

Abstract
FRET measurements were used to determine the domain-specific topography of perfringolysin O, a pore-forming toxin, on a membrane surface at different stages of pore formation. The data reveal that the elongated toxin monomer binds stably to the membrane in an “end-on” orientation, with its long axis approximately perpendicular to the plane of the membrane bilayer. This orientation is largely retained even after monomer association to form an oligomeric prepore complex. The domain 3 (D3) polypeptide segments that ultimately form transmembrane β-hairpins remain far above the membrane surface in both the membrane-bound monomer and prepore oligomer. Upon pore formation, these segments enter the bilayer, whereas D1 moves to a position that is substantially closer to the membrane. Therefore, the extended D2 β-structure that connects D1 to membrane-bound D4 appears to bend or otherwise reconfigure during the prepore-to-pore transition of the perfringolysin O oligomer.
Keywords: membrane protein, toxin, fluorescence, membrane
Perfringolysin O (PFO), a cytolytic toxin from the pathogenic bacterium Clostridium perfringens, perforates cholesterol-containing eukaryotic cell membranes by forming large aqueous pores that measure up to 300 Å in diameter (1). PFO belongs to a large family of protein toxins termed the “cholesterol-dependent cytolysins” (CDCs) that serve as potent virulence factors for various pathogenic Gram-positive bacteria (2). Pore formation is accomplished by a complex mechanism that includes the stable binding of water-soluble PFO monomers to membranes with sufficient cholesterol, lateral diffusion of monomers on the membrane surface, the association of monomers into oligomeric prepore complexes that are composed of up to 50 polypeptides, and then the concerted insertion of a large oligomeric amphipathic β-barrel as the prepore complex enters the membrane bilayer to create a large aqueous pore (3, 4).
The transition of the water-soluble PFO monomer into a membrane-inserted oligomer involves extensive changes in protein conformation that have been the recent focal point of research. Several structural states and rearrangements have been identified, along with various protein-lipid and protein-protein interactions that mediate the conformational changes, by using the crystal structure of the PFO monomer (5), a repertoire of site-specifically mutagenized PFO derivatives, and multiple independent fluorescence techniques (6). Domain 4 (D4) is located at one end of the elongated PFO monomer (Fig. 1A) and is responsible for membrane recognition and initial binding (7), but only the tip of this domain is embedded in the nonpolar interior of the bilayer (8). D4-lipid interactions trigger conformational changes in the spatially distant D3 that expose a previously hidden interface for oligomerization and, hence, prepore complex formation (9). Two sets of three short α-helices in D3 then undergo an α-helix-to-β-sheet transition to create two transmembrane β-hairpins (TMHs), TMH1 and TMH2, per monomer that are then inserted into the bilayer (10, 11). This insertion also occurs in stages because the TMHs are not exposed to the nonpolar core of the bilayer in the prepore complex (12). Therefore, the conversion of PFO from a water-soluble monomer to a membrane-inserted oligomer involves the structural coupling of D3 and D4 (7, 9).
Fig. 1.

PFO domains and possible rearrangements. (A) Ribbon representation of the crystal structure of monomeric water-soluble PFO (5) shows locations of E167 in D1 and A215 in D3 that were substituted with Cys and labeled with BODIPY. A cross-bar indicates locations of the two residues that formed an intramolecular disulfide bond after substitution by Cys; a star indicates the location of the F318A mutation. The image was generated by using molscript. (B) Cartoons of potential domain rearrangements as PFO first binds to the membrane and then oligomerizes before forming the inserted pore complex.
The crystallized PFO monomer is ≈115 × 30 Å (Fig. 1 A) (5). Because D4 remains peripheral to the bilayer throughout pore formation and is oriented with its long axis nearly perpendicular to it (8), PFO may bind to the target membrane surface in an “end-on” orientation (Fig. 1B, i) and interact with the membrane only via the hydrophobic loops of D4. The polypeptide stretches in D3 that eventually form TMH1 and TMH2 would then be located >40 Å above the membrane surface before insertion (Fig. 1 A), and major conformational changes would be necessary to bring the TMHs in D3 to the bilayer surface for insertion. Such changes are compatible with atomic force microscopy (AFM) data that show a large difference in the height above the membrane surface of the prepore complex (113 ± 5 Å) and the inserted pore complex (73 ± 5 Å) (13). This study also indicated, consistent with earlier work (12), that the polypeptide segments destined to form the TMHs do not contact the bilayer at the prepore stage of pore formation. Therefore, the AFM data suggest that the PFO molecule undergoes a “vertical collapse” on the membrane surface during the prepore-to-pore transition as the TMHs are inserted into the membrane bilayer.
