

Abstract
Pivotal clinical trials of B-cell maturation antigen-targeted chimeric antigen receptor T (CART)-cell therapy in patients with relapsed/refractory multiple myeloma (MM) resulted in remarkable initial responses, which led to a recent US Food and Drug Administration approval. Despite the success of this therapy, durable remissions continue to be low, and the predominant mechanism of resistance is loss of CART cells and inhibition by the tumor microenvironment (TME). MM is characterized by an immunosuppressive TME with an abundance of cancer-associated fibroblasts (CAFs). Using MM models, we studied the impact of CAFs on CART-cell efficacy and developed strategies to overcome CART-cell inhibition. We showed that CAFs inhibit CART-cell antitumor activity and promote MM progression. CAFs express molecules such as fibroblast activation protein and signaling lymphocyte activation molecule family-7, which are attractive immunotherapy targets. To overcome CAF-induced CART-cell inhibition, CART cells were generated targeting both MM cells and CAFs. This dual-targeting CART-cell strategy significantly improved the effector functions of CART cells. We show for the first time that dual targeting of both malignant plasma cells and the CAFs within the TME is a novel strategy to overcome resistance to CART-cell therapy in MM.
Key Points
-
•
CAFs are central components of the TME, promote MM growth, and inhibit CART-cell functions.
-
•
Dual targeting of CAFs and tumor cells with CART cells significantly reverses TME-induced immunosuppression.
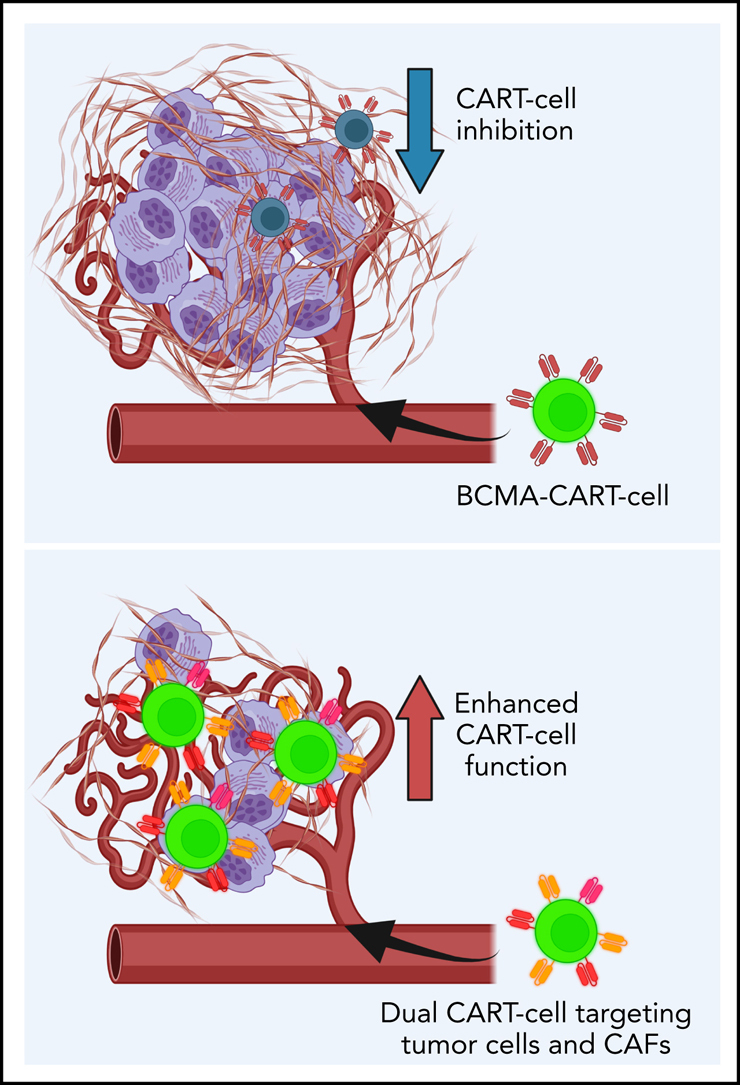
Introduction
Chimeric antigen receptor T (CART)-cell therapy has recently emerged as a potent and potentially curative therapy in hematologic malignancies.1,2 Unprecedented outcomes were reported after CD19-directed CART-cell therapy in B-cell malignancies. This led to the US Food and Drug Administration approval of several different CD19-directed CART-cell therapies since 2017. However, responses are transient in most patients, and loss of CART-cell persistence and function is the predominant mechanism of failure.2,3 An increasing body of literature suggests that the suppressive tumor microenvironment (TME) is significantly involved in the induction of CART-cell exhaustion and dysfunction.4,5,6
Multiple myeloma (MM) is an incurable B-cell lineage malignancy that accounts for 1.8% of all new cancer cases and 2.1% of cancer-related deaths.7 The outcomes of MM have significantly improved with the widespread use of bortezomib, immunomodulatory agents, and monoclonal antibodies (mAbs).8 However, the disease remains largely incurable, and patients ultimately fail to respond to, or become intolerant to, these therapies. Allogeneic hematopoietic stem cell transplantation is the only potentially curative treatment for patients with MM9 but is associated with high rates of nonrelapse mortality, highlighting the continued need for novel therapies in relapsed and refractory patients.
Recently, clinical trials showed that a single infusion of B-cell maturation antigen (BCMA)-targeted CART-cell therapy in patients with relapsed/refractory MM resulted in an initial complete remission (CR) rate of 80% to 100%.10,11,12 This led to the US Food and Drug Administration approval of BCMA-CART-cell therapy in March 2021. However, progression-free survival is short (<2 years), and most patients eventually relapse, mainly due to early dysfunction of CART cells and lack of persistence.10,11,13 MM is characterized by an immunosuppressive TME with an abundance of cancer-associated fibroblasts (CAFs). CAFs are a prominent component of the TME that have been shown to play a role in promoting tumor growth in different cancers.14,15 To reverse their impact on tumor progression, targeting CAFs with mAbs or with cellular therapy has been investigated as an independent strategy for the treatment of cancer. Preclinical studies have reported the efficacy of such approaches in inducing antitumor activity.16,17 Fibroblast activation protein (FAP), a protein overexpressed on the surface of CAFs, has been identified as a promising target for antibody or cellular immunotherapies.18
Given this information, we generated a unique hypothesis that CAFs induce CART-cell dysfunction that can be reversed by directly targeting CAFs. The current study investigated the interactions between CAFs and CART cells using MM as a model. We show that in MM, targeting both MM cells and CAFs with CART-cell therapy significantly overcomes CART-cell inhibition induced by the TME. Our studies illuminate a novel strategy to enhance CART-cell therapy that is applicable in MM and potentially in other human cancers.
