impact on key HBV dynamics. The role of CD4+ T cells, potentially crucial for the antiviral response, was not examined, highlighting a gap for future studies. Moreover, comparing KC-primed cells with HC-primed cells introduces variability not only in the nature of Ag-presenting cells but also in the magnitude and duration of Ag exposure.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 anti mouse TCF1/TCF7 | Cell Signaling Technology | Cat# 6444; RRID:AB_2797627 |
| Alexa Fluor 488 anti-mouse CD3 | BioLegend | Cat# 100210; RRID:AB_389301 |
| Alexa Fluor 488 anti-mouse CD62L | BioLegend | Cat# 104419; RRID:AB_493377 |
| Alexa Fluor 488, Chicken anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody | Thermo Fisher Scientific | Cat# A-21470; RRID: AB_2535873 |
| Alexa Fluor 568, Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody | Thermo Fisher Scientific | Cat# A-11077; RRID: AB_2534121 |
| Alexa Fluor 594 anti-mouse CD38 | BioLegend | Cat# 102725; RRID:AB_2566435 |
| Alexa Fluor 647 anti-human/mouse Granzyme B | BioLegend | Cat# 515406; RRID:AB_2566333 |
| Alexa Fluor 647 anti-mouse CD69 | BioLegend | Cat# 104517; RRID:AB_492848 |
| Alexa Fluor 700 anti-mouse CD107a (LAMP-1) | BioLegend | Cat# 121628; RRID:AB_2783063 |
| Alexa Fluor 700 Mouse Anti-Human IFN-γ | BD Biosciences | Cat# 557995; RRID:AB_396977 |
| Anti-Ki67 | Agilent | Cat# M7249; RRID:AB_2250503 |
| APC anti-mouse CD137 | BioLegend | Cat# 106109; RRID:AB_2564296 |
| APC anti-mouse CD94 | BioLegend | Cat# 105512; RRID:AB_2721459 |
| APC/Cyanine7 anti-mouse CD48 | BioLegend | Cat# 103432; RRID:AB_2561463 |
| BB515 Armenian Hamster Anti-ICOS (CD278) | BD Biosciences | Cat# 565880; RRID:AB_2744480) |
| BUV395 Mouse Anti-Mouse CD45.1 | BD Biosciences | Cat# 565212; RRID:AB_2722493 |
| BUV496 Mouse Anti-Rat CD90/Mouse CD90.1 | BD Biosciences | Cat# 741110; RRID:AB_2870702 |
| BUV563 Rat Anti-Mouse CD44 | BD Biosciences | Cat# 741227; RRID:AB_2870781 |
| BUV737 Mouse Anti-Human CD279 (PD-1) | BD Biosciences | Cat# 612791; RRID:AB_2870118 |
| BUV737 Rat Anti-Mouse CD279 (PD-1) | BD Biosciences | Cat# 749306; RRID:AB_2873680 |
| BUV805 Rat Anti-Mouse CD8a | BD Biosciences | Cat# 612898; RRID:AB_2870186 |
| BV711 anti-mouse CD186 (CXCR6) | BioLegend | Cat# 151111; RRID:AB_2721558 |
| BV785 anti-mouse CD223 (LAG-3) | BioLegend | Cat# 125219; RRID:AB_2566571 |
| BV605 anti-mouse TNF-alpha | BioLegend | Cat# 506329; RRID:AB_11123912 |
| BV650 Rat Anti-Mouse NKG2A/C/E | BD Biosciences | Cat# 740549; RRID:AB_2740251 |
| BV785 anti-mouse CD127 (IL-7Ralpha) | BioLegend | Cat# 135037; RRID:AB_2565269 |
| BV786 Rat Anti-Mouse CD134 | BD Biosciences | Cat# 740945; RRID:AB_2740573 |
| CD8a Monoclonal Antibody (53-6.7) | Thermo Fisher Scientific | Cat# 14-0081-82,;RRID:AB_467087 |
| Cleaved Caspase-3 (Asp175) Antibody | Cell Signaling Technology | Cat# 966; RRID:AB_2341188 |
| eFluo 450 anti mouse Ki-67 | Thermo Fisher Scientific | Cat# 48-5698-80; RRID:AB_11151155 |
| eFluo 660 anti-mouse TOX | Thermo Fisher Scientific | Cat# 50-6502-82; RRID:AB_2574265 |
| FITC anti human TNF-α | Miltenyi Biotec | Cat# 130-120-490; RRID:AB_2752116 |
| FITC anti-mouse CD45.2 | BioLegend | Cat# 109805; RRID:AB_313442 |
| GoInVivo Purified anti-mouse CD279 (PD-1) | BioLegend | Cat# 114112; RRID:AB_2566090 |
| Hepatitis B Virus Core Antigen | Agilent | Cat# B0586; RRID:AB_2335704 |
| InVivoMab anti-mouse L-Selectin (CD62L) | Bio X Cell | Cat# BE0021; RRID:AB_1107665 |
| InVivoMAb anti-mouse/human/rat/monkey ICOS (CD278) | Bio X Cell | Cat# BE0353; RRID:AB_2894772 |
| InVivoPlus anti-mouse CD137 (4-1BB) | Bio X Cell | Cat# BE0239; RRID:AB_2687721 |
| InVivoPlus anti-mouse CD16/CD32 | Bio X cell | Cat# BE0307; RRID:AB_2736987 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | Bio X Cell | Cat# BE0131; RRID:AB_10950184 |
| InVivoPlus anti-mouse LAG-3 | Bio X Cell | Cat# BE0174; RRID:AB_10949602 |
| InVivoPlus anti-mouse OX40 (CD134) | Bio X Cell | Cat# BE0031; RRID:AB_1107592 |
| Pacific Blue anti-mouse Ly108 | BioLegend | Cat# 134608; RRID:AB_2188093 |
| PE anti human CD4 | Miltenyi Biotec | Cat# 130-113-225; RRID:AB_2726036 |
| PE anti mouse CD197 (CCR7) | Thermo Fisher Scientific | Cat# 12-1971-80; RRID:AB_465904 |
| PE mouse anti-CD247, phospho (Tyr142) | BD Biosciences | Cat# 558448; RRID:AB_647237 |
| PE anti-mouse CD178 (FasL) | BioLegend | Cat# 106606; RRID:AB_313279 |
| PE anti-mouse CD200R (OX2R) | BioLegend | Cat# 123907; RRID:AB_2074081 |
| PE anti-mouse CD45.1 | BioLegend | Cat# 110707; RRID:AB_313496 |
| PE anti-mouse Granzyme A | BioLegend | Cat# 149703; RRID:AB_2565309 |
| PE Mouse Anti-Human Granzyme B | BD Biosciences | Cat# 561142; RRID:AB_10561690 |
| PE-CF594 Mouse Anti-Human CD3 | BD Biosciences | Cat# 562280; RRID:AB_11153674 |
| PE-CF594 Rat Anti-Mouse IFN-γ | BD Biosciences | Cat# 562333; RRID:AB_11154588 |
| PE-Cyanine7 anti human CD107 (LAMP-1) | Miltenyi Biotec | Cat# 25-1079-42; RRID:AB_2573384 |
| PE/Cyanine7 anti-mouse CCL5 (RANTES) | BioLegend | Cat# 149106; RRID:AB_2860706 |
| PE/Cyanine7 anti-mouse CD152 | BioLegend | Cat# 106313; RRID:AB_2564237 |
| PerCp anti human CD8 | Miltenyi Biotec | Cat# 130-113-160; RRID:AB_2725988 |
| PerCP/Cyanine5.5 anti-human CD223 (LAG-3) | BioLegend | Cat# 369312; RRID:AB_2629755 |
| Bacterial and virus strains | ||
| rLCMV-core/env | provided by D. Pinschewer | Bénéchet4 |
| Chemicals, peptides, and recombinant proteins | ||
| 4′,6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | Cat# D1306; RRID:AB_2629482 |
| BD GolgiPlug™ Protein Transport Inhibitor (Containing Brefeldin A) | BD Biosciences | Cat# 555029; RRID:AB_2869014 |
| BD GolgiStop™ Protein Transport Inhibitor (Containing Monensin) | BD Biosciences | Cat# 554724; RRID:AB_2869012 |
| Betaine | Sigma-Aldrich | Cat# 61962 |
| Bovine Serum Albumin (BSA) US Origin, Protease free, lyophilized powder | PAN Biotech | Cat# P06-139250 |
| Brilliant Stain Buffer | BD Biosciences | Cat# 566349; RRID:AB_2869750 |
| Collagenase | Sigma-Aldrich | Cat# 11088793001 |
| DMEM | Invitrogen | Cat# 41965-039 |
| DnaseI | Sigma-Aldrich | Cat# 04 536 282 001 |
