

Abstract
In recent years, there has been increased focus on exploring the role the non-protein-coding genome plays in Mendelian disorders. One class of particular interest is long non-coding RNAs (lncRNAs), which has recently been implicated in the regulation of diverse molecular processes. However, because lncRNAs do not encode protein, there is uncertainty regarding what constitutes a pathogenic lncRNA variant, and thus annotating such elements is challenging. The Developmental Genome Anatomy Project (DGAP) and similar projects recruit individuals with apparently balanced chromosomal abnormalities (BCAs) that disrupt or dysregulate genes in order to annotate the human genome. We hypothesized that rearrangements disrupting lncRNAs could be the underlying genetic etiology for the phenotypes of a subset of these individuals. Thus, we assessed 279 cases with BCAs and selected 191 cases with simple BCAs (breakpoints at only two genomic locations) for further analysis of lncRNA disruptions. From these, we identified 66 cases in which the chromosomal rearrangements directly disrupt lncRNAs. In 30 cases, no genes of any other class aside from lncRNAs are directly disrupted, consistent with the hypothesis that lncRNA disruptions could underly the phenotypes of these individuals. Strikingly, the lncRNAs MEF2C-AS1 and ENSG00000257522 are each disrupted in two unrelated cases. Furthermore, we experimentally tested the lncRNAs TBX2-AS1 and MEF2C-AS1 and found that knockdown of these lncRNAs resulted in decreased expression of the neighboring transcription factors TBX2 and MEF2C, respectively. To showcase the power of this genomic approach for annotating lncRNAs, here we focus on clinical reports and genetic analysis of seven individuals with likely developmental etiologies due to lncRNA disruptions.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00439-024-02693-y.
Introduction
Only ~ 2% of the human genome is represented in protein-coding messenger RNAs (mRNAs) (Mattick and Amaral 2022). The non-protein-coding genome comprises a diverse array of elements including those that transcribe long non-coding RNAs (lncRNAs), transcripts of at least 200 nucleotides in length that are not translated into proteins (Kopp and Mendell 2018). Current transcriptome annotations (Frankish et al. 2021) suggest that there are nearly 20,000 human lncRNAs, rivaling the number of protein-coding genes, and the expression of many of these lncRNAs is highly regulated. However, the biological roles of most lncRNAs remain to be determined, which is made challenging by the fact that lncRNAs can carry out a wide variety of different functions. For instance, some lncRNAs can act in trans to affect diverse biological processes at distant sites, whereas many other lncRNAs have been shown to act in cis to regulate target genes in a manner that depends upon the location from which the lncRNA is transcribed (Gil and Ulitsky 2020). Cis-acting lncRNAs can exist in many different genomic configurations with respect to their target gene, such as overlapping in the sense or antisense direction, sharing a promoter region, or encompassing enhancer elements that regulate the target gene (Statello et al. 2021; Ferrer and Dimitrova 2024). In some cases, the lncRNA locus exists at a substantial distance from its target gene when considering the linear nucleotide position, however it can be brought into proximity with its target by the three-dimensional chromatin conformation. Cis-acting lncRNAs can also carry out their roles in diverse ways, including through RNA-dependent mechanisms as well as through functions that do not require the lncRNA transcript itself, such as the act of transcription of the lncRNA locus (Gil and Ulitsky 2020). By regulating the expression of neighboring genes, cis-acting lncRNAs can play critical roles in modulating key biological processes.
As a testament to the importance of lncRNAs in normal development, deletions of certain lncRNAs have led to lethal phenotypes in mice as well as abnormal development of the neocortex, lung, gastrointestinal tract, and heart (Sauvageau et al. 2013; Feyder and Goff 2016; Andersen et al. 2019; Mattick et al. 2023). In addition, lncRNAs have been implicated in cancer and shown to affect cell division, metabolism, and tumor-host interactions. Thus, lncRNAs are essential to maintaining proper cellular homeostasis (Statello et al. 2021). A prior observation of a variant in a lncRNA causing human disease in a Mendelian fashion is a 27–63 kb deletion of a locus that encompasses a lncRNA upstream of the engrailed-1 gene (EN1) (MIM: 131290), which resulted in congenital limb abnormalities even though EN1 itself was not disrupted (Allou et al. 2021). Most recently, a pre-print manuscript (Ganesh et al. 2024) reported three individuals with deletions in CHASERR, a lncRNA proximal to CHD2 (MIM: 602119), a protein-coding gene that causes developmental and epileptic encephalopathy. Intriguingly, disruption of CHASERR leads to increased expression of CHD2 in cis, leading to a distinct clinical presentation compared to individuals with CHD2 haploinsufficiency.
Given the importance of lncRNAs to gaining a better understanding of developmental biology and improving clinical diagnoses, it is important to develop a better functional assessment of lncRNAs in the human genome. However, the methods of annotating protein-coding genes are not typically applicable to non-coding genes due to their fundamental differences (Mattick et al. 2023). For instance, single nucleotide variants (SNVs) can result in nonsense mutations that prematurely terminate proteins, and indels can cause translational frame shifts that alter the entire downstream amino acid sequence of a protein. However, because lncRNAs do not encode proteins, it is unclear what affect if any such mutations may have on lncRNAs. The study of lncRNAs remains to be elucidated with regard to definitive consensus over what constitutes a pathogenic lncRNA variant.
A prior landmark development in expanding a focus on DNA beyond protein-coding genes to the 3D genome was the discovery of topologically associated domains (TADs). TADs are megabase-sized genomic segments partitioning the genome into large regulatory units with frequent intra-domain chromatin interactions but relatively rare inter-domain interactions (Lupiáñez et al. 2015). Conserved across different cell types and species, they are considered crucial for spatiotemporal gene expression patterns. Topological boundary regions (TBRs) block interactions between adjacent TADs, and TBR disruption by chromosomal structural rearrangements can result in rewiring of genomic regulators leading to abnormal clinical phenotypes. Rigorous interpretation of clinical phenotypes requires assessment of the boundaries of TADs following a chromosomal structural rearrangement because complex phenotypes may be dissected from rearrangements that reposition lncRNAs with respect to the relevant protein-coding region. Such rearrangements can also disrupt or reposition important non-coding regulatory elements such as enhancers, and thus careful consideration of these various possibilities is required (D’haene and Vergult 2021). Along with lncRNAs, chromatin conformation, enhancer-associated chromatin modifications (e.g., H3K4me1 and H3K27Ac), and experimentally validated enhancer elements should all be analyzed when interpreting non-coding structural variants.
The Developmental Genome Anatomy Project (DGAP) (Higgins et al. 2008) and similar projects have historically explored balanced chromosomal rearrangements to establish possible relationships between genotypes and phenotypes through identifying nucleotide-level breakpoints via Sanger sequencing. Individuals with such rearrangements represent natural gene disruptions and dysregulations, and their chromosomal rearrangements can serve as ideal signposts for annotating the human genome. Unlike for protein-coding genes, it is hard to predict the pathogenicity of lncRNA variants because they are not translated and consequently frameshift and nonsense mutations may not disrupt their function. Here, we employ a foundational approach in human genetics using chromosomal rearrangements to interrogate potential phenotypic impacts of disrupted lncRNAs and their genomic repositioning resulting in dysregulation. Both disruption and dysregulation of lncRNAs therefore may increase the diagnostic yield of developmental disorders. We venture to make a call out to cytogeneticists to employ further the power of chromosomal rearrangements in yet another opportunity to contribute to annotating the genome, recognizing that there are many patients and families who still await diagnoses.
Methods
Human subjects
Study ID numbers are a consecutive alphanumeric list that are not known outside of the research group. The Partners HealthCare System Internal Review Board (IRB) gave ethical approval for this work under protocol number 1999P003090.
Breakpoint mapping
Genomic DNA from DGAP probands was sequenced to identify chromosomal breakpoints at nucleotide-level according to the previously published protocol (Talkowski et al. 2011; Hanscom and Talkowski 2014). Sanger sequencing results were aligned to the human genome using the UCSC Genome Browser BLAT tool (Kent 2002). Breakpoints were also compiled from previous publications (Talkowski et al. 2012b; Redin et al. 2017; Lowther et al. 2022). Breakpoint positions were converted from earlier genome builds to hg38 using the UCSC Genome Browser LiftOver tool (Kent et al. 2002).
Additional genetic analyses
To ensure that the patient phenotypes could not be attributed to other variants aside from the chromosomal rearrangements, whole exome sequencing was performed for a subset of cases by the Genomics Platform at the Broad Institute of MIT and Harvard (Cambridge, MA). Sequencing libraries were prepared from sample DNA (250 ng input) using the Twist Bioscience exome (~ 35 Mb target) assay (San Francisco, CA), which were then sequenced (150 bp paired end) on the Illumina NovaSeq platform (Illumina, San Diego, CA) to generate a coverage of > 85% of the target region at 20X read depth or greater. Sequencing data for each of the samples was processed through an internal pipeline using the BWA aligner for mapping to the human genome (GRCh38/hg38) and variant calling was performed using the Genome Analysis Toolkit (GATK) HaplotypeCaller package. Variants were then annotated and assessed for pathogenicity using the Seqr software (Pais et al. 2022). Variants with > 1% MAF were filtered out and variants in genes with an association with disease were prioritized for analysis. No candidate variants associated with the phenotypes were found.
