

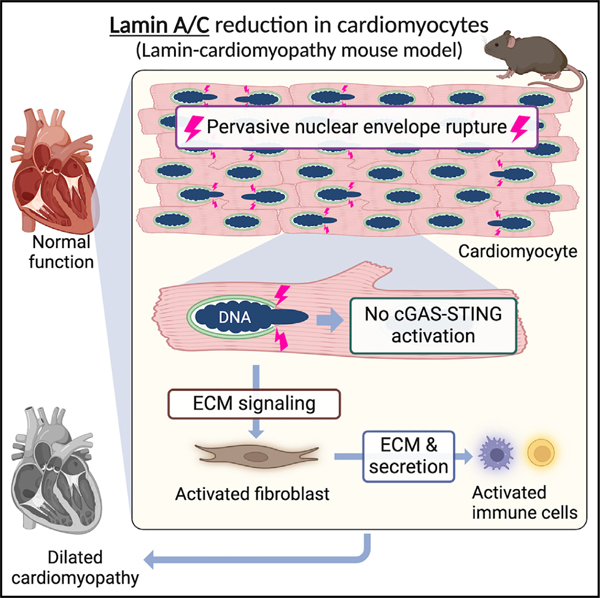
SUMMARY
Nuclear envelope (NE) ruptures are emerging observations in Lamin-related dilated cardiomyopathy, an adult-onset disease caused by loss-of-function mutations in Lamin A/C, a nuclear lamina component. Here, we test a prevailing hypothesis that NE ruptures trigger the pathological cGAS-STING cytosolic DNA-sensing pathway using a mouse model of Lamin cardiomyopathy. The reduction of Lamin A/C in cardiomyocyte of adult mice causes pervasive NE ruptures in cardiomyocytes, preceding inflammatory transcription, fibrosis, and fatal dilated cardiomyopathy. NE ruptures are followed by DNA damage accumulation without causing immediate cardiomyocyte death. However, cGAS-STING-dependent inflammatory signaling remains inactive. Deleting cGas or Sting does not rescue cardiomyopathy in the mouse model. The lack of cGAS-STING activation is likely due to the near absence of cGAS expression in adult cardiomyocytes at baseline. Instead, extracellular matrix (ECM) signaling is activated and predicted to initiate pro-inflammatory communication from Lamin-reduced cardiomyocytes to fibroblasts. Our work nominates ECM signaling, not cGAS-STING, as a potential inflammatory contributor in Lamin cardiomyopathy.
Graphical abstract
In brief
En et al. report pervasive nuclear envelope ruptures in cardiac myocytes in a mouse model of nuclear lamin-related cardiomyopathy. Surprisingly, nuclear envelope ruptures do not activate cGAS-STING cytosolic DNA-sensing innate immunity in hearts. Instead, ECM signaling from Lamin-mutant cardiomyocytes is predicted to initiate an inflammatory response.
INTRODUCTION
Lamin A/C (LMNA) are nuclear lamina proteins that provide structural integrity to the nuclear envelope (NE).1–4 Mutations in LMNA cause a spectrum of degenerative disorders, collectively called laminopathies, including frequent dilated cardiomyopathy (LMNA-related DCM).5–10 LMNA-related DCM is a prevalent adult-onset disease11,12 that accompanies cardiac conduction disease, fibrosis, heart failure, and mortality.13–16 LMNA-related DCM is predominantly caused by heterozygous loss-of-function LMNA mutations that cause Lamin A/C protein reduction most strongly in cardiomyocytes.16–20 Activation of stress-response-related signaling, such as mitogen-activated protein kinase signaling,21–24 mTOR signaling,25 and DNA damage response signaling,26,27 has been reported in LMNA-related DCM models. However, the direct pathological alterations caused by Lamin A/C reduction in cardiomyocytes remain undefined.
Reduction of nuclear lamins can cause ruptures of the NE (NE ruptures).28–34 Indeed, NE ruptures have been observed in patients,17,18,35–37 animal models,30,31,38–42 and cell culture models30,34 of LMNA-related DCM and laminopathies. However, the extent of NE ruptures in LMNA-related DCM and the contribution of this event to the pathogenesis of DCM remain undefined.
A prevailing hypothesis for LMNA-related DCM pathogenesis implicates the cGAS-STING pathway as a link between NE rupture, inflammatory signaling, and DCM pathogenesis.39,40,43–45 cGAS-STING is a cytosolic DNA-sensing innate immune mechanism, in which cGAS binds to cytosolic DNA and activates cell-autonomous STING-mediated interferon transcription.46,47 cGAS-STING pathway activation has been observed in a cellular model of progeria48 and Lmna-knockout developing hearts.27 cGAS-STING activation has also been reported in mouse models of other heart diseases.49–52 However, a direct link between NE ruptures and the cGAS-STING activation in LMNA-related DCM remains undefined. In this study, we report pervasive NE ruptures prior to DCM development and provide evidence against the hypothesis that cGAS-STING activation contributes to LMNA-related DCM. Instead, our study nominates a role for extracellular matrix (ECM) signaling from cardiomyocytes in this disease.
RESULTS
Lamin A/C reduction in cardiomyocytes causes DCM in adult mice
We generated a mouse model of LMNA-related DCM using cardiomyocyte-specific deletion of Lmna (LmnaF/F;Myh6-Mer-CreMer, called LmnaCKO hereafter) and littermate control wild-type mice (Lmna+/+;Myh6-MerCreMer) (Figures S1A and S1B). Lmna deletion in cardiomyocytes was induced by tamoxifen administration to adult mice at 6–8 weeks of age (Figure 1A). Lamin A and Lamin C proteins were 47% and 43% reduced, respectively, in cardiomyocytes of LmnaCKO mice at 2 weeks post-tamoxifen by immunoblots, consistent with the ~4-week half-life of Lamin A/C in mouse hearts53 (Figures 1B and S1C). Reduced, but persistent, Lamin A/C in individual cardiomyocytes was confirmed at 1, 2, and 3.5 weeks post-tamoxifen by tissue staining (Figure 1C). Thus, LmnaCKO mice reproduced the Lamin A/C protein insufficiency, not elimination, observed in cardiomyocytes of patients with LMNA-related DCM.18,20
Figure 1. Lamin A/C reduction in cardiomyocytes causes dilated cardiomyopathy in adult mice.
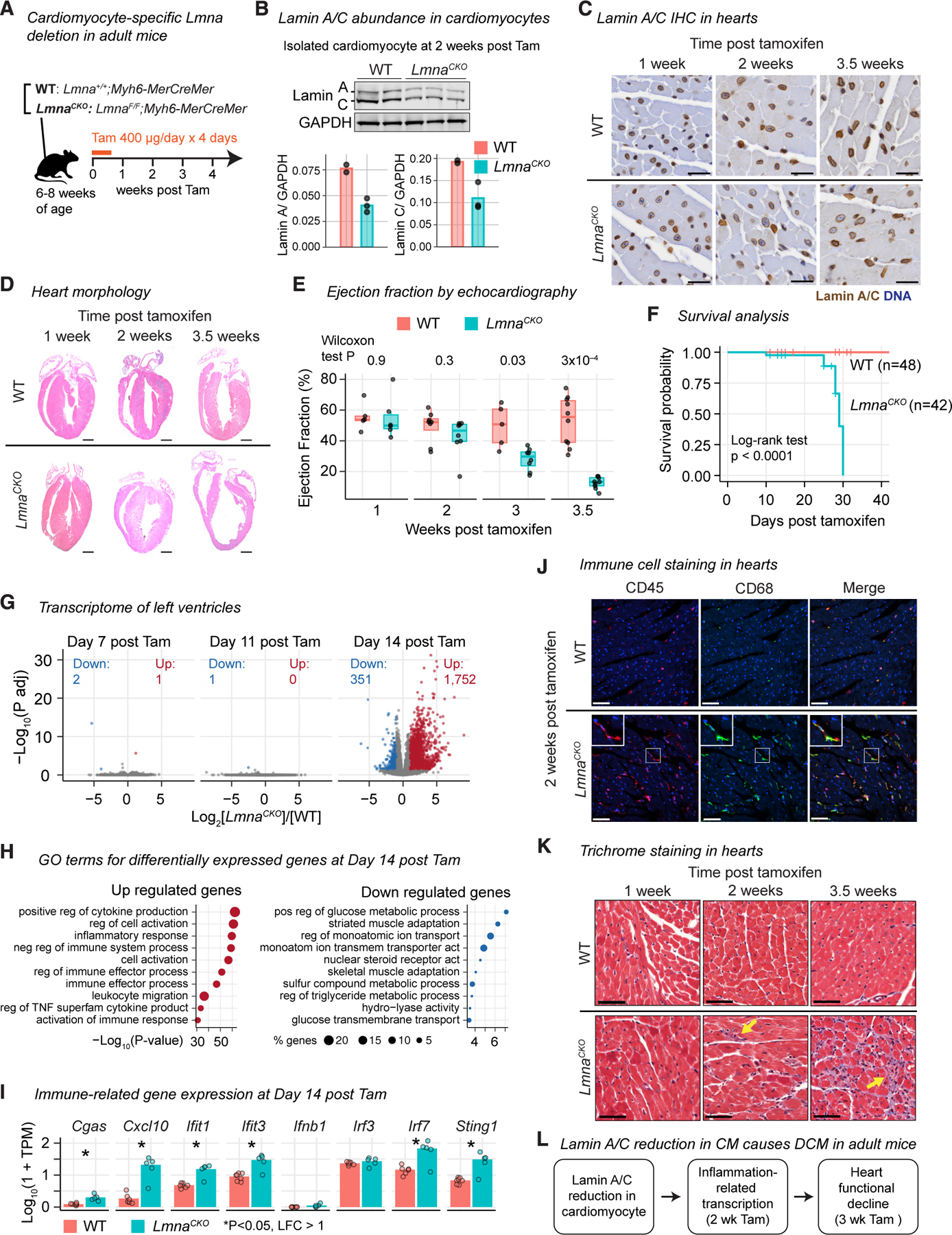
(A) Tamoxifen (Tam) induces Lmna deletion in cardiomyocytes in adult mice. See Figure S1 for related analyses.
(B) Lamin A/C immunoblot in isolated cardiomyocytes (top) with signal quantification (bottom). n = 2–3 mice/genotype.
(C) Lamin A/C immunohistochemistry (brown) with hematoxylin counterstaining (blue) in mouse heart tissues. Scale bar: 20 μm.
(D) Hematoxylin and eosin staining of hearts. Scale bar: 1 mm.
(E) Left ventricular ejection fraction with interquartile range (box) measured by echocardiography. p value, Wilcoxon test. n = 5–10 mice/genotype.
(F) Kaplan-Meier survival analysis. Wild type (WT) n = 48, LmnaCKO n = 42. p value, log-rank test.
(G) Volcano plot comparing RNA-seq read counts in LmnaCKO versus WT hearts. Number, upregulated or downregulated gene count. n = 3–7 mice/genotype.
(H) Top 10 Gene Ontology (GO) terms overrepresented among differentially expressed genes. p value is computed by Metascape.54
(I) Transcript abundance in hearts quantified by RNA-seq. TPM, normalized transcripts per million. Bar, mean. n = 5–7 mice/genotype. P, false discovery rate (FDR)-adjusted p value computed by a generalized linear model in DESeq2.55
(J) Immunofluorescence of heart tissue sections for CD45 (pan-leukocyte, red) and CD68 (macrophage, green). Scale bar: 20 μm.
(K) Masson’s trichrome staining of heart sections. Arrow, collagen deposition. Scale bar: 20 mm.
(L) Summary.
LmnaCKO mice developed progressive DCM as reported previously.38 Hearts of LmnaCKO mice were indistinguishable from wild-type hearts in gross morphology (Figure 1D) and contractile activity measured by echocardiography (Figure 1E) until 2 weeks post-tamoxifen. However, at 3 weeks post-tamoxifen, the left ventricular ejection fraction, an indicator of cardiac systolic function, significantly diminished (Figure 1E). At 3.5 weeks post-tamoxifen, LmnaCKO hearts became severely dilated, with a significant reduction of ventricular wall thickness and loss of systolic activity (Figures 1D, 1E, and S1D). LmnaCKO mice invariably died between 3.5 and 4.5 weeks post-tamoxifen (Figure 1F). Thus, the modest reduction of Lamin A/C in cardio-myocytes was sufficient to cause DCM that progressed to heart failure, recapitulating a crucial aspect of human LMNA-related DCM.
Lamin A/C reduction in cardiomyocytes activates inflammation-related transcription
To identify the onset of molecular changes preceding the functional decline of LmnaCKO hearts, we profiled the transcriptome of the heart at days 7, 11, and 14 post-tamoxifen by RNA-seq (Table S1). At days 7 and 11, LmnaCKO heart transcriptomes were almost identical to wild-type heart transcriptomes (Figure 1G). However, at day 14, LmnaCKO hearts exhibited strong transcriptional upregulation (1,751 genes) and modest downregulation (351 genes) (Figure 1G). The upregulated genes were highly overrepresented for Gene Ontology (GO) terms related to inflammatory responses (Figure 1H). These upregulated genes included innate immune-related genes such as Cxcl10, Ifit1, and Irf7 (Figure 1I), which can be activated by the cGAS-STING pathway. The downregulated genes were overrepresented for gene pathways for cardiomyocyte metabolism and function (Figure 1H).
