

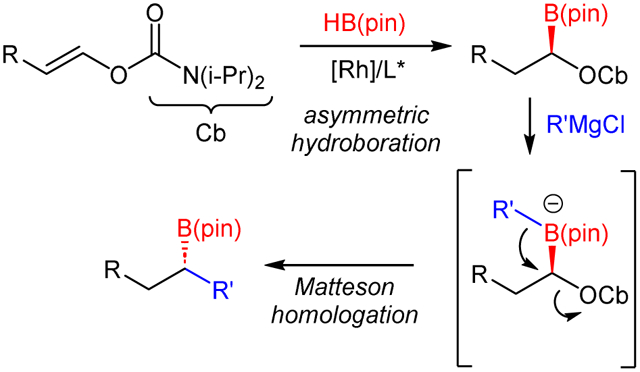
Abstract
Rh-catalyzed asymmetric hydroboration of enol carbamates yields α-boryl carbamates in good enantioselectivity. The enol carbamate starting materials can be prepared with moderate Z selectivity using a modified Juila olefination and used as an E/Z mixture, taking advantage of the faster reactivity of the major Z isomer in the directed hydroboration. Optically active α-boryl carbamates participate in a Matteson-type homologation with Grignard reagents in which the O-carbamate is substituted with high conservation of optical activity to provide enantioenriched secondary boronic esters.
Keywords: Hydroboration, Matteson homologation, Julia olefination, boronic esters, Rh catalysis
Graphical Abstract
The Matteson homologation remains a valuable synthetic transformation and the subject of continued development 60 years after its initial disclosure.1 The homologation involves a 1,2-metalate rearrangement of a boronate that features a leaving group alpha to boron (A, Scheme 1A). Noteworthy characteristics include 1) the ability to access the key intermediate A via addition of an organometallic to an α-functionalized boronic ester or via addition of a carbene equivalent (e.g. LiCH2Cl) to a boronic ester, 2) the stereospecificity of the reaction, both in terms of the migrating group (retention) and the carbon undergoing substitution (inversion), and 3) the synthetic utility of the resulting boronic ester, which can be converted to a wide range of useful functional groups.2,3
Scheme 1.
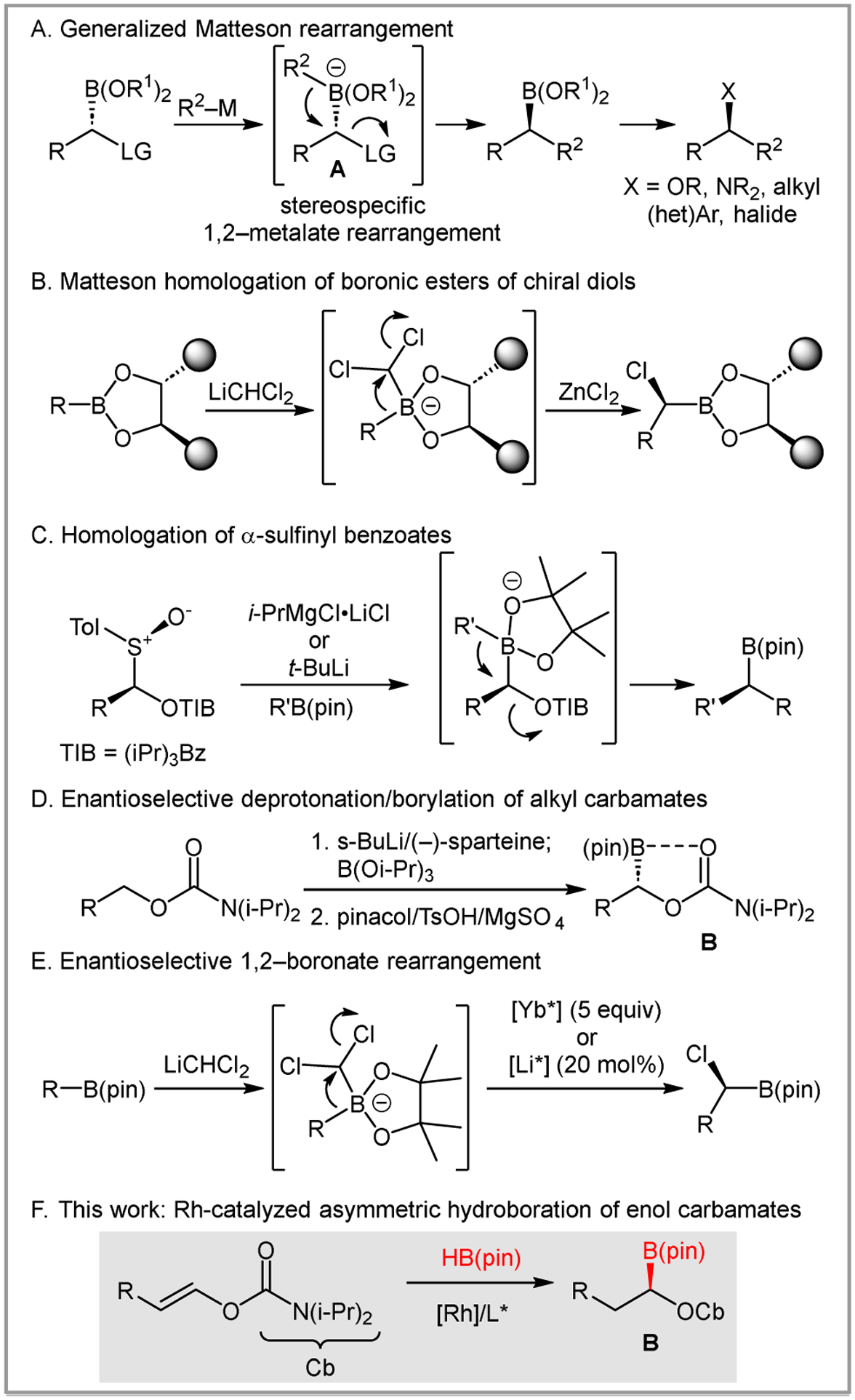
Approaches to stereoselective Matteson homologations.
