

Abstract
Functional innovation at the protein level is a key source of evolutionary novelties. The constraints on functional innovations are likely to be highly specific in different proteins, which are shaped by their unique histories and the extent of global epistasis that arises from their structures and biochemistries. These contextual nuances in the sequence-function relationship have implications both for a basic understanding of the evolutionary process and for engineering proteins with desirable properties. Here, we have investigated the molecular basis of novel function in a model member of an ancient, conserved, and biotechnologically relevant protein family. These Major Facilitator Superfamily sugar porters are a functionally diverse group of proteins that are thought to be highly plastic and evolvable. By dissecting a recent evolutionary innovation in an α-glucoside transporter from the yeast Saccharomyces eubayanus, we show that the ability to transport a novel substrate requires high-order interactions between many protein regions and numerous specific residues proximal to the transport channel. To reconcile the functional diversity of this family with the constrained evolution of this model protein, we generated new, state-of-the-art genome annotations for 332 Saccharomycotina yeast species spanning approximately 400 million years of evolution. By integrating phylogenetic and phenotypic analyses across these species, we show that the model yeast α-glucoside transporters likely evolved from a multifunctional ancestor and became subfunctionalized. The accumulation of additive and epistatic substitutions likely entrenched this subfunction, which made the simultaneous acquisition of multiple interacting substitutions the only reasonably accessible path to novelty.
Keywords: maltose, maltotriose, lager yeast, gene conversion, gene duplication, subfunctionalization, MFS, sugar transporter
INTRODUCTION
Many key evolutionary innovations arise from changes to protein sequences that alter their function (Cheng 1998; Zhang et al. 2002; Clark et al. 2003; Dorus et al. 2004; Lunzer et al. 2005; Nielsen et al. 2005; Hoekstra et al. 2006; Christin et al. 2007; Yokoyama et al. 2008; Voordeckers et al. 2012; Projecto-Garcia et al. 2013; Kaltenbach et al. 2018; Jabłońska and Tawfik 2022). Occasionally, these changes stem from dramatic mutational events, including the creation of highly novel coding sequences by gene conversion or ectopic recombination resulting in chimeric proteins (Long and Langley 1993; Nurminsky et al. 1998; Wang et al. 2000; Long et al. 2003; Patthy 2003; Zhang et al. 2004; Ciccarelli et al. 2005; Arguello et al. 2006; Rogers et al. 2010; Rogers and Hartl 2012; Leffler et al. 2017; Méheust et al. 2018; Baker and Hittinger 2019; Brouwers, Gorter de Vries, et al. 2019; Smithers et al. 2019; Baker et al. 2022). While gene conversion can theoretically accelerate the rate of evolution (or even enable adaptation altogether) by bypassing deleterious intermediates, this effect is primarily attributable to the presence of a rugged fitness landscape (Kauffman and Levin 1987; HANSEN et al. 2000; Cui et al. 2002; Bittihn and Tsimring 2017). Such rugged landscapes are manifestations of epistasis in the genotypic combinations underlying the phenotypic map and are prevalent in some empirical systems (Wright 1931; Wright 1932; Maynard Smith 1970; Weinreich et al. 2005; Weinreich et al. 2006; Gong et al. 2013; Weinreich et al. 2013; De Visser and Krug 2014; Sarkisyan et al. 2016; Starr and Thornton 2016; Wu et al. 2016; Pokusaeva et al. 2019; Yi and Dean 2019; Nishikawa et al. 2021; Park et al. 2022; Meger et al. 2024; Metzger et al. 2024). For other proteins, the fitness landscape may be much smoother, meaning that stepwise mutations with additive effects can underlie functional evolution (Lunzer et al. 2005; Bridgham et al. 2006; Weinreich et al. 2006; Poelwijk et al. 2007; Campbell et al. 2016; Kaltenbach et al. 2018; Srikant et al. 2020). In cases where novel protein function is linked to gene conversion events between homologs, these observations therefore raise a fundamental question: are such dramatic mutational events required to evolve new function, or are they probabilistic shortcuts in the evolutionary process whose prevalence is a predictable function of their combined effect size and relative mutation rate? Answering this question has significant implications for understanding and predicting evolutionary trajectories, as well as for designing and engineering novel proteins with desirable functions.
Recently, several remarkably parallel cases of functional innovation have been linked directly or speculatively to gene conversion events in an ecologically and biotechnologically relevant protein family: maltose transporters in Saccharomyces yeasts (Baker and Hittinger 2019; Brouwers, Gorter de Vries, et al. 2019; Hatanaka et al. 2022). This protein family consists of transporters similar to the Saccharomyces cerevisiae Mal31 protein, which has high specificity and high affinity for the disaccharide maltose, which contains two glucose moieties (Cheng and Michels 1991; Stambuk and Araujo 2001; Salema-Oom et al. 2005; Alves et al. 2008; Brown et al. 2010). Mal31-like proteins are encoded in nearly all genomes of Saccharomyces and some closely related species, and they are frequently encoded by multiple paralogs within each genome.
Maltose uptake is also mediated by a second family of proteins, which are related to S. cerevisiae Agt1. In contrast to the Mal31-like proteins, Agt1 is a generalist α-glucoside transporter with a broad substrate range, but it has generally lower affinity for those substrates (Han et al. 1995; Stambuk et al. 1999; Stambuk et al. 2000; Alves et al. 2008; Trichez et al. 2019). Notably, Agt1 can transport the glucose trisaccharide maltotriose, a molecule that is biochemically similar to maltose but contains a third glucose moiety. Although sometimes referred to as Mal11, Agt1 is a functionally distinct protein with ≈57% amino acid sequence identity to the Mal31-like proteins. In contrast to the Mal31-like proteins, Agt1-like proteins are rarer, both in presence and in paralog number, in the genomes of Saccharomyces yeasts and close relatives (Duval et al. 2010; Horak 2013).
The α-glucoside transporters (Agts) of Saccharomyces include the Agt1-like (“generalist”) and Mal31-like (“high-specificity”) proteins, as well as Mph2/3-like proteins (Day et al. 2002), which also have high specificity, albeit for the α-glucoside turanose (Brown et al. 2010). These Agts have been extensively studied due to their important role in the production of beer. Maltose and maltotriose are the two most abundant sugars in brewer’s wort (Meussdorfer and Zarnkow 2009), and their transport into the cell is the rate-limiting step in their fermentation (Zastrow et al. 2001; Horák 2013). The rarity of maltotriose transporters, such as Agt1, which almost always manifests as an inability to ferment this carbon source, therefore presents a barrier to the use of many non-domesticated yeasts in brewing applications.
This barrier is exemplified in Saccharomyces eubayanus, the wild, cold-tolerant parent of industrial lager-brewing hybrids (Libkind et al. 2011), whose development for commercial brewing is of great interest (Gibson et al. 2017; Hittinger et al. 2018; Cubillos et al. 2019). As almost all strains of S. eubayanus lack generalist Agts capable of transporting maltotriose (Brickwedde et al. 2018; Brouwers, Brickwedde, et al. 2019; Bergin et al. 2022), multiple attempts have been made to evolve maltotriose transporters de novo in S. eubayanus strains, using both mutagenesis (Brouwers, Gorter de Vries, et al. 2019) and adaptive laboratory evolution (Baker and Hittinger 2019). These experiments, performed independently in different backgrounds of S. eubayanus, yielded results that were as remarkable in their similarity as they were unexpected. In both cases, ectopic gene conversion between paralogous high-specificity (Mal31-like) maltose transporters without any native maltotriose transport capacity (Brickwedde et al. 2018; Baker and Hittinger 2019) resulted in chimeric proteins capable of transporting maltotriose.
Lending weight to the notion that recombination may be a common mechanism by which transporters in the high-specificity Agt family evolve new function, two newly discovered S. cerevisiae transporters (Hatanaka et al. 2022), as well as the Mty1 protein (Dietvorst et al. 2005; Salema-Oom et al. 2005), may possess signatures of more ancient gene conversion events (Brouwers, Gorter de Vries, et al. 2019). All these proteins transport maltotriose, but they cluster with Mal31-like proteins in phylogenetic analyses (Baker and Hittinger 2019; Hatanaka et al. 2022). Nonetheless, it remains unclear whether these dramatic mutational events are required for the evolution of novel function in this family or whether they are simply enriched due to the dynamic nature of the subtelomeric regions in which these genes reside (Mefford and Trask 2002; Fairhead and Dujon 2006; Gordon et al. 2009; Brown et al. 2010; Yue et al. 2017; Peter et al. 2018; Liu et al. 2019; O’Donnell et al. 2023).
The yeast α-glucoside transporters are H+ symporters belonging to the sugar porter family (TCDB: 2.A.1.1) of the Major Facilitator Superfamily (MFS), a vast, ubiquitous, and ancient group of transmembrane proteins present in all domains of life (Marger and Saier 1993; Pao et al. 1998; Saier 2000; Wang et al. 2020; Saier et al. 2021). Across great evolutionary distances, sugar porters share the highly characteristic MFS fold consisting of twelve transmembrane helices (TMHs) surrounding a hydrophilic central cavity that constitutes the transport channel (Abramson et al. 2003; Guan and Kaback 2006; Sun et al. 2012; Deng et al. 2014; Quistgaard et al. 2016; Bosshart and Fotiadis 2019; Kaback and Guan 2019; Paulsen et al. 2019; Drew et al. 2021). These TMHs are organized into two pseudosymmetrical six-helix bundles (N- and C-terminal), which are separated by a long intracellular linker (ICH domain). The transport channel is surrounded by four helices from each bundle, and TMHs stack tightly against their intra- bundle partners, with additional contacts between the N- and C-terminal domains at the inter-bundle interface. In S. cerevisiae Agt1, the sugar substrate and/or proton are thought to be bound primarily by charged residues projecting into this central cavity, which are conserved across fungal Agts (Henderson and Poolman 2017; Trichez et al. 2019). More generally, substrate affinity and specificity in MFS sugar transporters are mediated by extensive hydrogen bonding and occasionally by hydrophobic interactions between the sugar and the protein, as well as steric constraints that limit substrate accommodation; moreover, there is a growing appreciation for the fine-scale and occasionally cryptic contributions to affinity by residues within Van der Waals distance of the substrate (Kasahara et al. 1997; Kasahara and Kasahara 1998; Kasahara and Kasahara 2000; Guan and Kaback 2006; Kasahara et al. 2006; Guan et al. 2007; Kasahara et al. 2007; Kasahara et al. 2009; Kasahara and Kasahara 2010; Kasahara et al. 2011; Sun et al. 2012; Deng et al. 2014; Farwick et al. 2014; Deng et al. 2015; Bosshart and Fotiadis 2019; Kaback and Guan 2019; Drew et al. 2021; Guan and Hariharan 2021).
Nonetheless, the extensive and exquisite biochemical study of MFS sugar transporters has almost exclusively focused on the determinants of native substrate binding and affinity in extant proteins, while questions about how such proteins could evolve the capacity to transport a novel substrate de novo have been largely unaddressed. Understanding evolution-informed design principles in this protein family could enable the engineering of desirable properties in tractable proteins, with significant implications for industrial processes, including the fermentation of cellulosic and hemicellulosic biomass into next-generation biofuels and bioproducts (Ha et al. 2013; Farwick et al. 2014; Young et al. 2014; Turner et al. 2016; Hara et al. 2017; Oh et al. 2017; Casa-Villegas et al. 2018; Kim et al. 2018; Nijland et al. 2018; Nijland and Driessen 2020; Oh and Jin 2020; de Ruijter et al. 2020).
To this end, we aimed to dissect the molecular genetic basis of novel function in the chimeric S. eubayanus maltotriose transporter MalT434. MALT434 arose from an ectopic gene conversion event between genes encoding two paralogous maltose transporters, MalT3 and MalT4, which resulted in the replacement of approximately 230 base pairs of the MALT4 gene with the homologous portion of MALT3 (Baker and Hittinger 2019). Both MalT3 and MalT4 are members of the high-specificity maltose transporter family and incapable of transporting maltotriose (Brickwedde et al. 2018; Baker and Hittinger 2019), suggesting that intramolecular epistasis between their protein regions underlies the emergent maltotriose transport by MalT434. The translocated region of MALT3 encodes TMH 11 and portions of TMHs 10 and 12 (Fig. 1a), and it introduced 11 nonsynonymous mutations to the protein-coding sequence of MALT4 (Fig. 1b). All three proteins are predicted to have virtually identical structures across their entire folds (pairwise RMSD=0.955Å) and TMHs 10-12 (0.909Å, Fig. S1), suggesting that novel substrate transport might stem from a specific combination of substrate-interacting residues from distal protein regions in MalT434, rather than a global change to protein structure. In the simplest model, as few as a single interacting residue from each protein region could underlie the emergence of novel function, which would make the evolution of new function in this family predictable and tunable; in the most complex model, all 120 amino acid differences between the two parental transporters could contribute, which would render the evolution of new function incredibly difficult.
Figure 1.
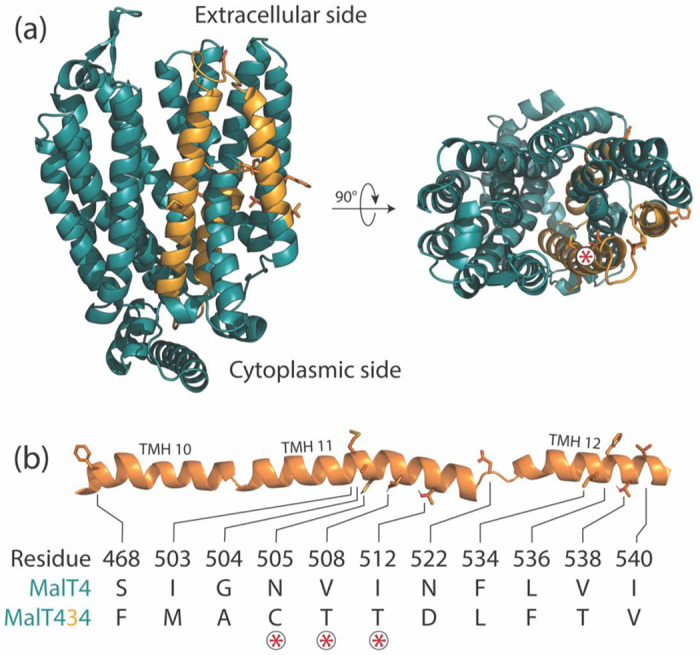
Architecture of a chimeric neofunctionalized α-glucoside transporter. (a) A structural model of the chimeric transporter MalT434 is shown from the side and top views, with alternating colors demarking regions contributed by different parental proteins. The top view is orientated looking down the transport channel. MalT3 side chains are drawn for the 11 substitutions between MalT4 and MalT434. The asterisk label marks the position of the three substitutions on a helical face that bounds the transport channel. (b) Schematic of mutations. The 11 substitutions between MalT4 and MalT434 are drawn as side chains along the cartoon secondary structure of the protein, with loops that connect transmembrane helices truncated for clarity. Polar hydrogens are shown. Asterisks mark the amino acids that face the transport channel.
