

Abstract
We model the competition between homologous recombination and point mutation in microbial genomes, and present evidence for two distinct phases, one uniform, the other genetically diverse. Depending on the specifics of homologous recombination, we find that global sequence divergence can be mediated by fronts propagating along the genome, whose characteristic signature on genome structure is elucidated, and apparently observed in closely related Bacillus strains. Front propagation provides an emergent, generic mechanism for microbial “speciation,” and suggests a classification of microorganisms on the basis of their propensity to support propagating fronts.
Keywords: evolution, horizontal gene transfer, microbial speciation, recombination
The transfer of genetic material between microbial cells plays a crucial role in their evolution, and poses fundamental questions to microbiology. Is there a tree of life for microbes (1-3)? Are there bacterial species (4, 5)? What are the mechanisms driving their diversification (3, 6-8)? These questions arise because genetic transfer couples the evolution of different genomes in a way that not only complicates their dynamics but obscures their very identity over time: the evolution is communal. Whereas the communality of genome evolution is restricted to species in sexual organisms, the major elements of microbial evolution, genetic transfer followed by illegitimate or homologous recombination, point mutations, genome rearrangements, do not apriori imply sharp genetic isolation boundaries. If there are none, notions such as species and speciation, despite being widely used heuristically, are misleading. Also, it is not clear whether there are classes of microbes with qualitatively different modes of communal evolution and what are the cellular properties that distinguish between them.
Gene transfer results when foreign DNA is taken up from the environment (transformation), delivered by a virus (transduction), or acquired through a direct cell to cell exchange (conjugation), and then permanently incorporated in the recipient genome by homologous or illegitimate recombination. Homologous recombination, mediated by dedicated cellular machinery, plays a vital error correction role in genome replication (9) but also allows a foreign DNA fragment to replace a sufficiently similar portion of the recipient genome. The probability of successful replacement in homologous recombination is proportional to the exponential of the number of sequence mismatches (10), the mechanism being organism-specific (11-13). Illegitimate recombination can be mediated by bacteriophage integrases, selfish genetic elements, or occur by chance DNA breakage and repair, and allows the acquisition of entirely novel traits from evolutionary distant organisms. Illegitimate genetic transfer, also known as horizontal gene transfer (HGT), can be inferred from the genome data through its atypical sequence composition (6) and the phylogenetic incongruences it causes (14). Although the extent of HGT is under heated debate (2), it is clear that it is much less frequent than homologous recombination. Relative rates of homologous recombination and point mutations in natural populations have been estimated by sequence diversity studies using multilocus sequence typing data in recently formed bacterial strains (15, 16). The probability that a gene changes as a result of homologous recombination can be many times higher than that for point mutations. Another manifestation of the pervasiveness of homologous recombination is that the evolution of strains within many named species cannot be represented by a phylogenetic tree (17-19). Although the importance of genetic transfer, and homologous recombination in particular, is firmly established (20), there are only a few sharp predictions about the resulting modes of microbial evolution. Relevant to our work is the observation of Lawrence (4) that HGT islands locally inhibit recombination. He concludes that global genetic isolation can be achieved through the gradual accumulation of hundreds of HGTs.
The purpose of this paper is to explore the emergent properties of the collective evolution of closely related bacterial genomes. We model the interplay of homologous recombination and point mutation in bacterial populations and show that elementary genome changes such as HGT, genome rearrangements, and insertions or deletions can trigger diversification fronts that in evolutionary short time propagate along the bacterial genomes and eventually lead to global sequence divergence of subpopulations. The diversification fronts can occur even in the absence of natural selection and demonstrate that fast neutral evolution can have nontrivial long-term evolutionary consequences. The robustness of this mechanism is sensitive to some of the details of homologous recombination, and suggests a way to classify the spectrum of evolutionary modes in bacteria based on specific details of their homologous recombination mechanisms. We establish a methodology for analyzing closely related genomes and give evidence for a large-scale step-like variation of homologous recombination rates in the Bacillus cereus group, which might be a signature of a diversification front. Finally, we discuss the biological implications of the propagation of diversification fronts, as a mechanism for speciation, a force favoring the formation of sharp genetic isolation boundaries, and a dynamical barrier for HGT and genome rearrangements.