Several strategies have been described to use the Matteson homologation to generate optically active boronic esters. Boronic acids derived from chiral non-racemic diols can lead to α-chloroboronic ester with high diastereoselectivity (Scheme 1B).2 These products can participate in a second, stereospecific homologation reaction (not shown). Alternatively, optically active α-sulfinyl benzoates can undergo metalation with retention of stereochemistry and subsequent trapping with boronic esters (Scheme 1C).4 Substitution of the benzoate provides enantioenriched boronic esters. Similarly, Hoppe demonstrated that directed, asymmetric lithiation of terminal carbamates provides a route to α-boryl carbamates (B), which can participate in Matteson homologation with Grignard reagents (Scheme 1D).5 Finally, Lewis acids complexed with chiral non-racemic ligands have been shown to promote the enantioselective Matteson rearrangement with lithium dichloromethane. A Yb(III)(bisoxazoline) complex was shown to work in super-stoichiometric quantities,6 while an in-situ generated Li salt was found to catalyze a highly enantioselective reaction (Scheme 1E).7,8
The Hoppe asymmetric lithiation/borylation strategy is attractive because it relies on readily available alcohol starting materials and gives rise to a reagent that can be used in asymmetric Matteson homologations.5 However, it requires stoichiometric quantities of sparteine, which has become challenging to access as the natural (−)-enantiomer9 while the unnatural (+)-enantiomer requires de novo synthesis.10 We reasoned that asymmetric hydroboration of enol carbamates could produce α-boryl carbamates with equal access to either enantiomer and the requirement for only catalytic quantities of the chiral controller (Scheme 1F). Here we describe the realization of this objective.
Asymmetric hydroboration has emerged as an effective strategy to synthetize optically active boronic esters.11 Directing groups near the olefin can impart high regioselectivity, as shown by Takacs with oximes and phosphonates.12 Takacs13 and Li14 also demonstrated that amides are also effective directing groups (Scheme 2A), while both Li15 and Tang16 have shown that enamides can direct asymmetric hydroboration (Scheme 2B). Since carbamates are effective directing groups for lithiation,17 we hypothesized that they might direct asymmetric hydroboration as well.18, 19
Scheme 2.
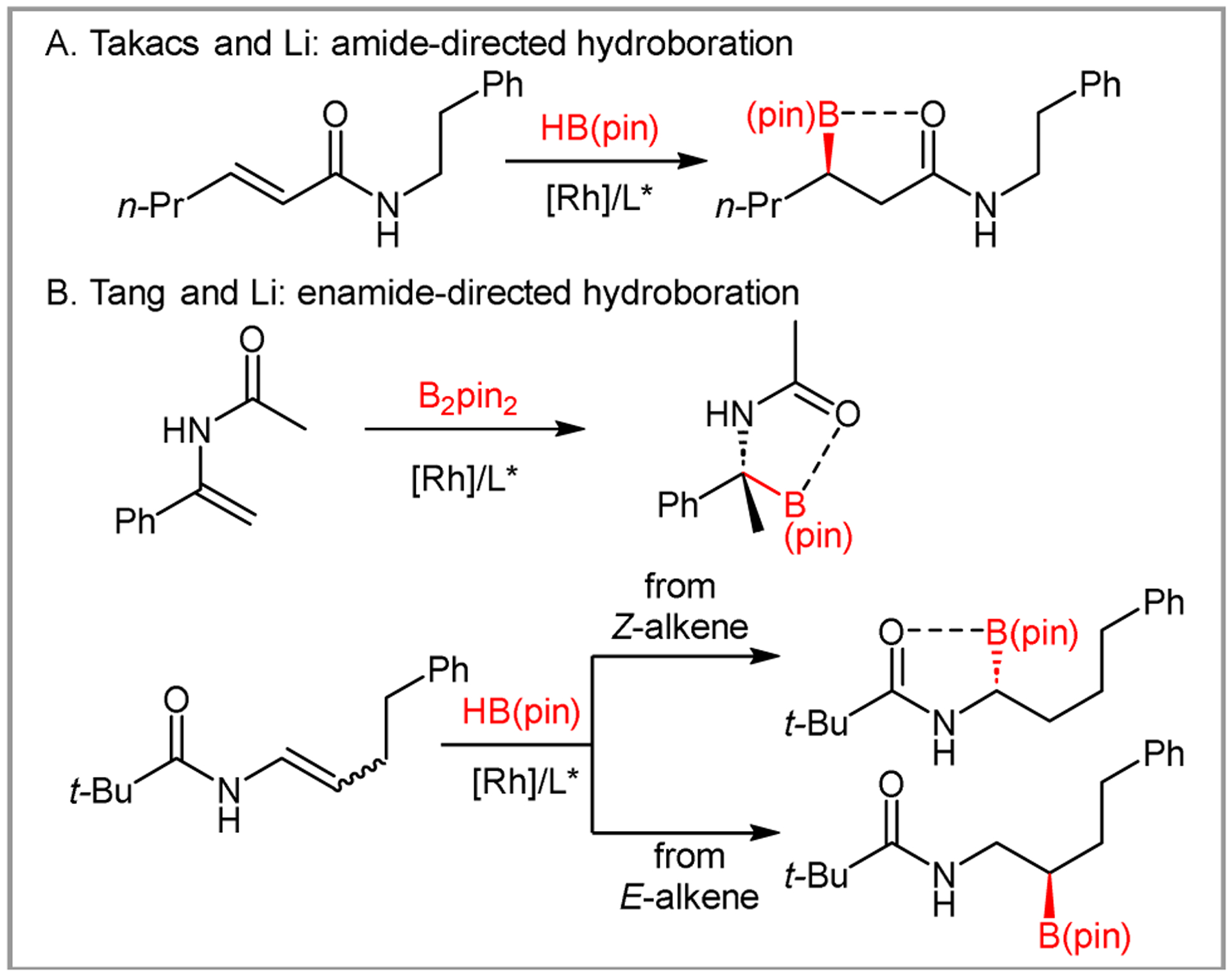
Carbonyl-directed catalytic asymmetric hydroborations.
