

Summary
High-grade ovarian cancer (HGOC) is a major cause of death in women. Early detection of HGOC usually leads to a cure, yet it remains a clinical challenge with over 90% HGOCs diagnosed at advanced stages. This is mainly because conventional biomarkers are not sensitive enough to detect the microscopic yet metastatic early lesions. In this study, we sequence the blood T cell receptor (TCR) repertoires of 466 patients with ovarian cancer and controls and systematically investigate the immune repertoire signatures in HGOCs. We observe quantifiable changes of selected TCRs in HGOCs that are reproducible in multiple independent cohorts. Importantly, these changes are stronger during stage I. Using pre-diagnostic patient blood samples from the Nurses’ Health Study, we confirm that HGOC signals can be detected in the blood TCR repertoire up to 4 years preceding conventional diagnosis. Our findings may provide the basis for future immune-based HGOC early detection criteria.
Graphical abstract
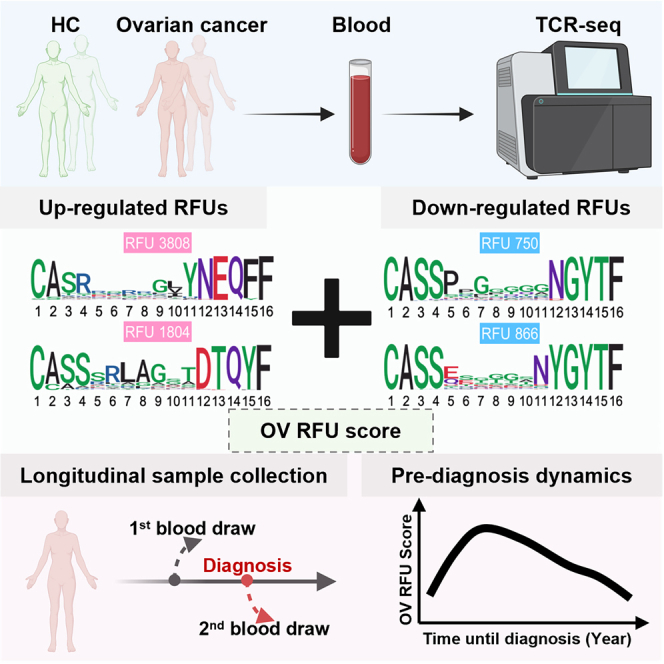
Highlights
-
•
Novel TCR repertoire analysis reveals T cell clones associated with HGOC
-
•
Transient immune response occurs during the early progression of HGOC
-
•
A TCR-based cancer score detects HGOC −4 to −2 years before conventional diagnosis
We made an unprecedented discovery that a strong and quantifiable change in the blood TCR repertoire that occurs 4 to 2 years before high-grade ovarian cancers could be diagnosed with conventional clinical tests. This finding provided insights for the development of immune-based biomarkers to detect early-stage ovarian cancers.
Introduction
Ovarian cancer represents 2.5% of all malignancies in women1 while causing 5% of cancer-related deaths.2 It consists of diverse histological subtypes, including serous, mucinous, clear cell, endometrioid, etc.3 High-grade ovarian cancer (HGOC), dominantly with serous histology,4 comprises over 70% of incident ovarian tumors1 and contributes to the disease’s high mortality rate. Ovarian tumors of other histological types are mostly low grade and much less lethal.5 Stage I high-grade serous carcinomas are confined within ovaries or fallopian tubes and are largely curable with complete surgical resection and chemotherapy (93%, 5-year relative survival).6 Conversely, patients with HGOC diagnosed at advanced stages have a 5-year survival of 31%.6 High-grade serous ovarian cancers frequently arise from a range of epithelial changes with p53 mutations, including serous tubal intraepithelial carcinoma (STIC) in the fallopian tube and atypical lesions in between p53 mutations and STIC.7 Current paradigms of serous carcinogenesis include a precursor lesion (STIC) with gradual progression to cancer and precursor metastasis into the peritoneal cavity.8,9 Unfortunately, conventional methods rarely facilitate the early detection or prevention of HGOCs, resulting in over 87% of cases being diagnosed at stage III or IV.
Given the apparent clinical benefit of detecting ovarian cancer early, noninvasive assays have been evaluated in large-scale, prospective screening trials, focusing on serum CA-125 levels10 and its changes over time,11 serum human epididymis protein 4,12,13 and transvaginal ultrasound.14 However, a recent longitudinal trial of over 200,000 subjects followed for more than 18 years showed no mortality benefit for HGOC among women routinely tested by one or a combination of these assays.15 Studies have further demonstrated that most conventional biomarkers have limited predictive ability until 6–12 months before diagnosis,16 possibly because HGOC primarily comprises microscopic lesions until late in its progression. Consequently, existing blood biomarkers or tumor imaging tests may lack sensitivity in detecting early-stage ovarian tumors.
Previously, we showed that early-stage cancers induce observable changes in the blood T cell receptor (TCR) repertoire.17 Although it remains unclear what causes these changes in patients with diverse genetic backgrounds, the concept of immunoediting18,19 may explain why signals emerge at early stages. Specifically, during the “elimination” phase, exposure to early tumor antigens could trigger a rapid expansion of cancer-associated T cells,20 leading to detectable signals in the TCR repertoire in circulating white blood cells. However, the vast diversity of the immune repertoire poses challenges in detecting such signals in large sample cohorts. Related to this purpose, we developed Geometric Isometry-based TCR AligNment Algorithm (GIANA)21 to perform isometric embedding for rapid TCR clustering. Although useful in finding disease-associated TCRs, GIANA embedding was incompatible with different TCR lengths and thus was not biologically relevant. Here, we introduced a trimer embedding framework to uniformly encode TCRs of different lengths based on sequence similarity, allowing quantitative dissection of TCR repertoire data into functional units. Subsequently, we collected preoperative blood samples from patients with ovarian tumors to identify TCR units that are enriched in patients with HGOC compared to women with benign ovarian tumors. Finally, we measured TCRs in pre-diagnostic blood specimens from patients diagnosed with ovarian cancer within 5 years after blood draw using samples from a large longitudinal cohort study and matched controls. Our analysis revealed transient but significant TCR repertoire changes occurring up to 4 years before conventional ovarian cancer diagnosis.
Results
Trimer embedding of TCRs and RFU definition
To quantitatively dissect the TCR repertoire data, we first obtained a numeric embedding of the β-chain complementarity-determining region 3 (CDR3β) region that preserved amino acid sequence similarity (Figure 1A). In brief, approximately 20 million TCRs from the public domain (Table S1) were clustered based on the variable gene (TRBV) and CDR3β sequences (Figure 1B) by GIANA21 to construct a trimer substitution matrix (Figures 1C and 1D). Approximated isometric embedding of each trimer was obtained using multidimensional scaling (Figure 1E), and the final embedding vector for each CDR3β was calculated by mean pooling of all the consecutive trimers in the amino acid sequence (Figure 1F).
Figure 1.

Trimer-guided embedding for TCRs and derivation of RFUs
(A) Method workflow. The first 3 steps describe the trimer-embedding Euclidean space, and the last two steps describe how repertoire functional units (RFUs) are defined.
(B) Massive clustering of TCRs from patients with diverse health conditions based on CDR3 amino acid sequence similarity.
(C) Illustration of replaceable trimers from small TCR clusters.
(D) Illustration of the trimer substitution matrix with each number represents the times a row trimer is replaced by the column trimer in a TCR cluster.
(E) Derivation of approximately isometric embedding for each trimer based on multidimensional scaling from the trimer substitution matrix in (D).
(F) Representation of each CDR3 sequence in the high-dimensional Euclidean space by averaging all the consecutive trimers.
(G) RFU definition by pooling 1.2 million TCRs from 120 individuals shown as t-distributed stochastic neighbor embedding plot. Colors denote distinct clusters with cluster centroids assigned by k-means.