We examined whether the upregulation of inflammation-related genes reflected an inflammatory response in LmnaCKO hearts. We observed that the CD45+CD68+ macrophage population increased in LmnaCKO hearts at 2 weeks post-tamoxifen (Figures 1J and S1E). We also observed increased interstitial collagen deposition at 2 weeks, which developed into extensive fibrosis by 3.5 weeks post-tamoxifen (Figure 1K). Thus, Lamin A/C reduction in cardiomyocytes resulted in extensive upregulation of inflammation-related genes, macrophage expansion, and the initial fibrotic response at 2 weeks post-tamoxifen, preceding the morphological and functional changes reflective of clinical DCM (Figure 1L).
LmnaCKO cardiomyocytes develop pervasive localized nuclear envelope (NE) ruptures
We examined whether Lamin A/C reduction caused NE ruptures in cardiomyocytes. Strikingly, we observed protrusion of DNA from nuclei inside the cardiomyocytes in LmnaCKO heart sections at 2 weeks post-tamoxifen, while this event was absent in wild-type hearts (Figure 2A; of note, 90% of cardiomyocytes are binucleated in adult mice56). Nuclei with protruded DNA were positive for Lamin A/C staining, indicating that partial reduction of Lamin A/C was sufficient to cause DNA protrusion (Figure 2A). Electron microscopy revealed a strong condensation of protruded DNA that appeared devoid of the surrounding nuclear membrane, suggesting NE ruptures (Figures 2B and S2A). We isolated cardiomyocytes from hearts at 2 weeks post-tamoxifen, immediately fixed them, and examined the nucleus. Lamin A/C signals were specifically lost at the tips of nuclei from which DNA protruded in LmnaCKO cardiomyocytes (Figure 2C). The Lamin A/C-lost locations also lost staining for PCM1, a perinuclear matrix protein localized at the cytoplasmic side of the outer nuclear membrane57 (Figure 2D). The loss of Lamin A/C and PCM1 occurred specifically at the tips of the elongated nuclei positioned along the longitudinal axis of the cardiomyocytes (Figures 2A–2D and S2A). The loss of NE proteins Lamin A/C and PCM1 at the nuclear tips suggested localized NE ruptures in LmnaCKO cardiomyocytes.
Figure 2. Lamin A/C reduction causes localized nuclear envelope rupture in cardiomyocytes.
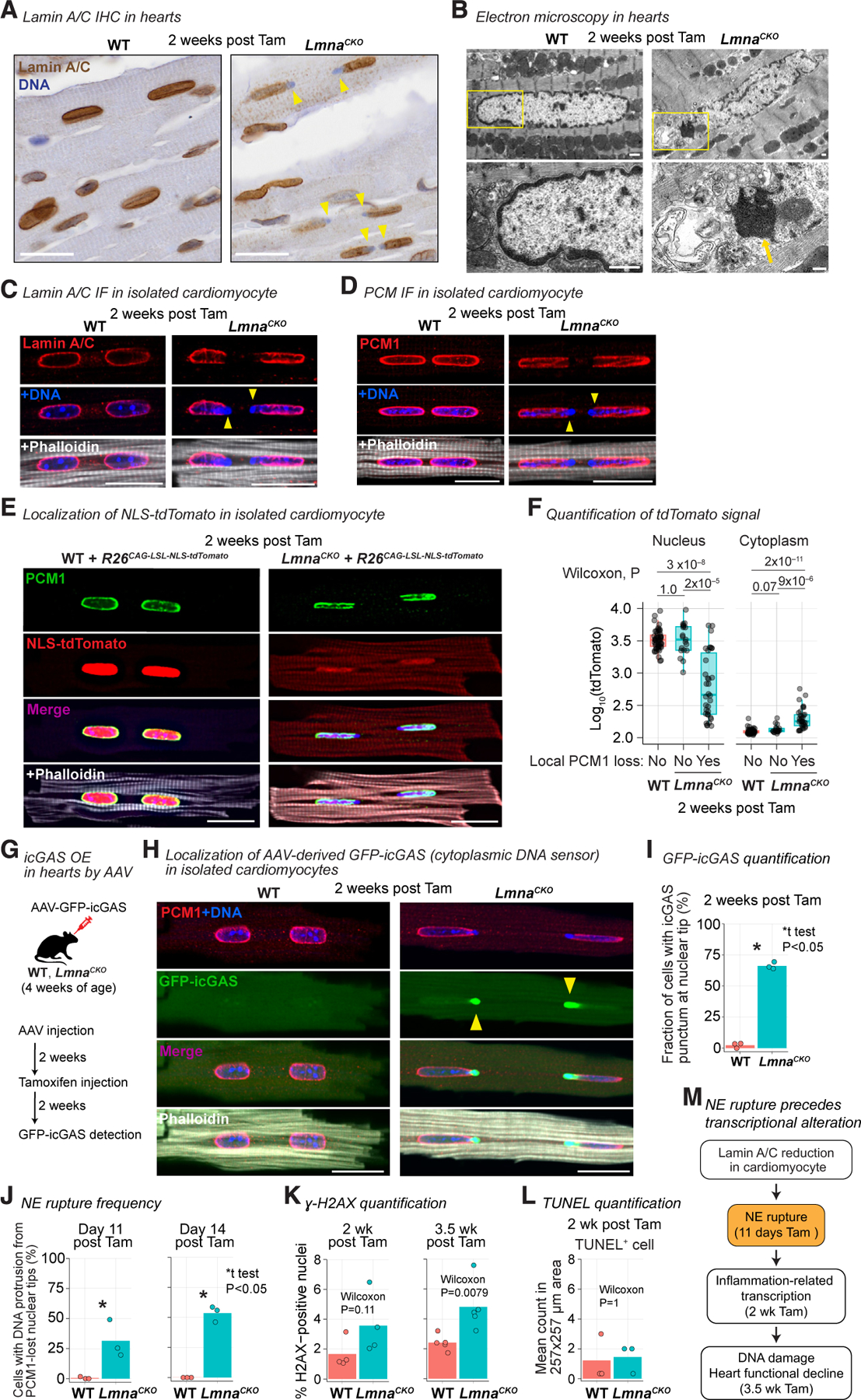
(A) Lamin A/C immunohistochemistry in heart tissue. Arrow, DNA protruded from nuclei. Scale bar: 20 μm. See Figure S2 for related analyses.
(B) Top: transmission electron micrograph of heart sections focusing on cardiomyocytes. Bottom: close-up image of the area indicated by rectangle in the top image. Arrow, protruded chromatin. Scale bar: 1 μm.
(C) Immunofluorescence for Lamin A/C in isolated cardiomyocytes. Phalloidin stains F-actin. Arrowhead, local loss of Lamin A/C with protruded DNA. Scale bar: 20 μm.
(D) Same as (C) but for PCM1 immunofluorescence.
(E) Native NLS-tdTomato signals in isolated cardiomyocytes with PCM-1 immunofluorescence. Scale bar: 20 μm.
(F) NLS-tdTomato signal intensity in nucleus and cytoplasm of cardiomyocytes (box, interquartile range). Cardiomyocytes are stratified by the presence or absence of local PCM-1 loss at the nuclear envelope. WT n = 51, LmnaCKO n = 53 (PCM1-loss 34, intact 19) cardiomyocytes. p value, Wilcoxon test.
(G) MyoAAV-mediated GFP-icGAS expression in cardiomyocytes in vivo.
(H) Native GFP-icGAS signals in isolated cardiomyocytes with PCM1 immunofluorescence. Scale bar: 20 μm.
(I) Percentage of cardiomyocytes with GFP-icGAS punctum at nuclear tip (bar, mean). n = 3 mice/genotype. p value, unpaired one-tailed Welch’s t test.
(J) Percentage of cardiomyocytes with DNA protrusion from PCM1-lost nuclear tip (bar, mean). n = 3 mice/genotype. p value, same as (I).
(K) Percentage of gamma-H2AX-positive nuclei in heart section (bar, mean). n = 4–5 mice/genotype. p value, same as (F).
(L) Number of TUNEL-positive cells in heart section (bar, mean). p value, same as (F). 3 mice/genotype.
(M) Summary.
To determine whether NE ruptures occurred, we examined the retention of a nucleus-localized fluorescent protein. We expressed nuclear localization signal (NLS)-fused tdTomato from the Rosa26CAG-LSL-tdTomato allele58 in cardiomyocytes and investigated tdTomato localization at 2 weeks post-tamoxifen. NLS-tdTomato was localized exclusively to the nucleus in wild-type cardiomyocytes as well as to the intact nuclei in LmnaCKO cardiomyocytes (Figures 2E and S2B). However, NLS-tdTomato signals significantly diminished in nuclei with local PCM1 loss in LmnaCKO cardiomyocytes (Figures 2E and 2F). Reciprocally, NLS-tdTomato signals increased in the cytoplasm of LmnaCKO cardiomyocytes with PCM1-lost nuclei. NLS-tdTomato leakage from the damaged nuclei indicated that the NE had ruptured.
To quantify NE rupture events, we expressed GFP-tagged catalytically inactive cGAS (icGAS) in cardiomyocytes in vivo using muscle-tropic adeno-associated virus (MyoAAV)59 (Figure 2G). icGAS is an ‘‘NE rupture marker’’ owing to the cGAS’s affinity to cytoplasmic DNA.60,61 Strong icGAS punctum appeared at every PCM1-lost site on the NE in LmnaCKO cardiomyocytes at 2 weeks post-tamoxifen (Figures 2H and S2C). 66% of LmnaCKO cardiomyocytes have at least one nucleus with icGAS puncta on the NE, while such puncta were virtually absent in wild-type cardiomyocytes (Figure 2I). Thus, NE ruptures were pervasive in LmnaCKO cardiomyocytes at 2 weeks post-tamoxifen.
To determine whether NE ruptures preceded the transcriptional change, we quantified NE ruptures at day 11 post-tamoxifen, when no transcriptional change was detected (Figure 1G). We used a local PCM1 loss with concomitant cytoplasmic DNA protrusion as a proxy for NE ruptures, as this feature strongly correlated with icGAS localization. At day 11, 31% of LmnaCKO cardiomyocytes had at least one ruptured nucleus (Figures 2J, S2D, and S2E). At day 14, this frequency increased to 54%, which was comparable to 66% based on the sensitive icGAS-based quantification (Figure 2J). Thus, about one-third of LmnaCKO cardiomyocytes developed NE ruptures as early as day 11 post-tamoxifen, preceding the earliest transcriptional changes, with more than 50% of cells presenting ruptures by day 14.
We investigated whether the pervasive NE ruptures in LmnaCKO cardiomyocytes accompanied DNA damage accumulation. We did not find a statistically significant increase of gamma-H2AX-positive nuclei indicative of DNA double-strand breaks in LmnaCKO hearts at 2 weeks post-tamoxifen (Figures 2K and S2F). In addition, there was no indication of increased cell death at this time point (Figures 2L and S2G). However, at 3.5 weeks post-tamoxifen, gamma-H2AX-positive nuclei significantly increased. Thus, pervasive NE ruptures did not cause immediate DNA damage accumulation or cell death at the time of strong inflammation-related transcriptional upregulation. Instead, DNA damage accumulated when the heart structure and function began to deteriorate (Figure 2M).
LmnaCKO cardiomyocytes do not activate cGAS-STING-related transcription
Given the pervasive NE ruptures, we examined whether the cGAS-STING cytosolic DNA-sensing pathway was activated within LmnaCKO cardiomyocytes. In the cGAS-STING pathway, cGAS detection of cytoplasmic DNA activates STING-mediated interferon transcription within cells bearing the cytoplasmic DNA.46,47 This pathway has been implicated in Lamin A/C-based cardiomyopathy in a previous study.27 We observed cGAS and STING protein expression in the heart, but interestingly, their expression within cardiomyocytes was very low in both wild-type and LmnaCKO backgrounds, by immunoblots (Figures 3A, S3A, and S3B). To test whether cGAS-STING-downstream genes were upregulated within cardiomyocytes of LmnaCKO hearts, we distinguished cardiomyocyte and non-cardiomyocyte transcriptomes in hearts using SLAM-IT-seq.62 SLAM-IT-seq labels transcripts in Cre-positive cardiomyocytes with 4-thiouracil (4sU) and leaves non-cardiomyocyte transcripts unlabeled (Figure S3C). As expected, cardiomyocyte-specific genes were specifically 4sU labeled in wild-type hearts (Figure S3D). We then assessed the labeling state of the 1,020 upregulated genes in LmnaCKO hearts and found that 135 genes were 4sU labeled (i.e., cardiomyocyte derived) and 885 genes were not 4sU labeled (i.e., non-cardiomyocyte derived) in LmnaCKO hearts at 2 weeks post-tamoxifen (Figure 3B). The cardiomyocyte-originated upregulated genes were not overrepresented for inflammation-related GO terms and did not include cGAS-STING downstream genes (Figures 3B and S3E; Table S2). Instead, they were overrepresented for the ECM-related GO terms such as ‘‘proteoglycans in cancer.’’ On the other hand, non-cardiomyocyte-originated upregulated transcripts were enriched for inflammation-related GO terms. These data suggested that the cGAS-STING pathway was not activated within LmnaCKO cardiomyocytes and that the inflammation-related transcriptional activation originated from non-cardiomyocytes in LmnaCKO hearts.