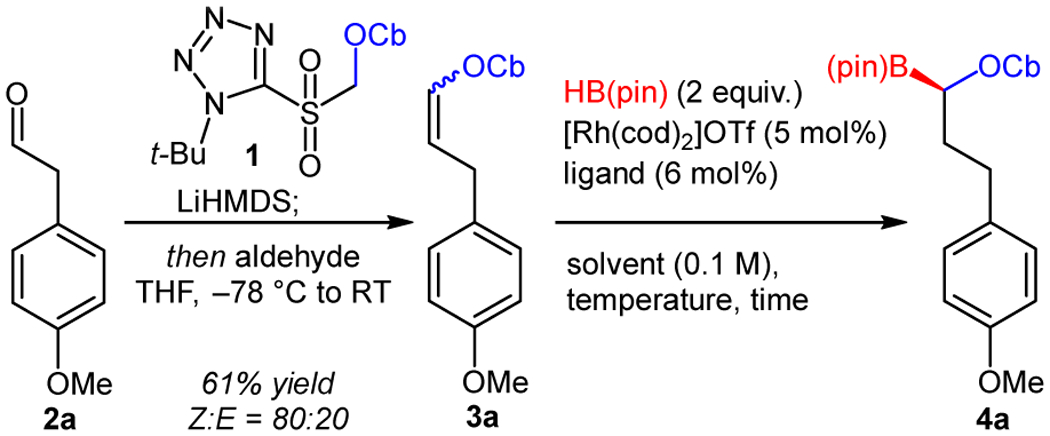
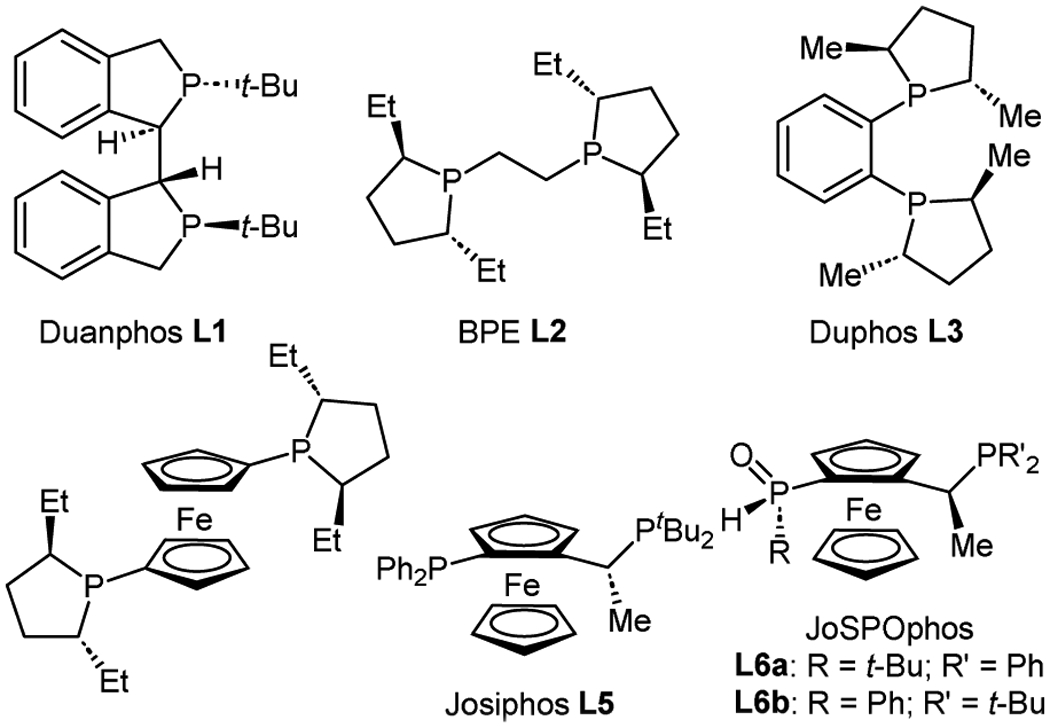
We started our investigation by examining the hydroboration of enol carbamate 3a, which was prepared with modest E/Z selectivity using a modified Julia-Kocienski olefination (Table 1).20, 21 A survey of ligands identified bisphosphines as a promising class to pursue, selected examples of which are shown in Table 1. Additional optimization studies shown in the supporting information. While Duanphos, BPE and Duphos gave poor reactivity and selectivity (Entries 1–3), ferrocene-containing ligands showed encouraging results. Ferrocelane gave modest yield and selectivity (Entry 4) while Josiphos-type ligands showed good reactivity but poor selectivity (Entry 5). JoSPOphos ligand L6b, featuring an alkyl phosphine and an aryl secondary phosphine oxide, combined high enantioselectivity with promising reactivity. By contrast, the isomeric ligand L6a did not support catalysis (Entries 6, 7). An evaluation of solvents revealed trifluorotoluene as optimal (see supporting information for more details) for reactivity. Whereas reactions in THF consumed only the Z isomer, the hydroboration in trifluorotoluene converted both olefin isomers. Since enantioselectivity dropped in this experiment, we speculated that E and Z olefin isomers could react with different rates and enantioselectivities (Entry 8). A control reaction with a ~1:1 E/Z mixture further depressed enantioselectivity and suggested that the E isomer might be converted with lower selectivity than the Z isomer (Entry 9 and see below). Finally, shorter reaction times or lower reaction temperatures selectively consumed the Z isomer and provided the hydroboration product in good yield and excellent enantioselectivity (Entries 10, 11).
