

Summary
Tumor-infiltrating regulatory T cells (TI-Tregs) elicit immunosuppressive effects in the tumor microenvironment (TME) leading to accelerated tumor growth and resistance to immunotherapies against solid tumors. Here, we demonstrate that poly-(ADP-ribose)-polymerase-11 (PARP11) is an essential regulator of immunosuppressive activities of TI-Tregs. Expression of PARP11 correlates with TI-Treg cell numbers and poor responses to immune checkpoint blockade (ICB) in human patients with cancer. Tumor-derived factors including adenosine and prostaglandin E2 induce PARP11 in TI-Tregs. Knockout of PARP11 in the cells of the TME or treatment of tumor-bearing mice with selective PARP11 inhibitor ITK7 inactivates TI-Tregs and reinvigorates anti-tumor immune responses. Accordingly, ITK7 decelerates tumor growth and significantly increases the efficacy of anti-tumor immunotherapies including ICB and adoptive transfer of chimeric antigen receptor (CAR) T cells. These results characterize PARP11 as a key driver of TI-Treg activities and a major regulator of immunosuppressive TME and argue for targeting PARP11 to augment anti-cancer immunotherapies.
Keywords: PARP11, Treg cells, tumor microenvironment, immunotherapy, PARP11 inhibitor, ITK7
Graphical abstract
Highlights
-
•
Tumor-derived factors upregulate PARP11 in the tumor-infiltrating Treg cells
-
•
PARP11 supports the immunosuppressive properties of Treg cells
-
•
Pharmacologic inhibition of PARP11 inactivates intratumoral Treg cells
-
•
PARP11 inhibitor augments the efficacy of immunotherapies
Basavaraja et al. demonstrate that induction of PARP11 in the intratumoral regulatory T (Treg) cells is required for their regulatory functions and contributes to the immunosuppressive tumor microenvironment. The selective inhibitor of PARP11 ITK7 inactivates tumor Treg cells and improves the efficacy of immunotherapies against tumors.
Introduction
Immunotherapeutic approaches including immune checkpoint blockade (ICB) and adoptive transfer of chimeric antigen receptor (CAR)-bearing T cells have emerged to revolutionize anti-cancer treatment and have greatly benefited some patients across many types of malignancies. Regrettably, these benefits are not observed in at least two-thirds of all treated patients who fail to display a complete response to ICB.1,2 The success rate of CAR T cell therapy is also limited, especially in solid tumors.3 The immunosuppressive tumor microenvironment (TME) of solid tumors is central to their ability to escape from immune surveillance and to resist ICB and adoptive cell transfer therapy (ACT).4,5 Within the TME, there are numerous cellular and acellular factors that attenuate anti-tumor immune responses. These factors including lactate, transforming growth factor β (TGF-β), adenosine, prostaglandin E2 (PGE2), interleukin-10 (IL-10), immunosuppressive myeloid cells, and regulatory T (Treg) cells promote tumor growth and confer resistance to immune therapies.4,5,6,7
Treg cells are CD4+ T cells characterized by constitutive expression of the scurfin transcription factor (also termed forkhead box P3, FOXP3). These cells are pivotal for restraining immune responses and preventing autoimmune disorders and immunopathology.8,9,10,11 Treg cells elicit immunosuppression through many mechanisms that involve contact with effector T cells or antigen-presenting cells or result from the secretion of immunosuppressive soluble factors such as PGE2, adenosine, TGF-β, and IL-10.10,12 Whereas factors in the TME stimulate the recruitment and differentiation of Treg cells, the tumor-infiltrating Treg cells (TI-Tregs) enable immunotolerance of cancer cells and thus promote the growth and progression of solid tumors.11,13,14,15 Accordingly, these TI-Tregs are instrumental in eliciting immune suppression in the TME, and their presence is associated with unfavorable prognosis for multiple cancers.16,17,18 Diverse mechanisms are also implicated in TI-Tregs’ role in resistance to immunotherapies. For example, anti-programmed death 1 (PD1) treatment stimulates the immunosuppressive activities of TI-Tregs.19 Furthermore, a byproduct generation of CAR-bearing Treg cells notably impedes the efficacy of CAR T therapies.20,21
Therapeutic targeting of Treg cells is complicated by the paucity of TI-Treg-specific agents and by the autoimmune or/and immunopathological side effects associated with systemic Treg inhibition.22,23,24 For example, the clinical efficacy of mogamulizumab that depletes CCR4+ Tregs is compromised by the concurrent elimination of anti-tumor central memory CD8+ T cells, which also exhibit low levels of CCR4 expression.25 One strategy is to identify and inhibit the TME pathways that selectively support suppressive activities of TI-Tregs without deleterious systemic inhibition of Treg cells. Additional benefits could be gained from the use of agents both interfering with TI-Treg and preventing the inhibition of the CD8+ cytotoxic T lymphocytes (CTLs) in the TME.
Our previous work identified the poly-(ADP-ribose)-polymerase (PARP) family member PARP11 as a pivotal mediator of CTL suppression by the tumor-derived factors in the TME.26 Here, we investigate whether PARP11 regulates the immunosuppressive activities of TI-Treg cells. We demonstrate that PARP11 is induced in the TI-Tregs, and its expression is associated with poor responses to immunotherapies in mice and in human patients with cancer.
PARP11 catalyzes “MARylation,” i.e., the transfer of a mono-ADP-ribose moiety from NAD+ onto diverse proteins27 including beta-transducin repeats-containing protein (β-TrCP) E3 ubiquitin ligase.28,29 MARylation of β-TrCP protects this protein from self-ubiquitination and subsequent proteasomal degradation. As a result, stabilized β-TrCP facilitates accelerated ubiquitination and degradation of its substrate proteins.28,29 Among those are critical regulators of the Treg cell function. For example, β-TrCP facilitates ubiquitination and degradation of IκBα, an inhibitor of the nuclear factor κB (NF-κB) pathway,30 which plays a key role in enabling Treg-suppressive activities.31 Conversely, β-TrCP-dependent proteolysis of β-catenin,30 a regulator of the WNT pathway which suppresses the Treg function,32,33 should increase the intratumoral immunosuppression by TI-Tregs. Similarly, β-TrCP facilitates ubiquitination and degradation of the interferon alpha/beta receptor 1 (IFNAR1) chain of type I interferon (IFN1) receptor and inactivates the IFN1 pathway,34 which otherwise inhibits TI-Treg activities and induces their fragility.35,36
Here, we demonstrate the pivotal role of PARP11 in controlling the suppressive functions of TI-Tregs without a detectable systemic effect on the Treg cells outside of the TME. We also provide a pre-clinical characterization of the selective small-molecule inhibitor of PARP11, ITK7.37 We demonstrate that this agent is well tolerated yet it robustly blocks the immune-suppressive activities of TI-Tregs, prevents the generation of CAR-bearing Treg cells, and enhances the efficacy of ICB and CAR T immunotherapies.
Results
Expression of PARP11 is upregulated in TI-Tregs and associated with failure of ICB therapies
We sought to determine whether expression of PARP11 is associated with survival outcomes of immunotherapies in patients with cancer.38 Analysis of The Cancer Genome Atlas database revealed that high expression of PARP11 was associated with poor survival outcomes for either pembrolizumab-treated patients with bladder cancer (Figure 1A) or for patients with melanoma receiving nivolumab (Figure 1B). Other reports highlighted a similar association in patients with glioblastoma multiforme.39 Conversely, lower levels of PARP11 expression were found in patients with melanoma who responded to pembrolizumab therapy40 compared to either partial responders or non-responders (Figure 1C). These findings link PARP11 expression in tumor tissues to a lack of responsiveness and/or resistance to ICB therapy.
Figure 1.

Expression of PARP11 is upregulated in TI-Tregs and associated with failure of ICB therapies
(A) Association between PARP11 expression and overall survival in patients with bladder cancer treated with pembrolizumab.
(B) Association between PARP11 expression and overall survival in patients with metastatic melanoma treated with nivolumab.
(C) PARP11 expression in patients with metastatic melanoma treated with pembrolizumab and grouped by indicated therapeutic outcomes: complete response (CR), partial response (PR), or non-responsive (NR).
(D) Association between PARP11 expression with tumor-infiltrating Treg cells (TI-Tregs) and FOXP3 expression in human melanoma (skin cutaneous melanoma), pancreatic (pancreatic adenocarcinoma), and colon (colon adenocarcinoma) cancers.
(E) qPCR analysis of Parp11 expression in Tregs cells isolated from tumors (TI-Treg, TU) or spleens (SP) from the Foxp3-YFP mice bearing MC38 s.c. tumors (n = 4).
(F) qPCR analysis of Parp11 expression in iTregs exposed to B16F10 tumor conditioned media (TCM) or control serum-free media (SFM) for 12 h.
(G) qPCR analysis of Parp11 expression in iTregs treated with vehicle or tumor-derived factors including PGE2 (1 μM), VEGFA (20 nM), or adenosine (1 mM) for 12 h (n = 4).
(H) qPCR analysis of Parp11 expression in iTregs treated with forskolin (FRS; 10 μM), adenosine (ADO; 1 mM), or prostaglandin E2 (PGE2; 1 μM) with or without PKA inhibitor H89 (10 μM) for 6 h (n = 4).
(I) Levels of βTrCP in the splenic Tregs or TI-Tregs isolated from MC38 s.c. tumor-bearing mice (n = 4).
Data are presented as mean ± SEM. Statistical analysis was performed using 1-way ANOVA with Tukey’s multiple comparison test (C, G, and H) or two-tailed unpaired Student’s t test (E, F, and I). A p value of less than 0.05 was considered as statistically significant for all.
Analysis of data mined from a human melanoma single-cell RNA sequencing database41 indicates that PARP11 is expressed in all types of immune cells infiltrating human cutaneous melanoma tumors including TI-Treg cells (Figure S1A). TI-Treg cells exhibited high levels of the NF-κB-inducible genes (e.g., NFKB1 and BIRC3) and low levels of IFN-stimulated (IRF7, IFITM1, and CH25H) and WNT/β-catenin-driven (DKK1/2, AXIN2, and TCF7) genes (Figure S1A). Within human melanoma tumors, PARP11 expression correlated with the expression of FOXP3, a marker of Treg cells, and with numbers of intratumoral Treg cells. Similar results were observed in colorectal adenocarcinoma and pancreatic ductal adenocarcinoma (PDAC) tumors (Figure 1D). In all, these findings are consistent with the ability of human TI-Treg cells to express PARP11.
Next, we aimed to determine whether PARP11 expression in TI-Treg cells can be recapitulated in mouse models. To this end, we inoculated transgenic mice expressing yellow fluorescent protein (YFP) under the control of FOXP3 promoter with subcutaneous syngeneic MC38 colon adenocarcinoma tumors. We isolated YFP+ Treg cells from either tumor or splenic tissues and analyzed the levels of Parp11 mRNA. Increased expression of Parp11 mRNA was detected in TI-Tregs compared to Treg cells isolated from the splenic tissues (Figure 1E).
In the TME, TI-Treg cells are exposed to diverse tumor-derived factors such as adenosine, PGE2, vascular endothelial growth factor (VEGF), and others.5 We further determined the effects of the tumor-derived factors on the induced Treg cells (iTregs) produced by differentiation in vitro. Treatment of these cells with media conditioned by B16F10 tumor cells significantly increased Parp11 expression (Figure 1F). Similar results were achieved after treatment with adenosine or PGE2 (Figure 1G), suggesting that some tumor-derived factors can upregulate Parp11 in Treg cells.
