

Hemoglobinopathies including thalassemias are among the most frequent genetic disorders worldwide. Primarily, these entities result from germline variants in the globin gene clusters and their cis-acting regulatory elements,1 and thus the World Health Organization classifies thalassemias as inherited diseases.2 Non-inherited disorders of globin chain synthesis mimicking the phenotype of thalassemias have also been described and are referred to as acquired thalassemias. These forms mainly affect the a-globin genes and are observed at much lower frequencies.3 Acquired a-thalassemias are associated mainly with myelodysplastic neoplasms (MDS) and are caused either by somatic deletions of the a-globin gene cluster on chromosome 16p13.3 limited to the hematological clone or, more commonly, by inactivating somatic variants of the trans-acting regulatory factor, ATRX, leading to downregulation of a-globin gene expression.3 In contrast, acquired somatic genetic variants causing reduced β-globin production and leading to a β-thalassemic phenotype are a rarity, and only a few, single cases have been described so far.4-7
Here, we describe a patient presenting with thalassemic erythrocyte parameter changes, i.e., microcytic, hypochromic anemia and a rather high erythrocyte count. Further evaluation including hemoglobin capillary electrophoresis and molecular genetic analyses exhibited clonal hematopoiesis with large deletions in the short and long arm of one chromosome 11 restricted to the neoplastic clone. The loss in the chromosomal band 11p15.4 harboring the complete β-globin gene cluster resulted in an acquired εγδβ-thalassemia which we documented at the molecular level.
Our laboratory received a peripheral blood sample from a 34-year-old woman with microcytic, hypochromic anemia for further investigation after exclusion of iron deficiency. Blood count measurement at our laboratory confirmed a mild microcytic, hypochromic anemia (hemoglobin [Hb] 11.0 g/dL, mean corpuscular volume [MCV] 72.3 fL, mean corpuscular hemoglobin 22.0 pg, mean corpuscluar hemoglobin concentration [MCH] 30.5 g/dL, red cell distribution width [RDW] 19.2 %, red blood cell [RBC] count 4.99x1012/L). The white blood cell count was normal (7.97x109/L), and the thrombocyte count was slightly increased (513x109/L). A peripheral blood smear showed polychromatic erythrocytes with basophilic stippling and target cells (Figure 1), and a normal leukocyte morphology and differential blood count (57% segmented neutrophils, 30% lymphocytes, 7% monocytes, and 6% eosinophils).
Further hemoglobinopathy work-up using capillary electrophoresis exhibited normal Hb fractions with HbA 96.8%, HbA2 2.7% and HbF 0.5%; no Hb variants were detected. Molecular genetics applying gap-polymerase chain reaction, multiplex-ligation-dependent probe amplification and targeted next generation sequencing (NGS) of HBA1, HBA2 and HBB revealed no aberration of the α-globin gene locus, but a loss of the complete β-globin gene cluster with an allele frequency of about 50% in peripheral blood DNA (Figure 2).
In order to delimit the precise breakpoints of the deletions and to identify potential further genomic structural variants, whole genome sequencing (WGS) of DNA from peripheral blood leukocytes showed large deletions of 3.8 Mbp at chromosome 11p15.5-4 (GRCh37/hg19; chr11_2207001::5984000) including the β-globin cluster, of 2.9 Mbp at 11p14.3-1 (chr11_24706001:: 27629000), and of 6.2 Mbp at chr11q22.3-q23.2 (chr11_108103001::114299000) involving the AT M gene (Figure 3A). Single nucleotide variants clinically significant in development of hematologic neoplasms were not detected.
Whole genome sequencing (WGS) findings were in line with chromosome banding analysis from peripheral blood showing a derivative chromosome 11 with the expected deletions in the p- and q-arm: 46,XX,der(11)del(11)(p15p14) del(11)(q22q23)[16]/46,XX[8]. The aberrant clone was found in phytohemagglutinin-stimulated cultures as well as in cultures stimulating the myeloid lineage. Interestingly, the aberrant karyotype was detected in only 16 of 24 metaphases and additional interphase-fluorescence in situ hybridisation analyses with probes for the NUP98 gene (11p15.4) and the ATM gene (11q22.3) confirmed the deletions in 85 of 100 and 80 of 100 cells, respectively, indicating that the deletions were not of germline origin but acquired in the hematopoietic cells. In a next step, we thus investigated DNA derived from oral mucosa of the patient as a germline surrogate by WGS to have a direct comparison with blood cell WGS results. The deletions on chromosome 11 were absent in the oral mucosa DNA of the patient (Figure 3B). In summary, we confirmed the somatic origin restricted to a hematopoietic clone of the deletion affecting the b-globin cluster at a molecular level. The patient has given informed consent to publish the findings, and the study has been approved by the institutional review board.