Although the AFM data reveal the existence of major structural changes in PFO during the prepore-to-pore transition, the domain specificity of the rearrangement was not addressed. Moreover, the AFM study did not examine the topography of membrane-bound PFO before oligomerization. Thus, we sought here to provide specific information about the nature of the PFO-membrane complex before oligomerization, as well as to monitor the topography of two PFO domains above the membrane surface at every step of the pore formation process. Specifically, what is the orientation of PFO when it first binds to the membrane surface? Does it bind to the membrane surface in an end-on or a “flat” orientation (Fig. 1B, i or ii)? What domain-specific conformational changes occur during the transition between the membrane-bound monomer and the membrane-bound oligomer? How far from the membrane surface are the D3 TMHs before insertion? To address these questions, we used a FRET approach that monitors the distance between the membrane surface and different domains of PFO at each stage of pore formation.
Methods
Fluorescent rPFO Derivatives. DNA encoding PFO(C459A), the cysteine-less derivative of PFO (termed rPFO; ref. 9), was site-specifically mutated by using QuikChange according to the manufacturer's protocol (Stratagene). Substituting Cys for residues 319 and 334 yielded a derivative with an intramolecular disulfide bond, termed rPFODS. The primary sequence of each construct was confirmed by DNA sequencing. The rPFO derivatives, overexpressed in Escherichia coli BL21(DE3)pLysS (Invitrogen), were purified and labeled with N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-S-indacene-3-yl)methyl)iodoacetamide (BODIPY; Molecular Probes) (10). BODIPY-labeled rPFO was separated from free dye by gel filtration (Sephadex G-50; i.d., 1.5 × 25 cm) in buffer A (50 mM Hepes, pH 7.5/100 mM NaCl) and stored in buffer A plus 10% (vol/vol) glycerol at -80°C. Labeling efficiency was 25-60% with molar absorptivity coefficients of 74,260 M-1cm-1 at 280 nm and 76,000 M-1cm-1 at 502 nm for PFO and BODIPY, respectively.
Vesicles. Liposomes containing 45 mol percentage of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids) and 55 mol percentage of cholesterol (Steraloids) were prepared by extrusion as described in ref. 12. For acceptor-containing vesicles, 1, 1.5, or 2 mol percentage of the total lipid was replaced with lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Rh-PE; Molecular Probes). The surface density of Rh-PE molecules in acceptors per square angstrom, σ, was calculated by assuming that each cholesterol and phospholipid molecule occupies 37 and 48 Å2 of surface area, respectively, in an equimolar cholesterol-phospholipid bilayer (14). This lipid mixture is in a homogeneous liquid-ordered phase at 25°C (34).
Spectral Measurements. Steady-state emission intensity, anisotropy, FRET, and lifetime measurements were done at 25°C in buffer A, as described (15, 16). The λex and λem values were 490 nm (2 nm bandpass) and 510 nm (4 nm) for BODIPY-labeled rPFO, and 570 nm (2 nm) and 590 nm (2 nm) for Rh-PE. The quantum yield, QD, of the donor dye (D) in the absence of the acceptor dye (A) was determined experimentally as described in refs. 15 and 16 by using a 460-nm excitation and corrected emission spectra (470-700 nm), yielding values of 0.65 and 0.74 for water-soluble and oxidized rPFODS(E167C-BODIPY) and rPFODS(A215C-BODIPY), respectively. Because the fluorescence lifetimes of the BODIPY-labeled rPFO derivatives (5.0 ± 0.2 ns) did not change appreciably after incubation with cholesterol-containing membranes, the quantum yields of the BODIPY dyes attached to these positions do not change as the protein progresses through all stages of pore formation. The spectral overlap integral, JDA, was determined to be 2.05 × 1015 M-1cm-1nm4 (15, 16) by using a molar extinction coefficient of 80,000 M-1cm-1 at 567 nm for Rh-PE, the Rh-PE absorbance spectrum in cholesterol-containing membranes, and the corrected emission spectrum of the BODIPY dye in rPFODS(A215C-BODIPY) in buffer A. The emission spectra of all BODIPY-labeled rPFO derivatives had the same shape (λmax = 511 nm; Fig. 2) and yielded the same JDA values at all stages of pore formation. R0, the distance between D and A that yields a FRET efficiency of 50%, equals [(8.79 × 10-5)QDJDAn-4κ2]1/6, where R0 is in angstroms, n is the refractive index of the medium between donor and acceptor, and κ2 is a geometric factor that depends on the relative orientation of the D and A dyes. R0 was 52 or 53 Å for FRET from BODIPY at rPFO residue 167 or 215, respectively, to Rh-PE in cholesterol-containing membranes, assuming values of 1.4 for n and 2/3 for κ2 (15-18).
Fig. 2.

Spectral overlap. The corrected emission spectrum of 6 nM rPFODS(A215C-BODIPY) (solid line) and the absorbance spectrum of Rh-PE (2 mol percentage) (dotted line) in cholesterol-containing vesicles (0.5 mM total lipid) are shown.
