

Abstract
OBJECTIVES:
A novel longitudinal clustering technique was applied to comprehensive autoantibody data from a large, well-characterized, multinational inception systemic lupus erythematosus (SLE) cohort to determine profiles predictive of clinical outcomes.
METHODS:
Demographic, clinical, and serological data from 805 SLE patients obtained within 15 months of diagnosis and at three- and five-year follow-up were included. For each visit, sera were assessed for 29 ANA immunofluorescence patterns and 20 autoantibodies. K-means clustering on principal component analysis-transformed longitudinal autoantibody profiles identified discrete phenotypic clusters. One-way ANOVA compared cluster enrolment demographics and clinical outcomes at ten-year follow-up. Cox proportional hazards model estimated the hazards ratio (HR) for survival adjusting for age of disease onset.
RESULTS:
Cluster 1 (n=137, high frequency of anti-Sm, anti-U1RNP, AC-5 (large nuclear speckled pattern), and high ANA titres) had the highest cumulative disease activity and immunosuppressants/biologics use at year ten. Cluster 2 (n=376, low anti-dsDNA and ANA titres) had the lowest disease activity, frequency of lupus nephritis, and immunosuppressants/biologics use. Cluster 3 (n=80, highest frequency of all five antiphospholipid antibodies) had the highest frequency of seizures and hypocomplementemia. Cluster 4 (n=212) also had high disease activity and was characterized by multiple autoantibody reactivity including to anti-histone, -dsDNA, -ribosomal P, -SSA/Ro60, -SSB/La, -Ro52/TRIM21, -PCNA, and -centromere B). Clusters 1 (adjusted HR 2.60 [95%CI: 1.12–6.05], p=0.03) and 3 (adjusted HR 2.87 [95%CI: 1.22–6.74], p=0.02) had lower survival compared to Cluster 2.
CONCLUSION:
Four discrete SLE patient longitudinal autoantibody clusters were predictive of long-term disease activity, organ involvement, treatment requirements, and mortality risk.
Introduction
Systemic Lupus Erythematosus (SLE) is a complex disease that is challenging to diagnose, prognosticate, and effectively treat due to substantial disease heterogeneity. It can affect virtually any organ system at different time points during its course. To better understand SLE’s underlying biology and how it relates to prognosis, attempts have been made to stratify SLE patients into endotypes including patient clusters based on common autoantibody profiles.1–11 SLE pathogenesis is multifaceted, but certain autoantibodies are a hallmark of SLE and have proven useful as diagnostic and predictive biomarkers for disease manifestations and activity.
Past machine learning (ML) analyses to define SLE clusters were cross-sectional, studied patients seen at single centres and assessed relatively few SLE-related autoantibodies.1–11 Over 200 different autoantibodies have been described in SLE, but only 10–20 are widely available through clinical diagnostic laboratories and utilized by clinicians and researchers.12 Furthermore, there are no reports of ML approaches to study longitudinal autoantibody data in SLE to date. Previous evaluations of antinuclear antibodies (ANA) and SLE-related autoantibodies using traditional statistical methods suggest that a patient’s antibody status can change from positive to within the normal range and vice versa during the disease course.13–16 However, factors influencing changes in autoantibody status over time are also poorly understood.
ML is inherently flexible and can identify patterns and interactions in large datasets and distinguish multiple clinical factors and autoantibody status that would otherwise be challenging to ascertain within the modeling assumptions and restrictions of traditional statistical methods. For this reason, ML techniques have been applied to stratify SLE patients into distinct phenotypes associated with different clinical outcomes including organ damage.1–11, 17, 18 Here we apply a ML clustering technique to a longitudinal comprehensive autoantibody panel to identify distinct subgroups of SLE patients that are predictive of future clinical outcomes.
Methods
Study Population
Between 1999 and 2011, 1827 patients fulfilling the 1997 Updated ACR SLE Classification Criteria for definite SLE19 within 15 months of diagnosis from 31 medical centres in 11 countries were enrolled into the Systemic Lupus International Collaborating Clinics (SLICC) inception cohort (https://sliccgroup.org).20 Sera, clinical and demographic data were collected at enrolment and annually thereafter. Of the 1827 patients, 1432 (78.4%) were followed for ≥ four years; of these 1432 patients, we included the 805 patients who provided an enrolment and two additional serum samples within five years of enrolment, with the third sample being ≥ four years after enrolment. Although we were not able to include the entire cohort, we demonstrated in a prior study that the 805 patients were similar to the 627 patients who provided ≥ four years of data, but did not have three available serial serum samples.21 The study was approved by the Institutional Review Board at each SLICC site. Permission from the SLICC Biological Material and Data Utilization Committee was obtained to access the required data and biobanked serum samples.
Clinically Defined Samples
Demographic and clinical data at enrolment included patient age, sex, disease duration, race/ethnicity, lupus nephritis (LN) (defined as fulfilling the ACR criterion for renal disease or if a renal biopsy was performed prior to cohort entry), ACR Classification Criteria fulfilled (total and individual), Systemic Lupus Erythematosus Disease Activity Index – 2000 (SLEDAI-2K) (global score and organ system scores),22 SLEDAI-2K adjusted mean score (AMS, measurement of lupus disease activity over time or area under the curve of SLEDAI-2K over time by adding the area of each of the blocks of visit interval divided by the length of time for the entire period)23, SLICC/ACR Damage Index (SDI),24 medication use (current and ever use of glucocorticoids, antimalarials, and immunosuppressive agents including biologics), and survival. Longitudinal data on nephritis, SLEDAI-2K, SDI, and medication use at three, five, and ten years after enrolment were also obtained. These demographic and clinical variables are described in greater detail in Supplemental Table 1.
ANA and Autoantibody Testing
Aliquots of the 805 SLICC patient sera at 1) enrolment (sample #1); 2) two to four years after enrolment (sample #2); and 3) four to ten years after enrolment (sample #3) were stored at −80°C until required for immunoassays and analyzed at MitogenDx (Calgary, Canada). Hereafter, samples #1 – 3 are referred to as enrolment, year three, and year five, respectively. Indirect immunofluorescence assay (IFA) using HEp-2 substrate (NovaLite,Werfen, San Diego, USA), was performed on all samples. In accord with the manufacturers’ directions, a positive test was defined as a titer of ≥1:80. IFA results (titres and patterns) were initially read by an automated digital IFA microscope (NovaView, Werfen) and then visually validated by a technologist with >15 years of experience. For any inconsistency or questionable patterns, a second individual (MJF) with >40 years of experience reviewed and reached a consensus. ANA IFA patterns were classified according to the most recently updated International Consensus on ANA Patterns recommendations (http://www.anapatterns.org/index.php).25 A quality assurance step was performed by repeating all ANA that were within the normal range (titer <1:80) and a random selection of the ANA-positive samples. The lab also participates in ICAP and College of American Pathologists ANA survey for quality assurance.
Anti-dsDNA and titers were detected by chemiluminescence immunoassay test (CIA) (Werfen). A cut-off of ≥27 IU/mL was utilized, where 27–35 IU/mL was indeterminate (borderline), and >35 IU/mL was positive. All samples were also tested for autoantibodies by an ALBIA (FIDIS Connective13: TheraDiag, Paris, France) on a Luminex 200 flow luminometer (BioRad, Hercules, CA USA) focusing on SLE-related analytes that included ribosomal P, Ro52/Tripartite Motif Protein 21 (TRIM21), SSA/Ro60, SSB/La, Sm, U1-RNP, Jo-1, centromere B, PCNA, and histones. The manufacturer’s recommended cut-off of >40 median fluorescence units, which is >2 standard deviations above the mean of internal controls, was considered positive.
Anti-phospholipid antibodies (APLAs) IgG and IgM anti-cardiolipin and IgG and IgM anti–β2-glycoprotein-1 (β2GP1) were measured using ELISA (Werfen). Using the revised Sapporo antiphospholipid syndrome classification criteria,26 a cut-off of >40 units for IgG/IgM anti-cardiolipin was considered medium to high positive while a cut-off of ≥20 units (>99th percentile) was considered positive for IgG/IgM anti–β2GP1. Non-criteria APLAs IgG and IgM anti-PS/PT (phosphatidyl serine/prothrombin complex) and anti-β2GP1-Domain 1 were tested using ELISA (QUANTA Lite, Werfen) and CIA (QUANTA Flash, Werfen) respectively. The cut-offs used were as recommended by the manufacturer and sensitivity and specificity confirmed by internal quality assurance and external quality assurance (EQA). All autoantibodies were measured at MitogenDx except for lupus anticoagulant, which was measured at the Oklahoma Medical Foundation (Oklahoma City, OK), by previously reported methods.27
All samples were tested for the presence of anti-DFS70 (dense fine speckled 70/lens epithelium derived growth factor) antibodies by CIA (Werfen). The assay used purified full length human recombinant DFS70 coated onto paramagnetic beads. The established cut-off for anti-DFS70 antibodies was >20 chemiluminescent units (CU).