Both adenosine and PGE2 act via cognate G protein-coupled receptors that signal through the generation of cyclic AMP, activation of protein kinase A (PKA), and ensuing phosphorylation and transactivation of the cAMP response element-binding protein (CREB) transcription factor.12 Intriguingly, numerous CREB-binding sites were predicted within the promoter and introns of the PARP11 gene (Figure S1B). Moreover, treating iTreg cells with either adenosine or PGE2 increased phosphorylation of CREB (Figure S1C) and its ability to bind to the Parp11 promoter region (Figure S1D) in a PKA-dependent manner. Furthermore, while the activator of PKA, forskolin, increased phospho-CREB1 binding to DNA (Figure S1D) and upregulated Parp11 mRNA in iTreg cells, pre-treatment of these cells with PKA inhibitor H89 prevented the induction of Parp11 in response to either adenosine or PGE2 (Figure 1H). These results demonstrate that tumor-derived factors can induce PARP11 in Treg cells and implicate the PKA pathway in this induction.
Previous studies demonstrated that PARP11 MARylates and stabilizes β-TrCP.29 Accordingly, treatment of iTreg cells with tumor-conditioned media in vitro also upregulated β-TrCP in these cells (Figure S1E) and induced the NF-κB-driven genes (Adoar1 and Cxcl13) while decreasing the expression of either IFN1-stimulated (Stat1, Irf1, and Irf7) or the WNT/β-catenin-dependent (Dkk1, Tbox3 and Axin2) genes (Figure S1F). Importantly, TI-Tregs from MC38 tumors displayed greater levels of β-TrCP compared to the splenic Treg cells from the same mice (Figure 1I). These results suggest that tumor-derived factors upregulate PARP11 and β-TrCP levels in TI-Treg cells.
PARP11 supports the immune-suppressive activities of TI-Tregs
To assess the significance of PARP11 induction in the function of Treg cells, we employed Parp11−/− knockout mice. As described previously, these mice exhibited teratozoospermia and male infertility but otherwise developed normally and did not show obvious signs of pathology26,42 such as deleterious autoimmune phenotypes, which were seen in FOXP3-deficient scurfy mice.8,9 Naive splenic Treg cells from these mice did not significantly differ from their wild-type (WT) counterparts in either frequency or expression of immunosuppressive markers such as TGF-β, IL-10, CD39, and CD73 (Figures S2A and S2B). Furthermore, no difference in frequency or number of the splenic Treg cells was observed in MC38 tumor-bearing mice (Figures S2C and S2D). These results suggest that inactivation of PARP11 does not elicit a systemic perturbation in Treg homeostasis.
To evaluate the roles of PARP11 in the immunosuppressive functions of TI-Treg cells, we used an MC38 colon adenocarcinoma mouse model. In line with our previous report,26 the growth of MC38 tumors was decelerated in the syngeneic Parp11 knockout mice (Figure 2A). PARP11-deficient TI-Treg cells from MC38 tumors displayed a decrease in frequency and numbers (Figures 2B and S2C). Similar results were observed in experiments using additional tumor models including MH6419c5 PDAC (Figures S2E and S2F) and B16F10 melanoma (Figures S2G and S2H).
Figure 2.

PARP11 supports immune-suppressive activities of TI-Tregs
(A) Volume and mass (on day 21 after inoculation) of s.c. MC38 tumors growing in WT or Parp11 knockout mice (n = 4).
(B) Frequencies (percentage of CD45+ cells) and absolute numbers (per gram of tumor tissue) of TI-Tregs isolated from MC38 tumor-bearing mice as indicated in (A).
(C–I) Levels of Ki67, IL-10, TGFβ, CD39, CTLA4, NRP1, and TBET in the splenic Tregs (SP) and TI-Tregs (TU) isolated from WT (blue symbols) or Papr11−/− (red symbols) mice described in (A).
(J) Flow cytometry analysis of percentage of IFN-γ+ Tregs isolated from mice described in (A).
Data are presented as mean ± SEM. Statistical analysis was performed using 1-way ANOVA with Sidak’s multiple comparison test (C–I) or log rank test (A) or two-tailed unpaired Student’s t test (A, B, and J). A p value of less than 0.05 was considered as statistically significant for all.
Whereas splenic WT and Parp11−/− Treg cells did not significantly differ in the levels of Ki67 proliferation marker, a decrease in Ki67 was observed in PARP11-deficient TI-Tregs (Figure 2C). Importantly, ablation of PARP11 led to a notable downregulation of immunosuppressive mediators such as IL-10, TGF-β, CD39, and CTL-associated protein 4 (CTLA-4), as well as the regulator of Treg fragility neuropilin 1 (NRP1)43,44 in TI-Tregs but not in the splenic Tregs (Figures 2D–2H). Importantly, PARP11-deficient TI-Treg cells exhibited increased T helper type 1 (Th1) markers including TBET and IFN-γ (Figures 2I and 2J). These results suggest that PARP11 supports the immune-suppressive activities of TI-Treg cells.
To directly assess the significance of PARP11 in the regulatory functions of Treg cells, we treated naive mouse CD4+ T cells with TGF-β to generate in-vitro-differentiated iTregs, which themselves produce TGF-β, IL-10, adenosine, and other immunosuppressive mediators.45 Such a population prepared from Parp11−/− cells exhibited lower levels of FOXP3 and Ki67 than the WT population (Figures S3A and S3B). Therefore, we have conducted subsequent experiments by normalizing the input of iTregs (WT or Parp11 knockout) using equal numbers of FOXP3+ cells in all experimental groups. Under these conditions, knockout of PARP11 in iTreg cells led to a partial loss of β-TrCP levels and an increase in cell surface IFNAR1 levels (Figure 3A). Accordingly, compared to WT iTreg cells, PARP11-deficient iTregs displayed an increase in expression of IFN1-stimulated and WNT-stimulated genes and downregulation of NF-κB-driven genes (Figure 3B). Furthermore, WT iTreg cells displayed greater levels of immunosuppressive mediators and regulators such as CD39, CD73, and NRP1 than PARP11-deficient iTregs (Figures S3C–S3E). Conversely, PARP11-null iTreg cells expressed higher levels of the Th1 marker TBET (Figure S3F) and greater levels of IFN-γ (Figure S3G).
Figure 3.

Loss of PARP11 is associated with reduced immunoregulatory functions of Tregs
(A) Levels of intracellular β-TrCP and cell surface IFNAR1 in iTregs derived from WT or Parp11−/− mice and normalized per FOXP3 levels (n = 4).
(B) qPCR analysis of expression of genes representing IFN1 pathway (Stat1, Irf1, Irf7, Isg15, Ifitim1, and Ch25h), WNT pathway (Tcf1, Tcf4, lef1, Dkk1, TBox3, and Axin2), and NF-κB pathway (Nfkb1, Adoar1, Il6, and Cxcl13) in iTregs derived from WT or Parp11−/− mice and normalized per FOXP3 levels (n = 5).
(C) Flow cytometry analysis and quantification of CD8+ T cell proliferation index in vitro. Activated WT CD8+ T cells stained with CellTrace Violet were co-cultured for 72 h with or without WT and Parp11−/− iTregs in the ratio 2:1 (n = 4).
(D) Lysis of MC38OVA-luc cells by OT1 CTLs pre-incubated for 24 h with or without WT or Parp11−/− iTregs (OT1: iTreg = 3:1; OT1: MC38OVA-luc = 10:1; n = 6).
(E) Lysis of MC38OVA-luc cells by OT1 CTLs pre-incubated for 24 h with or without TI-Treg cells isolated from MC38 tumors growing in WT or Parp11−/− mice (OT1: Treg = 2:1; OT1: MC38OVA-luc = 10:1; n = 6).
(F) Cell surface levels of CD80 and CD86 on CD11c+MHCII+ dendritic cells upon being co-cultured (1:1) with WT or Parp11−/− iTregs for 18 h (n = 3). Representative raw data are shown in Figure S3H.
(G) Schematic depiction of experiment to test comparative immunosuppressive activities of WT or Parp11−/− iTregs (2.5×106 cells/mouse) administered into MC38OVA tumor-bearing immunocompromised host (Rag1−/−) in vivo before adoptive transfer of OT1 CTLs (5 × 106 cells/mouse) (n = 3–5).
(H) Volume of MC38OVA s.c. tumors in mice described in (G).
(I) Kaplan-Meier analysis of survival of MC38OVA tumor-bearing mice described in (G).
(J) Schematic depiction of experiment to test comparative immunosuppressive activities of WT or Parp11−/− iTregs (1×106 cells/mouse) administered into C57BL/6 hosts before inoculation of MC38 tumors (1×106 cells/mouse, n = 4–5).
(K) Volume of s.c. MC38 tumors in mice described in (J).
(L) Kaplan-Meier analysis of survival of MC38 tumor-bearing mice as described in (J) (n = 4–5).
Data are presented as mean ± SEM. Statistical analysis was performed using 2-tailed unpaired Student’s t test (A and B), 1-way ANOVA with Tukey’s multiple comparison test (C, D, E, F, H, and K), or log rank test (I and L). A p value of less than 0.05 was considered as statistically significant for all.
We further compared the regulatory functions of iTreg cells derived from WT or Parp11−/− mice. First, we tested the ability of these cells to suppress in vitro proliferation of activated CD8+ T cells assessed by dilution of the CellTrace Violet dye. WT but not Parp11−/− iTreg cells significantly suppressed this proliferation (Figure 3C). Second, we used an in vitro tumoricidal assay that detects killing of luciferase-expressing MC38OVA tumor cells by antigen-specific OT1 CTLs.26 Under these conditions, neither WT nor PARP11-deficient iTregs elicited cytotoxic effects alone (Figure 3D). When co-incubated with OT1 CTLs, WT iTreg cells exhibited a notable suppression of tumor cell killing. Importantly, PARP11 knockout rendered iTreg cells inactive under these conditions (Figure 3D). We corroborated this conclusion using in vitro generated cells by studies, in which TI-Treg cells were isolated from MC38 tumors grown in WT or Parp11−/− mice. Under these conditions, knockout of Parp11−/− in TI-Tregs significantly diminished their ability to inhibit the cytotoxic activity of OT1 CTLs (Figure 3E). These results further demonstrate that PARP11 promotes the immune-suppressive activities of Treg cells.
Besides inactivating CTLs, Treg cells can suppress other immune cells.13,46 For example, Treg cells elicit their regulatory activities by CTLA-4-dependent removal of CD80 and CD86 regulators from the surface of the antigen-presenting cells via the processes of trans-endocytosis or/and trogocytosis.47,48 This removal is important for the tumor-promoting function of TI-Tregs.49,50 We compared the ability of WT and Parp11−/− iTregs to downregulate CD80 and CD86 from the surface of dendritic cells upon co-incubation. Whereas WT iTreg cells decreased CD80 and CD86 levels, PARP11-deficient iTregs did not display this activity (Figures 3F and S3H), further suggesting the importance of PARP11 in supporting the regulatory functions of Treg cells.
We next sought to determine whether PARP11 regulates Treg cells in vivo. To this end, we used two independent yet complementary approaches. First, we examined the ability of iTreg cells to temper the anti-tumor effects of adoptively transferred OT1 CTLs against MC38OVA tumors in Rag1-null mice (Figure 3G). As expected, the transfer of OT1 cells notably decelerated the growth of MC38OVA tumors and prolonged the survival of host mice (Figures 3H and 3I). This therapeutic effect was prevented by administration of WT but not Parp11−/− iTregs (Figures 3H and 3I), indicating the functional deficiency of iTreg cells lacking PARP11.
