

Abstract
Chronic kidney disease (CKD) impacts about 1 in 7 adults in the United States, but African Americans (AAs) carry a disproportionately higher burden of disease. Epigenetic modifications, such as DNA methylation at cytosine-phosphate-guanine (CpG) sites, have been linked to kidney function and may have clinical utility in predicting the risk of CKD. Given the dynamic relationship between the epigenome, environment, and disease, AAs may be especially sensitive to environment-driven methylation alterations. Moreover, risk models incorporating CpG methylation have been shown to predict disease across multiple racial groups. In this study, we developed a methylation risk score (MRS) for CKD in cohorts of AAs. We selected nine CpG sites that were previously reported to be associated with estimated glomerular filtration rate (eGFR) in epigenome-wide association studies to construct a MRS in the Hypertension Genetic Epidemiology Network (HyperGEN). In logistic mixed models, the MRS was significantly associated with prevalent CKD and was robust to multiple sensitivity analyses, including CKD risk factors. There was modest replication in validation cohorts. In summary, we demonstrated that an eGFR-based CpG score is an independent predictor of prevalent CKD, suggesting that MRS should be further investigated for clinical utility in evaluating CKD risk and progression.
Keywords: Chronic kidney disease, eGFR, Methylation risk score, Epigenetics
Subject terms: DNA methylation, Epidemiology, Chronic kidney disease
Introduction
More than 37 million adults (~ 15%) in the US are estimated to have chronic kidney disease (CKD), and ~ 800,000 have kidney failure1. There are significant racial disparities within CKD and kidney failure, particularly among African American (AA) adults, who experience a disproportionately higher burden of disease. AA adults are more likely to have CKD and develop organ failure at a rate nearly four-fold higher than white adults2. Additionally, while CKD often progresses asymptomatically in early disease, it is an independent risk factor for cardiovascular disease (CVD)2. Structural inequities contribute to these disparities (e.g., access to healthcare, socioeconomic status), as well as individual stress due to discrimination and racism. Yet the observed disparities are not fully explained by these factors, highlighting the need to understand the contributing biological influences on CKD3–5.
Heritability estimates of kidney traits are high, e.g., 44% for estimated glomerular filtration rate (eGFR) in a cohort of adults in the Netherlands6–8. Genetic markers associated with kidney function have been identified, and risk variants in some of these loci have been shown to have a higher prevalence in populations of African ancestry (e.g., APOL1). Still, known genetic variants account for < 10% of CKD phenotypic variation7,9,10. Furthermore, polygenic risk scores (PRS) that can incorporate up to millions of genetic variants associated with kidney disease—both known and unknown—are in development; yet, these risk models similarly explain a small fraction of the variation of kidney disease11–15. Therefore, it is important to consider the role of non-genetic heritable factors, such as epigenetics, in the unexplained, or “missing” heritability of CKD.
Epigenetic modifications are chemical alterations to DNA that can impact gene expression without changing the DNA sequence. These modifications are dynamic and may serve as biomarkers for the mechanisms by which one can understand health disparities due to harmful exposures. DNA methylation, a type of epigenetic modification, has been linked to CKD and other kidney traits in epigenome-wide association studies (EWAS)16–20. Moreover, EWAS have identified altered DNA methylation patterns at cytosine-phosphate-guanine (CpG) sites in association with stressful life events, air pollution, and neighborhood factors21–24. Notably, a study in 1.5 million individuals showed that DNA methylation explains a higher proportion of the heritability of kidney disease than gene expression25. And a Mendelian randomization analysis in ~ 35,000 adults identified multiple CpG sites that causally affected kidney function20. Thus, as a consequence of structural racism and social stress, AAs may be uniquely susceptible to epigenome-modifying exposures that increase CKD risk. Yet AAs are understudied with respect to CKD epigenomics.
Furthermore, while individual CpG sites may explain a small fraction of variance of their respective traits, studies have shown that incorporation of multiple methylation markers—similar to PRS—into risk algorithms may provide clinical utility26,27. Methylation risk scores (MRS) are an emerging tool unique from PRS given the potential for environmental exposures that may alter methylation levels. By computing risk based on the methylation level of relevant CpG sites, MRS can capture the potential impact of the exposome on gene expression. Because DNA methylation has been linked to CKD, MRS may be an important method of capturing both heritable and environmentally-driven risk for disease. To this end, we sought to develop and validate MRS for CKD. Because previous literature suggests that some social determinants of health (SDOH) may be linked to modifications in DNA methylation, we also assessed relationships between the CpG sites that comprised the optimized MRS and SDOH28–30.
Results
Figure 1 provides an overview of the study. Baseline characteristics of the study cohorts are presented in Table 1. In HyperGEN, the prevalence of CKD was 6%; in validation cohorts, CKD prevalence ranged from ~ 1 to 8%. The proportion of Stage G2 individuals ranged from 3 to 31%. HyperGEN EWAS summary statistics are presented in Supplemental Table 1. Selected CpG sites for each MRS are presented in Supplemental Table 2, and performance metrics of all MRS are summarized in Supplemental Table 3. The following CpG sites were used to construct the optimal MRS: cg02090160, cg11098259 (AQP9), cg12116137 (PRPF8), cg17944885 (ZNF788; ZNF20), cg00994936 (DAZAP1), cg02304370 (PHRF1), cg04460609 (LDB2), cg00501876 (CSRNP1), and cg04864179 (IRF5). MRS weights, selected from published eGFR EWAS, are summarized in Table 2.
Figure 1.

Study overview.
Table 1.