Table 1.
| ||||||
| Entry | L | Solvent | Temp (°C) | Time (h) | Yield (%)c | Ee (%)d |
|---|---|---|---|---|---|---|
| 1 | L1 | THF | 50 | 12 | <5 | 7 |
| 2 | L2 | THF | 50 | 12 | 13 | 34 |
| 3 | L3 | THF | 50 | 12 | 21 | n.d. |
| 4 | L4 | THF | 50 | 12 | 52 | 65 |
| 5 | L5 | THF | 50 | 12 | 69 | 10 |
| 6 | L6a | THF | 50 | 12 | 0 | n.d. |
| 7 | L6b | THF | 50 | 12 | 50 | 91 |
| 8e | L6b | PhCF3 | 50 | 12 | 80f | 78 |
| 9g | L6b | PhCF3 | 50 | 12 | 77 | 70 |
| 10e | L6b | PhCF3 | 35 | 18 | 65f | 97 |
| 11e | L6b | PhCF3 | rt | 48 | 67f | 97 |
| ||||||
Reaction carried out on a 4.0 mmol scale in THF (0.1 M) using Julia reagent 1 (1.5 equiv), LiHMDS (1.7 equiv) and aldehyde 2a (1.0 equiv).
Reactions were run on a 0.05 mmol scale using enol carbamate 3a (1.0 equiv), [Rh(cod)2]OTf (5 mol%), ligand (6 mol%) and HBpin (2.0 equiv) unless noted otherwise.
Yields determined by 1H NMR.
Ee values determined by HPLC (n.d. = not determined).
0.35 mmol scale.
Isolated yield.
3a with Z : E = 55 : 45 was used.
Two optimized conditions emerged from these studies. Conditions A were generally best for aryl-substituted olefins (35 °C, 18h, 5 mol% Rh), while Conditions B (rt, 48h, 10 mol% Rh) were usually preferred for alkyl-substituted olefins (Scheme 3).22
Scheme 3.
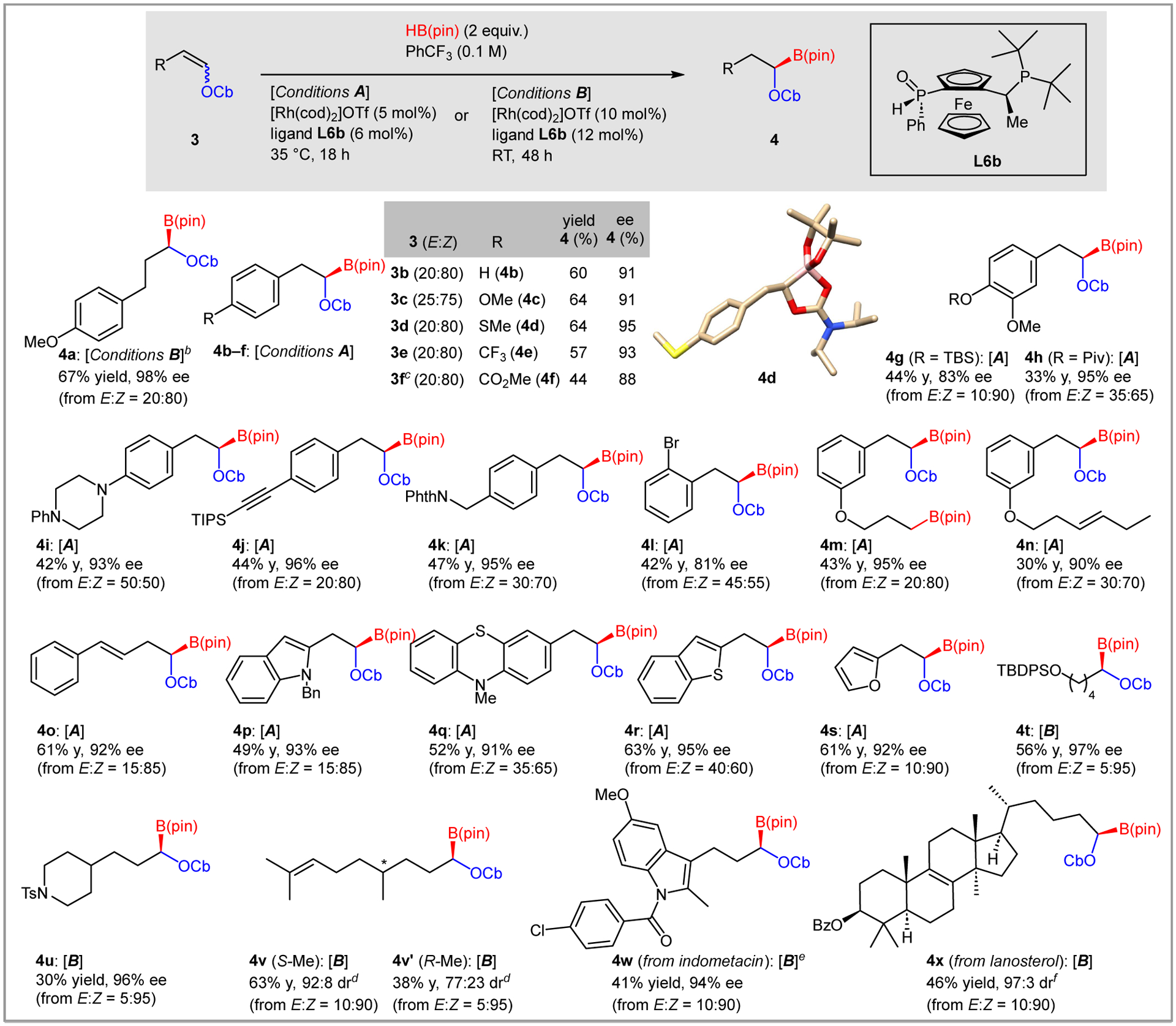
Substrate scope for the asymmetric hydroboration. aAll reactions were run in PhCF3 (0.10 M) using enol carbamate 3 (1.0 equiv), [Rh(cod)2]OTf (5–10 mol%), ligand L6b (6–12 mol%) and HB(pin) (2.0 equiv). Starting material, reagents and solvent were mixed in a N2-filled glove box. Yields of isolated products. Enantiomeric excess (ee) values were determined by HPLC. bWith [Rh(cod)2]OTf (5 mol%) and ligand L6b (6 mol%). cWith [Rh(cod)2]OTf (10 mol%) and ligand L6b (12 mol%). dDiastereomeric ratio (dr) was determined by 13C NMR. eReaction time = 14 h. fDiastereomeric ratio (dr) was determined by HPLC.