Second, we compared the effects of adoptive transfer of WT versus Parp11−/− iTregs on tumor growth in an immunocompetent model (Figure 3J). Administration of WT iTreg cells significantly accelerated the growth of MC38 tumors and decreased the lifespan of syngeneic WT mice, while Parp11−/− iTregs failed to display such activity (Figures 3K and 3L). Taken together, these data suggest that the induction of PARP11 in TI-Treg cells supports their immunosuppressive activities and may help them to promote tumor growth.
Selective PARP11 inhibitor ITK7 disrupts the immune-suppressive activities of TI-Tregs and activates the immune pathways in the TME
To validate and complement findings from genetic experiments involving Parp11−/− mice, we employed a pharmacologic approach using ITK7, a selective and potent small-molecule inhibitor of PARP11.37 Treatment of naive CD4 T cells differentiated into iTregs with ITK7 modestly yet significantly decreased the yield of FOXP3+ cells (Figure 4A). When equal numbers of FOXP3+ cells were analyzed, we observed that ITK7 treatment resulted in downregulation of intracellular β-TrCP (Figure 4B) and upregulation of cell surface IFNAR1 levels (Figure S4A). In addition, iTreg cells exposed to ITK7 displayed an increased expression of IFN1-stimulated and WNT/β-catenin-stimulated genes (Figure S4B).
Figure 4.

PARP11-specific inhibitor ITK7 mimics the loss of PARP11 phenotype on Tregs suppressive function
(A) Frequencies of CD25+FOXP3+ cells (percentage of CD4+ cells) within the WT iTreg population pre-exposed or not to ITK7 (5 nM) for 48 h (n = 4).
(B–D) Levels of β-TrCP, IL-10, and TGF-β in iTreg cells treated as described in (A).
(E) Flow cytometry analysis and quantification of CD8+ T cell proliferation index in vitro. Activated WT CD8+ T cells stained with CellTrace Violet were co-cultured for 72 h with or without WT iTreg cells (Treg:CD8 = 1:2) pre-treated or not with ITK7 (5 nM, 48 h) (n = 5).
(F) Lysis of MC38OVA-luc cells by OT1 CTLs pre-incubated (OT1:iTreg = 3:1; OT1:MC38OVA-lu = 10:1; n = 6) for 48 h with iTregs derived from mice of indicated genotype (WT, Parp11−/−, or Ifnar1−/−). These iTreg cells were pre-exposed to ITK7 (5 nM, 48 h) or vehicle.
Data are presented as mean ± SEM. Statistical analysis was performed using 2-tailed unpaired Student’s t test (A–D) and 1-way ANOVA with Tukey’s multiple comparison test (E and F). A p value of less than 0.05 was considered as statistically significant.
Importantly, iTreg cells treated with ITK7 expressed decreased levels of IL-10 (Figure 4C), TGF-β (Figure 4D), and NRP1 (Figure S4C), and a trend for decreased CD39 (Figure S4D), suggesting that pharmacologic inhibition of PARP11 may undermine the regulatory activities of Treg cells. Indeed, ITK7 treatment of fully differentiated WT iTreg cells attenuated their ability to reduce proliferation of CD8+ T cells (Figure 4E) or conventional CD4+ T cells (Figure S4E).
Furthermore, ITK7 treatment significantly attenuated the ability of iTregs to limit the killing of MC38OVA-luc cells by OT1 CTLs (Figure 4F). Effects of ITK7 were not seen in iTreg cells derived from the Parp11 knockout mice indicating target specificity of this agent (Figure 4F). A modest yet significant effect of ITK7 was also seen in IFNAR1-null iTreg cells (Figure 4F), suggesting potential importance of pathways in addition to those involving IFN1. In all, these results suggest that inhibition of PARP11 catalytic activity with ITK7 can disrupt the immunosuppressive activities of Treg cells.
We next profiled gene expression in the subcutaneous (s.c.) MC38 tumors from mice treated or not with ITK7 in vivo using RNA sequencing. Expression of many genes was either upregulated or downregulated in these tumors (Figures 5A and S5A). Gene signatures associated with the synthesis of cholesterol and other sterols were among those decreased upon ITK7 treatment (Figure 5B). Conversely, pathways associated with stimulation of the immune system including humoral immune response, adaptive immune response, and others were notably activated in tumors harvested from the ITK7-treated mice (Figure 5B). Accordingly, ITK7 treatment promoted immune gene expression signatures such as regulation of lymphocyte-mediated immunity, T cell proliferation, and T cell activation (Figure 5C) and regulation of immune effector process and activation of immune response (Figure S5B). These results are consistent with the hypothesis that ITK7 can reactivate immune responses in the TME in vivo.
Figure 5.

Selective PARP11 inhibitor ITK7 disrupts the immune-suppressive activities of TI-Tregs and activates the immune pathways in the TME
(A) Enhanced volcano plot of differentially expressed genes in MC38 s.c. tumors from mice administered with ITK7 (100 μg/mouse intraperitoneally [i.p.]) on days 7, 10, 13, 16, and 19 and harvested on day 20 after tumor inoculation (n = 2).
(B) Top 20 significantly enriched suppressed and activated gene set enrichment analysis (GSEA) pathways in MC38 tumors described in (A). Color indicates the adjusted p values and dot size indicates the number of genes within a particular pathway.
(C) GSEA and Kyoto Encyclopedia of Genes and Genomes plots of indicated signatures detected in MC38 tumors described in (A).
(D) Growth s.c. MC38 tumors and TI-Tregs frequencies (percentage of CD45+) and numbers (per gram of tumor tissue) in mice treated with vehicle or ITK7 (100 μg/mouse) as shown in the schema (n = 5).
(E) Levels of β-TrCP on TI-Tregs isolated from MC38 tumors described in (D).
(F) Levels of IL-10, TGF-β, and CD39 in TI-Tregs from tumors described in (D).
(G) Flow cytometry analysis of percentage of Ki-67+ and IFN-+ Tregs isolated from MC38 tumors described in (D) (n = 5).
Data are presented as mean ± SEM. Statistical analysis was performed with 2-tailed unpaired Student’s t test (D–G) or 1-way ANOVA with Tukey’s multiple comparison test (for tumor volumes shown in D). A p value of less than 0.05 was considered as statistically significant for all.
To further test this hypothesis, we analyzed immune cells from these MC38 tumors (Figure S5C) and found that administration of ITK7 decelerated tumor growth and decreased the numbers of TI-Tregs but not splenic Tregs (Figures 5D and S5D). Furthermore, ITK7 downregulated βTrCP (Figure 5E) and immunosuppressive mediators including IL-10, TGF-β, and CD39 in TI-Tregs from MC38 tumors (Figure 5F). No statistically significant changes in the levels of βTrCP, IL-10, TGF-β, or CD39 were found in the splenic Treg cells of mice treated with ITK7 (Figure S5E). We also noted that TI-Tregs from mice administered with ITK7 displayed decreased levels of Ki67 and increased levels of IFN-γ (Figure 5G), suggesting that ITK7 inactivates TI-Treg cells. Similar changes or trends in the numbers and IFN-γ expression of TI-Treg cells were observed in experiments wherein ITK7 was administered into mice bearing either subcutaneous (Figure S5F) or orthotopically grown MH6419c4 PDAC tumors (Figure S5G).
ITK7 reactivates CTLs and elicits anti-tumor effects alone and in combination with immunotherapies
PARP11 also plays an important role in attenuation of activities of the intratumoral CTLs.26 Accordingly, a greater frequency of intratumoral CD8+ CTLs was found in MC38 tumors from mice administered with ITK7 (Figure 6A). These CTLs exhibited increased levels of Ki67 and IFN-γ (Figure 6B). Similarly, increased numbers and IFN-γ expression were seen in the CTLs isolated from the subcutaneous MH6499c4 PDAC tumors (Figures S6A and S6B).
Figure 6.

ITK7 reactivates CTLs and elicits anti-tumor effects alone and in combination with immunotherapies
(A) Frequencies (percentage of CD45+) and numbers (per gram of tumor) of CD8+ cells from s.c. MC38 tumors described in 5D (n = 5).
(B) Flow cytometry analysis of percentage of Ki67+ and IFN-γ+ among the CD8+ CTLs T cells from MC38 tumors described in 5D (n = 5).
(C) Frequencies (percentage of CD45+) and numbers (per gram of tumor) of CD8+ cells from orthotopic PDAC MH6499c4 tumors (n = 5).
(D) Flow cytometry analysis of percentage of IFN-γ+ and granzyme B among the CD8+ CTLs T cells from orthotopic PDAC MH6499c4 tumors (n = 5).
(E) Schematic illustration and tumor mass of testing the anti-tumor efficacy of ITK7 (100 μg/mouse) against orthotopic MH6499C4 tumors in syngeneic WT mice. Mice were sacrificed on day 20 (n = 5).
(F) Volume of s.c. MH6499c4 tumors in WT, Parp11−/−, or Rag1−/− mice treated with either vehicle or ITK7 (100 μg/mouse i.p.) on days 4, 7, 10, 13, and 16 after tumor inoculation (n = 5).
(G) Schematic illustration of testing the anti-tumor effects of anti-PD1 (200 μg/mouse) and ITK7 (100 μg/mouse) against s.c. cold MH6419c5 tumors in syngeneic WT mice (n = 3–6).
(H) Volume of s.c. MH6419c5 tumors described in (G).
(I) The Kaplan-Meier survival analysis of MH6419c5 tumor-bearing mice described in (G). Mice were sacrificed when the tumor volume reached 1,000 mm3.
(J) Schematic illustration of testing the anti-tumor effects of anti-PD1 (200 μg/mouse) and ITK7 (100 μg/mouse) against orthotopic MH6499C4 tumors in syngeneic WT mice (n = 6).
(K) The Kaplan-Meier survival analysis of MH6499C4 tumor-bearing mice described in (J). Mice were sacrificed when they became moribund.
Data are presented as mean ± SEM. Statistical analysis was performed with 2-tailed unpaired Student’s t test (A–E), 1-way ANOVA with Tukey’s multiple comparison test (F and H) or log rank test (I and K). A p value of less than 0.05 was considered as statistically significant for all.
We next examined the CTLs isolated from the MH6499c4 PDAC tumors orthotopically growing in the pancreas. This analysis revealed that ITK7 treatment increased the presence of tumor-infiltrating CTLs (Figure 6C) and their expression of cytotoxic markers such as IFN-γ and granzyme B (Figure 6D). In all, these data suggest that, while inactivating TI-Treg cells, ITK7 can reactivate the intratumoral CTLs.
Treatment of MC38 colon adenocarcinoma cells with ITK7 in vitro led to a modest decrease in the rate of their proliferation or ability to form colonies (Figures S6C and S6D). When administered to naive or tumor-bearing mice, ITK7 was very well tolerated and did not cause overt pathology in any of the examined internal organs (Table S1). Mice administered with ITK7 did not differ from the vehicle-treated animals in body weight or weight of internal organs including the spleen, kidney, liver, and heart (Figure S6E). This observation encouraged us to examine the therapeutic effects of ITK7.
As was seen from Figure 5D, mice treated with ITK7 exhibited a significantly decelerated growth of s.c. MC38 tumors. A similar anti-tumor effect of ITK7 was observed against either orthotopic (Figure 6E) or s.c. (Figure 6F) MH6499c4 PDAC tumors growing in the WT mice. Although tumor growth was accelerated in lymphocyte-deficient Rag1-null mice and decelerated in Parp11−/− mice, administration of ITK7 did not significantly alter MH6499c4 PDAC tumor growth in either of these hosts (Figure 6F). These results suggest that ITK7 acts specifically via inhibiting PARP11 and elicits the anti-tumor effects in the manner that requires adaptive immunity.