| DTT | Invitrogen | Cat# 18064-014 |
| Dulbecco’s Phosphate-Buffered Saline, 1X without calcium and magnesium | Corning | Cat# 21-031-CV |
| EDTA | Westburg | Cat# 51234 |
| Elution Buffer | Qiagen | Cat# 19086 |
| Eosin | Sigma-Aldrich | Cat# 115935 |
| Ethanol | Carlo Erba | Cat# 414605 |
| Fetal Bovine Serum, 500 mL, Regular, South American Origin | Corning | Cat# 35-079-CV |
| FluorSave Reagent | Millipore | Cat# 345789 |
| Gentamicin | Gibco | Cat# 15710-049 |
| HBcAg peptide sequence 93-100 [MGLKFRQL] | Proteogenix | Custom made |
| HBsAg peptide sequence 28-39 [IPQSLDSWWTSL] | Proteogenix | Custom made |
| HBSS (Hank’s Balanced Salt Solution) | Corning | Cat# 20-021-CV |
| Hematoxylin | Sigma-Aldrich | Cat# 105174 |
| LIVE/DEAD™ Fixable Far Red Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34973 |
| Magnesium chloride MgCl2 | Sigma-Aldrich | Cat# M8266 |
| Ms H-2Kb/Ig Recom DimerX Pure | BD Biosciences | Cat# 550750; RRID:AB_2868900 |
| Ms H-2Ld/Ig Recom DimerX Pure | BD Biosciences | Cat# 550751; RRID:AB_2868901 |
| O.C.T. | Bio Optica | Cat# 05-9801 |
| Paraformaldehyde 10% | EMS | Cat# 15712 |
| Phosflow Lyse/Fix Buffer | BD Biosciences | Cat# 558049; RRID:AB_2869117 |
| Phosflow Perm/Wash Buffer I | BD Biosciences | Cat# 557885; RRID:AB_2869104 |
| RPMI 1640 | Gibco | Cat# 52400-025 |
| Treg-Protector (anti-ARTC2 Nanobody) | BioLegend | Cat# 149803; RRID:AB_2819899 |
| Tritonx100 | Sigma | Cat# T9284 |
| Viobilit 405/520 fixable dye | Miltenyi Biotec | Cat# 130-109-816 |
| YCO0206985 - Olig0-dT30VN | Qiagen | Cat# 339411 |
| YCO0206986 - ISPCR oligo | Qiagen | Cat# 339411 |
| YCO0206987 - TSO | Qiagen | Cat# 339412 |
| Zinc Formalin Fixative | Sigma-Aldrich | Cat# Z2902 |
| Zombie Aqua™ Fixable Viability Kit | BioLegend | Cat# 423101 |
| Critical commercial assays | ||
| Agencourt Ampure XP beads | Beckman Coulter | Cat# A63881 |
| Anti-FITC MicroBeads | Miltenyi Biotec | Cat# 130-048-701; RRID:AB_244371 |
| CellTrace™ Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34571 |
| Chromium Next GEM Chip G Single Cell Kit 48 rxns | Carlo Erba | Cat# FC51000120 |
| Chromium Next GEM Single Cell 3' Kit v3.1 dual index | Carlo Erba | Cat# FC51000268 |
| DIG-High Prime DNA Labeling and Detection Starter Kit II | Roche | Cat# 11585614910 |
| Dual Index Kit TT set A | Carlo Erba | Cat# FC51000215 |
| EasySep™ Mouse Naïve CD8+ T Cell Isolation Kit | Stem Cell technologies | Cat# 19858 |
| First Strand Buffer | Invitrogen | Cat# 18064-014 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat# 5067- 5592 |
| High Sensitivity RNA ScreenTape | Agilent | Cat# 5067- 5579 |
| KAPA HiFi HotStart ReadyMix | KAPA Biosystems | Cat# KK2601 |
| Nextera XT 24-index kit | Illumina | Cat# FC131-1001 |
| Nextera XT DNA sample preparation kit | Illumina | Cat# FC131-1024 |
| Qubit dsDNA HS Buffer | Thermo Fisher Scientific | Cat# Q32851 |
| Recombinant RNase inhibitor | Promega | Cat# N2611 |
| Relia Prep RNA Cell Miniprep System | Promega | Cat# Z6011 |
| SPRIselect 5ml | Beckman Coulter | Cat# B23317 |
| Superscript II reverse transcriptase | Invitrogen | Cat# 18064-014 |
| TURBO DNA-free™ Kit | Thermo Fisher Scientific | Cat# AM1907 |
| Deposited data | ||
| Bulk RNAseq data | This manuscript | GEO: GSE263460 |
| Single-cell RNAseq data | This manuscript | GEO: GSE240036 |
| Single-cell RNAseq data | Miller et al.26 | GEO: GSE122713 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | Stock No: 007914 |
| Mouse: B6.Cg-Ptprca Pepcb Tg(TcraBC10,TcrbBC10)3Chi/J (lineage BC10.3) | M. Isogawa; Isogawa et al.3 | Internal colony |
| Mouse: B6.Cg-Tg(CAG-DsRed∗MST)1Nagy/J | The Jackson Laboratory | Stock No: 006051 |
| Mouse: B6.FVB(129)-Tg(Alb1-cre)1Dlr/J | The Jackson Laboratory | Stock No: 016832 |
| Mouse: Balb/c | Charles River | Balb/c colony |
| Mouse: C.Cg-Tg(Tcra6C2,Tcrb6C2)16Chi/J (lineage 6C2.36) | M. Isogawa; Isogawa et al.3 | Internal colony |
| Mouse: C57BL/6 | Charles River | C57BL/6 colony |
| Mouse: C57BL/6-Tg(CAG-EGFP)1Osb/J | The Jackson Laboratory | Stock No: 003291 |
| Mouse: CBy.PL(B6)-Thya/ScrJ | The Jackson Laboratory | Stock No: 005443 |
| Mouse: lineage 1.3.32 | Francis V. Chisari, L. G. Guidotti.; Guidotti et al.24 | Internal colony |
| Mouse: MUP-core 50 [MC50] | Francis V. Chisari, L. G. Guidotti.; Guidotti et al.23 | Internal colony |
| Software and algorithms | ||
| Adobe Illustrator | Adobe | www.adobe.com |
| BD FACSDiva V8 | BD Biosciences | http://www.bdbiosciences.com/us/instruments/research/software/flow-cytometry-acquisition/bd-facsdiva-software/m/111112/overview |
| CytExpert | Beckman Coulter | https://www.beckman.com/flow-cytometry/instruments/cytoflex/software |
| edgeR | Robinson et al.41 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Enrichr | Kuleshov et al.42 | http://amp.pharm.mssm.edu/Enrichr/ |
| featureCounts | Liao et al.43 | https://www.rdocumentation.org/packages/Rsubread/versions/1.22.2/topics/featureCounts |
| fGSEA | Korotkevich et al.44 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Fiji-Imagej | Schneider et al.45 | https://imagej.net/Fiji/Downloads |
| FlowJo V10.10 | FlowJo | https://www.flowjo.com/ |
| GeneOntology | Ashburner et al.46 | http://geneontology.org/ |
| Ggplot2 | Wickham et al.47 | https://ggplot2.tidyverse.org |
| GseaPreranked | Subramanian et al.48 | https://gsea-msigdb.github.io/gseapreranked-gpmodule/v6/index.html |
| Ilastik | Berg et al.49 | https://www.ilastik.org/ |
| ImageScope | Leica Biosystem | N/A |
| Imaris bitplane | Imaris | https://imaris.oxinst.com/products/imaris-for-cell-biologists?gclid=Cj0KCQiAgomBBhDXARIsAFNyUqOQMD64vZvZMyBoHWFOYRm_ZPxHWLb_tWDl0pGjii8ZVNDkW-UNtRgaAnhfEALw_wcB |
| Leica Application Suite X (LAS X) | Leica Microsystem | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| LIMMA R package | Ritchie et al.50 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| pheatmap R | Kolde51 | https://www.rdocumentation.org/packages/pheatmap/versions/1.0.12/topics/pheatmap |
| Prism 10 | GraphPad software | https://www.graphpad.com/scientific-software/prism |