Baseline characteristics of MRS development and validation cohorts.
| N (%)/Mean (SD) | Development | Validation | |||||
|---|---|---|---|---|---|---|---|
| HyperGEN* (N = 608) |
GOLDN (N = 993) |
JHS* (N = 1709) |
MESA-AA* (N = 182) |
WHI-AS311 (N = 145) |
WHI-BAA23 (N = 2049) |
WHI-EMPC (N = 1172) |
|
| Age | 48.4 (11.1) | 49.4 (15.9) | 56.2 (12.3) | 60.9 (9.6) | 66.3 (7.1) | 64.5 (7.1) | 62.5 (6.9) |
| Sex, Male | 204 (33.6) | 474 (47.7) | 637 (37.3) | 77 (42.3) | 0 (0.0%) | ||
| Race, Black | 608 (100) | 0 (0) | 1,709 (100) | 181 (100) | 51 (35.2) | 665 (32.5) | 416 (35.5) |
| Current Smoker | 191 (31.4) | 81 (8.2) | 243 (14.2) | 34 (18.7) | 25 (17.2) | 208 (10.2) | 112 (9.6) |
| BMI | 32.4 (8.2) | 28.3 (5.5) | 32.0 (7.4) | 30.6 (5.6) | 29.8 (7.3) | 29.8 (6.1) | 29.8 (5.9) |
| Diabetes | 122 (20.1) | 77 (7.8) | 394 (23.1) | 61 (33.5) | 14 (9.7) | 210 (10.2) | 82 (7.0) |
| Hypertension | 460 (75.7) | 256 (25.8) | 1,002 (58.6) | 105 (57.7) | 31 (21.4) | 451 (22.0) | 181 (15.4) |
| CKD | 37 (6.1) | 6 (0.6) | 142 (8.3) | 15 (8.2) | 0 (0) | 19 (0.9) | 7 (0.6) |
| Stage G2 | 173 (28.5) | 127 (12.8) | 449 (26.3) | 56 (30.8) | 5 (3.4) | 119 (5.8) | 60 (5.1) |
| eGFR | 102.4 (29.5) | 123.0 (34.8) | 92.1 (25.4) | 90.9 (25.9) | 134.4 (31.1) | 136.5 (33.6) | 136.7 (30.0) |
*Indicates only AAs included. In GOLDN, all participants were EA. In WHI, selected participants were AA, EA, or Hispanic/Latino. CKD indicates individuals with eGFR < 60. Stage G2 indicates individuals with (60 ≤ eGFR < 90).
Table 2.
Overview of eGFR CpG sites selected for optimized MRS.
| Lead author (EWAS N) | CpG | Chr | Gene | Location | MRS weight | Gene function |
|---|---|---|---|---|---|---|
| Qiu (181)31 | cg02090160 | 2 | – | shore | − 0.499 | |
| cg11098259 | 15 | AQP9 | open sea | 0.501 | Aquaporin, H2O & urea transport, osmotic stress response | |
| Chu (4859)16 | cg12116137 | 17 | PRPF8 | open sea | 0.515 | RNA splicing, mRNA processing, proteolysis |
| cg17944885 | 19 |
ZNF788; ZNF20 |
shelf | − 0.484 | Pseudogene; transcription factor binding | |
| cg00994936 | 19 | DAZAP1 | island | 0.515 | RNA binding, placental development & spermatogenesis, cell proliferation | |
| Schlosser (33,605)20 | *cg02304370 | 3 | PHRF1 | shore | 1.09e−04 | Enables RNA polymerase binding; SLE, preeclampsia, fasting glucose |
| *cg04460609 | 2 | LDB2 | open sea | − 1.34e−04 | Transcription complex, embryo development | |
| *cg00501876 | 3 | CSRNP1 | shore | − 8.98e−05 | Transcription factor for Wnt signaling; apoptosis | |
| *cg04864179 | 7 | IRF5 | shore | − 8.58e−05 | Transcription factor for interferon activity; SLE & IBD |
*Causally associated w/ eGFR in Mendelian randomization. Direction refers to the direction of the association of the CpG methylation with the respective kidney trait in prior studies.
In HyperGEN, CKD cases had higher MRS than controls (p = 3.28E − 05, Fig. 2). In the continuous model, a 1 standard deviation (sd) increase in MRS was associated with 5.07 (95% CI 2.48–10.38, p < 0.0001) greater odds of CKD. Additionally, the prevalence of CKD increased with higher thresholds of MRS (Supplemental Fig. 1). Odds ratios showed even further stratification upon exclusion of G2s (Supplemental Table 4), and associations were robust after accounting for CKD risk factors (Supplemental Table 5). Results were consistent for eGFR, such that a 1 sd increase in MRS was associated with a 6.98 ± 1.75 mL/min/1.73 m2 decline in baseline eGFR (Supplemental Table 6). In the full model, the MRS explained 1.6% of the variance in CKD and 5.7% when G2s were excluded. Similarly, inclusion of the MRS improved the covariate model AUC from 0.88 to 0.92, with the MRS-only model boasting an AUC of 0.71.
Figure 2.

Distribution of optimized MRS in HyperGEN.
