

Summary
Recurrent high-grade gliomas (rHGGs) have a dismal prognosis, where the maximum tolerated dose (MTD) of IV terameprocol (5 days/month), a transcriptional inhibitor of specificity protein 1 (Sp1)-regulated proteins, is 1,700 mg/day with median area under the plasma concentration-time curve (AUC) of 31.3 μg∗h/mL. Given potentially increased efficacy with sustained systemic exposure and challenging logistics of daily IV therapy, here we investigate oral terameprocol for rHGGs in a multicenter, phase 1 trial (GATOR). Using a 3 + 3 dose-escalation design, we enroll 20 patients, with median age 60 years (range 31–80), 70% male, and median one relapse (range 1–3). Fasting patients tolerate 1,200 mg/day (n = 3), 2,400 mg/day (n = 6), 3,600 mg/day (n = 3), and 6,000 mg/day (n = 2) oral doses without major toxicities. However, increased dosage does not lead to increased systemic exposure, including in fed state (6,000 mg/day, n = 4), with maximal AUC <5 μg∗h/mL. These findings warrant trials investigating approaches that provide sustained systemic levels of transcription inhibitors to exploit their therapeutic potential. This study was registered at ClinicalTrials.gov (NCT02575794).
Keywords: EM-1421, tetra-o-methyl nordihydroguaiaretic acid, NDGA, Sp1 transcription factor, glioblastoma, brain tumor, CNS cancer, brain malignancy, glioma (MeSH), astrocytoma (MeSH)
Graphical abstract
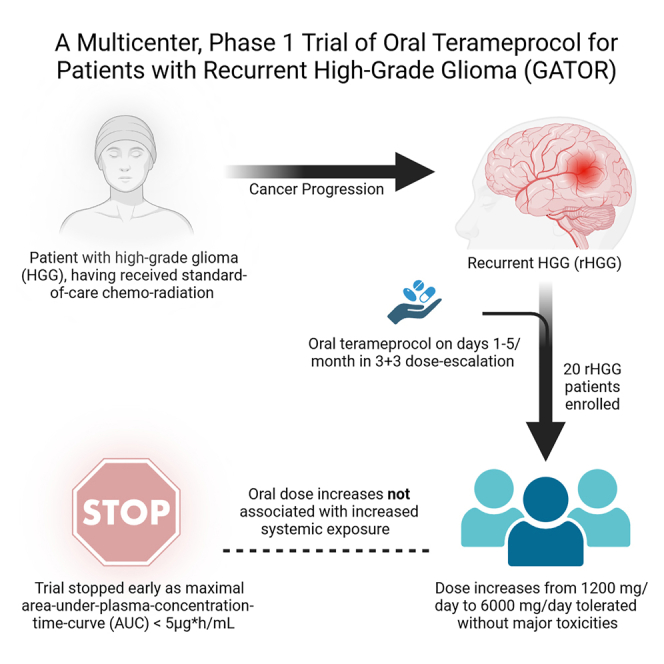
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.xcrm.2024.101630.
Highlights
-
•
Multicenter, phase 1 trial of oral terameprocol for recurrent high-grade glioma
-
•
Drug well tolerated in 3 + 3 dose-escalation design, but bioavailability was poor (∼2%)
-
•
Higher oral doses in patients not associated with increased systemic exposure
-
•
Trials investigating sustained systemic levels of transcription inhibitors are needed
Patients with recurrent high-grade glioma (rHGG) have a dismal prognosis, with prior reported feasibility of transcriptional inhibitor terameprocol when administered intravenously five days/month. Ahluwalia et al. report findings from a multicenter, phase 1 trial via the NCI Adult Brain Tumor Consortium, with a 3 + 3 dose-escalation design, of oral terameprocol for patients with rHGG.
Introduction
The most common malignant brain tumors in adults are World Health Organization (WHO) grade 3 and 4 gliomas, highly aggressive tumors with poor prognoses.1 Despite optimal treatment with radiation, surgery, temozolomide, and/or tumor-treating fields, most patients typically succumb to their disease within two years.2 Following disease progression, most chemotherapeutic agents have minimal activity, and none have improved survival, despite the thousands of patients accrued to neuro-oncology trials in the past three decades.3 Thus, testing novel treatment approaches is critical to improving clinical outcomes in patients with primary brain tumors.1,4,5
Terameprocol, a global transcriptional inhibitor, is a compound derived from the creosote bush Larrea tridentata, whose extracts have been used by Native Americans to treat various medical disorders.6,7,8 The active agent in the resin from the leaves is meso-nordihydroguaiaretic acid (NDGA), a lipophilic antioxidant.9 Most of the therapeutic effects of L. tridentata have been attributed to NDGA, whose synthetic meso-tetra-O-methyl derivative is called terameprocol (also known as EM1421 or M4N). Terameprocol has been demonstrated to possess antiviral, antiangiogenic, and antineoplastic activities. The compound has been shown to act against the human immunodeficiency virus (HIV), herpes simplex virus (HSV), pox virus, and human papilloma virus (HPV), among others.10,11,12,13,14,15,16 The drug’s activity has been demonstrated across numerous cancer cell lines and human tumor xenografts.17,18,19,20 Additionally, terameprocol induces reversible G2/M cell-cycle arrest in mammalian cell lines without significant cytotoxicity and is selectively tumoricidal in animal cancer models.10,21 The cell cycle blockade appears to be related to inhibiting the synthesis of cyclin-dependent kinase Cdc2 (also known as Cdk1 or p34), which is a primary regulator of the G2/M transition of the cell cycle.8,22,23 Its AKT inhibitor role has also been investigated, particularly with other agents acting on the PI3K/AKT/mTOR pathway.19,24 These effects likely stem from terameprocol’s transcriptional inhibition of proteins regulated by specificity protein 1 (Sp1).10,11,12,22,25 Expression of proteins regulated by Sp1 transcription factor, such as survivin (BIRC-5), Cdk1/cdc2, and vascular endothelial growth factor (VEGF), is known to be increased in cancer cells.26,27 Terameprocol has been demonstrated to inhibit the production and activation of survivin, an Sp1-regulated inhibitor of apoptosis protein.23,28,29
A phase 1 clinical trial of terameprocol (NCT00404248) was conducted by the National Cancer Institute (NCI) Adult Brain Tumor Consortium (ABTC) in 35 heavily pretreated patients with recurrent high-grade gliomas (rHGGs).30 The maximum tolerated dose (MTD) was established at 1,700 mg/day using a poly (ethylene glycol)-free formulation as a short intravenous infusion once daily for five consecutive days every 4 weeks, with median area under the plasma concentration–time curve (AUC) of 31.3 μg∗h/mL and mean clearance of nearly 54 L/h. Dose-limiting toxicities (DLTs) in that trial were grade 4 hypoxia and grade 2 interstitial nephritis, which occurred in one patient each. Stable disease was noted in 9 of 32 (38%) evaluable patients, with no objective responses observed. ABTC investigators also found that the concurrent administration of enzyme-inducing anti-epileptic drugs did not have a clinically significant effect on the serum pharmacokinetics, nor did terameprocol cause significant myelosuppression.30 While the study did establish the safety and therapeutic feasibility of terameprocol for rHGGs, efficacy was perceived to have been limited by the drug being given intravenously five days per month, instead of being received continuously, given terameprocol’s mechanism of transcriptional inhibition.