| RSEM tool | Li and Dewey52 | https://deweylab.github.io/RSEM/ |
| scVelo | Bergen et al.53 | https://scvelo.readthedocs.io |
| Seurat (v4.0.2) | Stuart et al.54 | https://satijalab.org/seurat/ |
| STAR aligner | Dobin et al.55 | https://github.com/alexdobin/STAR |
| Velocyto | La Manno et al.27 | http://velocyto.org |
| Other | ||
| Aperio GT 450 DX | Leica Biosystem | N/A |
| Cytek Aurora Spectral Analyzer | Cytekbio | N/A |
| CytoFLEX LX | Beckman Coulter | N/A |
| FACSAria Fusion | BD Bioscience | N/A |
| FACSymphony A5 | BD Bioscience | N/A |
| Illumina HiSeq 4000 system | Illumina | N/A |
| Infinite M Nano | Tecan | N/A |
| Inverted 2-Photon microscope TriM Scope II | LaVision Biotec | N/A |
| MACSQuant Tyto Cell Sorter | Miltenyi Biotec | N/A |
| MoFlo Legacy | Beckman Coulter | N/A |
| SP8 confocal microscope | Leica Biosystem | N/A |
| Tape Station 2100 | Agilent | N/A |
| UVItec | Eppendorf | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matteo Iannacone (iannacone.matteo@hsr.it).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The bulk RNA and scRNAseq datasets have been deposited in the Gene Expression Omnibus (GEO) repository and will be publicly available upon publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Mouse lines
C57BL/6, CD45.1 (inbred C57BL/6), Balb/c, Thy1.1 (CBy.PL(B6)-Thya/ScrJ), β-actin-GFP [C57BL/6-Tg (CAG-EGFP) 1Osb/J], Ai14 (RCL-tdT)-D [B6.Cg-Gt (ROSA) 26Sortm14 (CAG-tdTomato) Hze/J], β-actin-DsRed [B6.Cg-Tg (CAG-DsRed∗MST) 1Nagy/J] mice were purchased from Charles River or The Jackson Laboratory. MUP-core transgenic mice (lineage MUP-core 50 [MC50], inbred C57BL/6, H-2Kb), that express the HBV core protein in 100% of the hepatocytes under the transcriptional control of the mouse major urinary protein (MUP) promoter, have been previously described.23 HBV replication-competent transgenic mice (lineage 1.3.32, inbred C57BL/6, H-2Kb), that express all of the HBV Ags and replicate HBV in the liver at high levels without any evidence of cytopathology, have been previously described.24 In indicated experiments, MUP-core and HBV replication-competent transgenic mice were used as C57BL/6 x Balb/c H-2bxd F1 hybrids. Cor93 TCR transgenic mice (lineage BC10.3, inbred CD45.1), in which > 98% of the splenic CD8+ T cells recognize a H-2Kb-restricted epitope located between residues 93-100 in the HBV core protein (MGLKFRQL), have been previously described.3 Env28 TCR transgenic mice (lineage 6C2.36, inbred Thy1.1 Balb/c), in which >85% of the splenic CD8+ T cells recognize a H-2Ld-restricted epitope located between residues 28–39 of HBsAg (IPQSLDSWWTSL), have been previously described.3 For intravital imaging experiments, Cor93 TCR were crossbred with the CAG-dsRed lineage for homozygous CAG-dsRed expression. Furthermore, Alb1-Cre- MUP-core+/- mT/mG+/+ and MUP-core-/- mT/mG+/+ (control) mice were generated by crossing mT/mG (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) and MUP-core transgenic mice.
Mice were housed under specific pathogen-free conditions and entered experiments at 8-10 weeks of age. Mice of both sexes were used. In all experiments, mice were matched for age, sex and (for the 1.3.32 animals) serum HBeAg levels before experimental manipulations. All experimental animal procedures were approved by the Institutional Animal Committee of the San Raffaele Scientific Institute and are compliant with all relevant ethical regulations.
Viruses and viral vectors
The replication-incompetent LCMV-based vector encoding HBV core protein (rLCMV-core) has been previously described.4 Mice were injected intravenously with 2.5x105 infectious units of rLCMV-core 4 h before CD8+ T cells transfer unless otherwise indicated. In accordance with institutional guidelines, all infectious work was carried out in designated BSL-2 or BSL-3 workspaces.
Human samples
A total of 8 patients with HBeAg-positive chronic HBV infection were enrolled at the Unit of Infectious Diseases and Hepatology of the Azienda Ospedaliero-Universitaria of Parma, Italy. Diagnosis was based on serum HBsAg and HBeAg positivity lasting more than six months and normal ALT levels. Patient age ranged from 21 to 42 (participants clinical parameters, age, ethnicity and sex are reported in in Table S4). No patient was under antiviral therapy. All patients were negative for anti-hepatitis C virus, delta virus, human immunodeficiency virus type 1 (HIV-1) and type 2 (HIV-2) antibodies and for other markers of viral or autoimmune hepatitis. The influence of sex/gender, age or ethnicity on the therapeutic potential of co-signaling receptor modulation in hepatitis B is not known, and our study is not powered enough to address this aspect. Data presented in this study derive from a non-profit clinical project approved by the Ethic Committee Area Vasta Emilia Nord (record n. 730/2020/TESS/UNIPR).
Method details
Naïve T cell isolation, adoptive transfer, and in vivo treatments
Recipient mice were adoptively transferred with 2x104 or 106 HBV-specific naïve CD8+ TCR transgenic T cells isolated from the spleens of Cor93 and/or Env28 TCR transgenic mice, as described.4 The appropriate amount of TCR transgenic CD8+ T cells was confirmed by dimer FACS staining as described.4 The indicated amount cells were injected intravenously (i.v.) into recipient mice in 200 μl HBSS. In selected experiments, mice were splenectomized and injected intraperitoneally (i.p.) with 100 μg of anti-CD62L monoclonal antibody (clone MEL-14, BioXcell #BE0021) in PBS 4 h prior to cell injection. Splenectomy was performed according to standard protocols.56 In indicated experiments, recipient mice were injected i.p. with wither 100μg of anti-mouse PD-1 (clone RMP1-14, Biolegend #114114), anti-mouse LAG-3 (clone C9B7W, BioXcell #BE0174), anti-mouse CTLA-4 (clone 9H10, BioXcell #BE0131), anti-mouse ICOS (clone C398.4A, BioXcell #BE0353), anti-mouse OX40 (clone OX-86, BioXcell #BE0031) or anti-mouse 4-1BB (clone 3H3, BioXcell #BE0239) antibodies at saturating concentration one day after T cell transfer, unless otherwise indicated.