Here, we show that the basis of maltotriose transport is remarkably complex in this model neofunctionalized transporter. Novel function is shaped by a combination of additive and non-additive interactions between as many as seven regions in the MalT4 backbone and six substitutions across TMHs 10 and 11. At one critical site, very few amino acids can support novel function, which further limits the evolutionary paths available to the wild-type protein; at other sites, these requirements are less stringent. We propose that, overall, novel substrate transport is enabled by widening the transport channel while simultaneously creating a favorable electrostatic environment for the bulkier trisaccharide molecule. Finally, we reconstruct the evolutionary history of the high-specificity and generalist yeast Agts and their relationships to other sugar porters; unexpectedly, we show that the specialist maltose transporters are likely derived and subfunctionalized from a generalist ancestor. This specialization likely involved a gradual refinement of the transport channel to specifically accommodate maltose with higher affinity, which makes the reacquisition of ancestral generalist function difficult to achieve. While our results indicate that rational engineering for novel substrate transport in this protein family is likely to be difficult, they also highlight the abundance and diversity of transporters in biotechnologically relevant yeast species, which could be readily mined for desirable functions that have been exquisitely refined over billions of years of evolution, as well as perhaps recombined into new functions.
RESULTS
High-order intramolecular interactions are required to evolve a novel function in maltose transporters
We first investigated the scope and complexity of intramolecular interactions shaping the emergence of novel function in MalT434. We coarsely defined functional protein regions as the twelve transmembrane helices (TMHs), the intracellular (ICH) domain, and the partially unstructured intracellular N- and C-terminal regions. We iteratively constructed novel chimeric genes encoding transporters from MalT3 and MalT4 components and tested their ability to support growth on maltotriose when expressed from the native MALT4 locus (Fig 2). Unsurprisingly, the C-terminal portion of MalT4 present in MalT434 was neither necessary (construct 1) nor sufficient (construct 17) for maltotriose transport; indeed, its replacement with the corresponding region of MalT3 improved growth on maltotriose by 15.3% (p = 5.3x10−4, Mann-Whitney U test). By contrast, replacement of TMHs 8 and 9 and the N-terminal half of TMH 10 with their MalT3 counterparts (construct 2) reduced growth by 11.6% compared to MalT434 (p = 0.184), while still supporting robust growth. Dissection of the region N-terminal to TMH 8 revealed that the key interaction enabling maltotriose transport occurs between TMHs 10 and 11 of MalT3 and TMH 7 from MalT4. While necessary, this region alone was not sufficient to enable maltotriose transport in every protein context. In addition to the epistatic interaction between TMHs 7, 10, and 11, growth on maltotriose required the presence of TMHs 1 and 2 from MalT4 in combination with the ICH domain from MalT3 (construct 7), or alternatively, one or more of TMH 5, TMH 6, and the ICH domain from MalT4 (construct 15).
Figure 2.
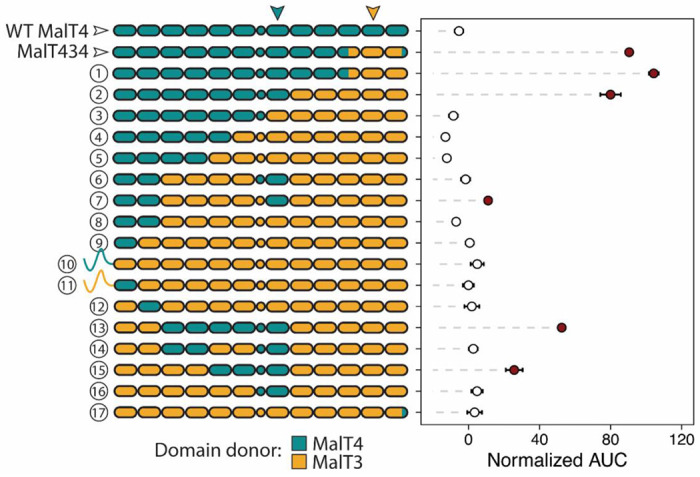
High-order intramolecular interactions are required to evolve a novel function in chimeric α-glucoside transporters. Points and bars show mean +/− SEM of normalized growth on maltotriose (AUC, area under the curve) of strains expressing chimeric transporters or wild-type MalT4 (top row). Filled circles denote growth significantly greater than the negative control (p < 0.01, Mann-Whitney U test with Benjamini-Hochberg correction). The architecture of each tested transporter is depicted as a cartoon on the y-axis, where rounded rectangles represent each of the twelve transmembrane helices and circles represent the intracellular ICH domain that links the N- and C-terminal six-helix bundles; regions are colored by parental protein identity. In almost every case, the N- and C-terminal intracellular regions have the same parental protein identity as the neighboring transmembrane helix and are omitted for clarity; the two exceptions are depicted. Inverted arrows indicate the location and identity of protein regions underlying the largest detected intramolecular interaction.
For chimeric constructs containing potentiating sequences at TMHs 5-7 and 10-12, growth on maltotriose generally increased additively with the number of MalT4 regions incorporated (linear regression, p < 2.2x10−16). Nonetheless, we found significant support (ANOVA, p < 2.2x10−16) for pairwise epistasis between the tested protein regions, including in the sign of the effects of the ICH domain and the C-terminal region (residues 541-613). For example, the addition of TMH 3 and TMH 4 from MalT4 in conjunction with MalT4 TMH 7 only increased growth on maltotriose if TMH 5 and TMH 6 from MalT4 were also present; similarly, the addition of TMH 1, TMH 2, and the ICH domain from MalT4 in conjunction with TMH 7 did not improve growth (construct 6 vs. 16, Fig. 2) unless in the presence of TMHs 3-6 from MalT4 (construct 2 vs. 13, 52% increase, p = 2.4x10−4). Along the quantitative functional spectrum of MalT3/4 chimeric proteins enabling growth on maltotriose, we therefore detected a complex combination of additive and epistatic intramolecular interactions among at least six protein regions.
Numerous substitutions are required to evolve a novel function in maltose transporters
We next dissected the contributions of the 11 substitutions in MalT434 relative to MalT4 (Fig. 1b) by introducing subsets of these to the gene encoding the native MalT4 protein (Fig. 3). We first tested the effect of a pair of suggestive substitutions, S468F and N522D, which were both unique in their location in the 3D structure and differed notably in side-chain chemistry. Nonetheless, this pair of mutations was insufficient for novel function in MalT4, so we coarsely tested the effect of the sets of mutations occurring before and after the end of TMH 11. Introduction of the five substitutions from residues 522-540, which span an extracellular loop and the majority of TMH 12, was insufficient to confer any growth on maltotriose. By contrast, the six mutations affecting TMHs 10 and 11 were sufficient to confer growth on maltotriose, and even improved it by 13.3% relative to MalT434 (p = 5.6x10−7, Mann-Whitney U test). Within this contiguous patch of substitutions, however, the contribution of individual amino acids to novel function was remarkably complex. Reversion of the six mutations singly to their MalT4 identity revealed that each had a significant effect on maltotriose growth, ranging from a 23.5% reduction (A504G, p = 2x10−6) to its complete abrogation (C505N, p = 5.2x10−11), with an average effect of 57.1%. We detected significant (p < 2.2x10−16) evidence of pairwise epistasis between substitutions, regardless of whether we considered all 11 sites or only the 6 on TMHs 10 and 11. Epistatic effects were notably non-uniform among tested combinations: for example, two single reversion mutations (M503I and T508V) had similar effects of 49.1% (p = 3.2x10−7) and 44.1% (p = 9.1x10−13) when introduced in the six-substitution background that supported robust growth on maltotriose. By contrast, when introduced in a four-mutation background with reduced ability to support growth on maltotriose (M503 C505 T508 T512), the effect of M503I remained large (42.6%, p = 0.002), while T508V effected only a small further reduction (4.97%, p = 0.8). Overall, we found that establishing novel function in MalT4 required a combination of three amino acid substitutions only accessible through a minimum of four non-consecutive nucleotide substitutions to the wild-type gene: N505C (2 nucleotide substitutions), I512T (1 substitution), and one of I503M (1 substitution) or V508T (2 substitutions).
Figure 3.
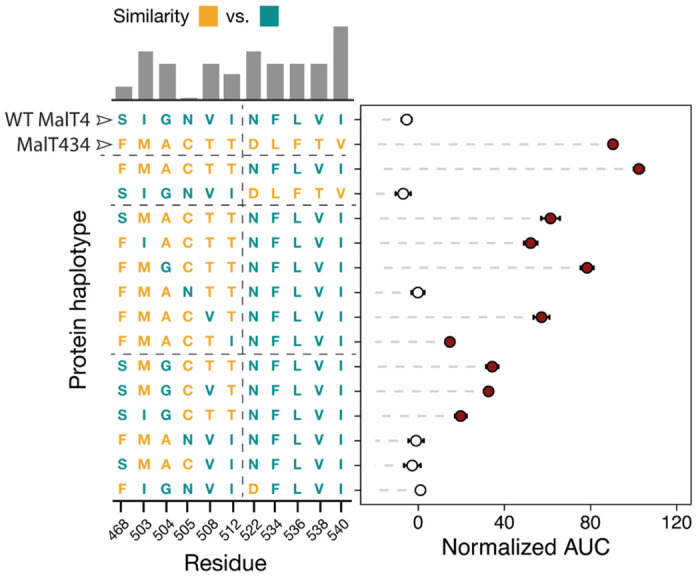
Numerous substitutions are required to evolve a novel function in a maltose transporter. Points and bars show mean +/− SEM of normalized growth on maltotriose (AUC, area under the curve) of strains expressing MalT4 variants. The genotype of each protein at the 11 sites that differ between MalT4 (top row) and MalT434 (second from top row) is depicted on the Y-axis. Filled circles denote growth significantly greater than the negative control (p < 0.01, Mann-Whitney U test with Benjamini-Hochberg correction). The bar chart shows rescaled BLOSUM similarity between the MalT4 and MalT3 residue at that site, with a higher bar indicating a more conservative substitution. Horizontal dotted lines in the protein haplotype grid separate related groups of genotypes. The vertical dotted line demarcates the substitutions that are sufficient (left) to impart novel function to MalT4 and those that are insufficient (right).
Granular mapping of epistasis between distal protein regions
Given the size of interacting protein regions and the complexity of their contributions to novel function, we sought to identify the key difference in amino acid sequence responsible for the large epistatic effect of transmembrane helix 7. The two parental transporters differ at six sites along TMH 7 (Fig. S2a): two neighboring substitutions (K357C and V358I, expressed relative to MalT4) occur at the intracellular C-terminal end, while two (A371I, V375T) are located approximately halfway along the helix and likely to be embedded in the plasma membrane. Two (A378T, S379Q) project into or neighbor the transport channel, differ in size and/or polarity, and are in close three-dimensional proximity to mutated residues on TMH 11 in MalT434 (Fig. 4a, Fig. S2b). We reasoned that one or both of A378T and S379Q might have a large effect on the interaction between TMH 7 and the translocated region of MalT3 present in functional chimeric transporters. To test these hypotheses, we mutated each of these residues to their MalT3 identity, singly and in combination, in a gene encoding the MalT4 transporter harboring the six mutations on TMHs 10 and 11 that conferred maximal maltotriose transport (Fig. 4b). While the A378T mutation did not affect growth on maltotriose, S379Q abolished it completely. The large epistatic interaction between TMH 7 and TMH 11 can thus be attributed to a single amino acid.
Figure 4.
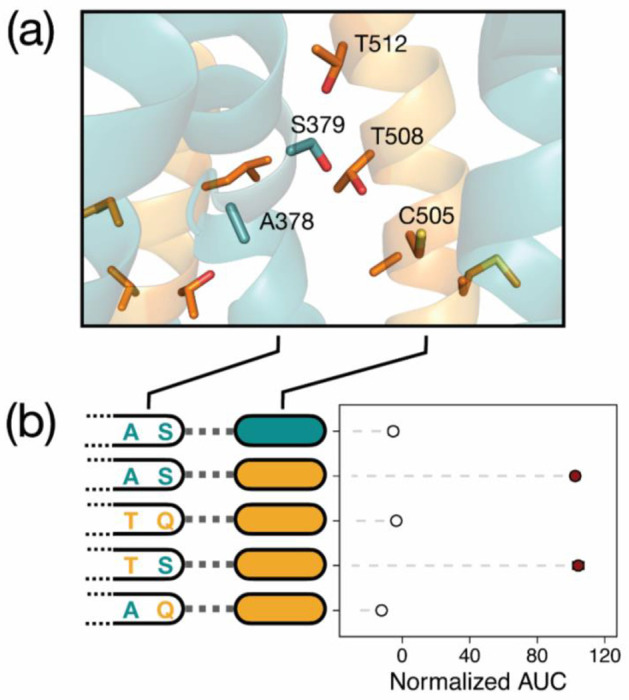
A single amino acid underlies a large epistatic effect. (a) Structural model of MalT434 with helices colored as in Fig. 1. Side chains are drawn for amino acids on transmembrane helices 7, 11, and 12 that are polymorphic between MalT3 and MalT4, and those that are proximal to or project into the transport channel are labeled. (b) Points and bars show mean +/− SEM of normalized growth on maltotriose (AUC, area under the curve) of strains expressing transporter variants. Filled circles denote growth significantly greater than the negative control (p < 0.01, Mann-Whitney U test with Benjamini-Hochberg correction). For each transporter, the parental protein identity at transmembrane helix 11 (filled rectangular ovals) and residues 378 and 379 in transmembrane helix 7 is depicted.