TAD analysis and visualization of chromatin interactions
TAD boundary positions previously identified by Dixon and colleagues (Dixon et al. 2012) were converted from hg18 to hg38 using the UCSC Genome Browser LiftOver tool (Kent et al. 2002). BEDTools was used to identify TADs that included DGAP breakpoints (Quinlan and Hall 2010). The USCS Genome Browser (Kent et al. 2002) was used to display these TAD regions. Along with the genes within these regions, we also display chromatin interactions identified through micro-C studies from H1-hESCs (Krietenstein et al. 2020). Furthermore, we display layered tracks showing the enhancer-associated chromatin modifications H3K4me1 and H3K27Ac based on ChIP-seq analyses (ENCODE Project Consortium 2012). These ChIP-seq datasets were generated from seven human cell lines representing a variety of cell types (GM12878 lymphoblastoid cells, H1-hESCs, human skeletal muscle myoblasts, human umbilical vein endothelial cells, K562 chronic myelogenous leukemia cells, normal human epidermal keratinocytes, and normal human lung fibroblasts) by the Bernstein Lab at the Broad Institute. We also show VISTA-validated enhancer elements that demonstrated reproducible patterns of activity through in vivo transgenic mouse assays (Visel et al. 2007).
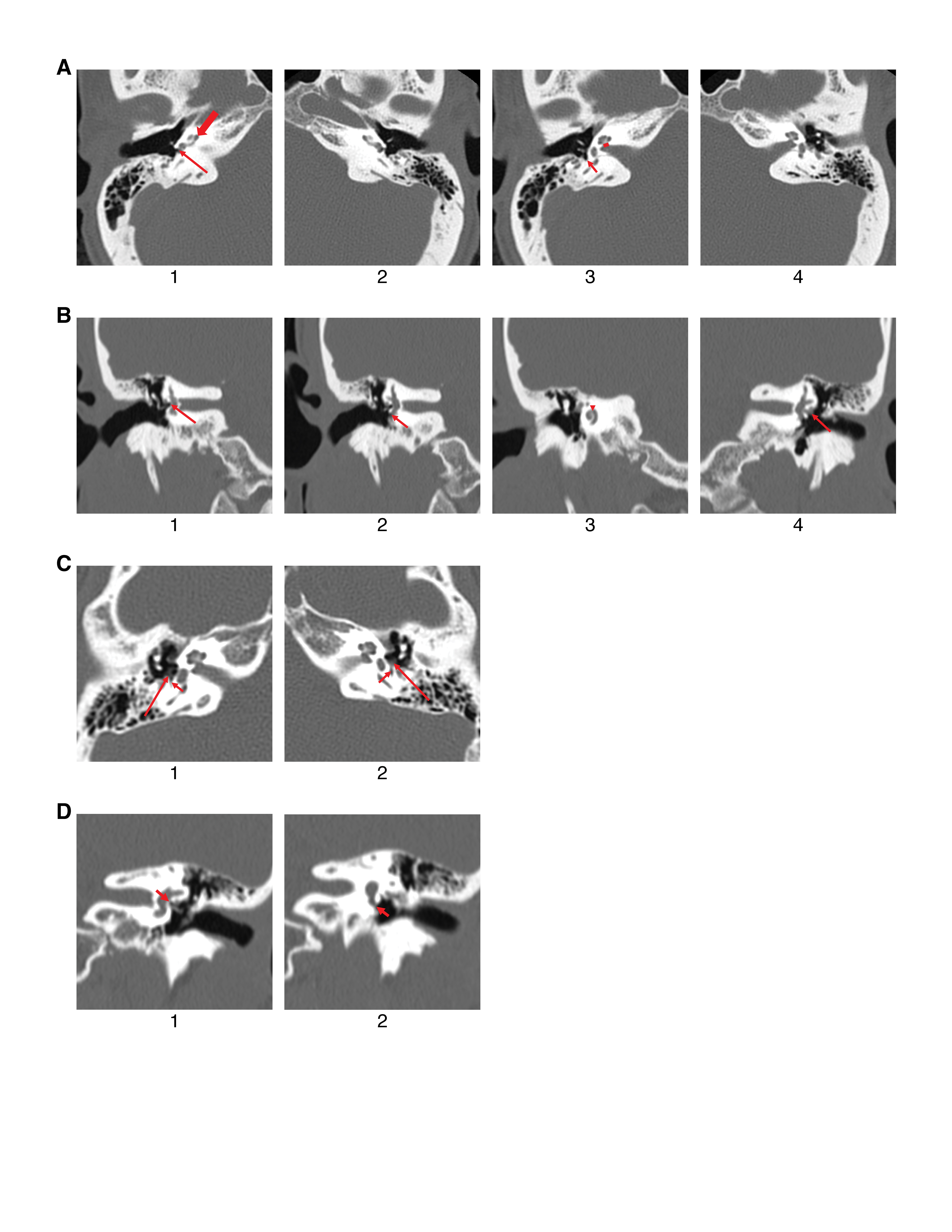
Temporal bone computerized tomography (CT)
Axial temporal bone CT without contrast for Fig. S1A-B consisted of helical images with the following parameters: Discovery STE system (General Electric Healthcare, Waukesha, WI); 0.625 mm slice thickness, effective mAs of 17, mA of 158, rotation time of 2161 milliseconds, pitch of 0.5625 and kvp of 140. Images were viewed in a plane parallel to that of the horizontal semicircular canal. Coronal reformatted images were obtained in a plane perpendicular to the axial images at 0.74 mm thickness.
Axial temporal bone CT without contrast for Fig. S1C-D consisted of helical images with the following parameters: Discovery STE system (General Electric Healthcare, Waukesha, WI); 0.625 mm slice thickness, effective mAs of 27, mA of 246, rotation time of 2161 milliseconds, pitch of 0.5625 and kvp of 100. Images were viewed in a plane parallel to that of the horizontal semicircular canal. Coronal reformatted images were obtained in a plane perpendicular to the axial images at 0.70 mm thickness.
Knockdown experiments
Target lncRNAs were knocked down in the human Lenti-X™ 293T Cell Line (Takara # 632180) using Silencer® Select siRNAs from ThermoFisher Scientific (see Table S4 for siRNA details). When available, siRNA sequences from previous publications were used (Zhang et al. 2016; Modi et al. 2023). For each sample, 12.5pmol of siRNA was reverse-transfected into 200,000 cells using 3uL of Lipofectamine™ RNAiMAX Transfection Reagent (ThermoFisher Scientific #13778150) in a final culture volume of 250uL. After 24 h, RNA was collected using TRI Reagent and stored at -80 °C. RNA was later purified using the Direct-zol RNA Microprep Kit (Zymo Research # R2061). For gene expression analysis, cDNA synthesis was performed using SuperScript™ IV VILO™ Master Mix (ThermoFisher Scientific #11756050), and digital droplet PCR (ddPCR) was performed using QX200™ ddPCR™ EvaGreen Supermix (Bio-Rad #1864033), the Automated Droplet Generator (Bio-Rad #1864101), and the QX200 Droplet Reader (Bio-Rad #1864003). For each sample, expression was normalized using the internal control gene HPRT1 (Anderson et al. 2015). See Table S4 for ddPCR primer details.
Results
Identification of human subjects with disrupted lncRNAs
We evaluated 279 cases of balanced chromosomal abnormalities and selected 191 cases with resolved breakpoints indicating a “simple” rearrangement (i.e., breakpoints at only two genomic locations and no significant genomic imbalance) for further analysis (Table S1). Using the most recent Human Gencode Reference, Release 45, GRCh38.p14 (Frankish et al. 2021), we then identified 66 cases in which at least one breakpoint overlapped a lncRNA (Table S2 and Table S3). Overall, 79 unique lncRNAs were directly disrupted in these cases, and four lncRNAs including MEF2C-AS1 and ENSG00000257522 were each disrupted in two unrelated individuals. In 30 of the cases, no genes of any other class aside from lncRNAs were directly disrupted by the breakpoints. In this report, we present seven cases disrupting five different lncRNAs as examples of the potential value of assessing lncRNAs as diagnostic etiologies (Table 1).
Table 1.