Therefore, ABTC investigators hypothesized that the utility of the drug, as a global transcriptional inhibitor, may be improved further potentially through continuous administration. Given the logistical constraints of extended intravenous dosing, an oral formulation of terameprocol was developed to allow the dosing schedule to be extended beyond 5 days a month if found safe and tolerable. Preclinical toxicokinetic data from dogs supported this hypothesis (Tables S1–S3), where oral administration in a continuous 28-day schedule was well tolerated and an increase from 100 mg/kg/day of oral terameprocol up to 300 mg/kg/day levels in males and 1,000 mg/kg/day in females resulted in increased systemic exposure.31
Therefore, this pre-registered, multicenter, phase 1 clinical trial was conducted to investigate the bioavailability, safety, tolerability, MTD, and pharmacokinetics of oral terameprocol administered five days per month to patients with high-grade gliomas (HGGs) in a 3 + 3 dose-escalation design (part 1). If the 5-day-per-month oral approach was deemed successful, then this trial aimed to determine the intratumoral drug concentration (part 2), followed by the determination of the maximum schedule for safe continuous administration (part 3). These objectives were in accordance with the recommendations by prior expert panels on the design of clinical trials for CNS drug delivery,3,5 with the trial protocol available in Supplemental Methods S1.
Results
The trial began enrolling patients in May 2018 and ended in March 2021. A total of 20 patients were enrolled in this phase 1 trial across five dose cohorts, with their baseline characteristics reported in Table 1. The median age of trial participants was 60 years (range 31–80), median Karnofsky Performance Status was 90 (range 60–90), and median number of relapses was 1 (range 1–3). 70% of the participants were male, 90% were White, and 85% had grade 4 gliomas. All participants had received prior radiotherapy. All patients had received systemic temozolomide with radiotherapy previously as per standard-of-care treatment for HGG in the first-line setting.
Table 1.
Patient and disease characteristics at baseline in the GATOR trial
| Characteristics | Total no. of patients (N = 20) |
|---|---|
| Age, median (range) | 60 (31–80) |
| Gender, N (%) | |
| Male | 14 (70%) |
| Female | 6 (30%) |
| Race/White, n (%) | |
| White | 18 (90%) |
| Non-White | 2 (10%) |
| KPS, median (range) | 90 (60–90) |
| Anticonvulsant usage, N (%) | 15 (75%) |
| Prior radiotherapy, N (%) | 20 (100%) |
| Prior gross total resection, N (%) | 15 (75%) |
| Prior number of relapses, median (range) | 1 (1–3) |
| Prior number of surgical procedures, median (range) | 1 (1–3) |
| Baseline measurable tumor, N (%) | 20 (100%) |
| Steroid usage, N (%) | 7 (35%) |
| Histological diagnosis, N (%) | |
| Glioblastoma (grade 4) | 17 (85%) |
| Grade 3 glioma | 3 (15%) |
KPS, Karnofsky Performance Status.
Overall, there were 3, 7, 3, 3, and 4 patients in the 1,200 mg/day without meal (dose level [DL]1fasted), 2,400 mg/day without meal (DL2fasted), 3,600 mg/day without meal (DL3fasted), 6,000 mg/day without meal (DL4fasted), and 6,000 mg/day with meal (DL4fed) cohorts, respectively. All patients received oral terameprocol on days 1–5 every 28 days. One patient in the 2,400 mg/day dose cohort who did not receive a single treatment dose was excluded from the analysis and 19 patients were, therefore, evaluated.
This trial enrolled the first three patients at DL1fasted of 1,200 mg/day in whom no DLT was seen (Table 2). Per protocol, three patients were enrolled at DL2fasted of 2,400 mg/day. The first patient in the DL2fasted cohort experienced a DLT (a Common Terminology Criteria for Adverse Events (CTCAE) grade 3 QTc prolongation) possibly related to terameprocol. Meanwhile, the third patient in this cohort experienced a serious adverse event (a CTCAE grade 4 borderline EKG criteria for myocardial infarction and a grade 3 seizure, which required hospitalization). The toxicities in the third patient were deemed unlikely to be related to terameprocol and either probably or possibly related to disease progression (Table 3). The safety data from DL2fasted and all relevant documentation were sent to the external Data Safety Monitoring Committee, which reviewed and accepted the site investigator’s assessment that the third patient’s death was unlikely to be related to terameprocol and probably related to tumor progression. This patient ultimately passed away from his disease. Thus, 1/3 of patients experienced DLT in the DL2fasted cohort.
Table 2.
Incidence and frequency of dose-limiting toxicities in the GATOR trial
| Dose level (mg/day) | N | Grade 3–4 non-hematologic toxicity | Grade 3–4 seizure or intracranial hemorrhage | ANC <500/mm3 | Platelets <25,000/mm3 | Febrile neutropenia | <80% of expected terameprocol dose due to hematologic toxicity | % of patients with DLT per dose level |
|---|---|---|---|---|---|---|---|---|
| 1,200 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2,400 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 33 |
| 2,400- EXP | 4a | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3,600 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6,000 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6,000 with meal | 4b | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
ANC, absolute neutrophil count; EXP, expanded dose cohort
1 patient out of the first 3 enrolled in the expanded 2,400 mg/day dose cohort was excluded due to deterioration prior to receiving therapy. Therefore the patient was replaced.
1 patient replaced (did not receive 80% of expected total dose during evaluation period).
Table 3.
Adverse events that were possibly, probably, or definitely related to terameprocol administration across all cycles (evaluable number of patients, N = 19)
| No. of patients (%) | Grade 1 | Grade 2 | Grade 3 | Total |
|---|---|---|---|---|
| Alanine aminotransferase increased | 2 (10.5%) | – | – | 2 (10.5%) |
| Anemia | 4 (21.1%) | – | – | 4 (21.1%) |
| Anorexia | 2 (10.5%) | 1 (5.3%) | – | 3 (15.8%) |
| Arthralgia | 1 (5.3%) | – | – | 1 (5.3%) |
| Aspartate aminotransferase increased | 1 (5.3%) | – | – | 1 (5.3%) |
| Blood bilirubin increased | 1 (5.3%) | – | – | 1 (5.3%) |
| Constipation | 1 (5.3%) | – | – | 1 (5.3%) |
| Diarrhea | 2 (10.5%) | – | – | 2 (10.5%) |
| Electrocardiogram QT corrected interval prolonged | – | – | 1 (5.3%) | 1 (5.3%) |
| Fatigue | 2 (10.5%) | 2 (10.5%) | – | 4 (21.1%) |
| Fever | 1 (5.3%) | – | – | 1 (5.3%) |
| Gastrointestinal pain | 1 (5.3%) | – | – | 1 (5.3%) |
| Hyperglycemia | 1(5.3%) | – | – | 1 (5.3%) |
| Hyperkalemia | – | 1 (5.3%) | – | 1 (5.3%) |
| Hypernatremia | 1 (5.3%) | – | – | 1 (5.3%) |
| Hypertension | 1 (5.3%) | – | – | 1 (5.3%) |
| Hypocalcemia | 1 (5.3%) | – | – | 1 (5.3%) |
| Hypokalemia | 1 (5.3%) | – | – | 2 (10.5%) |
| Hypomagnesemia | 3 (15.8%) | 1 (5.3%) | – | 4 (21.1%) |
| Hyponatremia | 3 (15.8%) | – | – | 3 (15.8%) |
| Hypophosphatemia | 1 (5.3%) | – | – | 1 (5.3%) |
| Lymphocyte count decreased | – | 1 (5.3%) | 1 (5.3%) | 2 (10.5%) |
| Mouth sores | 1 (5.3%) | – | – | 1 (5.3%) |
| Muscle cramp | 1 (5.3%) | – | – | 1 (5.3%) |
| Muscle weakness lower limb | 1 (5.3%) | – | – | 1 (5.3%) |
| Nausea | 3 (15.8%) | – | – | 3 (15.8%) |
| Platelet count decreased | 1 (5.3%) | – | – | 1 (5.3%) |
| Rash, maculopapular | 1 (5.3%) | – | – | 1 (5.3%) |
| Vomiting | 2 (10.5%) | – | – | 2 (10.5%) |
| Weight loss | 1 (5.3%) | – | – | 1 (5.3%) |
No grade 4 or grade 5 toxicity was found to be associated with terameprocol.