Given that ITK7 inhibits TI-Treg cells and reactivates the intratumoral CTLs, we sought to determine whether inclusion of ITK7 into the anti-cancer immunotherapeutic regimens may increase their effectiveness. The combination of ITK7 with anti-PD1 ICB treatment elicited a significantly greater anti-tumor therapeutic effect against either “cold” s.c. PDAC MH6419c5 tumors (Figures 6G–6I), orthotopic MH6499c4 PDAC tumors (Figures 6J and 6K), or s.c. MC38 tumors (Figures S6F and S6G).
Given that the generation of CAR-bearing Treg cells impedes CAR T therapies,20,21 we next sought to determine whether the use of ITK7 while manufacturing CAR T cells can increase their effectiveness. Treatment with ITK7 did not affect the expression of anti-hCD19 CAR transduced into human T cells (Figure S7). Yet, this treatment significantly decreased the expression of the mediator of adenosine production CD39 (Figure 7A). In addition, ITK7 prevented further increase in CD39 levels in CAR T cells treated with adenosine (Figure 7A). Furthermore, ITK7 inhibited adenosine-induced increase in the levels of lymphocyte-activation gene 3 (LAG3) and PD1 exhaustion markers on CAR T cells (Figures 7B and 7C).
Figure 7.

ITK7 improves the efficacy of CAR T therapy
(A–C) Levels of CD39, LAG3, and PD1 in CAR T cells after their pre-incubation with either a vehicle or ITK7 (10 nM) for 72 h, followed by additional treatment with or without adenosine (ADO, 1 μM, 24 h; n = 4).
(D) Lysis of hCD19-B16F10 cells by hCD19 CAR T cells treated as in (A) (n = 6).
(E) Schematic illustration of experiments combing CD19 CAR T cell therapy with ITK7 (100 μg/mouse) against s.c. hCD19-B16F10 tumors growing in Rag1−/− mice.
(F) Volume of hCD19-B16F10 s.c. tumors growing in Rag1−/− mice treated as described in (E). Treatments included anti-hCD19 CAR T cells (1.5×106/mouse, intravenously), pre-treated or not in vitro with ITK7 (10 nM; 72 h; n = 6), vehicle (DMSO, n = 3), ITK7 (100 μg/mouse, i.p., n = 3), or combination of ITK7 and CAR T cells administered into mice separately (n = 5).
(G) The Kaplan-Meier analysis of survival of mice described in (F). Mice were sacrificed when the tumor volume reached 1,000 mm3.
Data are presented as mean ± SEM. Statistical analysis was performed using 1-way ANOVA with Tukey’s multiple comparison test (A–F) or log rank test (G). A p value of less than 0.05 was considered as statistically significant for all.
Accordingly, pre-treatment of anti-hCD19 CAR T cells with ITK7 significantly increased their ability to kill B16F10 mouse melanoma cells expressing human CD19 (B16F10-hCD19) in vitro (Figure 7D). Furthermore, while pre-treatment of CAR T cells with immunosuppressive mediator adenosine protected B16F10-hCD19 cancer cells from lysis, the addition of ITK7 restored the killing phenotype (Figure 7D).
Genetic studies have demonstrated that ablation of PARP11 in mouse or human CAR T cells increases their anti-tumor efficacy.26 Thus, we next tested the effect of ITK7 on the efficacy of adoptive transfer of anti-hCD19 CAR T cells in the model of B16F10hCD19 tumors grown in the Rag1-null mice that lack native lymphocytes (Figure 7E). Under these conditions, administration of ITK7 alone had a modest effect on tumor volume and animal survival (Figures 7F and 7G), perhaps reflecting the importance of lymphocytes in responses to ITK7. Whereas adoptive transfer of anti-hCD19 CAR T cells significantly decelerated the growth of B16F10hCD19 tumors, co-administration of ITK7 in vivo notably stimulated the anti-tumor effects. Furthermore, merely pre-incubating CAR T cells with ITK7 ex vivo before the adoptive transfer of these cells significantly increased their therapeutic effects as manifested by suppression of tumor growth (Figure 7F) and increased animal survival (Figure 7G). Taken together, these results demonstrate that ITK7 improves CAR T cells’ fitness and activities and optimizes the efficacy of CAR T cell-based adoptive therapy.
Discussion
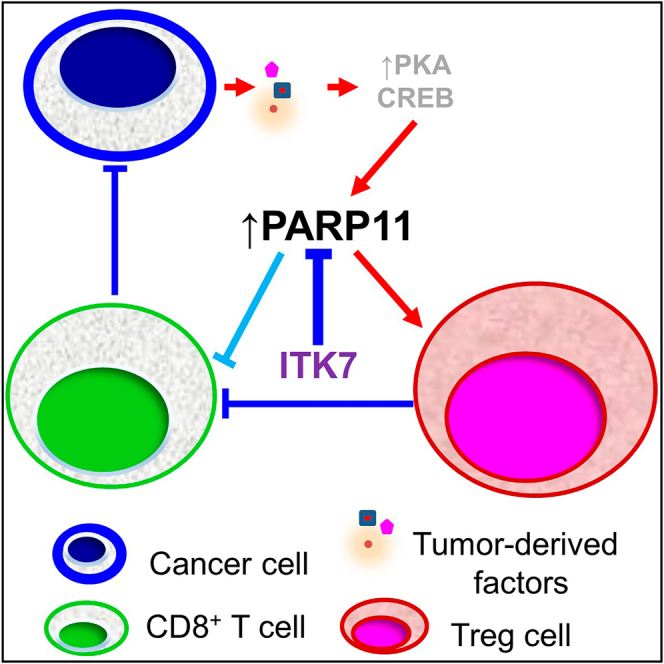
In this study, we demonstrate that expression of PARP11 is linked with TI-Tregs tumor infiltration and poor responses to ICB therapy in human patients with cancer. Induction of PARP11 by tumor-derived factors in TI-Treg cells is accompanied by upregulation of β-TrCP and altered expression of β-TrCP-regulated genes within the IFN1, NF-κB, and WNT/β-catenin pathways. Genetic and pharmacologic studies reveal that PARP11 is dispensable for homeostasis of Treg cells in normal tissues yet is essential for the regulatory functions of TI-Treg cells. The selective PARP11 inhibitor ITK7 inactivates TI-Treg cells, elicits anti-tumor activities, and increases the efficacies of ICB and CAR T cell immunotherapies.
Upregulation of PARP11 in TI-Treg cells
Knockout of PARP11 did not significantly affect splenic Treg cells but decreased the numbers and activities of TI-Treg cells (Figure 2). We attribute this phenomenon to dependence of TI-Tregs on the PARP11-dependent mechanisms that protect them from destabilization and inactivation that can occur in the sites of inflammation,51 including the inflammatory TME.52 Perhaps to ensure such protection, PARP11 expression is increased in TI-Tregs. Tumor-derived factors such as adenosine and PGE2 induce Parp11 in Treg cells (Figure 1). Given that functional Treg cells themselves produce these immunosuppressive mediators,12 it is plausible that upregulation of PARP11 is important for sustaining and perpetuating the ability of intratumoral Tregs to elicit their regulatory functions.
At least in part, activation of PKA by adenosine and PGE2 mediates the increase in PARP11 expression (Figure 1). Future studies will determine whether other agonists of the G protein-coupled receptors that are capable of activating PKA can also upregulate PARP11 in either TI-Treg cells or other cell types within the TME. Importantly, many of these agonists are known to suppress inflammation and restrict tissue damage,53 and some—such as sphingosine 1-phosphate—can stimulate the expansion of Treg cells.54 Plausibly, additional yet-to-be determined mechanisms unrelated to PKA activation can also be involved in PARP11 upregulation in TI-Treg cells.
PARP11 and regulatory functions of TI-Tregs
Loss of PARP11 appears to disrupt multiple regulatory properties of TI-Treg cells including production of immunosuppressive cytokines (IL-10 and TGF-β) and the adenosine-generating enzymes (CD39 and CD73), as well as the expression of NRP1 and CTLA-4. Accordingly, PARP11 knockout attenuates the ability of iTreg cells to strip the antigen-presenting cells from CD80 and CD86, interfere with CTL proliferation or cytotoxic activities, and promote tumor growth (Figure 4).
It is plausible that these biological phenomena can be mechanistically attributed to PARP11-dependent MARylation and the stabilization of β-TrCP in TI-Treg cells. Such stabilization is expected to accelerate the degradation of IFNAR1, β-catenin, and IκBα. Indeed, we observed changes in gene expression consistent with an increase in β-TrCP function including stimulation of the NF-κB pathway,30 which plays a key role in enabling Treg-suppressive activities.31 In addition, we detected inhibition of the WNT/β-catenin and IFN1 pathways, which act as negative regulators of Treg function.32,33,35,36 However, given that PARP11 can MARylate many other proteins besides β-TrCP,27 we cannot exclude additional mechanisms underlying PARP11-dependent maintenance of the regulatory function of TI-Tregs.
Immunosuppressive and pro-tumorigenic roles of PARP11 in the TME and the limitations of our studies
Knockout of PARP11 in the host mice significantly decelerates the growth of implanted colon adenocarcinoma, PDAC, and melanoma tumors (Figures 2 and S2). A similar phenotype was observed in WT syngeneic mice treated with PARP11 inhibitor ITK7 (Figure 6). Inactivation of PARP11 reinvigorates the anti-tumor immune responses (Figures 5 and 6). In all, these results suggest that PARP11 plays an important role in generating immunosuppressive TME and stimulating tumorigenesis.
It is highly likely that more than one cell type and more than one mechanism are involved in PARP11 induction to aid immunosuppression and promote tumor growth. Genetic evidence suggests the importance of PARP11 for inactivation of the intratumoral CTLs.26 Here, we show that induction of PARP11 supports the immune-suppressive activities of TI-Tregs. Given that induction of PARP11 accelerates IFNAR1 downregulation that supports the suppressive function of the granulocytic myeloid-derived suppressor cells,55 additional studies focused on the role of PARP11 in the intratumoral myeloid cells are warranted. Furthermore, given that loss of IFNAR1 is essential for efficient VEGF-driven angiogenesis56 and for stromagenesis driven by activated fibroblasts,57,58 the role of PARP11 in non-immune mechanisms stimulating tumor growth and progression cannot be ruled out and should be elucidated.
To this end, future studies should overcome the limitations associated with the use of the whole-body conventional Parp11 knockout and involve the generation of the conditional alleles followed by a compartment-specific ablation of PARP11 in diverse cell types within the TME. Furthermore, a complementary gain-of-function approach could utilize conditional overexpression of PARP11 in diverse cell types to mimic the situation in the TME. Future studies delineating specific PARP11 function in the different cellular compartments besides CTLs and Treg cells within the TME are warranted.
Therapeutic targeting of PARP11 for treatment of cancer
The importance of PARP11 acting on more than one cell type within the TME to promote tumor growth and suppress anti-tumor immune responses potentially renders this regulator a vulnerability that can be targeted for treatment of solid tumors.59 It has been noted that a non-selective Food and Drug Administration-approved drug rucaparib, which targets PARP1/260 but is also capable of inhibiting PARP11,29 prevented adenosine-induced upregulation of β-TrCP and downregulation of IFNAR1 and increased the efficacy of immunotherapies.26 However, given a broad substrate specificity of rucaparib, these phenotypes could not be specifically attributed to PARP11 inhibition.