Materials and methods
T-cell functional experiments
Intracellular cytokine analysis and T-cell degranulation assays were performed after incubation of CART cells with targets and BM-CAFs at the indicated ratios for 4 hours, in the presence of monensin (BioLegend, San Diego, CA), human CD49d (BD Biosciences, San Diego, CA), and human CD28 (BD Biosciences). After 4 hours, cells were harvested, and intracellular staining was performed after surface staining, followed by fixation and permeabilization with Fixation Medium A and B (Life Technologies, Oslo, Norway). For proliferation assays, CART cells labeled with carboxyfluorescein diacetate succinimidyl ester (Life Technologies), irradiated target cells, and bone marrow–derived CAFs (BM-CAFs) were cocultured at the indicated ratios. Cells were cocultured for 5 days, as described in the specific experiments, and then cells were harvested, and surface staining with APC-H7 anti-human CD3 (eBioscience, San Diego, CA), Brilliant Violet 421 anti-human CD45 (BioLegend), and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Invitrogen, Carlsbad, CA) was performed. Phorbol myristate acetate and ionomycin (MilliporeSigma, Burlington, MA) were used as positive nonspecific stimulants of T cells, at different concentrations as indicated in the specific experiments.
For killing assays, the BCMA+SLAMF7+ luciferase+ MM cell line OPM-2 or MM1.S cell lines were used. Luciferase+ control target cells were incubated at the indicated ratios with effector T cells for 24, 48, or 72 hours as indicated in the specific experiment. The killing was calculated by bioluminescence imaging (BLI) on a Xenogen IVIS-200 Spectrum camera (PerkinElmer, Hopkinton, MA) as a measure of residual live cells. For killing experiments, samples were treated with 1 µL d-luciferin (30 µg/mL) per 100 µL sample volume (Gold Biotechnology, St. Louis, MO) for 10 minutes before imaging.
Cytokine analysis
Cytokine analysis was performed on 72-hour cell supernatant. Debris was removed from the supernatant by centrifugation at 10 000g for 5 minutes. Supernatant was then diluted 1:2 with assay buffer before following the manufacturer's protocol for a Milliplex Human Cytokine/Chemokine MAGNETIC BEAD Premixed 38 Plex Kit (HCYTMAG-60K-PX38; MilliporeSigma). For transforming growth factor-β (TGF-β) analysis, 72-hour cell supernatant was also used. Supernatant was centrifuged at 10 000g for 5 minutes to remove the debris. Supernatant was then diluted 1:2 with assay buffer before following the manufacturer's protocol for Milliplex TGF-β 1 MAGNETIC BEAD Kit (TGFBMAG-64K-01; MilliporeSigma). Data were collected by using a Luminex (MilliporeSigma).
Bone marrow–derived cancer-associated fibroblasts
BM-CAFs were isolated as follows. BM aspirates of de-identified patients with newly diagnosed or relapsed MM (n = 19) were obtained from the Mayo Clinic MM biobank (supplemental Table 1, available on the Blood Web site) under a Mayo Clinic Institutional Review Board–approved protocol (IRB #521-93). CD138 was used as a marker to identify malignant plasma cells. The CD138– fractions were isolated by magnetic sorting using the fully automated RoboSep cell separation kit (EasySep Human Whole Blood and BM CD138+ Selection Kit; Stemcell Technologies, Vancouver, British Columbia, Canada). BM stromal cells were obtained after the adhesion of CD138– BM mononuclear cells to polystyrene flasks and cultured in Iscove modified Dulbecco medium (MilliporeSigma) with the addition of 10% fetal bovine serum (MilliporeSigma). Fibroblasts were purified from BM stromal cells through antifibroblast magnetic microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) that recognize fibroblast/epithelial cell marker (D7-FIB).19,20 Fibroblasts were cultured in Iscove modified Dulbecco medium containing 20% fetal bovine serum media and used for experiments as indicated in the specific experimental design. Neutralizing antibody clone 1D11 (monoclonal mouse immunoglobulin G) was used to interrogate the specific effects of TGF-β on CART cells. We confirmed that this antibody neutralizes TGF-β effectively (Figure 2B).
Figure 2.

CAFs inhibit BCMA-CART cells through direct and indirect interactions. (A) BCMA-CART-cell proliferation assay with BM-CAFs and anti–TGF-β was performed in transwell or non-transwell plates. Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled BCMA-CART cells were cocultured with lethally irradiated MM1.S for 5 days with BM-CAFs and anti–TGF-β. Absolute numbers of BCMA-CART cells were analyzed by using flow cytometry. BCMA-CART cells were defined as CD3-APC-H7–positive and CD45-BV421–positive cells (mean and standard error of the mean [SEM]; ***P < .001, one-way analysis of variance; n = 3, two replicates). (B-C) BCMA-CART cells were cocultured with irradiated MM1.S and BM-CAFs for 3 days, and supernatants were analyzed via singleplex or multiplex for cytokines/chemokines (mean and SEM; *P < .05, **P < .005, ***P < .0005, ****P < .0001, two-tailed Mann-Whitney U test; n = 2, two replicates). (D) BCMA-CART cells were cocultured with OPM-2 in the presence or absence of BM-CAFs. At 24 hours, cytotoxicity was assessed by luminescence relative to controls (*P < .05, ****P < .0001, two-way analysis of variance; n = 3, two replicates). (E) The evaluation of inhibitory receptors on BCMA-CART cells when they were cocultured with BCMA+SLAMF7+OPM-2 for 5 days in the presence or absence of BM-CAFs (mean and SEM; **P < .005, ****P < .0001, unpaired, two-sided t test; n = 2, two replicates). (F) The evaluation of inhibitory ligands on BM-CAFs when they were cocultured with BCMA-CART cells and irradiated MM1.S for 5 days; BM-CAFs were defined as FSP-1+CD38– fractions. (Mean and SEM; ****P < .0001, unpaired, two-sided t test; n = 3, two replicates). E:T, effector-to-target ratio; n.s., not significant.
OPM-2 and MM1.S xenograft and MM-TME mouse model
Male and female 8- to 12-week-old NSG mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained in an animal barrier space that is approved by the Institutional Biosafety Committee (IBC) for Biosafety Level 2 Plus (BSL2+) level experiments (IBC #HIP00000252.20). The BCMA+ SLAMF7+luciferase+ MM OPM-2 cell line, BCMA+SLAMF7+ luciferase+ MM MM1.S cell line, and BM-CAFs obtained from patients with newly diagnosed or relapsed MM were used to establish OPM-2, MM1.S xenografts, or MM-TME models, as specified in each experiment, under an Institutional Animal Care and Use Committee–approved protocol (IACUC #A00003102-17). Twenty-one to 28 days after injection, mice were imaged with a bioluminescent imager using a Xenogen IVIS-200 Spectrum camera (PerkinElmer) to confirm engraftment. Imaging was performed 10 minutes after the intraperitoneal injection of 10 µL/g d-luciferin (15 mg/mL; Gold Biotechnology). Mice were then randomized based on their BLI to receive different treatments as outlined in the specific experiments.
