

Abstract
Fabry disease is an X-linked hereditary disorder caused by deficient α-galactosidase A (GLA) activity. Patients with Fabry disease are often treated with enzyme replacement therapy (ERT). However, ERT often induces the formation of neutralizing antidrug antibodies (ADAs), which may impair the therapeutic efficacy. Here, we report the case of a 32-year-old man with Fabry disease and resultant neutralizing ADAs who was treated by switching from agalsidase-α to agalsidase-β. We monitored biomarkers, such as plasma globotriaosylsphingosine (lyso-Gb3), urinary globotriaosylceramide (Gb3), urinary mulberry bodies, renal and cardiac parameters, and disease severity during the treatment period. Although plasma lyso-Gb3 and urinary Gb3 levels quickly decreased within two months after the initiation of ERT with agalsidase-α, they gradually increased thereafter. The urinary mulberry bodies continued to appear. Both the ADA titer and serum mediated GLA inhibition rates started to increase after two months. Moreover, 3.5 years after ERT, the vacuolated podocyte area in the renal biopsy decreased slightly from 23.1 to 18.9%. However, plasma lyso-Gb3 levels increased, and urinary Gb3, mulberry body levels, and ADA titers remained high. Therefore, we switched to agalsidase-β which reduced, but did not normalize, plasma lyso-Gb3 levels and stabilized renal and cardiac parameters. Disease severity was attenuated. However, urinary Gb3 and mulberry body levels did not decrease noticeably in the presence of high ADA titers. The kidneys take up a small amount of the administered recombinant enzyme, and the clearance of Gb3 that has accumulated in the kidney may be limited despite the switching from agalsidase-α to agalsidase-β.
Keywords: Fabry disease, Enzyme replacement therapy, Antidrug antibody, Globotriaosylsphingosine, Urinary globotriaosylceramide, Urinary mulberry bodies
Introduction
Fabry disease is an X-linked hereditary disorder caused by deficient α-galactosidase A (GLA) activity, which results in the accumulation of globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) [1]. Plasma lyso-Gb3 levels are considered better biomarkers for disease progression than plasma Gb3 levels [2, 3]. Urinary Gb3, which originates from tubular cells and the urinary collecting system, is also a diagnostic biomarker for Fabry nephropathy [4]. The detection of urinary mulberry bodies that originate from podocytes is highly accurate in diagnosing patients with clinically suspected Fabry disease [5, 6]. Patients with Fabry disease have often been treated with two different enzyme replacement therapies (ERT), including recombinant GLA, agalsidase-α (0.2 mg/kg biweekly) or agalsidase-β (1.0 mg/kg biweekly) [7]. However, ERT often induces the formation of neutralizing antidrug antibodies (ADAs), which may impair therapeutic efficacy [8].
We report the case of a 32-year-old man with Fabry disease in whom neutralizing ADAs were treated by switching from agalsidase-α to agalsidase-β. In addition to renal and cardiac involvement and disease severity, we closely monitored the changes in ADA titers and levels of biomarkers such as plasma lyso-Gb3, urinary Gb3, and urinary mulberry bodies during the treatment period.
Case report
A 32-year-old male was admitted to our hospital for acute gastroenteritis. He had a medical history of heatstroke during the summer months, fever of unknown origin, and peripheral limb pain since childhood. Urinary proteins have been detected for several years. He had no history of allergies, medications, or family history. On admission, his vital signs were as follows: blood pressure, 105/68 mmHg; temperature, 36.2 °C; pulse, 78/min; and respiratory rate, 16/min. Physical examination revealed no abnormalities or skin lesions. His serum creatinine (sCr) level was 0.69 mg/dL, which was unchanged from baseline. Urinalysis revealed proteinuria (1.8 g/day) and hematuria (sedimented red blood cells, 11–30 per high-power field). The levels of urinary beta-2 microglobulin (107 μg/L; normal, < 289 μg/L) and N-acetyl-beta-D-glucosaminidase (3.1 U/L; normal, < 11.5 U/L) were normal. Mulberry bodies were detected in urinary sediment samples (Fig. 1a). Electrocardiography revealed left ventricular (LV) hypertrophy (Fig. 1b). Echocardiography revealed LV hypertrophy with normal systolic function (Fig. 1c). Based on these results, Fabry disease was suspected.