Per protocol, dose expansion at DL2fasted (2,400 mg/day) for three additional patients was carried out with no additional DLTs occurring. One patient completed enrollment in DL2fasted (2,400 mg/day) but was unable to participate in the study. Subsequent dose escalation resulted in three patients being treated at each of DL3fasted (3,600 mg/day) and DL4fasted (6,000 mg/day) with no DLTs, both groups receiving medications without meals. One patient in DL4fasted experienced an SAE with a grade 3 seizure, grade 2 fatigue, generalized muscle weakness, and gait disturbance, all deemed unlikely to be related to the study drug and probably related to their disease (Table 3). Based on safety data alone, the next step was to inititate DL5fasted (7,500 mg/day).
The results of the concurrent pharmacokinetic studies on study participants, however, demonstrated that systemic drug exposure remained consistently low despite increasing oral dosage (Table 4). Maximal systemic exposure, as measured by peak plasma concentration (Cmax, mean ± SD) for single dose (cycle 1 day 1), increased only from 0.03 ± 0.01 μg/mL (DL1fasted) to 0.20 ± 0.12 μg/mL (DL4fasted) and for multiple doses (cycle 1 day 4) from 0.03 ± 0.03 μg/mL (DL1fasted) to <0.20 μg/mL (DL4fasted). Due to extremely poor absorption, the sampling schema was inadequate to determine the half-life (T1/2) and area under the plasma concentration–time curve to infinity (AUCINF) for the majority of patients due to poor r2 values or large percent extrapolation. Among patients with available data, there was a slight increase in total exposure (as measured by AUCINF) through 3,600 mg/day dosing with a plateau at 6,000 mg/day after either single or multiple doses. AUCINF for cycle 1 day 1 of oral dose for DL1fasted was 0.1 μg∗h/mL (data from 1 single patient reportable) and for DL4fasted was 1.9 μg∗h/mL (data from 1 single patient reportable). For cycle 1 day 4 dosage, AUCINF could not be determined for DL1fasted, DL2fasted, and DL4fasted, while it was 2.5 ± 1.7 μg∗h/mL (three patients evaluable) for DL3fasted. The bioavailability was determined to be consistently around 2% (Table 4).
Table 4.
Plasma pharmacokinetic parameters of patients administered oral terameprocol in the GATOR trial
| Dose | Cmax (μg/mL) | Tmax (h) | AUCINF (μg∗h/mL) | F (%) | T1/2 (h) |
|---|---|---|---|---|---|
| Cycle 1 day 1 | |||||
| 1,200 mg | 0.03 ± 0.01 (3) | 1.0 (1.0–2.0; 3) | 0.1 (1) | 0.1 (1) | 1.9 (1) |
| 2,400 mg | 0.10 ± 0.05 (5) | 2.1 (1.9–4.0; 5) | 0.4, 0.7 (2) | 0.4, 0.6 (2) | 1.9, 6.2 (2) |
| 3,600 mg | 0.28 ± 0.19 (3) | 4.0 (2.0–6.0; 3) | 2.6 ± 2.3 (3) | 2.3 ± 2.1 (3) | 6.0 ± 0.6 (3) |
| 6,000 mg fasted | 0.20 ± 0.12 (3) | 4.0 (2.0–4.0; 3) | 1.9 (1) | 1.7 (1) | 8.0, 14.9 (2) |
| 6,000 mg fed | 0.33 ± 0.31 (4) | 4.1 (1.2–6.1; 4) | 2.4 ± 1.8 (3) | 2.2 ± 1.6 (3) | 6.6 ± 2.2 (3) |
| Cycle 1 day 4 | |||||
| 1,200 mg | 0.03 ± 0.03 (3) | 3.9 (2.0–4.1; 3) | NR (0) | NR (0) | 2.6, 3.1 (2) |
| 2,400 mg | 0.10 ± 0.06 (5) | 2.0 (1.9–4.1; 5) | NR (0) | NR (0) | 24.8 ± 6.2 (3) |
| 3,600 mg | 0.25 ± 0.14 (3) | 4.0 (2.0–6.0; 3) | 2.5 ± 1.7 (3) | 2.3 ± 1.5 (3) | 6.5 ± 0.8 (3) |
| 6,000 mg fasted | 0.1, 0.15 (2) | 2.0, 4.0 (2) | NR (0) | NR (0) | 14.9 (1) |
| 4,800 mg fed | 0.23 (1) | 5.9 (1) | NR (0) | NR (0) | 10.7 (1) |
| 6,000 mg fed | 0.60 ± 0.57 (3) | 8.0 (4.0–8.0; 3) | 2.2, 4.4 (2) | 2.0, 4.0 (2) | 5.2, 6.5 (2) |
Note: data are presented as the arithmetic mean ± SD (N). Tmax is presented as median (range; n). If n < 3, the actual values are reported; AUCINF, area under the plasma concentration-time curve to infinity; Cmax, peak plasma concentration; F, bioavailability; NR, not reportable; Tmax, time to peak concentration; T1/2, half-life.
The pharmacokinetic findings with oral dosing were discussed between the pharmaceutical sponsor and ABTC investigators, given that AUC with intravenous approach had reached 31.3 μg∗h/mL in previous studies. Oral administration of terameprocol was generally well tolerated. Toxicokinetic data from experiments in dogs were also considered, where administration of terameprocol, a highly lipophilic medication, under fed conditions resulted in increased systemic exposure with increasing oral doses (Tables S1–S3).31 Therefore, in an attempt to improve oral bioavailability, the protocol was amended to remove the requirement to take the oral drug on an empty stomach (Supplemental Methods S1). The participants were subsequently required to consume a meal within 30 min before or 30 min after taking the study drug. After protocol amendment, patients were then enrolled in the 6,000 mg/day with food (DL4fed) cohort. Four patients were included in DL4fed as the first patient had to be replaced due to noncompliance (6,000 mg/day was administered on days 1–3, then 4,800 mg on day 4, with none administered on day 5) and adverse events unrelated to the study drug. No DLTs occurred in the DL4fed cohort.
However, there was minimal improvement in the bioavailability between fasted and fed patient populations. Comparing cycle 1 day 1 measurement between DL4fasted and DL4fed cohorts, Cmax increased from 0.20 ± 0.12 μg/mL to 0.33 ± 0.31 μg/mL, while AUCINF slightly went up from 1.9 μg∗h/mL to 2.4 ± 1.8 μg∗h/mL. For cycle 1 day 4 measurements, AUCINF values for the two patients in the DL4fed cohort where data could be reported were 2.2 μg∗h/mL and 4.4 μg∗h/mL. These results were discussed by the ABTC central office with the pharmaceutical sponsor, and it was jointly decided, as part of standard operating procedure, to close the study due to the inability to reach the desired exposure of ∼31.3 μg∗h/mL with the oral formulation. Given the limited bioavailability and systemic exposure with oral dose, further investigations on dose distribution (part 2) and schedule expansion for continuous administration (part 3) were not pursued. In context of the limited systemic exposure, there were no partial or complete responses observed among the 19 participants, as expected. The median treatment duration was 8.1 weeks (2.3–176 weeks). Trial participants went off treatment either due to progressive disease (n = 17, 90%) or reasons other than toxicity. The best response to the treatment was stable disease among 6/19 patients (32%). The median overall survival was 8.1 months (95% confidence interval (CI): 2.8–10.8 months), while the median progression-free survival was 1.9 months (95% CI: 1.1–2.7 months) (Figure S1).
