

Abstract
Background and purpose
Two novel enzyme replacement therapies (ERTs), studied in phase 3 trials in late‐onset Pompe patients, reached marketing authorization by the European Medicines Agency in 2022 and 2023. The European Pompe Consortium (EPOC) updates and extends the scope of the 2017 recommendations for starting, switching and stopping ERT.
Methods
The European Pompe Consortium consists of 25 neuromuscular and metabolic experts from eight European countries. This update was performed after an in‐person meeting, three rounds of discussion and voting to provide a consensus recommendation.
Results
The patient should be symptomatic, that is, should have skeletal muscle weakness or respiratory muscle involvement. Muscle magnetic resonance imaging findings showing substantial fat replacement can support the decision to start in a patient‐by‐patient scenario. Limited evidence supports switching ERT if there is no indication that skeletal muscle and/or respiratory function have stabilized or improved during standard ERT of 12 months or after severe infusion‐associated reactions. Switching of ERT should be discussed on a patient‐by‐patient shared‐decision basis. If there are severe, unmanageable infusion‐associated reactions and no stabilization in skeletal muscle function during the first 2 years after starting or switching treatment, stopping ERT should be considered. After stopping ERT for inefficacy, restarting ERT can be considered. Six‐monthly European Pompe Consortium muscle function assessments are recommended.
Conclusions
The triple‐S criteria on ERT start, switch and stop include muscle magnetic resonance imaging as a supportive finding and the potential option of home infusion therapy. Six‐monthly long‐term monitoring of muscle function is highly recommended to cover insights into the patient's trajectory under ERT.
Keywords: enzyme replacement therapy, EPOC, late‐onset, Pompe disease, therapy switch
INTRODUCTION
Pompe disease, or glycogen storage disorder type II, is an inherited autosomal recessive multisystemic metabolic disorder caused by a deficiency of the lysosomal enzyme acid alpha‐glucosidase, resulting from disease‐associated sequence variations in the alpha‐glucosidase gene (GAA gene) of which more than 500 have been identified (www.pompe.variantdatabase.nl) to date. Pompe disease presents a spectrum of phenotypes, ranging from a rapidly fatal phenotype in infants (classic infantile Pompe disease) to more slowly progressive forms in older children and adults (late‐onset Pompe disease), in whom skeletal and respiratory muscles are mainly affected [1].
The European Pompe Consortium (EPOC), founded in September 2014 [2, 3], published its first recommendation on the start and stop criteria for enzyme replacement therapy (ERT) for late‐onset Pompe disease in this journal in 2017 [1]. Since then, two novel ERTs for late‐onset Pompe disease have been clinically evaluated in phase 3 trials and the first long‐term studies. Both novel ERTs reached marketing authorization by the European Medicines Agency and in several parts of the world in 2022 and 2023, respectively [4, 5, 6, 7, 8].
The 2017 EPOC recommendation was focused on the only licensed and worldwide available recombinant enzyme alglucosidase alfa [9]. It did not include switching criteria between products, as several new drugs were at the early stages of clinical investigation. At present/now, data from the recent phase 3 trials (COMET and PROPEL) [4, 7] and the first long‐term data [5, 6] are available, but it is still necessary to obtain real‐world, long‐term data for a final shared decision on switching therapy. A further relevant element for the patients living with Pompe disease for the newly licensed therapies is the option of home infusion treatment. Therefore, EPOC decided to update its 2017 recommendation, expand the scope to switching patients, and reconsider start, switch and stop criteria (triple‐S criteria) for enzyme replacement therapy of adult patients with Pompe disease.
METHODS
A consensus technique was adopted using the collective opinion of all EPOC panel members. Our structured method of developing consensus among the 25 expert panel members with long‐standing knowledge of diagnosing and treating Pompe disease from eight European countries started with an in‐person meeting in Rotterdam, the Netherlands, on 26–27 June 2023. This in‐person meeting was followed by three structured rounds of email editing and voting of all 25 panellists. A 100% consensus was reached on all criteria regarding diagnosis, starting, switching and stopping enzyme replacement therapy and monitoring in the real‐world setting in late‐onset Pompe disease. Our consensus was solely based on all publicly available clinical phase 2 and phase 3 ERT trial data and their extensions.