FRET Measurements. Four biochemically equivalent samples were prepared in parallel: sample D (D only) contained 6 nM BODIPY-labeled rPFO mutant, 94 nM corresponding unlabeled rPFO mutant, and cholesterol-containing vesicles lacking Rh-PE; sample DA (D plus A) contained the same protein mixture as in D and cholesterol-containing vesicles containing Rh-PE; sample A (A only) contained 100 nM unlabeled rPFO mutant and vesicles containing Rh-PE; and the blank (B) sample contained 100 nM unlabeled rPFO mutant and vesicles lacking Rh-PE. In all four samples, the total lipid concentration of the cholesterol-containing membranes was 100 μM, and the concentration of Rh-PE was the same in DA and A. All samples were incubated at 37°C for 15 min to permit complete binding of rPFO derivatives to (and, in some cases, insertion into) the cholesterol-containing membranes before spectral measurements at 25°C.
The net intensity of D, DA, or A (FD, FDA, and FA, respectively) was obtained by subtracting the B signal (FB). The B signal never exceeded 1% of the intensity of the D or DA samples. To correct for any signal in the DA sample caused by direct excitation of the acceptor, FA was subtracted from FDA. Making the reasonable assumption that the presence of D does not alter the absorptivity of the distant As, the ratio of the donor quantum yields in the absence and presence of the acceptor is then given by QD/QDA = FD/(FDA - FA).
Distance of Closest Approach. When the extent of energy transfer between Ds in one infinite plane and randomly and uniformly distributed As in a parallel infinite plane is small, L, the distance of closest approach between the Ds and As (i.e., the distance between the two planes) is given by the following (19):
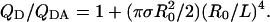 |
[1] |
The combined QD/QDA values from multiple independent experiments were plotted as a function of 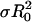 (Fig. 3), and linear least-squares regression analysis was used to determine the slope. Because use of Eq. 1 is justified only when L > R0 (20), the extent of energy transfer to As at the inner surface of the 50-Å-thick phospholipid bilayer is negligible here and has not been included in our calculations.
(Fig. 3), and linear least-squares regression analysis was used to determine the slope. Because use of Eq. 1 is justified only when L > R0 (20), the extent of energy transfer to As at the inner surface of the 50-Å-thick phospholipid bilayer is negligible here and has not been included in our calculations.
Fig. 3.

Dependence of energy transfer upon acceptor density. QD/QDA values for BODIPY-labeled D1 (A) or D3 (B) at different stages of pore formation and at three different acceptor densities. Best-fit lines were required to go through 0, 1. No line is shown for the pore complex data in B because L ≪ R0 and Eq. 1 does not apply.
Results
Rationale. D4 is responsible for membrane recognition and the initial binding of PFO to the membrane bilayer (7). Furthermore, D4 projects nearly perpendicularly from the bilayer surface in the membrane-inserted PFO oligomer (8). These results are consistent with the notion that the water-soluble monomer arrives end-on at the membrane surface and is initially anchored by D4 with the long axis of PFO perpendicular to the plane of the membrane bilayer, as shown in Fig. 1B (i). However, because the TMHs of PFO originate from the spatially distant D3, it is also possible that PFO might bind to the membrane so that D3 is positioned close to the membrane surface, as shown in Fig. 1B (ii). Moreover, these models are not mutually exclusive because there is a distinct possibility that PFO initially anchors to the membrane in an end-on orientation and then undergoes conformational changes that bring D3 into close proximity with the membrane surface for TMH insertion. Hence, to identify conformational changes in PFO during pore formation, we have directly determined the locations of D1 and D3 relative to the membrane surface at different stages of pore formation by using FRET.
FRET is an excellent method for identifying conformations and detecting conformational changes, as well as for measuring the magnitude of a conformational change within a distance range of 20-100 Å (21). The typical FRET experiment requires two fluorescent dyes, a D and an A, that are each located at a specific site in the same or different molecule(s). After excitation by the absorption of a photon, an excited D can nonradiatively transfer its excited-state energy to an A with appropriate spectral properties. The efficiency of this energy transfer depends primarily on the extent of the overlap between the emission of D and the absorption spectra of A (Fig. 2), the relative orientation of the D and A transition dipoles, and the distance between D and A. Donor emission intensity is reduced by FRET, and the magnitude of this decrease is used to measure the extent of energy transfer. FRET has been used primarily to measure point-to-point distances between two sites within a molecule or between two molecules (e.g., refs. 16 and 17).
A variation of the FRET technique allows one to measure the distance between two parallel planes. When a protein binds to a vesicle, the membrane surface forms one plane. Charged As can be localized at the aqueous-membrane interface by attachment to a phospholipid molecule (here, Rh-PE with a rhodamine attached to the headgroup of phosphatidylethanolamine). If all membrane-bound proteins adopt the same conformation, Ds covalently attached to the same single site on each protein will be located at the same height above the membrane for all proteins, thereby creating a second plane. The distance of closest approach between the two planes (i.e., the height of the D above the membrane surface) can then be quantified by using analytical expressions that integrate FRET from Ds in one plane to As that are distributed randomly and uniformly on the membrane surface. We used this approach to determine the locations above the membrane surface of the active sites of several membrane-bound blood-coagulation enzymes and to quantify coagulation cofactor-dependent alterations in the locations that appear to be functionally important (e.g., refs. 18, 22, and 23).