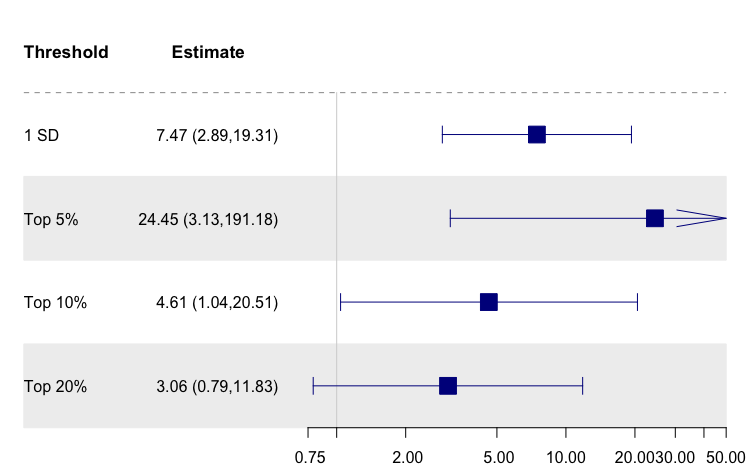
Among the validation cohorts, the continuous MRS was a modest predictor of CKD in JHS (OR[95% CI] 1.52[1.02,2.26], p = 0.038) and eGFR in WHI-AS311 (β(SE): − 8.84(4.06), p = 0.030) and WHI-BAA23 (β(SE): − 2.93(1.30), p = 0.024), excluding G2s. In MESA, GOLDN, and WHI-EMPC, MRS was not significantly associated with CKD or eGFR. In AA-only WHI models, MRS was not significantly associated with CKD or eGFR . Complete metrics of MRS performance among individual validation cohorts are summarized in Supplemental Table 7. In the meta-analysis of the validation cohorts, the MRS was significantly associated with CKD (OR[95% CI] 1.66[1.20,2.30], p = 0.003) and eGFR (β(SE): − 2.28(0.72), p = 0.002) for a 1 sd increase and at top 10 and 20% thresholds, but not the top 5% threshold (Fig. 3). Findings were consistent when we restricted analyses to AAs—OR[95% CI] 1.55[1.06,2.26], p = 0.029 for CKD and β(SE): − 2.19(1.17), p = 0.08 for eGFR for a 1 sd increase (Supplemental Table 7). We were unable to obtain threshold-specific estimates for the AA-only meta-analysis, as models for multiple cohorts did not converge in smaller strata.
Figure 3.

Forest plot of meta-analyzed MRS associations in validation cohorts. (A) Meta-analysis results for CKD associations in validation cohorts. (B) Meta-analysis results for eGFR associations in validation cohorts.
In secondary analyses of MRS associations with SDOH, three MRS CpG sites in JHS were marginally associated with individual and neighborhood-level factors: cg12116137 (PRPF8), cg17944885 (ZNF288; ZNF20), and cg00501876 (CSRNP1). Interestingly, the methylation pattern of cg17944885 (ZNF788; ZNF20) showed inverse association with “protective” SDOH—e.g., health insurance and higher SES—and positive associations with “detrimental” SDOH, e.g., violence and poverty. There was a similar pattern among associations with cg00501876 (CSRNP1). Results are summarized in Table 3.
Table 3.
Selected associations of SDOH with MRS CpG sites in JHS.
| Trait, B(SE) | cg12116137 (PRPF8) | cg17944885 (ZNF788; ZNF20) | cg00501876 (CSRNP1) |
|---|---|---|---|
| Individual | |||
| Alcohol use | n.s | n.s | − 0.0078 (0.0025) |
| Physical activity | n.s | n.s | 0.0034 (0.0015) |
| Insured | n.s | − 0.0056 (0.0028) | n.s |
| Depression | n.s | 0.0003 (0.0001) | − 0.0004 (0.0002) |
| Perceived stress | -0.0012 (0.0005) | n.s | n.s |
| Neighborhood | |||
| Problems | n.s | 0.0147 (0.0052) | n.s |
| SES | n.s | − 0.0007 (0.0003) | 0.0008 (0.0003) |
| Violence | n.s | 0.0203 (0.0077) | n.s |
| Poverty | n.s | 0.0170 (0.0075) | − 0.0226 (0.0095) |
| AA Density | n.s | 0.0195 (0.0036) | − 0.0098 (0.0045) |
Beta estimates presented for a CpG beta score ~ Trait. Linear mixed models were adjusted for age, sex, PC1-PC4, cell counts, and family (random). N.S.: not significant (p > 0.05).
Discussion
In this study, we utilized DNA methylation array data from 6,858 individuals who ranged from 18–85 years old, spanning from across the continental United States to develop MRS for CKD. Study-specific replication was not obtained in most of the validation cohorts, as the prevalence of CKD these cohorts (1–8%) is notably lower than national population estimates (~ 15%). Still, the meta-analyses suggests that the MRS is a significant predictor of prevalent CKD and eGFR for both AA and non-AA individuals. And to our knowledge, this study constitutes the first MRS developed for kidney function.
Unlike polygenic risk scores, which are commonly derived from genome-wide association study (GWAS) summary statistics using Bayesian methods and may include > 1 million variants, methods for developing MRS are less standardized and tend to apply a candidate-based approach for risk marker selection. For example, previous MRS in cancer studies have quantified absolute methylation at oncogenes via pyrosequencing of tumor tissue26,33,34. Another study derived CpG weights from model parameter estimates26. Other studies have utilized the least absolute shrinkage and selection operator (LASSO) penalized regression method and cross-validation to obtain MRS weights35–37. Furthermore, the type of methylation data used for MRS construction also varies from gene-based to CpG to mRNA methylation38. Still, the availability of array data (e.g., 450K and EPIC) allows CpG methylation to be one of the primary sources of MRS construction to date26,27,35–37,39–41.