For aryl substituted olefins, the electronic properties of the aryl ring had minimal impact, with strongly electron donating substituents like alkoxy (4c, 4g, 4h) and amino (4i) groups performing similarly as electron withdrawing -CF3 (4e) and esters (4f). Substitution was tolerated in the meta and para positions, and an ortho-Br (4l) potentially gives rise to various ortho substitution patterns. Non-basic heteroaryl rings were accommodated by the reaction, including indole (4p, 4w), phenothiazine (4q), benzothiophene (4r) and furan (4s). Alkyl-substituted enol carbamates also reacted selectively, with both linear (4t) and cyclic (4u) substrates providing the hydroboration product in high ee. Single crystals derived from thioether 4d were suitable for X-ray diffraction analysis (CCDC 226136). The crystal structure showed coordination of the carbamate carbonyl to the boron and established the absolute stereochemistry of 4d. Other products were assigned by analogy.
Collectively, the examples in Scheme 3 demonstrate the functional group compatibility of the reaction. Tolerated groups include esters (4f, 4h, 4x), silyl ethers (4g, 4t), imides, amides and sulfonamides (4k, 4u, 4w), and halides (4e, 4l, 4w). Basic amines and strongly coordinating heterocycles (e.g., pyridines) are not tolerated under the current reaction conditions. With regard to other unsaturated C-C bonds, alkyne 3j, conjugated diene 3o, and the trisubstituted olefin derived from citronellal (3v) showed group-selective reduction of the enol carbamate. By contrast, a terminal olefin reacted at similar rates as the enol carbamate, giving diborated product 4m exclusively. Disubstituted olefins appear to react slightly slower than enol carbamates, as 4n was isolated with the unactivated olefin intact, albeit in reduced yield due to partial double hydroboration. Finally, hydroboration of the enol carbamates derived from citronellal demonstrated a matched/mismatched effect wherein the S-methyl isomer was formed with substantially higher ee than the R-methyl isomer.
During optimization studies with a 20:80 E:Z mixture of 3a, we noticed that ee was dependent on conversion. In particular, high ee was obtained when only the Z isomer was consumed, but it decreased as the E isomer reacted. Many of the enol carbamates could not be isolated as pure olefin isomers, but ester 3f could be resolved flash chromatography, allowing us to investigate the issue more carefully (Scheme 4). Purified E-3f reacted slowly under the reaction conditions, providing <20% of the hydroboration product. The main product in this reaction results from hydrogenation, which proved impossible to separate from the hydroboration product. By contrast, Z-3f reacted cleanly and provided the hydroboration product in 96% ee. For comparison, a 20:80 E:Z mixture generated the 4f in 88% ee. Estimates based on ee and conversion indicate that the E isomer generates the same major enantiomer, albeit in substantially reduced selectivity and rate. Thus, high ee can generally be obtained by monitoring the reaction for conversion of the Z isomer.
Scheme 4.
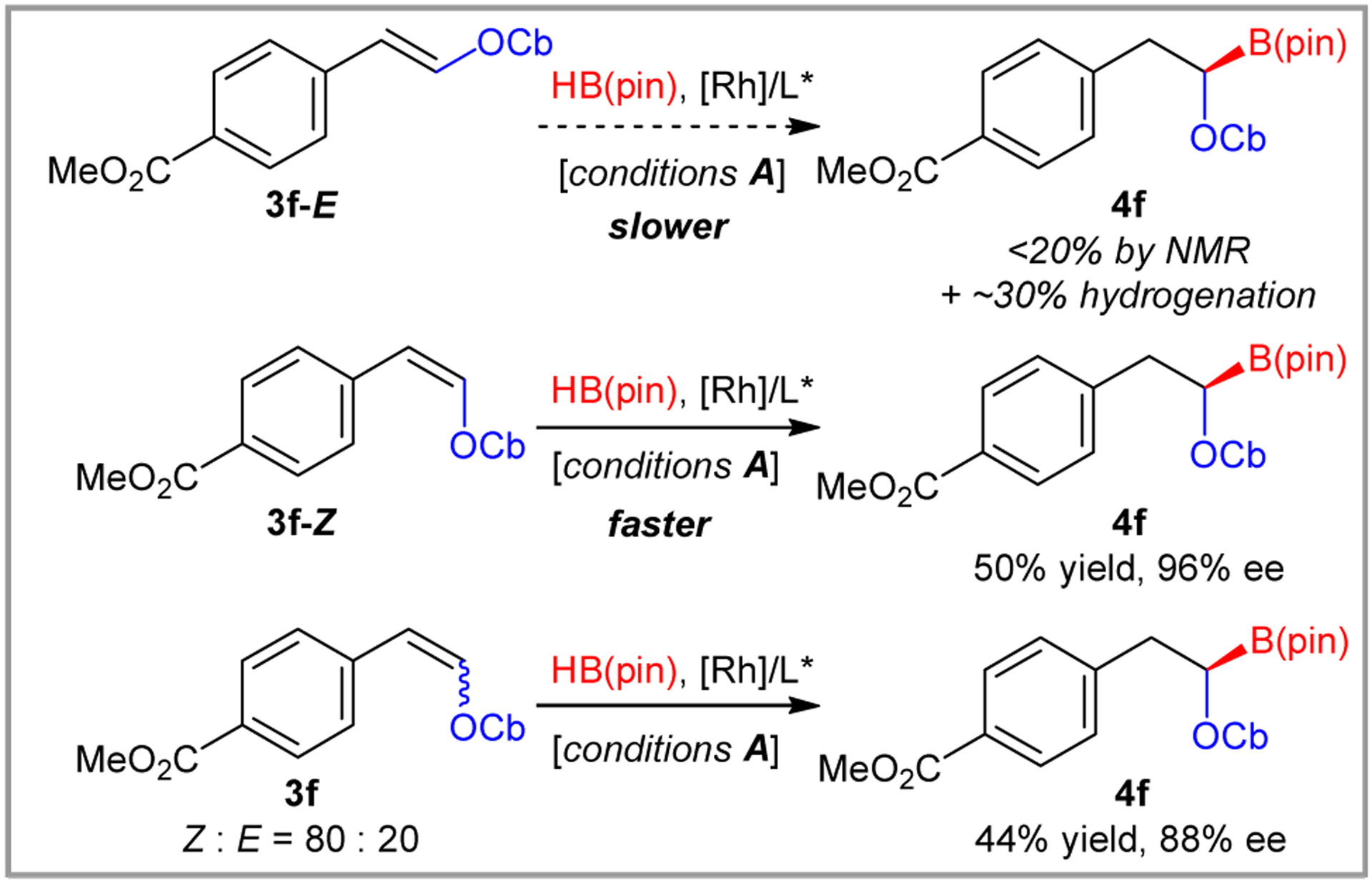
Impact of olefin stereochemistry on the asymmetric hydroboration. See Scheme 3 for reaction conditions.