Conventional cell sorting
Single-cell suspensions from livers were obtained as described.4 Briefly, mice livers were perfused with PBS, processed through mechanical and enzymatic digestion, and isolated cellular fractions were purified using differential centrifugation and Percoll gradient. Cells were finally stained with Viobility 405/520 fixable dye (Miltenyi), with PB-conjugated anti-CD8α (clone 53-6.7) and PE-conjugated anti-CD45.1 (clone A20) antibodies. Live CD8+ CD45.1+ cells were sorted on a MoFlo Legacy (Beckman Coulter) cell sorter in a buffer containing PBS with 2% FBS. Cell purity was greater than 98%. For scRNAseq experiments, single-cell suspensions from livers were stained with Zombie Aqua Fixable Viability Kit (BioLegend, #423101), APC-conjugated anti-CD8 (clone 53-6.7) and PE-conjugated anti-CD45.1 (clone A20) antibodies. Live CD8+ CD45.1+ cells were sorted on a BD FACSAria III cell sorter in a buffer containing PBS with 2% FBS.
For sequential adoptive transfer experiments, single-cell suspensions from livers were stained with Zombie Aqua Fixable Viability Kit (BioLegend, #423101), PB-conjugated anti-CD8α (clone 53-6.7), APC-Cy7 conjugated anti-CD48 (clone HM48-1), and APC conjugated anti-Slamf6 (clone 330-AJ) antibodies. Live CD8+ CD45.1+ and SLAMF6+ cells, or SLAMF6- CD48+ or CD48- cells were sorted on a BD FACSAria III cell sorter.
MACSQuant Tyto cell sorting
To obtain highly pure and viable Cor93 T cells for the experiments depicted in Figure 5G, single-cell suspensions isolated from the livers of CD45.2 HBV Tg mice, previously transferred with 106 naive Cor93 TN cells, underwent initial enrichment through Magnetic-Activated Cell Sorting (MACS) followed by subsequent Fluorescence-Activated Cell Sorting (FACS). For that, single-cell suspensions were stained with FITC-labeled anti-mouse CD45.2 antibody (clone 104), followed by anti-FITC antibody-coated micromagnetic beads (Miltenyi Biotech) and passed through a magnetic isolation column. Negative fraction containing CD45.1+ cells was obtained. Following this, cells were stained with PB-conjugated anti-SLAMF6 (330-AJ), PE-conjugated anti-CD8α (clone 53-6.7), and AlexaFluor647-conjugated anti-CD45.1 (clone A20). Total Cor93 T cells (CD45.1+ CD8+), Dys-TSL (CD45.1+ CD8+ SLAMF6+), and Dys-TRM (CD45.1+ CD8+ SLAMF6-) were sorted at a rate of 4 mL/h with ∼140 mbar pressure using a MACSQuant Tyto Sorter (Miltenyi Biotec) in a buffer containing PBS with 2% FBS plus 1mM EDTA. Cell debris, doublets, and dead cells were gated out during sorting. Following sorting, the purity of the target cells was verified by flow cytometry, ensuring a purity close to 99%.
Flow cytometry
For all experiments where mice were sacrificed 14 days or longer after T cell transfer, mice were injected in the tail vein with 50 μg of Treg-Protecto (anti-ARTC2 Nanobody, Biolegend #149802) 30 min before sacrifice. Staining of chemokine receptors was carried out at 37°C, 5% CO2 for 30 minutes in the presence of an anti-CD16/32 blocking antibody (Fc-block, BD Biosciences #553142). Staining of cell surface markers was performed with Brilliant Stain buffer (BD Biosciences #566349) according to manufacturer instructions. Stimulation and analyses of intracellular cytokines was performed as described.4 Briefly, cells were plated in at 106 per well in a 96-well U-bottom tissue culture plate with cognate peptide (2μg/ml) for 4 hours in incubator in complete RPMI medium containing Brefeldin-A (10μg/ml) and IL-2 (EL-4 supernatant, 10 U/ml). At the end of the stimulation cells were washed and stained as described above.
To stain intranuclear proteins, cells were processed using FoxP3/transcription Factor Staining Buffer Set (Thermo Fisher Scientific #00-5523-00) following manufacturer instructions. Cell viability was assessed by staining with Viobility 405/520 fixable dye (Miltenyi, #130-109-816) or LIVE/DEAD Fixable Far-Red dye (Invitrogen, # L34973), according to manufacturer instructions.
Abs used included: Anti-CD45.1 (clone A20, BD Biosciences #565212), anti-CD90.1 (cloneOX-7, BD Biosciences #741110), anti-CD44 (clone IM7, BD Biosciences #741227), anti PD-1 (clone EH12.1, BD Biosciences #612791), anti-CD8a (clone 53-6.7, BD Biosciences #612898), anti-OX40 (clone OX-86, BD Biosciences #740945), anti-ICOS (clone C398.4A, BD Biosciences #565881), anti-LAG-3 (clone 11C3C65, #369312), anti-CTLA-4 (clone UC10-4B9, #106314), anti-4-1BB (clone 17B5, #106110), anti-IFN-γ (clone XMG1.2, BD Biosciences #562333), anti-GranzymeB (clone GB11, # 515406), anti-CXCR6 (clone SA051D1, BD Bioscences #151111), anti-CD69 (clone: H1.2F3, # 104517), anti-PD1 (clone RMP1-30, BD Bioscences #749306), anti-LAG3 (clone C9B7W, # 125219), anti-NKG2A/C/E (clone 20d5, BD Bioscences #740549), anti-CD94 (clone 18d3, # 105511), anti-Tox (clone TXRX10, Invitrogen #50-6502-82), anti-CCL5 (clone 2E9/CCL5, #149106), anti-Granzyme A (clone 3G8.5, # 149704), anti-Granzyme B (clone GB11, BD Biosciences #752045), anti-SLAMF6 (clone 330-AJ, # 134608), anti-TCF1 (clone C63D9, Cell Signaling Tech #6444), anti-CD48 (clone HM48-1, #103432), anti-CD62L (clone MEL-14, #104420), anti-CD127 (clone A7R34, BD Bioscences #135037), anti-CCR7 (clone 4B12, eBioscience #12-1971-80), anti-Ki67 (clone SolA15, Thermo Fisher #48-5698-80), anti-CD200R (clone OX-110, #123908), anti-CD178 (FASL, clone MFL3, #106606), anti-CD107a (LAMP-1, clone 1D4B, #121628), anti-TNF-a (clone MP6-XT22, #506329), anti-CD3 (clone 17A2, #100210). All Abs were purchased from BioLegend, unless otherwise indicated.
Recombinant dimeric H-2Ld:Ig and H-2Kb:Ig fusion proteins (BD Biosciences) complexed with peptides derived from HBsAg (Env28-39) or from HBcAg (Cor93-100), respectively, were prepared according to the manufacturer instructions.
Flow cytometry analyses were performed either on a FACS CANTO II (BD Bioscience), CytoFLEX LX (Beckman Coulter), FACSymphony A5 (BD Biosciences) or Cytek Aurora (CytekBio) and analyzed with FlowJo software (Treestar).
TCR phosphoprotein signaling analysis
106 Cor93 TN cells were adoptively transferred into WT mice (C57BL/6 background) previously transduced with rLCMV-HBc 4 hours before (KC-primed ) or into HBV transgenic mice (lineage MUP-core). Livers were collected after 5 days, and intrahepatic leukocytes (IHL) were isolated. Every step has been performed at room temperature and cells have been serum-starved for 2 hours in incubator in controlled atmosphere (37°C, 5% CO2). Cells were stimulated with 10μg/ml of Cor93-100 cognate peptide for 10 minutes in incubator. At the end of stimulation, cells were washed, fixed with BD Phosflow Lyse/Fix buffer (BD Bioscience #558049) and permeabilized with Phosflow Perm Buffer I (BD Bioscience #557885), following the manufacturer’s instructions. Cells were finally stained with anti-mouse CD3ζ /CD247 (pY142)-PE (clone K25-407.69) and analyzed by flow cytometry (Cytek Aurora).