Details regarding the breakpoints and the disrupted genes for the cases highlighted in this manuscript. Genomic coordinates refer to GRCh38/hg38
| Subject_ID | break | chr | break_start | break_end | gene_start | gene_end | strand | gene_name | gene_type |
|---|---|---|---|---|---|---|---|---|---|
| DGAP148 | 1 | chrX | 39882591 | 39882592 | . | . | . | . | . |
| DGAP148 | 2 | chr11 | 127040509 | 127040509 | 127021736 | 127172363 | + | ENSG00000255087 | lncRNA |
| DGAP191 | 1 | chr5 | 89411065 | 89411070 | 88883328 | 89466398 | + | MEF2C-AS1 | lncRNA |
| DGAP191 | 2 | chr7 | 94378248 | 94378255 | 94278680 | 94395608 | - | ENSG00000285090 | lncRNA |
| DGAP218 | 1 | chr5 | 24272189 | 24272193 | . | . | . | . | . |
| DGAP218 | 2 | chr5 | 89105026 | 89105031 | 88883328 | 89466398 | + | MEF2C-AS1 | lncRNA |
| DGAP245 | 1 | chr3 | 36927650 | 36927959 | 36826819 | 36945057 | - | TRANK1 | protein_coding |
| DGAP245 | 2 | chr14 | 29276109 | 29276117 | 28936889 | 29372406 | + | ENSG00000258028 | lncRNA |
| DGAP245 | 2 | chr14 | 29276109 | 29276117 | 29210986 | 29392048 | - | ENSG00000257522 | lncRNA |
| DGAP353 | 1 | chr14 | 65855359 | 65855360 | . | . | . | . | . |
| DGAP353 | 2 | chr17 | 61393812 | 61393841 | 61361668 | 61400243 | - | ENSG00000267131 | lncRNA |
| DGAP353 | 2 | chr17 | 61393812 | 61393841 | 61393455 | 61411555 | - | TBX2-AS1 | lncRNA |
| DGAP355 | 1 | chr3 | 181488756 | 181489591 | 180989762 | 181836880 | + | SOX2-OT | lncRNA |
| DGAP355 | 2 | chr9 | 74712100 | 74713321 | . | . | . | . | . |
| NIJ1 | 1 | chr8 | 78898169 | 78898172 | 78805293 | 78956082 | + | MITA1 | lncRNA |
| NIJ1 | 2 | chr14 | 29296328 | 29296331 | 28936889 | 29372406 | + | ENSG00000258028 | lncRNA |
| NIJ1 | 2 | chr14 | 29296328 | 29296331 | 29210986 | 29392048 | - | ENSG00000257522 | lncRNA |
TBX2-AS1 is a candidate lncRNA for an association with hearing loss
The proband DGAP353 was diagnosed during gestation when her 24-year-old healthy mother underwent amniocentesis following an abnormal maternal serum screen for an elevated risk for trisomy 21. An apparently balanced translocation was detected in the female fetus between the long arms of chromosomes 14 and 17. Parental chromosome analyses revealed maternal inheritance and apparent structural identity to the maternal t(14;17) rearrangement. The daughter’s G-banded karyotype is described as 46,XX,t(14;17)(q24.3;q23)mat and the mother’s karyotype is 46,XX,t(14;17)(q24.3;q23). No clinical abnormalities were observed in the fetus and the pregnancy was continued. The daughter began developing signs of hearing loss at around the age of 10 years, and her hearing loss was found to be primarily sensorineural with a conductive element. By the age of 12 years, surgery was performed to rectify the conductive abnormalities, but the sensorineural hearing loss remained. The mother began wearing hearing aids at around the age of 40 years after a gradual decline in hearing for an unspecified time period. Both the daughter and her mother were otherwise healthy, typical of nonsyndromic deafness of unknown genetic etiology. Computerized tomography (CT) imaging of the temporal bones of the mother and daughter revealed abnormalities such as unusually small sinus tympani and narrowing of the round and oval windows (Fig. S1).
Both mother and daughter (DGAP353) harbor a translocation between chromosomes 14 and 17 with a 7 base-pair (bp) insertion of DNA of non-templated origin at the breakpoint in the der(17) chromosome (Fig. 1A). Following revision of suggested nomenclature (Ordulu et al. 2014), the next-generation cytogenetic nucleotide level research rearrangement is described below in a single string.
Fig. 1.

The lncRNA TBX2-AS1 is a candidate for an association with hearing loss. (A) Chromosome diagrams depict the translocation between 14q23.3 and 17q23.2 in DGAP353. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 14q23.3 breakpoints in DGAP353. (C) Expanded view of the genomic region surrounding the 17q23.2 breakpoints in DGAP353. The directly disrupted lncRNA TBX2-AS1 is highlighted in red. ENSG00000267131 has been identified as an isoform of TBX2-AS1 by LNCipedia (Volders et al. 2019)
46,XX,t(14;17)(q24.3;q23)mat.seq[GRCh38] t(14;17)(14pter→14q23.3(+)(65,855,3{58–60})::17q23.2(+)(61,393,84{1–3})→17qter;17pter→17q23.2(+)(61,393,812)::TATATAC::14q23.3(+)(65,855,359)→14qter)mat
The DGAP353 breakpoints do not overlap any genes on chromosome 14 (Fig. 1B); however, this translocation results in the direct disruption of the lncRNA TBX2-AS1 from chromosome 17 (Fig. 1C). The Gencode annotation also lists the lncRNA ENSG00000267131 as a separate gene that is disrupted by these breakpoints, however this has been identified as an isoform of TBX2-AS1 (called TBX2-AS1:3) on the basis of shared exonic sequences by LNCipedia (Volders et al. 2019) (Fig. S2). While little is known regarding the biological role of TBX2-AS1, particularly in the context of hearing, the orthologous mouse lncRNA (2610027K06Rik) has been detected in the cochlear and vestibular sensory epithelium of embryonic and postnatal mice (identified as “XLOC_007930”) (Ushakov et al. 2017). Using the Gene Expression Analysis Resource (gEAR) portal (Orvis et al. 2021), we further found that while Tbx2-as1 is detected in supporting cell types (pillar and Deiters cells), it is predominantly expressed by sensory inner hair cells, as determined through cell-type-specific RNA-seq (Liu et al. 2018). Thus, the expression pattern of Tbx2-as1 is consistent with the finding that the hearing loss demonstrated by DGAP353 and her mother is primarily sensorineural.
The lncRNA gene TBX2-AS1 exists in a divergent configuration with the transcription factor TBX2 (MIM: 600747). Divergent lncRNAs have transcriptional start sites (TSSs) within 5 kb of another gene and are transcribed in the opposite direction in a “head-to-head” configuration. Importantly, the DGAP353 translocation does not disrupt the shared TBX2/TBX2-AS1 promoter region, but rather removes the 3’ end of TBX2-AS1, suggesting a potential lncRNA-dependent mechanism. Divergent lncRNAs have generally been associated with positive regulation of their neighboring gene, particularly when the neighbor is a transcription factor (Luo et al. 2016; Wang et al. 2020). Some divergent lncRNAs have also been found to modulate the downstream functions of the protein generated by the neighboring gene. Thus, knowledge regarding the function of the protein-coding member of a divergent pair can provide insights into the potential biological role of the lncRNA partner.
Intriguingly, TBX2 has previously been linked with hearing and inner ear development. In mice, Tbx2 has been associated with otocyst patterning in inner ear morphogenesis, as mouse models in which Tbx2 was conditionally knocked out exhibit cochlear hypoplasia (Kaiser et al. 2021). Previous studies have also shown that deletions encompassing TBX2 and TBX2-AS1 are found in individuals with hearing loss, albeit in conjunction with other deleted genes (Ballif et al. 2010; Nimmakayalu et al. 2011; Schönewolf-Greulich et al. 2011). In addition, a recent study has shown that Tbx2 is required for inner hair cell and outer hair cell differentiation, demonstrating that it is a master regulator of hair cell fate (García-Añoveros et al. 2022). Therefore, we suggest that the translocation disrupting TBX2-AS1 in DGAP353 and her mother may lead to altered expression or function of TBX2, ultimately resulting in the phenotype of hearing loss.
Recurrent disruptions of the lncRNA MEF2C-AS1 in individuals with neurological phenotypes
We additionally identified two cases, DGAP191 and DGAP218, with chromosomal rearrangements that disrupt the lncRNA MEF2C-AS1. The next-generation cytogenetic nucleotide level research rearrangements are described below in single strings.
DGAP191: 46,XY,t(5;7)(q14.3;q21.3)dn.seq[GRCh38] t(5;7)(5pter→5q14.3(+)(89,411,06{3–5})::7q21.3(+)(94,378,2{48–50})→7qter;7pter→7q21.3(+)(94,378,25{3–5})::5q14.3(+)(89,411,07{0–2})→5qter)dn
DGAP218: 46,XX,inv(5)(p12q13.1)dn.seq[GRCh38] inv(5)(pter→p14.2(+)(24,272,19{3})::q14.3(-)(89,105,02{6})→p14.2(-)(24,272,189)::TATTTATATGACAAG::q14.3(+)(89,105,031)→qter)dn
In both cases, the 5q14.3 breakpoints directly disrupt the lncRNA MEF2C-AS1. In DGAP191, the 7q21.3 breakpoints additionally overlap the lncRNA ENSG00000285090, but no protein-coding genes are directly disrupted (Fig. 2). In DGAP218, MEF2C-AS1 is the only gene of any class that is directly disrupted (Fig. 3).
Fig. 2.

The lncRNA MEF2C-AS1 is disrupted in multiple individuals with neurological phenotypes, as shown here for DGAP191. (A) Chromosome diagrams depict the translocation between 5q14.3 and 7q21.3 in DGAP191. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 5q14.3 breakpoints in DGAP191. The directly disrupted lncRNA MEF2C-AS1 is highlighted in red. (C) Expanded view of the genomic region surrounding the 7q21.3 breakpoints in DGAP191. The directly disrupted lncRNA ENSG00000285090 is highlighted in red
Fig. 3.