Figure 3. cGAS and STING are not required for inflammation-related gene expression and DCM in LmnaCKO mice.
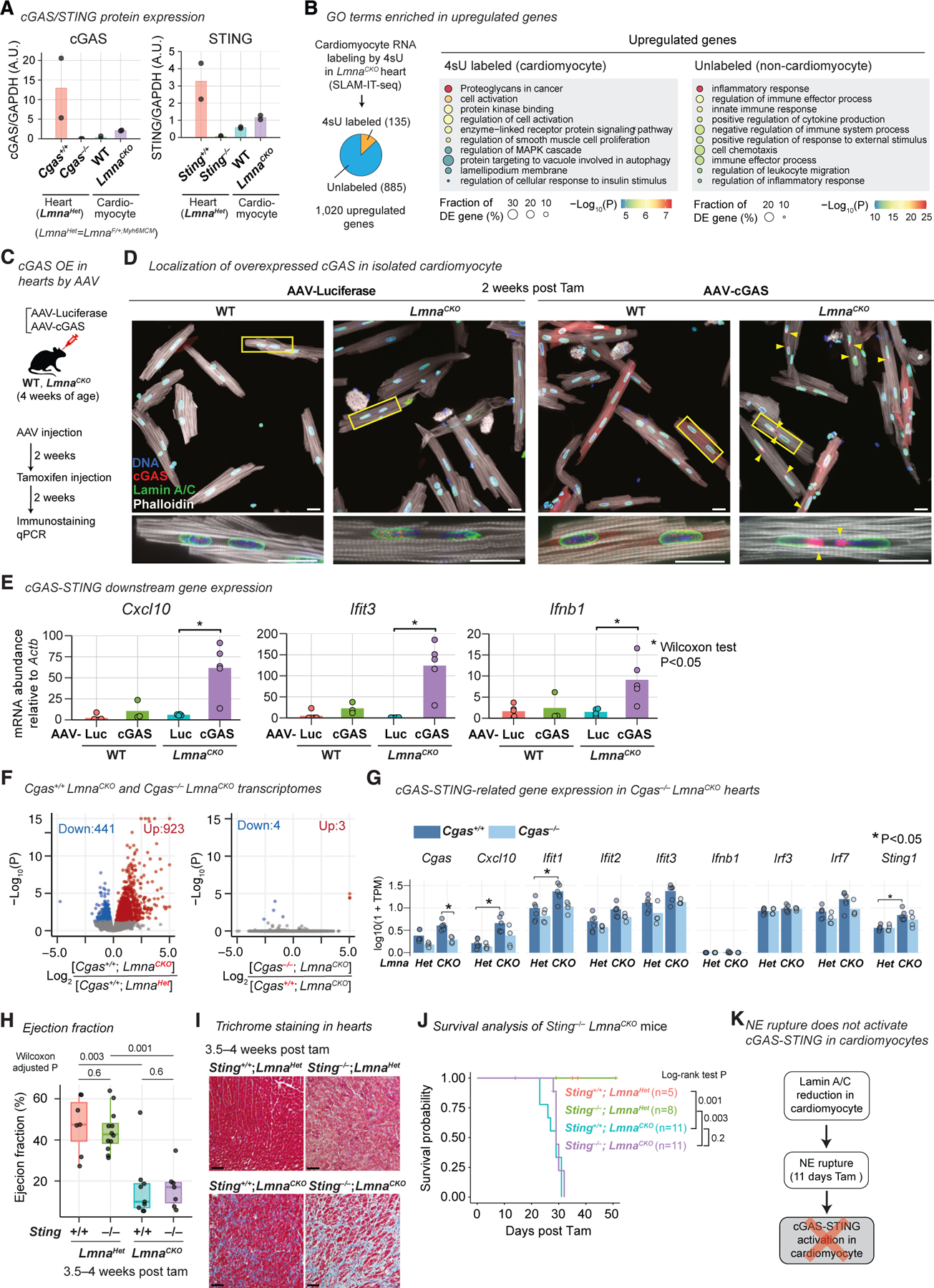
(A) Normalized cGAS and STING abundance quantified by immunoblot shown in Figures S3A and S3B (bar, mean). n = 2 mice/genotype.
(B) Left: classification of upregulated genes in LmnaCKO hearts at 2 weeks post-tamoxifen into cardiomyocyte-derived or non-cardiomyocyte-derived genes based on 4sU labeling in SLAM-IT-seq (n = 3–8 mice/genotype). Right: top 10 GO terms overrepresented in each class of upregulated genes. p value is computed by Metascape.54
(C) MyoAAV-mediated cGAS overexpression in cardiomyocytes in vivo.
(D) Top: anti-cGAS and Lamin A/C immunofluorescence in isolated cardiomyocytes. Bottom: close-up image of the area indicated by rectangle in the top image. Arrowhead, cGAS at nuclear tip. Scale bar: 20 μm.
(E) Transcript abundance in isolated cardiomyocytes normalized by Actb abundance quantified by quantitative PCR. Bar, mean. n = 3–5 mice/genotype. p value, Wilcoxon test.
(F) Volcano plot comparing RNA-seq read counts. Left: Cgas+/+;LmnaCKO (n = 6) versus Cgas+/+;LmnaHet (n = 6). Right: Cgas−/−;LmnaCKO (n = 4) versus Cgas+/+;LmnaCKO (n = 6).
(G) Transcript abundance quantified by RNA-seq. Bar, mean. P, FDR-adjusted p value computed by a generalized linear model in DESeq2.55
(H) Left ventricular ejection fraction measured by echocardiography (box, interquartile range). n = 6–12 mice/genotype. p value, same as (E).
(I) Masson’s trichrome staining of heart sections. Scale bar: 20 μm.
(J) Kaplan-Meier survival analysis. n = 5–11 mice/genotype. p value, log-rank test.
(K) Summary.
We hypothesized that the near absence of cGAS expression prevented cGAS-STING activation within LmnaCKO cardiomyocytes. We tested this hypothesis by overexpressing wild-type cGAS in cardiomyocytes in vivo with MyoAAV (Figure 3C). Without cGAS overexpression, endogenous cGAS was undetectable at NE rupture sites (Figure 3D), and the well-established cGAS-STING downstream genes Cxcl10, Ifit3, and Ifnb163–66 were transcriptionally silent in LmnaCKO cardiomyocytes (Figure 3E). However, cGAS overexpression resulted in strong cGAS accumulation at NE rupture sites (Figure 3D) and strong upregulation of Cxcl10 (10-fold), Ifit3 (128-fold), and Ifnb1 (6-fold) in LmnaCKO cardiomyocytes (Figure 3E). In wild-type mice, cGAS overexpression did not upregulate cGAS-down-stream genes, confirming the NE-rupture dependency of cGAS-downstream activation upon cGAS overexpression. Taken together, these data suggested that LmnaCKO cardiomyocytes did not activate the cGAS-STING pathway despite pervasive NE ruptures, likely due to the low cGAS expression in adult cardiomyocytes.
Cgas and Sting are not required for inflammation-related transcription and DCM in LmnaCKO mice
We examined the genetic requirement of Cgas and Sting, the essential mediators of the cGAS-STING pathway, for the inflammation-related transcriptional activation and DCM in LmnaCKO hearts. We validated the germline Cgas−/− allele67 crossed into LmnaCKO mice or control LmnaHet mice (LmnaF/+;Myh6Mer-CreMer) (Figure S3A) and performed RNA-seq in hearts at 2 weeks post-tamoxifen. We first confirmed the expected strong upregulation of inflammation-related genes in Cgas+/+;LmnaCKO hearts relative to Cgas+/+;LmnaHet hearts (Figures 3F and S3F; Table S1). We then compared Cgas−/−;LmnaCKO hearts with Cgas+/+;LmnaCKO hearts. There was almost no difference in gene expression (Figure 3F), including representative cGAS-STING-downstream genes (Figures 3G and S3G), between them. These data suggested that the inflammation-related transcription in LmnaCKO hearts was independent of cGAS.
We next combined germline Sting−/− mice68 with LmnaCKO mice (Figure S3B) and investigated the heart phenotype. By 3.5 to 4 weeks post-tamoxifen, Sting−/−;LmnaCKO mice exhibited a reduction of ejection fraction (Figure 3H) and fibrosis (Figure 3I) as severely as the littermate control Sting+/+;LmnaCKO mice. Likewise, Cgas−/−;LmnaCKO hearts developed severe fibrosis (Figure S3H). Moreover, both Sting−/−;LmnaCKO mice and Cgas−/−;LmnaCKO mice died as early as Sting+/+;LmnaCKO or Cgas+/+;LmnaCKO mice (Figures 3J and S3I). Sting−/−;LmnaHet and Cgas−/−;LmnaHet mice maintained normal hearts and remained healthy. Taken together, our data suggested that the cGAS-STING pathway did not mediate inflammatory transcription and the pathophysiologic features of DCM in LmnaCKO hearts (Figure 3K).
Cardiomyocytes, fibroblasts, and immune cells are transcriptionally altered in LmnaCKO hearts
We explored alternative mechanisms by which Lamin A/C reduction in cardiomyocytes caused DCM. To identify intercellular signaling initiated by LmnaCKO cardiomyocytes, we defined the cell-type-resolved transcriptional changes using single-nucleus (sn) RNA-seq (Table S3). We obtained transcriptomes for 14,111 nuclei from either wild-type or LmnaCKO hearts at 2 weeks post-tamoxifen (n = 3; Figure 4A). These nuclei were grouped into cardiomyocytes (CM1 and CM2), fibroblasts (Fib1 and Fib2), macrophages (Mac1 and Mac2), T cells, endothelial cells (coronary, lymphatic, and other), pericytes, or neurons, based on their transcriptomes (Figure S4A). Among these, CM2, Fib2, Mac2, and T cells were highly overrepresented by LmnaCKO heart-derived cells (Figures 4B and S4B). CM2 were characterized by high Rtn4 expression, a feature also reported in Lmna-KO myotubes69 and Lamin A/C-independent heart disease70–73 (Figure S4A). Fib2 were characterized by myofibroblast marker Postn,74 Mac2 by circulating monocytic receptor Ccr2,75 and T cells by T cell adaptor Skap1.76 We found extensive differential gene expression within cardiomyocytes (CM1 and CM2 combined), fibroblasts (Fib1 and Fib2 combined), and immune cells (Mac1, Mac2, and T cells combined), while other cell types exhibited almost no expression differences (Figures 4C, 4D, and S4C; Table S4). The upregulated genes in cardiomyocytes were not overrepresented for the cytosolic DNA-sensing pathway and did not include cell-death-related genes but did include some DNA-damage-repair-related genes, as expected (Figures S4D–S4F).
Figure 4. ECM-mediated signaling from LmnaCKO cardiomyocytes is predicted to activate fibroblasts.
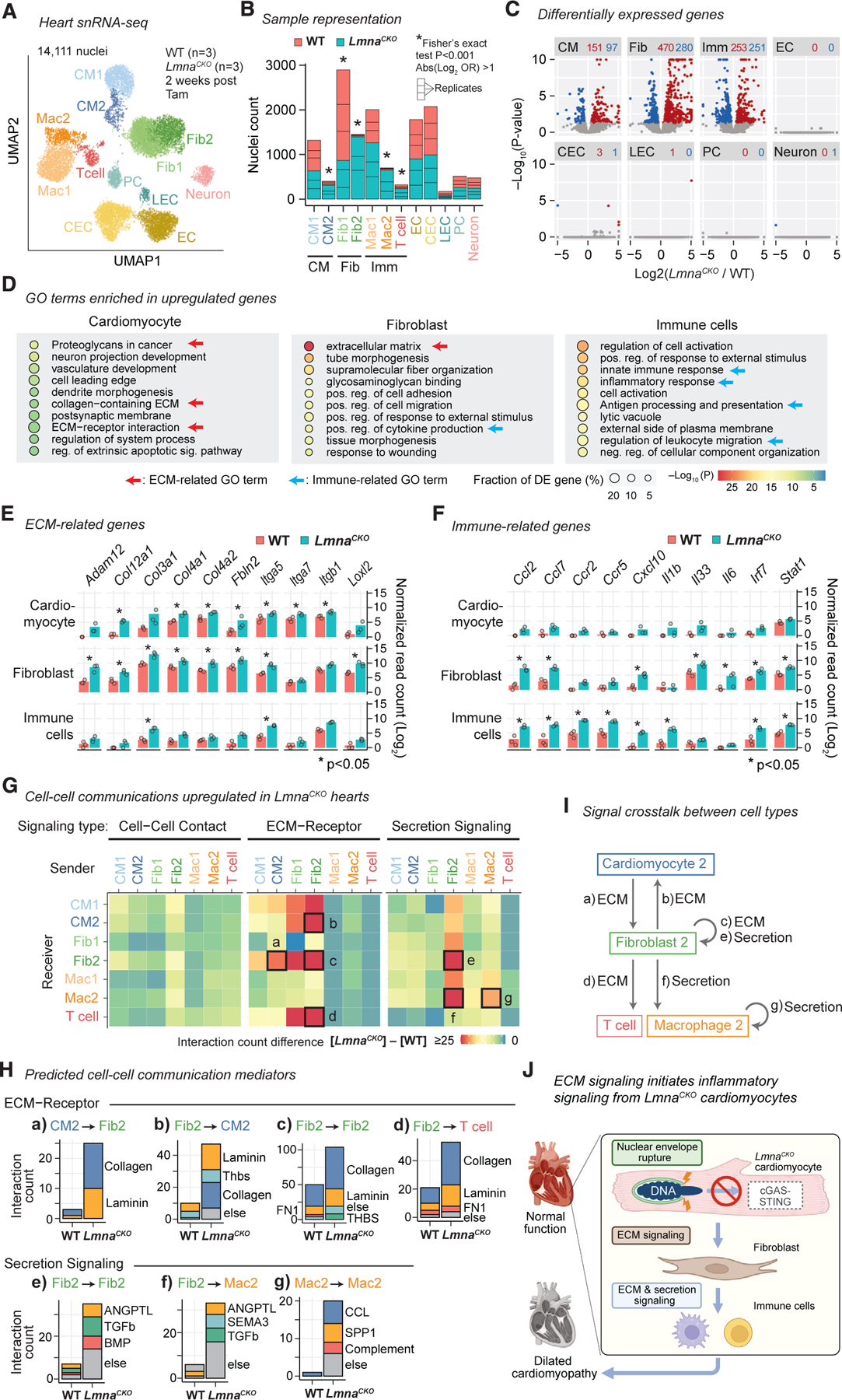
(A) snRNA-seq uniform manifold approximation and projection (UMAP) plot for WT and LmnaCKO heart nuclei. CM, cardiomyocyte; Fib, fibroblast; Mac, macrophage; EC, endothelial cell; CEC, coronary EC; LEC, lymphatic EC; PC, pericyte. See Figure S4 for additional analyses.