Although MRS development methods are becoming more sophisticated—e.g., machine-learning and pruning and thresholding (P + T)—a consensus on methodological frameworks does not yet exist, in contrast with PRS41–44. Therefore, in the absence of these recommendations, we attempted to construct MRS using markers with substantial statistical and functional support. First, the selected CpG sites were among the top findings in their discovery EWAS and had to have a consistent direction of association in the development cohort. The only exceptions were CpG sites that had been causally linked to kidney function via Mendelian randomization; and even among those four markers, only one (cg00501876, CSRNP1) did not meet the first two criteria. While filtering CpG sites on the basis of similar effect direction in HyperGEN can contribute to overfitting, we also considered additional support and/or biological plausibility for top candidates for MRS construction: replication in at least 2 EWAS in the literature, causal effect on kidney function in Mendelian randomization, location near genes linked to kidney function, and/or significant association with traits or diseases that contribute to kidney function (e.g., aging, diabetes) in prior EWAS. Second, the published effect estimates served as CpG weights because of the large sample sizes of the discovery EWAS (N ~ 5000 to 35,000) compared to the MRS development cohort (N ~ 600). Third, we derived multiple MRS and applied additional sensitivity analyses to obtain the optimal combination of CpG sites. The optimized risk model remained a significant predictor of CKD and eGFR in a large validation sample (N > 6,000).
There is a dynamic relationship between the environment, epigenome, and biological function. And previous studies have identified associations between psychosocial stressors and aberrant DNA methylation near genes relevant to cardiovascular disease and other metabolic processes29,30,46. Similarly, studies have demonstrated a link between neighborhood-level factors—e.g., violence and social disadvantage—and DNA methylation variations21,23. In JHS, we detected modest associations between health behaviors and SDOH and CpG sites included in our MRS, although these associations were not significant after accounting for multiple comparisons. Notably, cg00501876 (CSRNP1) has been causally linked to eGFR in prior studies, and its methylation was associated with most of the factors we assessed. The CpG site is located ~ 2 Kb upstream from a CpG island in an enhancer region. Further, cg12116137 (PRPF8) and cg17944885 (ZNF788; ZNF20) are in enhancer and alternative splicing regions, respectively; and both sites are mapped to genes involved in DNA transcription and mRNA processing. Thus, their methylation may contribute to altered gene expression in kidney disease. Additional studies, such as mediation analyses, are needed to ascertain causal and functional implications of these regions.
To our knowledge, this study is among the first to develop a methylation algorithm for CKD. Strengths of this study include the large sample size of the validation population, as well as the robustness of our findings in a multi-racial sample with significant representation of AAs, who are disproportionately burdened with CKD. Additionally, although prior EWAS weights were based on associations with the 2009 eGFRcr which contains a race coefficient, we optimized the MRS using the updated race-free equation. Furthermore, for multiple cohorts included in this analysis, severe kidney disease was an exclusion criterion of the parent studies, resulting in the smaller sample sizes and that affected the power of the analysis of CKD. We attempted to redress this by meta-analyzing validation cohort results, as well as assessing MRS associations with eGFR as a continuous trait. And we observed consistent directions of effect in both overall and AA-specific meta-analyses. Still, previously established methylation associations with kidney traits should be reevaluated in the context of the new eGFR equations, as well as in studies with a kidney disease prevalence that is comparable to national estimates.
Future studies should evaluate the capacity of MRS to predict incident outcomes (e.g., end-stage renal disease). These algorithms should also be compared to existing clinical algorithms, such as the Kidney Failure Risk Eq. 47 And inclusion of other ‘-omics’ data, such as PRS, with MRS may also improve risk prediction. This multi-pronged approach may aid in the improvement of prevention of kidney failure and reduce disparities.
Methods
Study populations
The cohorts used for this study are summarized in Table 1. Complete descriptions of study design, enrollment, DNA methylation arrays, and quality control (QC) procedures are detailed in the Supplemental Methods. Briefly, a subset of AA participants who had been selected from the extremes of left ventricular mass for epigenomic profiling as part of ancillary study in the Hypertension Genetic Epidemiology Network (HyperGEN) were used to develop the MRS and optimize risk models. Participants from the Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN), Jackson Heart Study (JHS), Multi-Ethnic Study of Atherosclerosis (MESA), and Women’s Health Initiative (WHI) with methylation data were used for external validation of the MRS.
The studies included in this analysis were conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board (IRB) of their respective institutions. Written informed consent to participate in epigenetic studies was obtained from all participants involved in all studies. Additionally, the specific use of these data was reviewed by the IRB of the University of Alabama at Birmingham (15 April 2021) and determined to be not human subjects research.
CKD phenotyping
Prevalent CKD status was defined as eGFR < 60 mL/min/1.73 m2, which is concordant with CKD Stage 3 or higher, as defined by the National Kidney Foundation’s (NKF) Kidney Disease: Improving Global Outcomes (KDIGO) guidelines48. We used the 2021 CKD Epidemiology Collaboration (CKD-EPI) equations to calculate eGFR as a function of participant age, sex, and serum creatinine at baseline visit49.