Statistical Analysis and Machine Learning
Principal component analysis (PCA) was applied to reduce the high dimensionality of longitudinal ANA and autoantibody profiles (results of 71 variables including positivity and titres of ANA and each autoantibody repeated over three visits: enrolment, year three, and year five). Cumulative variance explained was used to select the number of PCs, ensuring that the number of components chosen explains a significant proportion of the total variance, typically at least 70% to 80%, while avoiding overfitting. Then, K-means clustering algorithm on the PCA transformed ANA and autoantibody data was used. The optimal number of clusters was chosen using the elbow method.28 To evaluate cluster robustness, the PCA transformation and K-means clustering were repeated five times with different random seeds. We compared cluster demographic and clinical outcomes, including longitudinal disease activity (total SLEDAI-2K and AMS), SDI and organ-specific domains, and SLE therapies at ten-years post-enrolment, using one-way ANOVA test and a Benjamini-Hochberg correction with false discovery rate alpha = 0.05. Chi-square pairwise comparisons were performed to study differences in outcomes between pairs of clusters (e.g., frequency of LN between clusters and its association with anti-dsDNA positivity). Results were visualized using t-distributed stochastic neighbor embedding (t-SNE). Multivariable logistic regression, adjusted for age of disease onset, was used to determine if clusters were predictive with mortality at year ten. Survival curves were also constructed using Kaplan-Meier methods. Finally, a multivariable Cox proportional hazards model was fitted to estimate the adjusted hazards for survival, accounting for age of disease onset. For missing data (0.33% of the entire dataset), we used multiple imputation, where for each missing feature in the longitudinal data, the missing value was replaced by the mean of the other observed values for that time point. Python 3.7, scikit-learn, R 4.1.1 and STATA 15.1 software were used.
Results
Enrolment and Year Five Patient Clinical Characteristics
The 805 patients included in the study had a mean age at diagnosis of 35.2 years (SD 13.6), 88.7% (714/805) were female and 47.7% (384/805) were of race/ethnicity other than White (Supplemental Table 2). At enrolment, the disease duration was 0.58 years (SD 0.49), the frequency of nephritis was 28.9% and the mean total SLEDAI-2K score was 5.4 (SD 5.3). SLE medications at enrolment included 70.1% on antimalarials, 69.6% on glucocorticoids, and 41.0% on immunosuppressants. The changes in clinical characteristics of the patients from enrolment to year five have been described previously.16
Enrolment and Year Five Patient ANA and Autoantibody Profile
The most common autoantibodies at enrolment were anti-SSA/Ro60 (42.5%) followed by anti-Ro52/TRIM21 (37.5%), PS/PT IgG/IgM (36.3% either isotypes or 20.0% IgG, 26.6% IgM), anti-dsDNA (34.2%), anti-histones (31.3%), anti-U1RNP (28.2%), anti-Ribosomal P (24.3%) and anti-Sm (22.7%). The frequency of most SLE-related autoantibodies decreased at year five compared to enrolment (Table 1). The most common ANA patterns were AC-4 representing nuclear fine speckled (39.4%), AC-1 nuclear homogeneous (34.9%), AC-5 nuclear large speckled (34.4%), AC-19 cytoplasmic dense fine speckled (13.8%), AC-20 cytoplasmic fine speckled (12.4%), and AC-10 punctate nucleolar (7.2%). The frequency of ANA patterns at enrolment compared to year five did not change significantly for most patterns.
Table 1.
Patient ANA and autoantibody profile at enrolment and year five (n=805)
| Enrolment | Year 5 | Difference1 (95% CI) | |
|---|---|---|---|
| ANA ICAP Pattern | % | % | |
| AC-0 (No staining) | 1.7 | 1.1 | −0.6 (−1.9, 06) |
| AC-1 | 34.9 | 30.1 | −4.8 (−8.4, −1.3) |
| AC-2 | 1.1 | 1.2 | 0.1 (−0.8, 1.1) |
| AC-3 | 1.1 | 1.4 | 0.2 (−0.6, 1.1) |
| AC-4 | 39.4 | 47.5 | 8.1 (3.5, 12.6) |
| AC-5 (includes nuclear matrix pattern) | 34.4 | 36.1 | 1.7 (−2.2, 5.6) |
| AC-6 | 0.6 | 0.5 | −0.1 (−1.0, 0.7) |
| AC-7 | 4.0 | 5.7 | 1.7 (−0.2, 3.7) |
| AC-8 | 2.6 | 3.2 | 0.6 (−1.1, 2.3) |
| AC-9 | 0.5 | 1.2 | 0.7 (−0.2, 1.7) |
| AC-10 | 7.2 | 8.4 | 1.2 (−1.3, 3.8) |
| AC-11 | 0.1 | 0.4 | 0.2 (−0.3, 0.9) |
| AC-12 | 1.5 | 1.1 | −0.4 (−1.6, 0.9) |
| AC-13 | 0.2 | 0.2 | 0 (−0.6, 0.6) |
| AC-14 | 0 | 0 | 0 (−0.1, 0.1) |
| AC-15 | 0.2 | 0 | −0.2 (−0.7, 0.2) |
| AC-16 | 0.2 | 0.4 | 0.1 (−0.5, 0.7) |
| AC-17 | 0.2 | 0.2 | 0 (−0.5, 0.5) |
| AC-18 | 1.5 | 1.1 | −0.4 (−1.5, 0.7) |
| AC-19 | 13.8 | 17.5 | 3.7 (0.6, 6.9) |
| AC-20 | 12.4 | 12.2 | −0.2 (−3.5, 3.0) |
| AC-21 | 5.8 | 4.7 | −1.1 (−3.0, 0.8) |
| AC-22 | 0.2 | 0.1 | −0.1 (−0.7, 0.4) |
| AC-23 | 0.1 | 0.2 | 0.1 (−0.4, 0.7) |
| AC-24 | 2.0 | 3.6 | 1.6 (0.1, 3.1) |
| AC-25 | 0.1 | 0.1 | 0 (−0.1, 0.1) |
| AC-26 | 0.4 | 0.6 | 0.2 (−0.6, 1.1) |
| AC-27 | 1.5 | 0.9 | −0.6 (−1.7, 0.4) |
| AC-28 | 0.2 | 0.2 | 0 (−0.6, 0.6) |
| AC-29 | 0.1 | 0 | −0.1 (−0.5, 0.2) |
| Autoantibodies, % | |||
| dsDNA2 | 34.2 | 29.1 | −5.1 (−8.7, −1.6) |
| Ribosomal P | 24.3 | 20 | −4.3 (−7.8, −0.9) |
| Ro52/TRIM21 | 37.5 | 37.4 | −0.1 (−3.4, 3.2) |
| SSA/Ro60 | 42.5 | 42.0 | −0.5 (−3.7, 2.7) |
| SSB/La | 20.7 | 16.3 | −4.5 (−7.5, −1.5) |
| Sm | 22.7 | 14.7 | −8.1 (−11.1, −5.0) |
| U1RNP | 28.2 | 23.0 | −5.2 (−8.5, −2.0) |
| Histones | 31.3 | 22.7 | −8.6 (−12.1, −5.0) |
| Jo-1 | 1.5 | 3.7 | 2.2 (0.7, 3.7) |
| Centromere B | 2.7 | 5.5 | 2.7 (0.9, 4.5) |
| PCNA | 15.8 | 18.4 | 2.6 (−0.9, 6.1) |
| DFS70 | 6.1 | 6.0 | −0.1 (−1.3, 1.1) |
| Cardiolipin IgG/IgM3 | 20.5 | 16.4 | −4.0 (−7.5, −0.4) |
| β2GP1 IgG/IgM3 | 19.9 | 12.9 | −7.0(−9.9, −4.1) |
| Lupus anticoagulant4 | 19.5 | 14.2 | −5.3 (−8.3, −2.2) |
| PS/PT IgG/IgM | 36.3 | 26.0 | −10.3 (−13.9, −6.7) |
| β2GP1-Domain 1 | 10.3 | 7.8 | −2.5 (−4.8, −0.2) |
Abbreviations: AC, anti-cellular pattern according to ICAP nomenclature; ACR, American College of Rheumatology; ANA, anti-nuclear antibodies; β2GP1, β2-glycoprotein-1; CI, confidence interval; DFS, dense fine speckled; dx, diagnosis; dsDNA, double-stranded DNA; ICAP, International Consensus on ANA Patterns; IgG/M, immunoglobulin G/immunoglobulin M; Jo-1, histidyl tRNA synthetase; PCNA, proliferating cell nuclear antigen; PS/PT, phosphatidyl serine-prothrombin complex; RNP, ribonucleoprotein; SD, standard deviation; SLEDAI-2K, systemic lupus erythematosus disease activity index-2000; SDI, SLICC Damage index; Sm, Smith antigen (U2-U6 RNP); SSA, Sjögren syndrome antigen A or Ro60: SSB, Sjögren syndrome antigen B or La: TRIM21, Tripartite Motif Protein (TRIM) 21; yrs, years.