Thus, the trial dose escalation was terminated prematurely based on pharmacokinetics data, available for 18 and 17 patients on days 1 and 4, respectively (Table 4). Planned parts 2 and 3 evaluating intratumoral drug concentrations and safety of dosage extension from 5 to 30 days per month were not carried out. No patients were off the treatment due to toxicity. Overall, adverse events that were considered possibly, probably, or definitely related to terameprocol are described in Table 3. CTCAE grade 3 or 4 adverse events considered unrelated or unlikely to be related to terameprocol are described in Table S4.
Discussion
Thousands of patients have been enrolled in neuro-oncology trials in the past three decades, with limited results, indicating a general failure to meaningfully improve upon existing drug therapies, barring a few rare successes.3 These failures should inform the baseline requirements for investigations of novel agents in phase 2/3 trials, as was carried out here.3 Clinical trials of intravenous terameprocol conducted so far for various non-CNS tumors provided a substantial rationale for further exploring its effects as an anticancer drug, particularly through dosing strategies permitting continuous administration in a more feasible fashion.32 However, treating patients with primary brain tumors presents unique challenges and considerations.3,4,5 The initial trial of intravenous terameprocol in patients with rHGGs, reported by Grossman and colleagues in 2012, demonstrated the safety and feasibility of the drug, although the 5-day-per-month dosing of terameprocol, a transcriptional inhibitor, potentially inhibited activity. Given the intravenous route, daily administration was not feasible, and hence a trial was warranted utilizing drug delivery approaches permitting continuous administration.30 In this work, we report the results of a phase 1 dose-escalation and drug-distribution study of oral terameprocol in patients with rHGGs, i.e., WHO grade 3 and 4 gliomas. In the current study, dose escalation from 1,200 mg/day to 6,000 mg/day did not lead to improved oral bioavailability with saturable absorption at 6,000 mg. Hence, the decision to administer the drug with a meal to potentially increase bioavailability was taken through a protocol amendment, as justified by the preclinical data. However, the pharmacokinetic data of study participants showed minimal improvement in the bioavailability between fasted (a maximum of 2.5%) and fed (a maximum of 4.0%) patient populations. These data were discussed with the investigating team and the pharmaceutical sponsor, and the study was closed due to the inability to reach the desired bioavailability with the oral formulation.
Prior trials have reported promising activity of terameprocol when drug delivery has been adequate. Dunphy et al., in 2004, had reported a phase 1 trial of intralesional injection of terameprocol in three patients with refractory head and neck cancer.33 They had administered it intratumorally every week and demonstrated total necrosis of the injection site in 2/3 of patients who received three weekly doses at 10–15 mg/cm.33 Goel et al., in 2007, had reported the results of a dose-escalation trial of intravenous terameprocol in 29 advanced refractory solid tumors. They demonstrated DLT at 3,300 mg/day, along with partial response in 1/29 and stable disease in 6/29 patients.34 More recently, Tibes et al., in 2015, reported findings from a phase 1 trial of intravenous terameprocol in 16 patients with advanced acute myeloid leukemia or myelodysplastic syndrome.32 Enrolling four, five, and six patients in 1,000, 1,500, and 2,200 mg/day dose cohorts, they found no DLT, along with partial remission in one patient and stable disease in five individuals.
Terameprocol’s potential antitumor mechanistic activity has been well demonstrated. While NDGA has been historically used for various medicinal purposes,7 its derivative terameprocol has multipronged action in targeting the expression of Sp1-regulated genes, especially survivin and Cdc2/Cdk1.19,23,26,27,28 Sun et al. validated terameprocol’s inhibition of survivin and Cdk1 and also demonstrated its radiosensitizer action in non-small-cell lung cancer cell lines.35 Along with its antiviral action against HIV, HPV, and HSV, recent reports have also indicated terameprocol’s activity against West Nile virus and Zika virus.10,11,12,13,14,36 Antiviral activity has been reported to be secondary to disruption of cellular lipid metabolism, likely through inhibiting the sterol regulatory element-binding protein pathway.36 Targeting of survivin, which is highly expressed in HGG cells, through peptide vaccine conjugate has also resulted in promising clinical data published recently,37 and potential remains for combinatorial therapy. Future trials of terameprocol for glioblastoma may consider utilizing phenylalanine as a biomarker for better assessment of drug activity, as described recently through a combination of Raman microscopy and computational analysis to identify structural changes in glioblastoma bio-signatures.38
In conclusion, while oral terameprocol was found safe in patients with rHGG, poor absorption resulted in inadequate systemic exposure, thus limiting interpretations related to its potential efficacy. These findings also demonstrate the pharmacokinetic challenges of oral terameprocol and warrant trials investigating approaches that provide continually high systemic drug levels to fully exploit the therapeutic potential of transcriptional inhibitors.
Limitations of the study
First, the trial had to be closed early due to the failure to reach increased systemic exposure with increasing oral dosage of terameprocol in part 1 of the study. Thus, interpretations regarding clinical efficacy remain limited since adequate systemic exposure could not be achieved. Additionally, limited details were available regarding the tumor molecular profiling, such as isocitrate dehydrogenase (IDH) mutation and O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation status, as testing for these characteristics at baseline was not considered a part of the standard-of-care HGG workup at the participating North American institutions at the time the trial was designed in 2014. Extended time was later needed to obtain support to conduct the trial with NCI support. The trial ultimately enrolled patients from May 2018 to March 2021. Information collection related to molecular characteristics not being specified in the protocol and study closure at all participating sites precluded investigators from obtaining data on IDH mutation and MGMT promoter methylation status after the completion of the trial.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Terameprocol (Study Intervention/Drug) | Erimos Pharmaceuticals | EM-1421 |
| Biological samples | ||
| Individual Patient Data in GATOR trial | This paper | To be made available upon request to Manmeet S Ahluwalia, MD (manmeetA@baptisthealth.net) |
| Deposited data | ||
| Preclinical toxicokinetic data | Charles River Associates | Figshare data: https://doi.org/10.6084/m9.figshare.25966069 |
| Software and algorithms | ||
| SAS | SAS Institute (Cary, NC, US) | RRID:SCR_008567; https://www.sas.com/en_us/home.html |
| Phoenix WinNonlin | Certara (Radnor, PA, US) | RRID:SCR_024504; https://www.certara.com/software/phoenix-winnonlin/ |
| Other | ||
| NCI Protocol Identifier | National Cancer Institute | ABTC-1401 |
| Trial Registration | ClinicalTrials.Gov | NCT02575794 |
Resource availability
Lead contact
Further information and requests for individual patient data (IPD), especially for IPD meta-analysis, should be directed to and will be fulfilled by the lead contact, Manmeet S. Ahluwalia, MD, MBA (manmeetA@baptisthealth.net), upon reasonable request, completion of data-sharing agreements with ABTC, and approval by all regulatory authorities.
Materials availability
This study did not generate new or unique reagents.
