

Abstract
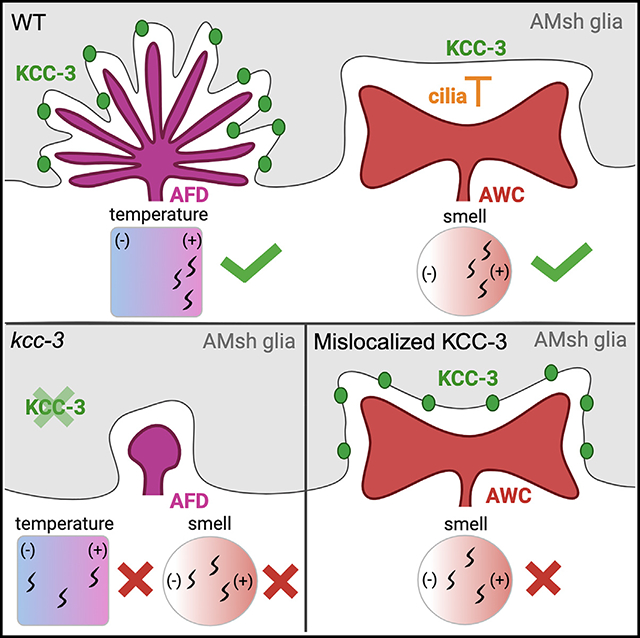
Glia interact with multiple neurons, but it is unclear whether their interactions with each neuron are different. Our interrogation at single-cell resolution reveals that a single glial cell exhibits specificity in its interactions with different contacting neurons. Briefly, C. elegans amphid sheath (AMsh) glia apical-like domains contact 12 neuron-endings. At these ad-neuronal membranes, AMsh glia localize the K/Cl transporter KCC-3 to a microdomain exclusively around the thermosensory AFD neuron to regulate its properties. Glial KCC-3 is transported to ad-neuronal regions, where distal cilia of non-AFD glia-associated chemosensory neurons constrain it to a microdomain at AFD-contacting glial membranes. Aberrant KCC-3 localization impacts both thermosensory (AFD) and chemosensory (non-AFD) neuron properties. Thus, neurons can interact non-synaptically through a shared glial cell by regulating microdomain localization of its cues. As AMsh and glia across species compartmentalize multiple cues like KCC-3, we posit that this may be a broadly conserved glial mechanism that modulates information processing across multimodal circuits.
In brief
Ray et al. show that a single glia creates distinct molecular microdomains of regulatory cues around individual neurons. One cue, the cation co-transporter KCC-3, is constrained to the AFD thermosensory neuron by its C-terminal region and cilia of non-AFD neurons. Aberrant KCC-3 localization affects both AFD and non-AFD neuron function.
Graphical Abstract
INTRODUCTION
Bilaterian nervous systems have two major cell types: glia and neurons. Glia physically contact neurons to regulate neuron shape, function, and animal behavior.1,2 Across both central and peripheral nervous systems (CNS and PNS, respectively), each glia contacts multiple neurons.3 This raises a fundamental logic question: does one glia modulate all of its contacting neurons similarly or through distinct mechanisms? If differently, then are different glia-neuron interactions interdependent or independently controlled within each glia? Decoding this logic of specificity in glia-neuron interactions molecularly is critical to understand the organizational principle of the nervous system.
This is relevant for both CNS and PNS/sensory biology. In the CNS, each astrocyte glia can interact with an estimated more than 1,000 neurons4 and can regulate excitatory and inhibitory neurons differently through distinct molecular regulators.3,5,6 Similarly, in sense organs like the retina, different glia contact multiple neurons, including rod and cone photoreceptors (retinal pigment epithelia, or RPE, glia-like cells) as well as interneurons (Müller glia).7 Likewise, in the tongue, type I support cells contact different taste receptor cell types.8 Functional studies suggest that there is likely specificity in these glia-neuron interactions. In murine astrocytes, intracellular calcium (Ca2+ dynamics vary with different neuron circuit activities, suggesting that glia functionally differentiate between different neuron inputs.9 In C. elegans, ablation of a single glial cell has differential effects on different sensory neurons.10 Finally, in adaptive experience-dependent myelin remodeling, mouse oligodendrocytes exhibit bias toward specific neuron classes.11 However, across all instances, molecular specificity of individual glia-neuron interactions remains to be explored.