Difference between enrolment and year 5 visit
Complete data available for n=798 patients
Complete data available for n= 800
Complete data available for n=282
ANA and Autoantibody Clusters
Four unique patient clusters (Figure 1) were identified using longitudinal trajectories of each autoantibody (Figure 2 and 95% confidence intervals shown in Supplementary Figure 1) and ANA pattern (Figure 3). These clusters were associated with clinical factors such as age of onset, race/ethnicity, BMI, and predicted disease activity, organ involvement, and treatment course at ten years of follow-up, and mortality (Table 3 and Supplemental Table 3). Of the 805 patients, date of death or follow-up data up to ten-years were available for 581 patients, of whom 71 died (12.2%). There were no significant differences in baseline demographic or clinical characteristics between the subset of 581 patients used to examine ten-year clinical outcomes and the 224 patients who did not provide ten-year clinical data (Supplemental Table 4).
Figure 1. Four autoantibody cluster groups identified among 805 SLE patients followed from enrolment through years 3 and 5.
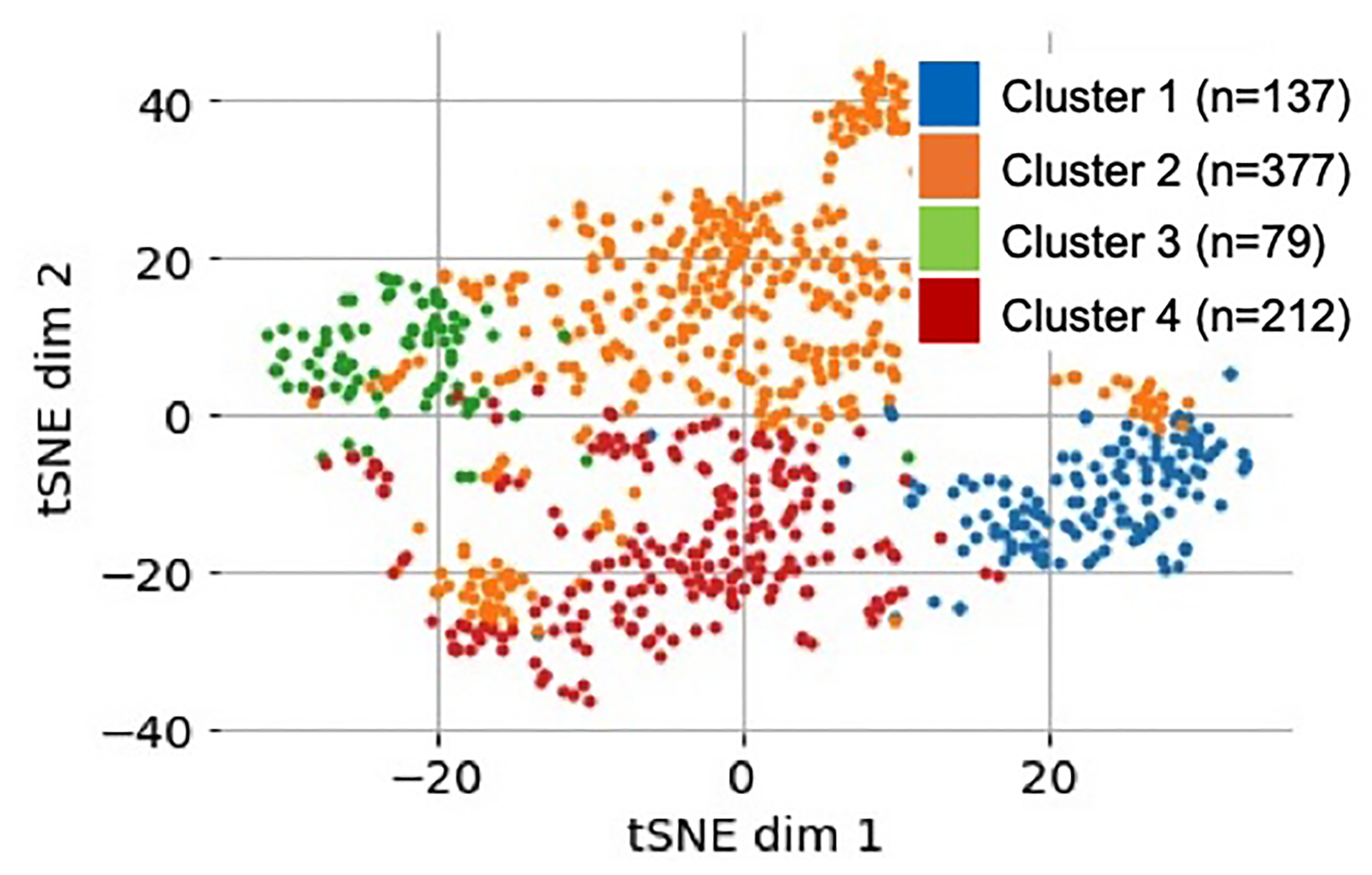
Latent space visualized using a t-distributed stochastic neighbor embedding (t-SNE) with colors based on cluster labels.
Figure 2. Autoantibody profile of 805 SLE patients in order of most prevalent autoantibodies in A) Cluster 1, B) Cluster 2, C) Cluster 3, D) Cluster 4.
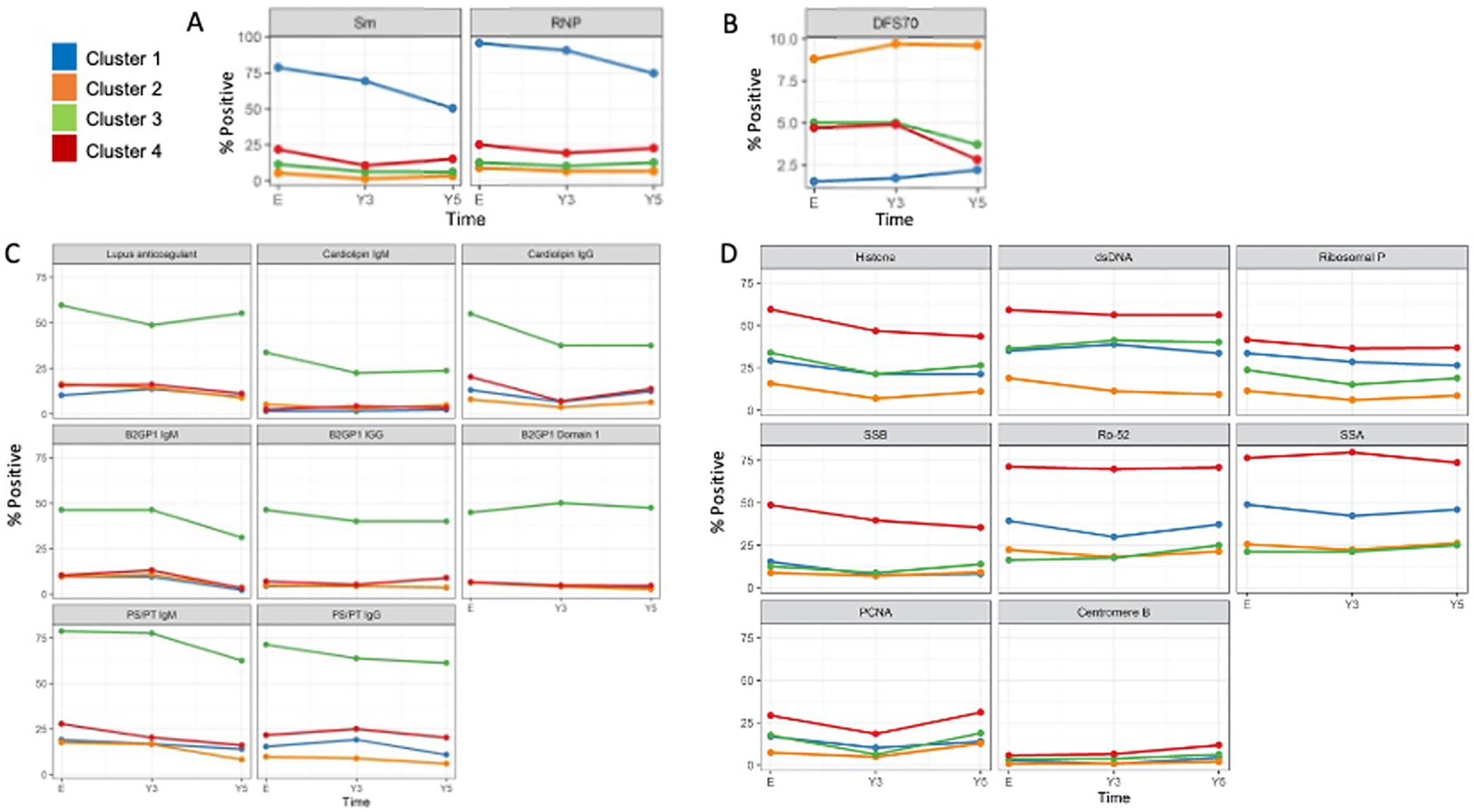
Standard deviation bars have been removed to make graphs easier to visualize.