Here, we provide a pre-clinical characterization of a highly selective and potent PARP11 inhibitor ITK7.37 ITK7 destabilized WT iTreg cells and attenuated their regulatory functions in vitro. In vivo experiments demonstrate that ITK7 is well tolerated and does not cause either systemic dysfunction of Treg cells or detectable autoimmune or immunopathological effects. Treatment with ITK7 changes gene expression within tumors to boost the immune response. ITK7 significantly enhances the presence of reinvigorated intratumoral CTLs, which display greater activity and effector function and less exhaustion. Finally, ITK7 notably decelerated growth of tumors in WT mice.
Importantly, ITK7 did not elicit any inhibition of the PARP11-deficient Treg cells in vitro. Furthermore, administration of ITK7 did not elicit any additional inhibition of tumor growth in Parp11−/− mice in vivo. These results indicate that ITK7 acts as a highly selective PARP11 inhibitor.
The use of ITK7 in syngeneic tumor mouse models in our study validates PARP11 as a promising therapeutic target for treating solid tumors. Importantly, administration of ITK7 elicited little (if any) effect on tumor growth in Rag1-null immunodeficient mice. The findings from our preclinical mouse studies suggest that ITK7 indeed effectively reawakens anti-tumor immunity. Including ITK7 in immunotherapeutic regimens significantly increased the efficacies of ICB and CAR T cell ACT immunotherapies in mice. Thus, small-molecule inhibition of PARP11 shows promise as a strategy for improving the immune response to cancer.
Intriguingly, treatment of CAR T cell formulations with ITK7 prior to ACT attenuated activities of CAR Treg cells and notably improved anti-tumor activities of CAR T cells. These results suggest that further evaluation of PARP11 inhibitors should include testing their ability to improve the manufacturing of CAR T products in vitro. Furthermore, given that a recently developed inhibitor of PARP7 elicited the anti-tumor effects in a manner depending on its ability to stimulate the production of IFN1,59,61 and PARP11 inhibitor stabilizes the receptor for these cytokines, it might be of further advantage to develop dual inhibitors of PARP11 and PARP7.
Limitations of the study
Genetic or pharmacologic inactivation of PARP11 decreases the numbers of TI-Treg cells while increasing the numbers of intratumoral CTLs. However, these findings do not differentiate between changes in T cell movement and infiltration versus changes of T cell proliferation in situ. Additional experiments will be needed to further uncover the mechanistic foundations underlying these phenotypes.
Furthermore, future studies should overcome the limitations associated with the use of the whole-body conventional Parp11 knockout and involve the generation of the conditional alleles followed by a compartment-specific ablation of PARP11 in diverse cell types within the TME. Furthermore, a complementary gain-of-function approach could utilize conditional overexpression of PARP11 in diverse cell types to mimic the situation in the TME. Future studies delineating specific PARP11 function in the different cellular compartments besides CTLs and Treg cells within the TME are warranted.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 FITC Clone 30-F11 |
Biolegend | (Cat# 103107; RRID:AB_312972) |
| CD45 APC Clone 30-F11 |
Biolegend | (Cat# 103112; RRID:AB_312977) |
| CD45 APC/cy7 Clone 30-F11 |
Biolegend | (Cat# 103116; RRID:AB_312981) |
| CD3 APC/Cy7 Clone 17A2 |
Biolegend | (Cat# 100222; RRID:AB_2242784) |
| CD8 APC Clone 53-6.7 |
Biolegend | (Cat# 162305; RRID:AB_2927947) |
| CD8 PE/Cy7 Clone 53-6.7 |
Biolegend | (Cat# 162311; RRID:AB_3083284) |
| CD8 Alexa Fluor 700 Clone 53-6.7 |
Biolegend | (Cat# 155022; RRID:AB_2890707) |
| CD8 APC/cy7 Clone 53-6.7 |
Biolegend | (Cat# 155015; RRID:AB_2890704) |
| FOXP3 PE Clone MF-14 |
Biolegend | (Cat# 118904; RRID:AB_2936574) |
| FOXP3 BV421 Clone MF-14 |
Biolegend | (Cat# 126419; RRID:AB_2565933) |
| NRP1 BV421 Clone 3 E−112 |
Biolegend | (Cat# 145209; RRID:AB_2562358) |
| CTLA4 BV605 Clone UC10-4B9 |
Biolegend | (Cat# 106323; RRID:AB_2566467) |
| CD39 PE dazzle Clone Duha59 |
Biolegend | (Cat# 143817; RRID:AB_2924475) |
| CD73 BV605 Clone TY/11.8 |
Biolegend | (Cat# 117205; RRID:AB_3083107) |
| TBET BV421 Clone 4B10 |
Biolegend | (Cat# 644815; RRID: AB_10896427) |
| IL10 APC Clone JES5-16E3 |
Biolegend | (Cat# 505009; RRID:AB_315363) |
| Ki67 PE dazzle Clone 16A8 |
Biolegend | (Cat# 151219; RRID:AB_2910306) |
| LAP TGFβ BV421 Clone TW7-16B4 |
Biolegend | (Cat# 141408; RRID:AB_2650898) |
| LAP TGFβ PerCP Clone TW7-16B4 |
Biolegend | (Cat# 141410; RRID: AB_2561592) |
| IFN-γ PE/CY7 Clone XMG1.2 |
Biolegend | (Cat# 505826; RRID: AB_2295770) |
| LAP TGFβ APC Clone TW7-16B4 |
Biolegend | (Cat# 141406; RRID: AB_10898159) |
| LAP TGFβ Biotin Clone TW7-16B4 |
Biolegend | (Cat# 141411; RRID:AB_2563131) |
| IFN-γ BV785 Clone XMG1.2 |
Biolegend | (Cat# 505837; RRID:AB_11219004) |
| Granzyme B AF6700 Clone QA18A28 |
Biolegend | (Cat# 154303; RRID:AB_2721462) |
| CD11c FITC Clone N418 |
BioLegend | (Cat# 117306; RRID:AB_313775) |
| CD80 PE Clone 16-10A1 |
Biolegend | (Cat# 104707; RRID:AB_313128) |
| CD86 APC Clone GL-1 |
Biolegend | (Cat# 105012; RRID:AB_493342) |
| Phospho-Creb1 PE Clone D3E6 |
Cell Signaling Technology | (Cat# 14228; RRID:AB_2798432) |
| βTrCP PE Clone C-6 |
Santacruz | (Cat# sc-390629 PE; RRID:AB_3096233) |
| CD4 PE/Cy5.5 Clone RM4-5 |
Invitrogen | (Cat# 35-0042-82; RRID:AB_11218300) |
| CD19-APC Clone 4G7 |
Biolegend | (Cat# 392504; RRID: AB_2728416) |
| CD19 CAR-PE Clone Y45 |
Acrobiosystems | (Cat#FM3-HPY53; RRID:AB_2921284) |
| Brilliant Violet 785™ Streptavidin | Biolegend | (Cat# 405249) |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Mouse IL-2 | Bio Legend | Cat# 575404 |
| ITK7 | Sigma-Aldrich | SML2669-25MG |
| PGE2 | Sigma-Aldrich | P0409; CAS: 363-24-6 |
| Mouse Recombinant TGF-β | R&D | 240-B; GenPept: P01137 |
| Adenosine | Sigma | Cat# A4036 |
| H89 | Cayman Chemical | CAS 130964-39-5 |
| Forskolin | Cayman Chemical | CAS: 66575-29-9 |
| DMSO | Sigma | Cat# 20-139 |
| VEGF165 | Preprotech | CAS: 450-32 |
| Ovalbumin (257–264) chicken | Sigma | Cat# S7951 |
| Golgi stop | BD bioscience | Cat# BDB554724 |
| PMA | Sigma | Cat#P8139 |
| Ionomycin | Stem cell technologies | Cat#73724 |
| DNase I | Roche | Cat# 10104159001 |
| Collagenase D | Roche | Cat# 11088882001 |
| Percoll | Sigma | Cat# 17-0891-01 |
| Lymphoprep™ | Stem cell technologies | Cat# 07851 |
| Dynabeads™ Mouse T-Activator CD3/CD28 | Gibco | Cat# 11453D |
| UltraComp eBeads™ Compensation Beads | Invitrogen | Cat# 01-2222-42 |
| CountBright™ Absolute Counting Beads | Invitrogen | Cat# C36950 |
| Fetal Bovine Serum | Hyclone | Cat# SH30071.03 |
| High-Capacity RNA-to-cDNA™ Kit | Applied Biosystems | Cat# 4387406 |
| Gibco™ L-Glutamine | Gibco | Cat# 25-030-081 |
| Intracellular Fix & Perm Buffer set | eBioscience | Cat# 88-8824 |
| Lipofectamin2000 | Invitrogen | Cat# 52887 |
| Retronectin | Takara | Cat# T100B |
| RBC lysis buffer | Biolegend | Cat# 420302 |
| Cell Activation Cocktail (with Brefeldin A) | Cat# 423303 | |
| Anti-CD3 | Biolegend | Cat# 100340; RRID: AB_11149115 |
| Anti-CD28 | Biolegend | Cat# 102116; RRID: AB_11147170 |
| Purified anti-mouse CD16/32 Antibody | Biolegend | Cat# 101302 |
| Anti-PD-1 | Bio X cell | Cat# BE0146 Clone: RMP1–14; RRID:AB_10949053 |
| CellTrace Violet | Thermo fisher | Cat#C34557 |
| CellTiter-Glo Viability Assay | Promega | Cat#G924A |
| Critical commercial assays | ||
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat# 4367659 |
| High-Capacity RNA-to-cDNA Kit | Applied Biosystems | Cat#4387406 |
| SimpleChIP Plus Enzymatic Chromatin IP Kit | Cell Signaling | Cat# 9005S |
| RNeasy Kits | Qiagen | Cat# 74004 |
| EasySep™ Mouse CD8+ T cell Isolation Kit | Stem cell technologies | Cat# 19853 |
| EasySep™ Mouse T cell Isolation Kit | Stem cell technologies | Cat# 19851 |
| EasySep™ Mouse CD4+ T cell Isolation Kit | Stem cell technologies | Cat# 19852 |
| ONE-Glo™ Luciferase Assay System | Promega | Cat# E6120 |
| ImmunoCult™ Mouse Treg Differentiation Supplement | Stem cell technologies | Cat# 10957 |
| EasySep™ Mouse CD4+CD25+ Regulatory T cell Isolation Kit II | Stem cell technologies | Cat# 18783 |
| EasySep™ Mouse Naive CD4+ T cell Isolation Kit | Stem cell technologies | Cat# 19765 |
| ONE-Glo™ Luciferase Assay System | Promega | Cat# E6120 |
| Deposited data | ||
| Bulk RNA-sequence data | This paper | GEO: accession number GSE249299 |
| Experimental models: Cell lines | ||
| Human: Phoenix-ECO | ATCC | Cat# CRL-3214™; RRID:CVCL_H717 |
| Mouse: MC38 | Kearfast | Cat# ENH204-FP; RRID:CVCL_B288 |
| Mouse: MC38 OVA | Katlinski et al.62 | RRID:CVCL_XJ96 |
| Mouse: MC38-OVA-Luciferase | Zhang et al.26 | N/A |
| Mouse: B16F10 | ATCC | Cat# CRL-6475™ |
| Mouse: MH6499c4 | Li et al.63 | N/A |
| Mouse: MH6499c5 | Li et al.63 | N/A |
| Human: HEK 293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Experimental models: Organisms/strains | ||
| Mouse: WT C57BL/6 | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | RRID:IMSR_JAX:003831 |
| Mouse: B6(Cg)-Ifnar1tm1.2Ees/J | Jackson Laboratory | RRID:IMSR_JAX:028288 |
| Mouse: B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J(Foxp3-Cre | Jackson Laboratory | RRID: IMSR_JAX:016959 |
| Mouse: B6.129S7-Rag1tm1Mom/J | Jackson Laboratory | RRID: IMSR_JAX:002216 |
| Mouse: C57BL/6- Parp11−/− | Zhang et al.26 | N/A |
| Oligonucleotides | ||
| Primers for q-PCR and ChiP qPCR | see Table S1 | N/A |
| Recombinant DNA | ||
| Retro-p-CD19-scFV-BBZ-MSGV | https://doi.org/10.1016/j.cell.2021.08.004 | N/A |
| Software and algorithms | ||
| FlowJo v10 | FlowJo, LLC | https://www.flowjo.com; RRID:SCR_008520 |
| GraphPad Prism v9.1.0 | GraphPad | https://www.graphpad.com; RRID:SCR_002798 |
| FACSDiva | BD Biosciences | N/A |
| R v 3.6.1 | The R Project for Statistical Computing | https://www.r-project.org/ |
| DESeq2 | Love et al.64 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA | BROAD Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| GseaVis | N/A | https://github.com/junjunlab/GseaVis |
| ClusterProfiler v3.10.1 | N/A | http://bioconductor.org/packages/clusterProfiler/ |
Resource availability
Lead contact
Additional information and requests for resources and reagents should be directed to the lead contact, Serge Y. Fuchs (syfuchs@vet.upenn.edu), who will fulfill these requests.