Fig. 1.

Findings of the renal and cardiac examinations. a Mulberry bodies in the urinary sediment. b Electrocardiography image showing left ventricular hypertrophy. c Echocardiography showing left ventricular hypertrophy with normal systolic function. d Cardiovascular magnetic resonance native T1 mapping reveals that native T1 values are diffusely reduced without pseudo-normalization
Renal biopsy revealed expanded glomerular podocytes with fine vacuolated changes (Fig. 2a). Toluidine blue staining showed lipid deposits in podocytes (Fig. 2b). Moreover, electron microscopy revealed myelin-like bodies in podocytes (Fig. 2c). These findings were consistent with those of Fabry nephropathy. Corneal verticillata was also observed. His leukocyte GLA activity was < 1 nmol/h/mg protein (controls: 17–65 nmol/h/mg protein). The plasma lyso-Gb3 level was apparently high (187 nmol/L; reference range: 0.35–0.71 nmol/L [9]). The urinary Gb3 level was also high (9.43 μg/mgCr, cut-off value indicating urinary Gb3-positive: > 0.1 μg/mgCr [10]). Genetic analysis revealed a duplication in exon 7 of the GLA gene (c.1109_1111dupCTT) (Fig. 3). Therefore, the patient was diagnosed with Fabry disease. After his diagnosis, genetic screening of his family revealed that his mother and older sister harbored the same GLA mutation.
Fig. 2.

Renal biopsy specimen by light microscopy at baseline (a–c) and after 3.5 years (d–f). a, d Expanded glomerular podocytes with fine vacuolated changes. Masson trichrome staining (400 × magnification), scale bar is 50 μm. The vacuolated podocyte area was 23.1 and 18.9%, representatively. b, e Lipid deposits in the podocytes. Toluidine blue staining (400 × magnification), scale bar is 50 μm. c, f Electron microscopy revealed myelin-like bodies in podocytes (1500 × magnification), scale bar is 50 μm
Fig. 3.
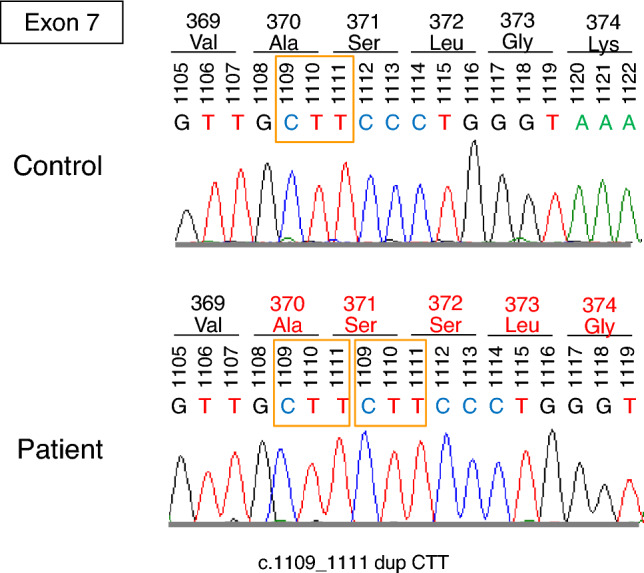
Genetic analysis revealed the duplication of CTT (c.1109_1111) in exon 7 of the GLA gene. GLA, α-galactosidase A
After the diagnosis was confirmed, ERT with agalsidase-α was initiated at a dose of 0.2 mg/kg biweekly. The time courses of urinary protein and sCr levels are presented in Fig. 4a. The urinary protein level fluctuated between 0.7 and 1.7 g/gCr. The sCr levels did not show an apparent increase (0.6–0.8 mg/dL). We monitored plasma lyso-Gb3 and urinary Gb3 levels, ADA titers, and serum-mediated GLA inhibition rates, as previously reported [9–11]. We also started to measure the number of urinary mulberry bodies using light microscopy on the whole field from 12 months after the initiation of ERT. The methodology to measure mulberry bodies of the urinary sediment specimen was as follows: urine (10 mL) was centrifuged at 500 g for 5 min. The pellet was resuspended in 200 μL of the supernatant, 100 μL of which was then mixed with 25 μL of the Sterheimer staining solution (ratio: 4:1). A 15 μL of the suspension was loaded on a glass slide covered by a cover glass (18 × 18 mm). We counted mulberry bodies, characteristic spiraling components that is grayish white and transparent.