Measurement of TCR affinity
The spleens from Cor93 TCR transgenic mice (lineage BC10.3) and Env28 TCR transgenic mice (lineage 6C2.36) were harvested and mechanically dissociated to obtain a single cell suspension, as described.4 Effector CD8+ T cells (TE) were generated in vitro as previously described.5 Briefly, splenocytes from BC10.3 mice were harvested, stimulated with cognate peptide for 1 hour, and then cultured in RPMI complete medium supplemented with IL-2 (EL-4 supernatant, 10 U/ml). Cells were split every two days over an 8-day period. TE were stimulated with the indicated amount of the cognate peptide and subsequently stained for intracellular IFN-γ production using flow cytometry, following the same protocol as described above.
In vitro killing assay
HBV-Tg mice (1.3.32 lineage) were adoptively transferred with 106 Cor93 TN and challenged 24 hours later with or without 100μg of 4-1BB agonist antibody. 5 days after T cell transfer, intrahepatic leukocytes (IHL) were isolated. For co-culture experiments, 2x105 Cor93 T cells were plated alongside 2x105 C57BL/6 splenocytes, which had been either pulsed with the cognate Cor93-100 peptide or left unpulsed. Peptide pulsing involved incubating the splenocytes with 5μg/ml of the Cor93-100 peptide for one hour at 37°C, followed by extensive washing to remove excess of peptide.
Before co-culture, unpulsed and peptide-pulsed splenocyte populations were labeled with distinct concentrations of cell trace violet (CTV, ThermoFisher #C34557) – low (500nM) for unpulsed and high (5μM) for pulsed cells. These populations were then mixed in a 1:1 ratio and co-cultured with Cor93 T cells for either 4 or 16 hours. Control conditions were obtained by culturing target cells alone, without Cor93 T cells. Cells were analyzed by flow cytometry and percentage of specific killing in the indicated conditions was calculated according to the following formula:
Southern blot analysis
Genomic DNA was isolated from frozen livers using the phenol/chloroform method and analyzed for intrahepatic HBV DNA contents by Southern blotting, as previously described.6 Briefly, genomic DNA was digested by Hind-III (NEB), separated in 1.4% agarose gel, and transferred to a positively charged nylon membrane (Roche). The membrane was hybridized with a DIG-dUTP–labeled, full-length HBV DNA fragment and detected by the alkaline phosphatase–labeled anti-DIG antibody according to the manufacturer’s instructions (DIG High Prime DNA Labeling and Detection Starter Kit II; Roche). The signals were acquired using Syngene Imaging System.
ScRNAseq library preparation
Single cells were processed on the Chromium platform (10x) using the Chromium Next GEM Single Cell 3’ v3.1 (Dual Index). After quality controls and quantification on TapeStation instrument (Agilent), libraries were sequenced on NovaSeq6000 platform (Illumina) generating around 25'000 reads/cell. Raw sequencing data were demultiplexed with the mkfastq application (Cell Ranger v.6.0.2). UMI-Tools (v.1.0.0) whitelist and extract commands were used to identify and select the number of cell barcodes to use in downstream analysis. Reads were mapped to the reference genome using STAR v.2.5.3a and assigned to genes with featureCounts v.1.6.4. GRCm38 was used as the reference genome. Bam files were sorted with samtools software (v1.9). Finally, Umi-Tools count was used to processing the UMIs aligned to each gene in each cell to find the number of distinct, error-corrected UMIs mapping to each gene. The UMI count tables of each cellular barcode were used for further analysis.
ScRNAseq bioinformatics analysis
Single cell data analysis was performed using Seurat (v4.0.2).54 Cells with sufficient bioinformatic quality were obtained after applying a filter of at least 200 genes expressed per cell and only genes expressed in at least 3 cells were retained. Moreover, cells with more than 10% of reads mapped to mitochondrial genes were also excluded from the analysis. UMI count matrix was further normalized and scaled following the standard Seurat workflow and Umap reduction was then applied on first 20 Principal Components after running PCA. Unbiased clustering was computed using the FindClusters function in Seurat with default parameters and a resolution value of 0.3. Specific markers for the different unbiased clusters were found using the function FindAllmarkers or FindMarkers in Seurat with default parameters. The plots showing normalized expression values with a color scale on top of Umap plots (on Figure 3F) and the Violin plots of specific genes were produced with FeaturePlot and VlnPlot Seurat functions, respectively. The max.cutoff parameter is set to “q95”. Raw count datasets from Miller et al.26 were downloaded from the Gene Expression Omnibus (GEO) database under accession no. GSE122713 and were re-analyzed following the abovementioned procedure. The gene signatures in Figure 5B were calculated with the AddModuleScore function in Seurat.
RNA velocity analysis
The velocyto python package27 was used to generate the loom file, a matrix containing information regarding spliced/unspliced RNA. Velocyto (version 0.17.17) was run through the command line interface (CLI) and the loom file was then used as input for scVelo (version 1.7.2), a scalable toolkit for RNA velocity analysis in single cells based on Bergen et al., 2020. After the standard pre-processing, RNA velocities were computed using the functions scv.tl.velocity (in dynamical mode) and scv.tl.velocity_graph. In order to visualize velocity fields across all genes and all cells the function scv.pl.velocity_embedding_stream has been used, built on the UMAP previously computed.
RNA Purification and RNAseq Library Preparation for bulk RNA sequencing
Between 40000 and 50000 Cor93 T cells in the indicated conditions were FACS-sorted as Live, Lineage- (CD19 / CD3 / Ly6G / NK1.1), CD8+ and CD45.1+. Total RNA was extracted using ReliaPrep RNA Cell Miniprep System (Promega Corporation, Madison Wisconsin, USA) according to manufacturer’s protocol. All Mouse RNAs were analyzed on Tape Station System, Automated Electrophoresis (Agilent, Santa Clara, CA, USA) for quality assessment with RNA Integrity Number (RIN) around 9. cDNA libraries were prepared using 4 ng of total RNA using the Smart-Seq v2 protocol (Picelli et al., 2014). The length distribution of the cDNA libraries was monitored using a High Sensitivity D5000 Screentape assay and analysis (Agilent, Santa Clara, CA, USA). All samples were subjected to an indexed sequencing run of 1x100nt of length through NovaSeq 6000 SP Reagent Kit (100 cycles).
RNAseq bioinformatics analysis
Raw reads were aligned to mouse genome build GRCm38 using STAR aligner (Dobin et al., 2013). Read counts per gene were then calculated using featureCounts based on GENCODE gene annotation version M16. Read counts were subject to log2 TPM (transcript per million) normalization to account for transcript length and library size.
Only genes with a CPM (Counts per Million) value higher than 1 in at least three samples were considered for following analysis. Differentially Expressed Genes (DEGs) between groups treated with 4-1BB agonist and PBS were identified by generating a linear model using LIMMA R package (Ritchie et al., 2015). Only DEGs with an adjusted P value < 0.05 (using Benjamini Hochberg correction method) were selected for further analysis.
Functional Enrichment Analysis
Of the 4,928 identified DEGs between 4-1BB and control (PBS) treated samples, 2,443 were up-regulated and 2,485 were down-regulated. The up-regulated ones were subject to a functional enrichment analysis using the EnrichR R package (Kuleshov et al., 2016). They were checked for any biological signature enrichment in the Gene Ontology Biological Process database (2021), BioPlanet (2019) and the MSigDB Hallmark database (2020). In order to select the top enriched terms, only those with a high Combined Score (-log(p-value) ∗ Odds Ratio) and an adjusted P value < 0.05 were considered.