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Bulk-seq data are accessible at GEO under accession number GSE249299.
-
•
No custom computer codes were generated in this study.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Animal studies
All mice were housed in a pathogen-free facility, and all procedures were performed in accordance with policies and guidelines outlined by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC, approved protocol # 803995). In vivo experiments carried out on mice of C57Bl/6 background (unless specified otherwise) were approved by the Institutional Animal Care and Use Committee of The University of Pennsylvania. C57BL/6 littermate Parp11+/+ (‘WT’) and Parp11−/− mice (Parp11 null) were described previously.42 All experimental mice were maintained single-sex cages, in an environment with a temperature of 20 ± 2°C and a 12 h light/12 h dark cycle, with lights turning on at 7:00 a.m. in accordance with American Association for Laboratory Animal Science guidelines. Littermate animals from different cages were randomly assigned into experimental groups, which were either co-housed or systematically exposed to other groups’ bedding to ensure equal exposure to the microbiota of all groups. The genotyping PCR primers were provided in Table S2.
Cell lines
MC38 cell line, derived from C57BL6 murine colon adenocarcinoma cells, was purchased from (Kearfast, ENH204-FP). The mouse melanoma cell lines B16F10 (ATCC CRL-6475) were purchased from ATCC. The mouse pancreatic tumor cell clone MH6499c565 was a kind gift from Dr. Ben Stanger at the University of Pennsylvania, Philadelphia, PA. Dr. Suzanne Ostrand-Rosenberg from the University of Maryland, Baltimore, USA, generously contributed mouse MC38OVA cell line. The B16F10 cells, modified to express human CD19 (hCD19-B16F10 – a gift from Andy Minn) and MC38OVA cells, were both engineered to stably express Firefly luciferase, have been previously described.26,36 All cells were cultured in DMEM (Gibco) with a supplement of 10% FBS (HyClone) and 100 U/mL Penicillin-Streptomycin (Gibco). All the cell lines were regularly tested for Mycoplasma.
Method details
Human dataset analysis
Survival curves for bladder cancer and melanoma patients treated with Anti-PD1 therapy were derived from NCBI Gene Expression Omnibus (GEO) repository. Their correlation with PARP11 mRNA expression and overall survival was analyzed using the KM Plotter database (http://kmplot.com/analysis). RNA data was derived from GEO database (GSE78220), which includes 28 patients with malignant melanoma who received anti-PD-1 treatment.40 According to the response to treatment, patients were divided into two groups: complete response, partial response, and non-response groups. After importing the data into R studio, extraneous details in sample names were removed, and patients were classified based on their observed response to treatment into three categories: Complete Response (CR), Partial Response (PR), and No Response (NR). Samples identified as outliers were excluded from further analyses. The expression data underwent log10 transformation for normalization. Pairwise comparisons between the response groups were conducted using the Tukey’s Honest Significant Difference (TukeyHSD) test. The results were visualized using violin plot.
For single cell analysis, single nuclei slide-tags datasets from human melanoma (SCP2171)41 were downloaded and processed from the Broad Institute Single Cell Portal. For correlation analysis using TCGA data from TIMER 2.0, we examined the relationship between PARP11 with FOXP3 expression, and tumor Treg infiltration, in Colon Adenocarcinoma (COAD), Skin Cutaneous Melanoma (SKCM), and Pancreatic Adenocarcinoma (PAAD).61
Tumor conditioned media (TCM) preparation
Tumor conditioned media (TCM) were prepared using B16F10 as outlined in a prior study.58 Briefly, B16F10 tumor cells were plated to reach 75–80% confluency the next day. Once they reached confluency, the media was removed, and the cells were washed 2–3 times with PBS to remove residual FBS. Then, the cells were cultured in serum free media (SFM) for 48 h. After that, the conditioned media was collected, centrifuged to remove cells and debris, and stored at −80°C until use. Our in vitro experiments with TCM involved either serum free media (SFM) or combination of TCM with complete media in a 3:1 ratio.
Pharmacological compounds and reagents
Adenosine (Sigma, CAS A4036), transforming growth factor β (TGFβ, R&D CAS 7666-MB-005/CF), prostaglandin E2 (PGE2, Sigma, CAS P0409), H89 (hydrochloride; CAS 130964-39-5; Cayman Chemical), ITK7 (Sigma, CAS SML2669), Recombinant Mouse Interleukin-2 (IL2, Biolegend, CAS 575404), Recombinant Murine VEGF165 (VEGFA, Preprotech, CAS 450-32) were purchased.
Flow cytometry analyses
All antibodies used for flow cytometry are listed in the key resources table. Single cells suspensions from tumors or spleens were prepared as previously described.26 Briefly, tissues were mechanically disassociated into small pieces followed by incubation with enzymatic dissociation solution (DMEM supplemented with, 1 mg/mL collagenase and 100 μg/mL of DNase I). The cells were then filtered through a 70 μm filter membrane to eliminate debris and resuspended in FACS buffer (PBS with 1% BSA, 1mM EDTA). Isolated cells were pre-incubated with anti-CD16/32 (Fc blocker) for 10min. Surface staining was performed using the fluorescent-conjugated antibodies for 30–45 min before washing and preparation for flow cytometry analysis. For intracellular staining, cells were fixed and permeabilized using the intra-nuclear staining kit (FoxP3 staining Buffer Set) according to manufacturer’s instructions (eBioscience). Fixed cells were then incubated with indicated antibodies for 1 h before washing and preparation for flow cytometry analysis. Intracellular cytokine analysis required cells to be stimulated with the Biolegend Activating Cocktail, for 5–6 h at 37 °C with 5% CO2 prior to the intracellular staining process. Cells acquisition was performed on a BD LSR II or FACSymphony using FACSDiva software (BD Biosciences), and analysis was performed using FlowJo software (Miltenyi). Fluorescence minus one (FMO) control were used to set the gates for positive staining.
Tumor growth studies
For subcutaneous tumor model, MC38 (0.5–1 × 106), MH6499c4 (1 × 106) MH6419c5 (0.75 x106), or B16F10 (0.5 -1 × 106) were inoculated subcutaneously into the right flanks of syngeneic C57BL/6 mice. Tumor size was measured every other day from day 4 using a caliper. Tumor volume was calculated using formula (width × width × length × 0.5). Studies using the PDAC orthotopic pancreatic tumor growth were carried out as described previously.63 Briefly, after anesthesia, a 1-cm nick was made in the abdominal wall and the pancreas was exteriorized and kept moist using PBS. MH6499c4(1.5 × 105) cells were injected into the pancreas using a 30-G needle and returned pancreas to the abdominal wall, and then the abdominal wall and skin were sutured carefully.
For immunotherapy experiments, 9–10 weeks-old male wild type mice were randomly divided into four groups: IgG/vehicle control, Anti-PD1, ITK7, and Anti-PD1 combined with ITK7. Each mouse was inoculated subcutaneously with MH6419c5 (0.5 × 106) or MC38 (0.5 × 106) or for orthotopically with MH6499c4 (0.2 × 106) cells. Starting on day 5 post-inoculation, treatments were administered as follows: ITK7 (100μg/mouse), anti-PD1 (BioXCell #BE0146, 200μg/mouse, intraperitoneally), or a combination of ITK7 and anti-PD1. For survival analysis, mice were tracked until tumor volume reached ∼1000 mm3 at which point the data were considered for Kaplan- Meier (KM) survival analysis. For orthotopic model, mice exhibiting signs of severe distress or morbidity, such as inability to move, hunched posture were considered to have reached endpoint for KM survival analysis.
ITK7 in vivo treatment
For in vivo therapeutical use, ITK7 was formulated as a 2 mg/mL solution in DMSO (Dimethyl sulfoxide). After formulation, ITK7 was stored at either −20oC for up to one month or at −80oC for long term use. Mice were administered ITK7 at a dose of 100 μg per mouse via intraperitoneal (i.p) injection following specified dosing regimen. To ensure the ITK7 safety, a comprehensive toxicology and pathological examination was performed involving healthy wild-type mice (Table S1).
Quantitative real-time PCR and chromatin immunoprecipitation assay
Total RNA was extracted either from cells or directly from snap-frozen tumor tissue using the RNA isolation kit (Qiagen, cat#74004). The concentration of RNA was then measured using a NanoDrop 2000 spectrophotometer. For cDNA synthesis, the Applied Biosystems High-Capacity RNA-to-cDNA Kit (Applied Biosystems#4387406) was employed. Real-time PCR assays were performed using the PowerUp SYBR Green Master Mix (Applied Biosystems #A25742). Gene expression levels were calculated based on the cycle threshold (ΔΔCT), which was set to the linear phase of DNA amplification. The raw data were normalized to Beta Actin, serving as a standard housekeeping gene. For ChiP-qPCR, iTregs were generated as described above. These cells were then cultured either with vehicle (DMSO), Forskolin (10μM), Adenosine (1 mM), PGE2 (1μM), or their combination with H89 (10μM) for 60 min. Post treatment, cells were collected, and chromatin immunoprecipitation was performed using the SimpleChIP Enzymatic Chromatin IP Kit. Immunoprecipitation was carried out with a phospho-CREB1 antibody at a 1:20 dilution. ChIP-qPCR for the Parp11 promoter was performed using oligonucleotide primers designed with Primer 3 software. The sequences of all oligonucleotide primers can be found in Table S2.
Tregs isolation and iTregs generation
In vitro generation of Tregs (iTregs) were carried as out previously described.36 Briefly, naive CD4+ T cells from WT or Parp11−/− mice spleen was isolated with EasySep mouse naive CD4+ T cell Isolation Kit (STEM CELL Technologies; Catalog # 319765). These cells were then differentiated with ImmunoCult Mouse Treg Differentiation Supplement as per the manufacturer’s recommendations (STEMCELL Technologies; Catalog# 10957). Post differentiation, small number of cells were collected for yield and purity assessment by the expression of CD4+CD25+FOXP3+ analyzed via flow cytometry.