Novel transporter function is constrained by specific biochemical requirements and context dependence
The mutational event that generated MalT434, as well as our experiments dissecting it, only sampled variation between two binary states: the specific amino acid residues of the parental proteins at each homologous site. In native contexts, however, many more amino acid substitutions are accessible in mutational space through single- or multi-nucleotide mutations; for example, seven amino acid substitutions require only a single nucleotide change from an asparagine codon, which is the wild-type amino acid at the crucial 505 site. While we found complex interactions between many sites to contribute to novel function in MalT4, the evolution of maltotriose transport would be far less constrained and more accessible through sequential point mutations if biochemically similar amino acids at key sites could enable a degree of novel function because it would increase the mutational target size and pool of mutations conferring a fitness benefit (Miyazaki and Arnold 1999; Podgornaia and Laub 2015).
We thus sought to clarify the biochemical requirements for maltotriose transport in a specific potentiated context: a MalT4 transporter harboring S379, F468, M503, A504, T508, and T512. In this state, amino acid identity at position 505 is crucial with the wild-type asparagine incapable of supporting growth on maltotriose and the recombinant cysteine supporting robust growth (Fig. 3). We successfully mutated this residue to 17 of the 20 possible amino acids, measured their ability to support growth on maltotriose, and used regression analyses to estimate the effect of side chain physicochemical properties on measured function (Fig. 5). Remarkably, only three substitutions supported any degree of statistically significant growth above baseline: serine, glycine, and cysteine. Side chain aromaticity, volume, composition, and hydropathy were all significant (p << 0.01) predictors of function, as was overall similarity to the wild-type residue asparagine. Even so, the strengths of these associations were almost entirely driven by the C505 variant: when these data were omitted, the global explanatory power was reduced dramatically (adjusted R2: 0.2263 vs. 0.8664; F-statistic: 9.533 vs. 242). Although some physicochemical properties remained statistically significant predictors of function, the strengths of these associations were generally weak (maximum | Kendall’s T|: 0.212).
Figure 5.
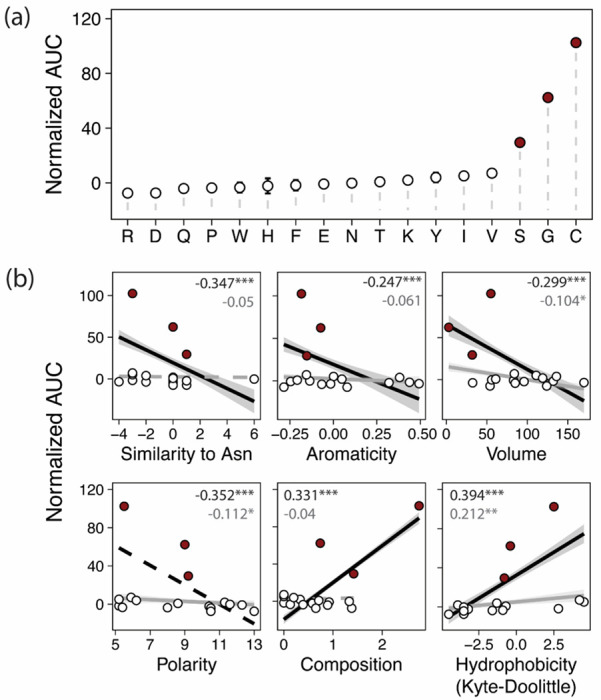
Physicochemical requirements constrain the evolution of novel function. (a) Points and bars show mean +/− SEM of normalized growth on maltotriose (AUC, area under the curve) of strains expressing MalT4 variants. The x-axis shows the amino acid identity at position 505; all variants share F468, M503, A504, T508, and T512. Filled circles denote growth significantly greater than the negative control (p<0.01, Mann-Whitney U test with Benjamini-Hochberg correction). (b) Correlations between growth and properties of the amino acid variant at position 505. Growth is plotted as in (a) against physicochemical property or overall similarity to the wild-type residue at position 505, asparagine. Lines and shaded ranges show regressions and 95% confidence intervals for significant (p < 0.05) regressions for all data (black) or after removing observations for C505 (gray). Dotted lines show regressions that are not statistically significant. Inset text shows Kendall’s T; ***p < 10−6, **p < 10−4, *p < 0.05.
Qualitatively, the fine-scale stringency of physicochemical requirements at position 505 was also noteworthy. Glycine, serine, and cysteine are three of the smallest amino acids, but amino acids with similar side chain volumes did not support growth on maltotriose. Serine and cysteine have side chains of similar size and structure capable of forming hydrogen bonds, but they differ in their polarity and hydrophobicity; nonetheless, residues similar to cysteine in both of these metrics did not support novel function. Indeed, C505’s ability to support novel function appeared to be the result of the specific combination of cysteine’s physicochemical properties (Fig. S3), albeit not due to its unique capacity to form disulfide bridges (Drew et al. 2021). Remarkably, this effect was dependent on positional context within the transporter: while substituting cysteine to serine at 505 reduced growth by 71.2% (p = 8.8x10−5), making the orthogonal serine to cysteine substitution at another key site, S379 (Fig. 4) reduced growth by 17.7% (p = 1.9x10−6) while still supporting robust growth (Fig. S4). Thus, while serine was largely unable to recapitulate the effect of cysteine at 505, the similarity between the two was sufficient to satisfy the requirements for novel function at position 379. The same was not true of two other hydrogen bond-competent residues, glutamic acid and glutamine, whose introduction at position 379 abolished growth (Fig. S4). This result suggests that, while serine and cysteine are interchangeable at this site, interactions between physical and chemical side chain properties still play a role. Finally, we found further evidence for these fine-scale requirements at position 512, where mutation of the permissive threonine to valine reduced growth by 34.5% (p = 7.4x10−9), while still supporting significantly improved growth over the wild-type MalT4 residue isoleucine (78.1% increase, p = 1.2x10−6). In summary, we find that the strengths, stringencies, and bases of physicochemical requirements all vary between sites that are critical for establishing novel function in MalT434. These results suggest that the serendipitous acquisition of a set of epistatically sufficient residues is highly improbable by point mutations alone (Lynch 2005).
High-specificity transporters are evolutionarily derived
The sum of our molecular analyses suggested that the acquisition of novel substrate transport by the high-specificity maltose transporter MalT4 is highly improbably and accessible only through the simultaneous acquisition of numerous interacting substitutions. This observation is consistent with previous failed attempts to establish a maltotriose transporter by introducing as many as 14 rational mutations to S. cerevisiae Mal61 (Hatanaka et al. 2022), a prototypical high-specificity maltose transporter closely related to MalT4. However, the presence of closely related generalist α-glucoside transporters, as typified by S. cerevisiae Agt1, suggests that this ability evolved at least once among yeast α-glucoside transporters. We sought to clarify the timing and mode of this historical evolutionary innovation by examining the phylogenetic relationships between the generalist and specialist α-glucoside transporters within Saccharomycotina yeasts, which have previously been assessed on only a few taxa (Brown et al. 2010; Cousseau et al. 2013; Baker and Hittinger 2019; de Ruijter et al. 2020; Hatanaka et al. 2022; Donzella et al. 2023).
We first generated high-quality protein-coding gene annotations for published genomes from 332 yeast species from the model subphylum Saccharomycotina, which spans more than 400 million years of evolution (X.-X. Shen et al. 2018). To formally test the expected monophyly of the α-glucoside transporters within the broader sugar porter family, we retrieved homologs of S. cerevisiae sugar porters from these predicted proteomes and constructed a comprehensive phylogeny of these 8,403 ecologically and biotechnologically relevant MFS proteins. This phylogeny split into several major clades, many of which contained at least one functionally characterized protein from S. cerevisiae or another species (Fig. S5). Both the high-specificity (Mal31- and Mph2/3-like) and generalist (Agt1-like) α-glucoside transporters clustered in a monophyletic group (“Agt clade”) that excluded other sugar porter families. All proteins in the Agt clade from the newly circumscribed order Saccharomycetales (Groenewald et al. 2023) grouped together with strong support (Fig. 6a). The monophyly of the Saccharomycetales Agts was interrupted in two cases: 1) a single protein from Ogataea naganishii sister to the Lachancea Agt1-like proteins; 2) and, more notably, a well- supported clade of Agts from Brettanomyces anomalus and Brettanomyces bruxellensis. The Brettanomyces species are documented recipients of numerous horizontal gene transfer events, including for genes involved in the metabolism of sucrose, an Agt1 substrate (Stambuk et al. 2000; Woolfit et al. 2007; Roach and Borneman 2020). Notably, B. bruxellensis is commonly associated with brewing environments, where its propensity to vigorously consume diverse sugars and independent evolution of aerobic fermentation make it a frequent contaminant and occasional desired contributor (Rozpedowska et al. 2011; Serra Colomer et al. 2019; Colomer et al. 2020).
Figure 6.
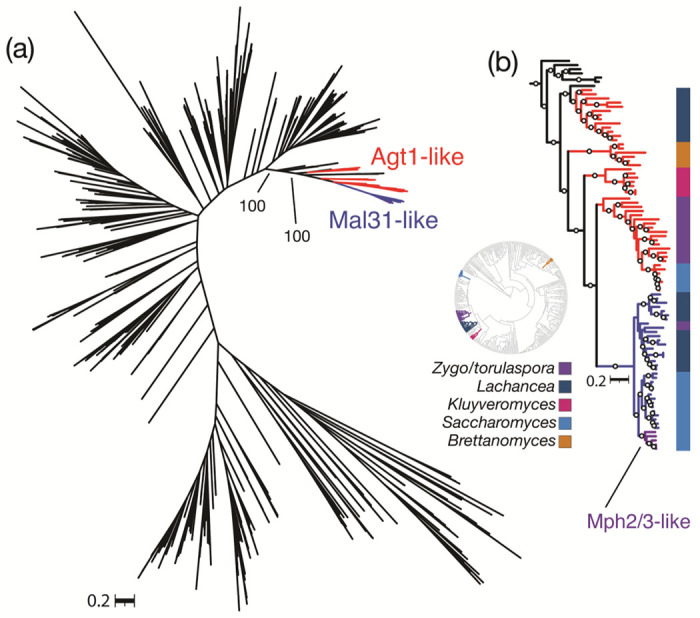
The high-specificity maltose transporters are evolutionarily derived and restricted to a subset of Saccharomycetales. (a) Consensus phylogeny of the α-glucoside transporter clade from 332 budding yeast genomes. Agt1-like and Mal31-like proteins from all Saccharomycetales are colored, as is the Saccharomyces-specific Mph2/3 clade. Bootstrap support is shown for two splits leading to the Saccharomycetales. (b) Rooted consensus tree of the clade containing Saccharomycetales α-glucoside transporters. Branches are colored as in (a) with the inclusion of a well-supported clade of Brettanomyces Agt1-like proteins that nests within the Saccharomycetales; the Saccharomyces-specific Mph2/3 clade is indicated. Circles denote branches with >90% bootstrap support. Colored bars outside the tree show genus-level taxonomic assignment, and the inset circular tree shows the Saccharomycotina species phylogeny (X.-X. Shen et al. 2018) with those genera colored; Zygo/torulaspora represents Zygosaccharomyces, Zygotorulaspora, and Torulaspora. The rooted maximum-likelihood tree can be found in Fig. S6. Newick-formatted trees are available in Data S2 and S3.
Surprisingly, the clade containing high-specificity Saccharomyces maltose transporters only included taxa from closely related species in the genera Saccharomyces and Lachancea, as well as one protein each from Zygotorulaspora florentina and Zygosaccharomyces kombuchaensis (Fig. 6b). Among the high-specificity Agts, the Mph2/3 clade was further restricted to Saccharomyces kudriavzevii, Saccharomyces mikatae, Saccharomyces paradoxus, and S. cerevisiae (Fig. 6b), which is consistent with an origin in the common ancestor of these species following their split from Saccharomyces arboricola and a recent segmental duplication in S. cerevisiae (Saccharomyces jurei is absent in this dataset). The sister clade to the high-specificity proteins contained generalist Agts from Saccharomyces, Torulaspora, and Zygotorulaspora species, with deeper branches to Kluyveromyces and Lachancea homologs (Fig. 6b). We thus conclude that the high-specificity transporters typified by S. cerevisiae Mal31, including S. eubayanus MalT4 and MalT3, form a clade restricted to Saccharomycetales.
Generalist-like transporters are quantitatively correlated with growth on α-glucosides
Our phylogenetic analyses suggested that the high-specificity Agts are evolutionarily and functionally derived from a generalist ancestor. In this model, the vast array of uncharacterized Agt-clade proteins encoded by diverse yeast species should include generalist transporters or transporters that became subfunctionalized following duplication of a generalist ancestor, and their presence should support growth on substrates of the generalist Agts. We collected quantitative growth measurements for 287 of the 332 species in our phylogenetic dataset on three sugars that are substrates of the generalist transporter S. cerevisiae Agt1 but not of the high-specificity transporters: maltotriose, trehalose, and methyl-α-glucoside (Han et al. 1995; Stambuk et al. 1999; Stambuk and Araujo 2001; Alves et al. 2008; Brown et al. 2010). We found many species across the Saccharomycotina to be capable of vigorous growth on these sugars as a sole carbon source (Fig. 7a). Growth on all three α-glucosides was nearly ubiquitous among Serinales, a speciose order with a high incidence of carbon niche-breadth generalists (Opulente et al. 2024). Most notably, growth on maltotriose was widespread across the yeast subphylum, in contrast to the documented rarity of this trait in the model genus Saccharomyces (Duval et al. 2010; Gallone et al. 2018; Langdon et al. 2020; Hutzler et al. 2021; Gyurchev et al. 2022; Peris et al. 2023). This metabolic deficiency was concomitant with the paucity of generalist-like Agt proteins encoded in Saccharomycetales genomes, which was similarly not representative of other yeast orders (Fig. 7b; p = 1.9x10−13). Indeed, patterns of α-glucoside growth qualitatively tracked the presence of genes encoding Agt proteins, with both subject to clear evolutionary shifts including losses (e.g. Saccharomycodales, Sporopachydermiales, and Trigonopsidales; Saturnispora, Zygosaccharomyces, Eremothecium, Kazachstania, Nakaseomyces, Naumovozyma, and Tetrapisispora spp.) and amplifications (Debayromyces, Metschnikowia, and Kuraishia spp.; subclades of Phaffomycetales, Dipodascales, Pichiales, and Lipomycetales). We used phylogenetically corrected least squares regressions (PGLS) to statistically test the strength of the correlation between Agt count and growth on each of the three tested Agt1 substrates (Fig. 7c). We detected significant positive correlations between Agt count and growth on each of the three α-glucosides (p ≤ 0.007). Thus, the generalist-like Agts detected in most Saccharomycotina genomes are likely to be true generalist transporters or recently subfunctionalized derivatives.