Results
CAFs, isolated from the BM of patients with MM, promote tumor growth and inhibit BCMA-directed CART cells
Before investigating the interactions between BM-CAFs and CART cells, we confirmed the strong preclinical activity of BCMA-CART cells against MM in vitro and in vivo. As expected,21,22,23 BCMA-CART cells resulted in potent effector functions against the BCMA+MM cell lines MM1.S and OPM-2 but not against the BCMA–K562 cells (supplemental Figure 1). Using OPM-2 xenografts, treatment with BCMA-CART cells resulted in complete eradication of the disease and long-term survival of the mice. Here, NOD/SCID γ chain-deficient (NSG) were injected with 1 × 106 luciferase+OPM-2 and assessed with BLI 4 weeks later to confirm engraftment (supplemental Figure 2A). To determine the most effective dose of BCMA-CART cells, we first performed a dose titration of BCMA-CART cells in vivo. Mice were randomized based on the tumor burden to receive different doses of BCMA-CART cells (0.25 × 106 to 1 × 106). Serial BLI revealed that 1 × 106 BCMA-CART cells were required to exhibit potent antitumor activity sufficient to induce disease remission (supplemental Figure 2B). We thus used 1 × 106 of BCMA-CART cells in subsequent experiments. To further test the efficacy of this dose of BCMA-CART cells, the OPM-2 xenograft mouse model was used. After engraftment of OPM-2, mice were randomized according to BLI-based tumor burden to receive either 1 × 106 untransduced T cells (UTD) or BCMA-CART cells. The BCMA-CART cells exhibited a potent antimyeloma effect and resulted in complete remission of the disease within 3 to 4 weeks after treatment (supplemental Figure 2C-D), long-term survival (supplemental Figure 2E), and infiltration of T cells into the tumor site (BM) (supplemental Figure 2F). In contrast, immunohistochemistry analysis of BM from the mice treated with UTD exhibited remarked infiltration of CD138+ malignant plasma cells (supplemental Figure 2G).
We next determined the impact of BM-CAFs on tumor growth and on BCMA-CART-cell functions. BM-CAFs from patients with newly diagnosed or relapsed/refractory MM were isolated (supplemental Table 1). We first studied the effect of BM-CAFs on MM tumor growth in vitro. BM-CAFs were cocultured with luciferase+MM1.S at a BM-CAF to MM.1S ratio of 0:1, 0.25:1, 0.5:1, or 1:1. Tumor growth was assessed by using BLI at multiple time points after culture. MM1.S proliferation was significantly enhanced in the presence of BM-CAFs (Figure 1A). Tumor growth was also promoted in vivo in the presence of BM-CAFs. Here, 0.25 × 106 luciferase+OPM-2 and 0.25 × 106 BM-CAFs were coinjected into the right flank of NSG mice. On day 12, tumor burden was assessed by using BLI. Mice engrafted with both OPM-2 and BM-CAFs exhibited significantly higher tumor burden compared with mice engrafted with OPM-2 only (Figure 1B). To establish a systemic model for BM-CAFs in MM,20 1 × 106 luciferase+OPM-2, in combination with either 1 × 106 BM-CAFs (MM-TME model) or phosphate-buffered saline (OPM-2 xenograft), were injected intravenously into NSG mice, and tumor burden was measured according to serial BLI. The MM-TME model showed significantly higher tumor burden (Figure 1C-D) and shorter overall survival (Figure 1E) compared with the OPM-2 xenograft. Five weeks after engraftment, BM examination of the MM-TME model confirmed trafficking of both MM and BM-CAFs to the BM (Figure 1F-G), whereas the OPM-2 xenograft did not show BM-CAFs in the BM (supplemental Figure 2G), suggesting this as a model to study the systemic interactions between MM and BM-CAFs in vivo. To further validate the utility of using BM-CAFs in a systemic MM model, we repeated the experiments using a different MM cell line, the MM1.S cells. Similarly, this model was created via injecting 1 × 106 of luciferase+ MM1.S and 1 × 106 of BM-CAFs intravenously into NSG mice. BLI revealed a significantly higher tumor burden when mice were injected with the combination of MM1.S and CAFs (supplemental Figure 3).
Figure 1.

BM-CAFs promote MM proliferation and impair BCMA-CART-cell function. (A) Luciferase+ BCMA+ SLAMF7+ MM1.S were cocultured with indicated ratios of BM-CAFs in vitro. The growth of MM1.S was assessed by BLI at 48 hours. (Mean and standard error of the mean [SEM]; **P < .005, ⋆⋆⋆P = .001, one-way analysis of variance; n = 3, two replicates). (B) Then, 2.5 × 105 luciferase+ BCMA+ SLAMF7+ OPM-2 were subcutaneously injected with or without 2.5 × 105 BM-CAFs into the right flank of NSG mice. Tumor growth was assessed by BLI three weeks after the injection (mean and SEM; ***P < .005, unpaired, two-sided t test; n = 6). (C and D) Then, 1 × 106 luciferase+ OPM-2 were intravenously injected with (MM-TME model) or without (OPM-2 xenograft) 1 × 106 BM-CAFs. Tumor growth was monitored by serial BLI (mean and SEM; **P < .005, ****P < .0001, unpaired, two-sided t test; n = 6). (E) Kaplan-Meier survival curve for MM-TME model and OPM-2 xenograft. MM-TME model vs OPM-2 xenograft hazard ratio, 35.68; 95% confidence interval, 2.687-473.6; **P = .0067 (log-rank test). (F) Immunohistochemical analysis of BM from MM-TME model. Magnification ×10. (G) Percentage of cells showing positive FSP-1 staining in BM from MM-TME model (***P < .005, unpaired, two-sided t test; n = 3). (H) Multiplex assay with the supernatant samples from the coculture of BCMA-CART cells and irradiated MM1.S, in the absence or presence of BM-CAFs (*P < .05, **P < .01, ***P < .005, unpaired, two-sided t test; n = 2, two replicates). FGF-2, fibroblast growth factor-2; GM-CSF, granulocyte macrophage colony-stimulating factor; GRO, growth-regulated oncogene; H&E, hematoxylin and eosin; IFN-γ, interferon-γ; sCD40L, soluble CD40 ligand; TNF, tumor necrosis factor.
Given the enhancement of MM growth in the presence of BM-CAFs in our preclinical studies, we aimed to determine the impact of BM-CAFs on CART-cell effector functions. BCMA-CART cells exhibited significantly lower proliferation (supplemental Figure 4A) and CD107a degranulation (supplemental Figure 4B) in the presence of BM-CAFs. Conversely, healthy donor BM-derived mesenchymal stem cells did not inhibit BCMA-CART-cell proliferation (supplemental Figure 5).
We next studied the specific effects of BM-CAFs on CART-cell cytokines. Here, lethally irradiated MM1.S were cocultured with BCMA-CART cells at a 1:1 ratio (CART-cell:MM1.S) in the presence or absence of BM-CAFs. On day 3, the supernatants were harvested and analyzed for a broad spectrum of human cytokines and chemokines. Inhibitory cytokines and growth factors, such as TGF-β, fibroblast growth factor-2, growth-regulated oncogene, interleukin (IL)-4, IL-5, monocyte chemotactic protein (MCP)-1, and MCP-2 were significantly increased while effector cytokines such as interferon-γ, granulocyte macrophage colony-stimulating factor, soluble CD40 ligand, tumor necrosis factor-α, tumor necrosis factor-β, and macrophage inflammatory protein 1-β were significantly decreased in the presence of BM-CAFs. Figure 1H presents data normalized to cytokines produced by BM-CAFs; raw data are shown in supplemental Figure 6. Similar results were seen when BCMA-CART cells were stimulated through a coculture with OPM-2 or the MM cell line ALMC-2 (supplemental Figure 7). This indicates a potential mechanism of CART-cell inhibition by BM-CAFs through the secretion of multiple suppressive cytokines.