For the isolation of tumor derived Tregs, mice (WT or Parp11−/− mice with Foxp3YFP−Cre background) bearing MC38 tumors were digested as previously described. Tumor Tregs were then enriched with EasySep Mouse CD4+CD25+ Regulatory T cell Isolation Kit II (STEMCELL Technologies, 18783). Subsequently, enriched Tregs were FACS sorted to select the YFP+ cells, which were then used for all for all downstream applications.
Tregs functional studies
Cytotoxicity assays were conducted to evaluate an ability of Tregs suppressive effects on CTL mediated tumor cell killing as previously described.26,36 Briefly, Tregs of different genotype (WT or Parp11−/−) or WT iTregs pretreated with 5–10nM ITK7 were co cultured with CTLs for 24 h. These cells were then co cultured with target MC38OVA cells at specific effector – to-target (E: T) ratios in 96 well. As controls, target cells alone measured spontaneous death luminescence (spontaneous death RLU) and water-lysed cells represented maximal killing (maximal killing RLU). After adding luciferase substrate (Bright-Glo; Promega, cat#E6110) and incubating 10 min at room temperature, luminescence was recorded using the EnVision (PerkinElmer) reader. Cell lysis percentage was calculated with a formula: % lysis = 100 × (spontaneous death RLU-tested RLU)/(spontaneous death RLU-maximal killing RLU).
The cell proliferation suppression assay was conducted as previously described36 with few modifications. Total CD8+ T-cells or CD4+ CD25− cells were isolated from naive WT mice spleens using EasySep Mouse CD8+ and CD4+CD25− T cell Isolation Kit and labeled with 5nM of Cell Trace Violet (CTV, Thermo Fisher, cat# C34557). These cells were cultured either alone or with Treg cells from specified sources in the presence of anti-CD3ε and anti-CD28. CTV staining was analyzed for proliferation using flow cytometry after 72 h. For the study of trogocytosis between Tregs and dendritic cells (DCs), bone marrow-derived dendritic cells (BMDCs) were generated as follows. Bone marrow cells were isolated from mice and cultured in dendritic cell differentiation medium consisting of RPMI-1640 supplemented with mouse recombinant granulocyte-macrophage colony-stimulating factor (10 ng/mL, Invitrogen) and mouse recombinant interleukin-4 (10 ng/mL, Invitrogen) for 7–8 days. Following culture, cells were then harvested and purified using the EasySep Mouse CD11c Positive Selection Kit II (StemCell Technologies, #18780), according to the manufacturer’s instructions. The isolated DCs were then pulsed with ovalbumin (OVA) peptide (50 μg/mL) for 8 h. Subsequently, these DCs were cocultured with iTregs at a 1:5 ratio, derived from wild-type (WT) and Par1-null mice, for 12 h. Flow cytometry analysis was then performed to measure the surface expression of CD80 and CD86 on the cocultured DCs.
For adoptive transfer, Rag1−/− mice were subcutaneously inoculated with MC38-OVA tumor cells (1 × 106). Upon confirmation of tumor growth, iTregs (either WT or Parp11−/− mice, 2.5 × 106/mouse) were intravenously transferred into MC38-OVA tumor-bearing mice on day 11. The following day, OT-I cells (5 × 106/mouse) were also administered. The tumor volume and survival of these mice was then closely monitored. For comparative immunosuppressive activities, WT or Parp11−/− mice derived iTregs (2.5 × 106/mouse) were intravenously administered into 8 weeks old WT male mice. This administration was performed prior to the s.c inoculation of MC38 cells (1 × 106/mice).
Mouse CAR T cells generation and functional studies
CAR T cell preparation and functional analysis were conducted as previously described.26 Briefly, total T cells were isolated from the spleen of WT (8–9-week-old) mice using T cells isolation kit (STEM cells technologies; cat #19851). These isolated T cells were activated with magnetic beads precoated with agonist antibodies against mouse CD3 and CD28 (Gibco; cat# 11456D) according to manufacturer’s instructions. On day 2, activated T cells underwent spin transduction with CD19-BBz CAR and were expanded for 4–5 days for purity assessment. Once after the confirmation of purity, CAR T cells were utilized for either in vitro or in vivo studies.
For the cytotoxicity assay, CAR T cells preincubated with either vehicle or ITK7, were co-cultured with target cells (hCD19-B16F10-luci) at indicated E/T ratio for 5 h. Cytotoxicity was then measured using luciferase-based cytotoxicity assay (Promega Bright-Glo, cat#E6120) as manufacture instructions. Luminescence was measured using the EnVision (PerkinElmer) plate reader. The percentage of lysis was calculated using the formula: % lysis = 100 × (spontaneous death RLU-tested RLU)/(spontaneous death RLU-maximal killing RLU).
For in vivo CAR T therapies, Rag1-null mice were subcutaneously inoculated with hCD19-B16F10 tumor cells (0.2×105 cells/mouse). Following tumor establishment, 9-week-old male mice were grouped into: Vehicle (n = 3), ITK7 (n = 3), CAR (n = 4), pre-ITK7 (n = 5), and CAR+ITK7 (n = 5). The vehicle and ITK7 groups received their respective treatments on days 6, 9, 12, and 15. The CAR group was administered with WT CD19-BBz CAR T cells. The pre-ITK7 group received CAR T cells pretreated with ITK7(10nM; 72 h; 2.5 × 106 cells/mouse on day 7 and 13). The CAR+ITK7 group got CAR T cells (2.5 × 106 cells/mouse on days 7 and 13) and ITK7 on days 6, 9, 12, and 15.
RNA sequencing
Bulk MC38 tumors samples collected from Vehicle or ITK7 treated mice were snap frozen. Total RNA was then extracted with RNeasy PlusMini Kit (QIAGEN). The extracted RNA samples were utilized RNA sequencing as previously described.36 The raw fastq files were aligned to the mouse genome mm39 by salmon 10.1 with standardized parameters. Normalization and differential expression (DE) analysis was conducted by DESeq2 v. 1.28.1. Significantly DE genes were defined as based on criteria of a Bonferroni-adjusted P-value <0.05 and a log2 fold change greater than 0.58 or less than −0.58. Gene set enrichment analysis (GSEA) was done using the pre-rank mode and gene list was ranked by the stat value from DESeq2 analysis. Further, significantly enriched pathways (P-value< 0.05) were determined through gene set enrichment analysis integrated in the R/Bioconductor packages cluster Profiler v. 3.14.3 and GseaVis in R. Data has been submitted to Gene Expression Omnibus (GSE number pending).
Statistics and reproducibility
All described results from both in vivo and in vitro experimental design are representative of at least three independent experiments. Statistical comparisons between two groups were made using two-tailed unpaired Student’s t-test. For multiple group comparisons, either one- or two-way ANOVA (mixed model) followed by Tukey’s post hoc test was employed. Tumor growth and survival curves was conducted using a repeated-measure two-way ANOVA (mixed model), followed by Tukey’s multiple comparisons test. Mouse survival data were represented using Kaplan-Meier curves. A P-value of less than 0.05 was considered statistically significant. Data are presented as the average ±S.E.M where applicable. All statistical analyses were performed using GraphPad Prism 10 software (GraphPad Prism Software Inc.).
Acknowledgments
This work was supported by NIH/NCI grants R01 CA285321 and R01 CA240814 to S.Y.F., R01 CA288849 to V.S.S. and S.Y.F., NIH/NINDS grant 2R01 NS088629 to M.S.C., R01 HD103027 to M.L.M.-F., and R15 HD100970 to R.G.M. We thank Dr. Hector Teran-Navarro for technical assistance, Drs. Suzanne Ostrand-Rosenberg (University of Utah), Ben Z. Stanger, and Andy Minn (University of Pennsylvania) for reagents, and the members of Fuchs, Guo, and Fan labs for critical discussion.
Author contributions
Conceptualization, R.B., W.G., Y.F., A.J.M., V.S.S., M.S.C., and S.Y.F.; methodology, R.B., H.Z., A.H., Z.L., E.R., and S.S.G.; reagents, M.L.M.-F. and R.G.M.; investigation, R.B., H.Z., A.H., Z.L., E.R., C.-A.A., S.S.G., V.C.N., and D.P.B.; visualization, R.B., H.Z., A.H., Z.L., E.R., and C.-A.A.; funding acquisition, M.L.M.-F., R.G.M., M.S.C., V.S.S., and S.Y.F.; project administration, M.S.C., V.S.S., and S.Y.F.; supervision, M.S.C., V.S.S., and S.Y.F.; writing – original draft, review, and editing, all authors.
Declaration of interests
M.S.C. is an inventor on an OHSU patent related to the general use of the ITK7 compound. M.S.C., R.B., and S.Y.F. are co-inventors on a pending patent from OHSU/University of Pennsylvania related to ITK7 use.
Published: July 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101649.