While it is molecularly enigmatic how glia interact with different contacting neurons, there is evidence that they interact non-uniformly with different contacting cell types through molecular asymmetry of contacting membranes. An emergent theme across glia is that polarity markers associated with canonical apical domains of polarized epithelia cells (e.g., PIP2, apical ßH-spectrin) localize to neuron-proximal (ad-neuronal) contacting membranes and basolateral domain polarity markers to membranes that appose the basal lamina/extracellular matrix (ECM) or endothelia (ab-neuronal).12 This is explicitly demonstrated for PNS Schwann cells and retinal RPE glia-like cells in mammals and ensheathing cells of the Drosophila CNS.13 Further, while mammalian CNS astrocytes do not have explicit apical-basal polarity, they also localize basolateral domainassociated AQP4/Aquaporins to endothelium-contacting endfeet and Ezrin, mGluR3/5, and GLT-1 to perisynaptic astrocytic processes at neuron contact sites.14 Whether ad-neuronal membranes contacting different neurons are further asymmetric remains to be determined.
To molecularly interrogate specificity in glia-neuron interactions at single-cell resolution, we exploited a single glia cell in C. elegans, the amphid sheath (AMsh) glia, as a powerful and genetically tractable experimental platform.15 This glia resides in the animal’s major sense organ at the anterior nose tip and offers four advantages for rapidly probing glia-neuron interactions definitively at single-cell and -gene resolution.15–17 One, each bilateral AMsh glia associates with 12 sensory neurons, specifically at their neuron-receptive endings (NREs), where they receive sensory input.18,19 Two, the identity, cell shape, and function of each NRE are invariant across animals. This allows reproducible inquiry into each of their individually distinct properties. Three, each NRE transduces a distinct sensory modality and animal behavior, each of which can be individually quantified in vivo. Finally, the animal’s optical transparency allows facile cell biology and functional studies in behaving animal nervous systems.
Of the 12 NREs contacting AMsh glia, eight extend ciliated NREs through an autotypic channel formed by AMsh glia (channel NREs), three are ramified cilia (wing NREs), and one is microvillar (AFD-NRE) (Figure 1A).15,19,20 Of these, AFD is the animal’s primary thermosensor, with the sensory transduction machinery located at the AFD-NRE embedded within AMsh glia.21,22 We have shown previously that AMsh glia regulate AFD-NRE shape and function dynamically throughout life through multiple regulatory modules.23–25 One glial regulatory cue identified was the potassium chloride co-transporter, KCC-3, whose loss causes defects in AFD-NRE shape, function, and associated animal thermosensory behavior.24,26