BM-CAF–induced inhibition of BCMA-CART cells is mediated through the secretion of inhibitory cytokines/chemokines
Having shown that BM-CAFs significantly promote tumor growth, inhibit BCMA-CART-cell functions, and are associated with high levels of suppressive cytokines and growth factors, we aimed to determine whether there were additional mechanisms by which BM-CAFs inhibit CART-cell functions. Here, we performed an antigen-specific proliferation assay using transwell plates to assess whether the inhibitory effects are caused by direct or indirect contact of CART cells and BM-CAFs. BCMA-CART cells were stimulated with lethally irradiated MM1.S with or without BM-CAFs in non-transwell (direct) or transwell (indirect) conditions. A ratio of CART cell:target:CAF of 1:1:0.1 was used. BM-CAFs significantly suppressed the expansion of CART cells in both non-transwell and transwell plates (Figure 2A). It has been shown that stimulated BM-CAFs produce high levels of TGF-β, an inhibitory cytokine with prominent immunosuppressive functions,24,25,26 and that TGF-β inhibits CART-cell functions.27,28 We therefore studied the specific effect of TGF-β depletion on tumor growth and on CART-cell functions in the presence of BM-CAFs to determine if this is the predominant mechanism of BM-CAF–mediated CART-cell inhibition. BM-CAFs produced TGF-β when cocultured with CART cells and MM1.S, and TGF-β was effectively neutralized by using anti-human TGF-β antibody (clone 1D11) (Figure 2B). TGF-β neutralization only improved CART-cell effector cytokine production in the presence of BM-CAFs (Figure 2C) but did not ameliorate the inhibition of antigen-specific proliferation (Figure 2A) or cytotoxicity of CART cells in vitro (Figure 2D; supplemental Figure 8). This indicated that BM-CAF–mediated CART-cell inhibition is not entirely TGF-β dependent.
Next, because BM-CAFs have been shown to exert their suppressive functions through inhibitory receptor/ligand interactions,29,30 we interrogated changes in the expression of inhibitory receptors and ligands on BM-CAFs and CART cells after the coculture. CART cells significantly upregulated programmed cell death protein 1 (PD-1) surface expression in the presence of BM-CAFs (Figure 2E); BM-CAFs upregulated the inhibitory ligand programmed cell death-ligand 1 (PD-L1) in the presence of activated CART cells (Figure 2F; supplemental Figure 9). PD-L2 was expressed on BM-CAFs regardless of T-cell activation status. To investigate the role of PD1/PD-L1 interaction on BM-CAF–mediated CART-cell suppression, we tested whether blocking the PD-1/PD-L1 axis with or without TGF-β neutralization reversed the negative effects of BM-CAFs. PD-L1 blockade with or without TGF-β neutralization did not reverse CAF-mediated inhibition of CART-cell antigen-specific proliferation (supplemental Figure 10).
Collectively, these data suggest that BM-CAF–induced CART-cell inhibition is not TGF-β and PD-1/PD-L1 axis dependent but mediated through the secretion of multiple inhibitory soluble factors.
Targeting BM-CAFs and malignant cells overcomes BM-CAF–induced CART-cell dysfunction
Because our experiments indicated that BM-CAFs induce CART-cell dysfunction and promote tumor growth, we aimed to develop a unique strategy to deplete these cells using CART-cell therapy that targets both the malignant plasma cells as well as BM-CAFs. First, we characterized the immunophenotype of BM-CAFs isolated from patients with MM. Consistent with prior literature,20,31,32 flow cytometric analysis showed high levels of FAP expression on the CD38–CD45–FSP-1+ cells isolated from the BM of patients with MM (Figure 3A; supplemental Figure 11). However, we also identified for the first time a significant expression of signaling lymphocyte activation molecule family-7 (SLAMF7) on BM-CAFs in patients with MM (Figure 3A; supplemental Figure 11; supplemental Figure 12), mostly on FAP+ BM-CAFs. BM-CAFs expressed high levels of FAP and SLMAF7, whereas there was very low or no expression of FAP or SLAMF7 on CD45– cells of healthy donors (Figure 3B). Supplemental Figure 13 depicts the expression of BCMA, FAP, and SLAMF7 on the cell lines used in this report (OPM-2, MM1.S, and ALMC-2).
Figure 3.

Targeting BM-CAFs and malignant cells overcome BM-CAF–induced CART-cell dysfunction. (A) CD45– CD38– FSP-1+ cells expressed SLAMF7 and FAP but not BCMA; n = 3. (B) SLAMF7 and FAP were not expressed in CD45– cells from healthy donor BM (*P < .05, unpaired, two-sided t test); n = 4. (C) FAP-CART cells were cocultured with luciferase+FAP+WI-38. At 24 hours, cytotoxicity was assessed by luminescence relative to controls. SLAMF7-CART cells were coincubated with luciferase+ MM1.S JeKo-1 or Jurkat (negative control) cell lines (mean and SEM; **P = .01, ****P < .0001, two-way analysis of variance; n = 3, two replicates). (D) Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled FAP- or SLAMF7-CART cells were cocultured with irradiated WI-38 (for FAP-CART cells), MM1.S (for SLAMF7-CART cells), K562 (negative control), or MM patient–derived BM-CAFs for 5 days. Medium and phorbol 12-myristate-13-acetate/ionomycin (PI) were used as negative and positive controls, respectively (n = 3, two replicates). (E) To assess degranulation and cytokine production, FAP or SLAMF7-CART cells were cocultured with WI-38 (for FAP-CART cells) or MM1.S (for SLAMF7-CART cells) for 4 hours. For negative controls, JeKo-1 (for FAP-CART cells) or Jurkat (for SLAMF7-CART cells) cells were used (mean and SEM; *P < .05, **P < .005, ***P < .001, ****P < .0001, one-way analysis of variance; n = 3, two replicates). FITC, fluorescein isothiocyanate; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-γ, interferon-γ; MFI, mean fluorescence intensity; n.s., not significant; PE, phycoerythrin.
We generated FAP-specific and SLAMF7-specific CART cells to target BM-CAFs (supplemental Figure 14). To confirm the specific effect of FAP-CART cells, we used the FAP+ cell line WI-38 and BM-CAFs derived from patients with MM to stimulate FAP-CART cells. The in vitro experiments show that both FAP+WI-38 and BM-CAFs induced comparable and potent activation of FAP-CART cells, as confirmed by their cytotoxic activity (Figure 3C), antigen-specific proliferation (Figure 3D), degranulation, and cytokine production (Figure 3E; supplemental Figure 15). Similarly, SLAMF7+MM1.S and BM-CAFs induced strong and comparable antigen-specific stimulation of SLAMF7-CART cells, as measured by antigen-specific lysis, degranulation, cytokine production, and proliferation (Figure 3C-E; supplemental Figure 16). To show the specificity of FAP- or SLAMF7-CART cells, FAP– JeKo-1 and SLAMF7– Jurkat cells were used as antigen-negative cell lines (Figure 3C-E). Finally, we confirmed that both FAP- and SLAMF7-CART cells have a potent cytotoxic effect against BM-CAFs (supplemental Figure 17).