Figure 1.
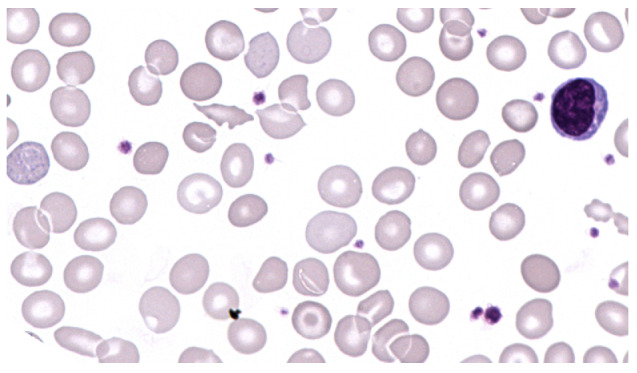
Peripheral blood smear. Peripheral blood smear showing microcytic and hypochrome erythrocytes, polychromasia, basophilic stippling and target cells (x400 magnification).
Figure 2.

Multiplex ligation-dependent probe amplification. Multiplex-ligation-dependent probe amplification revealing a loss of the complete b-globin gene cluster with an allele frequency of about 50% (red arrow).
In this case report, we describe an acquired thalassemic condition in clonal hematopoiesis resulting in a b-thalassemic phenotype and document it at the molecular level. In contrast to acquired forms of a-thalassemia that are an established observation in MDS, acquired b-thalassemia are extremely rare and described in only a few cases. So far, acquired gδb-thalassemias have been described in MDS,4,5,7 acute myeloid leukemia,8 juvenile myelomonocytic leukemia,6 and common variable immunodeficiency.9 In the present patient, bone marrow investigations would be necessary to differentiate between clonal cytopenia of unknown significance and a hematologic neoplasm, such as MDS, as final diagnosis. However, clonal hematopoiesis harboring a somatic 11p deletion as the pathophysiological cause of the acquired b-thalassemia could be reliably demonstrated. Previous studies have shown that genomic gains and losses are a frequent phenomenon in clonal hematopoiesis.10,11 This includes recurrent chromosome 11 aberrations, and loss of ATM due to chromosomal deletions as in the present case. Interestingly, ATM mutations and chromosome 11q deletions are recurrent aberrations in MDS,12,13 while deletions in the short arm of chromosome 11 are rather rare in clonal hematopoiesis.10,11
An increase in HbA2 above 3.6% with corresponding microcytic, hypochromic anemia with erythrocytosis is the hallmark phenotypical finding of inherited, heterozygous b-thalassemia. Interestingly, none of the previously described cases of acquired b-thalassemia exhibited an HbA2 increase suggesting the presence of deleterious aberrations of both the b- and δ-globin genes. However, most of those patients presented with an increase in HbF pointing to intact g-globin genes.5-8 Thus, these cases most likely represent δb-thalassemias caused by deletions in the b-globin gene cluster affecting the δ- and b-globin gene, but leaving the g-globin genes intact. Despite the phenotypic observations in those studies, the genetic correlates remain hypothetical as precise molecular experiments were lacking in most studies. In our case, we observed a normal distribution of the hemoglobin fractions without an increase in HbA2 or HbF. High resolution molecular genetic analyses documented on the sequence level that the absence of an HbA2 and HbF increase was due to the loss of the complete b-globin gene cluster in the neoplastic clone. The deletion includes the locus control region of the b-globin gene cluster in addition to the ee-, g-, δ and b-globin genes and is thus the documentation of an acquired form of egδb-thalassemia at the molecular level.
a to non-a globin ratio imbalance represents the pathophysiological source of the typical thalassemic phenotype of microcytic, hypochromic anemia.1 Despite its common genetic basis of variants disturbing regular globin gene transcription, the origin of those alterations vary and either represent germline variants that are inherited from generation to generation or are somatic mutations acquired in a hematopoietic cell of an individual. In most cases, thalassemias originate from inherited germline variants and are classified as disorders from the field of classical, non-malignant hematology. However, clinicians also need to consider acquired thalassemic syndromes in hematological malignancies, especially in cases of discrepancy between tentative diagnosis and corresponding blood count changes. For example, macrocytic anemia is a typical finding in MDS, and thus, additional conditions like iron deficiency or acquired thalassemia need to be ruled out when MDS is accompanied with microcytic hypochromic erythrocyte parameters. In those cases, molecular genetic analysis of the neoplastic cell clone is key to a precise diagnosis, which may be elusive when using standard tests like hemoglobin separation techniques used to screen for hereditary hemoglobinopathies.
Figure 3.