(B) WT and LmnaCKO nucleus count within each cell type cluster. Cell types combined in later analyses are indicated below the graph.
(C) snRNA-seq pseudo-bulk volcano plot comparing LmnaCKO versus WT heart nuclei. Red, upregulated genes. Blue, downregulated genes. The number of differentially expressed genes is indicated above the plot.
(D) GO terms enriched among upregulated genes within indicated cell type. p value is computed by Metascape.54
(E) Pseudo-bulk transcript abundance of ECM-related genes. Point, mean of normalized read count per replicate. Bar, mean across replicates. P, FDR-adjusted p value computed by a generalized linear model in DESeq2.55
(F) Same as (E) but for immune-related genes.
(G) Predicted signaling from sender cells (x axis) to receiver cells (y axis). Color: signaling gains in LmnaCKO hearts relative to WT hearts.
(H) Predicted signaling mediators for the signaling indicated by box in (G) with alphabetical labels.
(I) Predicted intercellular signaling indicated in (G) by alphabetical labels in LmnaCKO hearts.
(J) Model.
Interestingly, upregulated genes in LmnaCKO cardiomyocytes and those in fibroblasts were both most strongly overrepresented for ECM-related pathways (Figure 4D). The upregulated ECM-related genes included collagen and integrin genes (in cardiomyocytes and fibroblasts), and ECM proteinase and cross-linker genes (specifically in fibroblasts) (Figure 4E). On the other hand, fibroblasts and immune cells, but not cardiomyocytes, commonly upregulated immune-related genes including cytokine, cytokine receptor, and cytokine transcription factor genes (Figures 4D and F). These results suggested potential signal crosstalk between cardiomyocytes and fibroblasts and between fibroblasts and immune cells in LmnaCKO hearts.
ECM-mediated signaling from LmnaCKO cardiomyocytes is predicted to activate fibroblasts
We investigated intercellular signal crosstalk in LmnaCKO hearts by applying the CellChat program77 to the snRNA-seq data. This analysis predicted ECM-receptor signaling from CM2 to Fib2 as the only strongly upregulated signaling originating from CM2, the population predominantly consisting of LmnaCKO cardiomyocyte nuclei (Figure 4G, box a). Collagens and laminins were predicted to mediate this CM2-to-Fib2 signaling (Figure 4H). CM2 were not predicted to signal directly to immune cell populations. Instead, Fib2 were predicted to send ECM-mediated signals to CM2 (box b), Fib2 (box c), and T cells (box d) via collagens, laminins, thrombospondins, and fibronectins (Figures 4G and 4H). Fib2 were also predicted to send secretion-mediated signals to Fib2 (box e) and Mac2 (box f) via ANGPTL (angiopoietin-like), transforming growth factor beta, BMP, and SEMA3 (semaphorin3). Fib2 might therefore act as a central signaling hub in LmnaCKO hearts, receiving signals from CM2 and sending signals to immune cells (Figure 4I). Finally, Mac2 were predicted to send secretion-mediated signals to Mac2 themselves (box g) via CCL (chemokine ligands), SPP1 (osteopontins), and complement factors. This signaling might mediate the macrophage infiltration observed in LmnaCKO hearts (Figure 1J). Taken together, our results suggested that LmnaCKO cardiomyocytes activated fibroblasts via ECM-mediated signaling, not cytokine-mediated signaling such as cGAS-STING, and that fibroblasts activated and recruited immune cells through both ECM signaling and secretion signaling to orchestrate inflammatory signaling in the heart (Figure 4J).
DISCUSSION
We report that frequent localized NE ruptures precede transcriptional changes, DNA damage accumulation, and heart functional decline in LmnaCKO hearts. Given our observation that a 50% reduction of Lamin A/C proteins is sufficient to induce pervasive NE ruptures in cardiomyocytes, NE ruptures might be more prevalent than previously anticipated in patients with LMNA-related DCM. NE ruptures might have been overlooked due to the specific location of localized NE ruptures at the tips of elongated nuclei within cardiomyocytes. The mechanisms underlying NE ruptures and the causal role of NE ruptures in DCM remain to be investigated.
Our data indicate that the cGAS-STING pathway does not contribute to LMNA-related DCM in the adult mouse model investigated herein. This suggests that pharmacologically reducing cGAS-STING activity for treating LMNA-related DCM, an idea previously discussed,27,78 may be ineffective. Our conclusion diverges from a previous report that Cgas deletion can delay early post-natal cardiomyopathy caused by Lmna deletion in embryonic hearts.27 This difference may be due to the near absence of cGAS expression preventing cGAS-STING activation in adult cardiomyocytes, whereas the basal cGAS-STING activity may be higher in early post-natal cardiomyocytes.79,80 The lack of robust cGAS expression may reduce pathogen-derived DNA sensing in adult cardiomyocytes, similar to hepatocytes that also lack functional cGAS-STING.81 Many reports suggest an association between DNA damage and cGAS-STING activation.27,82–85 Our data that LmnaCKO cardiomyocytes accumulate DNA damage without activating cGAS-STING suggest that DNA damage and cGAS-STING can be uncoupled.
Our study nominates ECM signaling as a mechanism by which Lamin A/C-reduced cardiomyocytes initiate an inflammatory response by activating fibroblasts. A recent study reported a similar observation in which NE rupture induces ECM remodeling through unknown mechanisms that require DNA damage.86 Further study on the potential roles of ECM signaling in LMNA-related DCM is required.
Limitations of the study
We reduced Lamin A/C exclusively within the cardiomyocytes of adult mice. Whether Lamin A/C reduction in other cell types or at other stages causes NE ruptures and contributes to DCM through cGAS-STING activation remains an open question.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kohta Ikegami (Kohta.Ikegami@cchmc.org).
Materials availability
All mice are available at the sources specified in Key resources table. Plasmids produced in this study are provided upon request from the lead contact after material transfer agreements. Any information required to reanalyze the data reported in this paper is available from the lead contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-Lamin A/C | Abcam | Cat # Ab26300; RRID: AB_775965 |
| Anti-Lamin A/C | Santa Cruz | Cat # sc-376248; RRID: AB_10991536 |
| Anti-Lamin A/C | Santa Cruz | Cat # sc-20681; RRID:AB_648154 |
| Anti-CD45 antibody | R&D Systems | Cat # AF114-SP; RRID: AB_442146 |
| Anti-CD68 antibody | Cell Signaling | Cat # 97778; RRID: AB_2928056 |
| Anti-γ-H2A.X antibody | Cell Signaling | Cat # 9718; RRID: AB_2118009 |
| Anti-desmin antibody | Invitrogen | Cat # PA5-19063; RRID: AB_10977860 |
| Anti-PCM-1 antibody | Sigma | Cat # HPA023370; RRID: AB_1855072 |
| Anti-tdTomato antibody | OriGene | Cat # AB8181; RRID: AB_2722750 |
| Anti-COX4 antibody | R&D Systems | Cat # MAB6980; RRID: AB_10973832 |
| Anti-cGAS antibody | Cell Signaling | Cat # 31659; RRID: AB_2799008 |
| Anti-STING antibody | Cell Signaling | Cat # 13647; RRID: AB_2732796 |
| Anti-GAPDH antibody | ABclonal | Cat # AC001; RRID: AB_2619673 |
| Alexa Fluor Plus 647 Phalloidin | ThermoFisher | Cat # A30107 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tamoxifen | Sigma | Cat #T5648 |
| Collagenase Type II | Worthington | Cat # LS004177 |
| Polyethylenimine, Linear, MW 25000, Transfection Grade (PEI 25K™) | Fisher Scientific | Cat # 23966-100 |
| Target Retrieval Solution | DAKO | Cat #S2367 |
| Corn oil | Sigma | Cat #C8267 |
| Phenol/Chloroform/Isoamyl alcohol | Invitrogen | Cat # 15593031 |
| Trizol LS | Invitrogen | Cat # 10296010 |
| Heparin | Sagent Pharmaceuticals. | Cat # 25021-400-10 |
| VectaStain ELITE ABC Kits | Vector Laboratories | Cat # PK-6100 |
| DAB | DAKO | Cat #K3468 |
| ProLong Glass Antifade Mountant | Invitrogen | Cat #P36984 |
| Normal donkey serum | Jackson ImmunoResearch | Cat # 017-000-121 |
| Sudan Black B | Electron Microscopy Slides | Cat # 21610 |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | Cat #E2621 |
| Dulbecco’s modified Eagle medium | Fisher Scientific | Cat # 11965-092 |
| penicillin/streptomycin | Invitrogen | Cat # 105727 |
| OptiPRO™-SFM | Thermo Fisher | Cat # 12309019 |
| Fetal Bovine Serum | Sigma | Cat #F1051 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Masson’s Trichrome Kit | Thomas Scientific LLC | Cat # KTMTRPT |
| Click-iT™ Plus TUNEL Assay Kits for In Situ Apoptosis Detection; Alexa Fluor™ 488 | Invitrogen | Cat #C10617 |
| Luna Universal One-Step RT-qPCR Kit | New England Biolabs | Cat #E3005 |
| NEBNext UltraII Directional RNA Library Prep Kit | New England Biolabs | Cat #E7760 |
| Quant-seq 3ʹ mRNA-seq FWD kit | Lexogen | Cat #K01596 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq raw and analyzed data | This paper | GEO: GSE241590 |
| Lmna-LoxP mice | Jackson Laboratory | JAX stock No: 026284 |
| Myh6-MerCreMer transgene mice | Jackson Laboratory | JAX stock No: 005657 |
| Rosa26CAG-LSL-tdTomato mice | Jackson Laboratory | JAX stock No: 025106 |
| Cgas-null mice | Jackson Laboratory | JAX stock No: 026554 |
| CAG-Lox-Stop-Lox-Uprt transgene mice | Jackson Laboratory | JAX stock No: 021469 |
| Sting-flox mice | Jackson Laboratory | JAX stock No: 031670 |
| Mice carrying the Mef2C-AHF-Cre transgene allele | Verzi et al.87 | https://doi.org/10.1016/j.ydbio.2005.08.041 |
| Sting-null (generated from Sting-LoxP mouse) | This study | N/A |
| AAVpro 293T cell | TaKaRa | Cat # 632273 |
|
| ||
| Oligonucleotides | ||
|
| ||
| DNA oligonucleotide KI279 for Lmna genotype primer | Table_S3_oligonucleotides | Ikegami lab ID: KI279 |
| DNA oligonucleotide KI280 for Lmna genotype primer | Table_S3_oligonucleotides | Ikegami lab ID: Ki280 |
| DNA oligonucleotide EA062 for EGFP cloning | Table_S3_oligonucleotides | Ikegami lab ID: EA062 |
| DNA oligonucleotide EA064 for EGFP with GS linker cloning | Table_S3_oligonucleotides | Ikegami lab ID: EA064 |
| DNA oligonucleotide EA086 for icGAS (human cGAS with E225A/D227A amino acid substitutions) | Table_S3_oligonucleotides | Ikegami lab ID: EA086 |
| DNA oligonucleotide EA087 for mouse cGAS | Table_S3_oligonucleotides | Ikegami lab ID: EA087 |
| DNA oligonucleotide EA096 for Cxcl10 qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: EA096 |
| DNA oligonucleotide EA097 for Cxcl10 qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: EA097 |
| DNA oligonucleotide EA098 for Ifnb1 qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: EA098 |
| DNA oligonucleotide EA099 for Ifnb1 qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: EA099 |
| DNA oligonucleotide EA100 for Ifit3 qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: EA100 |
| DNA oligonucleotide EA101 for Ifit3 qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: EA101 |
| DNA oligonucleotide KI444 for Actb qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: KI444 |
| DNA oligonucleotide KI445 for Actb qPCR primer | Table_S3_oligonucleotides | Ikegami lab ID: KI445 |
| DNA oligonucleotide KI466 for AAV titering primer | Table_S3_oligonucleotides | Ikegami lab ID: KI466 |
| DNA oligonucleotide KI467 for AAV titering primer | Table_S3_oligonucleotides | Ikegami lab ID: KI467 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pAAV:cTNT::Luciferase | Lin et al.88 | pAAV:cTNT::Luciferase; Addgene plasmid #69915 |
| pAAV:cTNT::GFP-GS linker | This paper | Ikegami lab ID: bAE263_1 |