Fig. 7.
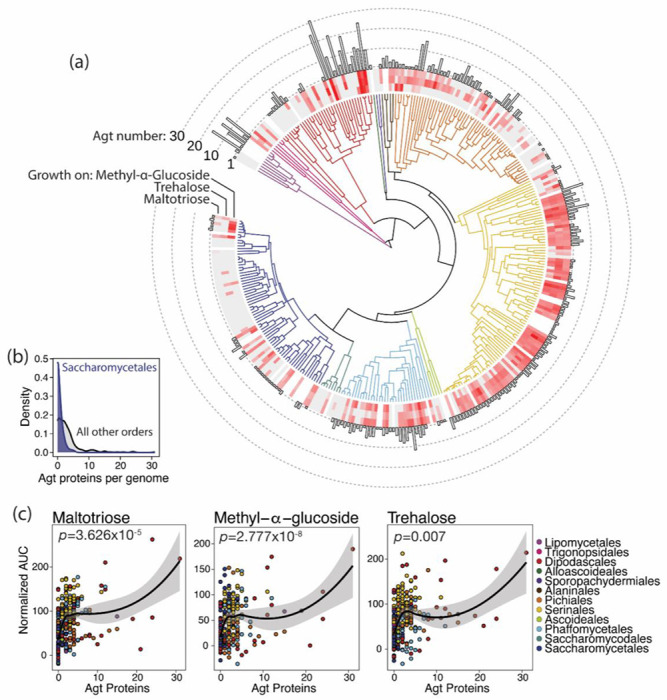
Species with Agt proteins grow on Agt1-specific substrates. (a) Time-calibrated phylogeny of 332 Saccharomycotina species (X.-X. Shen et al. 2018) with branches colored (key in panel c) by taxonomic order (Groenewald et al. 2023). Heatmaps around the tree show growth (normalized area under the curve) on α-glucosides: methyl-α-glucoside (inner ring), trehalose (middle ring), and maltotriose (outer ring). Gray boxes denote no growth above background; white boxes represent unsampled species. The bar chart shows the number of proteins in the α-glucoside transporter clade for each genome. (b) Generalist Agt content of Saccharomycetales genomes is not representative. Density plots show distributions of the number of Agt-clade proteins per genome for Saccharomycetales species (blue density) and species from all other orders (gray). (c) Scatterplots of Agt-clade transporter count versus growth on each α-glucoside. Each species is represented by a point, colored by taxonomic order. Lines and shaded regions are loess-smoothed regressions of the untransformed data; inset p-values are from phylogenetically corrected regressions (PGLS).
DISCUSSION
In the present work, we sought to understand how novel function could evolve in a model yeast α-glucoside transporter. To this end, we dissected the molecular basis of maltotriose transport in MalT434, which represents one of the most evolutionarily recent functional innovations in this family. We found that, in this chimeric protein, novel function is an emergent property of extensive additive and non-additive interactions between multiple protein regions and multiple residues on TMHs 7, 10, and 11 (Figs. 2–4). We observed that even conservative amino acid changes, as well as residues not predicted to interact with the substrate, had significant and unexpected effects on maltotriose transport (Fig. 3, Fig. 5). We also found evidence that the stringency of side chain physicochemical requirements likely differs substantially between crucial residues (Fig. 5, Fig. S4). Taken together, these results demonstrate that the evolution of novel function in a high-specificity Agt is highly constrained, which is consistent with recent observations (Hatanaka et al. 2022). In this model, the evolution of novel function in this family by gene conversion may indeed be the only remotely probable way that all the necessary interacting residues can readily be assembled in a single molecule, even if paralogs are free to sample neutral or deleterious mutational steps.
The gene conversion events leading to novel function in high-specificity yeast Agts share striking parallelism at both the sequence and structural scales. For example, the portions of Mty1 inferred to derive from different parental proteins encompass many of the same regions that we identified as having crucial interactions in MalT434 (Fig. S7a). Even more strikingly, the homologous residues at five of the seven sites that affect maltotriose transport in MalT434 are conserved in Mty1 (Fig. S7b). At the other two sites, Mty1 possesses amino acids that support reduced, but significant, growth in MalT434 (C505S and T512I). While many of the same sites likely contribute to novel function in both of these recombinant transporters, specific amino acids at key sites are still likely context-dependent, which makes functional evolution both more difficult to predict and to engineer in this family.
Compounding this difficulty is the cryptic nature of sites that we empirically determined to influence maltotriose transport but which are unlikely to interact with the substrate (Fig. 1). These substitutions may effect subtle changes to the overall conformation of the transporter, especially where they have the potential to interact with other protein regions that are proximal in tertiary space (e.g. F468). Moreover, there is a growing appreciation that, in yeast monosaccharide sugar porters, the fine-scale environment around the substrate binding site plays a surprisingly large role in sugar recognition and specificity, both by shaping an accommodating binding pocket and through interactions between substrate-interacting and non-interacting residues within van der Waals distance (Kasahara et al. 2009; Drew et al. 2021).
In MalT434, more concrete hypotheses can be made about the molecular contributions of other sites important for novel substrate transport. Molecular docking analyses place the maltotriose ligand in close proximity to the key sites on TMH 7 and TMH 11 (Fig. S8), with several of the sugar hydroxyl groups capable of engaging in a hydrogen-bonding network with the side chains of polar amino acid residues at those sites. Of the substitutions in MalT434 that face the transport channel, all three have polar and hydrogen bond-competent side chains of small-to-medium size; in wild-type MalT4, the residues at these sites have bulkier and/or hydrophobic side chains. Similarly, at the crucial 379 site on TMH 7, the permissive serine has a much smaller side chain than the prohibitive glutamine. Either of the prohibitive residues at 379 and the other crucial site 505 might introduce steric clashes with the terminal glucopyranose moiety of maltotriose (Fig. S8c), even though they themselves are likely capable of hydrogen-bonding with the substrate. Notably, the residue at position 379 may be involved in coupling substrate binding to gating during the transition to the occluded state (Drew et al. 2021), a key determinant of substrate recognition that involves more tightly embedding the sugar molecule in its binding site within the transport channel. In wild-type MalT4, position 379 has the smaller serine residue, while sites along TMH 11 have bulkier amino acids; in wild-type MalT3, position 379 has the larger glutamine residue, but TMH 11 has smaller, hydrophilic residues. Thus, in each native maltose transporter, the steric constraint of the transport channel may be finely tuned at co- evolving sites along TMH 7 and TMH 11 to accommodate maltose with higher affinity and specificity, which occur at the expense of steric exclusion of other substrates, such as maltotriose (Fig. S8e). This model is consistent with the crucial role of amino acid side chain length in shaping substrate specificity in some monosaccharide sugar porters (Kasahara et al. 2011; Drew et al. 2021), notwithstanding that we also detected a complex interaction between size and biochemical properties at the key 505 site.
The difficulty of functional innovation in the high-specificity Agts begs the question of how the related generalist Agts are capable of transporting not only maltose and maltotriose, but a diverse range of substrates. If the generalist transporters had evolved from a more specific ancestor, as has been suggested (Pougach et al. 2014), their extant substrate range would imply multiple bouts of highly constrained functional evolution. To determine when and how this broad substrate specificity may have evolved in the generalist Agts, we reconstructed the yeast sugar porter phylogeny from 332 newly annotated, representative Saccharomycotina genomes encompassing more than 400 million years of evolution (Fig. S5). This analysis showed that, somewhat unexpectedly, the high-specificity Agts are a derived clade within the generalist-like Agts (Fig. 6a). The copy number of these putative generalist Agts encoded by yeast genomes is strongly predictive of growth on Agt1-exclusive substrates (Fig. 7), which further supports the conclusion that these proteins are likely bona fide generalists. The evolution of maltotriose transport by high-specificity Agts is thus better regarded as a reacquisition of ancestral function than the de novo evolution of a truly novel function within this protein family.
It remains subject to debate whether the general trend of protein evolution is directional: from less to more intrinsically specific (Bridgham et al. 2006; Tawfik 2010; Copley 2012; Steindel et al. 2016; Wheeler et al. 2016; Wheeler and Harms 2021). Multiple lines of evidence now suggest that this mode is dominant in genes involved in α-glucoside metabolism in yeasts. In addition to the α-glucoside transporters, both the α-glucosidases of S. cerevisiae and the transcriptional activators that regulate the structural metabolic genes likely evolved from promiscuous ancestral proteins that optimized subfunctions following duplication events, rendering them specific for different α-glucosides (Brown et al. 2010; Voordeckers et al. 2012; Pougach et al. 2014). The extent of intramolecular epistasis apparent in the high-specificity Agts, which may arise both from intra-protein and protein-substrate interactions, may provide an explanation for the inherent difficulty of re-evolving maltotriose transport in these proteins. Functional entrenchment by historical contingency and epistasis is well documented, and the irreversibility of evolutionary trajectories at the molecular level may be a widespread phenomenon (Ortlund et al. 2007; Bridgham et al. 2009; Soylemez and Kondrashov 2012; Harms and Thornton 2014; Bank et al. 2015; Podgornaia and Laub 2015; Shah et al. 2015; Starr and Thornton 2016; Starr et al. 2017; Starr et al. 2018; Ben-David et al. 2020; Xie et al. 2021; Park et al. 2022). Although not directly tested here, there may be inherent tradeoffs between specificity and substrate affinity in yeast Agts (Stambuk and Araujo 2001; Salema-Oom et al. 2005; Hatanaka et al. 2022), which would suggest that walking back through the accumulated mutations that led to higher specificity in the Mal31-like transporters would be likely to incur an immediate functional tradeoff and therefore fitness cost. The recurrent gene conversion events that enable maltotriose transport among members of this family may, therefore, represent the only meaningfully accessible route to bypass these deleterious intermediates, but the high degree of context-dependence for mutational effects makes the prediction or engineering of this novel function difficult (Hatanaka et al. 2022).
Might the evolution of yeast sugar porters more broadly be organized along an axis of increasing specialization and specificity? This family encompasses functionally diverse transporters with varying specificities for different mono- and di-saccharides and sugar alcohols; notably, functionally similar proteins are not monophyletic across the family (Donzella et al. 2023). Our phylogenetic analysis of these proteins places the Agts, which may retain some glucose transport capacity (Wieczorke et al. 1999), as a deeply branching sister clade to most of the broader family (Fig. S5). These results imply multiple bouts of functional specialization from a highly promiscuous ancestor, in some cases starting from partially subfunctionalized ancestral proteins, with the Agts perhaps remaining the most representative of the ancestral multifunctionality. While the extant diversity of yeast sugar porters has generally been regarded as an example of functional diversification (i.e. highly plastic gains of novel substrate affinity; (Brown et al. 2010; Hatanaka et al. 2022; Donzella et al. 2023)), the evolution of this important gene family may have followed a very different mode. In the former model, functional diversification by neofunctionalization follows duplication of ancestral transporter genes, whereas our analyses suggest that duplications in this gene family may be primarily followed by subfunctionalizing escapes from adaptive conflict (Hughes 1994; Hittinger and Carroll 2007; Des Marais and Rausher 2008), wherein transporters can gain increased specificity and affinity for a narrow substrate range at the expense of other ancestral ligands.
These two models have distinct implications for the myriad biotechnological applications predicated upon sugar consumption by yeasts, which might be targets for improvement by protein engineering. If extant transporters are indeed highly plastic and evolvable, shifting or expanding their substrate range should be relatively simple. If, on the other hand, they have undergone entrenched specialization, they may be inherently less evolvable (Bridgham et al. 2009; Starr et al. 2018; Wheeler and Harms 2021). Results here and elsewhere (Hatanaka et al. 2022) support the latter corollary. However, this model also implies that reconstructed ancestral proteins, or even generalist extant proteins from this clade, might both possess desirable properties and be inherently highly amenable to engineering, mutagenesis, or directed evolution approaches.
METHODS
Strains and cultivation conditions
S. eubayanus strains, plasmids, and oligonucleotides used in this work are listed in Tables S1 and S2. Yeasts were propagated on YPD medium (1% yeast extract, 2% peptone, 2% glucose) supplemented with 400mg/L G418 and/or 50mg/L Nourseothricin (CloNAT) as appropriate and cryopreserved in 15% glycerol at −80° for long-term storage.
Transformation of S. eubayanus was performed by the PEG/LiAc/carrier DNA method (Gietz and Schiestl 2007) with minor modifications (Baker and Hittinger 2019). CRISPR-mediated gene deletions and insertions were achieved by co-transformation of pXIPHOS vectors (Kuang et al. 2018) and repair templates for homologous recombination. Repair templates were purified PCR products consisting of single linear fragments, multiple linear fragments for in vivo assembly, or recombinant amplicons generated by overlap extension PCR, depending on the application. All repair templates were amplified using Phusion polymerase (New England Biolabs) per the manufacturer’s instructions and purified using QiaQuick or MinElute spin columns (Qiagen).
We assessed transporter function via expression from the native MALT4 locus in yHJC207, a haploid derivative of the wild strain yHKS210 that was constructed as previously described (Crandall et al. 2023). Because the MALT2 and MALT4 loci are recent duplicates and almost identical at the nucleotide level, transporter variants were inserted into both loci out of necessity. Both MALT2 and MALT4 were simultaneously deleted using CRISPR-Cas9 and replaced with kanMX. Novel transporter variants, as well as MALT434 and S. eubayanus AGT1 positive controls, were subsequently inserted into both loci by co-transformation with a pXIPHOS vector expressing Cas9 and a gRNA targeting kanMX (Lee et al. 2021). Transformants were cured of plasmids, and the inserted alleles were sequenced.