The lncRNA MEF2C-AS1 is disrupted in multiple individuals with neurological phenotypes, as shown here for DGAP218. (A) Chromosome diagrams depict the inversion between 5p14.2 and 5q14.3 in DGAP218. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 5p14.2 breakpoints in DGAP218. (C) Expanded view of the genomic region surrounding the 5q14.3 breakpoints in DGAP218. The directly disrupted lncRNA MEF2C-AS1 is highlighted in red
We previously reported both of these individuals as part of a larger set of cases with breakpoints in 5q14.3 (Redin et al. 2017). This region is of particular interest due to 5q14.3 microdeletion syndrome, which is characterized by neurological phenotypes including intellectual disability and epilepsy (Zweier and Rauch 2012). This syndrome is now recognized to be driven by decreased expression of MEF2C (MIM: 600662), either through direct disruption of MEF2C or due to distal mutations (Zweier and Rauch 2012). Indeed, when we previously described DGAP191 and DGAP218 (Redin et al. 2017), we noted that their phenotypes were similar to individuals with direct MEF2C disruptions. Furthermore, we determined that levels of MEF2C expression were reduced by ~ 30% in lymphoblastoid cell lines from both DGAP191 and DGAP218 (Redin et al. 2017); however, no mention was made of MEF2C-AS1. Recent studies have further elucidated the functional effects of altering MEF2C or its topological organization (Mohajeri et al. 2022), but the potential role of MEF2C-AS1 remains unclear.
While there is still little known regarding the function of MEF2C-AS1, it has recently been found that overexpression of MEF2C-AS1 can increase the levels of MEF2C in human cervical cancer cell lines by serving as a microRNA sponge (Guo et al. 2022). Interestingly, MEF2C-AS1 is transcribed through multiple putative enhancers of MEF2C (D’haene et al. 2019), providing another potential mechanism for this lncRNA to regulate expression of its neighboring gene, as has previously been described for lncRNAs such as Bendr (Engreitz et al. 2016) and Uph (Anderson et al. 2016). Thus, for DGAP191 and DGAP218 we now propose that the disruption of MEF2C-AS1 leads to decreased expression of MEF2C, resulting in neurological phenotypes.
The lncRNA ENSG00000257522 is recurrently disrupted in individuals with microcephaly
Our analysis further identified two cases, DGAP245 and NIJ1, with chromosomal rearrangements that disrupt the lncRNA ENSG00000257522 (Figs. 4 and 5). These individuals exhibit shared phenotypes including microcephaly and defects of the corpus callosum (Table S1). The next-generation cytogenetic nucleotide level research rearrangements are described below in single strings.
Fig. 4.

The lncRNA ENSG00000257522 is disrupted in multiple individuals with microcephaly, as shown here for DGAP245. (A) Chromosome diagrams depict the translocation between 3p22.2 and 14q12 in DGAP245. Above, large regions containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. The region shown surrounding 14q12 is a TAD, with its borders previously defined in (Dixon et al. 2012). No TAD was defined surrounding 3p22.2, so instead the region including 1 Mb on either side of the breakpoints is displayed. Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene (B) Expanded view of the genomic region surrounding the 3p22.2 breakpoints in DGAP245. (C) Expanded view of the genomic region surrounding the 14q12 breakpoints in DGAP245. The directly disrupted lncRNAs ENSG00000258028 and ENSG00000257522 are highlighted in red
Fig. 5.

The lncRNA ENSG00000257522 is disrupted in multiple individuals with microcephaly, as shown here for NIJ1. (A) Chromosome diagrams depict the translocation between 8q21.13 and 14q12 in NIJ1. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 8q21.13 breakpoints in NIJ1. (C) Expanded view of the genomic region surrounding the 14q12 breakpoints in NIJ1. The directly disrupted lncRNAs ENSG00000258028 and ENSG00000257522 are highlighted in red
DGAP245: 46,XY,t(3;14)(p23;q13)dn.seq[GRCh38] t(3:14)(3qter→3p22.2(-)(36,927,959)::CATTTGTTCAAATTTAGTTCAAATGA::14q12(+)(29,276,117)→14qter;14pter→14q12(+)(29,276,10{8–9})::3p22.2(-)(36,927,6{49–50})→3pter)dn
NIJ1: 46,XX,t(8;14)(q21.2;q12)dn.seq[GRCh38] t(8;14)(8pter→8q21.12(+)(78,898,16{9})::14q12(+)(29,296,33{1})→14qter;14pter→14q12(+)(29,296,328)::AAAT::8q21.12(+)(78,898,172)→8qter)dn
In both cases, the 14q12 breakpoints directly disrupt the lncRNA ENSG00000257522 as well as the overlapping antisense lncRNA ENSG00000258028. In DGAP245, the 3p22.2 breakpoints additionally disrupt the protein-coding gene TRANK1 (MIM: 619316) (Fig. 4B), however this gene is not predicted to be haploinsufficient (pHaplo = 0.29) (Collins et al. 2022) and it has not been implicated in any human phenotypes by OMIM. In NIJ1, the 8q21.12 breakpoints disrupt the lncRNA MITA1 (Fig. 5B). Given that the only shared disruptions between these cases are to the lncRNAs ENSG00000257522 and ENSG00000258028, we focused on these for further analysis.
Using the GTEx database (Lonsdale et al. 2013), we found that ENSG00000258028 is not readily detected in neural tissue, and thus it is unlikely to cause the patient phenotypes. In contrast, ENSG00000257522 is primarily expressed in neural tissue (Fig. S3A), suggesting that it could play an important neurological role. Moreover, ENSG00000257522 exists within the same TAD as the protein-coding gene FOXG1 (MIM: 164874), which similarly exhibits a predominantly neural expression pattern (Fig. S3B). Disruptions in FOXG1 have been associated with a variant of Rett syndrome (MIM: 613454) (Ariani et al. 2008) as well as FOXG1 syndrome (Kortüm et al. 2011). Core phenotypes of these syndromes include microcephaly and corpus callosum defects, implicating FOXG1 dysregulation as the underlying genetic etiology in DGAP245 and NIJ1. Thus, we sought to identify potential regulatory elements that could be disrupted by the chromosomal rearrangements in these cases, and found three regions with prominent H3K4me1 chromatin modification (Fig.S3C), which is associated with enhancer activity (ENCODE Project Consortium 2012). Notably, one of these regions also exhibited H3K27Ac modification, which is also associated with enhancer activity (ENCODE Project Consortium 2012). Furthermore, these three regions each include VISTA enhancers that have been demonstrated to drive reporter expression in neural tissue in vivo in transgenic mice (hs566, hs1539, and hs1168) (Visel et al. 2007), and thus these regions exert experimentally validated enhancer activity.
Strikingly, all three of these enhancers exist within the lncRNA ENSG00000257522. While the most distal enhancer is partially disrupted by the breakpoints in DGAP245, the other two enhancers remain in the appropriate position relative to FOXG1. In NIJ1, all three of the enhancers are proximal to the breakpoints and are not separated from FOXG1. Thus, these enhancers are not directly disrupted by the chromosomal rearrangements, and instead their activity could be impaired due to the disruption of the lncRNA in which they are embedded. Indeed, transcription of lncRNAs through enhancers is a well-documented mechanism through which lncRNAs can regulate gene expression (Statello et al. 2021). Thus, we propose that the lncRNA ENSG00000257522 regulates the expression of FOXG1 through its effects on the embedded enhancers. This is consistent with previous findings that several lncRNAs function to modulate the expression of transcription factors and that this tight regulation is essential for maintaining proper functions of the transcription factors, particularly for pioneer factors such as FOXG1 (Ferrer and Dimitrova 2024).
Further supporting this, we also identified an individual with a complex de novo rearrangement that similarly disrupts ENSG00000257522. This individual, DGAP246, exhibits consistent phenotypes including microcephaly (Redin et al. 2017). The complex rearrangement in DGAP246 consists of 14 pairs of breakpoints, including eight breakpoints in 14q12. Overall, this results in the direct disruption of the lncRNA ENSG00000257522 while leaving the two most proximal enhancer elements in their correct position relative to FOXG1 (Fig. S3C). Taken together, these three cases implicate the lncRNA ENSG00000257522 in the regulation of FOXG1. Additionally, previous studies have reported several individuals with FOXG1 syndrome that harbor disruptions in this region, including a translocation in “Patient 1” that directly disrupts ENSG00000257522 (Mehrjouy et al. 2018). Similarly, a recent case report described another individual with FOXG1 syndrome whose balanced translocation had breakpoints mapping within the ENSG00000257522 lncRNA (Craig et al. 2020). Thus, we propose that disruptions of this lncRNA can cause phenotypes including microcephaly and defects of the corpus callosum, consistent with FOXG1 syndrome.
Potential regulation of KIRREL3 by its neighboring lncRNA ENSG00000255087
We previously described DGAP148 as an individual with a neurodevelopmental disorder including attention deficits and difficulty with spatial coordination (Talkowski et al. 2012b). We have recently received updated information from the referring clinical geneticist indicating that this individual is in overall good health but continues to receive medication for attention-deficit/hyperactivity disorder (ADHD). She was not able to complete regular high school, however she works as a helper in a veterinary clinic. While she lives with her father, she is autonomous for tasks of everyday living, including meals, laundry, exercise, and driving. She is also described as very sociable.