Figure 3. ANA titres and patterns for each cluster.
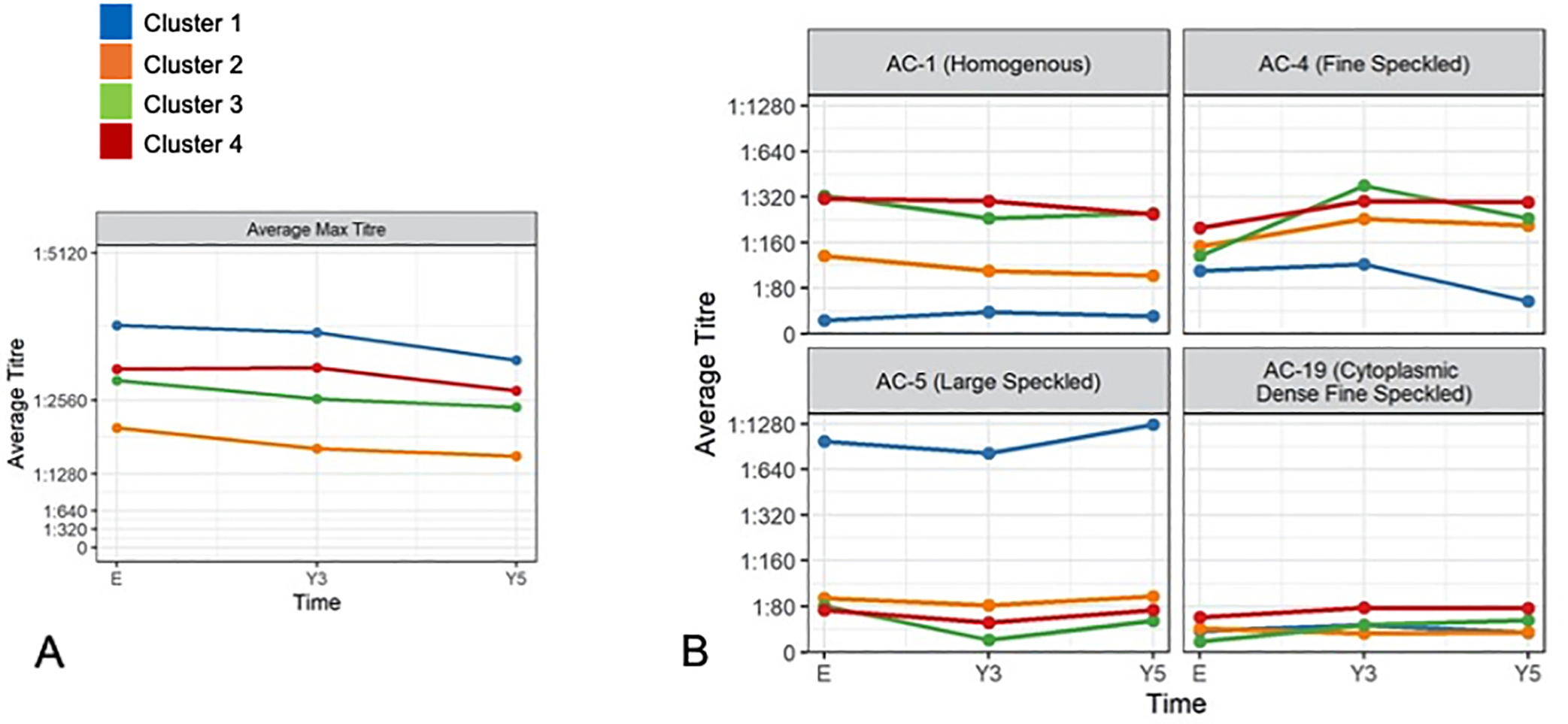
A) Mean maximum ANA titers slightly decreased over time. Cluster 1 (anti-Sm/RNP) highest mean maximum ANA titer. Cluster 2 (low anti-dsDNA) lowest mean maximum ANA titer. B) AC-1 (homogeneous pattern associated with anti-dsDNA, histones), AC-4 (fine speckled associated with anti-SSA/Ro60, anti-SSB/La), and AC- 19 (cytoplasmic dense fine speckled associated with anti-ribosomal P) correspond to the autoantibody profile observed in cluster 4. High titres of AC-5 (large specked associated with anti-Sm and anti-U1RNP antibodies) correspond to cluster 1 antibody profile. Remaining AC patterns had mean titers <1:80 at all three visits for all cluster groups.
Table 3.
Demographic and clinical characteristics that were statistically significant1 at enrolment and ten-year follow-up between the four SLE longitudinal autoantibody clusters
| Group 1 (n=137) | Group 2 (n=376) | Group 3 (n=80) | Group 4 (n=212) | p-value | FDR | |
|---|---|---|---|---|---|---|
| Enrolment Demographics | ||||||
| Mean Age of Diagnosis (SD), yrs | 31.5 (10.8) | 36.5 (13.9) | 32.5 (13.9) | 34.4 (14.1) | <0.001 | 0.014 |
| % Ethnicity | ||||||
| White | 32.1 | 61.7 | 68.8 | 42.5 | <0.001 | <0.001 |
| Asian | 30.7 | 20.5 | 13.8 | 31.1 | <0.001 | 0.014 |
| African | 27.0 | 9.6 | 6.2 | 14.6 | <0.001 | <0.001 |
| Mean BMI (SD), kg/m 2 | 24.3 (4.9) | 25.6 (6.1) | 26.1 (5.8) | 24.1 (4.8) | 0.003 | 0.029 |
| Clinical Characteristics at Year 10 Follow-Up | ||||||
| % Nephritis 2 | 56.2 | 32.1 | 50.9 | 46.9 | <0.001 | 0.001 |
| Mean SLEDAI-2K Score (SD) | ||||||
| Total Score3 | 3.2 (3.2) | 2.3 (2.9) | 3.0 (2.1) | 3.5 (3.4) | 0.002 | 0.020 |
| Adjusted Mean Score4 | 4.2 (2.7) | 2.6 (2.2) | 3.5 (1.7) | 3.9 (2.2) | <0.001 | <0.001 |
| Immunological Subscale | 1.64 (1.60) | 0.90 (1.31) | 1.96 (1.33) | 2.01 (1.61) | <0.001 | <0.001 |
| Low Complement | 0.85 (0.99) | 0.50 (0.87) | 1.19 (0.99) | 0.97 (1.00) | <0.001 | <0.001 |
| Mean SLICC Damage Index (SD) | ||||||
| Seizures | 0.01 (0.10) | 0.02 (0.16) | 0.11 (0.31) | 0.01 (0.8) | <0.001 | 0.004 |
| Skin Domain | 0.25 (0.53) | 0.08 (0.29) | 0.07 (0.32) | 0.07 (0.28) | <0.001 | 0.002 |
| Alopecia | 0.14 (0.35) | 0.04 (0.19) | 0.04 (0.19) | 0.04 (0.19) | <0.001 | 0.004 |
| Medications Ever | ||||||
| % Immunosuppressives/Biologics | 83.8 | 63.8 | 68.4 | 73.8 | 0.002 | 0.014 |
| % Azathioprine (Imuran) | 55.2 | 34.6 | 45.6 | 43.1 | 0.003 | 0.025 |
| % Mycophenolic Acid | 46.7 | 27.6 | 28.1 | 33.8 | 0.005 | 0.031 |
| % Belimumab | 14.3 | 4.1 | 5.3 | 6.3 | 0.005 | 0.031 |
| Medication Current | ||||||
| % Immunosuppressives/Biologics | 72.3 | 45.5 | 47.4 | 53.8 | <0.001 | 0.001 |
| % Azathioprine (Imuran) | 33.3 | 15.0 | 15.8 | 13.1 | <0.001 | 0.001 |
| % Mycophenolic Acid | 30.5 | 14.2 | 19.3 | 22.5 | 0.005 | 0.031 |
Comparison between cluster groups using one-way ANOVA test (null hypothesis that there is no difference between the means of the groups) and a Benjamini-Hochberg correction with false discovery rate (FDR) alpha = 0.05
LN was diagnosed by renal biopsy or fulfillment of the renal item on the ACR classification criteria.