Hoppe originally reported that α-boryl carbamates undergo stereospecific substitution with Grignard reagents.6 We confirmed this reactivity with products derived from asymmetric hydroboration, but also revealed some limitations (Scheme 5). Boronic ester 4a underwent substitution with a secondary (5a), aryl (5b) and alkenyl (5c) Grignard reagent in reasonable yields and high conservation of optical purity. However, reactions with linear or methyl Grignards were not successful, and PhLi and MeLi did not provide the desired product. Boronic ester 4c likewise reacted cleanly with PhMgCl, but alkyl and alkenyl Grignard reagents gave complex reaction mixtures. In general, the reactions were rapid, and performed best when the boronate was formed at −78 °C and then quickly placed into an ice bath. Prolonged stirring at −78 °C appeared to lead to racemization whereas starting the reaction at 4 °C resulted in messier reaction mixtures. Freshly prepared Grignard reagents performed most effectively, although the identity of the halogen (RMgBr vs RMgCl) did not appear to have a significant impact. Under the optimal conditions, several different boronate/Grignard combinations proved successful.23
Scheme 5.
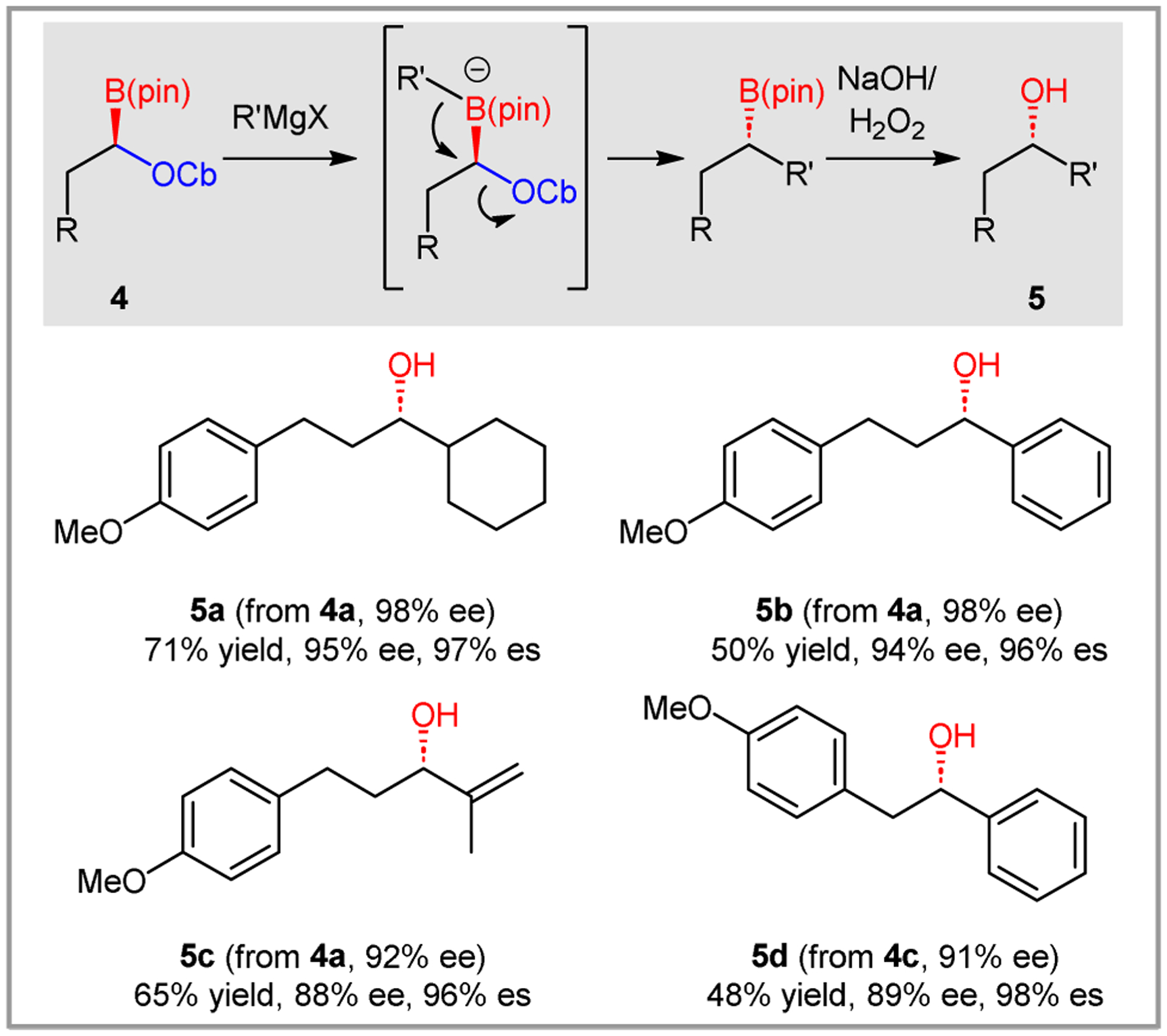
Reactions of hydroboration products with Grignard reagents. Reaction conditions: Et2O (0.1 M), Grignard reagent (1.4 equiv.), −78 to 4 °C, 2 h. Yields are given for products isolated after oxidation. Enantiomeric excess (ee) values were determined by HPLC. Enantioselectivity (es) = ee of product / ee of starting material.