Data and code availability
The methodology and findings of the preclinical toxicokinetic study are available on Figshare31 and Tables S1–-S3, respectively. The final amended version of the clinical protocol is available in the Supplemental Methods S1. This paper does not report any original code. The IPD data reported based on the clinical trial cannot be deposited in a public repository secondary to patient privacy concerns. The comparatively low incidence of high-grade gliomas overall, the IND status and non-standard-of-care-usage of trial medication (Terameprocol), and the limited sample size of the trial – all of these factors allow patients to be potentially deidentified from online publicly available datasets. For access to the data, please submit a request for access jointly to the lead contact (manmeetA@baptisthealth.net), the Adult Brain Tumor Consortium (ABTC) Manager (jfisher@jhmi.edu), and the ABTC data coordinator (ndanda1@jhmi.edu). The ABTC Central Office, located at Johns Hopkins University, Baltimore, MD, USA, has the primary responsibility of data storage and management. Upon receiving a request, the ABTC Central Office will review the request and if approved, work with the requestor to securely transmit the clinical trial data in a HIPAA-compliant manner. Detailed questions on submitting a request can also be directed to the ABTC manager or the data coordinator. The requestor must describe the objectives of the research project for which the data will be used. Data access will be considered for research purposes and non-commercial use only. In order to ensure patient privacy, access to personally identifiable information or sensitive clinical information will not be provided, and requests for data access must rigorously adhere to the consent agreements established with study participants. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Trial participants
Eligible study participants were at least 18 years of age with histologically confirmed supratentorial HGG (grade III or IV glioma) that had been progressive or recurrent following radiation therapy and chemotherapy. All subjects had histological confirmation of HGG (glioblastoma, anaplastic astrocytoma, anaplastic oligodendroglioma, high-grade astrocytoma, NOS [nomenclature according to the 2016 World Health Organization grading of gliomas] by either biopsy or resection. Patients were required to have measurable contrast-enhancing disease by MRI imaging within 21 days of starting treatment and to have recovered from any severe toxicity of prior therapy. Normal hematologic, renal, and hepatic function were required, along with a Karnofsky performance status of ≥60%, a Mini-Mental State Examination (MMSE) score of ≥15, and the ability to provide written informed consent. All patients with the potential for pregnancy or impregnating their partner had agreed to follow acceptable birth control methods, and women of childbearing potential were required to have a negative pregnancy test result. Patients on either enzyme-inducing or non-enzyme inducing antiepileptic drugs were eligible (30). Patients were excluded if they had a serious concurrent medical illness, infection, or malignancy; if they had a known sensitivity to terameprocol or any formulation excipients; if they were pregnant or breastfeeding; if they if they had a QTc(F) ≥450 mS; or if they were receiving recent anti-cancer therapy for their brain tumor (within 2–12 weeks, depending upon therapy) or specific hepatic enzyme inducers or inhibitors (Supplemental Methods S1). Participants were included after protocol approval by local Institutional Review Boards (IRBs) of all involved trial sites (Johns Hopkins Medicine IRB, Cleveland Clinic IRB, University of Pennsylvania IRB, Henry Ford Hospital IRB, Memorial Sloan Kettering Cancer Center IRB, University of Alabama at Birmingham IRB, University of Pittsburgh IRB, Wake Forest Health Sciences IRB), fulfillment of all regulatory requirements, and obtention of informed consent for participation from participants or their surrogate decision-makers. Demographic information and baseline clinical characteristics of included participants are described in Table 1.
Method details
Study design and reporting
The present work was a prospectively registered, phase 1, open-label, multicenter trial of oral terameprocol in patients with recurrent HGGs. The study was conducted through the National Cancer Institute (NCI) Adult Brain Tumor Consortium (Reference Code: ABTC-1401), whose predecessor was the New Approaches for Brain Tumor Therapy (NABTT) Consortium.39,40 The current work was conducted and reported following the Consolidated Standards Of Reporting Trials (CONSORT) guidance.41
Study phases and objectives
The work was planned to be conducted in three sequential parts. For part 1 of the study, the primary objective was to estimate the maximum tolerated dose (MTD) of terameprocol given orally on days 1–5 every 28 days in patients with recurrent HGG, i.e., a classic dose-escalation design. If the oral dosing for terameprocol was found to be tolerable and provided reasonable bioavailability in part 1 of the trial, then part 2 of the study aimed to determine the drug’s intratumoral concentration. Part 3 then aimed to determine the maximum schedule for safe continuous oral administration, from 5-day-a-month to potentially once daily, as further pre-specified in the clinical trial protocol (Supplemental Methods S1).
Study intervention
Erimos Pharmaceuticals (Houston, TX, US) provided formulated terameprocol in the form of 1-mL soft-gelatin capsules, with each capsule containing 300 mg of drug. In the NABTT-0503 trial (NCT00404248) in high-grade gliomas by Grossman et al., the MTD of IV terameprocol given once a day for five consecutive days monthly had been established at 1,700 mg/day.30 To estimate an initial safe dose for the oral formulation, the drug was administered orally once a day for five consecutive days each month at an initial dose of 1200 mg daily in this trial.
Trial conduct and assessment
Part 1 of the present trial aimed to define the safety, tolerability and MTD for terameprocol administered orally five days per month, using a standard 3 + 3 design for dose-finding. The starting dose of oral terameprocol of 1200 mg/day was self-administered daily for 5 days every 28 days on an empty stomach. This was concordant with prior dog toxicokinetic data and Grossman et al.’s prior trial, being ∼71% of the MTD of 1700 mg/day in the trial for IV formulation. Notably, tumor molecular characteristics, including MGMT promoter methylation and IDH mutation status, were planned to be evaluated in later phases of the trial, as these were not considered a part of standard-of-care HGG workup at the time of trial design at the participating institutions.
The standard 3 + 3 design was planned for dose finding in part 1. Up to six pre-specified dose levels of terameprocol were planned to be tested. If the MTD was not reached at the highest dose being tested, the highest dose was considered safe to take into Parts 2 and 3 of the study. Dose level (DL) 1 (starting dose) was 1200 mg/day with, DL2 was 2400 mg/day, DL3 was 3600 mg/day, DL4 was 6000 mg/day, DL5 was 7500 mg/day, DL6 was 9000 mg/day. Doses were scheduled to be escalated until the MTD was established based on the number of dose-limiting toxicities (DLTs), defined as clinically significant adverse event or abnormal laboratory values assessed as unrelated to disease progression, intercurrent illness, or concomitant medications and meeting any of the criteria below.
A. Criteria for hematological DLTs included an absolute neutrophil count of <500/μL, a platelet count of <25,000/μL, febrile neutropenia, any hematological toxicity preventing administration of >80% of planned terameprocol dose for the first cycle. Grade 3 or 4 lymphopenia was not considered DLT.
B. Criteria for non-hematological DLT included grade 3–4 seizure or intracranial hemorrhage; grade 3–4 severity except alopecia; grade 3 hyperglycemia; nausea/vomiting/diarrhea without sufficient prophylaxis lasting ≤3 days; grade 3 electrolyte disturbances that are asymptomatic and that respond to replacement therapy within 3 days; the first episode of deep venous thrombosis or pulmonary embolism; grade 3 QT interval corrected with Fridericia formula (QTcF). All other grade 3 neurological toxicities that responded to steroids within 3 days, anticonvulsants, or electrolyte correction were not considered DLT.
All patients who received at least one dose of terameprocol were evaluable for toxicity assessment. Patients who went off treatment for reasons other than toxicity and patients who were administered less than 80% of their expected dose of terameprocol in the study cohort were not evaluable for DLT. The targeted DLT rate was 33% and the dose escalation took a stepwise fashion. However, the dose determination was not only based on the safety, but also the drug concentration level in the plasma during the trial. The toxicity evaluation period was the first 4 weeks of treatment. Any DLT (as defined above) causing delay in treatment of over 21 days without recovery to ≤ grade 1 or baseline status resulted in taking the patient off treatment.