DGAP148 has a de novo translocation (Fig. 6A), and the next-generation cytogenetic nucleotide level research rearrangement is described below in a single string.
Fig. 6.

Disruption of the lncRNA ENSG00000255087 was identified in an individual with a neurodevelopmental disorder. (A) Chromosome diagrams depict the translocation between Xp11.4 and 11q24.2 in DGAP148. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the Xp11.4 breakpoints in DGAP148. (C) Expanded view of the genomic region surrounding the 11q24.2 breakpoints in DGAP148. The directly disrupted lncRNA ENSG00000255087 is highlighted in red
46,X,t(X;11)(p11.2;q23.3)dn.seq[GRCh38] t(X;11)(Xqter→Xp11.4(-)(39,882,592)::TCACTGTACAG::11q24.2(+)(127,040,509)→11qter;11pter→11q24.2(+)(127,040,509)::CTC::Xp11.4(-)(39,882,591)→Xpter)dn
No genes are directly disrupted by the Xp11.4 breakpoints (Fig. 6B), whereas the 11q24.2 breakpoints disrupt the lncRNA ENSG00000255087 (Fig. 6C).
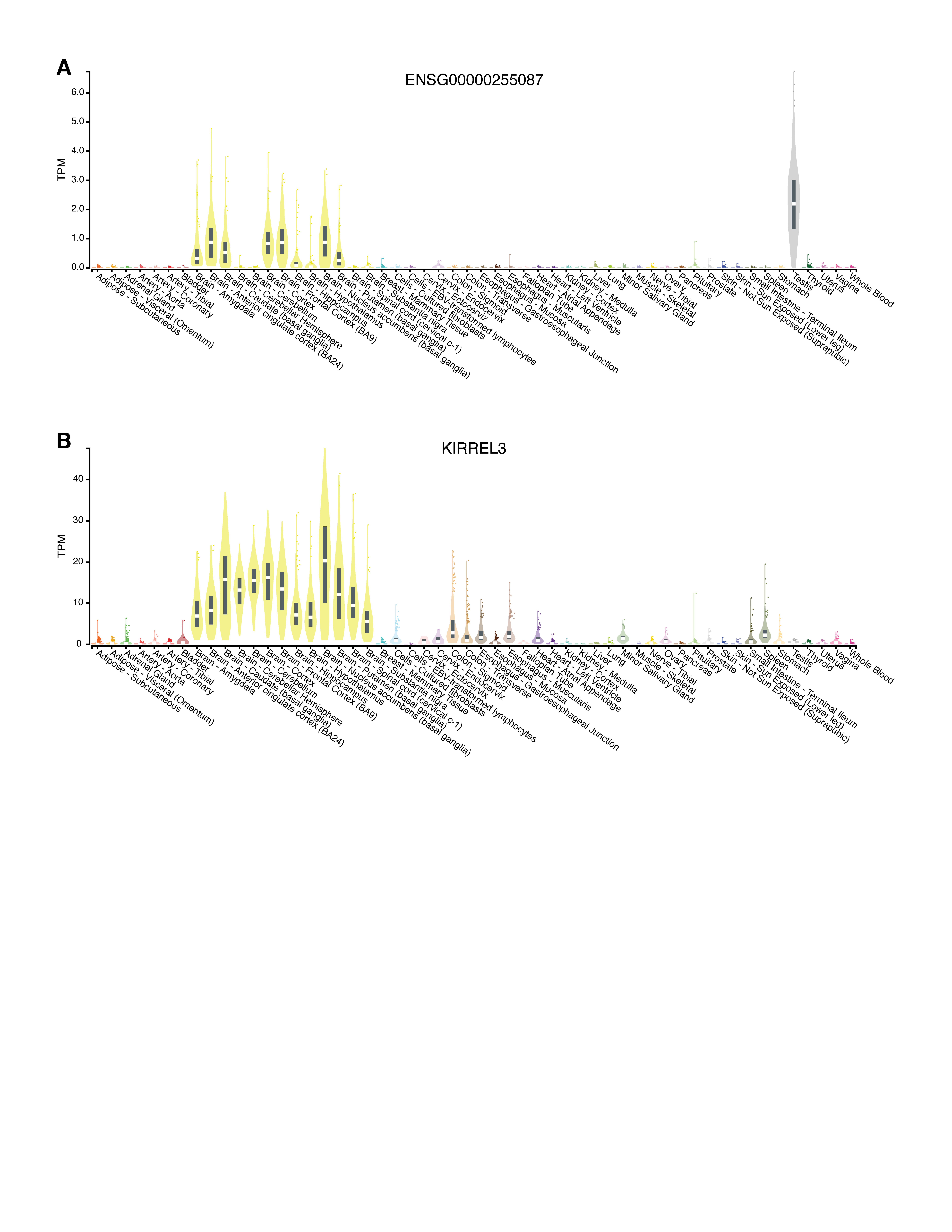
At the time of our initial report of DGAP148 (Talkowski et al. 2012b), we were unaware of the lncRNA ENSG00000255087, which still lacks any PubMed publications. However, ENSG00000255087 is approximately 20 kb upstream of the protein-coding gene KIRREL3 (MIM: 607761), which has been associated with neurodevelopmental phenotypes including attention deficits (Ciaccio et al. 2021; Querzani et al. 2023). We previously found that expression of KIRREL3 was reduced by ~ 30% in DGAP148 (Talkowski et al. 2012b), but the potential mechanism underlying this was unclear. Upon reanalyzing this case and determining that the lncRNA ENSG00000255087 is directly disrupted, we used the GTEx database (Lonsdale et al. 2013) to assess the expression of ENSG00000255087. We find that ENSG00000255087 is predominantly expressed in neural tissue (Fig. S4A), similar to the KIRREL3 expression pattern (Fig. S4B) and consistent with a potential neurodevelopmental role. Considering that the translocation in DGAP148 directly disrupts ENSG00000255087 and that DGAP148 exhibits decreased KIRREL3 levels, we suggest that the lncRNA ENSG00000255087 is a candidate for regulating expression of its neighboring gene KIRREL3. Furthermore, we propose that disruption of ENSG00000255087 can thus lead to the neurodevelopmental phenotypes described for DGAP148.
The lncRNA SOX2-OT is implicated in an individual with epilepsy and autism spectrum disorder
DGAP355 is a nonverbal individual with global developmental delay, autism spectrum disorder (ASD), seizures, and epilepsy, whose mother has a history of three miscarriages. This individual has a de novo translocation between chromosomes 3 and 9 (Fig. 7A), and the next-generation cytogenetic nucleotide level research rearrangement, as solved by liWGS, is described below in a single string.
Fig. 7.

The lncRNA SOX2-OT is implicated in an individual with epilepsy and autism spectrum disorder. (A) Chromosome diagrams depict the translocation between 3q26.33 and 9q21.13 in DGAP355. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 3q26.33 breakpoints in DGAP355. The directly disrupted lncRNA SOX2-OT is highlighted in red. (C) Expanded view of the genomic region surrounding the 9q21.13 breakpoints in DGAP355
46,XX,t(3;9)(q26.3;q21.1)dn.seq[GRCh38] t(3;9)(3pter→3q26.33(+)(181,488,756)::9q21.13(+)(74,713,321)→9qter;9pter→9q21.13(+)(74,712,100)::3q26.33(+)(181,489,591)→3qter)dn
The 3q26.33 breakpoints occur within the lncRNA SOX2-OT (MIM: 616338) (Fig. 7B), which is the only gene directly disrupted by this translocation (Fig. 7C). SOX2-OT consists of dozens of isoforms that together span a nearly 850 kb genomic region. The protein-coding gene SOX2 (MIM: 184429) exists entirely within an intron of SOX2-OT and is transcribed in the same direction. SOX2 is a transcription factor that serves as a crucial regulator of the potency and self-renewal capacity of several progenitor cell types (Arnold et al. 2011), and in particular SOX2 is known to play important roles in neural progenitor cells (Graham et al. 2003). SOX2-OT exhibits a similar expression pattern to SOX2, with both genes primarily expressed in neural tissue (Fig. S5). Recently, SOX2-OT has been found to affect SOX2 expression in varying ways in different contexts (Shahryari et al. 2015; Knauss et al. 2018; Li et al. 2020; Yin et al. 2020). Thus, we propose that the translocation in DGAP355 that disrupts the lncRNA SOX2-OT may lead to dysregulated expression of SOX2, resulting in neurodevelopmental phenotypes including ASD and epilepsy.
Knockdown of the lncRNAs TBX2-AS1 and MEF2C-AS1 results in decreased expression of their neighboring transcription factors
We sought to directly test the roles of implicated lncRNAs using the experimentally tractable human 293 cell line. We found that the lncRNAs TBX2-AS1 and MEF2C-AS1 were both expressed in this cell line, so we selected these candidates for further studies. Based on the genetic analyses described above, we proposed that these lncRNAs may regulate the expression of their neighboring transcription factors (TBX2 and MEF2C, respectively). Thus, we performed siRNA-mediated knockdown experiments to determine whether depleting these lncRNA transcripts leads to altered expression levels of their neighbors.