Figure 1. KCC-3 localizes specifically around AFD-NRE.
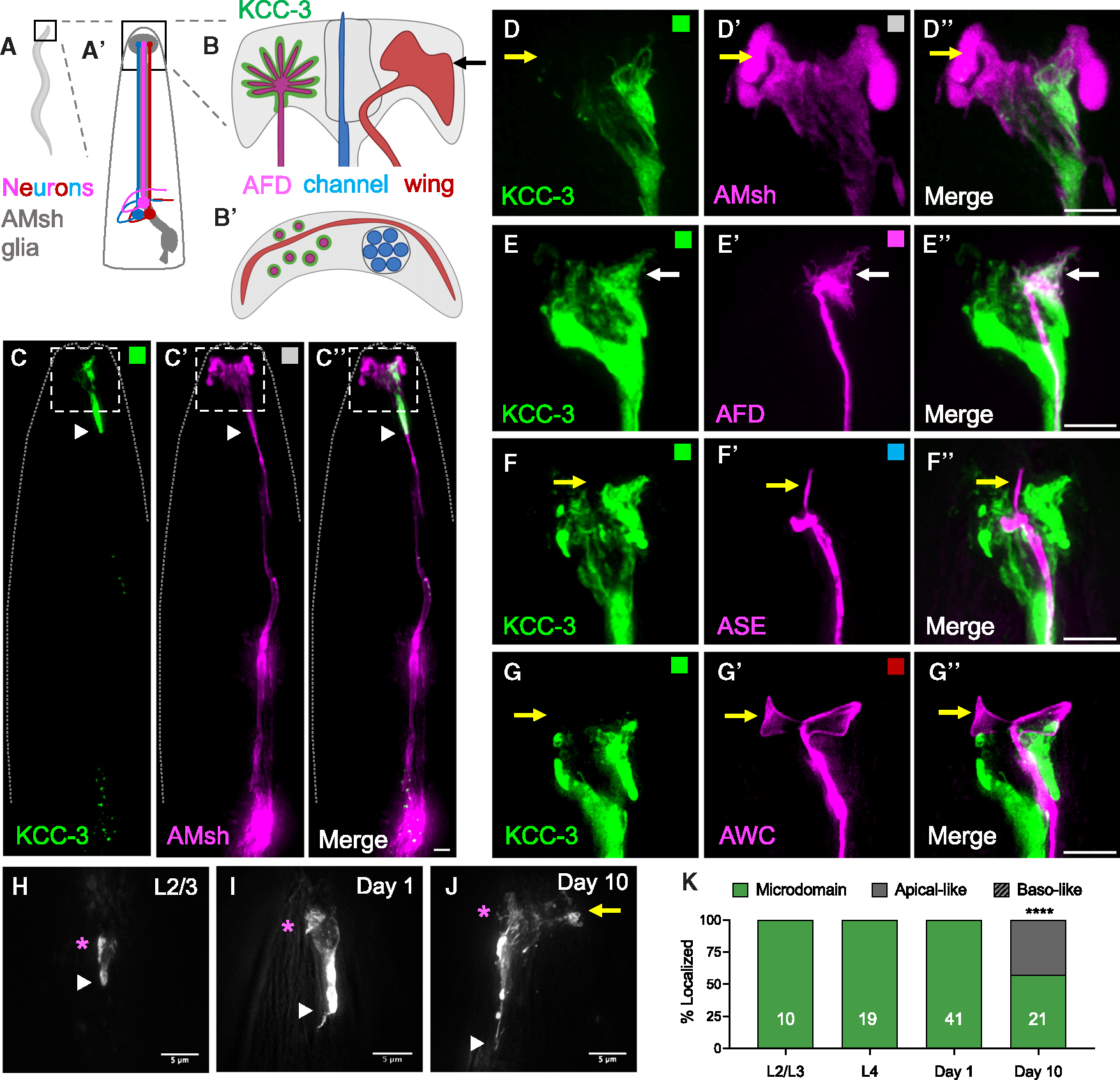
(A and A′) Schematic of a whole C. elegans (A) with a magnified region of the head (A′) showing a schematic of AMsh glia (gray) and three contacting neurons (magenta, red, and blue).
(B and B′) Schematic of AMsh glial contact sites with microvilli of AFD (magenta), channel (blue), and embedded wing (red) neurons as side profiles (B) and a top-down orthogonal view (B′) at the plane denoted by the arrow line in (B). Only one of the bilateral glia-neuron pairs is shown.
(C–C″) Fluorescent images of AMsh KCC-3 (C, green), AMsh glia (C′, magenta), and merge (C″, 8/8 animals). A dashed line indicates the outline of the animal.
(D–G″) Magnification the region in the white dotted boxes in C–C″.
(D–D″) Fluorescence images of AMsh KCC-3 (D, green), AMsh glia (D′, magenta), and merge (D″) showing restricted microdomain localization of KCC-3 (38/38 animals). Yellow arrows denote the region of AMsh glia that lacks KCC-3.
(E–E″) Fluorescence images of AMsh KCC-3 (E, green), an AFD neuron (E′, magenta), and merge (E″) showing KCC-3 localization to AFD-NREs (white arrows, 30/30 animals).
(F–F″) Fluorescence images of AMsh KCC-3 (F, green), an ASE neuron (F′, magenta), and merge (F″) showing lack of KCC-3 localization to ASE-NREs (yellow arrows, 15/15 animals).
(G–G″) Fluorescence images of AMsh KCC-3 (F, green), AWC neuron (F′, magenta), and merge (F″) showing lack of KCC-3 localization to AWC-NREs (yellow arrows, 20/20 animals).
(C–G′) Colored boxes correspond to schematic colors in (B).
(H–J) Fluorescence images of KCC-3 expressed under the AMsh-specific promoter (PF53F4.13) in L2/L3 larvae (H), day 1 adults (I), and day 10 adults (J). Magenta asterisks denote regions of AFD enrichment. A yellow arrow denotes expansion of KCC-3 beyond the microdomain. White arrowheads denote the GAB.
(K) Quantification of KCC-3 localization with age. n = number of animals on graph. ****p < 0.0001 compared with day 1 adults (Fisher’s exact test). Scale bars: 5 μm. All imaging data were gathered across multiple days/biological replicates. See also Figure S1.