To evaluate if trimer embedding could reflect TCR antigen specificity, we benchmarked this method using 1,031 TCRs with known specificity to 10 common immunogenic epitopes22 (Table S2). Specifically, we obtained the numeric embedding of each TCR and calculated the Euclidean distances for each pair of TCRs. This distance was used to predict if the pair of TCRs were specific to the same antigen. Indeed, we observed an area under the receiver operative characteristic curve (AUC) of 0.64, with better specificity at a lower-distance cutoff (Figures S1A and S1B). At a high specificity of 90%, this method reached a sensitivity of 30%, comparable to the state-of-art methods based on TCR sequence similarity.21,23,24,25 Importantly, as a similarity measure, trimer embedding preserves the “local specificity” of TCRs, i.e., if the distance of two TCRs continuously decreases to 0, then the probability that they share antigen specificity will approach 1. This property is guarded by the fact that TCR sequence similarity can be used as a surrogate for shared antigen specificity.23
With this property, we defined the “neighborhood” in the embedding space as local TCR clusters that likely recognize the same antigens. Such neighborhoods may carry disease-specific information. For example, a simple comparison between a B cell lymphoma sample and healthy control26 revealed several TCR neighborhoods enriched or depleted in a patient with lymphoma, each characterized by conserved CDR3 motifs (Figures S1C and S1D). Systematic investigation by pooling over 1 million TCRs from a healthy sample cohort27 revealed reproducible TCR clusters seen in genetically unrelated donors (Figure S1E). This observation indicated that the composition of the TCR repertoire might be conserved among different individuals. We thus divided the TCR space into 5,000 groups (Figure 1G), with over 84% of TCRs located within 0.018 Euclidean distance to the centroid, which is the cutoff of 90% specificity in the benchmark (Figure S1F). The group centroid was defined as a “repertoire functional unit” (RFU). Under this definition, TCRs assigned to each RFU have a 90% chance to recognize the same antigens, and thus RFUs can be viewed as the “genes” of a repertoire in the sense of antigen recognition. This approach allowed us to transform each TCR repertoire sample into a fixed-length numeric vector, i.e., the normalized TCR count of each RFU.
TCR repertoire landscape in patients with HGOC
We prospectively collected a discovery cohort of preoperative peripheral blood mononuclear cell samples from 213 women, including 67 patients with high-grade serous cancer, 49 with other types of histology (all low grade), and 97 with benign ovarian tumors. TCR repertoire sequencing data were obtained for each sample. Despite attempts to frequency match on age, the patients with cancer were significantly older than benign controls (Table S3). Therefore, we first investigated the impact of age over RFUs in the healthy individuals using publicly available TCR sequencing (TCR-seq) cohorts. We first analyzed the Emerson et al. cohort,27 which contained blood TCR repertoires of 666 healthy donors collected before 2017. We observed that the majority of RFUs were not age related, yet a small subset of RFUs showed strong negative correlations with age (Figure S2A). This observation was further confirmed using another large healthy cohort with 1,414 subjects28 (Figure S2B). Importantly, the RFUs with strong age associations in both cohorts were highly reproducible (Figure S2C), indicating that age-related RFUs are conserved in the general population. In addition to multivariable regression, these results warranted the direct exclusion of related RFUs to control for age in the downstream analysis.
We next visualized the TCR repertoires of all 213 individuals using the top 1,500 most variable RFUs (ranked by standard deviation) to obtain an overview of RFU distributions across different disease groups. Unsupervised hierarchical clustering revealed a distinguishable separation between HGOC and benign samples (Figure 2A), suggesting a global difference in the immune repertoire between these two conditions. Principal-component analysis (PCA) of the RFU matrix confirmed that PC1 is partially driven by disease categories (Figures 2B and 2C). In contrast, PC2 is influenced by race, with African American patients showing the largest separation from Asian patients (Figures 2D and 2E). To systematically investigate the differences of TCR repertoire between patients with HGOC and benign patients and identify RFUs as independent markers for HGOCs, we performed logistic regression adjusted for patient age and race for all 1,500 RFUs and observed significant results at false discovery rate (FDR) ≤0.2 (Figure 2F). In contrast, the comparisons between HGOC vs. low grade and low grade vs. benign yielded no significant RFUs, potentially due to the limited sample size and closer immunological backgrounds (Figure S2D). We then visualized the CDR3 motifs of the top up-/down-regulated ones in patients with HGOC (Figure 2G). We noted a conservative “RLAG” pattern at the 6th–9th positions of RFU 1804. CDR3s with this pattern, combined with the use of joining gene TRBJ2-3∗01 (DTQYF), have been reported to recognize the ELAGIGLTV epitope from melanoma antigen MART-1,29 which is reportedly expressed in ovarian neoplasms.30 No known cancer antigens were associated with the other three RFUs.
Figure 2.

Characterization of TCR repertoire landscape in patients with ovarian cancer
(A) Heatmap showing the distribution of the top 1,500 most variable RFUs of high-grade, low-grade, and benign patients.
(B) Distribution of patients with ovarian cancer and benign patients on the PCA plot calculated from the RFU-by-patient matrix.
(C) Violin plot showing the differences of PC1 across disease categories. Statistical significance was evaluated using one-way ANOVA.
(D) Distribution of patient races on the PCA plot.
(E) Violin plot showing the differences of PC2 across patient races. Statistical significance was evaluated using one-way ANOVA.
(F) Volcano plot showing the log odds ratio vs. FDR adjusted by Benjamini-Hochberg method. The odds ratio is estimated from logistic regression with disease status as a binary outcome, with each RFU being the covariate and adjusted for age and race. Blue: down-regulated; Red: up-regulated.
(G) Sequence logo analysis of selected top up-/down-regulated RFUs. Scale of logo height (y axis) was measured by bits ranging from 0 to 4.
RFU as a risk marker for HGOC
The above results indicated that selected RFUs are significantly altered in the blood repertoire of patients with HGOC compared to benign controls. We therefore proceeded to select a subset of RFUs to evaluate the risk of HGOC. First, we observed that although some RFUs reached high odds ratios (ORs) in the logistic regression, there was no difference in the median levels of these RFUs between patients with HGOC and benign patients (Figure 3A), suggesting the potential influence of outliers in parametric analysis. After the removal of such RFUs, we then defined the top 2 up- and down-regulated RFUs based on the ORs, which included RFUs 750, 866, 3808, and 1804. Notably, none of these RFUs were age related (Figure S3A), indicating that there is no need to correct age in a regression model. Hence, we no longer considered age as a confounder in the following analysis and arithmetically combined these RFUs to construct a predictor. We then surveyed the distributions of these RFUs across a wide spectrum of human cancers (Table S1). Interestingly, in addition to HGOC, RFU 750 was also down-regulated in melanoma and kidney cancer, where RFU 866 showed a similar yet insignificant trend (p = 0.24) (Figure 3B). On the other hand, RFU 1804 was also up-regulated in lung cancer, while RFU 3808 was higher in head and neck cancer (Figure S3B). Notably, for all four RFUs, healthy control samples from both children and adult cohorts had similar distributions to benign patients.
Figure 3.

Selected RFUs as biomarkers to distinguish HGOCs from benign ovarian lesions
(A) Selection criteria for the top informative RFUs. Odds ratios and ratios of medians (HGOC vs. benign) are displayed in a scatterplot. Odds ratios were calculated from the logistic regression described in Figure 2F. Blue color indicates ratio median <0.7 or >1.3.
(B) Boxplot showing the distributions of selected RFUs across multiple cancer types. Red arrow indicates ovarian cancer group. All analysis was performed using blood TCR repertoire samples from the public domain.
(C and D) Receiver operating characteristic (ROC) curves showing the prediction accuracy of up- or down-regulated RFUs to predict HGOCs against benign patients.
(E) Combination of up- and down-regulated RFUs as a joint biomarker: the OV RFU score. Statistical significance was evaluated using two-sided Wilcoxon test.
(F) Prediction accuracy of the OV RFU score illustrated by ROC curves.
(G) Donut plot showing the sample composition in the validation cohort, with total n = 97. The inner ring visualizes the percentage of patients with HGOC vs. benign patients, while the outer ring indicates the tumor stage for patients with HGOC and histological subgroups for benign patients.
(H) Performance of OV RFU score in the validation cohort.
(I) Violin plot showing the distributions of OV RFU scores across benign, stage I HGOC, and advanced HGOCs. Statistical significance was evaluated using Wilcoxon test.
(J) ROC curve for OV RFU score as a predictive biomarker for patients with stage I HGOC vs. benign patients.
We next investigated the human leukocyte antigen (HLA) allele associations for each of the four RFUs. We collected TCR repertoire samples from 1,208 individuals with HLA genotype information (Table S1) and performed sequence clustering using GIANA.21 For each TCR cluster, we tested if it was enriched for an HLA allele using Fisher’s exact test. 240,438 TCRs with significant HLA enrichment were identified at FDR = 0.05. Among these TCRs, the HLA-enriched TCRs belonging to these four RFUs were selected. To select the most enriched HLA alleles, we performed another enrichment analyses for each RFU. Specifically, for each allele and each RFU, we counted the number of TCRs specific to the HLA allele that have been assigned to this RFU and estimated the OR (Figure S3C). We made a cutoff at OR = 2 and selected the top enriched HLA alleles. Interestingly, each RFU was associated with at least 2 alleles, which made up a sizable fraction of the total population (Figure S3D).
We then tested the performance of the 4 RFUs as a potential risk predictor for HGOC against benign ovarian tumors. We directly used the sum of down-regulated RFUs (750 and 866) or up-regulated RFUs (3808 and 1804) as predictors and observed moderate predictive accuracy with an AUC slightly above 0.7 (Figures 3C and 3D). We combined the signals by using the up- minus the down-regulated RFU sums (1804 + 3808–866 – 750) as “OV RFU score.” As expected, this score is significantly higher in the HGOC vs. the benign group (Figure 3E), with an improved AUC of 0.77 (Figure 3F).