To knock down TBX2-AS1, we transfected 293 cultures with two separate siRNAs that had demonstrated strong efficiency in a previous study (Modi et al. 2023). After 24 h, we collected RNA from these samples and found that the siRNAs led to a 67.5% decrease in TBX2-AS1 expression (p < 0.0001) (Fig. 8A). Furthermore, this resulted in a small (8.6%) but statistically significant (p = 0.0367) decrease in the expression of TBX2 (Fig. 8B). Given the variety of TBX2-AS1 transcript isoforms (Fig. S2), it was not possible to target all of these isoforms with the siRNAs, which could contribute to the relatively small change in TBX2. We similarly used two custom siRNAs to knock down MEF2C-AS1 in 293s, which resulted in a 32.4% decrease in MEF2C-AS1 levels (p < 0.0001) (Fig. 8C). Moreover, this led to a 15.1% decrease in MEF2C (p = 0.0013) (Fig. 8D). These results demonstrate that depleting the transcripts of these lncRNAs is alone sufficient to decrease the expression of their neighboring transcription factors. Many lncRNAs have been found to “fine-tune” the expression of transcription factors, and even small changes in transcription factor levels can lead to substantial phenotypic consequences (Ferrer and Dimitrova 2024); thus, these experiments further implicate the lncRNAs TBX2-AS1 and MEF2C-AS1 in rare germline disorders.
Fig. 8.

Knockdown of the lncRNAs TBX2-AS1 and MEF2C-AS1 results in decreased expression of their neighboring genes. (A) Relative expression of TBX2-AS1 upon transfection with siRNAs targeting TBX2-AS1, as compared to negative control (Neg. Ctrl.) siRNAs. **** p < 0.0001. (B) Relative expression of TBX2 upon transfection with siRNAs targeting TBX2-AS1. * p = 0.0367. (C) Relative expression of MEF2C-AS1 upon transfection with siRNAs targeting MEF2C-AS1. **** p < 0.0001. (D) Relative expression of MEF2C upon transfection with siRNAs targeting MEF2C-AS1. ** p = 0.0013. All experiments were performed in human Lenti-X™ 293T cultures with two separate negative control and targeting siRNAs, with the results from siRNA #1 shown in blue and from siRNA #2 in red. Statistical analyses were performed using unpaired t tests
Several lncRNAs are directly disrupted in DGAP cases
Additional DGAP cases in which lncRNAs are directly disrupted are listed in Table S2. These lncRNAs warrant further consideration, particularly for cases in which no other genes are directly disrupted. Given the abundance of lncRNAs throughout the human genome, it is not rare for chromosomal rearrangements to disrupt lncRNAs, and yet this class of gene has remained largely overlooked. As updated human genome annotations continue to include new lncRNAs, it is increasingly likely to identify lncRNAs disrupted by chromosomal rearrangements. The cases described here emphasize the importance of carefully considering such disrupted lncRNAs when evaluating potential genetic etiologies underlying patient conditions.
Discussion
By virtue of their noncoding nature, it is difficult to assess the pathogenicity of lncRNA variants based on standards for protein-coding genes. As such, we propose a novel framework to implicate lncRNAs based on chromosomal rearrangements that disrupt the lncRNA. Importantly, chromosomal rearrangements can also separate non-coding regulatory elements from the genes they modulate, complicating the analysis of non-coding structural variants (D’haene and Vergult 2021). While all of the cases we discuss involve BCAs that directly disrupt lncRNAs, it remains possible that other non-coding elements such as enhancers may play a role in these cases as well. Thus, for all cases we have included data depicting chromatin conformation (Krietenstein et al. 2020), the enhancer-associated chromatin modifications H3K4me1 and H3K27Ac (ENCODE Project Consortium 2012), and VISTA-validated enhancer elements (Visel et al. 2007). We also draw particular attention to these features when they are especially relevant to certain cases. Overall, however, we selected these cases because the BCAs have breakpoints that occur within lncRNAs. Taken together, the cases presented here strongly suggest that the disruption of lncRNAs can result in rare germline disorders.
We first propose that disruption of the lncRNA TBX2-AS1 causes human disease in a Mendelian fashion in a familial case of hearing loss. This lncRNA is disrupted by a balanced chromosomal rearrangement that segregated with deafness/hard-of-hearing (DHH) from mother to daughter. Little is currently known about human TBX2-AS1 other than that it is divergent to TBX2. It is not listed currently in OMIM, and available databases including gnomAD (Karczewski et al. 2020) and DECIPHER (Firth et al. 2009) cannot be used to determine the level of constraint in the human genome pool or the tolerance to haploinsufficiency for lncRNAs because these metrics are defined specifically for protein-coding genes. TBX2, however, has been linked to hearing and inner ear development, including through the identification of deletions encompassing TBX2 and TBX2-AS1 (among other genes) that were found in individuals with hearing loss (Ballif et al. 2010; Nimmakayalu et al. 2011; Schönewolf-Greulich et al. 2011).
In the DGAP353 proband presented herein, the breakpoint did not affect TBX2 itself but interrupted TBX2-AS1. It has been shown that TBX2 maps to the edge of a TAD and is linked to TBX2-AS1 as a bi-directionally transcribed topological anchor point (tap)RNA (Amaral et al. 2018; Decaesteker et al. 2018). Expression levels of such lncRNAs have been found to be highly correlated with those of their nearest protein-coding genes, and this has also been observed between TBX2 and TBX2-AS1, suggesting that TBX2-AS1 and TBX2 may be connected on a regulatory level (Wansleben et al. 2014; Decaesteker et al. 2018). Moreover, we found that siRNA-mediated knockdown of TBX2-AS1 resulted in decreased expression of TBX2 (Fig. 8B). It is also possible that TBX2-AS1 could affect the function of the TBX2 protein, as has been demonstrated for other divergent lncRNAs (Rapicavoli et al. 2011; Vance et al. 2014; Pavlaki et al. 2018) and has recently been suggested for TBX2-AS1 (Modi et al. 2023). Thus, we propose the lncRNA TBX2-AS1 as a candidate for an association with hearing loss. To assess fully whether TBX2-AS1 disruption is the causal agent for their hearing loss, a mouse model in which Tbx2-as1 is knocked down while TBX2 remains intact is under development. Additional cases of hearing loss with deleterious TBX2-AS1 variants will be valuable to confirm the proposed association.
Furthermore, we describe recurrent disruptions of the lncRNAs MEF2C-AS1 and ENSG00000257522, and we propose that these disruptions lead to altered expression of the neighboring transcription factors MEF2C and FOXG1, respectively. Both of these lncRNAs are transcribed through VISTA-validated enhancer elements (Visel et al. 2007), suggesting a potential mechanism through which these lncRNAs could affect the regulation of their neighboring transcription factors. Regarding MEF2C, we previously reported multiple cases with BCAs that separated MEF2C from putative enhancer elements (Redin et al. 2017). This set of cases included the two cases we focus on in this manuscript, DGAP191 and DGAP218. Thus, the separation of these distal enhancers from MEF2C could certainly contribute to the phenotypes of these individuals. However, we also demonstrated that knockdown of the lncRNA MEF2C-AS1 is alone sufficient to cause decreased expression of MEF2C (Fig. 8D), suggesting that the direct disruption of this lncRNA also plays a role in the decreased expression of MEF2C that we previously reported in these individuals (Redin et al. 2017). With respect to FOXG1, we identified three VISTA-validated enhancer elements within the recurrently disrupted lncRNA ENSG00000257522. In one of the individuals (NIJ1), the rearrangement occurred distal to all three of these enhancers, leaving their positions unchanged relative to FOXG1 (Fig.S3C). While even farther distal enhancers could also play a role, the recurrent disruptions of the lncRNA ENSG00000257522 support its proposed involvement in the regulation of FOXG1.
Additionally, we suggest that the lncRNAs ENSG00000255087 and SOX2-OT may regulate their neighboring genes KIRREL3 and SOX2, respectively. These lncRNAs both exhibit expression patterns that are similar to their neighbors, with expression predominantly detected in neural tissue (Fig.S4-S5). While we previously reported decreased expression of KIRREL3 upon disruption of ENSG00000255087 in DGAP148 (Talkowski et al. 2012b), the function of this lncRNA has yet to be experimentally validated. Meanwhile, SOX2-OT has been shown to affect the expression of SOX2 in different ways depending on the context (Shahryari et al. 2015; Knauss et al. 2018; Li et al. 2020; Yin et al. 2020), which would benefit from further characterization. Thus, future experiments in human neural models including monolayer cultures and three-dimensional organoids could be highly valuable for elucidating the biological roles of these lncRNAs.
Taken together, the seven cases we describe here implicate lncRNAs in several phenotypes. We have also identified many additional disrupted lncRNAs that warrant further investigation (TableS2). All of these lncRNAs were identified due to BCAs that would disrupt the lncRNA transcripts, so it is possible that these lncRNAs function through RNA-dependent mechanisms. Some of these lncRNAs are also transcribed through enhancer elements (e.g., MEF2C-AS1 and ENSG00000257522) or other genes (e.g., TBX2-AS1 and SOX2-OT), and the act of transcription itself may be an important part of how these lncRNAs could regulate neighboring gene expression, either in tandem with or independently from RNA-based functions (Andergassen and Rinn 2022). Thus, complete characterization of lncRNA mechanisms generally requires the combination of multiple approaches (Kopp and Mendell 2018), including RNA-targeting methods such as siRNAs or CRISPR-Cas13 (Konermann et al. 2018) as well as strategies to disrupt transcription or to replace the lncRNA transcript with a reporter (Andergassen and Rinn 2022).