MRS development
We compiled published EWAS of CKD, eGFR, albuminuria, and urine albumin-to-creatinine ratio (UACR), conducted between 2014 and 202116–18,20,31,32,50,51. We then conducted EWAS for these traits in HyperGEN (Supplemental Table 1). The criteria for selection in the MRS were as follows: if published CpG sites (1) were available in both Illumina 450K and EPIC methylation arrays; (2) demonstrated the same directions of effect in HyperGEN EWAS at p < 0.1; and/or (3) had been causally linked to kidney function in prior Mendelian randomization analyses (n = 19 CpGs). We further prioritized CpG sites that had additional statistical support and/or biological plausibility: replication in multiple EWAS, location near genes linked to kidney function, and/or significant association with non-kidney traits or diseases that may contribute to kidney function (e.g., aging, diabetes) (n = 11 CpGs). We then derived multiple MRS as a weighted sum () of the published parameter estimate for the CpG association multiplied by the beta score of the respective CpG for each participant (Supplemental Table 2). For parameter estimates that were generated from models in which CpG methylation levels had been transformed (e.g., M-value), the estimates were back-transformed to reflect a beta score weight. We obtained MRS using effect estimates from EWAS for CKD or eGFR (Supplemental Table 3). For MRS based on eGFR EWAS weights, we multiplied the MRS by − 1 to reflect a higher MRS association with decreasing eGFR. We then log-transformed MRS to have a normal distribution.
MRS optimization
We fit logistic and linear mixed effect regression models in SAS (version 9.4, ‘proc glimmix’ function) to evaluate associations between MRS and prevalent CKD and baseline eGFR in HyperGEN, respectively. In HyperGEN, eGFR was calculated from creatinine-only (2021 eGFRcr) equation. Models were adjusted for age, sex, recruitment center, left ventricular mass index, the first four principal components (PCs) of ancestry, and Houseman-estimated cell counts—CD4 T lymphocytes, CD8 T lymphocytes, natural killer (NK) cells, B cells, and monocytes with granulocytes as the reference—as fixed effects, as well as family relatedness as a random effect52. In sensitivity analyses, we further adjusted models for CKD risk factors: obesity, as measured by body mass index (BMI); smoking status; hypertension; and diabetes. We also excluded KDIGO CKD Stage G2—participants with 60 ≤ eGFR < 90 with mild kidney impairment who may already be on the pathway to severe disease—from the control group and refit the models with the same covariates as described.
For each MRS we developed, we compared the distribution of the score between cases and controls. We compared the liability R2—the variance of the outcome explained by the predictor that also accounts for discrepancies between cohort and population-level disease prevalence. We fit covariate-only models and calculated the area under the curve (AUC) to evaluate the extent to which inclusion of the MRS with known risk factors improved risk prediction. Finally, we calculated the sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). Additionally, PPV and NPV were adjusted for population prevalence of CKD (aPPV, aNPV), which was obtained from the United States Renal Data System (USRDS). USRDS prevalence was based on estimates from the National Health and Nutrition Examination Survey between 2017 and March 20202.
MRS validation
The optimal MRS was selected based on the liability R2 and area under the curve (AUC) in HyperGEN. In validation cohorts, the MRS was computed using the weighted sum equation as previously described. CpG sites that were not available in validation datasets were excluded from the MRS calculation. eGFR was calculated using the 2021 eGFRcr equation. GOLDN and JHS models were adjusted for the same covariates as the primary HyperGEN model. However, in MESA and WHI, we performed fixed effect logistic and linear regression (‘proc logistic’ and ‘proc genmod’ in SAS) because there were no related individuals in the methylation cohorts and thus, no random effect. Further, in MESA and WHI models, we conducted exact logistic regression due to small sample size and/or very low CKD prevalence. In all WHI analyses, we fit models without sex as a covariate (as all participants were female). We included study-specific covariates, e.g., study arm (clinical trial vs. observational study), as needed. Further, we subset WHI cohorts to obtain AA-specific estimates of MRS association with kidney function.
Once we obtained study-specific estimates for CKD and eGFR in the validation cohorts, we meta-analyzed the associations using the METASOFT program, totaling 6,250 individuals. We also evaluated AA-specific associations in a second meta-analysis, totaling 3,023 individuals from JHS, MESA, and WHI. We applied random-effects models to account for between-study heterogeneity, and we evaluated the MRS associations at 1 sd and thresholds of 5%, 10%, and 20%, as in the primary model. Studies whose models did not converge for a given outcome and/or threshold were excluded, and if more than two of the eligible cohorts were excluded, we did not conduct meta-analysis for that threshold. Due to these criteria, we only report 1 sd associations for the AA-specific meta-analysis.
Secondary analyses
In secondary analyses, we evaluated associations with individual-level factors (alcohol use, physical activity, depression, insurance status, perceived daily discrimination, and perceived stress) and neighborhood-level factors (socioeconomic status (SES); social cohesion; favorable food stores; problems, e.g., excessive noise; violence; poverty rate; and density of AA residents) in JHS. We fit linear mixed models adjusted for age, sex, PC1-PC4, Houseman-estimated cell counts, and family relatedness (random). Neighborhoods were defined according to the 2000 US Census tract at baseline visit. SES was calculated according to the Diez-Roux et al. method, which is a composite of housing values, education, and income levels55. Social cohesion was calculated based on self-reported perceptions of close knitness, trust, values, and safety among neighbors. Favorable food stores represented the number of supermarkets and fruit and vegetable markets within 3 miles. The problems domain included self-reported noise, traffic, lack of access to shopping and parks, litter, and sidewalk condition. The violence domain was similarly based on self-reported frequency of violent activity. Poverty rate was calculated as the percent of persons living below the federal poverty level within a census tract, and density of AA residents was defined as the percentage of Black residents within a census tract.
Supplementary Information
{kind=link}
Acknowledgements
The authors thank the other investigators, the staff, and the participants of the HyperGEN, GOLDN, JHS, MESA, and WHI studies and TOPMed Consortium for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org. The authors gratefully acknowledge the resources provided by the University of Alabama at Birmingham IT-Research Computing group for high performance computing (HPC) support and CPU time on the Cheaha compute cluster.