Exploiting KCC-3 as a molecular tool, we report here that AMsh glia regulate associated NREs differently and uncover the underlying molecular mechanism. Briefly, glial KCC-3 affects the shape of only AFD-NRE of the 12 AMsh glia-associated NREs. In accord, KCC-3 localizes to a microdomain within the glia’s ad-neuronal membrane, specifically at AFD-NRE contacts, revealing that these membranes are molecularly asymmetric. KCC-3 N-terminal sequences localize it to neuron-proximal glial regions. Refinement to a microdomain apposing AFD-NRE requires KCC-3 C-terminal regions and cilia NREs of at least two non-AFD neurons, the olfactory AWC and gustatory ASE neurons. Strikingly, glial KCC-3 localization to AFD-NRE also impacts AWC neuron activity but not shape. Thus, compartmentalization of a glial cue to the thermosensory neuron AFD informs the fidelity of the animal’s sensory perception across modalities. This also reveals a mechanism by which neurons can coordinate non-synaptically by modulating the molecular property of a shared glial cell. Finally, we report at least three molecular microdomains in AMsh glia whose maintenance is largely independent. Glia across species express and localize cues like KCC-3 and interact with different neuron types. We therefore posit that regulating glial cue localization may be a general mechanism by which glia functionally enable cross-circuit information processing.
RESULTS
AMsh glia localize the K/Cl transporter KCC-3 to a microdomain around only AFD-NRE
We have reported previously that the AMsh glia uses the KCC-3 cation chloride co-transporter KCC-3 to regulate AFD-NRE shape and animal thermosensory behavior.24 Intriguingly, a translational kcc-3:GFP reporter does not localize uniformly on AMsh glial membranes (Figure S1A).24 To examine this further, we engineered a double-reporter transgenic animal, where KCC-3 driven under the AMsh-expressing PF53F4.13 promoter was tagged with mScarlet, and the AMsh glia was labeled with cytosolic CFP. Analyses of this strain using wide-field and 3D-structured illumination superresolution microscopy confirmed that KCC-3 localizes to the anterior region of AMsh glia, where the glia contacts associated NREs, including the AFD-NRE (Figures 1A–1C″). Further, within the anterior region, KCC-3 does not localize uniformly but is constrained to a subdomain (Figures 1B–B′ and 1D–D″). We call this subcellular KCC-3 localization a “glial molecular microdomain.”
The pattern of KCC-3 enrichment was architecturally reminiscent of the shape of AFD-NRE (Figure 1D). We therefore generated animal strains that co-labeled fluorescent reporter-tagged KCC-3 with reporters marking individual AMsh glia-associated neurons. We found that KCC-3 localizes to glial membranes proximal to AFD-NRE but not to any other glia-associated NREs (Figures 1E–1G″, S1B, and S1C″). Thus, AMsh glia differentiate between NREs of individual neurons and restrict KCC-3 to a single neuron contact site.
In addition to the microdomain, we noted that KCC-3 localization terminates sharply in the proximal glial process ~50μm away from the animal nose tip (Figure 1C, white arrowhead). For reasons explained below, we refer to this boundary as the “glial ad-neuronal boundary” (GAB).
Examination of kcc-3:GFP through development showed that KCC-3 anterior localization and GAB restriction are apparent in developing 3-fold stage embryos, shortly after glia are born (Figures S1D and S1E). Longitudinal examination of KCC-3 under the AMsh glia-specific promoter showed that the microdomain was apparent from the earliest stage examined (L2 larva onward, once PF53F4.13 turns on), is maintained through animal life, and deteriorates with animal age (Figures 1H–1K and S1F). We noted that, while microdomain localization was compromised with age, the KCC-3 GAB boundary maintained its integrity throughout (Figure 1J).
KCC-3 localizes to an apical-like ad-neuronal microdomain in AMsh glia
KCC-3 localization suggested that it was a facile molecular tool to probe specificity in glia-neuron interactions. To do so, we sought to first define the molecular identity of the KCC-3-bearing glial membrane. Glial membranes are proposed to exhibit epithelium-like apical-basal polarity, with apical domains facing neurons.27 To confirm this, we re-engineered previously reported SAX-7/L1CAM-based polarity markers in AMsh glia under PF53F4.13.27,28 Full-length SAX-7 has been reported to localize basolaterally (BasoRed) and truncated SAX-7 to apical membranes (ApiGreen).27 Our AMsh-expressed ApiGreen and BasoRed constructs phenocopied this, with BasoRed localizing to the cell body, process, and membranes where AMsh glia contacts epithelia, while the ApiGreen reporter localized to anterior regions of glia (Figures 2A–2C). Co-labeling both markers confirmed that the marked membranes were distinct (Figures S2A–A″; Video S1). Interestingly, the ApiGreen, but not BasoRed, reporter terminated at the GAB (Figures 2B and 2C). To confirm that these localization patterns are not artifacts of overexpressing the cell adhesion molecule SAX-7/L1CAM, we also generated an independent apical membrane reporter in AMsh glia, PH-PLCδ:GFP.29 PH-PLCδ:GFP phenocopied ApiGreen in labeling the ad-neuronal membrane and terminating at the GAB (Figure 2D). Thus, AMsh glia have molecularly asymmetrical membranes at neuron-contacting (ad-neuronal) versus epithelium-contacting (ab-neuronal) sites.