To evaluate the reproducibility of this score, we collected an independent validation cohort with 33 patients with HGOC and 64 benign patients (Figure 3G; Table S3). All blood samples were collected before surgery for TCR-seq data generation. We directly applied the above 4 RFU markers and calculated the OV RFU score. It separated patients with HGOC from benign patients with lower accuracy (AUC = 0.66) (Figure 3H). However, unlike the discovery cohort, the validation cohort included 5 stage I HGOC patient samples (Table S3). We investigated the distributions of RFU scores within stage I tumors and observed significantly higher scores than controls (Figure 3I). As a predictor, the RFU score reached an AUC = 0.81 for stage I HGOC vs. control (Figure 3J). Interestingly, the scores of late-stage HGOCs were lower than stage I tumors, although statistical significance was not reached due to the small sample size. These results indicated that the TCR repertoire may undergo nonlinear dynamic changes that peak during the early progression of ovarian malignancies.
Transient TCR repertoire changes in pre-diagnosis samples from patients with ovarian cancer
The above findings hold promise in early ovarian cancer detection, yet further evaluation using more HGOC samples is challenging due to the rarity of stage I patients at diagnosis. To address this issue, we utilized blood samples collected from the Nurses’ Health Studies (NHS/NHSII). These studies, with over 280,000 participants, have collected blood samples from over 60,000 women primarily in the 1990s and early 2000s and followed women for diagnosis of ovarian cancer within 5 years after blood draw.31 A subset of over 34,000 women gave two blood draws approximately 10–15 years apart. Among them, we identified 40 patients with ovarian cancer (33 patients with HGOC) with two blood draws before diagnosis, one remote (≥10 years) and one recent (≤5 years). We also assayed 38 healthy controls matched on age at the 1st and 2nd blood draws (Figure 4A; Table S4). All 156 NHS samples were sequenced for their TCR repertoires using the same commercial platform as the discovery and validation cohorts. We confirmed that within-individual dynamics is smaller than cross-individual variation,32 with the 2nd blood draw mostly similar to the 1st draw from the same person (Figures S4A and S4B). Given the higher RFU scores observed in patients with stage I HGOCs (Figure 3I), we hypothesized that a transient change may occur in the adaptive immune repertoire within 5 years prior to the conventional diagnosis, when the tumor is still at an early stage.
Figure 4.

Dynamic changes of blood TCR repertoire prior to conventional ovarian cancer diagnosis
(A) Diagram illustration of blood samples collected at 1st and 2nd time points for both subjects with cancer and control subjects.
(B) PCA analysis of samples at 1st or 2nd blood draw.
(C) Violin plot showing the distribution of PC1 or PC2 scores across putative ovarian risk factors. Statistical significance was evaluated using two-sided Wilcoxon test.
(D) Scatterplot showing the pre-diagnostic dynamics of OV RFU scores up to 5 years before conventional diagnosis. Loess smooth line was performed using only HGOC samples. Statistical significance was evaluated using permutation test.
(E) Boxplot showing the distributions of OV RFU scores in healthy controls and pre-diagnostic patients. Statistical significance was evaluated using two-sided Wilcoxon test. Red arrow indicates the location of 2–4 years before diagnosis. Adjusted p values for multiple hypothesis testing (padj) were performed using the Benjamini-Hochberg approach.
(F) Paired boxplots showing the increments of OV RFU scores (2nd time point – 1st time point) in both patient and control samples. Statistical significance was evaluated using paired two-sample Wilcoxon test.
(G) Scatterplot showing the pre-diagnostic dynamics of incremental OV RFU scores.
(H and I) Prediction accuracy of OV RFU scores or increment scores for pre-diagnostic patients with HGOC against healthy controls illustrated by ROC curves.
First, PCA plot of all samples at the 1st blood draw revealed no difference between patients with cancer (10–15 years before diagnosis) and healthy controls. At the 2nd blood draw, there is a slight yet nonsignificant difference at PC2 (p = 0.17, Figure 4B), suggesting that the cancer-induced changes were subtle, not driving global alterations in the TCR repertoire. We next examined the impacts of known ovarian cancer risk factors33,34 on the immune repertoire. There was no difference between patients with cancer and controls for menopausal status, tubal ligation, parity, or mycoplasma infection, with all p values exceeding 0.05 (Figure 4C). To avoid potential confounding effects, we removed all subjects with a family history of ovarian cancer in the downstream analysis.
We proceeded to investigate the dynamics of OV RFU scores in the pre-diagnostic samples. RFU scores using the 4 RFUs described above were directly calculated for the 37 passed-filter patients with ovarian cancer at the 2nd time point. Interestingly, RFU scores displayed a significantly nonrandom (p = 0.032) dynamic curve prior to diagnosis (−5 to 0 years) that matched our expectation (Figure 4D). Specifically, the score rapidly increased −5 to −4 years and peaked around −3 years before slowly decreasing until the time of diagnosis. We dissected this period into three intervals based on the shape of the curve: uphill (−5 to −4), peak (−4 to −2), and downhill (−2 to 0). Direct comparison of the RFU scores of each group with the scores of healthy controls revealed that the peak group was significantly higher than both time points of the control cohort (Figure 4E).
Sampling two time points for both cancer and control cohorts allowed us to track the TCR repertoire changes over time. The RFU scores of patients within the −4- to −2-year window showed significant increases when compared to their matched 1st time point, where the RFU scores of healthy individuals remained stable (Figure 4F). The increment of RFU scores between the two time points (denoted by “Δ”) displayed a nonlinear trend (Figure 4G). These results strongly supported our hypothesis that transient but strong immune changes occurred during the early development of ovarian cancer. We therefore evaluated the OV RFU score as a potential biomarker to detect ovarian cancer prior to its conventional diagnosis. If the disease is tracked within the −4- to −2-year window, then the RFU score would reach an AUC of 0.73, with 33% sensitivity at 95% specificity (Figure 4H). In contrast, the increment between time points (Δ) performed worse (Figure 4I), potentially because the random fluctuations in the TCR repertoire over 10 years reduced the signal/noise ratio.
To further validate the pre-diagnostic dynamics of OV RFU scores, we profiled another set of blood TCR repertoire samples from 41 pre-diagnostic (one time point, <5 years before diagnosis) patients with ovarian cancer (25 patients with HGOC) in the NHS cohort (Table S4). Patients with a family history of ovarian cancer were excluded. OV RFU scores of 4 RFUs were calculated for each patient, and the score was compared with pre-diagnostic years. We observed a nonlinear curve that peaks around 3.5 years before conventional diagnosis (Figure S4C). This curve matched the shape of the previous observation (p = 0.038), thus confirming the dynamic TCR repertoire change in the pre-diagnostic patients with HGOC.
Discussion
In this work, we analyzed 466 blood TCR-seq samples from patients with ovarian cancer and healthy/benign controls. Our computational analysis relied on a TCR embedding method specifically designed to quantify TCR repertoires. The RFU markers predicted in the discovery cohort were independently validated in two uniformly generated sample cohorts with age-matched controls, mitigating potential batch effects or data leakage.
TCRs exhibit heavy cross-reactivity, with one TCR recognizing ∼106 different peptides bounded by divergent HLA alleles.35 Interestingly, TCR antigen specificity prediction methods solely based on the similarity of βCDR3 sequences demonstrated a high clustering specificity disregarding the HLA background.23 Previous studies also showed that TCR specificities remain unchanged despite conservative replacement at certain positions of the CDR3 loops.36 Therefore, our estimated specificity for RFU must be considered in this context, i.e., TCRs from the same RFU could recognize 90% of the same pool of antigens at the given distance cutoff.
We anticipate that 5,000 TCR clusters or RFUs do not completely cover the diversity of the immune repertoire. The RFUs defined in this work are enriched for those with shared motifs across multiple individuals. Further, since the RFUs were defined using healthy donors, it is possible that disease-specific TCRs are underrepresented. TCR cluster analysis could be conducted on patients with cancer to potentially identify RFUs that are specific to tumor antigens. Regarding ovarian cancer detection, with sufficiently more samples, RFUs can be redefined using patients with HGOC or even from ovarian tumor-infiltrating T cells. HGOC-redefined RFUs might enhance prediction power when applied to prospectively collected patient cohorts.