PFO Derivatives Trapped at Different Stages of Pore Formation. We have shown (9) that an intramolecular disulfide bond engineered between Cys residues substituted at positions 319 and 334 in D3 traps the PFO (termed rPFODS here to denote the presence of the intramolecular disulfide bond) as a membrane-bound monomer in the absence of reducing agent. To determine the locations of D1 and D3 relative to the membrane surface at the membrane-bound monomer stage of pore formation, we introduced into rPFODS a third Cys residue located either at position 167 in D1 or at position 215 in TMH1 of D3 (Fig. 1 A), and we labeled this third free Cys with BODIPY to yield rPFODS(E167C-BODIPY) or rPFODS(A215C-BODIPY), respectively. In the presence of the reducing agent DTT, these intramolecularly disulfide-bonded molecules readily formed pores in cholesterol-containing membranes (data not shown) and, hence, were fully functional in the absence of the disulfide bond.
To block PFO insertion into the bilayer at the prepore stage of pore formation, we used a point mutant in PFO (F318A) that prevented TMH insertion and, thereby, arrested the PFO oligomer in the prepore state (9). In the F318A background, a single Cys was introduced at either position 167 or 215 in two separate rPFO mutants and labeled with BODIPY to yield rPFO(F318A E167C-BODIPY) and rPFO(F318A A215C-BODIPY), respectively.
Topography of PFO as a Membrane-Bound Monomer. To determine whether the PFO monomer initially binds to the membrane in an end-on or a flat orientation (Fig. 1 B, i or ii), we measured FRET between Rh-PE molecules located at the membrane surface and rPFODS(E167C-BODIPY) that was trapped in the monomeric state after binding to the membrane. When this mutant was bound to Rh-PE-containing membranes in the absence of DTT, essentially no energy transfer was observed from position 167 in D1 to Rh-PE at the membrane surface even at high acceptor density (Fig. 3A). Thus, the distance of closest approach of the D1 BODIPY probe to the membrane surface, L, is >110 Å. Because a flat orientation (Fig. 1B, ii) would have positioned the D1 BODIPY dye much closer to the membrane surface where significant energy transfer would have been observed, the PFO molecule must be stably bound to the membrane surface as a monomer in an end-on orientation (Fig. 1B, i).
We then determined the location of the D3 TMH segments in the membrane-bound PFO monomer relative to the membrane surface. Data from collisional quenching experiments indicate that the TMH residues do not come into contact with the hydrophobic core of the bilayer even at the prepore stage of pore formation (12, 13). However, very little is known about the positioning of these TMH segments relative to the membrane surface before their insertion into the bilayer. To ascertain the location of the TMH segments before membrane insertion, we chose a location (A215C) that would ultimately place the donor probe on the hydrophobic side of the TM β-barrel in the inserted pore complex. We then measured a significant extent of energy transfer between unreduced rPFODS(A215C-BODIPY) and Rh-PE (Fig. 3B), and the height of this residue in TMH1 above the membrane surface was determined to be ≈72 Å in the membrane-bound monomer (Table 1). Because QD/QDA was found to be directly proportional to σR20 over a range of σ values, as indicated by linear plots in Fig. 3, the approximation of Dewey and Hammes (19) (Eq. 1) can be used to calculate L. Thus, the TMHs are located far from the membrane surface at the initial stage of pore formation.
Table 1. Distance of closest approach determined by FRET.
| Domain | Membrane-bound state | Donor | L,* Å |
|---|---|---|---|
| D1 | Monomer | Oxidized rPFODS(E167C-BODIPY) | —† |
| D1 | Prepore | rPFO(F318A E167C-BODIPY) | 99 ± 2 |
| D1 | Pore | Reduced rPFODS(E 167C-BODIPY) | ≈65‡ |
| D3 | Monomer | Oxidized rPFODS(A215C-BODIPY) | 72 ± 2 |
| D3 | Prepore | rPFO(F318A A215C-BODIPY) | 62 ± 1 |
| D3 | Pore | Reduced rPFODS(A215C-BODIPY) | ND |
ND, not determined.
L was calculated as described in Methods. Average values and standard deviations are shown for six to nine independent measurements by using Rh-PE as the acceptor. The uncertainty shown is the experimental uncertainty of multiple measurements and not the uncertainty in κ2 or R0.
No energy transfer was observed.
This approximate L value was determined by using a reduced σ (see text).