Figure 2. KCC-3 localizes to a glial apical microdomain in an age-dependent manner.
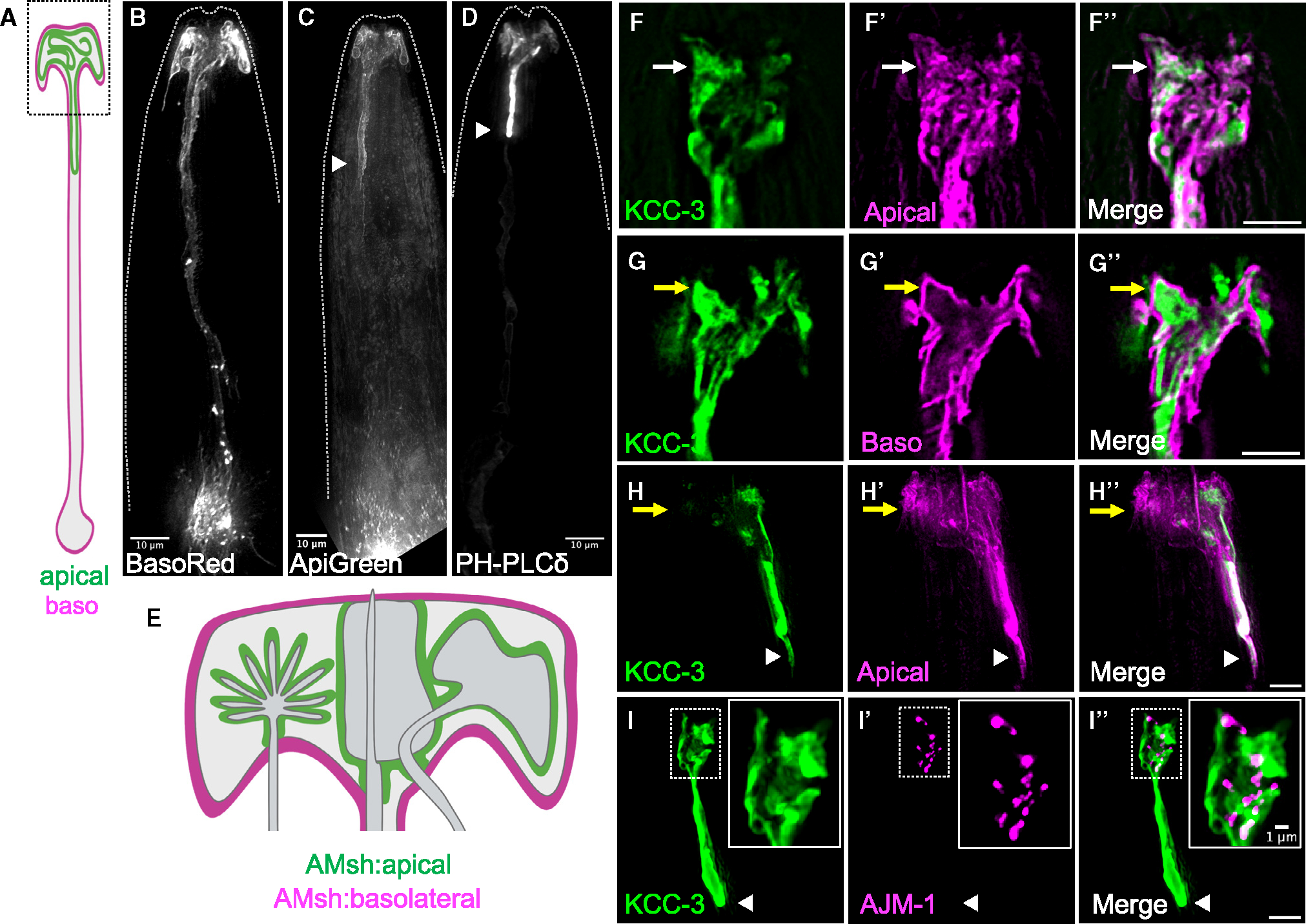
(A) Schematic of AMsh glia with apical and basolateral (baso) membranes marked.
(B–D) Fluorescence images of AMsh membranes tagged with BasoRed (B, 37/37 animals), ApiGreen (C, 24/24 animals), and a PH-PLC apical marker (D, 15/15 animals). Dashed lines indicate the outline of the animal.