Author contributions
Conceptualization: A.C.J., B.A.H., H.K.T., N.A.L., M.R.I. Methodology: A.C.J., A.P., V.S., B.A.H., H.K.T., N.A.L., N.D.A., M.R.I. Data curation: A.C.J., A.P., V.S., N.S.C., B.M., D.A., D.K.A., L.A.L., E.M.L., B.A.Y., C.J.D., S.S.R., J.C.M., J.I.R., K.D.T., H.J.K., R.P.T., P.D., S.K., T.L., Y.L., W.C.J., D.J.V.D.B., N.F., S.L., C.P.M., P.B., S.H., E.A.W., M.R.I. Analysis: A.C.J., A.P., V.S., H.K.T., N.D.A., N.S.C., B.M. Writing—draft preparation: A.C.J., M.R.I. Writing—review and editing: A.C.J., A.P., V.S., B.A.H., H.K.T., N.A.L., N.D.A., N.S.C., B.M., D.A., D.K.A., L.A.L., E.M.L., B.A.Y., C.J.D., S.S.R., J.C.M., J.I.R., K.D.T., H.J.K., R.P.T., P.D., S.K., T.L., Y.L., W.C.J., D.J.V.D.B., N.F., S.L., C.P.M., P.B., S.H., E.A.W., M.R.I. Supervision: B.A.H,. H.K.T., B.A.L., M.R.I. Funding: A.C.J., M.R.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) grant R35HL155466 (MRI). ACJ was also supported by the National Institute of Diabetes and Digestive and Kidney Diseases (F31DK128990 and T32DK116672). Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung, and Blood Institute (NHLBI). Methylomics for "NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis" (phs001416) was performed at Keck MGC (HHSN268201600034I). Support for methylomics for JHS, HyperGEN, GOLDN, and WHI are detailed in the study-specific acknowledgments. Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL120393; U01HL120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. Support for HyperGEN (R01HL055673) and GOLDN (R01HL104135 and U01HL72524) studies were provided by NIH/NHLBI (DKA). The Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I, and HHSN268201800012I) contracts from the NIH/NHLBI and the National Institute on Minority Health and Health Disparities (NIMHD). The MESA projects are conducted and supported by the NIH/NHLBI in collaboration with MESA investigators. Support for the Multi-Ethnic Study of Atherosclerosis (MESA) projects are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1TR001881, DK063491, and R01HL105756. The WHI program is funded by the NIH/NHLBI through 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, 75N92021D00005. WHI-AS311 was supported by American Cancer Society award 125299-RSG-13–100-01-CCE. WHI-EMPC was supported by the National Institute of Environmental Health Sciences (NIH/NIEHS) through R01ES020836 (EAW).
Data availability
The raw epigenotypic and phenotypic data for HyperGEN, GOLDN, and MESA can be accessed upon request via the TOPMed Consortium. JHS and WHI data can be accessed upon approval from the respective coordinating centers.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-68470-z.
References
- 1.Prevention, C. F. D. C. a. Chronic Kidney Disease in the United States, 2021. (Atlanta, GA, 2021).
- 2.System, U. S. R. D. 2020 USRDS Annual Data Report: Epidemiology of kidney disease in the United States. (National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD).
- 3.Laster, M., Shen, J. I. & Norris, K. C. Kidney disease among African Americans: A population perspective. Am. J. Kidney Dis.72, S3–S7. 10.1053/j.ajkd.2018.06.021 (2018). 10.1053/j.ajkd.2018.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vart, P., van Zon, S. K. R., Gansevoort, R. T., Bultmann, U. & Reijneveld, S. A. SES, chronic kidney disease, and race in the U.S.: A systematic review and meta-analysis. Am. J. Prev. Med.53, 730–739. 10.1016/j.amepre.2017.06.036 (2017). 10.1016/j.amepre.2017.06.036 [DOI] [PubMed] [Google Scholar]
- 5.Vart, P. et al. National trends in the prevalence of chronic kidney disease among racial/ethnic and socioeconomic status groups, 1988–2016. JAMA Netw. Open3, e207932. 10.1001/jamanetworkopen.2020.7932 (2020). 10.1001/jamanetworkopen.2020.7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang, J., Thio, C. H. L., Gansevoort, R. T. & Snieder, H. Familial aggregation of CKD and heritability of kidney biomarkers in the general population: The lifelines cohort study. Am. J. Kidney Dis.77, 869–878. 10.1053/j.ajkd.2020.11.012 (2021). 10.1053/j.ajkd.2020.11.012 [DOI] [PubMed] [Google Scholar]
- 7.Canadas-Garre, M. et al. Genetic susceptibility to chronic kidney disease—Some more pieces for the heritability puzzle. Front. Genet.10, 453. 10.3389/fgene.2019.00453 (2019). 10.3389/fgene.2019.00453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jefferis, J., Pelecanos, A., Catts, V. & Mallett, A. The heritability of kidney function using an older Australian twin population. Kidney Int. Rep.7, 1819–1830. 10.1016/j.ekir.2022.05.012 (2022). 10.1016/j.ekir.2022.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freedman, B. I., Limou, S., Ma, L. & Kopp, J. B. APOL1-associated nephropathy: A key contributor to racial disparities in CKD. Am. J. Kidney Dis.72, S8–S16. 10.1053/j.ajkd.2018.06.020 (2018). 10.1053/j.ajkd.2018.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pattaro, C. et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun.7, 10023. 10.1038/ncomms10023 (2016). 10.1038/ncomms10023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan, A. et al. Genome-wide polygenic score to predict chronic kidney disease across ancestries. Nat. Med.28, 1412–1420. 10.1038/s41591-022-01869-1 (2022). 10.1038/s41591-022-01869-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma, J., Yang, Q., Hwang, S. J., Fox, C. S. & Chu, A. Y. Genetic risk score and risk of stage 3 chronic kidney disease. BMC Nephrol.18, 32. 10.1186/s12882-017-0439-3 (2017). 10.1186/s12882-017-0439-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thio, C. H. L. et al. Evaluation of a genetic risk score based on creatinine-estimated glomerular filtration rate and its association with kidney outcomes. Nephrol. Dial. Transplant33, 1757–1764. 10.1093/ndt/gfx337 (2018). 10.1093/ndt/gfx337 [DOI] [PubMed] [Google Scholar]