The details of homologous recombination are by now reasonably well understood (10, 11). There are at least two common obstacles to successful integration of a DNA fragment. First, the end of the fragment must find a short region (≈20 bp) of sequence identity with the target genome to initiate the process. Second, the cell's mismatch repair system can abort the recombination process if it encounters mismatches between the fragment and the portion of the genome being replaced. Both of these obstacles lead to an exponential decrease of recombination with sequence divergence. There are also potentially important variations in the mechanism. Whereas sequence identity at only one end is required in Escherichia coli, very high sequence similarity at both ends is needed in Bacillus (11, 12) and mismatch repair seems less important. In Streptococcus, the effect of mismatch repair is intermediate in strength (13) but the overall dependence of sexual isolation on sequence divergence is very close to that in Bacillus. In addition, the underlying basis for distinguishing between donor and recipient DNA can differ. Do these differences in the details translate into qualitatively different evolutionary behavior? If so, then the details of the homologous recombination mechanism could be an important criterion for classifying bacteria. The computational studies described here clarify which details are the relevant determinants of the long-term evolutionary dynamics.
Models
Based on the above considerations, we construct sets of model rules that describe the interplay between homologous recombination and point mutations.
There are N circular strings of length L written in an alphabet of n symbols.
Each position in each genome is subject to point mutations with rate m. A point mutation changes a symbol to any other symbol with equal probability.
Each genome receives fragments at an average rate r. Each fragment of size F is derived from an arbitrary position from an arbitrary donor genome and attempts to recombine at the same genome position in the recipient.
To be considered for incorporation the fragment must find an identical segment of length M at an arbitrary chosen end (model I) or at both ends (model II).
The probability of incorporation is exp(-αd), where α is a coefficient expressing the strength of the mismatch repair system and d is the pointwise sequence difference, i.e., d counts the number of mismatches between the fragment and the genome sequence it is about to replace. We will also consider model III, where rule 4 is absent.
The genome strings can be thought of as representatives of different strains possessing at least partial ecological distinctiveness, so that random genetic drift is much stronger within strains than between strains. With this interpretation, we do not include random genetic drift but it can be straightforwardly added.
Propagation of Diversification Fronts
In these models, mutation and recombination play opposing roles: point mutations generate sequence diversity in the population, whereas recombination tends to make sequences more similar. At high recombination rates, an initially uniform population will remain close to uniform; at high mutation rates, all sequences will diverge from each other. An important property of homologous recombination is that the probability that a recombination event is successful decreases with sequence divergence, and becomes negligible, even for small levels of divergence (10).
These considerations suggest that the uniform phase is metastable: even when recombination is strong enough to maintain a state of near uniformity, it will not succeed in bringing together sufficiently diverged sequences. The diverged phase, on the other hand, is stable. If there is a boundary between a stable and a metastable phase, the generic expectation is that the stable phase will grow at the expense of the metastable one, as shown in Fig. 1. This will happen because homologous recombination is inhibited not only in the diverged phase but also in a finite region flanking it within the uniform phase. Mutations will accumulate in the flanking region, and as a result the diverged phase will grow. We will refer to the boundary between the uniform and diverged phases as a diversification front. Therefore, the system has the potential to sustain the propagation of diversification fronts. Such diversification fronts can be nucleated by processes that create regions of sequence difference between genomes in the population, such as HGT, genome rearrangements, and deletions or insertions and have important biological consequences for the evolution and diversification of microbes, as will be discussed later.
Fig. 1.

Schematic illustrating the process by which a diversification front propagates along a genome in a selection neutral situation. In the vicinity of the HGT island, recombination is suppressed relative to point mutations, allowing point differences to build up in the region flanking the HGT island. The newly accumulated sequence differences lead to the extension of the region where recombination is inhibited and, in turn, an accumulation of point differences further away from the HGT island. The process repeats itself.