(E) Magnification of the AMsh glia in the black dotted box in (A).
(F–F″) Fluorescence images of AMsh KCC-3 (F, green), truncated SAX-7 labeling AMsh apical membranes (F′, magenta), and merge (F″). White arrows denote overlay between KCC-3 and the AMsh apical marker (5/5 animals).
(G–G″) Fluorescence images of Pkcc-3:KCC-3:GFP (G, green), full-length SAX-7 labeling AMsh basolateral membranes (G′, magenta), and merge (G″). KCC-3 present outside of AMsh basolateral membranes is derived from other glia. Yellow arrows denote lack of co-localization between KCC-3 and AMsh basolateral membranes (4/4 animals).
(H–H″) Fluorescence images of AMsh KCC-3 (H, green), truncated SAX-7 labeling AMsh apical membranes (H′, magenta), and merge (H″). Yellow arrows denote the region of AMsh apical membranes that lacks KCC-3. KCC-3 and the AMsh apical marker co-localize at the GAB (3/3 animals).
(I–I″) Fluorescence images of AMsh KCC-3 (I, green), the tight junction protein AJM-1 (I′, magenta), and merge (I″). Note the lack of AJM-1 protein at the GAB (7/7 animals). Insets: magnification of the white dotted box.
White arrowheads denote the GAB. Scale bars: 5 μm unless otherwise noted. All imaging data were collected over multiple days/biological replicates. See also Figure S2.
Termination of KCC-3:mScarlet, ApiGreen, and PH-PLCδ:GFP at the GAB hinted that the KCC-3 microdomain may be within apical-like ad-neuronal glial membranes. To confirm this, we double-labeled AMsh glial polarity reporters with fluorescently labeled KCC-3. Indeed, KCC-3 co-localized with apical but not basolateral membrane markers (Figures 2E–2G″). Further, the GAB of KCC-3 and AMsh apical markers overlaid perfectly (Figures 2H–H″, white arrowhead). Finally, KCC-3 expression was restricted to a subset of membranes labeled with the apical marker, consistent with it being excluded from non-AFD contact sites (Figure 2H–H″, yellow arrows). Taken together, these data show that AMsh glia localize KCC-3 to a microdomain within apical-like ad-neuronal membranes at AFD-NRE contact sites. Corroborating this, unc-23/hBAG2 mutant animals, which, as we independently showed, have stretched apical-like glial membranes,28 also had expanded KCC-3 microdomains (Figure S2B).
Finally, while both KCC-3 and apical markers terminated at the GAB, three lines of evidence suggest that the GAB is not a canonical apical:basal boundary. One, epithelial apical-basal domain boundaries are decorated by tight junctions,30 but AMsh GAB was not limited by the junctional marker AJM-1/AJM31 (Figures 2I–I″). Instead, AJM-1 marks AMsh glia-NRE contacts anterior to the GAB, at the site where sensory neuron dendrites infiltrate glial sheath. Tight junctions assessed through electron micrographs, too, showed localization anterior to the GAB.18,19,27,28 Two, DLG-1/DiscsLarge, which recruits AJM-1 to epithelial tight junctions,32,33 was not expressed in adult AMsh glia, indicating that it was dispensable for GAB maintenance (Figures S2C–S2C″). Finally, orthogonal slices of co-labeled apical and basal polarity markers in vivo reveal a tube-within-tube configuration of the apical membrane within the basal membrane at the GAB (Figures S2D–S2F; Video S2). Thus, while AMsh glial membrane asymmetry is labeled by epithelial polarity markers, features like GAB are maintained by distinct molecular mechanisms.
C. elegans AMsh glia make distinct microdomains at different neuron contact sites
AMsh glia associate with 12 NREs. 8 of these traverse a channel made by the glia. Glial proteins that localize to and/or regulate this channel include DAF-6/Patched, CHE-14/SSD/Dispatched, VAP-1/CRISP2, and LIT-1/Nemo-like kinase.20,34 We examined KCC-3 localization in the context of these proteins. Co-labeling of VAP-1:sfGFP and KCC-3:mScarlet revealed distinct, non-overlapping localization within AMsh glia (Figures 3A–B″). Further, neither KCC-3 nor VAP-1 marked glial membranes around AWC-NRE (Figures 1G–G″ and 3C–C″). Thus, AMsh glia make at least three distinct molecular microdomains around different neuron contact-sites: around channel NREs (VAP-1 positive, KCC-3 negative), AFD-NRE (VAP-1 negative, KCC-3 positive), and AWC-NRE (VAP-1 negative, KCC-3 negative). This also indicated that microdomain localization is a property of AMsh glia, not of KCC-3 protein.