Topography of PFO in the Prepore Complex. To ascertain whether the topography of the membrane-bound PFO monomer changes significantly upon association with other monomers to form a prepore complex, we next measured the extent of energy transfer between Rh-PE and rPFO(F318A E167C-BODIPY) trapped at the prepore stage of pore formation. In this case, a small amount of energy transfer was observed (Fig. 3A), indicating that the location and/or orientation of D1 and its probe relative to the membrane surface had changed somewhat upon forming an oligomeric prepore complex. The increased FRET efficiency could be explained either by a translational movement by D1 toward the membrane surface and/or by a rotation of D1 and the BODIPY probe relative to the membrane surface that changes κ2 (see below). Whatever the case, the data indicate that the PFO molecule is still oriented in a largely end-on orientation (Fig. 1B, i) because the height of the BODIPY probe in D1 relative to the membrane surface is ≈99 Å even in the prepore complex (Table 1).
To determine whether monomer association and prepore formation cause the D3 TMH1 segment to move close to the membrane surface in preparation for pore formation, we measured the extent of energy transfer between rPFO(F318A A215C-BODIPY) and Rh-PE, and we found that rPFO(F318A A215C-BODIPY) had a higher FRET efficiency in the prepore oligomer than did rPFODS(A215C-BODIPY) in the monomeric membrane-bound state (Fig. 3B). By using Eq. 1, the probe in TMH1 was determined to be ≈62 Å above the membrane surface in the prepore complex (Table 1).
The increased FRET efficiency observed for the D3 probe upon PFO association to form the oligomeric prepore complex is most likely explained by a translational movement of the D3 TMHs toward the membrane surface because the anisotropy values for the BODIPY dye do not change significantly between the membrane-bound monomer and the prepore complex (rD = 0.21 and 0.20, respectively). Thus, in the prepore oligomer, the D3 TMH1 segment appears to be located closer to the membrane surface than in the membrane-bound monomer.
Topography of PFO in the Pore Complex. To estimate the location of D1 in the pore complex, we measured the extent of energy transfer between Rh-PE and rPFODS(E167C-BODIPY) under reducing conditions (+DTT) that readily allowed pore formation (9). Under these conditions, the measured FRET efficiency indicated that the D1 probes in the membrane-inserted pore complex and Rh-PE at the membrane surface (Fig. 3A) were separated by ≈74 Å.
However, the L value described above actually underestimates the extent of D1 movement toward the membrane because the Rh-PE is no longer distributed uniformly over the membrane after the pore is formed and the internal lipid “plug” is (by unknown mechanisms) lost to create the pore. A D in a membrane-inserted PFO oligomer will transfer energy only to Rh-PE molecules that are located within ≈110 Å (≈2 R0), and no Rh-PE will be present in the newly created 300-Å-diameter aqueous pore (1). If the D in D1 is positioned ≈65 Å above the surface (see below), only Rh-PE molecules directly below D at the surface within a circle of radius ≈90 Å will act as acceptors for FRET. To estimate the effect that pore formation would have on the surface density of Rh-PE around a D1 BODIPY probe in a pore complex, we calculated the fraction of the area of a circle of radius 90 Å that would overlap with a circle of radius 150 Å if D were located at the edge of the pore (i.e., the center of the smaller circle was assumed to lie on the circumference of the larger circle). The extent of overlap under these conditions was 44%. Therefore, it is reasonable to assume that the Rh-PE σ around each BODIPY-rPFO in an inserted pore complex is reduced on the order of 44%. By using this smaller σ value, L turns out to be ≈65 Å (Table 1). Although this number is only an estimate because of the assumptions made in its calculation, it is clear that the probe in D1 is located far from the membrane surface in the PFO pore complex (Table 1). It is also clear that there is a major change in D1 location relative to the membrane surface upon pore formation, because the D1 probe appears to move (assuming that the change in FRET efficiency is solely due to translational movement of D1) from ≈99 Å above the membrane surface to a height of ≈65 Å above the surface (Table 1).
As noted earlier, the location of residue 215 in the pore complex is fixed and known (11). Residue 215 in TMH1 faces the hydrophobic core of the membrane bilayer in the TM β-barrel and is located near the membrane surface. A D attached to 215 in the pore complex will be located within the bilayer, where L ≪ R0 and, hence, will transfer significant energy to As located on both membrane surfaces. Under these circumstances, the use of Eq. 1 is invalid. However, this limitation is unimportant here because our previous studies have determined where A215 is located in the pore complex.