In summary, Julia olefination of aryl and alkyl aldehydes with carbamate 1 offers a general route to enol carbamates with predominantly Z stereochemistry. Asymmetric hydroboration using a Rh(I)(JoSPOphos) complex selectively converted the Z-olefin to α-boryl carbamates with high enantioselectivity. Finally, the resulting α-boryl carbamates participate in Matteson-type homologation reactions with high stereospecificity, although opportunities exist to further improve the generality of this transformation.
Supplementary Material
Funding Information
Funding from Welch Foundation (I-1612) and the National Institutes of Health (RM1 GM142002).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References and Notes
- (1).Matteson DS; Mah RWH J. Am. Chem. Soc 1963, 85, 2599. [Google Scholar]
- (2).(a) Matteson DS J. Org. Chem 2013, 78, 10009. [DOI] [PubMed] [Google Scholar]; (b) Kinsinger T; Kazmaier UE J. Org. Chem 2022, 2022, e202200625. [Google Scholar]
- (3).(a) Casoni G; Kucukdisli M; Fordham JM; Burns M; Myers EL; Aggarwal VK J. Am. Chem. Soc 2017, 139, 11877. [DOI] [PubMed] [Google Scholar]; (b) Aiken SG; Bateman JM; Liao H-H; Fawcett A; Bootwicha T; Vincetti P; Myers EL; Noble A; Aggarwal VK Nat. Chem 2023, 15, 248. [DOI] [PubMed] [Google Scholar]
- (4).(a) Sandford C; Aggarwal VK Chemical Commun. 2017, 53, 5481. [DOI] [PubMed] [Google Scholar]
- (5).(b) Xu N; Liang H; Morken JP J. Am. Chem. Soc 2022, 144, 11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Beckmann E; Desai V; Hoppe D Synlett 2004, 2004, 2275. [Google Scholar]
- (7).Jadhav PK; Man H-WJ Am. Chem. Soc 1997, 119, 846. [Google Scholar]
- (8).Sharma HA; Essman JZ; Jacobsen EN Science 2021, 374, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang D; Zhou J; Hu Z; Xu TJ Am. Chem. Soc 2022, 144, 22870. [DOI] [PubMed] [Google Scholar]
- (10).Ritter SK Chem. Eng. News 2017, 95. [Google Scholar]
- (11).Tomioka K; Yamamoto Y; Yamada KI In Comprehensive Chirality, Carreira EM; Hisashi Y, Eds. Elsevier B.V.: 2012, 626. [Google Scholar]
- (12).(a) Crudden, Cathleen M; Edwards DE J. Org. Chem 2003, 2003, 4695. [Google Scholar]; (b) Geier SJ; Vogels CM; Melanson JA; Westcott SA Chem. Soc. Rev 2022, 51, 8877. [DOI] [PubMed] [Google Scholar]
- (13).(a) Chakrabarty S; Takacs JM ACS Catalysis 2018, 8, 10530. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bochat AJ; Shoba VM; Takacs JM Angew. Chem. Int. Ed 2019, 58, 9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Smith SM; Hoang GL; Pal R; Khaled MOB; Pelter LSW; Zeng XC; Takacs JM Chem. Commun 2012, 48, 12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhao W; Chen K-Z; Li A-Z; Li B-JJ Am. Chem. Soc 2022, 144, 13071. [DOI] [PubMed] [Google Scholar]
- (16).Bai X-Y; Zhao W; Sun X; Li B-JJ Am. Chem. Soc 2019, 141, 19870. [DOI] [PubMed] [Google Scholar]
- (17).Snieckus V Chem. Rev 1990, 90, 879. [Google Scholar]
- (18).Hu N; Zhao G; Zhang Y; Liu X; Li G; Tang WJ Am. Chem. Soc 2015, 137, 6746. [DOI] [PubMed] [Google Scholar]
- (19).Asymmetric hydroboration of enol ethers:; Dong W; Xu X; Ma H; Lei Y; Lin Z; Zhao W J. Am. Chem. Soc 2021, 143, 10902. [DOI] [PubMed] [Google Scholar]
- (20).Kocienski PJ; Bell A; Blakemore PR Synlett 2000, 2000, 365. [Google Scholar]
- (21).Representative procedure: Under an argon atmosphere, a solution of sulfone 1 (2.09 g, 6.0 mmol, 1.50 equiv) in anhydrous THF (40 mL) was cooled to −78 °C. LiHMDS (6.8 mL of 1.0 M in THF, 6.8 mmol, 1.7 equiv) was added dropwise, maintaining the temperature below −60 °C. The mixture was stirred with dry ice/acetone cooling for 40 min. Then a solution of 2-(4-methoxyphenyl)acetaldehyde 2a (0.60 g, 4.0 mmol, 1.00 equiv) in anhydrous THF (5.3 mL) was added dropwise, maintaining the temperature below −60 °C, and the mixture was stirred at ambient temperature overnight. Methanol (3 mL) was added to quench the reaction, and the volatiles were removed under vacuum. Subsequent flash chromatography on silica gel (hexanes/Et2O = 90:10) afforded 0.714 g (61%) of the product 3a as a yellow oil (Z : E = 80 : 20). 1H NMR (400 MHz, CDCl3) δ 7.19 – 7.09 (m, 3H, Z + 3H, E), 6.89 – 6.79 (m, 2H, Z + 2H, E), 5.43 (dt, J = 12.4, 7.5 Hz, 1H, E), 4.91 (td, J = 7.5, 6.4 Hz, 1H, Z), 4.11 (br s, 1H, Z + 1H, E), 3.81 (br s, 1H, Z + 1H, E), 3.79 (s, 3H, Z + 3H, E), 3.46 (dd, J = 7.5, 1.7 Hz, 2H, Z), 3.27 (dd, J = 7.4, 1.7 Hz, 2H, E), 1.30 – 1.19 (br m, 12H, Z + 12H, E). 13C NMR (151 MHz, CDCl3) δ 158.05, 153.05, 137.26, 135.63, 132.65, 132.57, 129.48, 129.31, 113.97, 111.35, 109.82, 55.41, 46.98, 45.82, 33.02, 30.46, 21.70, 20.51. ESI MS calcd for C17H26NO3+ [M+H]+ 292.2, found 292.2.