Clustering of up-regulated Terms
For visualization and analysis, the first 100 (ordered by adjusted P value) up-regulated terms were subject to a clustering algorithm, in order to identify the most prominent biological signatures. Briefly, a Jaccard Index Similarity score was calculated for each pair set of terms, based on the DEGs annotated for each term, using an in-house developed script. Next, terms were clustered using a hierarchical clustering method, using as distance measure the Pearson correlation between the calculated Jaccard Index Similarity scores. An arbitrary number of clusters was selected and manually annotated based on the terms present. To visualize the result, the pheatmap R package was used.
Intravital multiphoton microscopy
Anesthetics procedures and surgery for intravital microscopy were approved by local institutional animal care and use committee (IACUC) and were carried out as previously described.4 Briefly, mice were anesthetized using 5% isoflurane (1 L/min oxygen) delivered through an inhalation cone. A heparinized polyethylene catheter was then inserted into either the tail lateral vein or the hepatic vein, connected to an infusion pump that administers a 37°C saline solution at a rate of 0.5 ml/hour. This setup is crucial not only for hydrating the animals but also for administering Quantum dots to visualize blood flow and lymphocytes during the experiment. Surgery was conducted under 2% isoflurane anesthesia. Throughout the procedure, vital signs such as heart rate, respiratory rate, oxygen saturation, and vascular distension were continuously monitored in real-time using a MouseOx sensor (Starr Life Science Corp, Allison Park, PA, USA). This monitoring allows for the precise adjustment of anesthesia levels tailored to each animal, ensuring that oxygen saturation remains above 95% by varying the delivered oxygen amount. Intravital images were acquired with a LaVision BioTec TriMScope II coupled to a Nikon Ti-U inverted microscope enclosed in a custom-built environmental chamber (Life Imaging Services) that was maintained at 37°C during imaging using heated air. Fluorophores were excited with two tunable femtosecond-pulsed Ti:Sa lasers (Ultra II, Coherent) allowing 680–1080 nm wavelengths of excitations. Images were acquired using a water-immersion 25x objective with a high working distance (NA = 0.95, Olympus) and high sensitivity photomultiplier tubes (Hamamatsu H7422-40 GaAsP and Hamamatsu H7422-50 GaAsP red-extended PMT). Image stacks were acquired as 6-15 xy slices (squares of 150 μm x 150 μm to 300 μm x 300 μm) sampled with 4 μm z spacing every 20s for 10-60 min. Image stack sequences were rendered into four-dimensional videos with using Imaris software (Bitplane). Imaris was also used for semi-automated cell tracking and to manually separate intravascular and extravascular cell tracks. Data of cell centroids was used to derive key parameters of cell motility with the help of customized Matlab scripts (R2019b, MathWorks). The cell speed was calculated as total track length divided by the total track duration (μm/min). Arrest coefficients were calculated as the percentage of time a cell migrates below a motility threshold of 5 μm/min. The meandering index was derived by dividing track displacement by total track length. We further calculated corrected track straightness, which is defined as the meandering index multiplied with the square root of track duration.
Confocal Immunofluorescence Histology and Histochemistry
Confocal microscopy analysis of livers was performed as described.4 Briefly, the liver was fixed overnight in PBS with 4% paraformaldehyde and subsequently incubated for 24 hours in PBS with 30% sucrose. Next, liver lobes were embedded in O.C.T (Killik BioOptica) and cut at -20°C into 60 μm thick sections with a cryostat. Sections were blocked for 15 min with blocking buffer (PBS, 0.5% BSA, 0.3 % Triton) and stained for 1h at room temperature with anti-CD38 Alexa Fluor 594 (BioLegend, #102725) in wash/stain buffer (PBS, 0.2% BSA, 0.1% triton). Sections were then washed twice for 5 min, stained with DAPI (Sigma) for 5 min, washed again and mounted for imaging with FluorSave Reagent (Millipore). Image acquisition was performed with a 63x oil-immersion or 20x objective on an SP8 confocal microscope (Leica Microsystem). To minimize fluorophore spectral spillover, the Leica sequential laser excitation and detection modality was used. Where necessary to compensate for uneven slide illumination, fluorescent intensity of layers was normalized using Imaris normalize Layers tool. Where necessary, autofluorescence was filtered from the image by channel subtraction of a deep red autofluorescent channel from APC signal with the Imaris Channel Arithmetics tool.
For H&E, HBcAg and Caspase 3 immunohistochemistry, livers were perfused with PBS, harvested in Zn-formalin, and transferred into 70% ethanol 24 hours later. Subsequent to processing, the tissues were embedded in paraffin blocks and sectioned for staining. Primary antibodies included rat anti-Ki67 (clone TEC-3, Dako #M7249) polyclonal rabbit anti-HBcAg (Dako #B0586) and polyclonal rabbit anti-Cleaved Caspase-3 (Asp175, CST #9661). Primary antibodies were applied to the tissue sections at 1:500 dilution and allowed to incubate for 2 hours at room temperature to ensure optimal binding. This was followed by incubation with HRP-conjugated anti-rabbit secondary antibody for 30 minutes at room temperature. The antigen-antibody reaction was visualized using 3,3',5,5'-Tetramethylbenzidine (TMB) substrate, which provides a clear contrast for the identification of the target antigens. Bright-field images were acquired through an Aperio Scanscope System CS2 microscope and an ImageScope program (Leica Biosystem) following the manufacturer instructions.
Biochemical Analyses
The extent of hepatocellular injury was monitored by measuring serum alanine transaminase (sALT) activity at multiple time points after treatment, as previously described.5 Briefly, ALT liquid (Werfen #0018257440) was used for the quantitative determination of the serum ALT with an International Federation of Clinical Chemistry and laboratory medicine-optimized kinetic ultraviolet (UV) method in an ILab650 chemical analyzer (Instrumentation Laboratory). SeraChem control level 1 and level 2 (Werfen #0018162412 and #0018162512) were analyzed as quality control.
Analysis of human samples
Peripheral Blood Mononuclear Cell (PBMC) were isolated from fresh heparinized blood by Ficoll-Hypaque density gradient centrifugation and cryopreserved in liquid nitrogen until the day of analysis. Two mixtures of synthetic peptides were utilized for functional experiments, each consisting of 15-mers that overlap by ten amino acids and covering the complete sequence of either HBV core or polymerase proteins (Mimotopes, Victoria, Australia). Short-term T-cell lines were generated by 8-10 days PBMCs stimulation with HBV core or polymerase peptides (1μM), in the presence/absence of different compounds including anti-PD1 MAb (1, 5, 10 μg/ml, Thermo Fisher), OX40 ligand (30, 50, 100, 500, 1000 ng/ml, Sigma, Missouri, USA), 4-1BB ligand (0.5, 1, 2, 4 μg/ml, Enzo Life Sciences, New York, USA). All T-cell lines were supplemented with recombinant IL-2 on day 4 of culture, and the immunologic assays were performed on day 10.
T cell cytokine production (IFN-g, TNF-a, IL-2) and degranulation (CD107a) were detected by intracellular cytokine staining in treated or untreated T cell lines, upon stimulation with pools of 15-mer HBV peptides (1μM) and simultaneously stained with CD107a antibody. GolgiStop (BD Biosciences) and GolgiPlug (BD Biosciences) were added for 4 hours of incubation. After washing, cells were stained with surface markers for 15 min at room temperature. Cells were then fixed and permeabilized (Nordic MUbio, Susteren Netherlands) and stained with anti-IFN-g, anti-TNF-a monoclonal antibodies for 15 min at room temperature. Cells were finally washed and analyzed by flow cytometry. The antibodies used included anti-CD3-BD Horizon PE-CF594 (clone UCHT1), IFN-γ-AlexaFluor700 (Becton Dickinson, BD Biosciences, NJ, USA); TNF-α-FITC, CD4-PE, CD8-PerCP (Miltenyi Biotec, Bergisch Gladbach, Germany); CD107a-PE-Cy7 (ThermoFisher, Massachusetts, USA). For every stimulation, the dosage that resulted in the most significant enhancement for each cytokine was selected.