Fig. 4.

Time courses of a urinary protein and sCr levels, b plasma lyso-Gb3 level, c urinary Gb3 and mulberry body levels, d ADA titer and serum-mediated GLA inhibition rate in the Fabry disease patient following ERT. sCr, serum creatinine; lyso-Gb3, globotriaosylsphingosine; Gb3, globotriaosylceramide; ADA, antidrug antibody; GLA, α-galactosidase A; ERT, enzyme replacement therapy
The plasma lyso-Gb3 level quickly decreased within two months and stayed (70–80 nmol/L) until 21 months after the initiation of ERT (Fig. 4b). However, it gradually increased (120–143 nmol/L) afterwards. The urinary Gb3 level also quickly decreased within two months (Fig. 4c). However, it increased after three months and remained high afterwards (4–7 μg/mgCr). The urinary mulberry bodies continued to appear (Fig. 4c). Both the ADA titer (the cut-off value indicating antibody-positive: ΔOD 490 nm > 0.250 [11]) and the serum-mediated GLA inhibition rate (the cut-off value indicating inhibition-positive: > 49% [11]) started to increase after two months (Fig. 4d). They gradually increased up to ΔOD 490 nm = 2.66 and 79%, respectively, at 43 months.
Due to an insufficient response, a repeat renal biopsy was performed. The severity of podocyte vacuolation was assessed with the use of a previously reported method [6]. Before ERT initiation, the vacuolated podocyte area was 23.1% (Fig. 2a). After 3.5 years, this decreased to 18.9% (Fig. 2d, 2e), but myelin-like bodies in podocytes were still observed (Fig. 2f). Subsequently, we switched to agalsidase-β at a dose of 1.0 mg/kg biweekly. Although the ADA titer remained fairly high, the plasma lyso-Gb3 level quickly decreased from 133 to 95 nmol/L and remained relatively high (70–74 nmol/L) (Fig. 4b, 4d). Urinary Gb3 and mulberry body levels did not decrease after switching to agalsidase-β (Fig. 4c). After switching to agalsidase-β, the patient was pretreated with antihistamines for the prevention of infusion-associated reactions (IARs). IARs were not observed at all before and after switching.
Echocardiographic parameters at baseline and during treatment are presented in Table 1. Interventricular septum thickness, posterior LV wall thickness, and LV mass index did not significantly change during treatment. The global longitudinal strain (GLS) continued to slightly improve. LV ejection fraction remained within the normal range (≧60%). Cardiovascular magnetic resonance native T1 mapping revealed that the native T1 values were diffusely reduced without pseudo-normalization after a 5-year follow-up (Fig. 1d). The score obtained with the Fabry disease severity scoring system (DS3) at baseline was 8.6 (Table 1). The DS3 score was decreased to 3.7 after 5 years of observation.
Table 1.
The parameters of echocardiography and the DS3 for Fabry disease at baseline and during the treatment
| After ERT | ||||
|---|---|---|---|---|
| Baseline | Agalsidase-α | Agalsidase-β | ||
| 1 year | 3 year | 5 year (1-year follow- up after switching) | ||
| LVDd (mm) | 44.0 | 49.8 | 46.5 | 44.7 |
| LVDs (mm) | 26.1 | 32.2 | 29.0 | 26.1 |
| IVST (mm) | 9.3 | 11.5 | 11.8 | 12.3 |
| PWT (mm) | 10.7 | 11.5 | 11.8 | 12.5 |
| LVMI (g/m2) | 94 | 138 | 126 | 126 |
| LVEF (%) | 67.0 | 64.4 | 61.9 | 61.0 |
| GLS (%) | −14.1 | −14.6 | −15.7 | |
| BNP (pg/mL) | 18.5 | 14.4 | 32.6 | |
| DS3 score | 8.6 | 5.2 | 6.0 | 3.7 |
LVDd left ventricular diastolic dimension, LVDs left ventricular systolic dimension, IVST Inter ventricular septum thickness, PWT posterior left ventricular wall thickness, LVMI left ventricular mass index, LVEF left ventricular ejection fraction, GLS global longitudinal strain, BNP brain natriuretic peptide, ERT enzyme replacement therapy, DS3 disease severity scoring system
Discussion
In the present case, we switched from agalsidase-α to agalsidase-β because of the increase in biomarkers, such as plasma lyso-Gb3, urinary Gb3, and mulberry bodies, which were possibly affected by the formation of ADAs. We assessed the effects of switching from agalsidase-α to agalsidase-β on ADA titers, biomarkers, renal and cardiac parameters, and disease severity. This case provides important clinical suggestions regarding the usefulness of monitoring biomarkers and the type of treatment needed in case of ADA formation.