| pAAV:cTNT::GFP-icGAS | This paper | Ikegami lab ID: bAE299_1 |
| pAAV:cTNT:cGAS | This paper | Ikegami lab ID: bAE299_3 |
| pLJM1-EGFP | Sancak et al.89 | pLJM1-EGFP; Addgene plasmid #19319 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | N/A | https://imagej.net/ij/ |
| NIS Elements | Nikon Instruments Inc. | RRID:SCR_014329 |
| Odyssey CLx Imaging System | LI-COR Biosciences | https://www.licor.com/bio/odyssey-dlx/ |
| CellProfiler | Broad Institute | https://cellprofiler.org/ |
| R | R Core Team | https://www.R-project.org/ |
| survival package | Therneau and Grambsch90 | https://cran.r-project.org/web/packages/survival/index.html |
| survminer package | N/A | https://cran.r-project.org/web/packages/survminer/index.html |
| STAR version 2.7.9 | Dobin et al.91 | https://github.com/alexdobin/STAR |
| DESeq2 | Love et al.55 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Metascape | Zhou et al.54 | https://metascape.org/gp/index.html |
| sci-RNA-seq3 pipeline | Martin et al.92 | https://github.com/JunyueC/sci-RNA-seq3_pipeline |
| sci-RNA-seq analysis workflow | This paper | https://github.com/kohta-ikegami/En_CellReports_2024/ |
| SingleCellExperiment package | Amezquita et al.93 | https://www.bioconductor.org/packages/release/bioc/html/SingleCellExperiment.html |
| scDblFinder package | Germain et al.94 | https://bioconductor.org/packages/release/bioc/html/scDblFinder.html |
| scuttle package | McCarthy et al.95 | https://www.bioconductor.org/packages/release/bioc/html/scuttle.html |
| scran package | Lun et al.96 | https://bioconductor.org/packages/release/bioc/html/scran.html |
| scater package | McCarthy et al.95 | https://bioconductor.org/packages/release/bioc/html/scater.html |
| R packages GO.db | N/A | https://bioconductor.org/packages/release/data/annotation/html/GO.db.html |
| KEGGREST | N/A | https://bioconductor.org/packages/release/bioc/html/KEGGREST.html |
| CellChat package | Jin et al.77 | https://github.com/sqjin/CellChat |
| slamdunk package | Herzog et al.97 | https://github.com/t-neumann/slamdunk |
| countdata package | Pham et al.98 | https://www.rdocumentation.org/packages/countdata/versions/1.3 |
| ggplot2 package | Wickham99 | https://ggplot2.tidyverse.org/ |
|
| ||
| Other | ||
|
| ||
| Bio-Rad CFX96 Real-Time PCR Detection System | Bio-Rad | RRID:SCR_018064 |
| Yokogawa CSU-W1 Sora spinning disk confocal microscope | Nikon Instruments Inc. | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/csu-series/csu-w1 |
| Nikon A1R laser-scanning confocal microscope | Nikon Instruments Inc. | RRID:SCR_020317 |
| Automated Genotyping Services | Transnetyx, Inc. | https://www.transnetyx.com/ |
| Gencode M27 Basic gene annotation | The National Human Genome Research Institute | https://www.gencodegenes.org/mouse/release_M27.html |
Data and code availability
High-throughput sequencing data have been deposited at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are publicly available as of the date of publication. Accession numbers are listed in Key resources table.
This paper does not report original codes.
Any additional information required to reanalyze the data reported in this paper is available from Lead Contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mouse genetics and treatment
Lmna-LoxP mice100 were provided by Dr. Yixian Zheng at Carnegie Institution and are available at the Jackson Laboratory (JAX stock no: 026284). Myh6-MerCreMer (Myh6MCM) mice101 (JAX stock No: 005657), Rosa26CAG-LSL-tdTomato (‘‘Ai75’’) mice58 (JAX stock No: 25106), Cgas-null mice67 (JAX stock No: 026554), and CAG-Lox-Stop-Lox-Uprt mice102 (UprtTg; JAX stock No: 021469) were obtained from the Jackson Laboratory. Sting-null mice were generated by breeding Sting-flox mice,68 obtained from the Jackson Laboratory (JAX stock No: 031670), with Cre deleter mice (Mef2C-AHF-Cre mice87) provided by Dr. Brian L. Black at the University of California San Francisco. For cardiomyocyte-specific Lmna knockout experiments, LmnaF/F;Myh6MCMTg/0 (LmnaCKO) and Lmna+/+;-Myh6MCMTg/0 (wild-type control) mice were used. For the analysis of nuclear tdTomato intensity, LmnaF/F;Myh6MCMTg/0;Rosa26-CAG-LSL-tdTomato/+ and Lmna+/+;Myh6MCMTg/0;Rosa26CAG-LSL-tdTomato/+ mice were used. For SLAM-IT-seq experiments, LmnaF/F;Myh6MCMTg/0;UprtTg/0, Lmna+/+;Myh6MCMTg/0;UprtTg/0, LmnaF/F;Myh6MCMTg/0;Uprt0/0, and Lmna+/+;Myh6MCMTg/0;Uprt0/0 mice were used. For Cgas deletion experiments, Cgas−/−;LmnaF/F;Myh6MCMTg/0 mice, Cgas+/+;LmnaF/F;Myh6MCMTg/0, Cgas+/+; LmnaF/+;Myh6MCMTg/0, and Cgas−/−;LmnaF/+;Myh6MCMTg/0 mice were used. For Sting deletion experiments, Sting−/−;LmnaF/F; Myh6MCMTg/Tg mice, Sting+/+;LmnaF/F;Myh6MCMTg/Tg, Sting+/+;LmnaF/+;Myh6MCMTg/Tg, and Sting−/−; LmnaF/+;Myh6MCMTg/Tg mice were used. All mice used in experiments were administered with tamoxifen (Sigma, T5648) at 6–8 weeks of age via intraperitoneal injections (100 μL of 4 mg/mL solution per day for 4 consecutive days) dissolved in corn oil (Sigma, C8267). All mice were used in mixed genetic backgrounds and both sexes. All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Cincinnati Children’s Hospital under IACUC protocol 2021–0014 or at University of Chicago under IACUC protocol 71730–10. All procedures were performed in compliance with institutional and governmental regulations under PHS Animal Welfare Assurance number D16–00068 (Cincinnati Children’s) or D16–00322 (University of Chicago).
METHOD DETAILS
Mouse genotyping
For genotyping of the Lmna alleles, genomic DNA of isolated cardiomyocytes was extracted with Phenol/Chloroform/Isoamyl alcohol (Invitrogen,15593–031), treated with RNaseA and Proteinase K, and purified. Floxed, recombined, and wild type Lmna alleles were detected by PCR using the primers KI279 and KI280 (Table S3). Based on the agarose gel (1.5%) analysis of PCR-amplified products, recombination efficiency (%) was calculated by 153-bp band (Recombined) over 598-bp band (Floxed) intensities. Genotyping of Myh6-MerCreMer, Cgas, Sting, Uprt, and Rosa26::CAG-LSL-tdTomato alleles was performed at Transnetyx, Inc.
Echocardiography
The transthoracic echocardiography was performed using a Vevo 3100 LT (FUJI FILM VisualSonics) and a transducer of 50-MHz MX-700. Parasternal long-axis view and short-axis view at the papillary muscle level were imaged. Two-dimensional M-mode tracing was recorded at three or more consecutive cardiac cycles. Data were analyzed using Vevo LAB Software Package V3.2.6. Echocardiographic parameters between two groups were compared using Wilcoxon rank sum tests, and p-values were adjusted for multiple testing using the Benjamini-Hochberg procedure in R.
Survival analysis
Kaplan-Meier survival analyses were performed using the survival package90 and visualized using the survminer package in R (Key resources table). Log rank tests were performed using the survdiff function in the survival package.
Histological staining
Hearts were perfused with 100 mM KCl and fixed in 10% Formalin (Fisherbrand, 245–685). Fixed hearts were embedded in paraffin and sectioned to a thickness of 5 μm. Heart sections were stained with hematoxylin and eosin (H&E) or a Masson’s Trichrome Kit (Thomas Scientific LLC, KTMTRPT) according to the manufacturer’s protocol.
Cardiomyocyte isolation
Mice were administered with heparin (100 Units, NDC 25021–400-10), anesthetized with Isoflurane, and euthanized by cervical dislocation. Hearts were perfused with 100 mM KCl and moved to Tyrode’s solution (10 mM glucose, 5 mM HEPES, 5.4 mM KCl, 1.2 mM MgCl2, 150 mM NaCl and 2 mM sodium pyruvate, pH 7.35). Excised hearts were cannulated to the Langendorff retrograde perfusion system through the aorta and perfused with Base solution (Tyrode’s solution with 10 mM taurine and 12 mM 2,3-butanedione monoxime) and then digested with prewarmed Digestion buffer (Base solution with 180 U/mL Collagenase Type II (Worthington, LS004177) and 25 μM CaCl2) for 20 min at 37°C. Heart tissue was then isolated, gently minced in Base solution containing 5 mg/mL BSA. Cell suspension was filtrated through a 240 μm mesh. Cardiomyocytes settled to the bottom of conical tubes by gravity were used in experiments.
Immunohistochemistry
Deparaffinized heart tissue sections, that had been fixed with 10% formalin, were subjected to heat-induced antigen retrieval in Target Retrieval Solution (S2367, DAKO) for 5 min. Antigen-retrieved sections were incubated with anti-Lamin A/C antibody Ab26300 (rabbit, Abcam) (1:400) for 1 h at room temperature, followed by biotinylated anti-rabbit IgG antibody for 30 min at room temperature. The antigen-antibody binding was detected by Elite kit (PK-6100, Vector Laboratories) and DAB (DAKO, K3468) system. Sections were counterstained by hematoxylin.
Immunofluorescence on tissue sections
Deparaffinized heart tissue sections, that had been fixed with 10% formalin, were subjected to heat-induced antigen retrieval in 10 mM Tris EDTA buffer (pH 9.0) for 5 min. Antigen-retrieved sections were incubated overnight at 4°C with goat anti-CD45 antibody (R&D Systems, AF114-SP) and rabbit anti-CD68 antibody (Cell Signaling, 97778), or rabbit anti-γ-H2A.X antibody (Cell Signaling, 9718) and goat anti-desmin antibody (Invitrogen, PA5–19063), or rabbit anti-PCM1 antibody (Sigma, HPA023370), goat anti-TdTomato antibody (OriGene, AB8181), and mouse anti-Cox4 antibody (R&D Systems, MAB6980), followed by Alexa fluorophore-conjugated secondary antibodies for 1 h at 37°C. Cells were counterstained with DAPI (40,6-diamidino-2-phenylindole), submerged in 0.25% Sudan Black B (Electron Microscopy Slides, #21610) in 70% Isopropanol for 10 min, and then mounted with ProLong Glass Antifade Mountant (Invitrogen, P36984). Fluorescence signals and imaging were acquired using a Nikon A1R laser-scanning confocal microscope. CD45 and CD68 quantification of whole heart tissue was performed using Nikon NIS-Elements; each genotype had five biological replicates. CD45/CD68 positive cells were identified based on the overlap of the DAPI signal with CD45/CD68. Signal intensities between two groups were compared using Wilcoxon rank sum tests in R. For γ-H2A.X quantification, nuclei with DAPI were counted and those with at least one γ-H2A.X focus were quantified using CellProfiler. Five to eight images per mouse (374 3 374 mm2 per image) were used for the quantification.
Immunofluorescence on isolated cardiomyocytes
Isolated cardiomyocytes were fixed in 4% paraformaldehyde (Electron Microscopy Slides, #15710) in PHEM buffer (60 mM PIPES pH7.5, 25 mM HEPES pH7.5, 10 mM EGTA, 4 mM MgSO4) for 10 min at 37°C, then washed with PBS and attached to coverslips with Cell-Tak Cell Adhesive (Sigma Aldrich, 354240). Cells were blocked and permeabilized in a buffer containing 5% normal donkey serum (Jackson ImmunoResearch, 017–000-121), 1% non-fat milk, and 0.1% Triton X-100 in PBS for 1 h at 37°C. Permeabilized cells were incubated overnight at 4°C with mouse anti-Lamin A/C antibody (Santa Cruz, sc-376248), rabbit anti-PCM-1 antibody (Sigma, HPA023370), mouse anti-cGAS antibody (Cell Signaling, D3O8O). Cells were washed and incubated with Alexa fluorophore-conjugated secondary antibodies and Alexa Fluor Plus 647 Phalloidin for 1 h at 37°C. Cells were counterstained with DAPI and then mounted with ProLong Glass Antifade Mountant. Fluorescence signals were detected on Nikon A1R laser-scanning confocal microscope or Yokogawa CSU-W1 Sora spinning disk confocal microscope.