Quantitative growth measurements of S. eubayanus strains
Strains were streaked to single colonies on YPD plates, arrayed in 96-well plates in a randomized layout, and precultured in YPD at room temperature for 72 hours with gentle shaking. Precultures were serially diluted in minimal medium (0.5% ammonium sulfate, 0.017% Yeast Nitrogen Base) and inoculated into minimal medium containing 2% sugars in 96-well plates at a final dilution of 10−4. OD600 was measured every hour for 7 days using a SPECTROstar Omega plate reader (BMG Labtech) equipped with a microplate stacker. Raw growth data was summarized using GCAT (Bukhman et al. 2015). Area under the curve (AUC) measurements for growth on maltotriose, normalized to a common negative control within each experiment, were used as a response variable in linear models with protein identity (MalT3 or MalT4) at each domain or at key amino acid sites as categorical predictor variables. The effects of protein identity at some single regions and for many pairwise interactions could not be estimated due to singularities. We tested for evidence of epistasis by statistically comparing additive models and those with interaction terms (Li and Fay 2019). The amino acid properties compiled to test associations with transporter function included chemical composition, polarity, and volume (Grantham 1974), aromaticity (Xia and Li 1998), hydropathy (JANIN 1979; Kyte and Doolittle 1982; Hopp and Woods 1983; Eisenberg et al. 1984; Rose et al. 1985; Cornette et al. 1987; Engelman et al. 2003), and BLOSUM similarity (Henikoff and Henikoff 1992). Some matrices were compiled from Braun (Braun 2018). For dimensionality reduction, BLOSUM similarity was omitted.
Quantitative growth measurements of Saccharomycotina yeasts
Growth on α-glucosides was measured for the strains whose genome annotations were analyzed, which were primarily the type strains for their respective species. Strain information, including taxonomic order (Groenewald et al. 2023), major clade (X.-X. Shen et al. 2018), and updated annotation mapping, can be found in Table S3. Cryopreserved strains were inoculated directly to YPD in 96-well plates and incubated for 7 days at room temperature. Some slow-growing species failed to revive during this time frame and were removed from further analysis, and we did not phenotype opportunistic pathogens, ultimately resulting in data for 287 species. Precultures were inoculated to minimal medium with 1% sugar or no added carbon source using a pinning tool, incubated for 7 days at room temperature, and re-inoculated to new plates containing the same medium. OD600 of the second round of growth was measured every hour using a SPECTROstar Omega plate reader (BMG Labtech) equipped with a microplate stacker. The growth experiments were performed four times independently. Raw growth data was summarized using Growthcurver (Sprouffske and Wagner 2016). Wells with poor model fits were discarded, and each curve was manually inspected to identify species with unreliable growth curves (Opulente et al. 2024). Growth on each carbon source was normalized to the average growth of the same species in medium with no added carbon to control for background growth. Caper (cran.r-project.org/web/packages/caper/index.html) was used to fit phylogenetically corrected regressions (PGLS) to growth data and square-root transformed Agt number, using the rooted ML species phylogeny (X.X. Shen et al. 2018).
Structure prediction and analyses
Structural models for MalT434 were generated using four different software: AlphaFold2 (Jumper et al. 2021), Phyre2 (Kelley et al. 2015), I-TASSER (Yang et al. 2015), and SWISS-MODEL (A. Waterhouse et al. 2018). All gave extremely similar results across the structured region (mean and SD pairwise RMSD: 1.61±0.51Å), and AlphaFold2 models for all proteins of interest were generated and used for further analysis. Docking of maltotriose was performed using SwissDock (Grosdidier et al. 2011). Structure models and docking results were visualized in PyMol v2.5 (Schrödinger, LLC).
Genome annotation
To improve the quality of existing gene models, publicly available genome assemblies of 332 Saccharomycotina yeast species (X.-X. Shen et al. 2018) were re-annotated de novo. For consistency, we retained the assembly and species names, although some species have since been renamed; consult MycoBank (www.mycobank.org) for the most up-to-date taxonomic information. Repetitive sequences were softmasked with RepeatMasker v4.1.2, and protein-coding genes were annotated using ab inito predictors AUGUSTUS v3.4.0 (Stanke et al. 2008) and GeneMark-EP+ v4.6.1 (Brůna et al. 2020) in BRAKER (Brůna et al. 2021), with Saccharomycetes proteins in OrthoDB v10 (Kriventseva et al. 2019) as homology evidence and using the --fungus mode. Where applicable, the longest transcript of each gene was retained. BUSCO v5.7.0 (Manni et al. 2021) was used to assess the completeness of the new and preexisting genome annotations using single-copy yeast orthologs in OrthoDB v10 (R.M. Waterhouse et al. 2018).
This approach was chosen so as to generate a useful community resource in two ways: first, to enable direct comparisons with a larger, partially overlapping dataset of yeast genomes published recently (Opulente et al. 2024), which were annotated using identical methods; and second, to facilitate future studies by significantly improving the quality of annotations for the widely-used 332-genomes dataset. Median annotation completeness was increased from 94.6% to 98.8%, while the median percentage of missing BUSCO genes decreased to 0.9% from 4.6% (both p < 2.2x10−16, two-sided t-tests; Fig. S10). Table S4 documents BUSCO analyses of existing and updated annotations for all genomes. The full updated annotations in protein and nucleotide FASTA, GFF3, and GTF formats will be available on figshare (confidential link will be updated to a public link prior to publication).
Phylogenetic analyses
The amino acid translations of the newly predicted protein-coding genes were queried by BLASTp+ v2.9 (Camacho et al. 2009) using characterized Saccharomyces cerevisiae sugar transporters (Mal31, Agt1, Gal2, Hxt1-5, Hxt7) retrieved from SGD (Wong et al. 2023). BLAST subjects less than 400 or greater than 1000 amino acids in length were discarded to remove partial or fused annotations, based on distributions of sugar porter length in TCDB (Saier et al. 2006; Saier et al. 2021). Remaining proteins were annotated with their most similar S. cerevisiae homolog using a reciprocal BLASTp search against all translated ORFs in S. cerevisiae, which were retrieved from SGD. Protein sequences were aligned using the E-INS-i strategy of MAFFT v7.222 (Katoh et al. 2002; Katoh et al. 2005; Katoh and Standley 2013), and the alignment was trimmed with trimAL v1.4.22 (Capella-Gutiérrez et al. 2009) using the --gappyout parameter. The phylogeny was inferred using IQ-TREE v2.2.2.7 (Minh et al. 2020) with 1000 bootstraps (Hoang et al. 2018) and automatic substitution model selection (Kalyaanamoorthy et al. 2017). Due to the significant homology between MFS proteins, this dataset contained a small proportion of non-sugar porter MFS proteins, primarily belonging to the drug:proton antiporter family. These were retained in the alignment and tree inference to test the assumption of sugar porter monophyly. As expected, the sugar porters and non-sugar porter MFS proteins formed well-supported reciprocally monophyletic clades. The α-glucoside transporter phylogeny was refined by re-aligning the proteins from that clade and inferring the phylogeny as before, albeit with 10 independent runs of IQ-TREE with 10000 bootstrap replicates each and secondary branch support assessment by SH-aLRT tests. Trees were visualized and annotated in iTOL (Letunic and Bork 2021).
Supplementary Material
Table S1. S. eubayanus strains and plasmids used in this study.
Table S2. Oligonucleotides used in this study.
Table S3. Strain information for the 332 Saccharomycotina species. Column A (“Species name”) corresponds to Column C (“Species name”) of Table S1 from X.-X. Shen et al. 2018.
Table S4. Benchmark Universal Single-Copy Orthologs (BUSCO) statistics for existing and updated genome annotations of species in this study.
Data S1. Maximum likelihood phylogenetic trees of sugar porters and outgroup MFS proteins from Saccharomycotina genomes in Newick format.
Data S2. Consensus phylogenetic tree of Agt clade proteins in Newick format. Branch supports are from SH-aLRT test and ultrafast bootstrapping, respectively.
Data S3. Maximum likelihood phylogenetic tree of Agt clade proteins in Newick format. Branch supports are from SH-aLRT test and ultrafast bootstrapping, respectively.
ACKNOWLEDGMENTS
We are grateful to John F. Wolters for feedback on analyses and extensive curation of databases, Dana A. Opulente for advice on phenotyping, Kaitlin J. Fisher for sharing yeast strain copies that enabled high-throughput phenotyping, Xing-Xing Shen for advice on running IQ-TREE, and the Hittinger and Sato Labs for helpful discussion.
FUNDING
This material is based upon work supported by the National Institute of Food and Agriculture, United States Department of Agriculture, Hatch projects 1020204 and 7005101; the National Science Foundation under Grant Nos. DEB-2110403 and DEB-2110404, and in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE–SC0018409). Research in the Hittinger Lab is supported an H. I. Romnes Faculty Fellowship, supported by the Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. Research in the Rokas Lab is also supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (R01 AI153356) and the Burroughs Wellcome Fund. J.G.C. was supported by a Predoctoral Training Grant in Genetics funded by the National Institutes of Health under Grant No. T32GM007133 and by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1747503. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
COMPETING INTERESTS
The Wisconsin Alumni Research Foundation has filed a patent application on the technologies described herein with J.G.C. and C.T.H. as inventors. Strains are available for non-commercial, academic use under a material transfer agreement. A.R. is a scientific consultant for LifeMine Therapeutics, Inc.
DATA AVAILABILITY
New genome annotations for Saccharomycotina species are available on figshare (confidential link will be updated to a public link prior to publication). This confidential link is provided for review purposes and will be updated to a public link prior to publication. Other data underlying this article are available in the article and in its online supplementary material.
REFERENCES
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science (1979) [Internet] 301:610–615. Available from: 10.1126/science.1088196 [DOI] [PubMed] [Google Scholar]
- Alves SL, Herberts RA, Hollatz C, Trichez D, Miletti LC, de Araujo PS, Stambuk BU. 2008. Molecular Analysis of Maltotriose Active Transport and Fermentation by Saccharomyces cerevisiae Reveals a Determinant Role for the AGT1 Permease. Appl Environ Microbiol [Internet] 74:1494–1501. Available from: 10.1128/AEM.02570-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello JR, Chen Y, Yang S, Wang W, Long M. 2006. Origination of an X-Linked Testes Chimeric Gene by Illegitimate Recombination in Drosophila. PLoS Genet [Internet] 2:e77. Available from: 10.1371/journal.pgen.0020077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EP, Hittinger CT. 2019. Evolution of a novel chimeric maltotriose transporter in Saccharomyces eubayanus from parent proteins unable to perform this function.Zhang J, editor. PLoS Genet [Internet] 15:e1007786. Available from: 10.1371/journal.pgen.1007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EP, Sayegh R, Kohler KM, Borman W, Goodfellow CK, Brush ER, Barber MF. 2022. Evolution of host-microbe cell adherence by receptor domain shuffling. Elife [Internet] 11. Available from: https://elifesciences.org/articles/73330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank C, Hietpas RT, Jensen JD, Bolon DNA. 2015. A Systematic Survey of an Intragenic Epistatic Landscape. Mol Biol Evol [Internet] 32:229–238. Available from: 10.1093/molbev/msu301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David M, Soskine M, Dubovetskyi A, Cherukuri K-P, Dym O, Sussman JL, Liao Q, Szeler K, Kamerlin SCL, Tawfik DS. 2020. Enzyme Evolution: An Epistatic Ratchet versus a Smooth Reversible Transition.Barlow M, editor. Mol Biol Evol [Internet] 37:1133–1147. Available from: https://academic.oup.com/mbe/article/37/4/1133/5686393 [DOI] [PubMed] [Google Scholar]
- Bergin SA, Allen S, Hession C, Cinnéide EÓ, Ryan A, Byrne KP, Cróinín TÓ, Wolfe KH, Butler G, Morrissey J. 2022. Identification of European isolates of the lager yeast parent Saccharomyces eubayanus. FEMS Yeast Res [Internet] 22:1–9. Available from: https://academic.oup.com/femsyr/article/22/1/foac053/6874782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittihn P, Tsimring LS. 2017. Gene Conversion Facilitates Adaptive Evolution on Rugged Fitness Landscapes. Genetics [Internet] 207:1577–1589. Available from: https://academic.oup.com/genetics/article/207/4/1577/5930769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosshart PD, Fotiadis D. 2019. Secondary Active Transporters. In: Subcellular Biochemistry. Vol. 92. p. 275–299. Available from: 10.1007/978-3-030-18768-2_9 [DOI] [PubMed] [Google Scholar]
- Braun EL. 2018. An evolutionary model motivated by physicochemical properties of amino acids reveals variation among proteins. Bioinformatics 34:i350–i356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickwedde A, Brouwers N, Broek M van den, Gallego Murillo JS, Fraiture JL, Pronk JT, Daran JMG. 2018. Structural, physiological and regulatory analysis of maltose transporter genes in Saccharomyces eubayanus CBS 12357T. Front Microbiol 9:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgham JT, Carroll SM, Thornton JW. 2006. Evolution of hormone-receptor complexity by molecular exploitation. Science (1979) [Internet] 312:97–101. Available from: 10.1126/science.1123348 [DOI] [PubMed] [Google Scholar]