In male Fabry disease patients, the first ADAs mostly develop within 3–6 months after the initiation of ERT [7, 12, 13]. ADAs bind and neutralize ERT in the plasma, leading to the activation of macrophages that internalize ERT-ADA complexes, decreasing the cellular uptake of free ERT [7, 14]. Therefore, ADA formation may affect ERT efficacy. Plasma lyso-Gb3 is a useful specific biomarker for the diagnosing Fabry disease and monitoring the patient’s response to Fabry disease treatment [9]. The plasma lyso-Gb3 level has also been reported as a significant risk factor associated with clinical events and markers of disease progression [15]. In this case, the plasma lyso-Gb3 level quickly decreased within two months and increased thereafter, which may be due to the formation of ADAs. The urinary Gb3 level showed similar trends. Currently, there is no consensus on the treatment of patients with Fabry disease with neutralizing ADAs. Switching from agalsidase-α to agalsidase-β may attenuate the negative effects of ADAs by supersaturating ADAs and performing the catalytic function of excess enzymes [16]. It may further reduce the plasma lyso-Gb3 level but increase the antibody titer [16, 17].
Since we expected immune tolerance, we initially continued with the agalsidase-α treatment. However, because the plasma lyso-Gb3 level increased, and urinary Gb3 and mulberry body levels and the ADA titer remained high, we switched to agalsidase-β. In this case, ERT with agalsidase-β reduced, but not normalized, the plasma lyso-Gb3 level and stabilized renal and cardiac parameters. However, urinary Gb3 and mulberry body levels did not decrease noticeably in the presence of high ADA titers. This might be because the main origins of plasma lyso-Gb3 are thought to be the endothelium and the liver [18], and most of the agalsidase administered into the blood is taken up by the liver [19]. As the recombinant enzyme was switched, it was assumed that lyso-Gb3 accumulated in the liver was dissolved by the dosage effect of the recombinant enzyme, which was greater than the amount required for the saturation of ADAs. In contrast, the kidneys take up only a small amount of the administered recombinant enzyme [20], which may result in limited clearance of Gb3 deposition in the kidney, despite the switching from agalsidase-α to agalsidase-β.
Although the plasma lyso-Gb3 level is useful as both a diagnostic and treatment biomarker, it is unclear whether it reflects renal pathology [21]. There is no consensus on the correlation between urinary Gb3 levels and renal events [10, 22, 23]. Recently, it has been reported that urinary mulberry bodies might serve as useful biomarkers for the diagnosis and monitoring of ERT efficacy [6, 21, 24]. Urinary mulberry bodies are derived from podocytes containing lysosomes with Gb3 accumulation [6]. In our case, urinary mulberry body and urinary Gb3 levels showed the same trend, but there was no correlation between their values. The count of urinary mulberry bodies may vary depending on the ability and experience of the technicians. Therefore, the standardization of semiquantitative urinary mulberry body estimates is required. Recently, it was reported that the hematuria positivity rate was 31.0% in the biopsy-proven Fabry nephropathy in the Japan Renal Biopsy Registry [25]. In the present case, hematuria had disappeared after ERT.
Cardiac involvement is an important factor in the morbidity and mortality of patients with Fabry disease, with LV hypertrophy being the most common cause. Reduced GLS may be an early marker of Fabry cardiomyopathy [26]. In this case, the cardiac parameters of LV hypertrophy and GLS were stable over the five-year follow-up period after ERT initiation. Cardiovascular magnetic resonance native T1 mapping is a useful technique for detecting Gb3 accumulation and diffuse fibrosis [27]. After myocardial fibrosis, native T1 mapping may show pseudo-normalization; this was not observed in this case. Renal function was maintained after the initiation of ERT. DS3 for Fabry disease is useful for assessing and monitoring disease severity [28]. It includes five domains, with a maximum possible score of 32. The severity in this case decreased during the treatment period.