RESULTS
The new triple‐S criteria
Treatment should be started in patients who meet all the following criteria
Diagnostics
The patient should have a confirmed diagnosis of Pompe disease based on disease‐associated sequence variants on both alleles of the GAA gene and reduced enzyme activity demonstrated by glucosidase alpha enzyme activity testing. A positive dried blood spot as a first‐tier screening test should always be followed by molecular genetic analyses and/or enzyme activity testing in fibroblasts or other tissues to confirm the diagnosis. This is required to rule out a false positive dried blood spot test. Enzyme activity testing in patient‐derived fibroblasts is the most reliable way to measure residual GAA enzyme activity [10]. If the GAA enzyme activity is decreased under the disease threshold, all analytical efforts should be made to find both pathogenic GAA gene variants. This is required in cases where disease‐associated GAA variants cannot be identified using standard diagnostic analysis. Such variants may be present in introns, the promoter or mosaic form. For referencing known pathogenic variants of the GAA gene, see www.pompe.variantdatabase.nl.
Clinically symptomatic and supportive paraclinical sign
The patient should be symptomatic, that is, should have skeletal muscle weakness on clinical examination and a respiratory muscle involvement that can be demonstrated in pulmonary function tests (preferably performed in sitting and supine position). As a supportive paraclinical sign, it is recommended to perform muscle magnetic resonance imaging (MRI) in patients without obvious clinical signs. Muscle MRI findings showing substantial fat replacement of skeletal muscle tissue can support the decision to start treatment in a patient‐by‐patient scenario. A fat fraction of 20% or more in at least two muscles is considered as abnormal. This level can be detected visually without requiring software analysis [11].
Disease severity
The patient should have residual skeletal and respiratory muscle function, which is functionally relevant and clinically important for the patient to maintain or improve. The minimal residual functional state, where it is not recommended to start ERT, encompasses bedridden Medical Research Council grade <2; severe generalized muscle atrophy measured by clinical examination, ultrasound, or muscle MRI; maximally compromised or minimal residual spontaneous lung function with constant high CO2 levels, or complete dependence on invasive ventilation.
Advanced comorbidity
The patient should not have another life‐threatening illness that is in an advanced stage, where treatment to sustain life is inappropriate.
Commitment of the patient and clinician
The patient and clinician should commit to the recommended treatment schedule and regular monitoring. The patient will comply with regular ERT infusions and clinical assessments at least every 6–12 months, based on the center's practice, to evaluate responses to treatment.
Home infusion therapy
Starting home infusion therapy after the initial 3–12 month treatment and monitoring in the in‐patient clinic setting is possible. Careful monitoring during infusions is also warranted after transitioning to home infusion therapy.
Switching treatments can be considered for the following reasons
There is no indication that skeletal muscle and/or respiratory function have stabilized or improved during a minimum treatment period of 12 months.
The patient suffers from severe infusion‐associated reactions that cannot be adequately managed.
There is currently insufficient evidence to inform whether switching ERT treatment to one of the novel enzymes is beneficial. Switching should, therefore, be discussed on a patient‐by‐patient basis.
If a patient is switched to a new treatment, careful 3‐month monitoring with the advised EPOC assessments and a treatment period of at least 12 months is recommended.
Stopping treatment should be considered for any one of the following reasons
Infusion‐associated reactions
The patient suffers from severe infusion‐associated reactions that cannot be adequately managed by tapering the infusion rate and/or premedication with antihistamines or corticosteroids after a switch to another available treatment has been considered or attempted.
High‐neutralizing antidrug antibody titers
High‐neutralizing antidrug antibody titers that have resulted in a diminution of the clinical response to ERT can sometimes be detected. It is therefore recommended to investigate high‐neutralizing antidrug antibody titers in patients with rapidly declining motor or respiratory function. As the cut‐off definition of high sustained antibody levels is assay‐dependent, a threshold of 1:30,000–1:56,000 is suggested [12].