Figure 3. Microdomains as a general feature of glia.
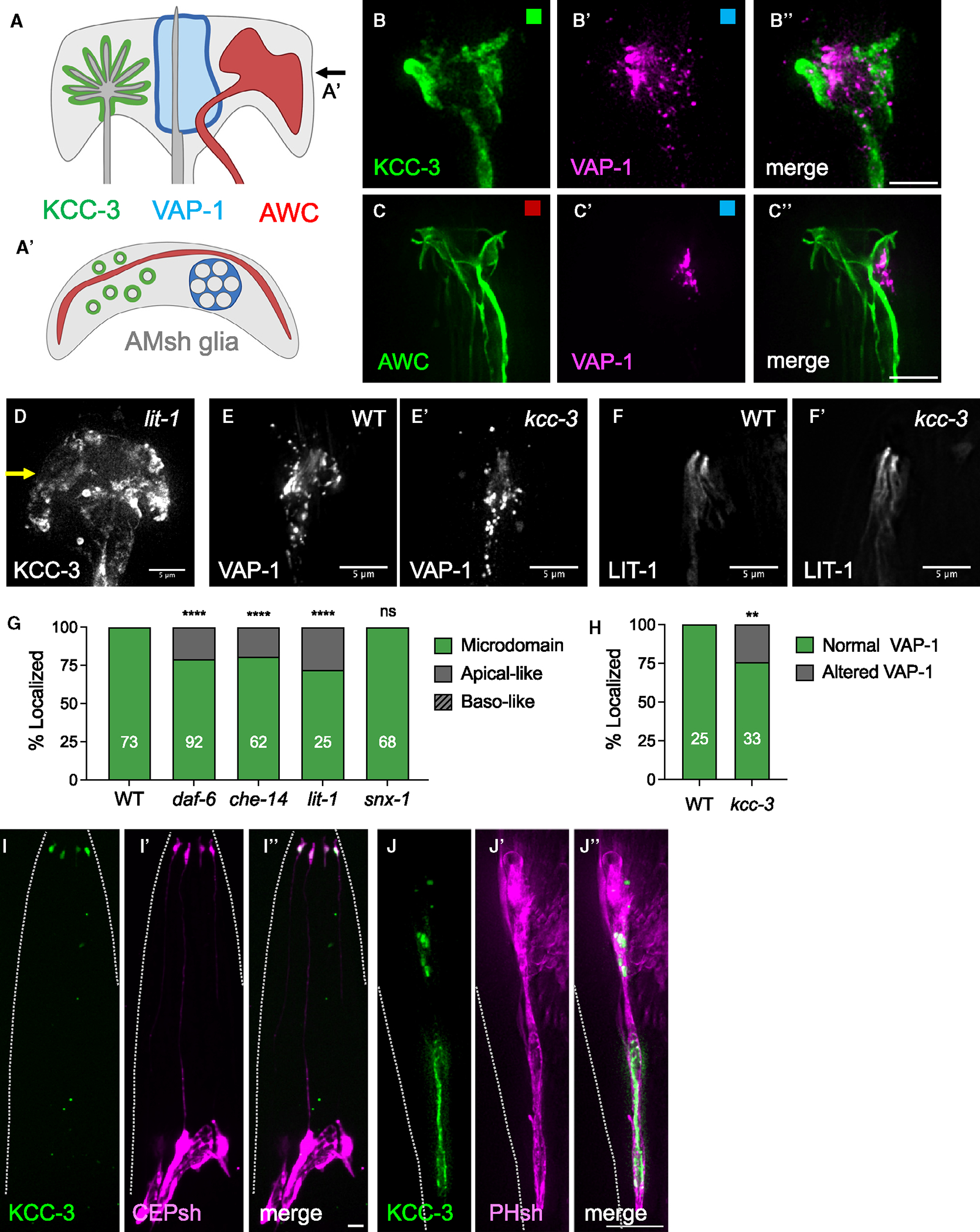
(A and A′) Schematic of multiple microdomains in AMsh glia as a side profile (A) and top-down orthogonal view (A′) at the plane denoted by the arrow in (A). The KCC-3 microdomain is shown in green, the VAP-1 channel microdomain in blue, and the AWC wing neuron microdomain in red.
(B–B″) Fluorescence images of KCC-3 (B, green), VAP-1 (B′, magenta), and merge (B″) denoting separate microdomains (5/5 animals).
(C–C″) Fluorescence images of AWC (C, green), VAP-1 (C′, magenta), and merge (C″) denoting separate microdomains (4/4 animals).