In conclusion, considering the various biomarkers and disease severity, switching from agalsidase-α to agalsidase-β may stabilize or delay the progression of renal and cardiac involvement in patients with Fabry disease, thereby forming neutralizing ADAs. However, because the kidneys take up only a small amount of the administered recombinant enzyme, the clearance of accumulated Gb3 from the kidneys may be limited despite the switching from agalsidase-α to agalsidase-β.
Acknowledgements
We would like to thank Editage for their assistance in editing the draft of this manuscript.
Funding
Hitoshi Sakuraba received a research grant from Sumitomo Pharma Co., Ltd. The funder played no role in the study design, data collection or analysis.
Declarations
Conflict of interest
The authors declare no conflicts of interest.
Human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional committee, and with the 1964 Helsinki Declaration and its later amendments and comparable ethical standards.
Informed Consent
Informed consent was obtained from the individual participant included in the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427–35. 10.1016/S0140-6736(08)61589-5 [DOI] [PubMed] [Google Scholar]
- 2.Togawa T, Kawashima I, Kodama T, Tsukimura T, Suzuki T, Fukushige T, et al. Tissue and plasma globotriaosylsphingosine could be a biomarker for assessing enzyme replacement therapy for Fabry disease. Biochem Biophys Res Commun. 2010;399:716–20. 10.1016/j.bbrc.2010.08.006 [DOI] [PubMed] [Google Scholar]
- 3.Nowak A, Mechtler TP, Desnick RJ, Kasper DC. Plasma LysoGb3: a useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol Genet Metab. 2017;120:57–61. 10.1016/j.ymgme.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 4.Schiffmann R, Waldek S, Benigni A, Auray-Blais C. Biomarkers of Fabry disease nephropathy. Clin J Am Soc Nephrol. 2010;5:360–4. 10.2215/CJN.06090809 [DOI] [PubMed] [Google Scholar]
- 5.Nakamura K, Mukai S, Takezawa Y, Natori Y, Miyazaki A, Ide Y, et al. Clinical utility of urinary mulberry bodies/cells testing in the diagnosis of Fabry disease. Mol Genet Metab Rep. 2023;36:100983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yonishi H, Namba-Hamano T, Hamano T, Hotta M, Nakamura J, Sakai S, et al. Urinary mulberry bodies as a potential biomarker for early diagnosis and efficacy assessment of enzyme replacement therapy in Fabry nephropathy. Nephrol Dial Transplant. 2021;37:53–62. 10.1093/ndt/gfaa298 [DOI] [PubMed] [Google Scholar]
- 7.Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–95. 10.1111/j.1523-1755.2004.00924.x [DOI] [PubMed] [Google Scholar]
- 8.Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E. Serum-mediated inhibition of enzyme replacement therapy in fabry disease. J Am Soc Nephrol. 2016;27:256–64. 10.1681/ASN.2014121226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakuraba H, Togawa T, Tsukimura T, Kato H. Plasma lyso-Gb3: a biomarker for monitoring fabry patients during enzyme replacement therapy. Clin Exp Nephrol. 2018;22:843–9. 10.1007/s10157-017-1525-3 [DOI] [PubMed] [Google Scholar]
- 10.Shiga T, Tsukimura T, Namai Y, Togawa T, Sakuraba H. Comparative urinary globotriaosylceramide analysis by thin-layer chromatography-immunostaining and liquid chromatography-tandem mass spectrometry in patients with Fabry disease. Mol Genet Metab Rep. 2021;29:100804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsukimura T, Tayama Y, Shiga T, Hirai K, Togawa T, Sakuraba H. Anti-drug antibody formation in Japanese Fabry patients following enzyme replacement therapy. Mol Genet Metab Rep. 2020;25:100650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deegan PB. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J Inherit Metab Dis. 2012;35:227–43. 10.1007/s10545-011-9400-y [DOI] [PubMed] [Google Scholar]