Simulations
To clarify this intuition, we performed a series of simulations of a population of interacting genomes, starting from two different initial conditions: (i) all sequences are the same, and (ii) all sequences are the same except for a strip, long compared with the typical size of recombining fragments, in which the sequences are random. We used three different models for the rules governing the dynamical behavior of homologous recombination: model I, requiring sequence identity at one end of the recombining fragment; model II, requiring sequence identity at both ends; and model III, with no requirement of sequence identity. The following central questions are addressed. Under what circumstances is there a well defined front propagation region; is it readily observable or is fine tuning of the parameters required? Do the three models differ qualitatively? To address these questions in a quantitative manner, we define an order parameter
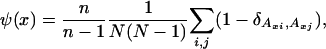 |
[1] |
where Axi denotes the letter at position x of genome i. The order parameter ψ measures the average difference in the population between the sequences at genome position x normalized so that ψ = 1 when the genomes are uncorrelated. This corresponds to the diverged phase of the system. In the opposite limit, ψ = 0, the genomes in the system are highly correlated, giving rise to the uniform phase of the system.
For each model, we studied the time evolution of the order parameter for different values of m/r and α. Typical values used for the other parameters are F = 500, M = 10, L = 10,000, N = 20, and n = 2. For each separate run, we measured ψ as a function of position within the genome and time. By varying α, we control the strength of the mismatch repair mechanism, and hence the success rate of recombination. The most important trend probed by our simulations is the behavior of the order parameter as a function of the ratio 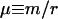 , the relative strength of point mutations versus recombination.
, the relative strength of point mutations versus recombination.
Results for Models I and III
For sufficiently low values of α, the equilibrium value of the order parameter varies gradually with μ = m/r, as shown in Fig. 2. The uniform and random strip initial conditions always relax to the same final state. The random strip simply dissolves, and no front propagation is observed. This situation arises when recombination is allowed almost regardless of the degree of sequence divergence.
Fig. 2.

The equilibrium value of the order parameter changes gradually with m/r in model I with α = 0, F = 500, M = 10, L = 10,000, N = 20, and n = 2. (Inset) A typical time evolution of the genome population. The vertical axis represents position along the genome and the color scale indicating the value of the order parameter (blue denoting uniform phase, red denoting diverged phase), whereas the horizontal axis is simulation time. A random strip dissolves without triggering a diversification front.
Above a threshold value of α, the uniform and diverged phases become distinct: for small values of μ, the order parameter is 0, and the system is genetically uniform. However, for large values of μ, the order parameter is close to unity, indicating that the system is genetically diverged. This transition appears to be sharp, as shown in Fig. 3. Furthermore, there is interesting dynamical behavior as a function of μ. For μ > μu, the uniform phase becomes unstable and the sequences diverge everywhere simultaneously. For μ < μs, the uniform phase is stable, and a finite region of diverged phase shrinks as a function of time, i.e., the uniform phase invades the diverged one. For μs < μ < μu, diversification proceeds through nucleation and growth of the diverged phase; in this parameter range, front propagation occurs.
Fig. 3.

Starting from a uniform state, the order parameter equilibrates to values close to 0 or 1 in model I with α = 0.4, F = 500, M = 10, L = 10,000, N = 20, and n = 2, indicating the existence of distinct uniform and diverged phases. The inset figures depict the genome population for the indicated value of m/r, as a function of time. The vertical axis represents position along the genome and the color scale indicates the value of the order parameter (blue denoting uniform phase, red denoting diverged phase), whereas the horizontal axis is simulation time. For μs < μ < μu, the random strip triggers a diversification front. For μ close to μu, spontaneous nucleation is possible.
From this behavior, we deduce the qualitative phase diagram presented in Fig. 4a. Model III, with no sequence identity requirement, shows qualitatively similar results (data not shown).
Fig. 4.

Phase diagram describing interplay between point mutation, recombination, and mismatch repair. (a) The phase diagram of models I and III. Distinct phases exist only above a threshold value of α and the width of the front propagation region, μu/μs, is <2. (b) The phase diagram of model II. Distinct phases exist for all values of α and the front propagation region is very wide: μu/μs > 100.