Next, we aimed to evaluate whether targeting SLAMF7 or FAP on BM-CAFs can inhibit the growth of CAF in vivo. Here, single-targeting SLAMF7- or FAP-CART cells were tested in our systemic MM-TME mouse model that incorporates BM-CAFs. Mice were randomized to treatment after high disease burden was established (supplemental Figure 18A). In this model with high tumor load, CART-cell therapy did not induce any antitumor activity, and mice from both groups died of tumor progression (supplemental Figure 18B-C). However, BM evaluation at the conclusion of this experiment showed no detection of FSP-1+ cells by immunohistochemistry staining (supplemental Figure 18D).
Dual-targeting BCMA-FAP- or BCMA-SLAMF7-CART cells show antitumor functions superior to BCMA-CART cells against MM in the presence of BM-CAFs in vitro
Next, we generated dual CART cells targeting both malignant plasma cells and BM-CAFs: BCMA-SLAMF7- and BCMA-FAP-CART cells (Figure 4A). These cells were generated through the dual transduction of two CAR vectors (supplemental Figure 14). Dual-targeting CART cells were able to overcome BM-CAF–mediated inhibition of BCMA-CART cells, as detailed in the following experiments.
Figure 4.

Dual-targeting BCMA-SLAMF7- and BCMA-FAP-CART cells overcomes the negative effects of BM-CAFs in vitro. (A) Representative flow plots of UTD, BCMA-CART-, BCMA-SLAMF7-CART-, and BCMA-FAP-CART cells. (B) UTD, BCMA-CART-, BCMA-SLAMF7-, or BCMA-FAP-CART cells were cocultured with luciferase+OPM-2 with BM-CAFs. At 24 hours, cytotoxicity was assessed by luminescence relative to controls (mean and standard error of the mean [SEM]; upper asterisks, BCMA-CART vs BCMA-FAP-CART cells; lower asterisks, BCMA-CART vs BCMA-SLAMF7-CART cells; *P < .05, **P < .005, ***P < .001, two-way analysis of variance [ANOVA]; n = 2, two replicates). (C) BCMA-, BCMA-SLAMF7-, or BCMA-FAP-CART cells were cocultured with irradiated MM1.S for 5 days with BM-CAFs (mean and SEM; *P < .05, ****P < .0001, one-way ANOVA; n = 3, two replicates). (D) BCMA-, BCMA-SLAMF7-, or BCMA-FAP-CART cells were cocultured with MM1.S for 4 hours with BM-CAFs (mean and SEM; *P < .05, **P < .005, ****P < .0001, one-way ANOVA; n = 3, two replicates). (E) BCMA-, BCMA-SLAMF7-, or BCMA-FAP-CART cells or UTD were cocultured with irradiated MM1.S for 3 days with BM-CAFs, and supernatants were analyzed via multiplex (n = 2, two replicates). E:T, effector-to-target ratio; FITC, fluorescein isothiocyanate; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-γ, interferon-γ; n.s., not significant; sCD40L, soluble CD40 ligand; TNF, tumor necrosis factor.
We cocultured dual- or single-targeting CART cells with OPM-2 cell lines in the presence of BM-CAFs. BCMA-FAP- or BCMA-SLAMF7-CART cells showed strong cytotoxicity, whereas BCMA-CART cells resulted in suboptimal killing effects in the presence of BM-CAFs (Figure 4B). Compared with single-targeting BCMA-CART-cell, dual-targeting BCMA-FAP- and BCMA-SLAMF7-CART cells displayed superior antigen-specific proliferation (Figure 4C), antigen-specific CD107a degranulation, and intracellular expression of key cytokines (interferon-γ and granulocyte macrophage colony-stimulating factor) when stimulated with MM1.S or OPM-2 in the presence of BM-CAFs (Figure 4D; supplemental Figure 19). Finally, when BCMA-FAP- or BCMA-SLAMF7-CART cells were stimulated with irradiated MM1.S or OPM-2 in the presence of BM-CAFs, they suppressed the secretion of BM-CAF–associated cytokines/chemokines such as TGF-β, IL-6, and MCP-1 (Figure 4E; supplemental Figure 20).
Having reported enhanced antitumor activity for dual BCMA-SLAMF7-CART cells, we aimed to determine if this is also due to dual targeting of BCMA and SLAMF7 on MM cells, independent of the effects on BM-CAFs. First, we compared the effector functions of dual BCMA-SLAMF7-CART vs single BCMA-CART cells in vitro, in the absence of BM-CAFs. There was no difference between the antigen-specific killing of dual BCMA-SLAMF7-CART and BCMA-CART cells against OPM-2 or MM1.S in vitro (supplemental Figure 21A-B). However, dual-targeting CART cells showed superior CD107a degranulation and cytokine production upon stimulation with either OPM-2 or MM1.S (supplemental Figure 21C-D). This action indicated enhanced antitumor activity due to dual targeting of SLAMF7 and BCMA on MM cells. However, the improvement in antigen-specific killing of BCMA-SLAMF7-CART cells, which was only observed in the presence of BM-CAFs, suggested an additional enhanced activity as a result of overcoming BM-CAF–mediated BCMA-CART cells inhibition (supplemental Figure 22).
We then tested the antimyeloma effect of dual-targeting CART cells in vivo. The OPM-2 xenograft was created by injecting 1 × 106 luciferase+OPM-2 via tail vein on day −28. In this model, mice were observed for an additional week to develop high disease burden before their treatment, to allow for the subsequent tumor relapse after treatment with CART cells. On day −1, BLI was performed, and mice were randomized according to tumor burden to receive the following: (1) UTD, (2) BCMA-CART cells, or (3) BCMA-SLAMF7-CART cells. Treatment with BCMA-SLAMF7-CART cells resulted in a more potent antitumor activity (supplemental Figure 21E-F) and prolonged overall survival compared with treatment with BCMA-CART cells (supplemental Figure 21G).
Dual targeting of BCMA-FAP- or BCMA-SLAMF7 with CART cells reverses BM-CAF–induced inhibition of BCMA-CART cells in a systemic MM-TME model
Finally, we investigated the impact of targeting both cancer cells and the TME to reverse the negative impacts of BM-CAFs in our systemic MM-TME model. This xenograft model was established by first injecting 1 × 106 luciferase+ OPM-2 and 1 × 106 BM-CAFs intravenously into NSG mice on day 0 (Figure 5A). Serial BLI exhibited tumor engraftment by day 21. Mice were observed for an additional week to develop high disease burden and to establish a relapse model. On day 28, mice were treated with 1 × 106 UTD, BCMA-, BCMA-FAP-, or BCMA-SLAMF7-CART cells. UTD or BCMA-CART cells failed to induce a discernible antitumor response in this immunosuppressive MM-TME model with relapsed MM. However, dual-targeting CART cells exhibited potent antitumor activity (Figure 5B-C) and resulted in a significantly prolonged overall survival (Figure 5D). Peripheral blood sampling 10 days after CART-cell treatment revealed significantly enhanced CART-cell expansion in mice treated with dual-targeting CART cells compared with mice treated with single-targeting BCMA-CART cells (Figure 5E).