- Bridgham JT, Ortlund EA, Thornton JW. 2009. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature [Internet] 461:515–519. Available from: https://www.nature.com/articles/nature08249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N, Brickwedde A, Gorter de Vries AR, van den Broek M, Weening SM, van den Eijnden L, Diderich JA, Bai FY, Pronk JT, Daran JMG. 2019. Himalayan saccharomyces eubayanus genome sequences reveal genetic markers explaining heterotic maltotriose consumption by saccharomyces pastorianus hybrids. Appl Environ Microbiol [Internet] 85. Available from: 10.1128/AEM.01516-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N, Gorter de Vries AR, van den Broek M, Weening SM, Elink Schuurman TD, Kuijpers NGA, Pronk JT, Daran J- MG. 2019. In vivo recombination of Saccharomyces eubayanus maltose-transporter genes yields a chimeric transporter that enables maltotriose fermentation.Zhang J, editor. PLoS Genet [Internet] 15:e1007853. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30946741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Murray AW, Verstrepen KJ. 2010. Rapid Expansion and Functional Divergence of Subtelomeric Gene Families in Yeasts. Current Biology [Internet] 20:895–903. Available from: 10.1016/j.cub.2010.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. 2021. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform [Internet] 3:1–11. Available from: 10.1093/nargab/lqaa108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brůna T, Lomsadze A, Borodovsky M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform [Internet] 2. Available from: 10.1093/nargab/lqaa026/5836691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhman Y V., DiPiazza NW, Piotrowski J, Shao J, Halstead AGW, Bui MD, Xie E, Sato TK. 2015. Modeling Microbial Growth Curves with GCAT. Bioenergy Res 8:1022–1030. [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: Architecture and applications. BMC Bioinformatics 10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell E, Kaltenbach M, Correy GJ, Carr PD, Porebski BT, Livingstone EK, Afriat-Jurnou L, Buckle AM, Weik M, Hollfelder F, et al. 2016. The role of protein dynamics in the evolution of new enzyme function. Nature Chemical Biology 2016 12:11 [Internet] 12:944–950. Available from: https://www.nature.com/articles/nchembio.2175 [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casa-Villegas M, Polaina J, Marín-Navarro J. 2018. Cellobiose fermentation by Saccharomyces cerevisiae: Comparative analysis of intra versus extracellular sugar hydrolysis. Process Biochemistry [Internet] 75:59–67. Available from: 10.1016/j.procbio.2018.09.005 [DOI] [Google Scholar]
- Cheng CHC. 1998. Evolution of the diverse antifreeze proteins. Curr Opin Genet Dev 8:715–720. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Michels CA. 1991. MAL11 and MAL61 encode the inducible high-affinity maltose transporter of Saccharomyces cerevisiae. J Bacteriol [Internet] 173:1817–1820. Available from: 10.1128/jb.173.5.1817-1820.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christin P-A, Salamin N, Savolainen V, Duvall MR, Besnard G. 2007. C4 Photosynthesis Evolved in Grasses via Parallel Adaptive Genetic Changes. Current Biology [Internet] 17:1241–1247. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0960982207015734 [DOI] [PubMed] [Google Scholar]
- Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P. 2005. Complex genomic rearrangements lead to novel primate gene function. Genome Res [Internet] 15:343–351. Available from: https://genome.cshlp.org/content/15/3/343.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Glanowski S, Nielsen R, Thomas PD, Kejariwal A, Todd MA, Tanenbaum DM, Civello D, Lu F, Murphy B, et al. 2003. Inferring Nonneutral Evolution from Human-Chimp-Mouse Orthologous Gene Trios. Science (1979) [Internet] 302:1960–1963. Available from: 10.1126/science.1088821 [DOI] [PubMed] [Google Scholar]
- Colomer MS, Chailyan A, Fennessy RT, Olsson KF, Johnsen L, Solodovnikova N, Forster J. 2020. Assessing Population Diversity of Brettanomyces Yeast Species and Identification of Strains for Brewing Applications. Front Microbiol [Internet] 11:495404. Available from: www.frontiersin.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley SD. 2012. Toward a systems biology perspective on enzyme evolution. Journal of Biological Chemistry [Internet] 287:3–10. Available from: http://www.jbc.org/article/S0021925820534856/fulltext [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornette JL, Cease KB, Margalit H, Spouge JL, Berzofsky JA, DeLisi C. 1987. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J Mol Biol 195:659–685. [DOI] [PubMed] [Google Scholar]
- Cousseau FEM, Alves SL, Trichez D, Stambuk BU. 2013. Characterization of maltotriose transporters from the Saccharomyces eubayanus subgenome of the hybrid Saccharomyces pastorianus lager brewing yeast strain Weihenstephan 34/70. Lett Appl Microbiol [Internet] 56:21–29. Available from: 10.1111/lam.12011 [DOI] [PubMed] [Google Scholar]
- Crandall JG, Fisher KJ, Sato TK, Hittinger CT. 2023. Ploidy evolution in a wild yeast is linked to an interaction between cell type and metabolism. PLoS Biol [Internet] 21:e3001909. Available from: 10.1371/journal.pbio.3001909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos FA, Gibson B, Grijalva-Vallejos N, Krogerus K, Nikulin J. 2019. Bioprospecting for brewers: Exploiting natural diversity for naturally diverse beers. Yeast [Internet] 36:383–398. Available from: 10.1002/yea.3380 [DOI] [PubMed] [Google Scholar]
- Cui Y, Wong WH, Bornberg-Bauer E, Chan HS. 2002. Recombinatoric exploration of novel folded structures: A heteropolymer-based model of protein evolutionary landscapes. Proc Natl Acad Sci U S A [Internet] 99:809–814. Available from: 10.1073/pnas.022240299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day RE, Higgins VJ, Rogers PJ, Dawes IW. 2002. Characterization of the putative maltose transporters encoded by YDL247w and YJR160c. Yeast [Internet] 19:1015–1027. Available from: 10.1002/yea.894 [DOI] [PubMed] [Google Scholar]
- Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, Ren W, Hirata K, Yamamoto M, Fan S, et al. 2015. Molecular basis of ligand recognition and transport by glucose transporters. Nature [Internet] 526:391–396. Available from: https://www.nature.com/articles/nature14655 [DOI] [PubMed] [Google Scholar]
- Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. 2014. Crystal structure of the human glucose transporter GLUT1. Nature [Internet] 510:121–125. Available from: https://www.nature.com/articles/nature13306 [DOI] [PubMed] [Google Scholar]
- Dietvorst J, Londesborough J, Steensma HY. 2005. Maltotriose utilization in lager yeast strains: MTTI encodes a maltotriose transporter. Yeast 22:775–788. [DOI] [PubMed] [Google Scholar]
- Donzella L, Sousa MJ, Morrissey JP. 2023. Evolution and functional diversification of yeast sugar transporters. Essays Biochem [Internet] 67:811–827. Available from: /essaysbiochem/article/67/5/811/232779/Evolution-and-functional-diversification-of-yeast [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorus S, Vallender EJ, Evans PD, Anderson JR, Gilbert SL, Mahowald M, Wyckoff GJ, Malcom CM, Lahn BT. 2004. Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell [Internet] 119:1027–1040. Available from: http://www.cell.com/article/S0092867404011432/fulltext [DOI] [PubMed] [Google Scholar]
- Drew D, North RA, Nagarathinam K, Tanabe M. 2021. Structures and General Transport Mechanisms by the Major Facilitator Superfamily (MFS). Chem Rev [Internet] 121:5289–5335. Available from: 10.1021/acs.chemrev.0c00983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval EH, Alves SL, Dunn B, Sherlock G, Stambuk BU. 2010. Microarray karyotyping of maltose-fermenting Saccharomyces yeasts with differing maltotriose utilization profiles reveals copy number variation in genes involved in maltose and maltotriose utilization. J Appl Microbiol 109:248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Schwarz E, Komaromy M, Wall R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179:125–142. [DOI] [PubMed] [Google Scholar]
- Engelman DM, Steitz TA, Goldman A. 2003. IDENTIFYING NONPOLAR TRANSBILAYER HELICES IN AMINO ACID SEQUENCES OF MEMBRANE PROTEINS. 10.1146/annurev.bb.15.060186.001541 15:321–353. [DOI] [PubMed] [Google Scholar]
- Fairhead C, Dujon B. 2006. Structure of Kluyveromyces lactis subtelomeres: duplications and gene content. FEMS Yeast Res [Internet] 6:428–441. Available from: 10.1111/j.1567-1364.2006.00033.x [DOI] [PubMed] [Google Scholar]
- Farwick A, Bruder S, Schadeweg V, Oreb M, Boles E. 2014. Engineering of yeast hexose transporters to transport d -xylose without inhibition by d -glucose. Proceedings of the National Academy of Sciences [Internet] 111:5159–5164. Available from: 10.1073/pnas.1323464111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallone B, Mertens S, Gordon JL, Maere S, Verstrepen KJ, Steensels J. 2018. Origins, evolution, domestication and diversity of Saccharomyces beer yeasts. Curr Opin Biotechnol [Internet] 49:148–155. Available from: 10.1016/j.copbio.2017.08.005 [DOI] [PubMed] [Google Scholar]
- Gibson B, Geertman J-MA, Hittinger CT, Krogerus K, Libkind D, Louis EJ, Magalhães F, Sampaio JP. 2017. New yeasts—new brews: modern approaches to brewing yeast design and development. FEMS Yeast Res [Internet] 17:1–13. Available from: 10.1093/femsyr/fox038 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. [DOI] [PubMed] [Google Scholar]
- Gong LI, Suchard MA, Bloom JD. 2013. Stability-mediated epistasis constrains the evolution of an influenza protein. Elife 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. 2009. Additions, Losses, and Rearrangements on the Evolutionary Route from a Reconstructed Ancestor to the Modern Saccharomyces cerevisiae Genome. PLoS Genet [Internet] 5:e1000485. Available from: 10.1371/journal.pgen.1000485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R. 1974. Amino Acid Difference Formula to Help Explain Protein Evolution. Science (1979) 185:862–864. [DOI] [PubMed] [Google Scholar]
- Groenewald M, Hittinger CT, Bensch K, Opulente DA, Shen XX, Li Y, Liu C, LaBella AL, Zhou X, Limtong S, et al. 2023. A genome-informed higher rank classification of the biotechnologically important fungal subphylum Saccharomycotina. Stud Mycol 105:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosdidier A, Zoete V, Michielin O. 2011. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res 39:W270–W277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L, Hariharan P. 2021. X-ray crystallography reveals molecular recognition mechanism for sugar binding in a melibiose transporter MelB. Commun Biol [Internet] 4:931. Available from: https://www.nature.com/articles/s42003-021-02462-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L, Kaback HR. 2006. LESSONS FROM LACTOSE PERMEASE. Annu Rev Biophys Biomol Struct [Internet] 35:67–91. Available from: 10.1146/annurev.biophys.35.040405.102005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L, Mirza O, Verner G, Iwata S, Kaback HR. 2007. Structural determination of wild-type lactose permease. Proceedings of the National Academy of Sciences [Internet] 104:15294–15298. Available from: https://www.pnas.org/content/104/39/15294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyurchev NY, Coral-Medina Á, Weening SM, Almayouf S, Kuijpers NGA, Nevoigt E, Louis EJ. 2022. Beyond Saccharomyces pastorianus for modern lager brews: Exploring non-cerevisiae Saccharomyces hybrids with heterotic maltotriose consumption and novel aroma profile. Front Microbiol 13:1025132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S-J, Kim H, Lin Y, Jang M-U, Galazka JM, Kim T-J, Cate JHD, Jin Y-S. 2013. Single Amino Acid Substitutions in HXT2.4 from Scheffersomyces stipitis Lead to Improved Cellobiose Fermentation by Engineered Saccharomyces cerevisiae. Appl Environ Microbiol [Internet] 79:1500–1507. Available from: http://dx.doi.org/10.1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han E-K, Cotty F, Sottas C, Jiang H, Michels CA. 1995. Characterization of AGT1 encoding a general alpha-glucoside transporter from Saccharomyces. Mol Microbiol [Internet] 17:1093–1107. Available from: 10.1111/j.1365-2958.1995.mmi_17061093.x [DOI] [PubMed] [Google Scholar]
- HANSEN TF, CARTER AJR, CHIU C-H. 2000. Gene Conversion may aid Adaptive Peak Shifts. J Theor Biol [Internet] 207:495–511. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022519300921891 [DOI] [PubMed] [Google Scholar]
- Hara KY, Kobayashi J, Yamada R, Sasaki D, Kuriya Y, Hirono-Hara Y, Ishii J, Araki M, Kondo A. 2017. Transporter engineering in biomass utilization by yeast. FEMS Yeast Res [Internet] 17:61. Available from: 10.1093/femsyr/fox061 [DOI] [PubMed] [Google Scholar]
- Harms MJ, Thornton JW. 2014. Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature [Internet] 512:203–207. Available from: https://www.nature.com/articles/nature13410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka H, Toyonaga H, Ishida Y, Mizohata E, Ono E. 2022. Functional diversity and plasticity in the sugar preferences of Saccharomyces MALT transporters in domesticated yeasts. FEMS Yeast Res [Internet] 22:1–10. Available from: 10.1093/femsyr/foac055 [DOI] [PubMed] [Google Scholar]
- Henderson R, Poolman B. 2017. Proton-solute coupling mechanism of the maltose transporter from Saccharomyces cerevisiae. Sci Rep [Internet] 7:14375. Available from: https://www.nature.com/articles/s41598-017-14438-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Carroll SB. 2007. Gene duplication and the adaptive evolution of a classic genetic switch. Nature [Internet] 449:677–681. Available from: https://www.nature.com/articles/nature06151 [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Steele JL, Ryder DS. 2018. Diverse yeasts for diverse fermented beverages and foods. Curr Opin Biotechnol [Internet] 49:199–206. Available from: 10.1016/j.copbio.2017.10.004 [DOI] [PubMed] [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol 35:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. 2006. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science (1979) [Internet] 313:101–104. Available from: 10.1126/science.1126121 [DOI] [PubMed] [Google Scholar]
- Hopp TP, Woods KR. 1983. A computer program for predicting protein antigenic determinants. Mol Immunol 20:483–489. [DOI] [PubMed] [Google Scholar]
- Horak J. 2013. Regulations of sugar transporters: insights from yeast. Curr Genet [Internet] 59:1–31. Available from: 10.1007/s00294-013-0388-8 [DOI] [PubMed] [Google Scholar]
- Hughes AL. 1994. The evolution of functionally novel proteins after gene duplication. Proc R Soc Lond B Biol Sci [Internet] 256:119–124. Available from: 10.1098/rspb.1994.0058 [DOI] [PubMed] [Google Scholar]
- Hutzler M, Michel M, Kunz O, Kuusisto T, Magalhães F, Krogerus K, Gibson B. 2021. Unique Brewing-Relevant Properties of a Strain of Saccharomyces jurei Isolated From Ash (Fraxinus excelsior). Front Microbiol [Internet] 12. Available from: 10.3389/fmicb.2021.645271/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabłońska J, Tawfik DS. 2022. Innovation and tinkering in the evolution of oxidases. Protein Science [Internet] 31:e4310. Available from: 10.1002/pro.4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANIN J. 1979. Surface and inside volumes in globular proteins. Nature 277:491–492. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 2021 596:7873 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback HR, Guan L. 2019. It takes two to tango: The dance of the permease. Journal of General Physiology [Internet] 151:878–886. Available from: 10.1085/jgp.201912377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach M, Burke JR, Dindo M, Pabis A, Munsberg FS, Rabin A, Kamerlin SCL, Noel JP, Tawfik DS. 2018. Evolution of chalcone isomerase from a noncatalytic ancestor. Nat Chem Biol [Internet] 14:548–555. Available from: 10.1038/s41589-018-0042-3 [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 2017 14:6 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara M, Shimoda E, Maeda M. 1997. Amino Acid Residues Responsible for Galactose Recognition in Yeast Gal2 Transporter. Journal of Biological Chemistry [Internet] 272:16721–16724. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925818392810 [DOI] [PubMed] [Google Scholar]