Plasmid construction
The expression vectors used in this study were derived from the pAAV:cTNT::Luciferase vector (gift from William Pu; Addgene plasmid # 69915).88 For cloning of pAAV:cTnT:cGAS and pAAV:cTnT::GFP vectors, we digested the pAAV:cTNT::Luciferase vector with NheI and NotI, and the luciferase gene was replaced with the cGAS or EGFP sequences using NEBuilder HiFi DNA Assembly Master Mix (NEB E2621). The pAAV:cTnT::GFP-icGAS vector was generated by inserting the catalytically-inactive cGAS (icGAS) sequence (Oligonucleotide ID: EA086, Table S3) into the pAAV:cTnT::GFP vector digested with NotI. The icGAS sequence was derived from the human cGAS protein sequence with an E225A/D227A amino acid substitution that abolishes enzyme activity and interferon production, but retains DNA binding ability and functions, used as an NE rupture marker previously.60 The DNA fragments for mouse wild-type Cgas (NM_173386.5) and icGAS were synthesized using gBlocks Gene Fragment synthesis service (Integrated DNA Technologies). EGFP with a flexible GS linker (GGGGS) at the C terminus103 was amplified from the pLJM1-EGFP vector (gift from David Sabatini; Addgene plasmid #19319)89 using the EA062 and EA064 primers (Table S3).
MyoAAV production and in vivo transduction
For myoAAV production, we used a published protocol with modifications.104 We co-transfected the pAAV-cTnT-transgene vector (see above), the pHelper vector (GenBank: AF369965.1), and the pRep/Cap 1A-MYO capsid vector59 into AAVpro 293T cells (TaKaRa, 632273) using polyethylenimine (PEI) reagent. The 1A-MYO capsid vector allows production of muscle-tropic AAV9 derivative (MyoAAV).59 The transfected cells were cultured in the OptiPRO-SFM medium (Thermo Fisher, 12309019) for 72 h at 37°C to produce the virus. Cells and the culture media were collected for AAV purification. AAV particles were extracted with chloroform, precipitated with polyethylene glycol, purified with DNase/RNase digestion followed by chloroform extraction, and concentrated in PBS with Amicon filters (Millipore), as detailed in a previous report.104 The viral genome copy numbers (vg) were estimated by qPCR using primers amplifying the cTnT promoter region (Oligonucleotide ID KI466 and KI467, Table S3). MyoAAV particles were administered to mice through retro-orbital venous sinus injection at a dose of 2×1011 vg per mouse at 4 weeks of age, which was 14–15 days prior to tamoxifen.
Image quantification related to nuclear envelope ruptures
For quantification of nuclei with PCM1-lost nuclear tips and DNA protrusion, we used immunofluorescent images of isolated cardiomyocytes stained for PCM1 and counter-stained with DAPI (DNA) and Phalloidin (F-actin). Rod-shaped cardiomyocytes that had a well-defined sarcomere structure based on Phalloidin staining and two or more nuclei were used for quantification. We quantified instances of DAPI signal protrusion from a nuclear envelope site at which PCM1 signals were specifically discontinued (‘‘local PCM1 loss’’). The percentage of cells with one or more nuclei with a local PCM1 loss and the number of such nuclei per cell were quantified manually post imaging. We obtained the percentage from >50 cardiomyocytes per animal with a total of 3 animals per genotype. The percentages of cardiomyocytes with PCM1-lost nuclei were compared between genotypes using unpaired one-tailed Welch’s t-tests in R.
For quantification of nuclei with icGAS puncta, we used isolated cardiomyocytes from wild-type or LmnaCKO mice treated with MyoAAV-GFP-icGAS. The percentage of cells with icGAS puncta was quantified manually post imaging. We obtained the percentage from >50 cardiomyocytes per animal with a total of 3 animals per genotype. The icGAS-positive cardiomyocyte percentages were compared between genotypes using an unpaired one-tailed Welch’s t test in R.
For quantification of NLS-tdTomato intensities, we used LmnaF/F;Myh6MCMTg/0;Rosa26CAG-LSL-tdTomato/+ and Lmna+/+; Myh6MCMTg/0;Rosa26CAG-LSL-tdTomato/+ mice. Isolated cardiomyocytes were fixed, immunostained for PCM1, and counterstained with DAPI. Quantification of NLS-tdTomato signals was performed in the Fiji image analysis software. Cardiomyocytes that had two or more nuclei were used for quantification. Nuclei with nuclear envelope ruptures were identified as described above. A freehand-drawn area was placed inside a nucleus for quantification of the nuclear tdTomato signal. A nucleus was selected for quantification using the following algorithm: If a cell had only ruptured nuclei, one ruptured nucleus was randomly selected for quantification; if a cell had both ruptured and unruptured nuclei, one ruptured nucleus was randomly selected for quantification; if a cell had only unruptured nuclei, one unruptured nucleus was randomly selected for quantification. For each nucleus, whether it had a local PCM1 loss with DNA protrusion was manually determined as described above. A second quantification area of the same size as the nuclear area was placed in the cytoplasm for cytoplasmic tdTomato signal quantification. For each of the two areas, a mean NLS-tdTomato pixel intensity was measured using the ROI manager function in Fiji. We performed this quantification process for three mice per genotype. In total, we had quantification information for 99 wild-type cardiomyocytes (all intact PCM1), 90 LmnaCKO cardiomyocytes with local PCM1 loss, and 36 LmnaCKO cardiomyocytes with intact PCM1. Signal intensities between two groups were compared using Wilcoxon rank sum tests, and p-values were adjusted for multiple testing using the Benjamini-Hochberg procedure in R.
TUNEL assay
TUNEL assays were performed on deparaffinized heart sections using a Click-iT Plus TUNEL Assay kit for In Situ Apoptosis Detection (Invitrogen, C10617), according to the manufacturer’s instruction. The samples were then co-stained with Alexa Fluor Plus 647 Phalloidin (Invitrogen, A30107) for 1 h at 37°C to visualize cells in tissue sections. For positive controls, sample tissues were treated with DNase I prior to the TUNEL assay. The number of cells with positive staining in the TUNEL assay (TUNEL+ cells) was determined using sections from three hearts per genotype. TUNEL positive cells in 2–3 images per mouse (257 × 257 μm2 per image) were counted manually and compared between two groups using Wilcoxon rank sum tests in R.
Immunoblot
Snap-frozen hearts or isolated cardiomyocytes were lysed in the RIPA buffer for cGAS and STING immunoblotting (20 mM Tris-HCl, 150 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM dithiothreitol, protease and phosphatase inhibitors) or the Urea buffer for Lamin A/C immunoblotting (20 mM HEPES pH 7.4, 1 M NaCl, 8 M urea, protease and phosphatase inhibitors). Proteins were extracted using pestle homogenization and sonication. Cell lysates were centrifuged at 13,000 rpm for 5 min at 4°C, and proteins in the supernatant were separated by SDS-PAGE. Proteins were transferred to a PVDF membrane. Membranes were blocked with nonfat milk. The primary antibodies were rabbit anti-Lamin A/C antibody (Santa Cruz, sc-20681, 1:1000), rabbit anti-cGAS antibody (Cell Signaling, #31659, 1:500), rabbit anti-STING antibody (Cell Signaling, #13647, 1:500), and rabbit anti-GAPDH antibody (ABclonal, AC001, 1:1000); Secondary antibodies were anti-rabbit or anti-mouse IgG (H + L) DyLight 680 and 800 (Cell Signaling, 1:5000). Signals were detected and quantified in the Odyssey CLx Imager (LI-COR). Gels after protein transfer were counter-stained with Coomassie to evaluate the loaded protein amount.
Electron microscopy
Heart tissues were dissected and fixed in 2.5% glutaraldehyde, post-fixed in 1% OsO4 for 1 h at 4°C, rinsed, dehydrated in ethanol, and infiltrated overnight. The semithin Epon sections were screened by light microscopy to select areas with the longitudinal orientation of myocytes that were subsequently processed to prepare thin sections. The sections were stained with 1% uranyl acetate, followed by lead citrate. The sections were imaged using Hitachi Model H-7650 Transmission Electron Microscope at Cincinnati Children’s Integrated Pathology Research Facility.
Quantitative reverse-transcriptase PCR (RT-PCR)
For RT-PCR analysis, we used wild-type treated with MyoAAV-Luciferase (n = 5) or MyoAAV-cGAS (n = 3) and LmnaCKO mice treated with MyoAAV-Luciferase (n = 4) or MyoAAV-cGAS (n = 5) at 2 weeks post tamoxifen. Total RNAs of isolated cardiomyocytes were extracted with Trizol LS (Invitrogen, 10296010), treated with DNaseI, and purified. Quantitative RT-PCR was conducted with the Luna Universal One-Step RT-qPCR Kit (NEB, E3005) on a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad) following the manufacturer’s instructions. The primers for mouse genes were: EA096 and EA097 for Cxcl10; EA098 and EA099 for Ifnb1; EA100 and EA101 for Ifit3; and KI444 and KI445 for Actb (Table S3). The delta-delta Ct method was used to quantify mRNA abundance with Actb as a normalization control. mRNA levels between two groups were compared using Wilcoxon rank sum tests, and p-values were adjusted for multiple testing using the Benjamini-Hochberg procedure in R.
RNA-seq
For RNA-seq comparing wild-type mice with LmnaCKO mice at different post tamoxifen times, we used wild-type (n = 5) and LmnaCKO mice (n = 4) at 1 week post tamoxifen, wild-type (n = 3) and LmnaCKO mice (n = 2) at 11 days post tamoxifen, and wild-type (n = 7) and LmnaCKO mice (n = 5) at 2 weeks post tamoxifen. For RNA-seq analysis of Lmna/Cgas double knockout mice, we used Cgas−/−; LmnaF/F;Myh6MCM mice (n = 4), Cgas+/+;LmnaF/F;Myh6MCM (n = 6), Cgas+/+;LmnaF/+;Myh6MCM (n = 6), and Cgas−/−;LmnaF/+;Myh6MCM (n = 4) for experiments. Hearts were perfused with 100 mM KCl and removed for RNA extraction. Total RNAs were extracted with Trizol LS (Invitrogen, 10296010), treated with DNaseI, and purified. For tamoxifen time course experiments, mRNA sequencing libraries were generated using the poly-A selection module in the NEBNext UltraII Directional RNA Library Prep Kit (NEB E7760) and sequenced on the Illumina HiSeq 2500 sequencer with single-end 50 cycles. For cGas/Lmna double knockout experiments, libraries were generated using QuantSeq 3’ mRNA-Seq Library Prep Kit FWD (Lexogen, K01596) and sequenced on the Illumina NovaSeq 6000 sequencer with single-end 100 cycles.
RNA-seq analysis
High-throughput sequencing reads were aligned to the mouse mm39 reference genome with the Gencode vM27 basic gene annotation using STAR version 2.7.9 91 with the default alignment parameters except using ‘‘clip3pAdapterSeq AGATCGGAA GAGCACACGTCTGAACTCCAGTCA’’. From raw read counts per transcript, TPMs (transcripts per million) were calculated in R as follows: TPM = 106 x RPK/sum(RPK), where RPK = read count/transcript length in kp, and sum(RPK) is the sum of RPKs for all transcripts. Raw read counts were used for differential gene expression analyses using DESeq255 in R. Protein-coding or lncRNA genes with adjusted p-value <0.05 and absolute log2 fold change >1 (NEB Directional RNA-seq) or with adjusted p-value <0.05 and absolute log2 fold change >0 (QuantSeq 3’ mRNA-seq) were considered differentially expressed genes (Table S1). Differentially expressed genes were analyzed for the enrichment of GO Biological Processes, GO Molecular Functions, GO Cellular Components, and KEGG pathway terms using Metascape54 with the default enrichment parameters.