Patient pharmacokinetics were assessed during the course of the trial to monitor for saturable absorption at higher doses of terameprocol and to ensure that the previously achieved median AUC (31.3 μg∗h/mL) from the 1700 mg/day MTD of IV terameprocol was not exceeded. Oral administration was initially planned in the fasted state. However, in-study observation of saturable absorption at higher oral doses and the results of a pharmacokinetic investigation in dogs (Tables S1–S3) where administration of doses of oral terameprocol shortly after feeding resulted in dose-related increases in systemic exposure of the lipophilic medication terameprocol resulted in a trial protocol amendment to administer oral terameprocol in the fed state in the current study. This amendment and results thereof are discussed further in the results section.
Provided the 5-day-per-month oral dosing for terameprocol was found to be tolerable and provided reasonable bioavailability in part 1 of the trial, then part 2 of the study aimed to determine the drug’s intratumoral concentration and part 3 aimed to determine the maximum schedule for the safe continuous oral administration, from 5-day-a-month to potentially once daily. However, parts 2 and 3 were ultimately not carried out due to insufficient bioavailability with oral dosing.
Quantification and statistical analysis
Pharmacokinetic analyses
Blood samples were collected on cycle 1 day 1 and 4 prior to terameprocol administration and at 0.5, 1, 2, 4, 6, 8, and 24 h after the dose. Samples were processed immediately by refrigerated (4°C) centrifugation for 10 min at 1800xg to obtain plasma. The plasma samples were stored at −70°C until analysis using a validated liquid chromatography-mass spectrometry assay over the range of 5–1000 ng/mL with dilutions of up to 1:10 (volume by volume) as previously described.42 Pharmacokinetic parameters were calculated from individual terameprocol concentration-time data using standard noncompartmental methods as implemented in Phoenix WinNonlin version 8.4 (Certara, Radnor, PA, US; RRID:SCR_024504). Bioavailability was compared to the median AUC value of 31.3 μg∗h/mL obtained with the 1700 mg IV dose. Pharmacokinetic parameters were summarized descriptively (Table 4).
Statistical analyses
Baseline patient and disease characteristics were summarized using descriptive summaries. All adverse events were graded according to Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. Adverse events that were deemed possibly, probably, or definitely attributed to terameprocol were summarized using descriptive statistics. The OS was defined as the time from the trial registration date to the date of death due to all causes or censored if the patient was last known alive before the data cut-off. The PFS was defined as the time from the trial registration to the date of disease progression or censored if the patient is still on treatment or censored at the date last known free of progressive disease. Median OS and PFS were estimated using the Kaplan–Meier method,43 while their confidence intervals were constructed by the method of Brookmeyer-Crowley.44 All statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC; RRID:SCR_008567). The graphical abstract was created using Biorender (https://biorender.com/).
Additional resources
The study medication had investigational new drug (IND) status by the Center for Drug Evaluation and Research (CDER) of the US Food and Drug Administration (IND number 127,108). The trial was prospectively registered on ClinicalTrials.Gov (NCT02575794), with the final amended protocol available in Supplemental Methods S1. The access website is as follows: https://clinicaltrials.gov/ct2/show/NCT02575794.
Acknowledgments
The authors thank the patients for participating in this trial and the staff at all participating institutions for their help. The authors specifically thank Serena Desideri, Neeraja Danda, Joy Fisher, Trisha Surakus, Brittni Griffin, and other staff at the Adult Brain Tumor Consortium (ABTC) office at the Johns Hopkins University for their administrative support. The authors note that the author M.R. passed away during the peer review of this manuscript.
This study was conducted by the US National Cancer Institute (NCI) Adult Brain Tumor Consortium (NCI Protocol no. ABTC-1401), supported by the National Institutes of Health (NIH grant UM1CA137443) and the Analytical Pharmacology Shared Resource of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30CA006973 and UL1TR003098 and the Shared Instrument Grants S10OD020091 and S10RR026824).
Trial Registration: The trial was prospectively registered on ClinicalTrials.gov prior to patient accrual (NCT02575794) and had a US FDA investigational new drug approval status (IND 127108).
Author contributions
Conceptualization, M.S.A., R.C.H., P.Y.W., and S.A.G.; methodology, M.S.A. and S.A.G.; clinical investigation and trial administration, M.S.A., M.H., F.S.L., A.F.P., B.N., A.D., G.L., D.M.P., P.Y.W., and S.A.G.; pharmacokinetic investigation, S.G. and M.R.; statistical analysis, X.Y.; writing – original draft preparation, M.S.A., A.O., X.Y., A.A.K., and S.A.G.; writing – review and editing, M.S.A., S.A.G., and A.O.; supervision, M.S.A., P.Y.W., and S.A.G. All authors reviewed the manuscript.
Declaration of interests
M.S.A. – grants: Seagen, AstraZeneca, BMS, Bayer, Incyte, Pharmacyclics, Novocure, MimiVax, and Merck. Consultation fees: Bayer, Novocure, Kiyatec, Insightec, GSK, Xoft, Nuvation, Celularity, SDP Oncology, Apollomics, Prelude Therapeutics, Janssen, Tocagen, Voyager Therapeutics, ViewRay, Caris Life Sciences, Pyramid Biosciences, Varian Medical Systems, Cairn Therapeutics, AnHeart Therapeutics, Menarini Ricerche, Sumitomo Pharma Oncology, Autem therapeutics, GT Medical Technologies, Allovir, Equillium Bio., QV Bioelectronics, and Theraguix. Scientific Advisory Board memberships: Cairn Therapeutics, Pyramid Biosciences, Bugworks, and Modifi Biosciences. Data Safety and Monitoring Committee membership: VBI Vaccines. Stock shareholder: MimiVax, CytoDyn, Trisalus Lifesciences, and MedInnovate Advisors, LLC.
D.M.P. – consulting or advisory role: Orbus Therapeutics, Sumitomo Dainippon Pharma Oncology, Inc., Stemline Therapeutics, and Novocure. Research funding: Pfizer (Inst), Novartis (Inst), NeOnc Technologies (Inst), Orbus Therapeutics (Inst), Bristol Myers Squibb (Inst), Genentech/Roche (Inst), Pharmacyclics (Inst), Bayer (Inst), Karyopharm Therapeutics (Inst), Apollomics (Inst), Vigeo Therapeutics (Inst), Global Coalition for Adaptive Research (Inst), MimiVax (Inst), Ono Pharmaceutical (Inst), and Mylan (Inst). Equity ownership/stock options: Pfizer (Pharmaceuticals) and Gilead (Pharmaceuticals).
M.H. – Data Safety Monitoring Board member: Parexel and Advarra. Institutional research funding: Novartis and Vanquish.
P.Y.W. – research support: AstraZeneca, Black Diamond, Bristol Meyers Squibb, Celgene, Chimerix, Eli Lily, Erasca, Genentech/Roche, Kazia, MediciNova, Merck, Novartis, Nuvation Bio, Servier, Vascular Biogenics, and VBI Vaccines. Advisory board/consultant: AstraZeneca, Black Diamond, Celularity, Chimerix, Day One Bio, Genenta, Glaxo Smith Kline, Merck, Mundipharma, Novartis, Novocure, Nuvation Bio, Prelude Therapeutics, Sapience, Servier, Sagimet, Vascular Biogenics, and VBI Vaccines.
R.C.H. has patents related to the drug terameprocol and is affiliated with Erimos Pharmaceuticals, which holds rights to the drug.
S.G. is an independent contractor/consultant to Erimos Pharmaceuticals, LLC with no ownership or other financial interest in Erimos Pharmaceuticals or its products.
Published: July 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101630.