Dye Orientation (κ2) Effects and Uncertainties in L. The R0 values were calculated by assuming that the transition dipoles of D and A are dynamically randomized during the excited-state lifetime of D (κ2 = 2/3). However, BODIPY attached to PFO and rhodamine attached to PE do not rotate with complete freedom, so there is uncertainty in R0 and, hence, L. Although κ2 cannot be determined experimentally in a nonrigid sample, the theoretical upper and lower limits of κ2 and, hence, R0 can be calculated from the measured anisotropy values of the membrane-bound donor (rD) and the acceptor (rA) that indicate the freedom of rotation of the dyes in the sample (24). For membrane-bound PFO in the monomer, prepore, and pore states, rD was 0.21, 0.20, and 0.22 at position 215 and 0.20, 0.15, and 0.12 at position 167, respectively (±0.01). Because rA was 0.26 ± 0.01, the maximum uncertainty in R0 due to κ2 is -22% to +27%. However, assuming a value of κ2 = 2/3 usually yields distances that differ by <10% from those determined by crystallography when such comparisons can be made. This conclusion is true for both point-to-point (e.g., refs. 17, 21, 25, and 26) and plane-to-plane (18, 23, 27, 28) FRET measurements. Furthermore, because Rh-PE is oriented randomly in the plane of the membrane in the plane-to-plane FRET measurements done here, the uncertainty in R0 due to orientation effects is further reduced.
Thus, the FRET-determined L values may be accurate or may differ slightly from the actual heights of D above the membrane surface at different stages of PFO pore formation because of the orientational uncertainty. Any nonuniformity in the distribution of Rh-PE at the membrane surface due to the high cholesterol content could also alter the measured L values. However, the same vesicles were added to each FRET sample, so if there was an unusual κ2 or σ value due to Rh-PE, all of the L calculations were affected in the same way. For that reason, we focused primarily on the relative, not absolute, distances, specifically on the changes in the locations of D1 and D3 during pore formation.
Discussion
Our FRET measurements have provided four important insights into the topography of membrane-bound PFO at different stages of pore formation. First, in the membrane-bound monomeric state, the PFO molecule is anchored to the membrane in an end-on orientation, with the long axis of the molecule nearly perpendicular to the plane of the bilayer. Second, monomer-monomer association and the formation of a prepore complex do not greatly alter the domain arrangement of individual PFO molecules relative to the membrane surface. Third, the D3 polypeptide segments that constitute the TMHs are far above the membrane surface even in the prepore complex, before insertion. Fourth, major changes in the overall topography and domain arrangement of the PFO molecule occur only during the prepore-to-pore transition of the oligomer.
FRET measurements between D1 of membrane-bound monomeric PFO and the membrane surface show conclusively that the elongated monomer is bound to the membrane in an end-on orientation. Despite the fact that PFO is anchored to the bilayer by only a few residues at the tip of D4 that are located at one end of the molecule (7, 8), D1 is stably positioned at the opposite end of the molecule >100 Å above the membrane surface. Also, FRET measurements between D3 of membrane-bound monomeric PFO and the membrane surface show that TMH1 is ≈72 Å above the membrane (Table 1). Thus, in the membrane-bound monomer stage of pore formation, the PFO molecule projects radially, or nearly radially, from the vesicle surface.
This structural arrangement, with an elongated protein projecting stably from a small “footprint” on the membrane surface, is unusual. However, it is not unprecedented. Proteins involved in effecting and regulating blood coagulation are elongated, bind reversibly to appropriate membranes with small footprints, and extend approximately perpendicularly from the membrane with the enzyme active sites located 70-95 Å above the surface (e.g., refs. 15, 18, 22, and 23).
The height of D1 above the membrane surface is slightly less in the oligomeric prepore complex than in the monomer. The increased FRET efficiency may be due to a more favorable alignment of the donor and acceptor transition dipoles and/or some D1 movement toward the membrane. Whatever the case, the small magnitude of the FRET efficiency demonstrates that even at the prepore stage of pore formation, D1 is still located far above the membrane surface, and PFO is bound in an end-on orientation in the prepore complex (Fig. 1B, i).
Because PFO is presumably poised in this obligatory prepore intermediate (12) to insert into the bilayer, an interesting and important structural question is where the D3 TMHs are located in the prepore complex. Are they positioned near or at the membrane surface just before insertion? Heuck et al. (12) showed that residues in the TMHs were not exposed to the nonpolar interior of the membrane, but they did not further characterize TMH positioning. Here, the FRET approach reveals that TMH1 is ≈62 Å above the membrane surface in the prepore complex. This substantial height above the surface reveals that the TMHs in the prepore complex, supposedly “poised” to insert into the bilayer, must still move a substantial distance to enter the membrane. For example, the fluorophore attached to TMH1 at position 215 must move from 62 Å above the membrane surface to a position located within the hydrophobic core of the bilayer. This major change in topography presumably explains, at least in part, why the prepore-to-pore conversion involves a substantial transition energy barrier that cannot be surmounted at low temperature (12) or with some PFO mutants (e.g., ref. 9). The energy is presumably necessary to power the large conformational rearrangements that move the D3 TMHs to the membrane surface and then into the bilayer during the prepore-to-pore transition.