Patient's wish
The patient wishes to stop ERT.
No treatment effect
If there is no indication that skeletal muscle function and/or respiratory function have stabilized or improved during the first 2 years after starting or switching treatment, it should be stopped. Therefore, regular 6‐monthly to yearly assessments using the minimal dataset (Figure 1) are strongly recommended.
FIGURE 1.
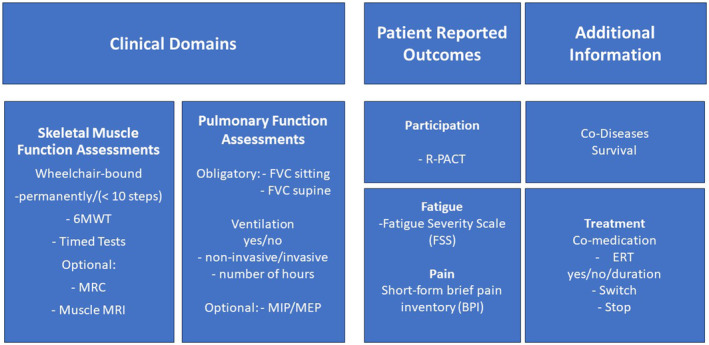
EPOC assessments in late‐onset Pompe disease. Domains affected in adults living with Pompe disease and minimal set of outcome measures. 6MWT, 6‐min walk test; BPI, Brief Pain Inventory; ERT, enzyme replacement therapy; FSS, Fatigue Severity Scale; FVC, Forced Vital Capacity; MIP/MEP, maximum inspiratory/expiratory pressure; MRC, the Medical Research Council grading scale (ranging from 0 to 5); R‐PACT, Rasch‐built Pompe‐specific activity scale; Timed tests, walking 10 m, climbing four steps, standing up from supine position and standing up from a chair.
If, after stopping treatment, the disease deteriorates faster than during treatment, restarting ERT can be considered.
End of life
The patient has another life‐threatening illness that is in an advanced stage, where treatment to sustain life is inappropriate. An early discussion and implementation of advanced care planning is highly recommended.
Lactation and pregnancy
Still, based on limited evidence, continuation of ERT can be considered during pregnancy and lactation.
DISCUSSION
This updated recommendation of ERT for persons living with late‐onset Pompe disease clarified the diagnostic criteria, included the value of muscle MRI as a supportive paraclinical finding for a treatment decision [11], and added home infusion therapy [13]. Switching treatment can be considered if there is no indication that skeletal muscle and/or respiratory function have stabilized or improved during a minimum standard ERT treatment period of 12 months. Another reason for switching is if the patient suffers from severe infusion‐associated reactions that cannot be adequately managed. However, for both issues, more scientific evidence must be gathered to determine whether switching is favorable and/or which enzyme treatment is preferable. Possible switching should, therefore, be discussed on a patient‐by‐patient shared‐decision basis. If a patient is switched to a new ERT treatment, careful 6‐monthly monitoring with the advised EPOC assessments (see Figure 1) and a treatment period of at least 12 months is recommended. Finally, stopping ERT treatment should be considered for patients suffering from severe infusion‐associated reactions that cannot be adequately managed, for whom switching does not seem appropriate, or who have already switched. It is also recommended to investigate high‐neutralizing antidrug antibody titers. The cut‐off definition of high antibody levels is proposed as 1:30,000–1:56,000 (assay dependent) [12]. Concurrence of the presence of high titer neutralizing antibodies and a diminution of the effect of ERT can be a reason to stop ERT. Finally, if there is no stabilization or improvement in skeletal muscle and/or respiratory function during the first 2 years after starting or switching treatment, it is recommended to stop the ERT [14, 15]. However, if, after stopping treatment, the disease deteriorates faster than during treatment, restarting ERT can still be considered. Therefore, regular 6‐monthly assessments using the EPOC assessments are strongly recommended.