The total score of SLEDAI-2K is the sum of all 24 descriptor scores. The total SLEDAI-2K score falls between 0 and 105, with higher scores representing higher disease activity.
A measurement of lupus disease activity over time determined by the calculation of the area under the curve of SLEDAI-2K over time by adding the area of each of the blocks of visit interval and then dividing by the length of time for the whole period.
Abbreviations: BMI, body mass index; SD, standard deviation; SLEDAI-2K, Systemic Lupus Erythematosus Disease Activity Index 2000; SLICC, Systemic Lupus International Collaborating Clinics; yrs, years.
Cluster 1 (n=137, 17.0%):
These patients were characterized by a high frequency of anti-Sm and anti-U1RNP antibodies (Table 2 and Supplemental Table 5). This group was the youngest at disease onset (31.5 years [SD 10.8]), had the highest proportion of African (27.0%) ancestry and lowest proportion of European ancestry (32.1%). At year ten, this cluster the highest cumulative disease activity (AMS 4.2 [SD 2.7]), mean SDI score for the skin domain (0.25 [SD 0.53]) and alopecia (0.14 [SD 0.35]), and frequency of immunosuppressant/biologic use (83.8% ever, 72.3% currently), particularly azathioprine (55.2% ever, 33.3% current), mycophenolic acid (46.7% ever, 30.5% current), and belimumab (14.3% ever, 6.7% current). Patients in this cluster also had the highest frequency of rituximab use (8.6% ever, 1.9% current), but this was not statistically significant. A complete list of immunosuppressant/biologics is available in Supplemental Table 3.
Table 2.
Frequency of Autoantibodies in Each Cluster Over Time
| Cluster 1 (n=137) | Cluster 2 (n=376) | Cluster 3 (n=80) | Cluster 4 (n=212) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antibody | Enrolment | Y3 | Y5 | Enrolment | Y3 | Y5 | Enrolment | Y3 | Y5 | Enrolment | Y3 | Y5 |
| Sm | 78.8 | 69.3 | 50.4 | 5.3 | 1.3 | 3.2 | 11.3 | 6.3 | 6.3 | 21.7 | 10.4 | 15.1 |
| U1RNP | 95.6 | 90.5 | 74.5 | 8.8 | 6.6 | 6.6 | 12.5 | 10.0 | 12.5 | 25.0 | 19.3 | 22.6 |
| DFS70 | 1.5 | 1.5 | 2.2 | 8.8 | 9.8 | 9.6 | 5.0 | 5.0 | 3.8 | 4.7 | 4.8 | 2.8 |
| β2GP1 IgG | 4.4 | 4.4 | 3.7 | 4.8 | 4.3 | 3.7 | 46.3 | 40.0 | 40.0 | 7.1 | 5.2 | 9.0 |
| β2GP1 IgM | 9.5 | 9.5 | 2.2 | 9.6 | 10.9 | 3.5 | 46.3 | 46.3 | 31.3 | 10.4 | 13.2 | 3.3 |
| Cardiolipin IgG | 13.1 | 6.6 | 12.4 | 8.0 | 3.7 | 6.4 | 55.0 | 37.5 | 37.5 | 20.3 | 7.1 | 13.7 |
| Cardiolipin IgM | 1.5 | 1.5 | 2.2 | 5.1 | 2.9 | 4.8 | 33.8 | 22.5 | 23.8 | 2.4 | 4.2 | 3.3 |
| β2GP1-Domain 1 | 6.6 | 4.4 | 3.7 | 6.4 | 4.0 | 2.7 | 45.0 | 50.1 | 47.5 | 6.6 | 4.7 | 4.7 |
| Lupus anticoagulant | 8.61 | 10.72 | 8.61 | 16.03 | 12.44 | 8.33 | 64.85 | 70.26 | 60.65 | 15.07 | 14.58 | 10.67 |
| PS/PT IgG | 15.3 | 19.0 | 10.9 | 9.8 | 8.8 | 5.9 | 71.3 | 63.8 | 61.3 | 21.7 | 25.0 | 20.3 |
| PS/PT IgM | 19.0 | 16.8 | 13.9 | 17.6 | 16.5 | 8.2 | 78.8 | 77.5 | 62.5 | 27.8 | 20.3 | 16.0 |
| dsDNA | 35.0 | 38.7 | 33.6 | 18.9 | 11.2 | 9.3 | 36.3 | 41.3 | 40.0 | 59.0 | 56.1 | 56.1 |
| Histone | 29.2 | 21.2 | 21.2 | 15.7 | 6.9 | 10.9 | 33.8 | 21.3 | 26.3 | 59.4 | 46.7 | 43.4 |
| PCNA | 16.8 | 10.2 | 13.9 | 7.4 | 4.8 | 12.8 | 17.5 | 6.3 | 18.8 | 29.2 | 18.4 | 31.1 |
| Ribosomal P | 33.6 | 28.5 | 26.3 | 11.4 | 6.1 | 8.5 | 23.8 | 15.0 | 18.8 | 41.5 | 36.3 | 36.8 |
| Ro52/TRIM21 | 39.4 | 29.9 | 37.2 | 22.3 | 18.1 | 21.3 | 16.3 | 17.5 | 25.0 | 71.2 | 69.8 | 70.8 |
| SSA/Ro60 | 48.9 | 42.3 | 46.0 | 25.5 | 22.3 | 26.3 | 21.3 | 21.3 | 25.0 | 76.4 | 79.7 | 73.6 |
| SSB/La | 15.3 | 7.3 | 8.0 | 8.8 | 6.9 | 9.0 | 12.5 | 8.8 | 13.8 | 48.6 | 39.6 | 35.4 |
| Centromere B | 2.9 | 0.7 | 4.4 | 0.8 | 1.1 | 2.1 | 3.8 | 3.8 | 6.3 | 5.7 | 6.6 | 11.8 |
| Jo-1 | 0.0 | 0.7 | 0.7 | 1.1 | 1.3 | 2.4 | 1.3 | 0.0 | 7.5 | 3.3 | 0.9 | 6.6 |
Darker red shading indicates higher frequency, lighter shading indicates lower frequency of autoantibody
n=116 complete data,
n=84,
n=337,
n=237,
n=71,
n=47,
n=180,
n=117
Cluster 2 (n=376, 46.7%):
This was the largest cluster and was characterized by low frequency of anti-dsDNA and relatively high frequency of anti-DFS70. Patients in this cluster were oldest at disease onset (36.9 years [SD 13.9]) and predominantly of European ancestry (61.7%). At year ten, this cluster had the lowest proportion of patients with nephritis (32.1%), the lowest disease activity (total SLEDAI-2K 2.3 [SD 2.9] and AMS 2.6 [SD 2.2]), lowest SLEDAI-2K score for immunological subscale (0.90 [SD 1.31]) including low complement levels (0.50 [0.87]), and lowest frequency of immunosuppressant/biologic use (63.8% ever, 45.5% currently), including azathioprine (34.6% ever, 15.0% current), mycophenolic acid (27.6% ever, 14.2% current), and belimumab (4.1% ever, 3.3% current).