It is unlikely that the four selected RFUs encompassed all the TCRs informative to HGOCs. The number of markers discovered in this study was limited by the small number of HGOC samples, particularly early-stage tumors. Further, all the patients with HGOC in the discovery cohort were diagnosed at an advanced stage, which, according to our observations above, yielded dampened immune response that reduced statistical power. More stage I HGOC samples with age-matched benign controls from future clinical studies would be ideal for uncovering informative RFUs and improving predictive accuracy. Furthermore, combining pre-diagnostic blood samples from multiple large prospective cohort studies could augment the power to identify early detection markers.37
Previous studies implied that the precursor lesion STIC develops approximately 6 years prior to HGOC.38,39 Accordingly, the 1st blood draw (>10 years) from the NHS cohort behaved similarly to the healthy controls (Figure 4B), while the 2nd (<5 years) exhibited dynamic changes that strikingly coincide with the immunoediting process.19 Specifically, during tumor initiation, recognition of tumor antigens results in a rapid expansion of the tumor-reactive T cells, creating the “uphill” part. Genome instability caused by p53 mutations likely produced more tumor neoantigens required for T cell recognition.40 The curve peaks when the tumor reaches equilibrium with the immune system. Further tumor progression creates a more immunosuppressive environment that slows down T cell expansion and causes immune exhaustion,41 ultimately leading to the contraction of tumor-reactive T cells in the blood. Despite these matched dynamics, validating the pre-diagnostic behavior of the immune repertoire in patients with ovarian cancer requires more clinical data from larger sample cohorts and more diverse populations.
Our analysis revealed different patterns in the TCR repertoire in HGOCs compared to low-grade and benign tumors. Unlike high-grade serous carcinomas with STIC origin, most low-grade tumors (mucinous, clear cell, endometrial, etc.) arise from the benign precursors.42 For example, while most mucinous cystadenoma are benign, approximately 10% of them become malignant borderline ovarian tumors (low grade). Hence, low-grade ovarian tumors may share a developmental lineage and environment with benign histology. In addition, as mentioned above, the mutation burden of HGOCs is much higher than low-grade tumors due to frequent p53 mutations, leading to more neoantigen presentation and elevated immune infiltration.43 These factors might collectively contribute to the distinct immunological landscape in HGOC tumors.
Together, our analyses support that ovarian tumor progression causes observable changes in the blood TCR repertoire, which are stronger at the early stage, when HGOC is more likely to be curable. This further suggests that immune biomarker discoveries in higher-stage tumors, where immunosuppression is prevalent, may not be efficient. Current testing approaches fail to detect HGOCs at an early stage.15 Therefore, immune-based approaches may offer advantages in detecting ovarian cancer within the −4- to −2-year window, crucial for HGOC early detection. The cost to generate the TCR-seq data for OV RFU score inference is approximately $200 per patient, close to a standard serum biomarker test.44 Finally, our study demonstrates the value of using prospectively collected samples in identifying biomarkers by quantifying these RFU changes, which may yield practical solutions for immune-based ovarian cancer early detection.
Limitations of the study
Our study remains exploratory in nature. First, although the pre-diagnostic curve of the RFU scores matched our findings in the validation cohort of cases and benign controls, there is no definitive evidence to support that women with samples collected at the peak phase had bona fide stage I HGOCs. Second, our analysis was performed at the RFU level, where the antigen specificity of individual TCRs was not investigated. This is mainly due to the lack of established T cell antigens from early-stage HGOCs, which can be improved with future immunogenomic research on ovarian cancers. Third, the association between TCR and disease likely depends on HLA genotype, which unfortunately is not available in our patient cohorts. Inclusion of HLA allele information in future investigations is expected to increase the prediction performance. Finally, as a screening biomarker, the sensitivity and specificity of the RFU score are far lower than what would be required to reach 10% positive predictive value,10 mainly due to the small sample size of this study. As a diagnostic tool, it is less accurate than established indices, such as Risk of Malignancy Algorithm (ROMA),45 Risk of Malignancy Index (RMI),46 or a recent approach based on metabolome of uterine fluid.47 Prospectively, the prediction accuracy of the OV RFU score might be improved by (1) including more TCR-seq samples from patients with early stage HGOC or (2) longitudinal sampling at smaller intervals before diagnosis, as TCR dynamics is more conserved within individuals. Future emphasis could be given to large clinical networks to recruit such patients or that have banked such samples.48
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kits | QIAGEN | Cat# 69504 |
| Deposited data | ||
| Processed RFU matrices | This paper | https://doi.org/10.5281/zenodo.11209912 |
| The top 10,000 TCRs from discovery and validation cohorts | This paper | https://doi.org/10.5281/zenodo.11204147 |
| Full list of public datasets used in this paper | N/A | Table S1 |
| TCR-seq data for Yellow Fever Virus samples | DeWittt et al.49 | PMID: 25653453 |
| TCR-seq data for Tuberculosis samples | DeWittt et al.50 | PMID: 29914888 |
| TCR-seq data for Multiple Sclerosis samples | Emerson et al.51 | PMID: 23428915 |
| TCR-seq data for Rheumatoid Arthritis samples | Savola et al.52 | PMID: 28635960 |
| TCR-seq data for Bladder Cancer samples | Snyder et al.53 | PMID: 28552987 |
| TCR-seq data for Lung Cancer and Brain Metastasis samples | Mansfield et al.54 | PMID: 29391594 |
| TCR-seq data for Breast Cancer samples | Page et al.55 | PMID: 27587469 |
| TCR-seq data for Cervical Cancer, Healthy Donor samples | Kuhs et al.56 | PMID: 29385144 |
| TCR-seq data for Healthy Donor samples | Nolan et al.28 | PMID: 32793896 |
| TCR-seq data for Colorectal Cancer samples | Le et al.57 | PMID: 28596308 |
| TCR-seq data for Melanoma samples | Robert et al.58 | PMID: 24583799 |
| TCR-seq data for Glioma samples | Robert et al.59 | PMID: 26968205 |
| TCR-seq data for Early-stage Breast Cancer samples | Beausang et al.60 | PMID: 29138313 |
| TCR-seq data for Breast Cancer samples | Page et al.61 | PMID: 31831558 |
| TCR-seq data for Healthy Elderly samples | Lindau et al.62 | PMID: 30541882 |
| TCR-seq data for Healthy Donor (GVHD patients) samples | Kanakry et al.63 | PMID: 27213183 |
| TCR-seq data for Ovarian Cancer, Pancreatic Cancer samples | Beshnova et al.17 | PMID: 32817363 |
| TCR-seq data for Leukemia samples | Suessmuth et al.64 | PMID: 25852054 |
| TCR-seq data for Lung Cancer samples | Formenti et al.65 | PMID: 30397353 |
| TCR-seq data for Lung Cancer samples | Reuben et al.66 | PMID: 32001676 |
| TCR-seq data for Melanoma samples | Robert et al.58 | PMID: 24583799 |
| TCR-seq data for Glioma samples | Robert et al.59 | PMID: 26968205 |
| TCR-seq data for Early-stage Breast Cancer samples | Beausang et al.60 | PMID: 29138313 |
| TCR-seq data for Breast Cancer samples | Page et al.61 | PMID: 31831558 |
| TCR-seq data for Healthy Elderly samples | Lindau et al.62 | PMID: 30541882 |
| TCR-seq data for Healthy Donor (GVHD patients) samples | Kanakry et al.63 | PMID: 27213183 |
| TCR-seq data for Ovarian Cancer, Pancreatic Cancer samples | Beshnova et al.17 | PMID: 32817363 |
| TCR-seq data for Leukemia samples | Suessmuth et al.64 | PMID: 25852054 |
| TCR-seq data for Melanoma samples | Valpione et al.67 | PMID: 32110781 |
| TCR-seq data for Ovarian Cancer samples | Emerson et al.68 | PMID: 24027095 |
| TCR-seq data for Pancreatic Cancer samples | Stromnes et al.69 | PMID: 29066497 |
| TCR-seq data for Melanoma samples | Tumeh et al.70 | PMID: 25428505 |
| TCR-seq data for Healthy Donor, Multiple Sclerosis samples | Ramien et al.71 | PMID: 31644905 |
| TCR-seq data for Renal Cell Carcinoma samples | Chow et al.72 | PMID: 32900949 |
| TCR-seq data for Healthy Donors samples | Emerson et al.73 (batch 1) | PMID: 28369038 |
| TCR-seq data for Healthy Donors samples | Emerson et al.73 (batch 2) | PMID: 28369038 |
| TCR-seq data for Hochkin lymphoma and healthy control samples | Cader et al.74 | PMID: 32778827 |
| TCR-seq data for Healthy Donor (Pre-adult) samples | Mitchell et al.75 | PMID: 35998036 |
| TCR-seq data for Lung Cancer samples | Creelan et al.76 | PMID: 34385708 |
| TCR-seq data for Head and Neck Cancer samples | Liu et al.77 | PMID: 34755131 |
| TCR-seq data for Type 1 diabetes samples | Mitchell et al.75 | PMID: 35998036 |
| TCR-seq data for Mycobacterium tuberculosis infection samples | Musvosvi et al.78 | PMID: 36604540 |
| TCR-seq data for Melanoma samples | Valpione et al.67 | PMID: 32110781 |
| Software and algorithms | ||
| R codes for RFU calculation | This paper | https://doi.org/10.5281/zenodo.11209912 |
| R version 4.3.2 | The R Foundation | https://www.r-project.org/ |
| BioRender | biorender.com | https://www.biorender.com/ |
Resources availability
Lead contact
Further information and requests for resources, reagents and codes should be directed to and will be fulfilled by the lead contact, Dr. Bo Li (lib3@chop.edu).