- 14.Yun, S. et al. Genetic risk score raises the risk of incidence of chronic kidney disease in Korean general population-based cohort. Clin. Exp. Nephrol.23, 995–1003. 10.1007/s10157-019-01731-8 (2019). 10.1007/s10157-019-01731-8 [DOI] [PubMed] [Google Scholar]
- 15.Zusi, C. et al. A renal genetic risk score (GRS) is associated with kidney dysfunction in people with type 2 diabetes. Diabetes. Res. Clin. Pract.144, 137–143. 10.1016/j.diabres.2018.08.013 (2018). 10.1016/j.diabres.2018.08.013 [DOI] [PubMed] [Google Scholar]
- 16.Chu, A. Y. et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun.8, 1286. 10.1038/s41467-017-01297-7 (2017). 10.1038/s41467-017-01297-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ammous, F. et al. Epigenome-wide association study identifies DNA methylation sites associated with target organ damage in older African Americans. Epigenetics16, 862–875. 10.1080/15592294.2020.1827717 (2021). 10.1080/15592294.2020.1827717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen, J. et al. Epigenetic associations with estimated glomerular filtration rate among men with human immunodeficiency virus infection. Clin. Infect Dis.70, 667–673. 10.1093/cid/ciz240 (2020). 10.1093/cid/ciz240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecamwasam, A. et al. DNA methylation profiling identifies epigenetic differences between early versus late stages of diabetic chronic kidney disease. Nephrol. Dial. Transplant10.1093/ndt/gfaa226 (2020). 10.1093/ndt/gfaa226 [DOI] [PubMed] [Google Scholar]
- 20.Schlosser, P. et al. Meta-analyses identify DNA methylation associated with kidney function and damage. Nat. Commun.12, 7174. 10.1038/s41467-021-27234-3 (2021). 10.1038/s41467-021-27234-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giurgescu, C. et al. Neighborhood environment and DNA methylation: Implications for cardiovascular disease risk. J. Urban Health96, 23–34. 10.1007/s11524-018-00341-1 (2019). 10.1007/s11524-018-00341-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Czamara, D. et al. Effects of stressful life-events on DNA methylation in panic disorder and major depressive disorder. Clin. Epigenetics14, 55. 10.1186/s13148-022-01274-y (2022). 10.1186/s13148-022-01274-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan, Q. et al. Exposure to violence, chronic stress, nasal DNA methylation, and atopic asthma in children. Pediatr. Pulmonol.56, 1896–1905. 10.1002/ppul.25372 (2021). 10.1002/ppul.25372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin, E. M. & Fry, R. C. Environmental influences on the epigenome: Exposure- associated DNA methylation in human populations. Ann. Rev. Public Health39, 309–333. 10.1146/annurev-publhealth-040617-014629 (2018). 10.1146/annurev-publhealth-040617-014629 [DOI] [PubMed] [Google Scholar]
- 25.Liu, H. et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat. Genet.54, 950–962. 10.1038/s41588-022-01097-w (2022). 10.1038/s41588-022-01097-w [DOI] [PubMed] [Google Scholar]
- 26.Hidalgo, B. A. et al. A 6-CpG validated methylation risk score model for metabolic syndrome: The HyperGEN and GOLDN studies. PLoS One16, e0259836. 10.1371/journal.pone.0259836 (2021). 10.1371/journal.pone.0259836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang, W. L. et al. A six-CpG panel with DNA methylation biomarkers predicting treatment response of chemoradiation in esophageal squamous cell carcinoma. J. Gastroenterol.52, 705–714. 10.1007/s00535-016-1265-2 (2017). 10.1007/s00535-016-1265-2 [DOI] [PubMed] [Google Scholar]
- 28.van der Laan, L. C. et al. Epigenome-wide association study for perceived discrimination among sub-Saharan African migrants in Europe—The RODAM study. Sci. Rep.10, 4919. 10.1038/s41598-020-61649-0 (2020). 10.1038/s41598-020-61649-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Barcelona Mendoza, V., Huang, Y., Crusto, C. A., Sun, Y. V. & Taylor, J. Y. Perceived racial discrimination and DNA methylation among african american women in the InterGEN study. Biol. Res. Nurs.20, 145–152. 10.1177/1099800417748759 (2018). 10.1177/1099800417748759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottschalk, M. G., Domschke, K. & Schiele, M. A. Epigenetics underlying susceptibility and resilience relating to daily life stress, work stress, and socioeconomic status. Front. Psychiatry11, 163. 10.3389/fpsyt.2020.00163 (2020). 10.3389/fpsyt.2020.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu, C. et al. Cytosine methylation predicts renal function decline in American Indians. Kidney Int.93, 1417–1431. 10.1016/j.kint.2018.01.036 (2018). 10.1016/j.kint.2018.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breeze, C. E. et al. Epigenome-wide association study of kidney function identifies trans-ethnic and ethnic-specific loci. Genome. Med.13, 74. 10.1186/s13073-021-00877-z (2021). 10.1186/s13073-021-00877-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmad, A. S. et al. A novel DNA methylation score accurately predicts death from prostate cancer in men with low to intermediate clinical risk factors. Oncotarget7, 71833–71840. 10.18632/oncotarget.12377 (2016). 10.18632/oncotarget.12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheol Kim, D. et al. Quantitative DNA methylation and recurrence of breast cancer: A study of 30 candidate genes. Cancer Biomark.11, 75–88. 10.3233/CBM-2012-0266 (2012). 10.3233/CBM-2012-0266 [DOI] [PubMed] [Google Scholar]