In conclusion, the new EPOC triple‐S criteria recommendation tries to provide physicians and persons living with Pompe disease guidance on starting, switching and stopping ERT for late‐onset Pompe disease.
AUTHOR CONTRIBUTIONS
Benedikt Schoser: Conceptualization; investigation; funding acquisition; writing—original draft; methodology; validation; visualization; writing—review and editing; software; formal analysis; project administration; resources; supervision; data curation. Nadine A. M. E. van der Beek: Conceptualization; investigation; writing—original draft; methodology; validation; writing—review and editing; formal analysis; supervision. Broomfield Alexander: Investigation; writing—original draft; validation; writing—review and editing; formal analysis. Esther Brusse: Investigation; writing—original draft; validation; writing—review and editing. Jordi Diaz‐Manera: Conceptualization; investigation; writing—original draft; methodology; validation; writing—review and editing; supervision; formal analysis. Andreas Hahn: Conceptualization; writing—original draft; validation; writing—review and editing; supervision; formal analysis. Thomas Hundsberger: Conceptualization; writing—original draft; validation; writing—review and editing; supervision; formal analysis. Cornelia Kornblum: Conceptualization; investigation; writing—original draft; validation; supervision. Michelle Kruijshaar: Conceptualization; investigation; writing—original draft; validation; writing—review and editing; supervision. Pascal Laforet: Conceptualization; writing—original draft; validation; writing—review and editing; supervision; formal analysis. Eugen Mengel: Investigation; writing—original draft; validation; writing—review and editing; formal analysis. Tiziana Mongini: Conceptualization; writing—original draft; writing—review and editing; validation; formal analysis. David Orlikowski: Investigation; writing—original draft; validation; writing—review and editing; formal analysis. Parenti Giancarlo: Investigation; writing—original draft; writing—review and editing; validation; formal analysis. W. W. M. Pim Pijnappel: Conceptualization; investigation; writing—original draft; validation; supervision; writing—review and editing; formal analysis. Roberts Mark: Conceptualization; investigation; writing—original draft; validation; writing—review and editing; supervision; formal analysis. Scherer Thomas: Investigation; writing—original draft; validation; writing—review and editing; formal analysis. Antonio Toscano: Conceptualization; investigation; writing—original draft; validation; writing—review and editing; formal analysis; supervision. John Vissing: Supervision; formal analysis; validation; writing—review and editing; conceptualization; investigation; writing—original draft. Johanna M. P. van den Hout: Investigation; writing—original draft; validation; writing—review and editing; formal analysis. Pieter A. van Doorn: Conceptualization; investigation; writing—original draft; methodology; validation; writing—review and editing; formal analysis; supervision; data curation. Stephan Wenninger: Investigation; writing—original draft; validation; writing—review and editing; formal analysis. Ans T. van der Ploeg: Conceptualization; investigation; writing—original draft; writing—review and editing; visualization; validation; methodology; formal analysis; software; project administration; data curation; supervision; resources.