Supplemental information
References
- 1.Bagchi S., Yuan R., Engleman E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021;16:223–249. doi: 10.1146/annurev-pathol-042020-042741. [DOI] [PubMed] [Google Scholar]
- 2.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kronig M.N., Wehrli M., Salas-Benito D., Maus M.V. Hurdles race for CAR T-cell therapy in digestive tract cancer. Immunol. Rev. 2023;320:100–119. doi: 10.1111/imr.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 5.Joyce J.A., Fearon D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 6.O'Donnell J.S., Teng M.W.L., Smyth M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019;16:151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 7.Smyth M.J., Ngiow S.F., Ribas A., Teng M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016;13:143–158. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 8.Brunkow M.E., Jeffery E.W., Hjerrild K.A., Paeper B., Clark L.B., Yasayko S.A., Wilkinson J.E., Galas D., Ziegler S.F., Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 9.Fontenot J.D., Gavin M.A., Rudensky A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 10.Plitas G., Rudensky A.Y. Regulatory T Cells: Differentiation and Function. Cancer Immunol. Res. 2016;4:721–725. doi: 10.1158/2326-6066.CIR-16-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakaguchi S., Yamaguchi T., Nomura T., Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Whiteside T.L., Jackson E.K. Adenosine and prostaglandin e2 production by human inducible regulatory T cells in health and disease. Front. Immunol. 2013;4:212. doi: 10.3389/fimmu.2013.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yano H., Andrews L.P., Workman C.J., Vignali D.A.A. Intratumoral regulatory T cells: markers, subsets and their impact on anti-tumor immunity. Immunology. 2019;157:232–247. doi: 10.1111/imm.13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dadey R.E., Workman C.J., Vignali D.A.A. Regulatory T Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020;1273:105–134. doi: 10.1007/978-3-030-49270-0_6. [DOI] [PubMed] [Google Scholar]
- 15.Buszko M., Shevach E.M. Control of regulatory T cell homeostasis. Curr. Opin. Immunol. 2020;67:18–26. doi: 10.1016/j.coi.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M., et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 17.Saito T., Nishikawa H., Wada H., Nagano Y., Sugiyama D., Atarashi K., Maeda Y., Hamaguchi M., Ohkura N., Sato E., et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016;22:679–684. doi: 10.1038/nm.4086. [DOI] [PubMed] [Google Scholar]
- 18.Turnis M.E., Sawant D.V., Szymczak-Workman A.L., Andrews L.P., Delgoffe G.M., Yano H., Beres A.J., Vogel P., Workman C.J., Vignali D.A.A. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44:316–329. doi: 10.1016/j.immuni.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamada T., Togashi Y., Tay C., Ha D., Sasaki A., Nakamura Y., Sato E., Fukuoka S., Tada Y., Tanaka A., et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA. 2019;116:9999–10008. doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Good Z., Spiegel J.Y., Sahaf B., Malipatlolla M.B., Ehlinger Z.J., Kurra S., Desai M.H., Reynolds W.D., Wong Lin A., Vandris P., et al. Post-infusion CAR T(Reg) cells identify patients resistant to CD19-CAR therapy. Nat. Med. 2022;28:1860–1871. doi: 10.1038/s41591-022-01960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haradhvala N.J., Leick M.B., Maurer K., Gohil S.H., Larson R.C., Yao N., Gallagher K.M.E., Katsis K., Frigault M.J., Southard J., et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 2022;28:1848–1859. doi: 10.1038/s41591-022-01959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shan F., Somasundaram A., Bruno T.C., Workman C.J., Vignali D.A.A. Therapeutic targeting of regulatory T cells in cancer. Trends Cancer. 2022;8:944–961. doi: 10.1016/j.trecan.2022.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tay C., Tanaka A., Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. 2023;41:450–465. doi: 10.1016/j.ccell.2023.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Dees S., Ganesan R., Singh S., Grewal I.S. Regulatory T cell targeting in cancer: Emerging strategies in immunotherapy. Eur. J. Immunol. 2021;51:280–291. doi: 10.1002/eji.202048992. [DOI] [PubMed] [Google Scholar]
- 25.Maeda Y., Wada H., Sugiyama D., Saito T., Irie T., Itahashi K., Minoura K., Suzuki S., Kojima T., Kakimi K., et al. Depletion of central memory CD8(+) T cells might impede the antitumor therapeutic effect of Mogamulizumab. Nat. Commun. 2021;12:7280. doi: 10.1038/s41467-021-27574-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H., Yu P., Tomar V.S., Chen X., Atherton M.J., Lu Z., Zhang H.G., Li S., Ortiz A., Gui J., et al. Targeting PARP11 to avert immunosuppression and improve CAR T therapy in solid tumors. Nat. Cancer. 2022;3:808–820. doi: 10.1038/s43018-022-00383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter-O'Connell I., Jin H., Morgan R.K., Zaja R., David L.L., Ahel I., Cohen M.S. Identifying Family-Member-Specific Targets of Mono-ARTDs by Using a Chemical Genetics Approach. Cell Rep. 2016;14:621–631. doi: 10.1016/j.celrep.2015.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo T., Liu J., Chen X., Jin L., Huang F., Zheng H. PARP11 regulates total levels of type-I interferon receptor IFNAR1. Nat. Microbiol. 2019;4:1771–1773. doi: 10.1038/s41564-019-0582-7. [DOI] [PubMed] [Google Scholar]
- 29.Guo T., Zuo Y., Qian L., Liu J., Yuan Y., Xu K., Miao Y., Feng Q., Chen X., Jin L., et al. ADP-ribosyltransferase PARP11 modulates the interferon antiviral response by mono-ADP-ribosylating the ubiquitin E3 ligase beta-TrCP. Nat. Microbiol. 2019;4:1872–1884. doi: 10.1038/s41564-019-0428-3. [DOI] [PubMed] [Google Scholar]
- 30.Fuchs S.Y., Spiegelman V.S., Kumar K.G.S. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 31.Hovelmeyer N., Schmidt-Supprian M., Ohnmacht C. NF-kappaB in control of regulatory T cell development, identity, and function. J. Mol. Med. (Berl.) 2022;100:985–995. doi: 10.1007/s00109-022-02215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quandt J., Arnovitz S., Haghi L., Woehlk J., Mohsin A., Okoreeh M., Mathur P.S., Emmanuel A.O., Osman A., Krishnan M., et al. Wnt-beta-catenin activation epigenetically reprograms T(reg) cells in inflammatory bowel disease and dysplastic progression. Nat. Immunol. 2021;22:471–484. doi: 10.1038/s41590-021-00889-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Loosdregt J., Fleskens V., Tiemessen M.M., Mokry M., van Boxtel R., Meerding J., Pals C.E.G.M., Kurek D., Baert M.R.M., Delemarre E.M., et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. 2013;39:298–310. doi: 10.1016/j.immuni.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Fuchs S.Y. Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J. Interferon Cytokine Res. 2013;33:211–225. doi: 10.1089/jir.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gangaplara A., Martens C., Dahlstrom E., Metidji A., Gokhale A.S., Glass D.D., Lopez-Ocasio M., Baur R., Kanakabandi K., Porcella S.F., Shevach E.M. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H., Tomar V.S., Li J., Basavaraja R., Yan F., Gui J., McBrearty N., Costich T.L., Beiting D.P., Blanco M.A., et al. Protection of Regulatory T Cells from Fragility and Inactivation in the Tumor Microenvironment. Cancer Immunol. Res. 2022;10:1490–1505. doi: 10.1158/2326-6066.CIR-22-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirby I.T., Kojic A., Arnold M.R., Thorsell A.G., Karlberg T., Vermehren-Schmaedick A., Sreenivasan R., Schultz C., Schüler H., Cohen M.S. A Potent and Selective PARP11 Inhibitor Suggests Coupling between Cellular Localization and Catalytic Activity. Cell Chem. Biol. 2018;25:1547–1553.e12. doi: 10.1016/j.chembiol.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Kovacs S.A., Fekete J.T., Gyorffy B. Predictive biomarkers of immunotherapy response with pharmacological applications in solid tumors. Acta Pharmacol. Sin. 2023;44:1879–1889. doi: 10.1038/s41401-023-01079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S., Zhang M., Li T., Chen X., Wu Q., Tian D., Granot Z., Xu H., Hao J., Zhang H. A Comprehensively Prognostic and Immunological Analysis of PARP11 in Pan-cancer. J. Leukoc. Biol. 2024 doi: 10.1093/jleuko/qiae030. [DOI] [PubMed] [Google Scholar]
- 40.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russell A.J.C., Weir J.A., Nadaf N.M., Shabet M., Kumar V., Kambhampati S., Raichur R., Marrero G.J., Liu S., Balderrama K.S., et al. Slide-tags enables single-nucleus barcoding for multimodal spatial genomics. Nature. 2024;625:101–109. doi: 10.1038/s41586-023-06837-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer-Ficca M.L., Ihara M., Bader J.J., Leu N.A., Beneke S., Meyer R.G. Spermatid head elongation with normal nuclear shaping requires ADP-ribosyltransferase PARP11 (ARTD11) in mice. Biol. Reprod. 2015;92:80. doi: 10.1095/biolreprod.114.123661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgoffe G.M., Woo S.R., Turnis M.E., Gravano D.M., Guy C., Overacre A.E., Bettini M.L., Vogel P., Finkelstein D., Bonnevier J., et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501:252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Overacre-Delgoffe A.E., Chikina M., Dadey R.E., Yano H., Brunazzi E.A., Shayan G., Horne W., Moskovitz J.M., Kolls J.K., Sander C., et al. Interferon-gamma Drives T(reg) Fragility to Promote Anti-tumor Immunity. Cell. 2017;169:1130–1141.e11. doi: 10.1016/j.cell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmitt E.G., Williams C.B. Generation and function of induced regulatory T cells. Front. Immunol. 2013;4:152. doi: 10.3389/fimmu.2013.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Togashi Y., Shitara K., Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019;16:356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 47.Qureshi O.S., Zheng Y., Nakamura K., Attridge K., Manzotti C., Schmidt E.M., Baker J., Jeffery L.E., Kaur S., Briggs Z., et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akkaya B., Oya Y., Akkaya M., Al Souz J., Holstein A.H., Kamenyeva O., Kabat J., Matsumura R., Dorward D.W., Glass D.D., Shevach E.M. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat. Immunol. 2019;20:218–231. doi: 10.1038/s41590-018-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bauer C.A., Kim E.Y., Marangoni F., Carrizosa E., Claudio N.M., Mempel T.R. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Invest. 2014;124:2425–2440. doi: 10.1172/JCI66375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang J.E., Hajdu C.H., Liot C., Miller G., Dustin M.L., Bar-Sagi D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017;20:558–571. doi: 10.1016/j.celrep.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawant D.V., Vignali D.A.A. Once a Treg, always a Treg? Immunol. Rev. 2014;259:173–191. doi: 10.1111/imr.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munn D.H., Sharma M.D., Johnson T.S. Treg Destabilization and Reprogramming: Implications for Cancer Immunotherapy. Cancer Res. 2018;78:5191–5199. doi: 10.1158/0008-5472.CAN-18-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohta A., Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y.N., Zhang H., Zhang L., Cai T.T., Huang D.J., He J., Ni H.H., Zhou F.J., Zhang X.S., Li J. Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: leading to poor survival in bladder cancer. Cell Death Dis. 2019;10:50. doi: 10.1038/s41419-018-1298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alicea-Torres K., Sanseviero E., Gui J., Chen J., Veglia F., Yu Q., Donthireddy L., Kossenkov A., Lin C., Fu S., et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat. Commun. 2021;12:1717. doi: 10.1038/s41467-021-22033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng H., Qian J., Carbone C.J., Leu N.A., Baker D.P., Fuchs S.Y. Vascular endothelial growth factor-induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood. 2011;118:4003–4006. doi: 10.1182/blood-2011-06-359745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cho C., Mukherjee R., Peck A.R., Sun Y., McBrearty N., Katlinski K.V., Gui J., Govindaraju P.K., Puré E., Rui H., Fuchs S.Y. Cancer-associated fibroblasts downregulate type I interferon receptor to stimulate intratumoral stromagenesis. Oncogene. 2020;39:6129–6137. doi: 10.1038/s41388-020-01424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gui J., Zahedi F., Ortiz A., Cho C., Katlinski K.V., Alicea-Torres K., Li J., Todd L., Zhang H., Beiting D.P., et al. Activation of p38α stress-activated protein kinase drives the formation of the pre-metastatic niche in the lungs. Nat. Cancer. 2020;1:603–619. doi: 10.1038/s43018-020-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks D.M., Anand S., Cohen M.S. Immunomodulatory roles of PARPs: Shaping the tumor microenvironment, one ADP-ribose at a time. Curr. Opin. Chem. Biol. 2023;77 doi: 10.1016/j.cbpa.2023.102402. [DOI] [PubMed] [Google Scholar]
- 60.Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34:360–394. doi: 10.1101/gad.334516.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gozgit J.M., Vasbinder M.M., Abo R.P., Kunii K., Kuplast-Barr K.G., Gui B., Lu A.Z., Molina J.R., Minissale E., Swinger K.K., et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell. 2021;39:1214–1226.e10. doi: 10.1016/j.ccell.2021.06.018. [DOI] [PubMed] [Google Scholar]
- 62.Katlinski K.V., Gui J., Katlinskaya Y.V., Ortiz A., Chakraborty R., Bhattacharya S., Carbone C.J., Beiting D.P., Girondo M.A., Peck A.R., et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell. 2017;31:194–207. doi: 10.1016/j.ccell.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J., Byrne K.T., Yan F., Yamazoe T., Chen Z., Baslan T., Richman L.P., Lin J.H., Sun Y.H., Rech A.J., et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49:178–193.e7. doi: 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li T., Fu J., Zeng Z., Cohen D., Li J., Chen Q., Li B., Liu X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48:W509–W514. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Bulk-seq data are accessible at GEO under accession number GSE249299.
-
•
No custom computer codes were generated in this study.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.