Materials availability
This study did not generate any new unique reagents or models.
Data and code availability
The TCR-seq datasets for the discovery and validation cohorts generated in this study are publicly available on Zenodo at https://doi.org/10.5281/zenodo.11204147. According to standard controlled access procedures, submitted written applications to request use of NHS/NHSII data/resources will be reviewed by External Collaborations Committee to verify that the proposed use maintains the protection of the privacy of participants and the confidentiality of the data. All investigators wishing to use NHS/NHSII data are asked to submit a brief description of the proposed project at the following URL: https://www.nurseshealthstudy.org/researchers. The lead contact author (B.L.) will assist the reader in communication with the NHS committee to access the data. R codes for RFU calculation are deposited in Zenodo at the following https://doi.org/10.5281/zenodo.11209912. The codebase is also available at GitHub: https://github.com/s175573/RFU. Processed RFU matrices for reproducing the results of this work are available on Zenodo at: https://doi.org/10.5281/zenodo.11209912. Any additional information is available from the lead contact author upon request.
Experimental model and study participant details
Ovarian cancer patient cohorts and Nurses’ Health Study samples
Women present with ovarian tumors were consented at Parkland Hospital at UT Southwestern Medical Center (UTSW) between 2019 and 2022. No stage-I HGOC patients were collected during this period. Blood samples were collected prior to surgeries and stored in EDTA tubes in −80° freezer. Tumor histology, including benign or malignant, was available after pathological verification. Sample collection was approved by the Institutional Review Boards (IRB) with protocol number STU-2020-442. Informed consent was obtained from all patients before sample collection. All samples collected at UTSW were used as the discovery cohort. Buffy coat samples of patients in the validation cohort were purchased from Accio Biobank Online in 2021.
Additional samples were obtained from the Nurses’ Health Studies (NHS/NHSII), two large prospective cohorts starting in 1976 (NHS) and 1989 (NHSII), with over 238,000 women. Between 1989 and 1990, 32,826 NHS participants donated self-collected blood samples, which were shipped on ice via courier where and processed into plasma, red blood cell, and white blood cell components; a second collection in 2000–2002 from over 19,000 of these women used similar protocols. Similarly, between 1996 and 1999, 29,611 NHSII participants donated blood samples; a second collection occurred from 2011 to 2014. Cases of ovarian cancer were identified via self-report on biennial questionnaires, report of family members, or via the National Death Index. Medical records or reports from cancer registries were used to confirm the diagnosis. Cases were matched to controls who were alive and had at least one ovary at the time of the case diagnosis and matched on age, menopausal status, date and time of blood collection, fasting status, and hormone therapy use. For this analysis, we assayed both the first and second blood draw samples from cases that were diagnosed within 5 years after the second blood draw and their matched controls. De-identified patient information, including age at blood draws, age at diagnosis, tubal ligation status, parity, menopausal status, and other ovarian cancer risk factors were provided for the analysis.
Method details
Description of TCR repertoire samples and preprocessing
All TCR repertoire sequencing samples that were not produced from this study were accessed from the immuneAccess database managed by Adaptive Biotechnology. These samples were profiled using the immunoSEQ platform developed by the company. Zip files were directly downloaded through the ‘Export’ function and selecting ‘v2’. Accession numbers for each cohort are available in Table S1. For each repertoire sample, sequences with missing variable genes or nonproductive CDR3 regions were removed. The top 10,000 TCRs with most abundant clonality were selected for RFU calculation. These preprocessing criteria were applied to all the TCR-seq samples throughout this study.
Genomic DNA isolation and TCR repertoire sequencing
Genomic DNA was isolated from 200 μL whole blood (from UTSW) or 10 μl buffy coat (from Accio Biobank or NHS) using the DNeasy Blood and Tissue Kit (Cat# 69504, Qiagen) following the manufacturer’s guidelines. gDNA concentration was measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). The purity of gDNA was determined by measuring the 260–280 nm absorbance ratio. Optimal purity was expected to be in the range of 1.7–2.0. The integrity of the gDNA samples was assessed for evidence of degradation using agarose gel electrophoresis. Appropriate quality gDNA was expected to migrate predominantly above 10 kb on agarose gels. All samples passed DNA purity and integrity quality controls. Twenty samples of gDNA were sent to Adaptive Biotechnology for targeted TCR β chain repertoire sequencing using immunoSEQ at survey sequencing depth. Raw TCR reads were processed with immunoSEQ Analyzer for CDR3 assembly, variable/joining gene calling, and clonal frequency estimations.
Repertoire functional unit method description
-
(i)
TCR embedding.
We applied GIANA21 to perform clustering of over 20 million TCRs using both the CDR3 sequences and variable gene alleles obtained from public domain (Figure 1B). These samples covered a wide spectrum of disease context, including healthy individuals and patients with cancer, autoimmune disorders as well as viral infections (Table S1). Previous work, including ours, have demonstrated that TCRs clustered using such strategy are highly specific (≥95%) to the same antigen epitopes,23,24,25 with smaller (n ≤ 5) clusters being more likely to share antigen-specificity.21
From GIANA output, we identified a total of 821K such clusters. An example of a typical cluster of two sequences, CSARQGARTYEQYF and CSARQGAYTYEQYF, bear a mismatch R/Y in position 8 (Figure 1C). We considered the amino acids flanking this mismatch and extracted the trimer sequences from both TCRs. As the two TCRs likely share antigen-specificity, the two trimers, ART and AYT, are thus considered ‘replaceable’ in the context of antigen recognition. We then traversed all 821K clusters and built the 8,000-by-8,000 trimer-substitution matrix (TSM) by calculating the number of replacements of each trimer pairs (Figure 1D). We calculated the Spearman’s correlation matrix using TSM and converted it into a Euclidean distance matrix (EDM). Next, similar as in GIANA, we obtained the isometric embedding vector for each of the trimers using multi-dimensional scaling based on the EDM. This approach allowed us to use a numeric vector to represent each trimer, with similar trimers located closely in the Euclidean space (Figure 1E). The embedding of each CDR3 sequence is then calculated as the average of all the vectors from consecutive trimers (Figure 1F). This embedding is a continuous representation of TCR similarity.
-
(ii)
Benchmark using antigen-specific TCRs.
We benchmarked the trimer-based embedding using 1,031 TCR sequences with known antigen-specificity to 9 epitopes (Table S2). This dataset has been used in our previous work to benchmark the specificity of TCR clustering.25 To avoid bias toward epitope(s) with excessive amount of TCRs, we restricted the antigens with <170 TCRs. Coordinates were calculated for each CDR3 sequence. For each pair of TCRs, we calculated the Euclidean distance as the predictor, with the response being if or not the two TCRs share the same antigen. From the total of half million comparisons, we excluded pairs with distances >0.025, based on the fact that trimer-based embedding is only powerful to identify shared antigen specificity among TCRs with similar sequences. ROC curve was generated with the above predictor and response variables, with AUC calculated using the curve.
-
(iii)
Definition of Repertoire Functional Units (RFU).
We pooled 1.2 million TCRs from 120 healthy donors from a previous study,27 and projected them onto the Euclidean space with trimer-based embedding. We divided the TCR sequences in this space into 5,000 groups with the k-means method. We referred the centroid of each group as a ‘Repertoire Functional Unit’, or RFU. To calculate the RFU vector of a new TCR repertoire sample, we first select the top 10,000 most abundant TCRs based on clonal frequencies. For each TCR, we calculate the embedding vector and assign it to the closest centroid from 5,000 RFUs. The value of each RFU is determined by the number of TCRs assigned to its centroid. We chose 5,000 as the group number so that the expected count for each RFU is 2.
-
(iv)
Calculation of OV RFU score.
The assignment of RFUs were based on the centroids of the TCR neighborhoods in the trimer embedding space. Since all of the four selected RFUs were not age-associated, when calculating the OV RFU scores, we simply added the upregulated RFUs and subtracted the downregulated ones. This calculation only involves the counts of the four given RFUs from an immune repertoire. OV RFU score is defined as RFU1804 + RFU3808 - RFU750 - RFU866. The first two terms are the ‘Up RFUs’ and the last two terms are the ‘Down RFUs’. The full score is thus Up RFUs – Down RFUs.