Quantification and statistical analysis
Results are expressed as mean ± SEM with the exception of pairwise comparisons and violin plots, in which only mean is displayed. Individual data points are presented unless the analysis includes a temporal component. All statistical analyses were performed in Prism (GraphPad Software), and details are provided in the figure legends. Normality of data distribution was tested in all graphs with a Shapiro-Wilk or D’Agostino & Pearson normality test and parametric tests were chosen only when normality could be confirmed for each dataset. One-tailed test was chosen over two-tailed test when basic biology dictates that the change between the control and treatment group can only occur into one direction.
For each analysis, statistical significance is indicated as follows: ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001. Data above p=0.05 threshold were considered not statistically significant and are not highlighted in the comparisons.
Sample sizes were determined according to standard guidelines to ensure sufficient statistical power, targeting 90% power level, and taking into account the expected effect size and data variability. The number of animals or patients is indicated as “n” in the corresponding figure legend.
For flow cytometry data, t-SNE was selected for its effectiveness in visualizing high-dimensional data in lower-dimensional spaces. However, for datasets coming from scRNAseq experiments, UMAP was chosen as it offers advantages due to its ability to handle larger datasets more efficiently.
Acknowledgments
We thank the members of the Iannacone laboratory for discussions and technical support and D. Pinschewer for providing rLCMV. M.I. is supported by the European Research Council (ERC) Advanced Grant 101141363; ERC Proof of Concept Grant 101138728; Italian Association for Cancer Research (AIRC) grants 19891 and 22737; Italian Ministry for University and Research grants PE00000007 (INF-ACT) and PRIN 2022FMESXL; and Funded Research Agreements from Asher Biotherapeutics, VIR Biotechnology, and BlueJay Therapeutics. F.A. is supported by the MERU Foundation. M.R. is supported by Cariplo (2021-1551). C.C.K. received an AIRC fellowship (25395). X.F. received an iCARE-2 co-funded by AIRC and Marie-Curie Fellowship (800924). K.K. received a Uehara Memorial Foundation fellowship. C.G.B. received an EMBO fellowship (ALTF180-2020). M.D.G. is supported by AIRC (27564), GAHF CDA, and ERC-StG-101116224. D.I. is supported by AIRC (26183) and Horizon-2020 (964481-IN2SIGHT). M. Isogawa is supported by the Research Program on Hepatitis from the Japan Agency for Medical Research and Development (23fk0310519). L.G.G. is supported by PRIN-20224NMLXK, PRIN-P2022Z8HNC, PE00000007 (INF-ACT), AIRC (22737), and a donation from Fondazione SAME.
Author contributions
F.A., C.L., M.R., C.C.K., X.F., K.K., and C.G.B. designed, performed, and analyzed experiments. F.A., C.L., C.C.K., and X.F. drafted the original manuscript; X.F., F.M., B.P., and G.N. performed the intravital imaging experiments; C.L. performed the bioinformatic analyses; B.P., G.N., V.F., P.D.L., C.G.B., E.B.B., L.G., C.P., S.R., and V.V. performed mouse experiments; F.A., I.M., C.F., C.B., and P.F. performed or analyzed the experiments on human samples; M.G. and R.O. helped with the scRNA-seq experiments; F.A., C.C.K., and C.L. drafted the figures; F.A. finalized the figures and drafted the revised manuscript. J.M.G.-M., E.L., D.I., M.D.G., M. Isogawa, and L.G.G. provided conceptual advice; M. Iannacone designed and coordinated the study, provided funding, and wrote the manuscript.
F.A., C.L., M.R., C.C.K., X.F., K.K., and C.G.B. have contributed equally to this work. The authors agree that the aforementioned individuals are entitled to reorder the sequence of authorship for this publication in their respective CVs.
Declaration of interests
M. Iannacone participates in advisory boards/consultantship for Asher Biotherapeutics, GentiBio, BlueJay Therapeutics, and Aligos Therapeutics. L.G.G. participates in boards/consultantship for Genenta Science, Epsilen Bio, Aligos Therapeutics, Medicxi, Chroma Medicine, and Ananda Immunotherapies.
Published: June 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2024.05.038.
Supplemental information
References
- 1.Iannacone M., Guidotti L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2022;22:19–32. doi: 10.1038/s41577-021-00549-4. [DOI] [PubMed] [Google Scholar]
- 2.Kawashima K., Andreata F., Beccaria C.G., Iannacone M. Priming and Maintenance of Adaptive Immunity in the Liver. Annu. Rev. Immunol. 2024 doi: 10.1146/annurev-immunol-090122-041354. Published online February 15, 2024. [DOI] [PubMed] [Google Scholar]
- 3.Isogawa M., Chung J., Murata Y., Kakimi K., Chisari F.V. CD40 Activation Rescues Antiviral CD8+ T Cells from PD-1-Mediated Exhaustion. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bénéchet A.P., De Simone G., Di Lucia P., Cilenti F., Barbiera G., Le Bert N., Fumagalli V., Lusito E., Moalli F., Bianchessi V., et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature. 2019;574:200–205. doi: 10.1038/s41586-019-1620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guidotti L.G., Inverso D., Sironi L., Di Lucia P., Fioravanti J., Ganzer L., Fiocchi A., Vacca M., Aiolfi R., Sammicheli S., et al. Immunosurveillance of the Liver by Intravascular Effector CD8+ T Cells. Cell. 2015;161:486–500. doi: 10.1016/j.cell.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simone G.D., Andreata F., Bleriot C., Fumagalli V., Laura C., Garcia-Manteiga J.M., Lucia P.D., Gilotto S., Ficht X., Ponti F.F.D., et al. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 2021;54:2089–2100.e8. doi: 10.1016/j.immuni.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreata F., Moynihan K.D., Fumagalli V., Di Lucia P.D., Pappas D.C., Kawashima K., Ni I., Bessette P.H., Perucchini C., Bono E., et al. CD8 cis-targeted IL-2 drives potent antiviral activity against hepatitis B virus. Sci. Transl. Med. 2024;16 doi: 10.1126/scitranslmed.adi1572. [DOI] [PubMed] [Google Scholar]
- 8.Gane E., Verdon D.J., Brooks A.E., Gaggar A., Nguyen A.H., Subramanian G.M., Schwabe C., Dunbar P.R. Anti-PD-1 Blockade with Nivolumab with and without Therapeutic Vaccination for Virally Suppressed Chronic Hepatitis B: A Pilot Study. J. Hepatol. 2019;71:900–907. doi: 10.1016/j.jhep.2019.06.028. [DOI] [PubMed] [Google Scholar]
- 9.Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van der Braber M., Rozeman E.A., Haanen J.B.A.G., Blank C.U., et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2018;176:775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brummelman J., Mazza E.M.C., Alvisi G., Colombo F.S., Grilli A., Mikulak J., Mavilio D., Alloisio M., Ferrari F., Lopci E., et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumorsStem-like. J. Exp. Med. 2018;215:2520–2535. doi: 10.1084/jem.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng C., Zheng L., Yoo J.-K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q., et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 12.Alfei F., Kanev K., Hofmann M., Wu M., Ghoneim H.E., Roelli P., Utzschneider D.T., von Hoesslin M., Cullen J.G., Fan Y., et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265–269. doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 13.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zander R., Schauder D., Xin G., Nguyen C., Wu X., Zajac A., Cui W. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. 2019;51:1028–1042.e4. doi: 10.1016/j.immuni.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hudson W.H., Gensheimer J., Hashimoto M., Wieland A., Valanparambil R.M., Li P., Lin J.-X., Konieczny B.T., Im S.J., Freeman G.J., et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic. Immunity. 2019;51:1043–1058.e4. doi: 10.1016/j.immuni.