Here, we used siRNAs to knock down two lncRNAs, which resulted in decreased expression of the neighboring transcription factors (Fig. 8). Many transcription factors are highly sensitive to small changes in expression, and minor disruptions can lead to meaningful phenotypic consequences (Ferrer and Dimitrova 2024). Thus, these experiments suggest that the RNA transcripts of these lncRNAs can play important biological roles; however, this does not preclude the possibility that they also function through RNA-independent mechanisms. We therefore propose that all of the lncRNAs discussed here warrant further experimental dissection in future studies. Human cell cultures, particularly from cell types that are relevant to the patient phenotypes, can provide a tractable model system. For lncRNAs that have mouse homologs such as TBX2-AS1, genetic mouse models can provide valuable insights into the roles of lncRNAs throughout development. Combining multiple model systems and experimental approaches will be especially powerful for characterizing intricate lncRNA functions.
The potential connections between the lncRNAs highlighted here and patient phenotypes were uncovered due to balanced chromosomal rearrangements in these loci; as such, we suggest that such rearrangements are an untapped resource to functionally annotate lncRNAs. We propose that geneticists pay special attention to potential dysregulation of lncRNAs in patients where balanced chromosomal rearrangements do not disrupt protein-coding genes in a manner consistent with the observed phenotypes. With an increasing number of chromosomal rearrangements mapped due to inexpensive whole genome sequencing and optical genome mapping, additional lncRNAs that underly developmental diseases await characterization.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
DGAP individuals, their families and their clinicians are acknowledged for participation in DGAP. We also acknowledge participation of the Harvard Medical School-affiliated DGAP PIs and members of their laboratories. This study was supported by the National Institute of General Medical Sciences T32 GM007748 (awarded to C.C.M. and funding provided to R.E.A) and P01 GM061354 (awarded to C.C.M). C.C.M. is also supported by the NIHR Manchester Biomedical Research Centre. C.A.W. is an Investigator of the Howard Hughes Medical Institute and is supported by the National Institute of Neurological Disorders and Stroke 5R01NS035129 and by the Allen Discovery Center for Human Brain Evolution through the Paul G. Allen Frontiers Program. E.L.M. is supported by the National Institute on Aging K99 fellowship K99AG081456.
Author contributions
All authors have no competing interests as defined by Springer, or other interests that might be perceived to influence the results and/or discussion reported in this paper. All authors reviewed the manuscript.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ibrahim F. Alkuraya and Abna Ajeesh with equal contributions.
References
- Allou L, Balzano S, Magg A et al (2021) Non-coding deletions identify Maenli lncRNA as a limb-specific En1 regulator. Nature 592:93–98. 10.1038/s41586-021-03208-9 10.1038/s41586-021-03208-9 [DOI] [PubMed] [Google Scholar]
- Amaral PP, Leonardi T, Han N et al (2018) Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci. Genome Biol 19:32. 10.1186/s13059-018-1405-5 10.1186/s13059-018-1405-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D, Rinn JL (2022) From genotype to phenotype: genetics of mammalian long non-coding RNAs in vivo. Nat Rev Genet 23:229–243. 10.1038/s41576-021-00427-8 10.1038/s41576-021-00427-8 [DOI] [PubMed] [Google Scholar]
- Andersen RE, Hong SJ, Lim JJ et al (2019) The long noncoding RNA pnky is a trans-acting Regulator of cortical development in vivo. Dev Cell 49:632–642e7. 10.1016/J.DEVCEL.2019.04.032 10.1016/J.DEVCEL.2019.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GW, Deans PJM, Taylor RDT et al (2015) Characterisation of neurons derived from a cortical human neural stem cell line CTX0E16. Stem Cell Res Ther 6:149. 10.1186/s13287-015-0136-8 10.1186/s13287-015-0136-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KM, Anderson DM, McAnally JR et al (2016) Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 539:433–436. 10.1038/nature20128 10.1038/nature20128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariani F, Hayek G, Rondinella D et al (2008) FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 83:89–93. 10.1016/j.ajhg.2008.05.015 10.1016/j.ajhg.2008.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Sarkar A, Yram MA et al (2011) Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9:317–329. 10.1016/j.stem.2011.09.001 10.1016/j.stem.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BC, Theisen A, Rosenfeld JA et al (2010) Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet 86:454–461. 10.1016/j.ajhg.2010.01.038 10.1016/j.ajhg.2010.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaccio C, Leonardi E, Polli R et al (2021) A missense De Novo variant in the CASK-interactor KIRREL3 gene leading to neurodevelopmental disorder with mild cerebellar hypoplasia. Neuropediatrics 52:484–488. 10.1055/s-0041-1725964 10.1055/s-0041-1725964 [DOI] [PubMed] [Google Scholar]
- Collins RL, Glessner JT, Porcu E et al (2022) A cross-disorder dosage sensitivity map of the human genome. Cell 185:3041–3055e25. 10.1016/j.cell.2022.06.036 10.1016/j.cell.2022.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig CP, Calamaro E, Fong C-T et al (2020) Diagnosis of FOXG1 syndrome caused by recurrent balanced chromosomal rearrangements: case study and literature review. Mol Cytogenet 13:40. 10.1186/s13039-020-00506-1 10.1186/s13039-020-00506-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’haene E, Vergult S (2021) Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet Med 23:34–46. 10.1038/s41436-020-00974-1 10.1038/s41436-020-00974-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’haene E, Bar-Yaacov R, Bariah I et al (2019) A neuronal enhancer network upstream of MEF2C is compromised in patients with rett-like characteristics. Hum Mol Genet 28:818–827. 10.1093/hmg/ddy393 10.1093/hmg/ddy393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaesteker B, Denecker G, Van Neste C et al (2018) TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat Commun 9:4866. 10.1038/s41467-018-06699-9 10.1038/s41467-018-06699-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380. 10.1038/nature11082 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. 10.1038/nature11247 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM et al (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539:452–455 10.1038/nature20149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer J, Dimitrova N (2024) Transcription regulation by long non-coding RNAs: mechanisms and disease relevance. Nat Rev Mol Cell Biol. 10.1038/s41580-023-00694-9 10.1038/s41580-023-00694-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyder M, Goff LA (2016) Investigating long noncoding RNAs using animal models. J Clin Invest 126:2783–2791. 10.1172/JCI84422 10.1172/JCI84422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP et al (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet 84:524–533. 10.1016/j.ajhg.2009.03.010 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Jungreis I et al (2021) GENCODE 2021. Nucleic Acids Res 49:D916–D923. 10.1093/nar/gkaa1087 10.1093/nar/gkaa1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh VS, Riquin K, Chatron N et al (2024) Novel syndromic neurodevelopmental disorder caused by de novo deletion of CHASERR, a long noncoding RNA. 10.1101/2024.01.31.24301497. medRxiv
- García-Añoveros J, Clancy JC, Foo CZ et al (2022) Tbx2 is a master regulator of inner versus outer hair cell differentiation. Nature 605:298–303. 10.1038/s41586-022-04668-3 10.1038/s41586-022-04668-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil N, Ulitsky I (2020) Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet 21:102–117. 10.1038/s41576-019-0184-5 10.1038/s41576-019-0184-5 [DOI] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, Pevny L (2003) SOX2 functions to maintain neural progenitor identity. Neuron 39:749–765. 10.1016/s0896-6273(03)00497-5 10.1016/s0896-6273(03)00497-5 [DOI] [PubMed] [Google Scholar]
- Guo Q, Zhang L, Zhao L et al (2022) MEF2C-AS1 regulates its nearby gene MEF2C to mediate cervical cancer cell malignant phenotypes in vitro. Biochem Biophys Res Commun 632:48–54. 10.1016/j.bbrc.2022.09.091 10.1016/j.bbrc.2022.09.091 [DOI] [PubMed] [Google Scholar]
- Hanscom C, Talkowski M (2014) Design of large-insert jumping libraries for structural variant detection using Illumina sequencing. Curr Protoc Hum Genet 80. 7.22.1–7.22.9 [DOI] [PMC free article] [PubMed]
- Higgins AW, Alkuraya FS, Bosco AF et al (2008) Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am J Hum Genet 82:712–722. 10.1016/j.ajhg.2008.01.011 10.1016/j.ajhg.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser M, Wojahn I, Rudat C et al (2021) Regulation of otocyst patterning by Tbx2 and Tbx3 is required for inner ear morphogenesis in the mouse. Development 148. 10.1242/dev.195651 [DOI] [PubMed]
- Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. 10.1038/s41586-020-2308-7 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12:656–664. 10.1101/gr.229202 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS et al (2002) The human genome browser at UCSC. Genome Res 12:996–1006. 10.1101/gr.229102 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauss JL, Miao N, Kim S-N et al (2018) Long noncoding RNA Sox2ot and transcription factor YY1 co-regulate the differentiation of cortical neural progenitors by repressing Sox2. Cell Death Dis 9:799. 10.1038/s41419-018-0840-2 10.1038/s41419-018-0840-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ et al (2018) Transcriptome Engineering with RNA-Targeting type VI-D CRISPR effectors. Cell 173:665–676e14. 10.1016/j.cell.2018.02.033 10.1016/j.cell.2018.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F, Mendell JT (2018) Functional classification and experimental dissection of long noncoding RNAs. Cell 172:393–407. 10.1016/j.cell.2018.01.011 10.1016/j.cell.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortüm F, Das S, Flindt M et al (2011) The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet 48:396–406. 10.1136/jmg.2010.087528 10.1136/jmg.2010.087528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietenstein N, Abraham S, Venev SV et al (2020) Ultrastructural details of mammalian chromosome Architecture. Mol Cell 78:554–565.e7 10.1016/j.molcel.2020.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P-Y, Wang P, Gao S-G, Dong D-Y (2020) Long noncoding RNA SOX2-OT: regulations, functions, and roles on Mental illnesses, cancers, and Diabetic complications. Biomed Res Int 2020:2901589. 10.1155/2020/2901589 10.1155/2020/2901589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chen L, Giffen KP et al (2018) Cell-specific transcriptome analysis shows that adult pillar and deiters’ cells Express genes encoding Machinery for specializations of Cochlear Hair cells. Front Mol Neurosci 11:356. 10.3389/fnmol.2018.00356 10.3389/fnmol.2018.00356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M et al (2013) The genotype-tissue expression (GTEx) project. Nat Genet 45:580–585. 10.1038/ng.2653 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther C, Mehrjouy MM, Collins RL et al (2022) Balanced chromosomal rearrangements offer insights into coding and noncoding genomic features associated with developmental disorders. medRxiv 2022.02.15.22270795 10.1101/2022.02.15.22270795
- Luo S, Lu JY, Liu L et al (2016) Divergent lncRNAs regulate Gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell 18:637–652. 10.1016/j.stem.2016.01.024 10.1016/j.stem.2016.01.024 [DOI] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V et al (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161:1012–1025. 10.1016/j.cell.2015.04.004 10.1016/j.cell.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J, Amaral P (2022) The Human Genome. RNA, the epicenter of genetic information. CRC, Boca Raton, pp 125–135 [PubMed] [Google Scholar]
- Mattick JS, Amaral PP, Carninci P et al (2023) Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol 24:430–447. 10.1038/s41580-022-00566-8 10.1038/s41580-022-00566-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrjouy MM, Fonseca ACS, Ehmke N et al (2018) Regulatory variants of FOXG1 in the context of its topological domain organisation. Eur J Hum Genet 26:186–196. 10.1038/s41431-017-0011-4 10.1038/s41431-017-0011-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi A, Lopez G, Conkrite KL et al (2023) Integrative Genomic Analyses Identify LncRNA Regulatory Networks across Pediatric Leukemias and solid tumors. Cancer Res 83:3462–3477. 10.1158/0008-5472.CAN-22-3186 10.1158/0008-5472.CAN-22-3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajeri K, Yadav R, D’haene E et al (2022) Transcriptional and functional consequences of alterations to MEF2C and its topological organization in neuronal models. Am J Hum Genet 109:2049–2067. 10.1016/j.ajhg.2022.09.015 10.1016/j.ajhg.2022.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmakayalu M, Major H, Sheffield V et al (2011) Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am J Med Genet A 155A:418–423. 10.1002/ajmg.a.33827 10.1002/ajmg.a.33827 [DOI] [PubMed] [Google Scholar]
- Ordulu Z, Wong KE, Currall BB et al (2014) Describing sequencing results of structural chromosome rearrangements with a suggested next-generation cytogenetic nomenclature. Am J Hum Genet 94:695–709. 10.1016/j.ajhg.2014.03.020 10.1016/j.ajhg.2014.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvis J, Gottfried B, Kancherla J et al (2021) gEAR: gene expression analysis resource portal for community-driven, multi-omic data exploration. Nat Methods 18:843–844. 10.1038/s41592-021-01200-9 10.1038/s41592-021-01200-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais LS, Snow H, Weisburd B et al (2022) Seqr: a web-based analysis and collaboration tool for rare disease genomics. Hum Mutat 43:698–707. 10.1002/humu.24366 10.1002/humu.24366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlaki I, Alammari F, Sun B et al (2018) The long non-coding RNA paupar promotes KAP1‐dependent chromatin changes and regulates olfactory bulb neurogenesis. EMBO J 37:e98219. 10.15252/embj.201798219 10.15252/embj.201798219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querzani A, Sirchia F, Rustioni G et al (2023) KIRREL3-related disorders: a case report confirming the radiological features and expanding the clinical spectrum to a less severe phenotype. Ital J Pediatr 49:99. 10.1186/s13052-023-01488-7 10.1186/s13052-023-01488-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. 10.1093/bioinformatics/btq033 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapicavoli NA, Poth EM, Zhu H, Blackshaw S (2011) The long noncoding RNA Six3OS acts in trans to regulate retinal development by modulating Six3 activity. Neural Dev 6:32. 10.1186/1749-8104-6-32 10.1186/1749-8104-6-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL et al (2017) The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet 49:36–45. 10.1038/ng.3720 10.1038/ng.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Goff LA, Lodato S et al (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2:e01749. 10.7554/eLife.01749 10.7554/eLife.01749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönewolf-Greulich B, Ronan A, Ravn K et al (2011) Two new cases with microdeletion of 17q23.2 suggest presence of a candidate gene for sensorineural hearing loss within this region. Am J Med Genet A 155A:2964–2969. 10.1002/ajmg.a.34302 10.1002/ajmg.a.34302 [DOI] [PubMed] [Google Scholar]
- Shahryari A, Jazi MS, Samaei NM, Mowla SJ (2015) Long non-coding RNA SOX2OT: expression signature, splicing patterns, and emerging roles in pluripotency and tumorigenesis. Front Genet 6:196. 10.3389/fgene.2015.00196 10.3389/fgene.2015.00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamou M, Ng S-Y, Brand H et al (2020) A balanced translocation in Kallmann Syndrome implicates a long noncoding RNA, RMST, as a GnRH neuronal Regulator. J Clin Endocrinol Metab 105:e231–e244. 10.1210/clinem/dgz011 10.1210/clinem/dgz011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statello L, Guo C-J, Chen L-L, Huarte M (2021) Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22:96–118. 10.1038/s41580-020-00315-9 10.1038/s41580-020-00315-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Ernst C, Heilbut A et al (2011) Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet 88:469–481. 10.1016/j.ajhg.2011.03.013 10.1016/j.ajhg.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Maussion G, Crapper L et al (2012a) Disruption of a large intergenic noncoding RNA in subjects with neurodevelopmental disabilities. Am J Hum Genet 91:1128–1134. 10.1016/j.ajhg.2012.10.016 10.1016/j.ajhg.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I et al (2012b) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149:525–537. 10.1016/j.cell.2012.03.028 10.1016/j.cell.2012.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushakov K, Koffler-Brill T, Rom A et al (2017) Genome-wide identification and expression profiling of long non-coding RNAs in auditory and vestibular systems. Sci Rep 7:8637. 10.1038/s41598-017-08320-3 10.1038/s41598-017-08320-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KW, Sansom SN, Lee S et al (2014) The long non-coding RNA paupar regulates the expression of both local and distal genes. EMBO J 33:296–311. 10.1002/embj.201386225 10.1002/embj.201386225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA (2007) VISTA enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res 35:D88–92. 10.1093/nar/gkl822 10.1093/nar/gkl822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volders P-J, Anckaert J, Verheggen K et al (2019) LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res 47:D135–D139. 10.1093/nar/gky1031 10.1093/nar/gky1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen S, Li W et al (2020) Associating divergent lncRNAs with target genes by integrating genome sequence, gene expression and chromatin accessibility data. NAR Genom Bioinform 2:lqaa019. 10.1093/nargab/lqaa019 10.1093/nargab/lqaa019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansleben S, Peres J, Hare S et al (2014) T-box transcription factors in cancer biology. Biochim Biophys Acta 1846:380–391. 10.1016/j.bbcan.2014.08.004 10.1016/j.bbcan.2014.08.004 [DOI] [PubMed] [Google Scholar]
- Yin J, Shen Y, Si Y et al (2020) Knockdown of long non-coding RNA SOX2OT downregulates SOX2 to improve hippocampal neurogenesis and cognitive function in a mouse model of sepsis-associated encephalopathy. J Neuroinflammation 17:320. 10.1186/s12974-020-01970-7 10.1186/s12974-020-01970-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Hong H, Sun X et al (2016) MicroRNA-10b regulates epithelial-mesenchymal transition by modulating KLF4/Notch1/E-cadherin in cisplatin-resistant nasopharyngeal carcinoma cells. Am J Cancer Res 6:141–156 [PMC free article] [PubMed] [Google Scholar]
- Zweier M, Rauch A (2012) The MEF2C-Related and 5q14.3q15 Microdeletion Syndrome. Mol Syndromol 2:164–170. 10.1159/000337496 10.1159/000337496 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.