Results for Model II
For Model II, with sequence identity requirement at both ends, we observe front propagation even for α = 0. Moreover, the width 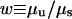 of the interval μs < μ< μu, where front propagation occurs, is very wide. Whereas we always observed w ≤ 2 for models I and III, for model II we could not even observe the point μu, and w > 100. This results in the phase diagram qualitatively represented on Fig. 4b. The front speed can be as high as several times the fragment size per average point mutation time near the transition to the diverged phase, and is a rapidly decreasing function of the recombination rate.
of the interval μs < μ< μu, where front propagation occurs, is very wide. Whereas we always observed w ≤ 2 for models I and III, for model II we could not even observe the point μu, and w > 100. This results in the phase diagram qualitatively represented on Fig. 4b. The front speed can be as high as several times the fragment size per average point mutation time near the transition to the diverged phase, and is a rapidly decreasing function of the recombination rate.
To summarize, there is a qualitative difference between the situation with no sequence identity requirement (model III) or sequence identity requirement at only one end (model I) and model II with sequence identity requirement at both ends. The difference is manifested in the phase diagram and the width of the front propagation region.
Microbe Classification
These theoretical predictions imply that we can classify microbial genomes according to the details of the recombination dynamics: class I, consisting of models I and III, and class II, consisting of model II. The distinguishing feature of the classes is whether the recombination dynamics requires sequence identity at both ends of the incorporated segment. For class II, as long as the uniformity of a population is maintained by homologous recombination, it will support propagating diversification fronts. For class I, diversification fronts are possible only within a narrow interval of the ratio of mutation to recombination rates and are therefore unlikely.
The existence of class I and class II indicates that the details of homologous recombination are important beyond the fact that the probability of recombination exponentially decreases with sequence divergence. Therefore, it is necessary to elucidate further the differences between homologous recombination mechanisms in different bacteria and work out their consequences for front propagation. For example, if mismatch repair is nick-directed and not methyl-directed (13), then more mismatches will be detected near the ends of the recombining fragments. This, in turn, will make front propagation more robust, because a greater fraction of the average homogenizing capability of recombination will be inhibited by a phase boundary. Also, if nonhomologous DNA loops formed during the recombination process are not corrected efficiently, then small deletions, insertions, slippage, and inversions would not trigger diversification fronts. Because micro rearrangements are presumably frequent, the efficiency of loop repair will be an important factor in determining the rate of nucleation of fronts. Finally, it is important to know whether and how the length of the incorporated fragments is dynamically dependent on the differences between the donor and recipient.
To seek evidence for the front propagation mechanism, we now compare available completely sequenced genomes of closely related microbes. The most direct evidence for front propagation from genome data alone would be an extended step-like pattern in the sequence divergence of closely related well aligned genomes, with the diverged region centered around a region of HGT, deletion, or genome rearrangement. The front profile reflects the different times after genetic isolation of different parts of the chromosome. Under conventional uniform molecular clock assumptions, it will be approximately linear, with a slope determined by the distance the front travels during the time it takes the sequences to fully diverge once recombination is inhibited. Slowly changing components of the sequence divergence, such as nonsynonymous substitutions, lead to more extended profiles.
Analysis of Genome Data
We consider the sequenced genomes in the genus Bacillus. It is in Bacillus that Majewski and Cohan (12) discovered the requirement for sequence identity at both ends, and our simulations indicate that front propagation is more likely to occur in such systems.
We obtained the complete genome sequences from the NCBI database, together with the positions and orientations of the known or predicted protein coding regions, tRNAs, and rRNAs. We globally aligned all pairs using the nucmer script of the mummer package (21) (nucmer -b 50 -g 300 -c 65 -mum), obtaining a list of well aligned regions for each pair. Three B. cereus strains (ATCC 10987, 14579, and ZK; refs. 22 and 23), three Bacillus anthracis strains (Ames, Ames Ancestor, and Sterne), and Bacillus thuringiensis serovar konkukian str. 97-27 genomes were close, highly colinear, and analyzed further. The three anthracis strains were practically identical, and only Ames was used in the analysis.