HLA association analysis
We collected four public TCR-seq sample cohorts27,79,80,81 with HLA genotype information available. We compiled them into a mixed cohort totaling 1,208 individuals. The top 30,000 TCRs of each sample were selected and pooled together to perform an HLA-enrichment analysis, thus incorporating a total of 36 million TCRs as the training data. We implemented a highly efficient TCR clustering method, GIANA to perform sequence-similarity-based TCR grouping.21 HLA alleles were analyzed at 2-digits resolution. For each GIANA clustered TCR group, we performed a Fisher’s exact test to evaluate its enrichment to all the HLA alleles in the patient samples. 240,438 significantly enriched TCRs were identified (at FDR = 0.05). Among these TCRs, the HLA-enriched TCRs belonging to these four RFUs were selected. To select the most enriched HLA alleles, we performed another enrichment analyses for each RFU. Specifically, for each allele and each RFU, we counted the number of TCRs specific to this allele that have been assigned to this RFU, and estimated the odds ratios. We made a cutoff at odds ratio = 2 and selected the top enriched HLA alleles. For each RFU, we calculated the percentage of individuals in the discovery cohort (n = 1,208) that carried the top enriched HLA alleles.
Quantification and statistical analysis
Computational and statistical analyses in this work were performed using the R programming language v4.3.0. Principal component analysis (PCA) was performed as a dimension reduction technique to visualize the samples in different groups (Figures 2B, 2D and 4D). Logistic regression adjusted for patient age and race: (Figure 2F) was implemented using the glm function. FDR control was using the Benjamini-Hochberg method. Sequence logos were generated using package ggseqlogo (v0.1), by performing multiple sequence alignment (msa, v1.32.0) using CDR3s with length 16. Donut plot (Figure 3) was generated using package webr (v0.1.5). ROC curves with 95 confidence intervals and AUC values were generated using package pROC (v1.18.2). Neighbor joining trees were calculated and visualized using R package ape (v5.7-1). Subpanels of main figures were produced using ggplot2 (v3.4.2). Permutation test in Figure 4D was performed as follows: with the goal of testing how significant the peak-like dynamics of prediagnostic curve, we randomly permuted the RFU scores for 10,000 times and recalculated the Loess smooth curve with default parameters (R function loess). For each permutation, we calculated the range of the curve (max - min), denoted as Dr. The range of the unshuffled curve is denoted as D0. p value was estimated as the number of permutations with Dr greater than D0 divided by 1,000.
Acknowledgments
This work is supported by NCI R01 grants CA258524 (B.L. and J.L.) and CA245318 (B.L.). Sample collection was partially supported by Department of Obstetrics and Gynecology, UTSW.
Author contributions
B.L. and J.L. conceived the project. B.L. developed the method and performed analysis with X.Y. M.P. performed HLA analysis. J.L. lead sample collection at UTSW. C.A.H. and S.S.T. provided NHS samples and data. J.Y. performed sample preparation. B.L. wrote the manuscript with X.Y. B.L. and J.L. supervised the study.
Declaration of interests
B.L. is the inventor of patent “TCR repertoire functional units”: (PCT) WO2023147530A1.
Published: June 14, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101612.
Contributor Information
Jayanthi Lea, Email: jayanthi.lea@utsouthwestern.edu.
Bo Li, Email: libo2@pennmedicine.upenn.edu.
Supplemental information
TCR-seq samples were used to generate the clusters for trimer substitution matrix, define RFU centroids, perform age association analysis, identify HLA allele enrichment, or compare selected RFU values across different cancer types.
References
- 1.Ovarian Cancer Research Alliance. Ovarian Cancer Research Alliance. https://ocrahope.org/.
- 2.Cancer Research UK Cancer Research UK. https://www.cancerresearchuk.org/.
- 3.Kaku T., Ogawa S., Kawano Y., Ohishi Y., Kobayashi H., Hirakawa T., Nakano H. Histological classification of ovarian cancer. Med. Electron. Microsc. 2003;36:9–17. doi: 10.1007/s007950300002. [DOI] [PubMed] [Google Scholar]
- 4.Kim J., Park E.Y., Kim O., Schilder J.M., Coffey D.M., Cho C.H., Bast R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers. 2018;10 doi: 10.3390/cancers10110433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gockley A., Melamed A., Bregar A.J., Clemmer J.T., Birrer M., Schorge J.O., Del Carmen M.G., Rauh-Hain J.A. Outcomes of Women With High-Grade and Low-Grade Advanced-Stage Serous Epithelial Ovarian Cancer. Obstet. Gynecol. 2017;129:439–447. doi: 10.1097/AOG.0000000000001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.American Cancer Society American Cancer Society. cancer.org.
- 7.McDaniel A.S., Stall J.N., Hovelson D.H., Cani A.K., Liu C.J., Tomlins S.A., Cho K.R. Next-Generation Sequencing of Tubal Intraepithelial Carcinomas. JAMA Oncol. 2015;1:1128–1132. doi: 10.1001/jamaoncol.2015.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weinberger V., Bednarikova M., Cibula D., Zikan M. Serous tubal intraepithelial carcinoma (STIC) - clinical impact and management. Expert Rev. Anticancer Ther. 2016;16:1311–1321. doi: 10.1080/14737140.2016.1247699. [DOI] [PubMed] [Google Scholar]
- 9.Crum C.P., Yoon J.Y., Feltmate C.M. Clinical commentary: Extra-uterine high-grade serous carcinoma: two pathways, two preventions? Gynecol. Oncol. 2023;169:1–3. doi: 10.1016/j.ygyno.2022.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs I., Bast R.C., Jr. The CA 125 tumour-associated antigen: a review of the literature. Hum. Reprod. 1989;4:1–12. doi: 10.1093/oxfordjournals.humrep.a136832. [DOI] [PubMed] [Google Scholar]
- 11.Skates S.J. Ovarian cancer screening: development of the risk of ovarian cancer algorithm (ROCA) and ROCA screening trials. Int. J. Gynecol. Cancer. 2012;22:S24–S26. doi: 10.1097/IGC.0b013e318256488a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J., Dowdy S., Tipton T., Podratz K., Lu W.G., Xie X., Jiang S.W. HE4 as a biomarker for ovarian and endometrial cancer management. Expert Rev. Mol. Diagn. 2009;9:555–566. doi: 10.1586/erm.09.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Gorp T., Cadron I., Despierre E., Daemen A., Leunen K., Amant F., Timmerman D., De Moor B., Vergote I. HE4 and CA125 as a diagnostic test in ovarian cancer: prospective validation of the Risk of Ovarian Malignancy Algorithm. Br. J. Cancer. 2011;104:863–870. doi: 10.1038/sj.bjc.6606092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Nagell J.R., Jr., Hoff J.T. Transvaginal ultrasonography in ovarian cancer screening: current perspectives. Int. J. Womens Health. 2013;6:25–33. doi: 10.2147/IJWH.S38347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menon U., Gentry-Maharaj A., Burnell M., Singh N., Ryan A., Karpinskyj C., Carlino G., Taylor J., Massingham S.K., Raikou M., et al. Ovarian cancer population screening and mortality after long-term follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2021;397:2182–2193. doi: 10.1016/S0140-6736(21)00731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terry K.L., Schock H., Fortner R.T., Hüsing A., Fichorova R.N., Yamamoto H.S., Vitonis A.F., Johnson T., Overvad K., Tjønneland A., et al. A Prospective Evaluation of Early Detection Biomarkers for Ovarian Cancer in the European EPIC Cohort. Clin. Cancer Res. 2016;22:4664–4675. doi: 10.1158/1078-0432.CCR-16-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beshnova D., Ye J., Onabolu O., Moon B., Zheng W., Fu Y.X., Brugarolas J., Lea J., Li B. De novo prediction of cancer-associated T cell receptors for noninvasive cancer detection. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aaz3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunn G.P., Bruce A.T., Ikeda H., Old L.J., Schreiber R.D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 19.Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 20.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 2020;20:651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H., Zhan X., Li B. GIANA allows computationally-efficient TCR clustering and multi-disease repertoire classification by isometric transformation. Nat. Commun. 2021;12:4699. doi: 10.1038/s41467-021-25006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dash P., Fiore-Gartland A.J., Hertz T., Wang G.C., Sharma S., Souquette A., Crawford J.C., Clemens E.B., Nguyen T.H.O., Kedzierska K., et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93. doi: 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glanville J., Huang H., Nau A., Hatton O., Wagar L.E., Rubelt F., Ji X., Han A., Krams S.M., Pettus C., et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547:94–98. doi: 10.1038/nature22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H., Liu L., Zhang J., Chen J., Ye J., Shukla S., Qiao J., Zhan X., Chen H., Wu C.J., et al. Investigation of Antigen-Specific T-Cell Receptor Clusters in Human Cancers. Clin. Cancer Res. 2020;26:1359–1371. doi: 10.1158/1078-0432.CCR-19-3249. [DOI] [PubMed] [Google Scholar]