Figure 5.

BCMA-SLAMF7- or BCMA-FAP-CART cells show a long-term durable response and improve overall survival in the MM-TME mouse model. (A-B) Four weeks after the injection of 1 × 106 OPM-2 and 1 × 106 BM-CAFs, mice were randomized into 4 groups: (1) UTD, (2) BCMA-CART cells, (3) BCMA-SLAMF7-CART cells, or (4) BCMA-FAP-CART cells. Tumor burden was assessed by using BLI (4 mice per group). (C) BLI curves of the MM-TME mouse models treated with UTD, single BCMA-CART-, dual-targeted BCMA-SLAMF7-, or BCMA-FAP-CART cells (mean and standard error of the mean; *P < .05, two-way analysis of variance at day 14). (D) Kaplan-Meier survival curves of the MM-TME mouse models treated with UTD, single BCMA-CART-, dual-targeted BCMA-SLAMF7-, or BCMA-FAP-CART cells (BCMA-CART- vs BCMA-SLAMF7-CART cells hazard ratio, 0.03020 [95% confidence interval, 0.001835-0.4969; *P = .01]; BCMA-CART- vs BCMA-FAP-CART cells hazard ratio, 0.03020 [95% confidence interval, 0.001835-0.4969; *P = .01]). (E-F) Mice were bled on day 10 after T-cell infusion. T-cell expansion (E) was analyzed with flow cytometry, and TGF-β levels were analyzed with singleplex (F). Absolute number of T cells per microliter was calculated with counting beads (mean and standard error of the mean; *P < .05, **P < .005, ****P < .0001, one-way analysis of variance). (G) Immunohistochemical analysis of BM samples from MM-TME mice treated with CART cells targeting BCMA, BCMA-SLAMF7, or BCMA-FAP (upper left, upper middle, lower left, and lower middle, magnification ×40; upper right and lower right, magnification ×20). n.s., not significant.
Day 10 serum TGF-β levels showed significant increases in the mice treated with BCMA-CART cells, whereas the mice treated with BCMA-SLAMF7- or BCMA-FAP-CART cells exhibited significant reductions in serum TGF-β levels (Figure 5F). To validate these findings, the experiment was repeated with a different MM model using the MM1.S cell line. The systemic MM1.S MM-TME mouse model was established through coimplantation of BM-CAFs (1 × 106) and luciferase+ MM1.S cells (1 × 106). Similar to the OPM-2 MM-TME model, dual-targeting BCMA-FAP- or BCMA-SLAMF7-CART cells resulted in improved antitumor activity and overall survival compared with single BCMA-CART cells (supplemental Figure 23).
At the conclusion of this experiment, mice were killed, femurs were harvested, and BM was analyzed by immunohistochemistry. Mice treated with single-targeting BCMA-CART cells did not display any CD3+ T-cell infiltration but showed a dominant population of CD138+ malignant plasma cells that were surrounded by FSP+CAFs in the BM. Conversely, dual-targeting BCMA-SLAMF7- or BCMA-FAP-CART-cell groups displayed a higher presence of CD3+T cells and reduced/absent CD138+ plasma cells and FSP-1+CAFs (Figure 5G), suggesting enhanced T-cell persistence in the BM.
Discussion
The goal of the current study was to better understand the mechanisms for the relative lack of efficacy of CART-cell therapy in certain hematologic malignancies and most solid tumors. Using MM as a model, we identified a novel mechanism for TME-induced resistance to CART-cell therapy through an abundance of BM-CAFs. The advances from this model further confirmed that BM-CAFs, isolated from the BM of patients with MM, promote tumor growth and suppress CART cells' effector functions and antitumor activity. With this knowledge, we targeted BM-CAFs with dual-targeting CART-cell strategies. Our preclinical studies showed that targeting BM-CAFs reverses CART-cell dysfunction and enhances antitumor activity. This study represents the first preclinical model that recapitulates the tumor and BM-CAFs in relation to CART-cell efficacy. Our MM-TME model is especially crucial in testing the capabilities of TME-targeting CART cells as a strategy to enhance CART-cell functions. This approach holds promise to further extend the use of CART-cell therapy in hematologic malignancies.
The TME is a complex biological structure that supports tumor cells and also appears to further suppress tumor immunosurveillance.14 In various types of cancers, the TME can be viewed as a composite of cancer cells and stromal cells such as CAFs, regulatory T cells, myeloid-derived suppressor cells, mesenchymal cells, and endothelial cells, as well as soluble growth factors (especially TGF-β, platelet-derived growth factor, epidermal growth factor, vascular endothelial growth factor, and fibroblast growth factor). CAFs are a heterogeneous group of cells that appear to regulate immune cells and promote tumor progression. They produce several growth factors, including TGF-β, which is a potent immunomodulatory molecule aberrantly expressed in various types of malignancies. Our data suggest that TGF-β secreted by BM-CAFs is not a predominant mechanism of CART-cell inhibition. BM-CAFs suppress CART cells through both contact-dependent and cytokine-mediated effects. When BCMA-CART cells were stimulated and cocultured with BM-CAFs, the surface expression of inhibitory receptors such as PD-1 was significantly upregulated on CART cells, whereas BM-CAFs simultaneously overexpressed inhibitory ligands such as PD-L1. This is consistent with prior reports of CAF-induced immune dysfunction through inhibitory ligand–receptor interactions and modulation of exhaustion pathways.33,34 In pancreatic carcinoma, CAFs induced the surface expression of immune checkpoints, such as PD-1, lymphocyte activation gene-3 (LAG-3), CTLA-4, and T-cell immunoglobulin-3 (TIM-3), and contributed to diminished immune functions, which were restored with anti–PD-1 inhibitor.35 Melanoma-associated fibroblasts directly suppress CD8+ T-cell proliferation and function by upregulating the expression of PD-L1.30 Our experiments further indicate that blocking PD1/PD-L1, or the combination of TGF-β neutralization and PD1/PD-L1 blockade, are insufficient strategies to overcome the negative effects of BM-CAFs.
As a more comprehensive approach, rather than targeting TGF-β or other inhibitory cytokines alone, we targeted the source of multiple mechanisms of immunosuppression (BM-CAFs) with CART cells. BCMA-FAP- or BCMA-SLAMF7-CART cells were used to target both malignant plasma cells and the TME. FAP-CART cell therapy has been investigated in preclinical studies, with evidence for both antitumor efficacy and unfortunate toxicities. CART cells targeting murine FAP (mFAP) have an antitumor effect on malignant pleural mesothelioma cell lines.36 Tran et al37 reported that targeting mFAP with CART-cell therapy resulted in lethal bone toxicity and cachexia. In contrast, another group showed that mFAP-CART cells partially depleted tumor-associated FAP+ cells while retaining antitumor efficacy but did not report severe side effects.36 However, it is unknown whether targeting human FAP would yield such toxicity. Schuberth et al38 designed and produced an anti-human FAP-CART cells that recognized FAP-expressing cell lines and showed cytotoxic activity. This group launched a phase 1 clinical trial of FAP-CART cell therapy for patients with malignant pleural mesothelioma (using FAP5, the same clone as our FAP-CAR; #NCT01722149). Encouragingly, there have been no severe adverse events reported to date, and according to their update, immune-mediated toxicity has not been observed.39 In addition, FAP-CART cells were recently used to decrease cardiac fibrosis and regenerate cardiac function in a mouse model of hypertensive heart failure.40 A comprehensive evaluation of the potential hematopoietic and off-target toxicities of our constructs is ongoing and will be reported in a follow-up article.