For each pair, we mapped the well aligned regions on one of the genomes, and constructed a series of coarse-grained profiles by sliding a window of width W along the genome while excluding nonaligned regions (resulting from insertions and deletions) from the averaging, as depicted graphically in Fig. 5. The profiles have gaps where the window covers less than a threshold fraction f of fW unambiguously aligned nucleotides. We used W in the range of 40,000 to 120,000 and f between 0.5 and 0.8. We looked at the coarse-grained profiles for the DNA point differences, as well as intergene, intragene, third codon, first and second codon, synonymous, and nonsynonymous (as defined in ref. 24) differences.
Fig. 5.

To construct the divergence profiles we first identify the well aligned regions (represented by color bars and arrows) using mummer, then map the differences (represented by red circles) onto the reference genome and slide a window of width W along the genome.
B. cereus ATCC 10987 exhibits a distinct step-like pattern of sequence difference when compared to B. cereus ZK (Fig. 6), B. anthracis Ames, and B. thuringiensis serovar konkukian str. 97-27. The pattern is also present in each of the other difference components: synonymous, nonsynonymous, gene, and intergene. What is the explanation for this pattern? Does it involve homologous recombination or not? Is it a result of a front propagation during the separation of B. cereus ATCC 10987 with the common ancestor of B. cereus ZK, B. anthracis Ames, and B. thuringiensis serovar konkukian str. 97-27?
Fig. 6.

The step-like profile of the sequence difference between B. cereus ATCC 10987 and B. cereus ZK obtained by sliding a 60,000 window with f = 2/3 along the genome.
To answer these questions, we first examined the variation of the nucleotide composition along the genome. Based on the GC and AT skews, the replication terminus is located at ≈2.6 Mb, away from the position of the difference profile step. The GC content varies smoothly along the genome and does not exhibit a step pattern. It has a minimum near the replication terminus.
The step pattern is partially correlated with the density of protein coding regions in the above genomes, the sequence differences being larger where the density is lower. However, because all difference components exhibit the pattern, it cannot be simply an artifact due to different proportions of gene and intergene regions with different mutation rates. Moreover, within the well aligned regions, the intergene regions are, on average, only ≈15% more divergent than protein coding regions and the gene density varies only in the 75-90% range. Therefore, the small differences in the proportions of sites with different mutation rates would have to have been somehow amplified if varying coding density were the underlying cause of the pattern. The nonaligned regions have a higher intergene fraction than aligned ones, suggesting a possible mechanism by which the density of protein coding regions can indirectly affect sequence divergence by a preferential accumulation of interstrain alignment gaps in intergene regions and a corresponding reduction of recombination rates.
Could it be that not just the proportion of site types, but the point mutation rates themselves vary gradually along the genome, leading to the above pattern? To answer this question, we turn to the distribution of lengths of maximal exact matches (DLMEM) between pairs of aligned sequences. If differences had accumulated by a Poisson mutational process, then we would expect an exponential distribution. Recombination, on the other hand, will lead to a broader distribution and, for example, a deviation from the Poisson statistics value (unity) for the ratio of the standard deviation and the mean (25).
Whether these deviations are statistically significant can be determined by comparing with the distribution of this ratio for the case without recombination.
We gathered DLMEM statistics for different well aligned regions. The ratio of the standard deviation and mean is significantly above 1, as shown in Fig. 7a. Moreover, there is a positive correlation between this ratio and the length of the uninterrupted well aligned regions, a trend that agrees with the notion that nonaligned parts inhibit recombination within the adjacent aligned regions.
Fig. 7.

DLMEM statistics resulting from the comparison of B. thuringiensis and B. cereus ATCC 10987. (a) The standard deviation and mean for the distribution of lengths of maximal exact matches within a well-aligned region is positively correlated with the length of the region. The actual data (blue dots) is contrasted with a null hypothesis with matched sequence difference for each region (red asterisk). (b) The standard deviation and mean DLMEM profile obtained by using a 120,000 window with f = 0.5 along B. thuringiensis exhibits a step-like pattern.