- 26.Cader F.Z., Hu X., Goh W.L., Wienand K., Ouyang J., Mandato E., Redd R., Lawton L.N., Chen P.H., Weirather J.L., et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020;26:1468–1479. doi: 10.1038/s41591-020-1006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emerson R.O., DeWitt W.S., Vignali M., Gravley J., Hu J.K., Osborne E.J., Desmarais C., Klinger M., Carlson C.S., Hansen J.A., et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 2017;49:659–665. doi: 10.1038/ng.3822. [DOI] [PubMed] [Google Scholar]
- 28.Nolan S., Vignali M., Klinger M., Dines J.N., Kaplan I.M., Svejnoha E., Craft T., Boland K., Pesesky M., Gittelman R.M., et al. A large-scale database of T-cell receptor beta (TCRβ) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Res. Sq. 2020 doi: 10.21203/rs.3.rs-51964/v1. [DOI] [Google Scholar]
- 29.Goncharov M., Bagaev D., Shcherbinin D., Zvyagin I., Bolotin D., Thomas P.G., Minervina A.A., Pogorelyy M.V., Ladell K., McLaren J.E., et al. VDJdb in the pandemic era: a compendium of T cell receptors specific for SARS-CoV-2. Nat. Methods. 2022;19:1017–1019. doi: 10.1038/s41592-022-01578-0. [DOI] [PubMed] [Google Scholar]
- 30.Yao D.X., Soslow R.A., Hedvat C.V., Leitao M., Baergen R.N. Melan-A (A103) and inhibin expression in ovarian neoplasms. Appl. Immunohistochem. Mol. Morphol. 2003;11:244–249. doi: 10.1097/00129039-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Bao Y., Bertoia M.L., Lenart E.B., Stampfer M.J., Willett W.C., Speizer F.E., Chavarro J.E. Origin, Methods, and Evolution of the Three Nurses' Health Studies. Am. J. Public Health. 2016;106:1573–1581. doi: 10.2105/AJPH.2016.303338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu N.D., Bi H.S., Emerson R.O., Sherwood A.M., Birnbaum M.E., Robins H.S., Alm E.J. Longitudinal immunosequencing in healthy people reveals persistent T cell receptors rich in highly public receptors. BMC Immunol. 2019;20:19. doi: 10.1186/s12865-019-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang S., Li J.Y., Wu J., Meng L., Shou C.C. Mycoplasma infections and different human carcinomas. World J. Gastroenterol. 2001;7:266–269. doi: 10.3748/wjg.v7.i2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reid B.M., Permuth J.B., Sellers T.A. Epidemiology of ovarian cancer: a review. Cancer Biol. Med. 2017;14:9–32. doi: 10.20892/j.issn.2095-3941.2016.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee C.H., Salio M., Napolitani G., Ogg G., Simmons A., Koohy H. Predicting Cross-Reactivity and Antigen Specificity of T Cell Receptors. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.565096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birnbaum M.E., Mendoza J.L., Sethi D.K., Dong S., Glanville J., Dobbins J., Ozkan E., Davis M.M., Wucherpfennig K.W., Garcia K.C. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. 2014;157:1073–1087. doi: 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Townsend M.K., Trabert B., Fortner R.T., Arslan A.A., Buring J.E., Carter B.D., Giles G.G., Irvin S.R., Jones M.E., Kaaks R., et al. Cohort Profile: The Ovarian Cancer Cohort Consortium (OC3) Int. J. Epidemiol. 2022;51:e73–e86. doi: 10.1093/ije/dyab211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shih I.M., Wang Y., Wang T.L. The Origin of Ovarian Cancer Species and Precancerous Landscape. Am. J. Pathol. 2021;191:26–39. doi: 10.1016/j.ajpath.2020.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conner J.R., Meserve E., Pizer E., Garber J., Roh M., Urban N., Drescher C., Quade B.J., Muto M., Howitt B.E., et al. Outcome of unexpected adnexal neoplasia discovered during risk reduction salpingo-oophorectomy in women with germ-line BRCA1 or BRCA2 mutations. Gynecol. Oncol. 2014;132:280–286. doi: 10.1016/j.ygyno.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghezelayagh T.S., Kohrn B.F., Fredrickson J., Manhardt E., Radke M.R., Katz R., Gray H.J., Urban R.R., Pennington K.P., Liao J.B., et al. Uterine lavage identifies cancer mutations and increased TP53 somatic mutation burden in individuals with ovarian cancer. Cancer Res. Commun. 2022;2:1282–1292. doi: 10.1158/2767-9764.crc-22-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blank C.U., Haining W.N., Held W., Hogan P.G., Kallies A., Lugli E., Lynn R.C., Philip M., Rao A., Restifo N.P., et al. Defining 'T cell exhaustion'. Nat. Rev. Immunol. 2019;19:665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dey P., Nakayama K., Razia S., Ishikawa M., Ishibashi T., Yamashita H., Kanno K., Sato S., Kiyono T., Kyo S. Development of Low-Grade Serous Ovarian Carcinoma from Benign Ovarian Serous Cystadenoma Cells. Cancers. 2022;14 doi: 10.3390/cancers14061506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H., Liu J., Yang J., Wang Z., Zhang Z., Peng J., Wang Y., Hong L. A novel tumor mutational burden-based risk model predicts prognosis and correlates with immune infiltration in ovarian cancer. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.943389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moss H.A., Berchuck A., Neely M.L., Myers E.R., Havrilesky L.J. Estimating Cost-effectiveness of a Multimodal Ovarian Cancer Screening Program in the United States: Secondary Analysis of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS) JAMA Oncol. 2018;4:190–195. doi: 10.1001/jamaoncol.2017.4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chudecka-Glaz A.M. ROMA, an algorithm for ovarian cancer. Clin. Chim. Acta. 2015;440:143–151. doi: 10.1016/j.cca.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 46.Javdekar R., Maitra N. Risk of Malignancy Index (RMI) in Evaluation of Adnexal Mass. J. Obstet. Gynaecol. India. 2015;65:117–121. doi: 10.1007/s13224-014-0609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang P., Ma J., Li W., Wang Q., Xiao Y., Jiang Y., Gu X., Wu Y., Dong S., Guo H., Li M. Profiling the metabolome of uterine fluid for early detection of ovarian cancer. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang C., Ma A., McNutt M.E., Hoyd R., Wheeler C.E., Robinson L.A., Chan C.H.F., Zakharia Y., Dodd R.D., Ulrich C.M., et al. A bioinformatics tool for identifying intratumoral microbes from the ORIEN dataset. bioRxiv. 2023 doi: 10.1101/2023.05.24.541982. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeWitt W.S., Emerson R.O., Lindau P., Vignali M., Snyder T.M., Desmarais C., Sanders C., Utsugi H., Warren E.H., McElrath J., et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J. Virol. 2015;89:4517–4526. doi: 10.1128/JVI.03474-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeWitt W.S., Yu K.K.Q., Wilburn D.B., Sherwood A., Vignali M., Day C.L., Scriba T.J., Robins H.S., Swanson W.J., Emerson R.O., et al. A Diverse Lipid Antigen-Specific TCR Repertoire Is Clonally Expanded during Active Tuberculosis. J. Immunol. 2018;201:888–896. doi: 10.4049/jimmunol.1800186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emerson R., Sherwood A., Desmarais C., Malhotra S., Phippard D., Robins H. Estimating the ratio of CD4+ to CD8+ T cells using high-throughput sequence data. J. Immunol. Methods. 2013;391:14–21. doi: 10.1016/j.jim.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Savola P., Kelkka T., Rajala H.L., Kuuliala A., Kuuliala K., Eldfors S., Ellonen P., Lagström S., Lepistö M., Hannunen T., et al. Somatic mutations in clonally expanded cytotoxic T lymphocytes in patients with newly diagnosed rheumatoid arthritis. Nat. Commun. 2017;8 doi: 10.1038/ncomms15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Snyder A., Nathanson T., Funt S.A., Ahuja A., Buros Novik J., Hellmann M.D., Chang E., Aksoy B.A., Al-Ahmadie H., Yusko E., et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017;14 doi: 10.1371/journal.pmed.