Although TMH1 moves >60 Å during the prepore-to-pore transition, D1 appears to move ≈34 Å closer to the membrane surface (Table 1). This number is only an estimate because the loss of As during pore formation complicates the determination of σ in the latter case. However, the FRET measurements agree very well with the heights of the prepore and pore complexes determined by using AFM. Czajkowsky et al. (13) reported a distance of 73 ± 5 Å between the top of the PFO pore complex and the membrane surface, which is very similar to the 65 Å distance of closest approach between the D1 fluorophore and the As at the membrane surface (Table 1). The observed changes in the height of the PFO complex during the prepore-to-pore transition were also similar: ≈40 Å by AFM (13) and ≈34 Å by FRET (Table 1). Thus, two independent approaches agree on the magnitude of the structural change in the overall height of the PFO complex during pore formation. Also, the FRET approach reveals that some PFO domain movements are significantly larger than the AFM-detected structural changes.
In summary, the water-soluble PFO monomer (Fig. 4, i) initially anchors itself to a cholesterol-containing target membrane in an end-on orientation, with its long axis perpendicular to the plane of the membrane bilayer (Fig. 4, ii). At this stage, TMH-forming D3 segments are located ≈72 Å above the membrane surface (Fig. 4, ii). Monomer-monomer association and subsequent prepore formation slightly lowers the heights of both D1 and D3 above the membrane surface (Fig. 4, iii). Concomitant with the prepore-to-pore transition, the molecule experiences a marked reduction in height above the membrane surface as D1 moves from ≈100 Å in the prepore complex to ≈65 Å in the membrane-inserted pore complex and D3 inserts into the nonpolar core of the bilayer as part of the TM β-barrel (Fig. 4, iv). Because D4 projects perpendicularly from the membrane even in the pore complex (8), the D2 β-structure presumably bends or reconfigures to allow D1 and D3 to move 40-60 Å toward and into the membrane. Thus, major topographical changes in PFO structure accompany pore formation.
Fig. 4.

FRET-detected changes in PFO topography. Based on domain-specific FRET measurements, the orientation of membrane-bound PFO at different stages before membrane insertion is shown and described in the text. For simplicity, only one monomer in the prepore (iii) and pore (iv) complexes is shown.
The changes in protein conformation that accompany the transition of other pore-forming toxins from a water-soluble to a membrane-inserted state appear to be relatively modest (29-31). For example, in the case of the Staphylococcus aureus α-hemolysin, the TM β-hairpin that each monomer contributes to the 14-stranded β-barrel originates simply as an extension of its core β-sheet, with minimal changes to either the secondary structure or the topography of its oligomeric prepore complex on the membrane surface (30, 32, 33). However, as shown here, the changes in both secondary structure and topography are substantial for PFO during pore formation, thereby demonstrating that the mechanism of CDC pore formation differs substantially from that described for other toxins.
Acknowledgments
We thank Dr. John Flanagan for early experiments. This work was supported by National Institutes of Health Grant AI 37657 and the Robert A. Welch Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PFO, perfringolysin O; CDC, cholesterol-dependent cytolysin; Dn, domain n; TMH, transmembrane β-hairpin; AFM, atomic force microscopy; BODIPY, N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-S-indacene-3-yl)methyl)iodoacetamide; Rh-PE, lissamine rhodamine 1,2, dihexadecanoyl-sn-glycero-3-phosphoethanolamine; D, donor dye; A, acceptor dye; DA, D plus A; B, blank.
References
- 1.Olofsson, A., Hebert, H. & Thelestam, M. (1993) FEBS Lett. 319, 125-127. [DOI] [PubMed] [Google Scholar]
- 2.Alouf, J. E. (1999) in The Comprehensive Sourcebook of Bacterial Protein Toxins, eds. Alouf, J. E. & Freer, J. H. (Academic, London), pp. 443-456.