Single-nucleus RNA-seq
For single-nucleus RNA-seq, we used wild-type (n = 3) and LmnaCKO mice (n = 3) at 2 weeks post tamoxifen. Combinatorial indexing-based single-nucleus RNA-seq was conducted according to the optimized sci-RNA-seq3 protocol developed previously.92 Briefly, mice were anesthetized with isoflurane, euthanized by cervical dislocation, and then hearts were perfused with 100 mM KCl. Removed hearts were diced into small pieces in cold PBS containing Diethyl pyrocarbonate (DEPC, Sigma D5758). Fresh heart pieces were dounce-homogenized to liberate nuclei in Hypotonic lysis buffer B (7.7 mM Na2HPO4, 4.5 mM NaH2PO4, 1.8 mM KH2PO4, 2.7 mM KCl, 10.3 mM NaCl, 3 mM MgCl2, 0.08% BSA, 0.025% Igepal CA-630, 1% DEPC) and passed through cell strainers. Nuclei were fixed in 1 mg/mL dithiobis(succinimidyl propionate) (DSP, Thermo Fisher 22585) for 15 min on ice, washed, and stored at −80°C until use. For each mouse, 50,000 nuclei were placed in each of 8 wells of a 96-well plate (a total 48 wells for 6 mice). The remaining 48 wells were filled with similarly processed mouse heart nuclei with comparable sample quality from an unrelated project. Nuclei were subjected to reverse transcription with 96-indexed poly-T RT primers that contained a unique molecular identifier (UMI) sequence (3lvl_mRNA_RT_plate_1; Table S3). Nuclei from all wells were combined and redistributed evenly to 96 wells. Redistributed cells were subjected to ligation with 96-indexed ligation primers in 96 wells (3lvl_mRNA_Lig_plate_1; Table S3). Nuclei from all wells were combined again and redistributed evenly to 96 wells at 1,000 nuclei per well. Redistributed cells underwent second strand synthesis, protease digestion, tagmentation using N7-loaded Tn5 transposase (Oligonucleotide ID KI455 and KI456, Table S3), and PCR-amplified using 96-indexed PCR P7 primers (PCR_P7_plate1; Table S3) and the unindexed P5 primer (Oligonucleotide ID KI454, Table S3), for 16 cycles. PCR amplicons were combined and size-selected for DNA fragments between 250 bp and 600 bp. Purified library DNA were sequenced for 50 bases on the paired-end mode (34 cycles on Read1 to sequence the ligation index, UMI, and the RT barcode; 10 cycles on Index1 to sequence the P7 index; and 48 cycles on Read2 to sequence the cDNA) using an Illumina NovaSeq6000 sequencer.
Single-nucleus RNA-seq analysis
Pre-processing of the single-nucleus RNA-seq data was performed using the published sci-RNA-seq3 pipeline.92 The sci-RNA-seq3 pipeline takes fastq files separated by the P7 index as input, aligns reads to the genome, and returns a gene-by-cell read count matrix. First, raw reads were trimmed to remove poly A tails, aligned to the mm39 mouse reference genome using STAR,91 and filtered for MAPQ greater than or equal to 30. Duplicate reads were removed based on UMI. Read counts per gene were computed for each cell defined by the unique combination of the RT index, ligation index, and P7 index. The gene-by-cell count matrix produced by the sci-RNA-seq3 pipeline was further processed in the SingleCellExperiment package framework in R.93 We used the scDblFinder package94 for doublet detection and the scuttle package95 for mitochondrial RNA quantification in R. We obtained 14,111 nuclei after filtering for nuclei with the scDblFinder doublet score less than 0.25 and mitochondrial RNA contamination percentage less than 2% of total RNAs per cell. Read counts for a gene were normalized by per-cell read depth and then log-transformed to obtain normalized expression values (‘‘logcounts’’). Logcounts were used to plot expression values in graphs. Principal component analysis (PCA) was performed on logcounts for top 2,000 highly variable genes across cells using the fixedPCA function in the scran package.96 The top 50 principal components were used in the runUMAP function in the scater package95 with n_neighbors = 20 and min_dist = 1 parameters to obtain UMAP dimensions. To cluster cells, logcounts were processed in the clusterCells function in the scran package using the top 50 principal components, with the number of nearest neighbors k = 12. Cell clusters were annotated manually with cell type names based on marker gene expression. Per-gene read counts were summed within cell groups and processed by the DESeq2 package55 to perform pseudo-bulk differential gene expression analysis. Genes with adjusted p-values smaller than 0.05 were defined as differentially expressed genes (Table S4). GO analysis of differentially expressed genes was performed as described in the RNA-seq analysis section using Metascape.54 For the analysis of the enrichment within selected GO and KEGG terms, all genes associated with the selected terms were obtained using the R packages GO.db and KEGGREST, and enrichment of differentially expressed genes among these genes was computed by Fisher’s exact test with p-values adjusted by the Benjamini-Hochberg procedure. Cell-cell communication analysis was performed using the CellChat package in R77 using the 12 cell type annotations and the default mouse database with a minimum of ten cells for the analysis. Positive communications were selected by the subsetCommunication function in CellChat with threshold p-value less than 0.05, and the reported interaction counts were used to plot data. All software and packages used in the analysis are listed in Key resources table.
SLAM-IT-seq
For SLAM-IT-seq experiments, we used LmnaF/F;Myh6MCMTg/0;UprtTg/0 (n = 6) and Lmna+/+;Myh6MCMTg/0;UprtTg/0 (n = 8) mice as experimental groups, and LmnaF/F;Myh6MCMTg/0;Uprt0/0 (n = 4) and Lmna+/+;Myh6MCMTg/0;Uprt0/0 (n = 3) mice as negative control groups that would not incorporate 4-thiouracil (4sU) due to the absence of the Uprt allele. All mice were administered with tamoxifen and used at 2 weeks post tamoxifen treatment. SLAM-IT-seq was conducted according to a previously established protocol.62 Briefly, mice were intraperitoneally injected with 4-thiouracil (4sU) dissolved in a DMSO/corn oil (1:3) solution at a dose of 10 mg per gram of body weight. Six hours after the injection, mice were sacrificed, and hearts were perfused with 100 mM KCl and removed. RNAs from the left ventricular free wall were extracted by Trizol LS, purified, and DNaseI-treated using Directzol RNA miniprep kit (Zymo, R2052). Purified RNAs (1.5 μg) were subjected to alkylation in the presence of 12 mM Iodoacetamide (IAA, Sigma I1149). High-throughput sequencing libraries were generated from the alkylated RNAs using Quant-seq 3’ mRNA-seq FWD kit (Lexogen K01596) and sequenced 100 bases on the single-end mode on an Illumina NovaSeq6000 sequencer.
SLAM-IT-seq analysis
We analyzed SLAM-IT-seq data according to the published SLAM-IT-seq data analysis pipeline that uses the slamdunk package97 and R functions.62 First, raw reads were processed by the slamdunk all function in the the slamdunk package with the following alignment parameters: –trim-5p 12 –topn 100 –multimap –max-read-length 101 with default –var-fraction (0.8) and default -c (1). This function aligned reads to 3’ UTRs of Gencode vM27 genes (total 208,307 UTRs from 37,065 unique genes) in the mm39 mouse reference genome. Aligned reads were processed by the alleyoop utrrates function in the slamdunk package with default parameters to quantify C-to-T conversion events. UTRs with at least one read coverage for all 21 samples were retained for further analyses (47,717 UTRs from 14,250 unique genes). Statistically significant C-to-T conversion events in each transcript (i.e., the likelihood of cardiomyocyte-derived transcripts) were identified by comparing the conversion events in mice carrying the Uprt allele with those in mice not carrying this allele using beta-binomial test with the bbtest function98 in the countdata package in R. P-values of the beta binomial test were adjusted with the Benjamini-Hochberg procedure for multiple tests (Table S2). A transcript with the lowest adjusted p-value was chosen to represent a gene if there were multiple transcripts per gene. Genes with adjusted p-values smaller than 0.05 were defined as originating from cardiomyocytes, whereas genes with adjusted p-values greater than or equal to 0.05 were defined as not originating from cardiomyocytes. All software and packages used in the analysis are listed in Key resources table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using the following procedures: Wilcoxon tests with a Benjamini-Hochberg-adjusted p-value threshold of 0.05 computed by base R functions (Figures 1E, 2F, 2K, 2L, 3E, 3H, S1D, and S1E); Log rank tests with a p-value threshold of 0.05 computed by the survival package in R90 (Figures 1F, 3J, and S3I); Generalized linear model with a Benjamini-Hochberg-adjusted p-value threshold of 0.05 computed by the DESeq2 package in R55 (Figures 1G, 1I, 3G, 4C, 4E, 4F, S3G, and S4E); Metascape54 (Figures 1H, 3B, 4D, S3F, S4C, and S4F); unpaired one-tailed Welch’s t test with a p-value threshold of 0.05 computed by base R functions (Figures 2I and 2J); Beta binomial test with a Benjamini-Hochberg-adjusted p-value threshold of 0.05 computed by base R functions (Figures S3D and S3E); and Fisher’s exact test with Benjamini-Hochberg-adjusted p-value threshold of 0.05 (Figure S4D).
Statistical details of experiments and sample sizes are described in STAR Methods and indicated in figures and/or figure legends. Data are plotted using the ggplot2 package in R.
Supplementary Material
Highlights.
Lamin A/C reduction causes pervasive nuclear envelope ruptures in cardiomyocytes
Nuclear envelope ruptures do not activate cGAS-STING pathway in cardiomyocytes
cGAS-STING does not contribute to Lamin A/C-dependent cardiomyopathy in adult mice
ECM signaling from Lamin-reduced cardiomyocytes may initiate inflammatory response
ACKNOWLEDGMENTS
We thank Melanie Gucwa, James Brainer, and core facilities for animal care, microscopy, genomics, pathology, and viral vectors at Cincinnati Children’s and the University of Chicago for their assistance. Some graphics are created with BioRender.com. This work is supported by NIH grant R21/R33 AG054770 (K.I.), Cincinnati Children’s Research Innovation and Pilot grant (K.I.), and NIH grants R01 HL163523, R01 HL124836, and R01 HL126509 (I.P.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114284.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Aebi U, Cohn J, Buhle L, and Gerace L (1986). The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323, 560–564. [DOI] [PubMed] [Google Scholar]
- 2.Goldman AE, Maul G, Steinert PM, Yang HY, and Goldman RD (1986). Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc. Natl. Acad. Sci. USA 83, 3839–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Röber RA, Weber K, and Osborn M (1989). Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105, 365–378. [DOI] [PubMed] [Google Scholar]
- 4.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Soli-mando L, and Goldman RD (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22, 832–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr., Spudich S, De Girolami U, et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med 341, 1715–1724. [DOI] [PubMed] [Google Scholar]
- 6.Bonne G, Di Barletta MR, Varnous S, Bécane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, et al. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet 21, 285–288. [DOI] [PubMed] [Google Scholar]
- 7.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, et al. (2003). LMNA mutations in atypical Werner’s syndrome. Lancet 362, 440–445. [DOI] [PubMed] [Google Scholar]
- 9.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, and Lévy N (2003). Lamin a truncation in Hutchinson-Gilford progeria. Science 300, 2055. [DOI] [PubMed] [Google Scholar]
- 10.Cao H, and Hegele RA (2000). Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet 9, 109–112. [DOI] [PubMed] [Google Scholar]
- 11.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, et al. (2008). Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am. Heart J 156, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hershberger RE, and Morales A (2008). LMNA-Related Dilated Cardiomyopathy. In GeneReviews®, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A, eds. (University of Washington, Seattle: ). [Google Scholar]
- 13.Taylor MRG, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, et al. (2003). Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol 41, 771–780. [DOI] [PubMed] [Google Scholar]
- 14.Sébillon P, Bouchier C, Bidot LD, Bonne G, Ahamed K, Charron P, Drouin-Garraud V, Millaire A, Desrumeaux G, Benaïche A, et al. (2003). Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J. Med. Genet 40, 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rijsingen IAW, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Grasso M, et al. (2013). Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail 15, 376–384. [DOI] [PubMed] [Google Scholar]
- 16.Charron P, Arbustini E, and Bonne G (2012). What Should the Cardiologist know about Lamin Disease? Arrhythm. Electrophysiol. Rev 1, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verga L, Concardi M, Pilotto A, Bellini O, Pasotti M, Repetto A, Tavazzi L, and Arbustini E (2003). Loss of lamin A/C expression revealed by immuno-electron microscopy in dilated cardiomyopathy with atrioventricular block caused by LMNA gene defects. Virchows Arch 443, 664–671. [DOI] [PubMed] [Google Scholar]
- 18.Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, and Tavazzi L (2002). Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J. Am. Coll. Cardiol 39, 981–990. [DOI] [PubMed] [Google Scholar]
- 19.Kato K, Ohno S, Sonoda K, Makiyama T, Ozawa T, and Horie M (2020). Splice site mutation of LMNA causes severe dilated cardiomyopathy via strong dominant reduction of total lamin expression. Eur. Heart J 41. 10.1093/ehjci/ehaa946.0333. [DOI] [Google Scholar]
- 20.Narula N, Favalli V, Tarantino P, Grasso M, Pilotto A, Bellazzi R, Serio A, Gambarin FI, Charron P, Meder B, et al. (2012). Quantitative expression of the mutated lamin A/C gene in patients with cardiolaminopathy. J. Am. Coll. Cardiol 60, 1916–1920. [DOI] [PubMed] [Google Scholar]