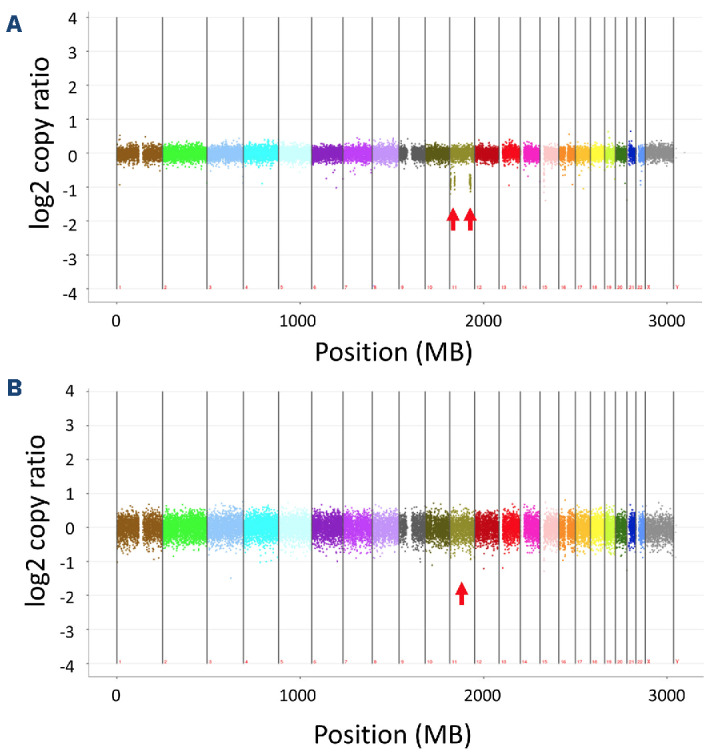
Copy number variation analysis of the entire genome by next generation sequencing. Copy number variant analysis of the entire genome by next generation sequencing indicating the presence of deletions on chromosome 11 (red arrows) in peripheral blood DNA (A) and their absence in oral mucosa DNA (B).
Data-sharing statement
Original data are available upon request in accordance with applying data protection rules.
References
- 1.Bain BJ. Haemoglobinopathy diagnosis. 3rd ed. Wiley Blackwell; 2020. [Google Scholar]
- 2.World Health Organization. Thalassaemia and other haemoglobinopathies. https://apps.who.int/gb/ebwha/pdf_files/EB118/B118_5-en.pdf Accessed January 01, 2024. [Google Scholar]
- 3.Steensma DP, Gibbons RJ, Higgs DR. Acquired alphathalassemia in association with myelodysplastic syndrome and other hematologic malignancies. Blood. 2005;105(2):443-452. [DOI] [PubMed] [Google Scholar]
- 4.Brunner AM, Steensma DP. Myelodysplastic syndrome associated with acquired beta thalassemia: “BTMDS”. Am J Hematol. 2016;91(8):E325-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoyagi Y, Akimoto H, Yamaoka K, et al. [Refractory anemia with ringed sideroblasts complicated with delta beta-thalassemia-like hemoglobinopathy]. Rinsho Ketsueki. 1989;30(5):674-679. [PubMed] [Google Scholar]
- 6.Honig GR, Suarez CR, Vida LN, Lu SJ, Liu ET. Juvenile myelomonocytic leukemia (JMML) with the hematologic phenotype of severe beta thalassemia. Am J Hematol. 1998;58(1):67-71. [DOI] [PubMed] [Google Scholar]
- 7.Hoyle C, Kaeda J, Leslie J, Luzzatto L. Acquired beta thalassaemia trait in MDS. Br J Haematol. 1991;79(1):116-117. [DOI] [PubMed] [Google Scholar]
- 8.Markham RE, Butler F, Goh K-O, Rowley PT. Erythroleukemia manifesting δβ-thalassemia. Hemoglobin. 2009;7(1):71-78. [DOI] [PubMed] [Google Scholar]
- 9.Belickova M, Schroeder HW, Guan YL, et al. Clonal hematopoiesis and acquired thalassemia in common variable immunodeficiency. Mol Med. 1994;1(1):56-61. [PMC free article] [PubMed] [Google Scholar]
- 10.Loh PR, Genovese G, McCarroll SA. Monogenic and polygenic inheritance become instruments for clonal selection. Nature. 2020;584(7819):136-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao T, Ptashkin R, Bolton KL, et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. 2021;12(1):338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stengel A, Kern W, Meggendorfer M, Haferlach T, Haferlach C. MDS with deletions in the long arm of chromosome 11 are associated with a high frequency of SF3B1 mutations. Leukemia. 2017;31(9):1995-1997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data are available upon request in accordance with applying data protection rules.