Supplemental information
References
- 1.Wen P.Y., Weller M., Lee E.Q., Alexander B.M., Barnholtz-Sloan J.S., Barthel F.P., Batchelor T.T., Bindra R.S., Chang S.M., Chiocca E.A., et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22:1073–1113. doi: 10.1093/neuonc/noaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Grossman S.A., Romo C.G., Rudek M.A., Supko J., Fisher J., Nabors L.B., Wen P.Y., Peereboom D.M., Ellingson B.M., Elmquist W., et al. Baseline requirements for novel agents being considered for phase II/III brain cancer efficacy trials: conclusions from the Adult Brain Tumor Consortium’s first workshop on CNS drug delivery. Neuro Oncol. 2020;22:1422–1424. doi: 10.1093/neuonc/noaa142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin V.A., Abrey L.E., Heffron T.P., Tonge P.J., Dar A.C., Weiss W.A., Gallo J.M. CNS Anticancer Drug Discovery and Development: 2016 conference insights. CNS Oncol. 2017;6:167–177. doi: 10.2217/cns-2017-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin V.A., Tonge P.J., Gallo J.M., Birtwistle M.R., Dar A.C., Iavarone A., Paddison P.J., Heffron T.P., Elmquist W.F., Lachowicz J.E., et al. CNS Anticancer Drug Discovery and Development Conference White Paper. Neuro Oncol. 2015;17:vi1–vi26. doi: 10.1093/neuonc/nov169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arteaga S., Carmona A., Luis J., Andrade-Cetto A., Cárdenas R. Effect of Larrea tridentata (creosote bush) on cholesterol gallstones and bile secretion in hamsters. J. Pharm. Pharmacol. 2005;57:1093–1099. doi: 10.1211/jpp.57.9.0004. [DOI] [PubMed] [Google Scholar]
- 7.Arteaga S., Andrade-Cetto A., Cárdenas R. Larrea tridentata (Creosote bush), an abundant plant of Mexican and US-American deserts and its metabolite nordihydroguaiaretic acid. J. Ethnopharmacol. 2005;98:231–239. doi: 10.1016/j.jep.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Lu J.M., Nurko J., Weakley S.M., Jiang J., Kougias P., Lin P.H., Yao Q., Chen C. Molecular mechanisms and clinical applications of nordihydroguaiaretic acid (NDGA) and its derivatives: an update. Med Sci Monit. 2010;16 RA93-100 878531 [pii] [PMC free article] [PubMed] [Google Scholar]
- 9.Floriano-Sanchez E., Villanueva C., Medina-Campos O.N., Rocha D., Sanchez-Gonzalez D.J., Cardenas-Rodriguez N., Pedraza-Chaverri J. Nordihydroguaiaretic acid is a potent in vitro scavenger of peroxynitrite, singlet oxygen, hydroxyl radical, superoxide anion and hypochlorous acid and prevents in vivo ozone-induced tyrosine nitration in lungs. Free Radic. Res. 2006;40:523–533. doi: 10.1080/10715760500419365. V68255712GH7416Q [pii] [DOI] [PubMed] [Google Scholar]
- 10.Chen H., Teng L., Li J.N., Park R., Mold D.E., Gnabre J., Hwu J.R., Tseng W.N., Huang R.C. Antiviral activities of methylated nordihydroguaiaretic acids. 2. Targeting herpes simplex virus replication by the mutation insensitive transcription inhibitor tetra-O-methyl-NDGA. J. Med. Chem. 1998;41:3001–3007. doi: 10.1021/jm980182w. [DOI] [PubMed] [Google Scholar]
- 11.Craigo J., Callahan M., Huang R.C., DeLucia A.L. Inhibition of human papillomavirus type 16 gene expression by nordihydroguaiaretic acid plant lignan derivatives. Antiviral Res. 2000;47:19–28. doi: 10.1016/s0166-3542(00)00089-9. S0166-3542(00)00089-9 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Hwu J.R., Tseng W.N., Gnabre J., Giza P., Huang R.C. Antiviral activities of methylated nordihydroguaiaretic acids. 1. Synthesis, structure identification, and inhibition of tat-regulated HIV transactivation. J. Med. Chem. 1998;41:2994–3000. doi: 10.1021/jm970819w. [DOI] [PubMed] [Google Scholar]
- 13.Gnabre J.N., Brady J.N., Clanton D.J., Ito Y., Dittmer J., Bates R.B., Huang R.C. Inhibition of human immunodeficiency virus type 1 transcription and replication by DNA sequence-selective plant lignans. Proc. Natl. Acad. Sci. USA. 1995;92:11239–11243. doi: 10.1073/pnas.92.24.11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park R., Giza P.E., Mold D.E., Huang R.C.C. Inhibition of HSV-1 replication and reactivation by the mutation-insensitive transcription inhibitor tetra-O-glycyl-nordihydroguaiaretic acid. Antiviral Res. 2003;58:35–45. doi: 10.1016/s0166-3542(02)00165-1. [DOI] [PubMed] [Google Scholar]
- 15.Khanna N., Dalby R., Tan M., Arnold S., Stern J., Frazer N. Phase I/II clinical safety studies of terameprocol vaginal ointment. Gynecol. Oncol. 2007;107:554–562. doi: 10.1016/j.ygyno.2007.08.074. [DOI] [PubMed] [Google Scholar]
- 16.Pollara J.J., Laster S.M., Petty I.T.D. Inhibition of poxvirus growth by Terameprocol, a methylated derivative of nordihydroguaiaretic acid. Antiviral Res. 2010;88:287–295. doi: 10.1016/j.antiviral.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Chang C.C., Liang Y.C., Klutz A., Hsu C.I., Lin C.F., Mold D.E., Chou T.C., Lee Y.C., Huang R.C.C. Reversal of multidrug resistance by two nordihydroguaiaretic acid derivatives, M4N and maltose-M3N, and their use in combination with doxorubicin or paclitaxel. Cancer Chemother. Pharmacol. 2006;58:640–653. doi: 10.1007/s00280-006-0214-9. [DOI] [PubMed] [Google Scholar]
- 18.Lambert J.D., Meyers R.O., Timmermann B.N., Dorr R.T. tetra-O-methylnordihydroguaiaretic acid inhibits melanoma in vivo. Cancer Lett. 2001;171:47–56. doi: 10.1016/s0304-3835(01)00560-2. [DOI] [PubMed] [Google Scholar]
- 19.Mak D.H., Schober W.D., Chen W., Heller J., Andreeff M., Carter B.Z. Tetra-O-methyl nordihydroguaiaretic acid inhibits growth and induces death of leukemia cells independent of Cdc2 and survivin. Leuk. Lymphoma. 2007;48:774–785. doi: 10.1080/10428190601186143. [DOI] [PubMed] [Google Scholar]
- 20.Meyers R.O., Lambert J.D., Hajicek N., Pourpak A., Kalaitzis J.A., Dorr R.T. Synthesis, characterization, and anti-melanoma activity of tetra-O-substituted analogs of nordihydroguaiaretic acid. Bioorg. Med. Chem. Lett. 2009;19:4752–4755. doi: 10.1016/j.bmcl.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heller J.D., Kuo J., Wu T.C., Kast W.M., Huang R.C. Tetra-O-methyl nordihydroguaiaretic acid induces G2 arrest in mammalian cells and exhibits tumoricidal activity in vivo. Cancer Res. 2001;61:5499–5504. [PubMed] [Google Scholar]
- 22.Smolewski P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. Idrugs. 2008;11:204–214. [PubMed] [Google Scholar]
- 23.Chang C.C., Heller J.D., Kuo J., Huang R.C.C. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc. Natl. Acad. Sci. USA. 2004;101:13239–13244. doi: 10.1073/pnas.0405407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao A., Lin C.Y., Wu R.C., Lee Y.S., Lee L.Y., Tsai C.L., Yang L.Y., Liu H., Chen S.J., Wang T.H., Lai C.H. The combination of everolimus and terameprocol exerts synergistic antiproliferative effects in endometrial cancer: molecular role of insulin-like growth factor binding protein 2. J. Mol. Med. 2018;96:1251–1266. doi: 10.1007/s00109-018-1699-5. [DOI] [PubMed] [Google Scholar]