- 35.Barbu, M. C. et al. Epigenetic prediction of major depressive disorder. Mol. Psychiatry26, 5112–5123. 10.1038/s41380-020-0808-3 (2021). 10.1038/s41380-020-0808-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu, X., Ma, X. & Wu, C. A methylomics-correlated nomogram predicts the recurrence free survival risk of kidney renal clear cell carcinoma. Math. Biosci. Eng.18, 8559–8576. 10.3934/mbe.2021424 (2021). 10.3934/mbe.2021424 [DOI] [PubMed] [Google Scholar]
- 37.Dugué, P. A. et al. Methylation scores for smoking, alcohol consumption, and body mass index and risk of seven types of cancer. Int. J. Cancer.10.1002/ijc.34513 (2023). 10.1002/ijc.34513 [DOI] [PubMed] [Google Scholar]
- 38.Zhou, J. et al. A risk score model based on nine differentially methylated mrnas for predicting prognosis of patients with clear cell renal cell carcinoma. Dis. Markers2021, 8863799. 10.1155/2021/8863799 (2021). 10.1155/2021/8863799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei, J. H. et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat. Commun.6, 8699. 10.1038/ncomms9699 (2015). 10.1038/ncomms9699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cappozzo, A. et al. A blood DNA methylation biomarker for predicting short-term risk of cardiovascular events. Clin. Epigenetics14, 121. 10.1186/s13148-022-01341-4 (2022). 10.1186/s13148-022-01341-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu, A. K. et al. DNA methylation signature aberration as potential biomarkers in treatment-resistant schizophrenia: Constructing a methylation risk score using a machine learning method. J. Psychiatr. Res.157, 57–65. 10.1016/j.jpsychires.2022.11.008 (2022). 10.1016/j.jpsychires.2022.11.008 [DOI] [PubMed] [Google Scholar]
- 42.Chen, J. et al. Pruning and thresholding approach for methylation risk scores in multi-ancestry populations. Epigenetics18, 2187172. 10.1080/15592294.2023.2187172 (2023). 10.1080/15592294.2023.2187172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hao, L. et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat. Med.10.1038/s41591-022-01767-6 (2022). 10.1038/s41591-022-01767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huls, A. & Czamara, D. Methodological challenges in constructing DNA methylation risk scores. Epigenetics15, 1–11. 10.1080/15592294.2019.1644879 (2020). 10.1080/15592294.2019.1644879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matías-García, P. R. et al. DNAm-based signatures of accelerated aging and mortality in blood are associated with low renal function. Clin. Epigenetics13, 121. 10.1186/s13148-021-01082-w (2021). 10.1186/s13148-021-01082-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalinowski, J. et al. Stress overload and DNA methylation in African American women in the intergenerational impact of genetic and psychological factors on blood pressure study. Epigenet Insights15, 25168657221126310. 10.1177/25168657221126314 (2022). 10.1177/25168657221126314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramspek, C. L. et al. Kidney failure prediction models: A comprehensive external validation study in patients with advanced CKD. J. Am. Soc. Nephrol.32, 1174–1186. 10.1681/ASN.2020071077 (2021). 10.1681/ASN.2020071077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levey, A. S. et al. Nomenclature for kidney function and disease: executive summary and glossary from a kidney disease: Improving global outcomes (KDIGO) consensus conference. Kidney Dis. (Basel)6, 309–317. 10.1159/000509359 (2020). 10.1159/000509359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inker, L. A. et al. New creatinine- and cystatin c-based equations to estimate GFR without race. N. Engl. J. Med.10.1056/NEJMoa2102953 (2021). 10.1056/NEJMoa2102953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smyth, L. J., McKay, G. J., Maxwell, A. P. & McKnight, A. J. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics9, 366–376. 10.4161/epi.27161 (2014). 10.4161/epi.27161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smyth, L. J. et al. Assessment of differentially methylated loci in individuals with end-stage kidney disease attributed to diabetic kidney disease: An exploratory study. Clin. Epigenetics13, 99. 10.1186/s13148-021-01081-x (2021). 10.1186/s13148-021-01081-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform.13, 86. 10.1186/1471-2105-13-86 (2012). 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol.14, R115. 10.1186/gb-2013-14-10-r115 (2013). 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, Y. et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun.8, 14617. 10.1038/ncomms14617 (2017). 10.1038/ncomms14617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diez Roux, A. V. et al. Neighborhood of residence and incidence of coronary heart disease. N. Engl. J. Med.345, 99–106. 10.1056/NEJM200107123450205 (2001). 10.1056/NEJM200107123450205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw epigenotypic and phenotypic data for HyperGEN, GOLDN, and MESA can be accessed upon request via the TOPMed Consortium. JHS and WHI data can be accessed upon approval from the respective coordinating centers.