We then looked for evidence of different rates of homologous recombination along the chromosome by studying the changes in the DLMEM statistics in a sliding window. There is again a step-like pattern for the ratio of the standard deviation and the mean, as shown in Fig. 7b.
Deviation of the ratio of the standard deviation and the mean of a DLMEM is a sign of clustering of the differences along the chromosome. Are there reasons for clustering that do not involve homologous recombination? If different genes have very different evolution rates, then this can lead to apparent clustering. For example, different gene expression levels can lead to different synonymous mutation rates and an apparent clustering of differences within the weakly expressed genes. To control for this, we compare the DLMEM for neutral mutations with a null model with matched neutral divergence of each protein coding region separately. The pattern is present in the real data but almost completely disappears in the control. The residue is due to correlations of the divergences of adjacent proteins which are expected in the presence of homologous recombination. Because, presumably, there is no reason apart for recombination for clustering of synonymous substitutions within each gene separately, this test not only rules out genes with different evolutionary rates as an explanation but also gives confidence that the standard deviation over mean deviations from unity are predominantly due to homologous recombination.
Further evidence supporting the homologous recombination interpretation of the ratio of the standard deviation and the mean of DLMEM comes from contrasting the above observations with the results of the comparison between the completely sequenced Buchnera aphidicola strains APS, BP, and SG. Because these are intracellular parasites lacking the RecA gene, we expect no homologous recombination. Indeed, we find that there is no statistically significant deviation from unity of the standard deviation over mean and a highly uniform difference profile.
In summary, the above data indicate that there are large-scale step-like variations of the rates of homologous recombination along the analyzed microbial genomes, apparently consistent with the hypothesis that diversification proceeded by front propagation.
Discussion
Here, we discuss the consequences of the front propagation mechanism for the fate of bacteria that have acquired useful skills through HGT or have undergone a large-scale genome rearrangement. We argue that the front propagation mechanism facilitates global genetic isolation between strains, and, as such, is a mechanism for what may be loosely termed “speciation.” On the other hand, the front propagation mechanism reduces the chances that chromosomal changes, such as incorporation of HGTs or rearrangements, will be evolutionary successful, thus creating a dynamical barrier to the accumulation of such mutations in evolutionary time.
A bacterium can acquire a new skill by means of HGT. This can lead to the extinction of those bacteria that do not possess the beneficial (under appropriate selection pressure) HGT fragment. Alternatively, HGT can allow the invasion or foundation of a new biochemical niche, while being disadvantageous in the former one, or lead to specialization within the old niche. [Indeed, ecological distinctiveness without spatial isolation is not unusual for microbes. Even in the simplest of environments (monoculture lab experiments) coexisting strains emerge spontaneously (26). However, the creation of coexisting genotypes by HGT cannot properly be termed speciation, because the genotypes are not genetically isolated with respect to homologous recombination, except for a small region surrounding the HGT.]
The front propagation mechanism makes local isolation unstable, because the HGT event nucleates a diversification front, leading eventually to a global isolation of the carriers of the HGT event from the rest of the population. Therefore, ecological distinctiveness accompanied by local isolation is enough to generate speciation, even when homologous recombination is not reduced by the ecological distinctiveness. Note that this outcome is different from the one proposed by Lawrence (4), who suggested that global isolation is only achieved through the accumulation of hundreds of HGTs. Our work has demonstrated that even a single HGT or genome rearrangement can lead to global sequence divergence.
It is difficult to apply the biological species concept to groups of strains that are isolated at some loci and not at others (27). Because of diversification front propagation, a community of bacteria in which pairs of bacteria are genetically isolated at some loci, but not others, is unstable and tends to partition itself into groups which are globally isolated from each other with respect to homologous recombination. This is because genetically isolated regions will suppress recombination and trigger fronts into neighboring nonisolated regions. This instability will be even stronger if the different genomes are not colinear or do not have the same set of genes. Therefore, well defined genetic isolation boundaries emerge spontaneously through the front propagation mechanism even if there is no functional barrier to gene transfer.