- (22).Enol carbamate 3a (102 mg, 0.35 mmol, 1.0 equiv) was weighed in a 2 dram screw-cap vial, which was then transferred into a N2-filled glove box. Inside a glove box ligand L6b (10 mg, 21 μmol, 6 mol%) was weighed in a 1 dram screw-cap vial. [Rh(cod)2]OTf (8.5 mg, 17.5 μmol, 5 mol%) was weighed in a separate 20-mL screw-cap vial equipped with a stir bar. Substrate 3a was dissolved in PhCF3 (2.8.0 mL), and the resulting solution was transferred to the vial with ligand (additional 0.7 mL PhCF3 was used for rinsing). The solution of substrate 3a and ligand L6b was then transferred to the vial with catalyst. HBpin (0.10 mL, 0.090 g, 0.70 mmol, 2.0 equiv) was then added, and the mixture was briefly stirred. The reaction vial was sealed with Teflon-coated screw cap. The reaction mixture was removed from the glove box and stirred at room temperature for 48 h. The mixture was concentrated, and pinacol was removed by transferring the crude product into a 250 mL round-bottom flask, dissolving in MeOH (2 mL), adding water (0.4–0.8 mL) until the solution became cloudy, and concentrating on a rotary evaporator. After 6–9 azeotropic evaporation cycles pinacol was removed completely. Flash chromatography on silica gel (hexanes/EtOAc = 70:30) afforded 98 mg (67%) of the product 4a as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.16 – 7.07 (m, 2H), 6.87 – 6.77 (m, 2H), 4.07 (hept, J = 6.9 Hz, 1H), 3.82 (dd, J = 10.6, 3.9 Hz, 1H), 3.78 (s, 3H), 3.76 (hept, J = 6.9 Hz, 1H), 2.79 (ddd, J = 14.7, 9.9, 5.3 Hz, 1H), 2.65 (ddd, J = 13.8, 9.4, 6.6 Hz, 1H), 2.03 – 1.93 (m, 1H), 1.92 – 1.80 (m, 1H), 1.28 – 1.20 (m, 12H), 1.17 (s, 12H). 13C NMR (101 MHz, CDCl3) δ 162.77, 157.66, 134.60, 129.42, 113.70, 79.74, 55.26, 48.44, 46.61, 33.61, 33.35, 25.30, 24.96, 20.62, 20.61, 20.35, 20.26 (the carbon attached to boron was not observed due to quadrupolar relaxation). 11B NMR (128 MHz, CDCl3) δ 12.86. ESI MS calcd for C23H39BNO5+ [M+H]+ 420.3, found 420.2. Chiral HPLC CHIRALCEL® OZ-3, 0.75% IPA in hexane, 1.0 mL/min, 220 nm: 98% ee (7.1 and 11.7 min).
- (23).At −78 °C and under argon, freshly c-HexMgCl (0.12 mmol, 1.4 equiv) was added dropwise (over 2 min) to a solution of 4a (35 mg, 0.083 mmol, 1.0 equiv, 98% ee) in anhydrous Et2O (1.0 mL). The mixture was stirred under argon in a cold room (4 °C) for 2 h. After that, insoluble solids were removed by filtration through a Pasteur pipette with a silica gel plug (ca. 1 mL of silica gel was used; additional 2-mL portion of Et2O was used for washing), and the filtrate was concentrated under vacuum. The crude secondary boronate was then redissolved in THF (1.0 mL), and the solution was cooled to 0 °C and treated with 3M NaOH (0.17 mL) and 30% aq. H2O2 (0.17 mL). The mixture was stirred at ambient temperature for 2 h. At 0 °C, sat. aq. Na2S2O3 (1 mL) was carefully added to quench the reaction, and the product was extracted with EtOAc (3 × 2 mL). Combined organic layers were dried over Na2SO4, filtered and concentrated. Subsequent flash chromatography eluting with hexanes/EtOAc = 85:15) afforded 15 mg (71%) of the 5a as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.18 – 7.08 (m, 2H), 6.88 – 6.79 (m, 2H), 3.79 (s, 3H), 3.38 (ddd, J = 8.9, 5.4, 3.2 Hz, 1H), 2.77 (ddd, J = 13.8, 9.9, 5.4 Hz, 1H), 2.59 (ddd, J = 13.8, 9.7, 6.7 Hz, 1H), 1.88 – 1.58 (m, 6H), 1.42 – 0.91 (m, 7H). 13C NMR (151 MHz, CDCl3) δ 157.83, 134.53, 129.43, 113.92, 75.72, 55.39, 43.90, 36.26, 31.56, 29.30, 27.91, 26.65, 26.46, 26.32. ESI MS calcd for C16H23O+ [M–OH]+ 231.17, found 231.2. Chiral HPLC CHIRALCEL® OD-H, 3.5% IPA in hexane, 1.5 mL/min, 220 nm: 95% ee, 97% es.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.