CONFLICT OF INTEREST STATEMENT
This article and its recommendations were developed independently of the pharmaceutical industry. Esther Brusse has no relevant conflicts of interest. Jordi Diaz‐Manera has received research grants from Spark, Sarepta, Sanofi and Boehringer Ingelheim and payments for consultancy or as speaker from Amicus Therapeutics, Astellas, Sanofi, Sarepta, Lupin and Boehringer Ingelheim and payments for consultancy or as a speaker from Amicus Therapeutics, Astellas, Sanofi, Sarepta, Lupin and Spark. He declares no stocks or shares. Andreas Hahn has received honoraria from Sanofi‐Aventis and Amicus as an advisory board member and for lectures. He declares no stocks or shares. Pascal Laforet has received research grants from Amicus Therapeutics Inc. and Sanofi, AMDA Foundation, Association Francophone des Glycogénoses, Vaincre les Maladies Lysosomales (VML) and speaker's honoraria from Amicus Therapeutics Inc. and Sanofi. He has participated as a scientific advisor for Amicus Therapeutics Inc., Sanofi and Spark Therapeutics. He declares no stocks or shares. Cornelia Kornblum has received speaker's honoraria and/or travel funding from Chiesi, Amicus Therapeutics, Fulcrum Therapeutics, Novartis, Sanofi and Santhera. She acknowledges financial support as an advisory board member and/or primary investigator for Amicus Therapeutics, Fulcrum Therapeutics, Hormosan, Reneo Pharmaceuticals, Roche Pharma AG, Sanofi and Stealth BioTherapeutics. She declares no stocks or shares. Eugen Mengel received research grants, speakers' fees and consulting honoraria from Sanofi, Orphazyme, Takeda, Cyclo Therapeutics, Idorsia, JCR, Denali, Amicus and Avrobio. Tiziana Mongini has received honoraria from Sanofi‐Aventis and Amicus as an advisory board member and for lectures. She received financial support for educational activities from Sanofi, Biogen and Roche. She declares no stocks or shares. David Orlikowski was an investigator for Amicus Therapeutics Inc. and Spark Therapeutics. He has participated as a scientific advisor for Spark Therapeutics. He declares no stocks or shares. W. W. M. Pim Pijnappel is an advisor of LentiCure B.V. Thomas Scherer has received an investigator‐initiated research grant from Sanofi and speaker's honoraria from Amicus Therapeutics and Sanofi. He has participated as a scientific advisor for Amicus Therapeutics and Sanofi. He declares no stocks or shares. Benedikt Schoser has received unrestricted research grants from Amicus, Astellas, Roche diagnostics, Marigold Foundation, AMDA Foundation and speaker's honoraria from Amicus Therapeutics Inc., Alexion, Kedrion and Sanofi. He has participated as a scientific advisor for Amicus, Argenx, Astellas, Bayer, Pepgen, Sanofi, Spark and Taysha. He declares no stocks or shares. Nadine A. M. E. van der Beek has participated in advisory board meetings for Sanofi and Bayer and has received speaker honoraria from Sanofi under agreements between Erasmus MC University Medical Center and the relevant industry. Pieter A. van Doorn has received unrestricted research grants from the Prinses Beatrix Spierfonds and Sanquin Blood Supply. He participated as a scientific advisor/steering committee member for Annexon, Argenx, Hansa, Octapharma, Roche, and Sanofi Genzyme, all for indications other than Pompe disease. All payments were made to Erasmus MC University Medical Center. He has no relevant conflicts of interest. Johanna van den Hout has participated in advisory board meetings for Sanofi and has received speaker honoraria from Sanofi and Amicus under agreements between Erasmus MC University Medical Center and the relevant industry. John Vissing has acted as a consultant for Amicus Therapeutics, Argenx BVBA, Arvinas, Atamyo Therapeutics, Biogen, Dyne Therapeutics, Fulcrum Therapeutics, Horizon Therapeutics, Lupin, ML Biopharma, Regeneron, Roche, Sanofi Genzyme, Sarepta Therapeutics and UCB Biopharma SPRL. He has received research grants, travel support, and/or speaker fees from Alexion, AstraZeneca Rare Disease, Edgewise Therapeutics, Fulcrum Therapeutics, Horizon Therapeutics, Sanofi Genzyme and UCB Biopharma SPRL. Ans T. van der Ploeg has acted as advisor and participated in clinical trials, registries and/or investigational projects for Amicus Therapeutics, Alexion, AskBio, Astellas, Bayer, Biogen, Biomarin, Chiesi, Denali, Dynacure, Pharming, Sarepta, Sanofi Genzyme, Ultragenix under agreements between the industry and Erasmus MC University Medical Center. All other authors declare no financial or other conflicts of interest.
ACKNOWLEDGMENTS
The authors thank all persons living with Pompe disease and their caregivers for their ongoing commitment to clinical trials. Open Access funding enabled and organized by Projekt DEAL.