What happens when a HGT or a rearrangement brings some advantage, but without enabling the recipient to adopt an entirely distinct ecological role? Achieving complete ecological distinctiveness might be a gradual process. In this case, the new genotype will be successful initially but not necessarily in the long run because it will be competing with other beneficial mutations at other loci that emerge throughout the population. Beneficial mutations trigger selective sweeps that can be either global, purging the diversity throughout some ecological niche or, because of homologous recombination, local, purging the diversity only around the locus of the beneficial mutation. In a population in which relative sequence uniformity is maintained by homologous recombination, local selective sweeps will be the norm. However, diversification fronts nucleated in the carriers of a HGT or a rearrangement will propagate by accumulation of neutral mutations and potentially lead to global genetic isolation of the carriers long before they have a chance to achieve a full ecological distinctiveness.
New strains are easily formed by readily absorbing foreign genetic material, rearranging the genomes, etc. However, they are typically short-lived entities, because they are excluded from the communal evolution following a diversification front propagation. Front propagation implies that the evolutionary rate of HGT accumulation is less than the rate suggested by looking at strains; this can be, in principle, tested against the data. This mechanism can also explain why gene order is highly conserved in some bacterial groups: there exists a dynamical barrier to the survival of rearranged genomes.
These considerations also have implications for the applicability of molecular phylogenetics and the ongoing debate about the nature of the impact of HGT on the tree of life. Front propagation limits the impact of HGT, reinforcing in a complementary way Woese's concept of a complexity barrier to HGT (1). Our argument is complementary because it does not rely on the nature of the interactions between the genes: there is a barrier to HGT arising from the population dynamics alone.
Our work leaves open a number of interesting issues related to the effect of highly conserved regions on front propagation. A large immutable region can present an impassable obstacle to front propagation. Candidates for such obstacles are rRNA operons, tRNA genes, and overlapping genes. Such regions lack the flexibility arising from the degeneracy of the genetic code. HGT islands inserted near front obstacles will lead to the diversification of a smaller fraction of the recipient genome, and have a greater chance to avoid extinction. Is there a correlation between evolutionary persistent HGTs and RNA gene positions? If a genome region is already diversified there is no penalty for the incorporation of another useful HGT island. Is there clustering of HGT islands? How is front propagation modified for clonal bacteria (19)? Finally, is front propagation beneficial? If front propagation obstacles are allowed to evolve or at least reposition themselves, what configuration of obstacles would result?
On the basis of computer simulations, we have suggested that the interplay between homologous recombination and point mutations can lead to propagating fronts, in whose wake a population of microbes becomes genetically diverse in evolutionary short time. Thus, even in the absence of selection pressure and ecological barriers to genetic exchange, gene-exchange boundaries can emerge as a statistical consequence of the detailed dynamics of recombination. We have presented a preliminary analysis of available genome data for the B. cereus group that is consistent with the presence of front propagation. These findings prompt speculations about the implications for the evolution and the classification of microbes.
Our model can be extended in a number of directions, including explicit accounting for the role of space, the existence of a nontrivial network of gene exchange connectivity, and the effects of sharing of beneficial mutations.
A promising approach to looking for diversification fronts is metagenomics data. Such data can give us a consensus genome for an ensemble of closely related organisms, inhabiting the same environment, and an estimate for the sequence diversity along the consensus genome (28). This diversity can be directly related to the order parameter ψ(x). A step-like variation in ψ(x) might be an indication of a diversification front.
Acknowledgments
We thank Phil Hugenholtz for bringing the work of Lawrence to our attention after the main results of our study had been obtained and Yoshi Oono for useful discussions. We also thank two anonymous referees for helpful suggestions that improved this work. This work was partially supported by National Science Foundation Grant NSF-EAR-02-21743.
Author contributions: K.V. and N.G. designed research; K.V. and N.G. performed research; K.V. and N.G. analyzed data; and K.V. and N.G. wrote the paper.
Abbreviations: HGT, horizontal gene transfer; DLMEM, distribution of lengths of maximal exact matches.