Cluster 3 (n=80, 9.9%):
This was the smallest cluster and had the highest frequency of both criteria (anti-cardiolipin IgG/IgM, anti-β2GP1 IgG/IgM, lupus anticoagulant) and non-criteria APLAs (PS/PT IgG/IgM and anti-β2GP1-Domain 1 IgG/IgM) over time. For most APLAs, titres were highest at enrolment and then decreased over time (Supplemental Table 6). They had the highest proportion of European ancestry (68.8%), lowest proportion of Asian (13.8%) and African (6.2%) ancestry, and highest mean body mass index (26.1 kg/m2 [SD 5.8]) at enrolment. At year ten, this cluster had the highest SLEDAI-2K subscale scores for low complement levels (1.19 [SD 0.99]) and the highest mean SDI scores for neuropsychiatric involvement (0.37 [SD 0.86]) including strokes (0.14 [SD 0.44]) and seizures (0.11 [SD 0.31]), however, only seizures were significantly different between clusters after correcting for multiple comparisons. Of note, the association between this cluster with neuropsychiatric involvement and strokes were statistically significant at year five (data not shown).
Cluster 4 (n=212, 26.3%):
This cluster was characterized by positivity to many autoantibodies including histone, dsDNA, ribosomal P, SSB/La, Ro52/TRIM21, anti-SSA/Ro60, PCNA, and centromere B. They had the highest proportion of patients of Asian ancestry (31.1%) and lowest mean body mass index (24.1 kg/m2 [SD 4.8]) at enrolment. At year ten, this cluster had the highest total SLEDAI-2K score (3.5 [SD 3.4]), particularly for the immunological subscale (2.01 [SD 1.61]).
The ANA patterns corresponded to the autoantibody profile of each cluster (Figure 3). Cluster 1 had the highest ANA titres over time and while Cluster 2 had the lowest. Cluster 1 had the highest mean maximum ANA titres for AC-5 (large speckled pattern, which is associated with anti-Sm and anti-RNP). Cluster 4 had higher mean maximum ANA titres for AC-1 (homogeneous pattern associated with anti-dsDNA, histones), AC-4 (fine speckled associated with anti-SSA/Ro60, anti-SSB/La), and AC-19 (cytoplasmic dense fine speckled associated with anti-ribosomal P), corresponding as well to its autoantibody profile. Remaining ANA patterns (AC-2, 3, 6–18, 20–29) had mean titers <1:80 at all three visits for all cluster groups.
Mortality
Cluster 3 had the highest proportion of patients who died (7.9%) at year ten, followed by Cluster 1 (4.7%), 4 (3.7%), and 2 (3.2%). The odds of survival at 10 years were significantly lower in patients in Cluster 3 compared to patients in Cluster 2 (adjusted odds ratio (OR) 0.28, 95%CI: 0.08–0.94). There were no statistical differences in odds of survival between the other clusters. The Kaplan-Meier survival curves are shown in Supplemental Figure 2. Hazards of survival, adjusted for age at disease onset in a multivariable Cox regression demonstrates that patients in Clusters 1 (adjusted hazards ratio (HR) 2.60 [95% CI: 1.12–6.05], p=0.03) and 3 (adjusted HR 2.87 [95% CI: 1.22–6.74], p=0.02) had lower survival compared to patients in Cluster 2.
Ten principal components were chosen to capture 75% of the cumulative explained variance of the dataset (Supplemental Figure 3) prior to k-mean clustering. Cluster robustness was high as the original four clusters presented above and the new clusters generated in the robustness evaluation agreed, as indicated by a high average adjusted Rand index (ARI) (ARI 0.971, where 1.0 represents identical clustering and 0 represents exact opposite).
Anti-dsDNA and LN
A comparison of LN between pairs of clusters demonstrated that only cluster 2 (lowest frequency of anti-dsDNA positivity) had significantly lower frequency of LN compared to cluster 1, 3, and 4 at year five (p =0.01, p=0.01, p=0.007, respectively) and year ten (p<0.001, p=0.01, p=0.004, respectively). There was no difference in LN frequency when clusters 1, 3 and 4 were compared to each other (data not shown). LN and anti-dsDNA frequency were strongly associated with each other by current anti-dsDNA positivity (p<0.0001), mean titre (p=0.0002), and ever positive (p<0.0001).
Discussion
SLE is a heterogeneous disease with respect to manifestations, progression, and treatment responses, but the presence of circulating autoantibodies points to a fundamental underlying mechanism of immune dysregulation and disease pathogenesis. Therefore, grouping SLE patients into autoantibody subsets to reconcile disease heterogeneity may elucidate this complex disease and identify more personalized monitoring and treatment plans, as well as distinguish those patients at higher risk for disease progression and organ damage. This is the first study to identify endotypes of SLE patients based on ML analysis using longitudinal autoantibody profiles (20 autoantibodies and 29 ANA pattern interpretations) over the first five years of disease from a large international, multicenter inception cohort. Four distinct serologic clusters were associated with clinical features such as age of onset, race/ethnicity, BMI, and predictive of long-term disease activity, organ involvement, treatment course, and mortality.
While similar clusters have been described, prior studies were based on smaller cohorts, single-centres, and/or cross-sectional analysis of only a limited set of autoantibodies, thereby limiting the generalizability of the results.1–11 This current study fulfilled a need for a in-depth analysis of more diverse SLE patients who were well characterized both at inception and in long-term follow-up, providing a comprehensive analysis of autoantibodies and ANA patterns, especially when over 200 SLE-related autoantibodies have been described.12 This study also analyzed several novel autoantibodies that have important clinical implications, including anti-DFS70,29 anti-PS/PT IgG/IgM,30 and anti-β2GP1-Domain1 IgG/IgM31. Unlike prior cross-sectional studies that examined associations, we used prospectively collected data in a protocolized fashion that demonstrated that clustering based on biomarker data within the first five years can add predictive value for clinically relevant outcomes at year ten and beyond, including the risk of mortality. All ANAs and other autoantibodies were tested in one accredited, central laboratory, thereby avoiding interlaboratory variation as a factor in ANA and autoantibody fluctuations over time. We also used assays that were CE marked, Health Canada and/or U.S. Food and Drug Administration approved, setting us apart from some studies that have used research use only or laboratory developed tests. Using this robust approach, we demonstrated that ANA titres and most autoantibodies decreased in frequency over the first five years of follow-up. To examine multiple autoantibody profiles and their potential evolution over time, considering their linkages and interactions, we leveraged ML to identify meaningful patterns and relationships with disease outcomes. We believe these are crucial strengths of this study that adds novel information to the current understanding of SLE heterogeneity.
While there is promise in incorporating these clusters into future personalized models of health care for patients, this study purposefully conducted extended longitudinal autoantibody profiling that may not be available at all centres. However, this detailed autoantibody testing approach allowed us to achieve a better understanding of SLE heterogeneity and disease pathogenesis. Two high risk clusters (1 and 4) characterized by multiple autoantibody reactivities, high disease activity and immunosuppressant/biologic use, were found more commonly among non-White races/ethnicities known to have more severe SLE.32 This is further evidence that genetic factors may be underpinning differences in immune dysregulation susceptibility that lead to increased autoantibody production, immune complex formation, inflammation, and eventual organ damage.
Epitope spreading in genetically susceptible individuals may also explain why these two high risk clusters (1 and 4) had distinct patterns of autoantibody reactivities.33, 34 For instance, autoantibodies to Sm and U1RNP, which were frequently observed in Cluster 1, are directed against distinct components of related macromolecular complexes. For example, U1RNP is one of several small nuclear ribonucleoprotein particles (snRNP), each consisting of a unique small nuclear RNA (U1-U6 RNAs), specific associated proteins, and common core Smith (Sm) proteins. An antibody response beginning with one particular epitope can then be followed by a spread of the immune response to other epitopes in the same polypeptide (intramolecular) and/or other distinct but structurally similar molecules (intermolecular). 35 Therefore, autoantibodies can exist in “linked” sets, a well described phenomena that helps explain co-prevalence of many autoantibodies.33, 34
Another high-risk profile cluster was cluster 3, which had multiple elevated APLAs and severe disease outcomes including seizures and mortality. In a prior SLICC study of lupus anticoagulant, anti-cardiolipin, and anti-ß2GP1 tested at baseline, only an association between lupus anticoagulant and increased risk of cerebrovascular disease (p= 0.04) could be detected.29 As APLAs are known to fluctuate over the disease course,30 serial measurements on all five APLAs were analyzed in this current study. We showed that when all five APLAs were persistently positive, this was predictive of the future occurrence of several severe SLE-related outcomes such as seizures and mortality. There was also an association between APLAs with strokes at year five and year ten, although it was not significant at year ten after adjustment for multiple comparisons. This may be related to APLAs titres declining over time and a higher frequency of strokes in other clusters, but due to non-SLE related atherosclerosis. Our study is also the first to examine longitudinal profiles of less commonly reported non-criteria APLAs. Both anti-PS/PT IgG/IgM and anti-β2GP1-Domain1 were identified in cluster 3. Recent studies have shown that aPS/PT antibodies are predictive of cardiovascular disease events in SLE including strokes, irrespective of a history of antiphospholipid syndrome.31 aPS/PT antibodies can additionally identify patients that are negative for the criteria aPLAs, thereby closing the seronegative gap, and are associated with increased risk of thrombosis that is additive to other criteria aPLAs.32 In our study, the frequency of aPS/PT antibodies IgG/IgM over the five years (36.3–26.0% for the presence of either isotype, 16.0–20.2% IgG only, 16.6–26.6% IgM only) was consistently higher than the frequency of the other APLAs, which is in keeping with other studies and suggesting they may be important biomarkers for SLE patients.33
The absence of specific autoantibodies in SLE or the presence of others may represent patients who are at lower risk of severe SLE. We demonstrated that SLE patients belonging to cluster 2 had a milder disease course characterized by low titre ANAs and lack of autoantibody reactivity, including anti-dsDNA. Accordingly, Cluster 2 also had the lowest frequency of LN at years 5 and 10, which is not surprising as we also demonstrated that the frequency of anti-dsDNA was strongly associated with LN in a univariate analysis. This group also had a relatively higher frequency of anti-DFS70 antibodies over time. In an earlier SLICC study, monospecific anti-DFS70 (no other detectable autoantibodies) at disease inception has been shown to be uncommon in SLE (1.1%).34 In this longitudinal study, the results of anti-DFS70 in cluster 2 suggest that it may be a good prognostic biomarker among those with established disease.