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansfield A.S., Ren H., Sutor S., Sarangi V., Nair A., Davila J., Elsbernd L.R., Udell J.B., Dronca R.S., Park S., et al. Contraction of T cell richness in lung cancer brain metastases. Sci. Rep. 2018;8:2171. doi: 10.1038/s41598-018-20622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Page D.B., Yuan J., Redmond D., Wen Y.H., Durack J.C., Emerson R., Solomon S., Dong Z., Wong P., Comstock C., et al. Deep Sequencing of T-cell Receptor DNA as a Biomarker of Clonally Expanded TILs in Breast Cancer after Immunotherapy. Cancer Immunol. Res. 2016;4:835–844. doi: 10.1158/2326-6066.CIR-16-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lang Kuhs K.A., Lin S.W., Hua X., Schiffman M., Burk R.D., Rodriguez A.C., Herrero R., Abnet C.C., Freedman N.D., Pinto L.A., et al. T cell receptor repertoire among women who cleared and failed to clear cervical human papillomavirus infection: An exploratory proof-of-principle study. PLoS One. 2018;13 doi: 10.1371/journal.pone.0178167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le D.T., Durham J.N., Smith K.N., Wang H., Bartlett B.R., Aulakh L.K., Lu S., Kemberling H., Wilt C., Luber B.S., et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robert L., Tsoi J., Wang X., Emerson R., Homet B., Chodon T., Mok S., Huang R.R., Cochran A.J., Comin-Anduix B., et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu M., Sedighim S., Wang T., Antonios J.P., Everson R.G., Tucker A.M., Du L., Emerson R., Yusko E., Sanders C., et al. TCR Sequencing Can Identify and Track Glioma-Infiltrating T Cells after DC Vaccination. Cancer Immunol. Res. 2016;4:412–418. doi: 10.1158/2326-6066.CIR-15-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beausang J.F., Wheeler A.J., Chan N.H., Hanft V.R., Dirbas F.M., Jeffrey S.S., Quake S.R. T cell receptor sequencing of early-stage breast cancer tumors identifies altered clonal structure of the T cell repertoire. Proc. Natl. Acad. Sci. USA. 2017;114:E10409–E10417. doi: 10.1073/pnas.1713863114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Page D.B., Pucilowska J., Sanchez K.G., Conrad V.K., Conlin A.K., Acheson A.K., Perlewitz K.S., Imatani J.H., Aliabadi-Wahle S., Moxon N., et al. A Phase Ib Study of Preoperative, Locoregional IRX-2 Cytokine Immunotherapy to Prime Immune Responses in Patients with Early-Stage Breast Cancer. Clin. Cancer Res. 2020;26:1595–1605. doi: 10.1158/1078-0432.CCR-19-1119. [DOI] [PubMed] [Google Scholar]
- 62.Lindau P., Mukherjee R., Gutschow M.V., Vignali M., Warren E.H., Riddell S.R., Makar K.W., Turtle C.J., Robins H.S. Cytomegalovirus Exposure in the Elderly Does Not Reduce CD8 T Cell Repertoire Diversity. J. Immunol. 2019;202:476–483. doi: 10.4049/jimmunol.1800217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanakry C.G., Coffey D.G., Towlerton A.M.H., Vulic A., Storer B.E., Chou J., Yeung C.C.S., Gocke C.D., Robins H.S., O'Donnell P.V., et al. Origin and evolution of the T cell repertoire after posttransplantation cyclophosphamide. JCI Insight. 2016;1 doi: 10.1172/jci.insight.86252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suessmuth Y., Mukherjee R., Watkins B., Koura D.T., Finstermeier K., Desmarais C., Stempora L., Horan J.T., Langston A., Qayed M., et al. CMV reactivation drives posttransplant T-cell reconstitution and results in defects in the underlying TCRβ repertoire. Blood. 2015;125:3835–3850. doi: 10.1182/blood-2015-03-631853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Formenti S.C., Rudqvist N.P., Golden E., Cooper B., Wennerberg E., Lhuillier C., Vanpouille-Box C., Friedman K., Ferrari de Andrade L., Wucherpfennig K.W., et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018;24:1845–1851. doi: 10.1038/s41591-018-0232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reuben A., Zhang J., Chiou S.H., Gittelman R.M., Li J., Lee W.C., Fujimoto J., Behrens C., Liu X., Wang F., et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Commun. 2020;11:603. doi: 10.1038/s41467-019-14273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Valpione S., Galvani E., Tweedy J., Mundra P.A., Banyard A., Middlehurst P., Barry J., Mills S., Salih Z., Weightman J., et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat. Cancer. 2020;1:210–221. doi: 10.1038/s43018-019-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emerson R.O., Sherwood A.M., Rieder M.J., Guenthoer J., Williamson D.W., Carlson C.S., Drescher C.W., Tewari M., Bielas J.H., Robins H.S. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J. Pathol. 2013;231:433–440. doi: 10.1002/path.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stromnes I.M., Hulbert A., Pierce R.H., Greenberg P.D., Hingorani S.R. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2017;5:978–991. doi: 10.1158/2326-6066.CIR-16-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J.M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramien C., Yusko E.C., Engler J.B., Gamradt S., Patas K., Schweingruber N., Willing A., Rosenkranz S.C., Diemert A., Harrison A., et al. T Cell Repertoire Dynamics during Pregnancy in Multiple Sclerosis. Cell Rep. 2019;29:810–815.e4. doi: 10.1016/j.celrep.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 72.Chow J., Hoffend N.C., Abrams S.I., Schwaab T., Singh A.K., Muhitch J.B. Radiation induces dynamic changes to the T cell repertoire in renal cell carcinoma patients. Proc. Natl. Acad. Sci. USA. 2020;117:23721–23729. doi: 10.1073/pnas.2001933117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emerson R.O., DeWitt W.S., Vignali M., Gravley J., Hu J.K., Osborne E.J., Desmarais C., Klinger M., Carlson C.S., Hansen J.A., et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 2017;49:659–665. doi: 10.1038/ng.3822. [DOI] [PubMed] [Google Scholar]
- 74.Cader F.Z., Hu X., Goh W.L., Wienand K., Ouyang J., Mandato E., Redd R., Lawton L.N., Chen P.H., Weirather J.L., et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020;26:1468–1479. doi: 10.1038/s41591-020-1006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mitchell A.M., Baschal E.E., McDaniel K.A., Simmons K.M., Pyle L., Waugh K., Steck A.K., Yu L., Gottlieb P.A., Rewers M.J., et al. Temporal development of T cell receptor repertoires during childhood in health and disease. JCI Insight. 2022;7 doi: 10.1172/jci.insight.161885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Creelan B.C., Wang C., Teer J.K., Toloza E.M., Yao J., Kim S., Landin A.M., Mullinax J.E., Saller J.J., Saltos A.N., et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med. 2021;27:1410–1418. doi: 10.1038/s41591-021-01462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu S., Knochelmann H.M., Lomeli S.H., Hong A., Richardson M., Yang Z., Lim R.J., Wang Y., Dumitras C., Krysan K., et al. Response and recurrence correlates in individuals treated with neoadjuvant anti-PD-1 therapy for resectable oral cavity squamous cell carcinoma. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Musvosvi M., Huang H., Wang C., Xia Q., Rozot V., Krishnan A., Acs P., Cheruku A., Obermoser G., Leslie A., et al. T cell receptor repertoires associated with control and disease progression following Mycobacterium tuberculosis infection. Nat. Med. 2023;29:258–269. doi: 10.1038/s41591-022-02110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Delmonte O.M., Oguz C., Dobbs K., Myint-Hpu K., Palterer B., Abers M.S., Draper D., Truong M., Kaplan I.M., Gittelman R.M., et al. Perturbations of the T-cell receptor repertoire in response to SARS-CoV-2 in immunocompetent and immunocompromised individuals. J. Allergy Clin. Immunol. 2023 doi: 10.1016/j.jaci.2023.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mitchell A.M., Baschal E.E., McDaniel K.A., Simmons K.M., Pyle L., Waugh K., Steck A.K., Yu L., Gottlieb P.A., Rewers M.J., et al. Temporal development of T cell receptor repertoires during childhood in health and disease. JCI Insight. 2022;7 doi: 10.1172/jci.insight.161885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Musvosvi M., Huang H., Wang C., Xia Q., Rozot V., Krishnan A., Acs P., Cheruku A., Obermoser G., Leslie A., et al. T cell receptor repertoires associated with control and disease progression following Mycobacterium tuberculosis infection. Nat. Med. 2023;29:258–269. doi: 10.1038/s41591-022-02110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TCR-seq samples were used to generate the clusters for trimer substitution matrix, define RFU centroids, perform age association analysis, identify HLA allele enrichment, or compare selected RFU values across different cancer types.
Data Availability Statement
The TCR-seq datasets for the discovery and validation cohorts generated in this study are publicly available on Zenodo at https://doi.org/10.5281/zenodo.11204147. According to standard controlled access procedures, submitted written applications to request use of NHS/NHSII data/resources will be reviewed by External Collaborations Committee to verify that the proposed use maintains the protection of the privacy of participants and the confidentiality of the data. All investigators wishing to use NHS/NHSII data are asked to submit a brief description of the proposed project at the following URL: https://www.nurseshealthstudy.org/researchers. The lead contact author (B.L.) will assist the reader in communication with the NHS committee to access the data. R codes for RFU calculation are deposited in Zenodo at the following https://doi.org/10.5281/zenodo.11209912. The codebase is also available at GitHub: https://github.com/s175573/RFU. Processed RFU matrices for reproducing the results of this work are available on Zenodo at: https://doi.org/10.5281/zenodo.11209912. Any additional information is available from the lead contact author upon request.