References
- 1.Woese, C. (2002) Proc. Natl. Acad. Sci. USA 99, 8742-8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurland, C., Canback, B. & Berg, O. (2003) Proc. Natl. Acad. Sci. USA 100, 9658-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gogarten, J., Doolittle, W. & Lawrence, J. (2002) Mol. Biol. Evol. 19, 2226-2238. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence, J. (2002) Theor. Pop. Biol. 61, 449-460. [DOI] [PubMed] [Google Scholar]
- 5.Cohan, F. (2001) Syst. Biol. 50, 513-524. [DOI] [PubMed] [Google Scholar]
- 6.Ochman, H., Lawrence, J. & Groisman, E. (2000) Nature 405, 299-304. [DOI] [PubMed] [Google Scholar]
- 7.Berg, O. & Kurland, C. (2002) Mol. Biol. Evol. 19, 2265-2276. [DOI] [PubMed] [Google Scholar]
- 8.Joyce, E., Chan, K., Salama, N. & Falkow, S. (2002) Nat. Rev. Genet. 3, 462-473. [DOI] [PubMed] [Google Scholar]
- 9.Radding, C. (2001) Proc. Natl. Acad. Sci. USA 98, 8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vulic, M., Dionisio, F., Taddei, F. & Radman, M. (1997) Proc. Natl. Acad. Sci. USA 94, 9763-9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majewski, J. & Cohan, F. (1998) Genetics 148, 13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majewski, J. & Cohan, F. (1998) Genetics 153, 1525-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majewski, J., Zawadzki, P., Cohan, F. & Dowson, C. (2000) J. Bacteriol. 182, 1016-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doolittle, W. (1999) Science 284, 2124-2128. [DOI] [PubMed] [Google Scholar]
- 15.Guttman, S. & Dykhuizen, D. (1994) Science 266, 1380-1383. [DOI] [PubMed] [Google Scholar]
- 16.Feil, E. & Spratt, B. (2001) Annu. Rev. Microbiol. 55, 561-590. [DOI] [PubMed] [Google Scholar]
- 17.Maynard Smith, J., Smith, N. H., O'Rourke, M. & Spratt, B. (1993) Proc. Natl. Acad. Sci. USA 90, 4384-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feil, E., Holmes, E., Bessen, D., Chan, M.-S., Dayi, N. P., Enright, M., Goldstein, R., Hood, D. W., Kaliai, A., Moore, C. E., Zhou, J. & Spratt, B. (2001) Proc. Natl. Acad. Sci. USA 98, 182-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milkman, R. (1997) Genetics 146, 745-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feil, E. (2004) Nat. Rev. Microbiol. 2, 483-495. [DOI] [PubMed] [Google Scholar]
- 21.Kurtz, S., Phillippy, A., Delcher, A., Smoot, M., Shumway, M., Antonescu, C. & Salzberg, S. (2004) Genome Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanova, N., Sorokin, A., Anderson, I., Galleron, N., Candelon, B., Kapatral, V., Bhattacharyya, A., Reznik, G., Mikhailova, N., Lapidus, A., et al. (2003) Nature 423, 87-91. [DOI] [PubMed] [Google Scholar]
- 23.Rasco, D., Ravel, J., Okstad, O. A., Helgason, E., Cer, R. Z., Jiang, L., Shores, K., Fouts, D., Tourasse, N., Angiuoli, S., et al. (2004) Nucleic Acids Res. 32, 977-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, W.-H. (1993) J. Mol. Evol. 36, 96-99. [DOI] [PubMed] [Google Scholar]
- 25.Sawyer, S. (1989) Mol. Biol. Evol. 6, 526-538. [DOI] [PubMed] [Google Scholar]
- 26.Treves, D., Manning, S. & Adams, J. (1998) Mol. Biol. Evol. 15, 789-797. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence, J. & Hendickson, H. (2003) Mol. Microbiol. 50, 739-749. [DOI] [PubMed] [Google Scholar]
- 28.Tyson, G., Chapman, J., Hugenholtz, P., Allen, E., Ram, R., Richardson, P., Solovyev, V., Rubin, E., Rokhsar, D. & Banfield, J. (2004) Nature 428, 37-43. [DOI] [PubMed] [Google Scholar]