We acknowledge some important limitations of this study. First, although this is the first to report longitudinal clusters, the duration of follow-up is rather short, which may explain why there were no differences observed for many comparisons. One previous single centre study showed reduced survival among SLE patients with APLAs after 20 years of follow-up.6 Future studies with longer follow-up data are underway which will allow examination of disease damage and survival. Second, as the enrolment visit could occur up to 15 months after diagnosis (although mean disease duration at enrolment was 0.58 years), most patients (>96%) had already been exposed to at least one immunomodulatory medication by enrolment, potentially influencing ANA and autoantibody results. We showed that although the frequency of most autoantibodies fluctuated over time, the autoantibody profiles of the clusters themselves remained stable. The clinical applicability of the results (i.e., value of monitoring an extended autoantibody profile over time) is also limited as most centres will not be able to perform serial measurements of all 20 autoantibodies included in the cluster analysis. Future studies to build and validate a panel of the key autoantibodies that can stratify patients into these clusters are needed, taking into consideration test availability and costs.
In summary, our ML analysis of comprehensive and longitudinal ANA and autoantibody signatures has identified four unique endotypes of SLE patients associated with important SLE outcomes. This suggests that early characterization of autoantibody profiles may be helpful in reconciling disease heterogeneity and understanding disease pathogenesis, which may guide clinical prognostication to identify those with more aggressive disease phenotypes and inform design of personalized diagnostic and treatment strategies. Future studies are required to determine whether other ‘omics’ biomarkers (exposome, epigenome, genome, transcriptome, microbiome, metabolome, proteome) with the aid of ML approaches can add value to the predictive power of autoantibodies demonstrated in this study. We anticipate that these clusters will become a benchmark to study other SLE-related outcomes, including potential use as a stratification factor for heterogeneous patient populations in clinical trials and for evaluating differential burden of health care resource utilization. Further validation studies may also inform clinical follow-up and therapeutic approaches after diagnosis.
Supplementary Material
KEY MESSAGES.
What is already known on this topic
To better understand systemic lupus erythematosus’ (SLE) underlying biology and disease heterogeneity, attempts have been made to stratify patients into endotypes based on common autoantibody profiles and using machine learning (ML).
However, past studies were cross-sectional, studied patients seen at single centres and assessed relatively few SLE-related autoantibodies.
What this study adds
A comprehensive panel of autoantibodies (20 autoantibodies and 29 antinuclear pattern interpretations) was evaluated in a large, well-characterized cohort of SLE patients using a longitudinal and machine learning approach.
We demonstrated that there were four distinct serologic clusters in the first five years of disease associated with clinical features such as age of onset, race/ethnicity, BMI, and predictive of disease activity, organ involvement, and treatment course at ten years of follow up, and mortality.
How this study might affect research, practice or policy
Early characterization of autoantibody profiles may be helpful in reconciling disease heterogeneity and understanding disease pathogenesis.
This may guide clinical prognostication and inform design of personalized diagnostic and treatment strategies.
Acknowledgements:
The authors are grateful for the technical assistance of Ms. Haiyan Hou, Meifeng Zhang (MitogenDx, University of Calgary), and Chynace Lambalgen.
Disclosure:
Dr. Choi has received consulting fees from Janssen, AstraZeneca, Mallinckrodt Pharmaceuticals, and MitogenDx (less than $10,000).
Dr. Clarke has received consulting fees, speaking fees, and/or honoraria from AstraZeneca, BristolMyersSquibb, and Glaxo Smith Kline (less than $10,000 each) and research support from Glaxo Smith Kline.
Dr. Fritzler is Director of Mitogen Diagnostics Corporation (Calgary, AB Canada) and a consultant to Werfen International (San Diego, CA, USA; Barcelona, Spain), Aesku Group (Wendelsheim, Germany) and Alexion Canada (less than $10,000).
Dr. Gordon has received consulting fees, speaking fees, and/or honoraria from Eli Lilly, UCB, GlaxoSmithKline, Merck Serono and BMS (less than $10,000 each) and grants from UCB. Grants from UCB were not to Dr. Gordon but to Sandwell and West Birmingham Hospitals NHS Trust.
Dr. Gladman received consulting fees, speaking fees, and/or honoraria from GlaxoSmithKline (less than $10,000).
Dr. Bruce has received consulting fees, speaking fees, and/or honoraria from Eli Lilly, UCB, Roche, Merck Serono, MedImmune (less than $10,000 each) and grants from UCB, Genzyme Sanofi, and GlaxoSmithKline.
Dr. Ginzler has paid consultation with investment analysts Guidepoint Global Gerson Lerman Group.
Dr. Kalunian has received grants from UCB, Human Genome Sciences/GlaxoSmithKline, Takeda, Ablynx, Bristol-Myers Squibb, Pfizer, and Kyowa Hakko Kirin, and has received consulting fees from Exagen Diagnostics, Genentech, Eli Lilly, Bristol-Myers Squibb, and Anthera (less than $10,000 each).
Dr. Costenbader has consulted for or collaborated on research projects with Janssen, Glaxo Smith Kline, Gilead, Exagen Diagnostics, Lilly, Merck, Astra Zeneca, Amgenand Neutrolis (less than $10,000 each).
The remainder of the authors have no disclosures.
Grant Support:
Dr. Choi is supported by the Lupus Foundation of America Gary S. Gilkeson Career Development Award and research gifts in kind from MitogenDx (Calgary, Canada).
Dr. Clarke holds The Arthritis Society Research Chair in Rheumatic Diseases at the University of Calgary.
Dr. Hanly’s work was supported by the Canadian Institutes of Health Research (research grant MOP-88526).
Dr. Gordon’s work was supported by Lupus UK, Sandwell and West Birmingham Hospitals NHS Trust and the NIHR/Wellcome Trust Clinical Research Facility in Birmingham.
Dr. Bae’s work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1A6A1A03038899).
The Montreal General Hospital Lupus Clinic is partially supported by the Singer Family Fund for Lupus Research.
Dr. Rahman and Dr. Isenberg are supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
The Hopkins Lupus Cohort is supported by NIH Grants AR043727 and AR069572
Dr. Fortin presently holds a tier 1 Canada Research Chair on Systemic Autoimmune Rheumatic Diseases at Université Laval, and part of this work was done while he was still holding a Distinguished Senior Investigator of The Arthritis Society.
Dr. Bruce is an NIHR Senior Investigator and is funded by Arthritis Research UK, the National Institute for Health Research Manchester Biomedical Research Centre and the NIHR/Wellcome Trust Manchester Clinical Research Facility. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Dr. Dooley’s work was supported by the NIH grant RR00046.
Dr. Ramsey-Goldman’s work was supported by the NIH (grants 1U54TR001353 formerly 8UL1TR000150 and UL-1RR-025741, K24-AR-02318, and P60AR064464 formerly P60-AR-48098).
Dr. Manzi is supported by grants R01 AR046588 and K24 AR002213
Dr. Ruiz-Irastorza is supported by the Department of Education, Universities and Research of the Basque Government.
Dr. Jacobsen is supported by the Danish Rheumatism Association (A1028) and the Novo Nordisk Foundation (A05990).