Schoser B, van der Beek NAME, Broomfield A, et al. Start, switch and stop (triple‐S) criteria for enzyme replacement therapy of late‐onset Pompe disease: European Pompe Consortium recommendation update 2024. Eur J Neurol. 2024;31:e16383. doi: 10.1111/ene.16383
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10‐year experience. Eur J Neurol. 2017;24(768):e31. [DOI] [PubMed] [Google Scholar]
- 2. Schoser B, Laforêt P, Kruijshaar ME, et al. European POmpe Consortium (EPOC). Minutes of the European POmpe Consortium (EPOC) Meeting March 27 to 28, 2015, Munich, Germany. Acta Myol. 2015;34(2–3):141‐143. [PMC free article] [PubMed] [Google Scholar]
- 3. Schoser B, Laforêt P, Kruijshaar ME, et al. 208th ENMC International Workshop: formation of a European Network to develop a European data sharing model and treatment guidelines for Pompe disease Naarden, The Netherlands, 26–28 September 2014. Neuromuscul Disord. 2015;25(8):674‐678. [DOI] [PubMed] [Google Scholar]
- 4. Diaz‐Manera J, Kishnani PS, Kushlaf H, et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late‐onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021;20(12):1012‐1026. Erratum in: Lancet Neurol. 2022;21(4):e4. [DOI] [PubMed] [Google Scholar]
- 5. Dimachkie MM, Barohn RJ, Byrne B, et al. Long‐term safety and efficacy of avalglucosidase alfa in patients with late‐onset Pompe disease. Neurology. 2022;99(5):e536‐e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kishnani PS, Diaz‐Manera J, Toscano A, et al. Efficacy and safety of avalglucosidase alfa in patients with late‐onset Pompe disease after 97 weeks: a phase 3 randomized clinical trial. JAMA Neurol. 2023;80(6):558‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schoser B, Roberts M, Byrne BJ, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late‐onset Pompe disease (PROPEL): an international, randomised, double‐blind, parallel‐group, phase 3 trial. Lancet Neurol. 2021;20(12):1027‐1037. Erratum in: Lancet Neurol. 2023;22(10):e11. [DOI] [PubMed] [Google Scholar]
- 8. Schoser B, Laforet P. Therapeutic thoroughfares for adults living with Pompe disease. Curr Opin Neurol. 2022;35(5):645‐650. [DOI] [PubMed] [Google Scholar]
- 9. van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late‐onset Pompe's disease. N Engl J Med. 2010;362(15):1396‐1406. [DOI] [PubMed] [Google Scholar]
- 10. Niño MY, Wijgerde M, de Faria DOS, et al. Enzymatic diagnosis of Pompe disease: lessons from 28 years of experience. Eur J Hum Genet. 2021;29(3):434‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nuñez‐Peralta C, Alonso‐Pérez J, Llauger J, et al. Follow‐up of late‐onset Pompe disease patients with muscle magnetic resonance imaging reveals increase in fat replacement in skeletal muscles. J Cachexia Sarcopenia Muscle. 2020;11(4):1032‐1046. Erratum in: J Cachexia Sarcopenia Muscle. 2021;12(5):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ditters IAM, van Kooten HA, van der Beek NAME, van der Ploeg AT, Huidekoper HH, van den Hout JMP. Are anti‐rhGAA antibodies a determinant of treatment outcome in adults with late‐onset Pompe disease? A systematic review. Biomolecules. 2023;13(9):1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ditters IAM, van Kooten HA, van der Beek NAME, et al. Home‐based infusion of alglucosidase alfa can safely be implemented in adults with late‐onset Pompe disease: lessons learned from 18,380 infusions. BioDrugs. 2023;37(5):685‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hundsberger T, Schoser B, Leupold D, Rösler KM, Putora PM. Comparison of recent pivotal recommendations for the diagnosis and treatment of late‐onset Pompe disease using diagnostic nodes—the Pompe disease burden scale. J Neurol. 2019;266(8):2010‐2017. [DOI] [PubMed] [Google Scholar]
- 15. van Kooten HA, Harlaar L, van der Beek NAME, et al. Discontinuation of enzyme replacement therapy in adults with Pompe disease: evaluating the European POmpe Consortium stop criteria. Neuromuscul Disord. 2020;30(1):59‐66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.