- 21.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, and Worman HJ (2007). Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest 117, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muchir A, Shan J, Bonne G, Lehnart SE, and Worman HJ (2009). Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet 18, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu W, Muchir A, Shan J, Bonne G, and Worman HJ (2011). Mitogen-Activated Protein Kinase Inhibitors Improve Heart Function and Prevent Fibrosis in Cardiomyopathy Caused by Mutation in Lamin A/C Gene. Circulation 123, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, and Worman HJ (2012). Abnormal p38a mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet 21, 4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, and Worman HJ (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med 4, 144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SN, Lombardi R, Karmouch J, Tsai J-Y, Czernuszewicz G, Taylor MRG, Mestroni L, Coarfa C, Gurha P, and Marian AJ (2019). DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated With LMNA (Lamin A/C) Mutations. Circ. Res 124, 856–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheedipudi SM, Asghar S, and Marian AJ (2022). Genetic Ablation of the DNA Damage Response Pathway Attenuates Lamin-Associated Dilated Cardiomyopathy in Mice. JACC. Basic Transl. Sci 7, 1232–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidson PM, and Lammerding J (2014). Broken nuclei–lamins, nuclear mechanics, and disease. Trends Cell Biol 24, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen NY, Yang Y, Weston TA, Belling JN, Heizer P, Tu Y, Kim P, Edillo L, Jonas SJ, Weiss PS, et al. (2019). An absence of lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc. Natl. Acad. Sci. USA 116, 25870–25879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho S, Vashisth M, Abbas A, Majkut S, Vogel K, Xia Y, Ivanovska IL, Irianto J, Tewari M, Zhu K, et al. (2019). Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 49, 920–935.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Earle AJ, Kirby TJ, Fedorchak GR, Isermann P, Patel J, Iruvanti S, Moore SA, Bonne G, Wallrath LL, and Lammerding J (2020). Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater 19, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatch EM, and Hetzer MW (2016). Nuclear envelope rupture is induced by actin-based nucleus confinement. J. Cell Biol 215, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vargas JD, Hatch EM, Anderson DJ, and Hetzer MW (2012). Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Vos WH, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, Manders EMM, Verstraeten VLRM, van Steensel MAM, Marcelis CLM, et al. (2011). Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol. Genet 20, 4175–4186. [DOI] [PubMed] [Google Scholar]
- 35.Sylvius N, Bilinska ZT, Veinot JP, Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan K-L, Tang ASL, et al. (2005). In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet 42, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siu C-W, Lee Y-K, Ho JC-Y, Lai W-H, Chan Y-C, Ng K-M, Wong L-Y, Au K-W, Lau Y-M, Zhang J, et al. (2012). Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 4, 803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan Y, Tan D, Zhang X, Song D, Chang X, Wang S, Yan H, Ge L, Yang H, Bö nnemann C, et al. (2020). Nuclear Factor-kB Pathway Mediates the Molecular Pathogenesis of LMNA-Related Muscular Dystrophies. Biochem. Genet 58, 966–980. [DOI] [PubMed] [Google Scholar]
- 38.Chai RJ, Werner H, Li PY, Lee YL, Nyein KT, Solovei I, Luu TDA, Sharma B, Navasankari R, Maric M, et al. (2021). Disrupting the LINC complex by AAV mediated gene transduction prevents progression of Lamin induced cardiomyopathy. Nat. Commun 12, 4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen R, Buchmann S, Kroth A, Arias-Loza A-P, Kohlhaas M, Wagner N, Grüner G, Nickel A, Cirnu A, Williams T, et al. (2023). Mechanistic Insights of the LEMD2 p.L13R Mutation and Its Role in Cardiomyopathy. Circ. Res 132, e43–e58. [DOI] [PubMed] [Google Scholar]
- 40.Kim PH, Chen NY, Heizer PJ, Tu Y, Weston TA, Fong JL-C, Gill NK, Rowat AC, Young SG, and Fong LG (2021). Nuclear membrane ruptures underlie the vascular pathology in a mouse model of Hutchinson-Gilford progeria syndrome. JCI Insight 6, e151515. 10.1172/jci.insight.151515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross JA, Arcos-Villacis N, Battey E, Boogerd C, Orellana CA, Marhuenda E, Swiatlowska P, Hodzic D, Prin F, Mohun T, et al. (2023). Lem2 is essential for cardiac development by maintaining nuclear integrity. Cardiovasc. Res 119, 2074–2088. 10.1093/cvr/cvad061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Ramirez-Martinez A, Chen K, McAnally JR, Cai C, Durbacz MZ, Chemello F, Wang Z, Xu L, Bassel-Duby R, et al. (2023). Net39 protects muscle nuclei from mechanical stress during the pathogenesis of Emery-Dreifuss muscular dystrophy. J. Clin. Invest 133, e163333. 10.1172/JCI163333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eissenberg JC, and Gonzalo S (2020). Pushing the limit on laminopathies. Nat. Mater 19, 378–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gauthier BR, and Comaills V (2021). Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. Int. J. Mol. Sci 22, 7281. 10.3390/ijms22147281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, and Adams PD (2021). Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184, 5506–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yum S, Li M, Fang Y, and Chen ZJ (2021). TBK1 recruitment to STING activates both IRF3 and NF-kB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 118, e2100225118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decout A, Katz JD, Venkatraman S, and Ablasser A (2021). The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol 21, 548–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, Dorsett D, Kubben N, Batista LFZ, and Gonzalo S (2018). A Cell-Intrinsic Interferon-like Response Links Replication Stress to Cellular Aging Caused by Progerin. Cell Rep 22, 2006–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.King KR, Aguirre AD, Ye Y-X, Sun Y, Roh JD, Ng RP Jr., Kohler RH, Arlauckas SP, Iwamoto Y, Savol A, et al. (2017). IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med 23, 1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, Chen ZJ, Gillette TG, and Hill JA (2018). Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation 137, 2613–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu D, Cui Y-X, Wu M-Y, Li L, Su L-N, Lian Z, and Chen H (2020). Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am. J. Physiol. Heart Circ. Physiol 318, H1525–H1537. [DOI] [PubMed] [Google Scholar]
- 52.Yan M, Li Y, Luo Q, Zeng W, Shao X, Li L, Wang Q, Wang D, Zhang Y, Diao H, et al. (2022). Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov 8, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasper J, Welle K, Swovick K, Hryhorenko J, Ghaemmaghami S, and Buchwalter A (2024). Long lifetime and tissue-specific accumulation of lamin A/C in Hutchinson-Gilford progeria syndrome. J. Cell Biol 223, e202307049. 10.1083/jcb.202307049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, et al. (2017). Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet 49, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srsen V, Fant X, Heald R, Rabouille C, and Merdes A (2009). Centrosome proteins form an insoluble perinuclear matrix during muscle cell differentiation. BMC Cell Biol 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018). A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, Krunnfusz A, Tavakoli S, Widrick JJ, Messemer KA, et al. (2021). Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 184, 4919–4938.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, and Lammerding J (2016). Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Duménil AM, Manel N, and Piel M (2016). ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362. [DOI] [PubMed] [Google Scholar]
- 62.Matsushima W, Herzog VA, Neumann T, Gapp K, Zuber J, Ameres SL, and Miska EA (2019). Sequencing cell-type-specific transcriptomes with SLAM-ITseq. Nat. Protoc 14, 2261–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuhl N, Linder A, Philipp N, Nixdorf D, Fischer H, Veth S, Kuut G, Xu TT, Theurich S, Carell T, et al. (2023). STING agonism turns human T cells into interferon-producing cells but impedes their functionality. EMBO Rep 24, e55536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Xu P, Rivara S, Liu C, Ricci J, Ren X, Hurley JH, and Ablasser A (2022). Clathrin-associated AP-1 controls termination of STING signalling. Nature 610, 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ikushima H, Negishi H, and Taniguchi T (2013). The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb. Symp. Quant. Biol 78, 105–116. [DOI] [PubMed] [Google Scholar]
- 66.Della Corte CM, Sen T, Gay CM, Ramkumar K, Diao L, Cardnell RJ, Rodriguez BL, Stewart CA, Papadimitrakopoulou VA, Gibson L, et al. (2020). STING Pathway Expression Identifies NSCLC With an Immune-Responsive Phenotype. J. Thorac. Oncol 15, 777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al. (2014). Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud A-L, Cambier JC, and Lenz LL (2011). MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J. Immunol 187, 2595–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cohen TV, Gnocchi VF, Cohen JE, Phadke A, Liu H, Ellis JA, Foisner R, Stewart CL, Zammit PS, and Partridge TA (2013). Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of Lap2a. Hum. Mol. Genet 22, 2852–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ortega A, Roselló-Lletí E, Tarazón E, Molina-Navarro MM, Martínez-Dolz L, González-Juanatey JR, Lago F, Montoro-Mateos JD, Salvador A, Rivera M, and Portolés M (2014). Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy. PLoS One 9, e107635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sasagawa S, Nishimura Y, Okabe S, Murakami S, Ashikawa Y, Yuge M, Kawaguchi K, Kawase R, Okamoto R, Ito M, and Tanaka T (2016). Downregulation of GSTK1 Is a Common Mechanism Underlying Hypertrophic Cardiomyopathy. Front. Pharmacol 7, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan P, Zhang L, Cheng T, Wang J, Zhou J, Zhao L, Hua C, and Xia Q (2021). MiR-590–5p inhibits pathological hypertrophy mediated heart failure by targeting RTN4. J. Mol. Histol 52, 955–964. [DOI] [PubMed] [Google Scholar]
- 73.Wehrens M, de Leeuw AE, Wright-Clark M, Eding JEC, Boogerd CJ, Molenaar B, van der Kraak PH, Kuster DWD, van der Velden J, Michels M, et al. (2022). Single-cell transcriptomics provides insights into hypertrophic cardiomyopathy. Cell Rep 39, 110809. [DOI] [PubMed] [Google Scholar]
- 74.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, and Conway SJ (2009). Origin of cardiac fibroblasts and the role of periostin. Circ. Res 105, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, and Mann DL (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 111, 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu C, Raab M, Gui Y, and Rudd CE (2023). Multi-functional adaptor SKAP1: regulator of integrin activation, the stop-signal, and the proliferation of T cells. Front. Immunol 14, 1192838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 12, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang H, Ren L, and Wu JC (2022). New Insights Into the Therapy for Lamin-Associated Dilated Cardiomyopathy. JACC. Basic Transl. Sci 7, 1246–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Long Z-J, Wang J-D, Xu J-Q, Lei X-X, and Liu Q (2022). cGAS/STING cross-talks with cell cycle and potentiates cancer immunotherapy. Mol. Ther 30, 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, Reinert LS, Dagnaes-Hansen F, Hollensen AK, Mikkelsen JG, et al. (2016). Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 64, 746–759. [DOI] [PubMed] [Google Scholar]
- 82.Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Willaume S, Rass E, Fontanilla-Ramirez P, Moussa A, Wanschoor P, and Bertrand P (2021). A Link between Replicative Stress, Lamin Proteins, and Inflammation. Genes 12, 552. 10.3390/genes12040552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rouhi L, Auguste G, Zhou Q, Lombardi R, Olcum M, Pourebrahim K, Cheedipudi SM, Asghar S, Hong K, Robertson MJ, et al. (2022). Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype. J. Cardiovasc. Aging 2, 30. 10.20517/jca.2022.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Y, Chen X, Zhao Y, Wang X-Y, Luo Y-W, Chen L, Wang W, Zhong S, Hu M, Dai Z, et al. (2023). Small cytosolic double-stranded DNA represses cyclic GMP-AMP synthase activation and induces autophagy. Cell Rep 42, 112852. [DOI] [PubMed] [Google Scholar]
- 86.Nader G.P.d.F., Agüera-Gonzalez S, Routet F, Gratia M, Maurin M, Cancila V, Cadart C, Palamidessi A, Ramos RN, San Roman M, et al. (2021). Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 184, 5230–5246.e22. [DOI] [PubMed] [Google Scholar]
- 87.Verzi MP, McCulley DJ, De Val S, Dodou E, and Black BL (2005). The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol 287, 134–145. [DOI] [PubMed] [Google Scholar]
- 88.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PRR, et al. (2014). Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res 115, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Therneau TM, and Grambsch PM (2013). Modeling Survival Data: Extending the Cox Model (Springer Science & Business Media; ). [Google Scholar]
- 91.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martin BK, Qiu C, Nichols E, Phung M, Green-Gladden R, Srivatsan S, Blecher-Gonen R, Beliveau BJ, Trapnell C, Cao J, and Shendure J (2023). Optimized single-nucleus transcriptional profiling by combinatorial indexing. Nat. Protoc 18, 188–207. 10.1038/s41596-022-00752-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlin-ger L, Marini F, Rue-Albrecht K, Risso D, Soneson C, et al. (2020). Orchestrating single-cell analysis with Bioconductor. Nat. Methods 17, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Germain P-L, Lun A, Garcia Meixide C, Macnair W, and Robinson MD (2021). Doublet identification in single-cell sequencing data using scDblFinder. F1000Res 10, 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McCarthy DJ, Campbell KR, Lun ATL, and Wills QF (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lun ATL, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, and Ameres SL (2017). Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pham TV, Piersma SR, Warmoes M, and Jimenez CR (2010). On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics. Bioinformatics 26, 363–369. [DOI] [PubMed] [Google Scholar]
- 99.Wickham H (2016). ggplot2 (Springer New York; ). [Google Scholar]
- 100.Kim Y, and Zheng Y (2013). Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun 440, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, and Molkentin JD (2001). Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res 89, 20–25. [DOI] [PubMed] [Google Scholar]
- 102.Gay L, Miller MR, Ventura PB, Devasthali V, Vue Z, Thompson HL, Temple S, Zong H, Cleary MD, Stankunas K, and Doe CQ (2013). Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev 27, 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen X, Zaro JL, and Shen W-C (2013). Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev 65, 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Negrini M, Wang G, Heuer A, Björklund T, and Davidsson M (2020). AAV Production Everywhere: A Simple, Fast, and Reliable Protocol for Inhouse AAV Vector Production Based on Chloroform Extraction. Curr. Protoc. Neurosci 93, e103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data have been deposited at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are publicly available as of the date of publication. Accession numbers are listed in Key resources table.
This paper does not report original codes.
Any additional information required to reanalyze the data reported in this paper is available from Lead Contact upon request.