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z., Ji Z., Ngiow S.F., Manne S., Cai Z., Huang A.C., Johnson J., Staupe R.P., Bengsch B., Xu C., et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity. 2019;51:840–855.e5. doi: 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Utzschneider D.T., Charmoy M., Chennupati V., Pousse L., Ferreira D.P., Calderon-Copete S., Danilo M., Alfei F., Hofmann M., Wieland D., et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 18.Kallies A., Zehn D., Utzschneider D.T. Precursor exhausted T cells: key to successful immunotherapy? Nat. Rev. Immunol. 2020;20:128–136. doi: 10.1038/s41577-019-0223-7. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy P.T.F., Sandalova E., Jo J., Gill U., Ushiro-Lumb I., Tan A.T., Naik S., Foster G.R., Bertoletti A. Preserved T-Cell Function in Children and Young Adults With Immune-Tolerant Chronic Hepatitis B. Gastroenterology. 2012;143:637–645. doi: 10.1053/j.gastro.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Mason W.S., Gill U.S., Litwin S., Zhou Y., Peri S., Pop O., Hong M.L.W., Naik S., Quaglia A., Bertoletti A., et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology. 2016;151:986–998.e4. doi: 10.1053/j.gastro.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Bert N.L., Gill U.S., Hong M., Kunasegaran K., Tan D.Z.M., Ahmad R., Cheng Y., Dutertre C.-A., Heinecke A., Rivino L., et al. Effects of Hepatitis B Surface Antigen on Virus-Specific and Global T Cells in Patients With Chronic Hepatitis B Virus infection. Gastroenterology. 2020;159:652–664. doi: 10.1053/j.gastro.2020.04.019. [DOI] [PubMed] [Google Scholar]
- 22.European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu. European Association. for the Study of the Liver EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017;67:370–398. doi: 10.1016/j.jhep.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 23.Guidotti L.G., Martinez V., Loh Y.T., Rogler C.E., Chisari F.V. Hepatitis B virus nucleocapsid particles do not cross the hepatocyte nuclear membrane in transgenic mice. J. Virol. 1994;68:5469–5475. doi: 10.1128/JVI.68.9.5469-5475.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guidotti L.G., Matzke B., Schaller H., Chisari F.V. High-level hepatitis B virus replication in transgenic mice. J. Virol. 1995;69:6158–6169. doi: 10.1128/JVI.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tse S.-W., Radtke A.J., Espinosa D.A., Cockburn I.A., Zavala F. The Chemokine Receptor CXCR6 Is Required for the Maintenance of Liver Memory CD8+ T Cells Specific for Infectious Pathogens. J. Infect. Dis. 2014;210:1508–1516. doi: 10.1093/infdis/jiu281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller B.C., Sen D.R., Al Abosy R.A., Bi K., Virkud Y.V., LaFleur M.W., Yates K.B., Lako A., Felt K., Naik G.S., et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.La Manno G.L., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisicaro P., Barili V., Rossi M., Montali I., Vecchi A., Acerbi G., Laccabue D., Zecca A., Penna A., Missale G., et al. Pathogenetic Mechanisms of T Cell Dysfunction in Chronic HBV Infection and Related Therapeutic Approaches. Front. Immunol. 2020;11:849. doi: 10.3389/fimmu.2020.00849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mieli-Vergani G., Bansal S., Daniel J.F., Kansu A., Kelly D., Martin C., Tizzard S., Wirth S., Zhou J., Vergani D. Peginterferon Alfa-2a (40KD) Plus Lamivudine or Entecavir in Children with Immune-tolerant Chronic Hepatitis B. J. Pediatr. Gastroenterol. Nutr. 2021;73:156–160. doi: 10.1097/MPG.0000000000003118. [DOI] [PubMed] [Google Scholar]
- 30.Rosenthal P., Ling S.C., Belle S.H., Murray K.F., Rodriguez-Baez N., Schwarzenberg S.J., Teckman J., Lin H.S., Schwarz K.B., Hepatitis B Research Network (HBRN) Combination of Entecavir/Peginterferon Alfa-2a in Children With Hepatitis B e Antigen–Positive Immune Tolerant Chronic Hepatitis B Virus Infection. Hepatology. 2019;69:2326–2337. doi: 10.1002/hep.30312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feld J.J., Terrault N.A., Lin H.S., Belle S.H., Chung R.T., Tsai N., Khalili M., Perrillo R., Cooper S.L., Ghany M.G., et al. Entecavir and Peginterferon Alfa-2a in Adults With Hepatitis B e Antigen–Positive Immune-Tolerant Chronic Hepatitis B Virus Infection. Hepatology. 2019;69:2338–2348. doi: 10.1002/hep.30417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fisicaro P., Barili V., Montanini B., Acerbi G., Ferracin M., Guerrieri F., Salerno D., Boni C., Massari M., Cavallo M.C., et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017;23:327–336. doi: 10.1038/nm.4275. [DOI] [PubMed] [Google Scholar]
- 33.Kawashima K., Isogawa M., Onishi M., Baudi I., Saito S., Nakajima A., Fujita T., Tanaka Y. Restoration of type I interferon signaling in intrahepatically primed CD8+ T cells promotes functional differentiation. JCI Insight. 2021;6 doi: 10.1172/jci.insight.145761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Y., Zhu Y.O., Becht E., Aw P., Chen J., Poidinger M., de Sessions P.F., Hibberd M.L., Bertoletti A., Lim S.G., et al. Multifactorial heterogeneity of virus-specific T cells and association with the progression of human chronic hepatitis B infection. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aau6905. [DOI] [PubMed] [Google Scholar]
- 35.Siddiqui I., Schaeuble K., Chennupati V., Marraco S.A.F., Calderon-Copete S., Ferreira D.P., Carmona S.J., Scarpellino L., Gfeller D., Pradervand S., et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity. 2019;50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 36.Jansen C.S., Prokhnevska N., Master V.A., Sanda M.G., Carlisle J.W., Bilen M.A., Cardenas M., Wilkinson S., Lake R., Sowalsky A.G., et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576:465–470. doi: 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichler A.C., Carrié N., Cuisinier M., Ghazali S., Voisin A., Axisa P.-P., Tosolini M., Mazzotti C., Golec D.P., Maheo S., et al. TCR-independent CD137 (4-1BB) signaling promotes CD8+-exhausted T cell proliferation and terminal differentiation. Immunity. 2023;56:1631–1648.e10. doi: 10.1016/j.immuni.2023.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee D.Y., Choi B.K., Lee D.G., Kim Y.H., Kim C.H., Lee S.J., Kwon B.S. 4-1BB Signaling Activates the T Cell Factor 1 Effector/β-Catenin Pathway with Delayed Kinetics via ERK Signaling and Delayed PI3K/AKT Activation to Promote the Proliferation of CD8+ T Cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0069677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melero I., Shuford W.W., Newby S.A., Aruffo A., Ledbetter J.A., Hellström K.E., Mittler R.S., Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 40.Claus C., Ferrara-Koller C., Klein C. The emerging landscape of novel 4-1BB (CD137) agonistic drugs for cancer immunotherapy. mAbs. 2023;15 doi: 10.1080/19420862.2023.2167189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkw377. W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 44.Korotkevich G., Sukhov V., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2019 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 45.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer Cham; 2016. [Google Scholar]
- 48.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berg S., Kutra D., Kroeger T., Straehle C.N., Kausler B.X., Haubold C., Schiegg M., Ales J., Beier T., Rudy M., et al. ilastik: interactive machine learning for (bio)image analysis. Nat Methods. 2019;16:1226–1232. doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- 50.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kolde R. CRAN; 2019. pheatmap: Pretty Heatmaps.https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf [Google Scholar]
- 52.Li B., Dewey C N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 54.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reeves J.P., Reeves P.A., Chin L.T. Survival Surgery: Removal of the Spleen or Thymus. Curr. Protoc. Immunol. 2001;Chapter 1:1.10.1–1.10.11. doi: 10.1002/0471142735.im0110s02. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The bulk RNA and scRNAseq datasets have been deposited in the Gene Expression Omnibus (GEO) repository and will be publicly available upon publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.