- Kasahara T, Ishiguro M, Kasahara M. 2006. Eight Amino Acid Residues in Transmembrane Segments of Yeast Glucose Transporter Hxt2 Are Required for High Affinity Transport. Journal of Biological Chemistry 281:18532–18538. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kasahara M. 1998. Tryptophan 388 in Putative Transmembrane Segment 10 of the Rat Glucose Transporter Glut1 Is Essential for Glucose Transport. Journal of Biological Chemistry 273:29113–29117. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kasahara M. 2000. Three Aromatic Amino Acid Residues Critical for Galactose Transport in Yeast Gal2 Transporter. Journal of Biological Chemistry 275:4422–4428. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kasahara M. 2010. Identification of a Key Residue Determining Substrate Affinity in the Yeast Glucose Transporter Hxt7. Journal of Biological Chemistry [Internet] 285:26263–26268. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925820595902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Maeda M, Boles E, Kasahara M. 2009. Identification of a key residue determining substrate affinity in the human glucose transporter GLUT1. Biochimica et Biophysica Acta (BBA) - Biomembranes 1788:1051–1055. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Maeda M, Ishiguro M, Kasahara M. 2007. Identification by Comprehensive Chimeric Analysis of a Key Residue Responsible for High Affinity Glucose Transport by Yeast HXT2. Journal of Biological Chemistry 282:13146–13150. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Shimogawara K, Kasahara M. 2011. Crucial Effects of Amino Acid Side Chain Length in Transmembrane Segment 5 on Substrate Affinity in Yeast Glucose Transporter Hxt7. Biochemistry [Internet] 50:8674–8681. Available from: 10.1021/bi200958s [DOI] [PubMed] [Google Scholar]
- Katoh K, Kuma KI, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma KI, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. MolBiolEvol 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S, Levin S. 1987. Towards a general theory of adaptive walks on rugged landscapes. J Theor Biol [Internet] 128:11–45. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022519387800292 [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Oh EJ, Lane ST, Lee WH, Cate JHD, Jin YS. 2018. Enhanced cellobiose fermentation by engineered Saccharomyces cerevisiae expressing a mutant cellodextrin facilitator and cellobiose phosphorylase. J Biotechnol 275:53–59. [DOI] [PubMed] [Google Scholar]
- Kriventseva EV., Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simao FA, Zdobnov EM. 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res [Internet] 47:D807–D811. Available from: 10.1093/nar/gky1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang MC, Kominek J, Alexander WG, Cheng J-F, Wrobel RL, Hittinger CT. 2018. Repeated Cis-Regulatory Tuning of a Metabolic Bottleneck Gene during Evolution.Wittkopp P, editor. Mol Biol Evol [Internet] 35:1968–1981. Available from: https://academic.oup.com/mbe/article/35/8/1968/5000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132. [DOI] [PubMed] [Google Scholar]
- Langdon QK, Peris D, Eizaguirre JI, Opulente DA, Buh K V, Sylvester K, Jarzyna M, Rodriguez ME, Lopes CA, Libkind D, et al. 2020. Postglacial migration shaped the genomic diversity and global distribution of the wild ancestor of lager-brewing hybrids. PLoS Genet [Internet] 16:e1008680. Available from: 10.1371/journal.pgen.1008680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Tremaine M, Place M, Liu L, Pier A, Krause DJ, Xie D, Zhang Y, Landick R, Gasch AP, et al. 2021. Crabtree/Warburg-like aerobic xylose fermentation by engineered Saccharomyces cerevisiae. Metab Eng 68:119–130. [DOI] [PubMed] [Google Scholar]
- Leffler EM, Band G, Busby GBJ, Kivinen K, Le QS, Clarke GM, Bojang KA, Conway DJ, Jallow M, Sisay-Joof F, et al. 2017. Resistance to malaria through structural variation of red blood cell invasion receptors. Science (1979) [Internet] 356:1140–1152. Available from: 10.1126/science.aam6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2021. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XC, Fay JC. 2019. Multiple changes underlie allelic divergence of CUP2 between Saccharomyces species. G3: Genes, Genomes, Genetics 9:3595–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libkind D, Hittinger CT, Valério E, Gonçalves C, Dover J, Johnston M, Gonçalves P, Sampaio JP. 2011. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proceedings of the National Academy of Sciences [Internet] 108:14539–14544. Available from: 10.1073/pnas.1105430108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Maclean CJ, Zhang J. 2019. Evolution of the Yeast Recombination Landscape. Mol Biol Evol [Internet] 36:412–422. Available from: 10.1093/molbev/msy233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Betrán E, Thornton K, Wang W. 2003. The origin of new genes: glimpses from the young and old. Nature Reviews Genetics 2003 4:11 [Internet] 4:865–875. Available from: https://www.nature.com/articles/nrg1204 [DOI] [PubMed] [Google Scholar]
- Long M, Langley CH. 1993. Natural Selection and the Origin of jingwei, a Chimeric Processed Functional Gene in Drosophila. Science (1979) [Internet] 260:91–95. Available from: 10.1126/science.7682012 [DOI] [PubMed] [Google Scholar]
- Lunzer M, Miller SP, Felsheim R, Dean AM. 2005. Evolution: The biochemical architecture of an ancient adaptive landscape. Science (1979) [Internet] 310:499–501. Available from: 10.1126/science.1115649 [DOI] [PubMed] [Google Scholar]
- Lynch M. 2005. Simple evolutionary pathways to complex proteins. Protein Science [Internet] 14:2217–2225. Available from: 10.1110/ps.041171805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. 2021. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol [Internet] 38:4647–4654. Available from: 10.1093/molbev/msab199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Marais DL, Rausher MD. 2008. Escape from adaptive conflict after duplication in an anthocyanin pathway gene. Nature 454:762–765. [DOI] [PubMed] [Google Scholar]
- Marger MD, Saier MH. 1993. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem Sci [Internet] 18:13–20. Available from: https://linkinghub.elsevier.com/retrieve/pii/096800049390081W [DOI] [PubMed] [Google Scholar]
- Maynard Smith J. 1970. Natural Selection and the Concept of a Protein Space. Nature 225:563–564. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Trask BJ. 2002. The complex structure and dynamic evolution of human subtelomeres. Nature Reviews Genetics 2002 3:2 [Internet] 3:91–102. Available from: https://www.nature.com/articles/nrg727 [DOI] [PubMed] [Google Scholar]
- Meger AT, Spence MA, Sandhu M, Matthews D, Chen J, Jackson CJ, Raman S. 2024. Rugged fitness landscapes minimize promiscuity in the evolution of transcriptional repressors. Cell Syst [Internet] 15:374–387.e6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2405471224000620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méheust R, Bhattacharya D, Pathmanathan JS, McInerney JO, Lopez P, Bapteste E. 2018. Formation of chimeric genes with essential functions at the origin of eukaryotes. BMC Biol [Internet] 16:30. Available from: 10.1186/s12915-018-0500-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger BP, Park Y, Starr TN, Thornton JW. 2024. Epistasis facilitates functional evolution in an ancient transcription factor. Elife [Internet] 12. Available from: https://elifesciences.org/articles/88737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meussdorfer F, Zarnkow M. 2009. Starchy Raw Materials. In: Esslinger HM, editor. Handbook of Brewing: Process, Technology, Markets. Weinheim: Wiley-VCH. p. 43–83. [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R, Teeling E. 2020. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37:1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Arnold FH. 1999. Exploring nonnatural evolutionary pathways by saturation mutagenesis: Rapid improvement of protein function. J Mol Evol [Internet] 49:716–720. Available from: 10.1007/PL00006593 [DOI] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, Fledel-Alon A, Tanenbaum DM, Civello D, White TJ, et al. 2005. A Scan for Positively Selected Genes in the Genomes of Humans and Chimpanzees. PLoS Biol [Internet] 3:e170. Available from: 10.1371/journal.pbio.0030170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijland JG, Driessen AJM. 2020. Engineering of Pentose Transport in Saccharomyces cerevisiae for Biotechnological Applications. Front Bioeng Biotechnol [Internet] 7:464. Available from: 10.3389/fbioe.2019.00464/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijland JG, Shin HY, de Waal PP, Klaassen P, Driessen AJM. 2018. Increased xylose affinity of Hxt2 through gene shuffling of hexose transporters in Saccharomyces cerevisiae. J Appl Microbiol [Internet] 124:503–510. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29240974 [DOI] [PubMed] [Google Scholar]
- Nishikawa KK, Hoppe N, Smith R, Bingman C, Raman S. 2021. Epistasis shapes the fitness landscape of an allosteric specificity switch. Nat Commun [Internet] 12:5562. Available from: https://www.nature.com/articles/s41467-021-25826-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurminsky DI, Nurminskaya M V., De Aguiar D, Hartl DL. 1998. Selective sweep of a newly evolved sperm-specific gene in Drosophila. Nature 1998 396:6711 [Internet] 396:572–575. Available from: https://www.nature.com/articles/25126 [DOI] [PubMed] [Google Scholar]
- O’Donnell S, Yue J-X, Saada OA, Agier N, Caradec C, Cokelaer T, De Chiara M, Delmas S, Dutreux F, Fournier T, et al. 2023. Telomere-to-telomere assemblies of 142 strains characterize the genome structural landscape in Saccharomyces cerevisiae. Nat Genet [Internet] 55:1390–1399. Available from: https://www.nature.com/articles/s41588-023-01459-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh EJ, Jin YS. 2020. Engineering of Saccharomyces cerevisiae for efficient fermentation of cellulose. FEMS Yeast Res 20. [DOI] [PubMed] [Google Scholar]
- Oh EJ, Kwak S, Kim H, Jin Y-S. 2017. Transporter engineering for cellobiose fermentation under lower pH conditions by engineered Saccharomyces cerevisiae. Bioresour Technol [Internet] 245:1469–1475. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0960852417308064 [DOI] [PubMed] [Google Scholar]
- Opulente DA, LaBella AL, Harrison M-C, Wolters JF, Liu C, Yonglin Li, Kominek J, Steenwyk JL, Stoneman HR, VanDenAvond J, et al. 2024. Genomic factors shape carbon and nitrogen metabolic niche breadth across Saccharomycotina yeasts. Science (1979) [Internet] 384. Available from: 10.1126/science.adj4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. 2007. Crystal Structure of an Ancient Protein: Evolution by Conformational Epistasis. Science (1979) [Internet] 317:1544–1548. Available from: 10.1126/science.1142819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH. 1998. Major Facilitator Superfamily. Microbiology and Molecular Biology Reviews [Internet] 62:1–34. Available from: 10.1128/MMBR.62.1.1-34.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Metzger BPH, Thornton JW. 2022. Epistatic drift causes gradual decay of predictability in protein evolution. Science (1979) [Internet] 376:823–830. Available from: 10.1126/science.abn6895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patthy L. 2003. Modular assembly of genes and the evolution of new functions. Genetica [Internet] 118:217–231. Available from: 10.1023/A:1024182432483 [DOI] [PubMed] [Google Scholar]
- Paulsen PA, Custódio TF, Pedersen BP. 2019. Crystal structure of the plant symporter STP10 illuminates sugar uptake mechanism in monosaccharide transporter superfamily. Nat Commun [Internet] 10. Available from: 10.1038/s41467-018-08176-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris D, Ubbelohde EJ, Kuang MC, Kominek J, Langdon QK, Adams M, Koshalek JA, Hulfachor AB, Opulente DA, Hall DJ, et al. 2023. Macroevolutionary diversity of traits and genomes in the model yeast genus Saccharomyces. Nat Commun [Internet] 14:690. Available from: https://www.nature.com/articles/s41467-023-36139-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, et al. 2018. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature [Internet] 556:339–344. Available from: https://www.nature.com/articles/s41586-018-0030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podgornaia AI, Laub MT. 2015. Pervasive degeneracy and epistasis in a protein-protein interface. Science (1979) [Internet] 347:673–677. Available from: 10.1126/science.1257360 [DOI] [PubMed] [Google Scholar]
- Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ. 2007. Empirical fitness landscapes reveal accessible evolutionary paths. Nature 2006 445:7126 [Internet] 445:383–386. Available from: https://www.nature.com/articles/nature05451 [DOI] [PubMed] [Google Scholar]
- Pokusaeva VO, Usmanova DR, Putintseva E V., Espinar L, Sarkisyan KS, Mishin AS, Bogatyreva NS, Ivankov DN, Akopyan A V., Avvakumov SY, et al. 2019. An experimental assay of the interactions of amino acids from orthologous sequences shaping a complex fitness landscape. PLoS Genet [Internet] 15:e1008079. Available from: 10.1371/journal.pgen.1008079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pougach K, Voet A, Kondrashov FA, Voordeckers K, Christiaens JF, Baying B, Benes V, Sakai R, Aerts J, Zhu B, et al. 2014. Duplication of a promiscuous transcription factor drives the emergence of a new regulatory network. Nat Commun [Internet] 5:1–11. Available from: 10.1038/ncomms5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Projecto-Garcia J, Natarajan C, Moriyama H, Weber RE, Fago A, Cheviron ZA, Dudley R, McGuire JA, Witt CC, Storz JF. 2013. Repeated elevational transitions in hemoglobin function during the evolution of Andean hummingbirds. Proc Natl Acad Sci U S A [Internet] 110:20669–20674. Available from: 10.1073/pnas.1315456110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistgaard EM, Löw C, Guettou F, Nordlund P. 2016. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol [Internet] 17:123–132. Available from: https://www.nature.com/articles/nrm.2015.25 [DOI] [PubMed] [Google Scholar]