- 13.Smid BE, Hoogendijk SL, Wijburg FA, Hollak CE, Linthorst GE. A revised home treatment algorithm for Fabry disease: influence of antibody formation. Mol Genet Metab. 2013;108:132–7. 10.1016/j.ymgme.2012.12.005 [DOI] [PubMed] [Google Scholar]
- 14.Lenders M, Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with fabry disease. J Am Soc Nephrol. 2018;29:2265–78. 10.1681/ASN.2018030329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak A, Beuschlein F, Sivasubramaniam V, Kasper D, Warnock DG. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry disease. J Med Genet. 2022;59:287–93. 10.1136/jmedgenet-2020-107338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenders M, Neusser LP, Rudnicki M, Nordbeck P, Canaan-Kuhl S, Nowak A, et al. Dose-dependent effect of enzyme replacement therapy on neutralizing antidrug antibody titers and clinical outcome in patients with fabry disease. J Am Soc Nephrol. 2018;29:2879–89. 10.1681/ASN.2018070740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenders M, Nordbeck P, Canaan-Kuhl S, Kreul L, Duning T, Lorenz L, et al. Treatment switch in Fabry disease- a matter of dose? J Med Genet. 2021;58:342–50. 10.1136/jmedgenet-2020-106874 [DOI] [PubMed] [Google Scholar]
- 18.Kok K, Zwiers KC, Boot RG, Overkleeft HS, Aerts JMFG, Artola M. Fabry disease: Molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules. 2021;11:271. 10.3390/biom11020271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azevedo O, Gago MF, Miltenberger-Miltenyi G, Sousa N, Cunha D. Fabry disease therapy: state-of-the-art and current challenges. Int J Mol Sci. 2020;22:206. 10.3390/ijms22010206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002;62:1933–46. 10.1046/j.1523-1755.2002.00675.x [DOI] [PubMed] [Google Scholar]
- 21.Shimohata H, Yamashita M, Ohgi K, Maruyama H, Takayasu M, Hirayama K, et al. Clinical course and pathological findings of two late-onset Fabry hemizygous patients including mulberry cell counts after enzyme replacement therapy. CEN Case Rep. 2020;9:237–42. 10.1007/s13730-020-00463-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moura AP, Hammerschmidt T, Deon M, Giugliani R, Vargas CR. Investigation of correlation of urinary globotriaosylceramide (Gb3) levels with markers of renal function in patients with Fabry disease. Clin Chim Acta. 2018;478:62–7. 10.1016/j.cca.2017.12.033 [DOI] [PubMed] [Google Scholar]
- 23.Burlina A, Brand E, Hughes D, Kantola I, Krӓmer J, Nowak A, et al. An expert consensus on the recommendations for the use of biomarkers in Fabry disease. Mol Genet Metab. 2023;139: 107585. 10.1016/j.ymgme.2023.107585 [DOI] [PubMed] [Google Scholar]
- 24.Aoyama Y, Ushio Y, Yokoyama T, Taneda S, Makabe S, Nishida M, et al. Urinary Mulberry Cells as a Biomarker of the Efficacy of Enzyme Replacement Therapy for Fabry Disease. Intern Med. 2020;59:971–6. 10.2169/internalmedicine.3813-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nasu M, Nakagawa N, Hara S, Yano J, Kurokawa Y, Nakamura N, et al. A nationwide cross-sectional analysis of biopsy-proven Fabry nephropathy: the Japan Renal Biopsy Registry. Clin Exp Nephrol. 2023;27:141–50. 10.1007/s10157-022-02287-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu DY, Huang WM, Wang WT, Hung SC, Sung SH, Chen CH, et al. Reduced global longitudinal strain as a marker for early detection of Fabry cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2022;23:487–95. 10.1093/ehjci/jeab214 [DOI] [PubMed] [Google Scholar]
- 27.Umer M, Kalra DK. Cardiac MRI in Fabry disease. Front Cardiovasc Med. 2022;9:1075639. 10.3389/fcvm.2022.1075639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giannini EH, Mehta AB, Hilz MJ, Beck M, Bichet DG, Brady RO, et al. A validated disease severity scoring system for Fabry disease. Mol Genet Metab. 2010;99:283–90. 10.1016/j.ymgme.2009.10.178 [DOI] [PubMed] [Google Scholar]