- 3.Heuck, A. P., Tweten, R. K. & Johnson, A. E. (2001) Biochemistry 40, 9065-9073. [DOI] [PubMed] [Google Scholar]
- 4.Tweten, R. K., Parker, M. W. & Johnson, A. E. (2001) Curr. Top. Microbiol. Immunol. 257, 15-33. [DOI] [PubMed] [Google Scholar]
- 5.Rossjohn, J., Feil, S. C., Mckinstry, W. J., Tweten, R. K. & Parker, M. W. (1997) Cell 89, 685-692. [DOI] [PubMed] [Google Scholar]
- 6.Heuck, A. P. & Johnson, A. E. (2002) Cell Biochem. Biophys. 36, 89-102. [DOI] [PubMed] [Google Scholar]
- 7.Heuck, A. P., Hotze, E. M., Tweten, R. K. & Johnson, A. E. (2000) Mol. Cell 6, 1233-1242. [DOI] [PubMed] [Google Scholar]
- 8.Ramachandran, R., Heuck, A. P., Tweten, R. K. & Johnson, A. E. (2002) Nat. Struct. Biol. 9, 823-827. [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran, R., Tweten, R. K. & Johnson, A. E. (2004) Nat. Struct. Mol. Biol. 11, 697-705. [DOI] [PubMed] [Google Scholar]
- 10.Shepard, L. A., Heuck, A. P., Hamman, B. D., Rossjohn, J., Parker, M. W., Ryan, K. R., Johnson, A. E. & Tweten, R. K. (1998) Biochemistry 37, 14563-14574. [DOI] [PubMed] [Google Scholar]
- 11.Shatursky, O., Heuck, A. P., Shepard, L. A., Rossjohn, J., Parker, M. W., Johnson, A. E. & Tweten, R. K. (1999) Cell 99, 293-299. [DOI] [PubMed] [Google Scholar]
- 12.Heuck, A. P., Tweten, R. K. & Johnson, A. E. (2003) J. Biol. Chem. 278, 31218-31225. [DOI] [PubMed] [Google Scholar]
- 13.Czajkowsky, D. M., Hotze, E. M., Shao, Z. & Tweten, R. K. (2004) EMBO J. 23, 3206-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lecuyer, H. & Dervichian, D. G. (1969) J. Mol. Biol. 45, 39-57. [DOI] [PubMed] [Google Scholar]
- 15.Mutucumarana, V. P., Duffy, E. J., Lollar, P. & Johnson, A. E. (1992) J. Biol. Chem. 267, 17012-17021. [PubMed] [Google Scholar]
- 16.Woolhead, C., McCormick, P. J. & Johnson, A. E. (2004) Cell 116, 725-736. [DOI] [PubMed] [Google Scholar]
- 17.Johnson, A. E., Adkins, H. J., Matthews, E. A. & Cantor, C. R. (1982) J. Mol. Biol. 156, 113-140. [DOI] [PubMed] [Google Scholar]
- 18.Yegneswaran, S., Wood, G. M., Esmon, C. T. & Johnson, A. E. (1997) J. Biol. Chem. 272, 25013-25021. [DOI] [PubMed] [Google Scholar]
- 19.Dewey, T. G. & Hammes, G. G. (1980) Biophys. J. 32, 1023-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baird, B. & Holowka, D. (1985) Biochemistry 24, 6252-6259. [DOI] [PubMed] [Google Scholar]
- 21.Stryer, L. (1978) Annu. Rev. Biochem. 47, 819-846. [DOI] [PubMed] [Google Scholar]
- 22.Husten, E. J., Esmon, C. T. & Johnson, A. E. (1987) J. Biol. Chem. 262, 12953-12961. [PubMed] [Google Scholar]
- 23.McCallum, C. D., Hapak, R. C., Neuenschwander, P. F., Morrissey, J. H. & Johnson, A. E. (1996) J. Biol. Chem. 271, 28168-28175. [DOI] [PubMed] [Google Scholar]
- 24.Dale, R. E., Eisinger, J. & Blumberg, W. E. (1979) Biophys. J. 26, 161-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu, P. & Brand, L. (1992) Biochemistry 31, 7939-7947. [DOI] [PubMed] [Google Scholar]
- 26.Yusupov, M. M., Yusupova, G. Z., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H. D. & Noller, H. F. (2001) Science 292, 883-896. [DOI] [PubMed] [Google Scholar]
- 27.Banner, D. W., D'Arcy, A., Chene, C., Winkler, F. K., Guha, A., Konigsberg, W. H., Nemerson, Y. & Kirchhofer, D. (1996) Nature 380, 41-46. [DOI] [PubMed] [Google Scholar]
- 28.Adams, T. E., Hockin, M. F., Mann, K. G. & Everse, S. J. (2004) Proc. Natl. Acad. Sci. USA 101, 8918-8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petosa, C., Collier, R. J., Klimpel, K. R., Leppla, S. H. & Liddington, R. C. (1997) Nature 385, 833-838. [DOI] [PubMed] [Google Scholar]
- 30.Montoya, M. & Gouaux, E. (2003) Biochim. Biophys. Acta 1609, 19-27. [DOI] [PubMed] [Google Scholar]
- 31.Lacy, D. B., Wigelsworth, D. J., Melnyk, R. A., Harrison, S. C. & Collier, R. J. (2004) Proc. Natl. Acad. Sci. USA 101, 13147-13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song, L., Hobaugh, M. R., Shustak, C., Cheley, S., Bayley, H. & Gouaux, J. E. (1996) Science 274, 1859-1866. [DOI] [PubMed] [Google Scholar]
- 33.Olson, R., Nariya, H., Yokota, K., Kamio, Y. & Gouaux, E. (1999) Nat. Struct. Biol. 6, 134-140. [DOI] [PubMed] [Google Scholar]
- 34.de Almeida, R. F. M., Fedorov, A. & Prieto, M. (2003) Biophys. J. 85, 2406-2416. [DOI] [PMC free article] [PubMed] [Google Scholar]