- Roach MJ, Borneman AR. 2020. New genome assemblies reveal patterns of domestication and adaptation across Brettanomyces (Dekkera) species. BMC Genomics [Internet] 21:1–14. Available from: 10.1186/s12864-020-6595-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Bedford T, Lyons AM, Hartl DL. 2010. Adaptive impact of the chimeric gene Quetzalcoatl in Drosophila melanogaster. Proceedings of the National Academy of Sciences [Internet] 107:10943–10948. Available from: https://www.pnas.org/content/107/24/10943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Hartl DL. 2012. Chimeric Genes as a Source of Rapid Evolution in Drosophila melanogaster. Mol Biol Evol [Internet] 29:517–529. Available from: 10.1093/molbev/msr184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. 1985. Hydrophobicity of Amino Acid Residues in Globular Proteins. Science (1979) 229:834–838. [DOI] [PubMed] [Google Scholar]
- Rozpedowska E, Hellborg L, Ishchuk OP, Orhan F, Galafassi S, Merico A, Woolfit M, Compagno C, Piškur J. 2011. Parallel evolution of the make-accumulate-consume strategy in Saccharomyces and Dekkera yeasts. Nature Communications 2011 2:1 [Internet] 2:1–7. Available from: https://www.nature.com/articles/ncomms1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter JC, Igarashi K, Penttilä M. 2020. The Lipomyces starkeyi gene Ls120451 encodes a cellobiose transporter that enables cellobiose fermentation in Saccharomyces cerevisiae. FEMS Yeast Res [Internet] 20. Available from: https://academic.oup.com/femsyr/article/20/3/foaa019/5822765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH. 2000. Families of transmembrane sugar transport proteins. Mol Microbiol [Internet] 35:699–710. Available from: 10.1046/j.1365-2958.2000.01759.x [DOI] [PubMed] [Google Scholar]
- Saier MH, Reddy VS, Moreno-Hagelsieb G, Hendargo KJ, Zhang Y, Iddamsetty V, Lam KJK, Tian N, Russum S, Wang J, et al. 2021. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res [Internet] 49:D461–D467. Available from: 10.1093/nar/gkaa1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Tran C V., Barabote RD. 2006. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res [Internet] 34:D181–D186. Available from: 10.1093/nar/gkj001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salema-Oom M, Valadão Pinto V, Gonçalves P, Spencer-Martins I. 2005. Maltotriose Utilization by Industrial Saccharomyces Strains: Characterization of a New Member of the α-Glucoside Transporter Family. Appl Environ Microbiol [Internet] 71:5044–5049. Available from: 10.1128/AEM.71.9.5044-5049.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisyan KS, Bolotin DA, Meer M V., Usmanova DR, Mishin AS, Sharonov G V., Ivankov DN, Bozhanova NG, Baranov MS, Soylemez O, et al. 2016. Local fitness landscape of the green fluorescent protein. Nature 2015 533:7603 [Internet] 533:397–401. Available from: https://www.nature.com/articles/nature17995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra Colomer M, Funch B, Forster J. 2019. The raise of Brettanomyces yeast species for beer production. Curr Opin Biotechnol 56:30–35. [DOI] [PubMed] [Google Scholar]
- Shah P, McCandlish DM, Plotkin JB. 2015. Contingency and entrenchment in protein evolution under purifying selection. Proc Natl Acad Sci U S A [Internet] 112:E3226–E3235. Available from: 10.1073/pnas.1412933112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh K V., Haase MAB, Wisecaver JH, Wang M, Doering DT, et al. 2018. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell [Internet] 175:1533–1545.e20. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867418313321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh K V., Haase MAB, Wisecaver JH, Wang M, Doering DT, et al. 2018. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 175:1533–1545.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithers B, Oates M, Gough J. 2019. ‘Why genes in pieces?’—revisited. Nucleic Acids Res [Internet] 47:4970–4973. Available from: https://academic.oup.com/nar/article/47/10/4970/5475075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soylemez O, Kondrashov FA. 2012. Estimating the Rate of Irreversibility in Protein Evolution. Genome Biol Evol [Internet] 4:1213–1222. Available from: 10.1093/gbe/evs096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprouffske K, Wagner A. 2016. Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikant S, Gaudet R, Murray AW. 2020. Selecting for Altered Substrate Specificity Reveals the Evolutionary Flexibility of ATP-Binding Cassette Transporters. Current Biology 30:1689–1702.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambuk BU, Araujo PS. 2001. Kinetics of active alpha-glucoside transport in Saccharomyces cerevisiae. FEMS Yeast Res [Internet] 1:73–78. Available from: 10.1111/j.1567-1364.2001.tb00015.x [DOI] [PubMed] [Google Scholar]
- Stambuk BU, Batista AS, De Araujo PS. 2000. Kinetics of active sucrose transport in Saccharomyces cerevisiae. J Biosci Bioeng [Internet] 89:212–214. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1389172300887423 [DOI] [PubMed] [Google Scholar]
- Stambuk BU, Silva MA, Panek AD, Araujo PS. 1999. Active α-glucoside transport in Saccharomyces cerevisiae. FEMS Microbiol Lett [Internet] 170:105–110. Available from: 10.1111/j.1574-6968.1999.tb13361.x [DOI] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D. 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics [Internet] 24:637–644. Available from: 10.1093/bioinformatics/btn013 [DOI] [PubMed] [Google Scholar]
- Starr TN, Flynn JM, Mishra P, Bolon DNA, Thornton JW. 2018. Pervasive contingency and entrenchment in a billion years of Hsp90 evolution. Proceedings of the National Academy of Sciences [Internet] 115:4453–4458. Available from: https://www.pnas.org/content/115/17/4453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr TN, Picton LK, Thornton JW. 2017. Alternative evolutionary histories in the sequence space of an ancient protein. Nature [Internet] 549:409–413. Available from: https://www.nature.com/articles/nature23902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr TN, Thornton JW. 2016. Epistasis in protein evolution. Protein Science [Internet] 25:1204–1218. Available from: 10.1002/pro.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steindel PA, Chen EH, Wirth JD, Theobald DL. 2016. Gradual neofunctionalization in the convergent evolution of trichomonad lactate and malate dehydrogenases. Protein Science [Internet] 25:1319–1331. Available from: 10.1002/pro.2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. 2012. Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature [Internet] 490:361–366. Available from: https://www.nature.com/articles/nature11524 [DOI] [PubMed] [Google Scholar]
- Tawfik OK and S D. 2010. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu Rev Biochem [Internet] 79:471–505. Available from: 10.1146/annurev-biochem-030409-143718 [DOI] [PubMed] [Google Scholar]
- Trichez D, Knychala MM, Figueiredo CM, Alves SL, da Silva MA, Miletti LC, de Araujo PS, Stambuk BU. 2019. Key amino acid residues of the AGT1 permease required for maltotriose consumption and fermentation by Saccharomyces cerevisiae. J Appl Microbiol [Internet] 126:580–594. Available from: https://academic.oup.com/jambio/article/126/2/580/6715112 [DOI] [PubMed] [Google Scholar]
- Turner TL, Kim H, Kong II, Liu J-J, Zhang G-C, Jin Y-S. 2016. Engineering and Evolution of Saccharomyces cerevisiae to Produce Biofuels and Chemicals. In: Advances in Biochemical Engineering/Biotechnology. Vol. 162. Springer, Cham. p. 175–215. Available from: 10.1007/10_2016_22 [DOI] [PubMed] [Google Scholar]
- De Visser JAGM, Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nature Reviews Genetics 2014 15:7 [Internet] 15:480–490. Available from: https://www.nature.com/articles/nrg3744 [DOI] [PubMed] [Google Scholar]
- Voordeckers K, Brown CA, Vanneste K, van der Zande E, Voet A, Maere S, Verstrepen KJ. 2012. Reconstruction of Ancestral Metabolic Enzymes Reveals Molecular Mechanisms Underlying Evolutionary Innovation through Gene Duplication. PLoS Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Davejan P, Hendargo KJ, Javadi-Razaz I, Chou A, Yee DC, Ghazi F, Lam KJK, Conn AM, Madrigal A, et al. 2020. Expansion of the Major Facilitator Superfamily (MFS) to include novel transporters as well as transmembrane-acting enzymes. Biochimica et Biophysica Acta (BBA) - Biomembranes [Internet] 1862:183277. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0005273620301085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhang J, Alvarez C, Llopart A, Long M. 2000. The Origin of the Jingwei Gene and the Complex Modular Structure of Its Parental Gene, Yellow Emperor, in Drosophila melanogaster. Mol Biol Evol [Internet] 17:1294–1301. Available from: http://academic.oup.com/mbe/article/17/9/1294/994535 [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Seppey M, Simao FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva E V., Zdobnov EM. 2018. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol Biol Evol [Internet] 35:543–548. Available from: 10.1093/molbev/msx319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Delaney NF, DePristo MA, Hartl DL. 2006. Darwinian Evolution Can Follow Only Very Few Mutational Paths to Fitter Proteins. Science (1979) [Internet] 312:111–114. Available from: 10.1126/science.1123539 [DOI] [PubMed] [Google Scholar]
- Weinreich DM, Lan Y, Wylie CS, Heckendorn RB. 2013. Should evolutionary geneticists worry about higher-order epistasis? Curr Opin Genet Dev 23:700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Watson RA, Chao L. 2005. PERSPECTIVE: SIGN EPISTASIS AND GENETIC COSTRAINT ON EVOLUTIONARY TRAJECTORIES. Evolution (N Y) [Internet] 59:1165–1174. Available from: 10.1111/j.0014-3820.2005.tb01768.x [DOI] [PubMed] [Google Scholar]
- Wheeler LC, Harms MJ. 2021. Were Ancestral Proteins Less Specific? Malik H, editor. Mol Biol Evol [Internet] 38:2227–2239. Available from: 10.1093/molbev/msab019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler LC, Lim SA, Marqusee S, Harms MJ. 2016. The thermostability and specificity of ancient proteins. Curr Opin Struct Biol 38:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorke R, Krampe S, Weierstall T, Freidel K, Hollenberg CP, Boles E. 1999. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett [Internet] 464:123–128. Available from: 10.1016/S0014-5793(99)01698-1 [DOI] [PubMed] [Google Scholar]
- Wong ED, Miyasato SR, Aleksander S, Karra K, Nash RS, Skrzypek MS, Weng S, Engel SR, Cherry JM. 2023. Saccharomyces genome database update: server architecture, pan-genome nomenclature, and external resources. Genetics [Internet] 224:191. Available from: 10.1093/genetics/iyac191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfit M, Rozpȩdowska E, Piškur J, Wolfe KH. 2007. Genome survey sequencing of the wine spoilage yeast Dekkera (Brettanomyces) bruxellensis. Eukaryot Cell [Internet] 6:721–733. Available from: 10.1128/ec.00338-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. 1931. EVOLUTION IN MENDELIAN POPULATIONS. Genetics [Internet] 16:97–159. Available from: 10.1093/genetics/16.2.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. 1932. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. Proceedings of the 6th International Congress of Genetics 1:356–366. [Google Scholar]
- Wu NC, Dai L, Olson CA, Lloyd-Smith JO, Sun R. 2016. Adaptation in protein fitness landscapes is facilitated by indirect paths. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Li WH. 1998. What amino acid properties affect protein evolution? J Mol Evol 47:557–564. [DOI] [PubMed] [Google Scholar]
- Xie VC, Pu J, Metzger BPH, Thornton JW, Dickinson BC. 2021. Contingency and chance erase necessity in the experimental evolution of ancestral proteins. Elife [Internet] 10:1–87. Available from: https://elifesciences.org/articles/67336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Dean AM. 2019. Adaptive Landscapes in the Age of Synthetic Biology. Mol Biol Evol [Internet] 36:890–907. Available from: 10.1093/molbev/msz004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Tada T, Zhang H, Britt L. 2008. Elucidation of phenotypic adaptations: Molecular analyses of dim-light vision proteins in vertebrates. Proc Natl Acad Sci U S A [Internet] 105:13480–13485. Available from: 10.1073/pnas.0802426105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EM, Tong A, Bui H, Spofford C, Alper HS. 2014. Rewiring yeast sugar transporter preference through modifying a conserved protein motif. Proceedings of the National Academy of Sciences [Internet] 111:131–136. Available from: 10.1073/pnas.1311970111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J-X, Li J, Aigrain L, Hallin J, Persson K, Oliver K, Bergström A, Coupland P, Warringer J, Lagomarsino MC, et al. 2017. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat Genet [Internet] 49:913–924. Available from: https://www.nature.com/articles/ng.3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zastrow CR, Hollatz C, de Araujo PS, Stambuk BU. 2001. Maltotriose fermentation by Saccharomyces cerevisiae. J Ind Microbiol Biotechnol [Internet] 27:34–38. Available from: https://academic.oup.com/jimb/article/27/1/34-38/5990354 [DOI] [PubMed] [Google Scholar]
- Zhang J, Dean AM, Brunet F, Long M. 2004. Evolving protein functional diversity in new genes of Drosophila. Proceedings of the National Academy of Sciences [Internet] 101:16246–16250. Available from: 10.1073/pnas.0407066101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang Y ping, Rosenberg HF. 2002. Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nature Genetics 2002 30:4 [Internet] 30:411–415. Available from: https://www.nature.com/articles/ng852 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. S. eubayanus strains and plasmids used in this study.
Table S2. Oligonucleotides used in this study.
Table S3. Strain information for the 332 Saccharomycotina species. Column A (“Species name”) corresponds to Column C (“Species name”) of Table S1 from X.-X. Shen et al. 2018.
Table S4. Benchmark Universal Single-Copy Orthologs (BUSCO) statistics for existing and updated genome annotations of species in this study.
Data S1. Maximum likelihood phylogenetic trees of sugar porters and outgroup MFS proteins from Saccharomycotina genomes in Newick format.
Data S2. Consensus phylogenetic tree of Agt clade proteins in Newick format. Branch supports are from SH-aLRT test and ultrafast bootstrapping, respectively.
Data S3. Maximum likelihood phylogenetic tree of Agt clade proteins in Newick format. Branch supports are from SH-aLRT test and ultrafast bootstrapping, respectively.
Data Availability Statement
New genome annotations for Saccharomycotina species are available on figshare (confidential link will be updated to a public link prior to publication). This confidential link is provided for review purposes and will be updated to a public link prior to publication. Other data underlying this article are available in the article and in its online supplementary material.