An additional novel finding of the current study is the significant expression of SLAMF7 on BM-CAFs. SLAMF7 is known to be expressed on MM cells and has been evaluated as a target using several modalities.41 SLAMF7-CART cells42 showed potent antimyeloma activity and are being tested in a clinical trial (#NCT03958656). The main concern for the clinical application of SLAMF7-CART cells therapy is “off-target” effects. Lymphocytes, including B, T, and natural killer cells, express SLAMF7 to a lesser extent than MM cells, and do not appear to be depleted by the SLAMF7 mAb elotuzumab. Elotuzumab has been tested in patients with MM, without unexpected adverse events; specifically, significant lymphopenia has not yet been reported.43 In this study, we observed no significant fratricide during SLAMF7- or BCMA-SLAMF7-CART cell production. Similar to SLAMF7-CART cells recently reported by Amatya et al,44 the CD4/CD8 ratio of SLAMF7- or BCMA-SLAMF7-CART cells was increased slightly compared with BCMA- or FAP-CART cells (supplemental Figure 24). We hypothesize that the known low expression of SLAMF7 on CART cells may be contributing to CART-cell stimulation during their expansion. Such observations have been reported by others when molecules on T cells are targeted with CART-cell therapy. In this report, expansion of CD5-directed CART cells was improved compared with control CART cells.45
Because our experiments revealed significant expression of SLAMF7 on BM-CAFs, targeting both BCMA and SLAMF7 represents another promising strategy for targeting MM cells and the TME.
Although dual-targeting BCMA-SLAMF7-CART cells has been shown by others to exhibit potent antitumor activity in vivo, they were only investigated in xenograft models lacking TME components.46 In MM, malignant plasma cells localize and develop in the BM by establishing bidirectional interactions with BM-CAFs and the extracellular matrix, which promotes tumor cell survival and treatment resistance, including adoptive immunotherapies.4,5,6 Our MM-TME mouse model is a unique and pathophysiologically relevant model of human MM. We have shown in this study that single-targeting BCMA-CART cells were inhibited in this MM-TME model, but dual-targeting CART cells directed at the MM cells and BM-CAFs were able to overcome resistance and effectively reduce tumor burden.
There are limitations to this study that must be considered. There were no significant differences between BCMA-SLAMF7- or BCMA-FAP-CART cells in terms of in vitro or in vivo activities in our MM-TME model. Because our understanding of the clinical safety profile and in vivo activity of multitargeted CART-cell therapy is limited, translation to the clinic must proceed with caution. For example, we and others have shown that multitargeted CART cells are more activated47,48,49 and thus might be associated with higher risks of life-threatening toxicities such as cytokine release syndrome or neurotoxicity. Therefore, when such therapies are applied in the clinic, appropriately designed phase 1 and 2 clinical trial protocols will need to include the vigilant monitoring of patients and preemptive treatment of toxicities with anticytokine-directed measures.50 In addition, because SLAMF7 is known to be expressed on mature immune cells,51 precaution should be taken as dual-targeting CART cells are translated into the clinic.
In summary, we report that dual targeting of malignant plasma cells and BM-CAFs with BCMA-SLAMF7- or BCMA-FAP-CART cells is able to overcome TME-induced CART-cell inhibition. We identify a novel mechanism of resistance to CART-cell therapy induced by BM-CAFs within the TME. This study illuminates a novel strategy to develop more potent CART cells that can be applied to MM and other malignancies to combat TME-mediated CART-cell resistance.
Acknowledgments
The authors thank Stephen J. Russell for his feedback and critical review. Figures were created with BioRender.com.
This work was supported through grants from Mayo Clinic Multiple Myeloma SPORE P50CA186781 (R.S.) and K12CA090628 (S.S.K.), the Mayo Clinic Cancer Center (S.S.K.), the Mayo Clinic K2R pipeline (S.S.K.), the National Comprehensive Cancer Network (S.S.K.), the Mayo Clinic Center for Individualized Medicine (S.S.K.), the Predolin Foundation (R.S. and S.S.K.), the Exact Foundation (S.S.K.), the Eagles Foundation (R.S.), and the Lu Foundation (Y.L. and S.S.K.).
Footnotes
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Chimeric antigen receptor T (CAR T) cell therapy shows great promise as a treatment for relapsed multiple myeloma, but T-cell exhaustion is limiting. Sakemura et al document that cancer-associated fibroblasts (CAFs) in the bone marrow niche inhibit standard CAR T-cell antimyeloma activity. They describe a new approach that eliminates CAFs. The authors demonstrate promising in vitro and preclinical in vivo efficacy of CAR T cells that dually target BCMA on myeloma cells and either SLAMF7 or fibroblast-activating protein on CAFs.
Authorship
Contribution: R.S. and S.S.K. formulated the initial concept and designed experiments; R.S., M.J.C., E.L.S., M.H., C.M.R., K.J.S., E.E.T., W.K.N., M.J.H., P.H., I.C., M.M.A., E.D.B., L.A.K.F., and M.W.R. performed CART-cell experiments; D.P.L. performed immunohistochemistry staining experiments; M.J.H. and M.H. performed mouse experiments; K.J.S., D.K.W., and D.F.J. provided samples and reagents; S.A.P., Y.L., N.E.K., and P.L.B. provided input; S.S.K. supervised the study; R.S. and S.S.K. wrote the manuscript; E.L.S. edited the manuscript; and all authors edited and approved the final version of the manuscript.
Conflict-of-interest disclosure: S.S.K. is an inventor on patents in the field of CAR immunotherapy that are licensed to Novartis (through an agreement between Mayo Clinic, University of Pennsylvania, and Novartis). R.S., M.J.C., and S.S.K. are inventors on patents in the field of CAR immunotherapy that are licensed to Humanigen (through Mayo Clinic). S.S.K. and M.H. are inventors on patents in the field of CAR immunotherapy that are licensed to Mettaforge (through Mayo Clinic). S.S.K. receives research funding from Kite, Gilead, Juno, Celgene, Novartis, Humanigen, MorphoSys, Tolero, Sunesis, and Lentigen. N.E.K. receives research funding from Acerta Pharma, Bristol Myers Squibb, Celgene, Pharmacyclics, Tolero Pharmaceuticals, MEI Pharma, Sunesis, and AbbVie; is a member of the Data Safety Monitoring Committee for Agios Pharm, AstraZeneca, Celgene, CytomX Therapeutics, MorphoSys, and Rigel; and is an advisory board member for CytomX Therapy, Pharmacyclics, Dava Oncology, Juno Therapeutics, AstraZeneca, and Oncotracker. Y.L. receives research funding from Janssen, Takeda, Merck, Kite, and Celgene/BMS; is a member of the Data Safety Monitoring Committee for Sorrento; and has participated in advisory board meetings of Kite, Gilead, Janssen, Bristol Myers Squibb, Celgene, JUNO, bluebird bio, Legend Biotech, and Gamida Cell (she was not personally compensated for her participation). S.A.P. receives research funding from Pharmacyclics, MorphoSys, Janssen, AstraZeneca, TG Therapeutics, Bristol Myers Squibb, Merck, AbbVie, and Ascentage Pharma; and has participated in advisory board meetings of Pharmacyclics, AstraZeneca, Genentech, Gilead, GlaxoSmithKline, Verastem Oncology, and AbbVie (he was not personally compensated for his participation). The remaining authors declare no competing financial interests.