Dr. Costenbader is supported by NIH K24 AR066109.
Footnotes
Patient and Public Involvement:
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
Data Sharing Statement:
All data relevant to the study are included in the article or uploaded as supplementary information.
References
- 1.Budde P, Zucht HD, Vordenbäumen S, Goehler H, Fischer-Betz R, Gamer M, et al. Multiparametric detection of autoantibodies in systemic lupus erythematosus. Lupus. 2016;25(8):812–22. [DOI] [PubMed] [Google Scholar]
- 2.Mizus M, Li J, Goldman D, Petri MA. Autoantibody clustering of lupus-associated pulmonary hypertension. Lupus Sci Med. 2019;6(1):e000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.To CH, Petri M. Is antibody clustering predictive of clinical subsets and damage in systemic lupus erythematosus? Arthritis Rheum. 2005;52(12):4003–10. [DOI] [PubMed] [Google Scholar]
- 4.Tápanes FJ, Vásquez M, Ramírez R, Matheus C, Rodríguez MA, Bianco N. Cluster analysis of antinuclear autoantibodies in the prognosis of SLE nephropathy: are anti-extractable nuclear antibodies protective? Lupus. 2000;9(6):437–44. [DOI] [PubMed] [Google Scholar]
- 5.Pacheco Y, Barahona-Correa J, Monsalve DM, Acosta-Ampudia Y, Rojas M, Rodríguez Y, et al. Cytokine and autoantibody clusters interaction in systemic lupus erythematosus. J Transl Med. 2017;15(1):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artim-Esen B, Çene E, Şahinkaya Y, Ertan S, Pehlivan Ö, Kamali S, et al. Cluster analysis of autoantibodies in 852 patients with systemic lupus erythematosus from a single center. J Rheumatol. 2014;41(7):1304–10. [DOI] [PubMed] [Google Scholar]
- 7.Ching KH, Burbelo PD, Tipton C, Wei C, Petri M, Sanz I, et al. Two major autoantibody clusters in systemic lupus erythematosus. PLoS One. 2012;7(2):e32001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guthridge JM, Lu R, Tran LT, Arriens C, Aberle T, Kamp S, et al. Adults with systemic lupus exhibit distinct molecular phenotypes in a cross-sectional study. EClinicalMedicine. 2020;20:100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diaz-Gallo LM, Oke V, Lundstrom E, Elvin K, Ling Wu Y, Eketjall S, et al. Four Systemic Lupus Erythematosus Subgroups, Defined by Autoantibodies Status, Differ Regarding HLA-DRB1 Genotype Associations and Immunological and Clinical Manifestations. ACR Open Rheumatol. 2022;4(1):27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciascia S, Radin M, Cecchi I, Bertolaccini ML, Bertero MT, Rubini E, et al. Identifying phenotypes of patients with antiphospholipid antibodies: results from a cluster analysis in a large cohort of patients. Rheumatology (Oxford). 2021;60(3):1106–13. [DOI] [PubMed] [Google Scholar]
- 11.Jung JY, Lee HY, Lee E, Kim HA, Yoon D, Suh CH. Three Clinical Clusters Identified through Hierarchical Cluster Analysis Using Initial Laboratory Findings in Korean Patients with Systemic Lupus Erythematosus. J Clin Med. 2022;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. 2015;14(1):75–9. [DOI] [PubMed] [Google Scholar]
- 13.Frodlund M, Wettero J, Dahle C, Dahlstrom O, Skogh T, Ronnelid J, et al. Longitudinal anti-nuclear antibody (ANA) seroconversion in systemic lupus erythematosus: a prospective study of Swedish cases with recent-onset disease. Clin Exp Immunol. 2020;199(3):245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tench CM, Isenberg DA. The variation in anti-ENA characteristics between different ethnic populations with systemic lupus erythematosus over a 10-year period. Lupus. 2000;9(5):374–6. [DOI] [PubMed] [Google Scholar]
- 15.Acosta-Merida A, Isenberg DA. Antinuclear antibodies seroconversion in 100 patients with lupus. Clin Exp Rheumatol. 2013;31(4):656. [PubMed] [Google Scholar]
- 16.Choi MY, Clarke AE, Urowitz M, Hanly J, St-Pierre Y, Gordon C, et al. Longitudinal analysis of ANA in the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann Rheum Dis. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figgett WA, Monaghan K, Ng M, Alhamdoosh M, Maraskovsky E, Wilson NJ, et al. Machine learning applied to whole-blood RNA-sequencing data uncovers distinct subsets of patients with systemic lupus erythematosus. Clinical & translational immunology. 2019;8(12):e01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kan H, Nagar S, Patel J, Wallace DJ, Molta C, Chang DJ. Longitudinal Treatment Patterns and Associated Outcomes in Patients With Newly Diagnosed Systemic Lupus Erythematosus. Clin Ther. 2016;38(3):610–24. [DOI] [PubMed] [Google Scholar]
- 19.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. [DOI] [PubMed] [Google Scholar]
- 20.Isenberg D, Ramsey-Goldman R, Gladman D, Hanly J. The Systemic Lupus International Collaborating Clinics (SLICC) group–It was 20 years ago today. Lupus. 2011;20(13):1426–32. [DOI] [PubMed] [Google Scholar]
- 21.Choi MY, Clarke AE, Urowitz M, Hanly J, St-Pierre Y, Gordon C, et al. Longitudinal analysis of ANA in the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann Rheum Dis. 2022;81(8):1143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–91. [PubMed] [Google Scholar]
- 23.Ibañez D, Urowitz MB, Gladman DD. Summarizing disease features over time: I. Adjusted mean SLEDAI derivation and application to an index of disease activity in lupus. J Rheumatol. 2003;30(9):1977–82. [PubMed] [Google Scholar]
- 24.Gladman D, Ginzler E, Goldsmith C, Fortin P, Liang M, Urowitz M, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39(3):363–9. [DOI] [PubMed] [Google Scholar]
- 25.Damoiseaux J, von Muhlen CA, Garcia-De La Torre I, Carballo OG, de Melo Cruvinel W, Francescantonio PL, et al. International consensus on ANA patterns (ICAP): the bumpy road towards a consensus on reporting ANA results. Auto Immun Highlights. 2016;7(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295–306. [DOI] [PubMed] [Google Scholar]
- 27.Hanly J, Urowitz M, Siannis F, Farewell V, Gordon C, Bae S, et al. Autoantibodies and neuropsychiatric events at the time of systemic lupus erythematosus diagnosis: results from an international inception cohort study. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology. 2008;58(3):843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syakur M, Khotimah B, Rochman E, Satoto BD, editors. Integration k-means clustering method and elbow method for identification of the best customer profile cluster. IOP conference series: materials science and engineering; 2018: IOP Publishing. [Google Scholar]
- 29.Hanly JG, Urowitz MB, Su L, Bae SC, Gordon C, Clarke A, et al. Autoantibodies as biomarkers for the prediction of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis. 2011;70(10):1726–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chighizola CB, Pregnolato F, Andrade D, Tektonidou M, Pengo V, Ruiz-Irastorza G, et al. Fluctuation of Anti-Domain 1 and Anti-Beta2 Glycoprotein I Antibody Titers Over Time in Patients with Persistently Positive Antiphospholipid Antibodies. Arthritis Rheumatol. 2023. [DOI] [PubMed] [Google Scholar]
- 31.Egri N, Bentow C, Rubio L, Norman GL, Lopez-Sanudo S, Mahler M, et al. Anti-Phosphatidylserine/Prothrombin Antibodies at Two Points: Correlation With Lupus Anticoagulant and Thrombotic Risk. Front Immunol. 2021;12:754469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sciascia S, Murru V, Sanna G, Roccatello D, Khamashta MA, Bertolaccini ML. Clinical accuracy for diagnosis of antiphospholipid syndrome in systemic lupus erythematosus: evaluation of 23 possible combinations of antiphospholipid antibody specificities. J Thromb Haemost. 2012;10(12):2512–8. [DOI] [PubMed] [Google Scholar]
- 33.Marchetti T, Ribi C, Perneger T, Trendelenburg M, Huynh-Do U, de Moerloose P, et al. Prevalence, persistence and clinical correlations of classic and novel antiphospholipid antibodies in systemic lupus erythematosus. Rheumatology (Oxford). 2018;57(8):1350–7. [DOI] [PubMed] [Google Scholar]
- 34.Choi MY, Clarke AE, Fritzler MJ. Do anti-DFS70 antibodies temper disease activity and progression in SLE? Lupus. 2021;30(5):852–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.