- 25.Lopez R.A., Goodman A.B., Rhodes M., Blomberg J.A.L., Heller J. The anticancer activity of the transcription inhibitor terameprocol (meso-tetra-O-methyl nordihydroguaiaretic acid) formulated for systemic administration. Anti Cancer Drugs. 2007;18:933–939. doi: 10.1097/CAD.0b013e32813148e0. [DOI] [PubMed] [Google Scholar]
- 26.Vizcaíno C., Mansilla S., Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015;152:111–124. doi: 10.1016/j.pharmthera.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 27.Beishline K., Azizkhan-Clifford J. Sp1 and the 'hallmarks of cancer. FEBS J. 2015;282:224–258. doi: 10.1111/febs.13148. [DOI] [PubMed] [Google Scholar]
- 28.Cheung C.H.A., Cheng L., Chang K.Y., Chen H.H., Chang J.Y. Investigations of survivin: the past, present and future. Front. Biosci. 2011;16:952–961. doi: 10.2741/3728. 3728 [pii] [DOI] [PubMed] [Google Scholar]
- 29.Ryan B.M., O'Donovan N., Duffy M.J. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev. 2009;35:553–562. doi: 10.1016/j.ctrv.2009.05.003. S0305-7372(09)00085-1 [pii] [DOI] [PubMed] [Google Scholar]
- 30.Grossman S.A., Ye X., Peereboom D., Rosenfeld M.R., Mikkelsen T., Supko J.G., Desideri S., Adult Brain Tumor Consortium Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro Oncol. 2012;14:511–517. doi: 10.1093/neuonc/nor230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charles-River A 28-day Study of Terameprocol by Oral Gavage in Dogs with a 14-day Recovery Period. 14 October 2016. 2016. [DOI]
- 32.Tibes R., McDonagh K.T., Lekakis L., Bogenberger J.M., Kim S., Frazer N., Mohrland S., Bassett D., Garcia R., Schroeder K., et al. Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Invest. New Drugs. 2015;33:389–396. doi: 10.1007/s10637-014-0198-y. [DOI] [PubMed] [Google Scholar]
- 33.Dunphy F.R., Dukelow K.K., Provenzal J., Crawford J. Phase I clinical results of intralesional injection of tetra-o-methy nordihydroguaiaretic acid (M4N) in refractory head and neck cancer. J. Clin. Oncol. 2004;22:5614. doi: 10.1200/jco.2004.22.90140.5614. [DOI] [Google Scholar]
- 34.Goel S., Burris H., Mendelson D., Gollamudi R., Stern J., Frazer N., Jones S., Gordon M., Mani S. A phase I study of intravenous tetra-O-methyl nordihydroguaiaretic acid in patients with refractory malignancy. J. Clin. Oncol. 2007;25:3584. doi: 10.1200/jco.2007.25.18_suppl.3584. [DOI] [Google Scholar]
- 35.Sun Y., Giacalone N.J., Lu B. Terameprocol (tetra-O-methyl nordihydroguaiaretic acid), an inhibitor of Sp1-mediated survivin transcription, induces radiosensitization in non-small cell lung carcinoma. J. Thorac. Oncol. 2011;6:8–14. doi: 10.1097/JTO.0b013e3181fa646a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merino-Ramos T., Jiménez de Oya N., Saiz J.C., Martín-Acebes M.A. Antiviral Activity of Nordihydroguaiaretic Acid and Its Derivative Tetra-O-Methyl Nordihydroguaiaretic Acid against West Nile Virus and Zika Virus. Antimicrob. Agents Chemother. 2017;6 doi: 10.1128/aac.00376-17. e00376–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahluwalia M.S., Reardon D.A., Abad A.P., Curry W.T., Wong E.T., Figel S.A., Mechtler L.L., Peereboom D.M., Hutson A.D., Withers H.G., et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023;41:1453–1465. doi: 10.1200/jco.22.00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manciu F.S., Guerrero J., Bennet K.E., Chang S.Y., Rahman M., Martinez Lopez L.V., Chantigian S., Castellanos M., Manciu M. Assessing Nordihydroguaiaretic Acid Therapeutic Effect for Glioblastoma Multiforme. Sensors. 2022;22 doi: 10.3390/s22072643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grossman S.A., Fisher J.D., Piantadosi S., Brem H. The New Approaches to Brain Tumor Therapy (NABTT) CNS Consortium: Organization, Objectives, and Activities. Cancer Control. 1998;5:107–114. doi: 10.1177/107327489800500201. [DOI] [PubMed] [Google Scholar]
- 40.Chang S.M., Lamborn K.R., Kuhn J.G., Yung W.K.A., Gilbert M.R., Wen P.Y., Fine H.A., Mehta M.P., DeAngelis L.M., Lieberman F.S., et al. Neurooncology clinical trial design for targeted therapies: Lessons learned from the North American Brain Tumor Consortium. Neuro Oncol. 2008;10:631–642. doi: 10.1215/15228517-2008-021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schulz K.F., Altman D.G., Moher D., CONSORT Group CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. Bmj. 2010;340 doi: 10.1136/bmj.c332. [DOI] [PubMed] [Google Scholar]
- 42.Anders N.M., Romo C.G., Hemingway A., Ahluwalia M.S., Rudek M.A. Quantitation of terameprocol in human plasma by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2022;209 doi: 10.1016/j.jpba.2021.114525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaplan E.L., Meier P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958;53:457–481. doi: 10.1080/01621459.1958.10501452. [DOI] [Google Scholar]
- 44.Brookmeyer R., Crowley J. A Confidence Interval for the Median Survival Time. Biometrics. 1982;38:29–41. doi: 10.2307/2530286. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The methodology and findings of the preclinical toxicokinetic study are available on Figshare31 and Tables S1–-S3, respectively. The final amended version of the clinical protocol is available in the Supplemental Methods S1. This paper does not report any original code. The IPD data reported based on the clinical trial cannot be deposited in a public repository secondary to patient privacy concerns. The comparatively low incidence of high-grade gliomas overall, the IND status and non-standard-of-care-usage of trial medication (Terameprocol), and the limited sample size of the trial – all of these factors allow patients to be potentially deidentified from online publicly available datasets. For access to the data, please submit a request for access jointly to the lead contact (manmeetA@baptisthealth.net), the Adult Brain Tumor Consortium (ABTC) Manager (jfisher@jhmi.edu), and the ABTC data coordinator (ndanda1@jhmi.edu). The ABTC Central Office, located at Johns Hopkins University, Baltimore, MD, USA, has the primary responsibility of data storage and management. Upon receiving a request, the ABTC Central Office will review the request and if approved, work with the requestor to securely transmit the clinical trial data in a HIPAA-compliant manner. Detailed questions on submitting a request can also be directed to the ABTC manager or the data coordinator. The requestor must describe the objectives of the research project for which the data will be used. Data access will be considered for research purposes and non-commercial use only. In order to ensure patient privacy, access to personally identifiable information or sensitive clinical information will not be provided, and requests for data access must rigorously adhere to the consent agreements established with study participants. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.