Supplementary Material
REFERENCES
- 1.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 5.Siska PJ, Beckermann KE, Mason FM, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2(12):93411. doi: 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakemura R, Cox MJ, Hefazi M, Siegler EL, Kenderian SS. Resistance to CART cell therapy: lessons learned from the treatment of hematological malignancies. Leuk Lymphoma. 2021;62(9):2052–2063. doi: 10.1080/10428194.2021.1894648. [DOI] [PubMed] [Google Scholar]
- 7.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 8.Kumar SK, Lee JH, Lahuerta JJ, et al; International Myeloma Working Group,. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study [published correction appears in Leukemia. 2012;26(5):1153]. Leukemia., 2012;26(1): 149-157. [DOI] [PMC free article] [PubMed]
- 9.Tricot G, Vesole DH, Jagannath S, Hilton J, Munshi N, Barlogie B. Graft-versus-myeloma effect: proof of principle. Blood. 1996;87(3):1196–1198. [PubMed] [Google Scholar]
- 10.Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–2280. doi: 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen AD, Garfall AL, Stadtmauer EA, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–2221. doi: 10.1172/JCI126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141. doi: 10.1186/s13045-018-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;10:1835. doi: 10.3389/fimmu.2019.01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 18.Fassnacht M, Lee J, Milazzo C, et al. Induction of CD4(+) and CD8(+) T-cell responses to the human stromal antigen, fibroblast activation protein: implication for cancer immunotherapy. Clin Cancer Res. 2005;11(15):5566–5571. doi: 10.1158/1078-0432.CCR-05-0699. [DOI] [PubMed] [Google Scholar]
- 19.Jones EA, Kinsey SE, English A, et al. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002;46(12):3349–3360. doi: 10.1002/art.10696. [DOI] [PubMed] [Google Scholar]
- 20.Frassanito MA, Rao L, Moschetta M, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia. 2014;28(4):904–916. doi: 10.1038/leu.2013.254. [DOI] [PubMed] [Google Scholar]
- 21.Friedman KM, Garrett TE, Evans JW, et al. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther. 2018;29(5):585–601. doi: 10.1089/hum.2018.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bu DX, Singh R, Choi EE, et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget. 2018;9(40):25764–25780. doi: 10.18632/oncotarget.25359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakao A, Afrakhte M, Morén A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389(6651):631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 25.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7(11):1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 27.Morgan MA, Schambach A. Engineering CAR-T cells for improved function against solid tumors. Front Immunol. 2018;9(2493):2493. doi: 10.3389/fimmu.2018.02493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen ML, Pittet MJ, Gorelik L, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci USA. 2005;102(2):419–424. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T cells to protect tumour cells. Nat Commun. 2018;9(1):948. doi: 10.1038/s41467-018-03347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalili JS, Liu S, Rodríguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18(19):5329–5340. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henry LR, Lee HO, Lee JS, et al. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin Cancer Res. 2007;13(6):1736–1741. doi: 10.1158/1078-0432.CCR-06-1746. [DOI] [PubMed] [Google Scholar]
- 32.Scanlan MJ, Raj BK, Calvo B, et al. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc Natl Acad Sci USA. 1994;91(12):5657–5661. doi: 10.1073/pnas.91.12.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghebeh H, Tulbah A, Mohammed S, et al. Expression of B7-H1 in breast cancer patients is strongly associated with high proliferative Ki-67-expressing tumor cells. Int J Cancer. 2007;121(4):751–758. doi: 10.1002/ijc.22703. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Chen L, Xiong Y, et al. Knockdown of PD-L1 in human gastric cancer cells inhibits tumor progression and improves the cytotoxic sensitivity to CIK therapy. Cell Physiol Biochem. 2017;41(3):907–920. doi: 10.1159/000460504. [DOI] [PubMed] [Google Scholar]
- 35.Gorchs L, Fernández Moro C, Bankhead P, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4+ and CD8+ T-cells. Front Immunol. 2019;10:847. doi: 10.3389/fimmu.2019.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran E, Chinnasamy D, Yu Z, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210(6):1125–1135. doi: 10.1084/jem.20130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuberth PC, Hagedorn C, Jensen SM, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. 2013;11:187. doi: 10.1186/1479-5876-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Curioni A, Britschgi C, Hiltbrunner S, et al. A phase I clinical trial of malignant pleural mesothelioma treated with locally delivered autologous anti-FAP-targeted CAR T-cells. Ann Oncol. 2019;30 v501. [Google Scholar]
- 40.Aghajanian H, Kimura T, Rurik JG, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430–433. doi: 10.1038/s41586-019-1546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chim CS, Kumar SK, Orlowski RZ, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018;32(2):252–262. doi: 10.1038/leu.2017.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gogishvili T, Danhof S, Prommersberger S, et al. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood. 2017;130(26):2838–2847. doi: 10.1182/blood-2017-04-778423. [DOI] [PubMed] [Google Scholar]
- 43.Neyer L, Ding H, Chen D, et al. Effect of elotuzumab on circulating lymphocytes, chemokines, and cytokines in multiple myeloma patients. Blood. 2010;116(21):4070. [Google Scholar]
- 44.Amatya C, Pegues MA, Lam N, et al. Development of CAR T cells expressing a suicide gene plus a chimeric antigen receptor targeting signaling lymphocytic-activation molecule F7. Mol Ther. 2021;29(2):702–717. doi: 10.1016/j.ymthe.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126(8):983–992. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen KH, Wada M, Pinz KG, et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia. 2018;32(2):402–412. doi: 10.1038/leu.2017.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Jiang P, Zhang X, et al. Anti-CD19/CD22 dual CAR-T therapy for refractory and relapsed B-cell acute lymphoblastic leukemia. Blood. 2019;134:284. suppl 1. [Google Scholar]
- 48.Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126(10):3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hegde M, Corder A, Chow KKH, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma [published correction appears in Mol Ther. 2014;22(5):1063] Mol Ther. 2013;21(11):2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709. doi: 10.1182/blood-2018-10-881722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boles KS, Stepp SE, Bennett M, Kumar V, Mathew PA. 2B4 (CD244) and CS1: novel members of the CD2 subset of the immunoglobulin superfamily molecules expressed on natural killer cells and other leukocytes. Immunol Rev. 2001;181:234–249. doi: 10.1034/j.1600-065x.2001.1810120.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.