(B–C′) Colored boxes in correspond to schematic colors in (A).
(D) Fluorescence image of KCC-3 in lit-1 mutants. A yellow arrow indicates expansion of KCC-3 beyond the microdomain.
(E and E′) Fluorescence images of VAP-1 in wild-type (E) and kcc-3 mutant (E′) backgrounds.
(F and F′) Fluorescence images of LIT-1–1 in wild-type (F) and kcc-3 mutant (F′) backgrounds.
(G) Quantification of KCC-3 localization in daf-6, che-14, lit-1, and snx-1 mutants. Data represent 2–4 biological replicates.
(H) Quantification of VAP-1 localization in the wild type (WT) and kcc-3 mutant. Data represent 3 biological replicates.
(I–I″) Fluorescence images of KCC-3 expressed under the CEPsh-specific Phlh-17 promoter (I, green), CEPsh glia (I′, magenta), and overlay (I″) denoting apical KCC-3 (5/5 animals).
(J–J″) Fluorescence images of KCC-3 expressed under the AMsh/PHsh-specific PF53F14.13 promoter (J, green), PHsh glia (J′, magenta), and overlay (J″) denoting apical KCC-3 (7/7 animals).
Dashed lines in (I–J″) indicate the outline of the animal. **p < 0.01, ****p < 0.0001. Fisher’s exact test. Scale bars: 5 μm. n = number of animals on graph. All imaging data were collected over multiple days/biological replicates.
Given this segregation, we asked whether KCC-3 and channel microdomain cues affect each other’s localization. For this, we examined whether mutations in one gene affected localization of the other. We found that KCC-3 localization was partially affected in animals mutant for daf-6, che-14, and lit-1 (Figures 3D and 3G). However, while SNX-1/Retromer antagonizes DAF-6 to regulate channel architecture,35 we found it to be dispensable for KCC-3 localization (Figure 3G). In corollary, kcc-3 mutants marginally impacted VAP-1 localization and did not affect LIT-1 localization (Figures 3E–F′ and 3H). Taken together, we concluded that AMsh glia maintain molecular microdomains largely independently, with some crosstalk.
Next, we probed whether restricted localization of cues was a feature of only AMsh or glia generally. KCC-3 expresses in many sheath glia.36,37 Cell-specific expression of KCC-3:mScarlet in two other glia, CEPsh (in the animal head) and PHsh (in the animal tail) glia, showed restricted localization of KCC-3 to anterior process ends around neuron contact sites (Figures 3I and 3J″), similar to AMsh. Thus, localization of KCC-3 to anterior microdomains is a general property across C. elegans glia.
The glial KCC-3 microdomain does not require canonical KCC regulators
We tested a role of known regulators of the cation chloride transporter (CCC) family of proteins in KCC-3 localization. WNK and the GCK/Ste20 kinases SPAK/PASK and OSR are major regulators of N/KCC transporters across species.38–42 The C. elegans genome encodes a single WNK ortholog (WNK-1).38 Neither the loss-of-function wnk-1(tm487) mutation nor wnk-1 RNAi altered AMsh glial KCC-3 microdomain localization (Figures S3A and S3B). Consistent with this, in silico analyses showed that C. elegans KCC-3 lacks conserved WNK or GCK kinase phosphorylation sites or binding motifs (Figures S3C–S3E). Loss of ARGK-1/creatine kinase, which co-localizes with KCC-3 in cultured cells,43,44 also did not disrupt KCC-3 localization (Figure S3A). Thus, microdomain localization of KCC-3 in AMsh glia is independent of previously described CCC regulators.
Different protein regions regulate KCC-3 targeting to distinct membrane regions
To define protein domains in KCC-3 that guide localization to a microdomain, we exploited the interspersed sequence similarity of KCC-3 coding regions with that of related KCC-1 and KCC-2 (Figure 4A). In silico analysis showed that dissimilarity between KCC-1/KCC-2 and KCC-3 coding sequences was largely confined to the N-terminal intracellular domain (amino acids [aa] 1–90), the large extracellular loop (LEL) between TM5 and TM6 (aa 265–397), and an 81-aa region of the C terminus (aa 915–996) (Figure 4B). Further, we found that, in striking contrast to KCC-3, fluorescently tagged KCC-1 and KCC-2 localized to basolateral ab-neuronal membranes in AMsh glia (Figures 4C–4F). Thus, sequences dissimilar between KCC-1/KCC-2 and KCC-3 drive both ad-neuronal and microdomain localization of KCC-3.
Figure 4. Glial KCC-3 localizes in a two-step process through two protein regions.
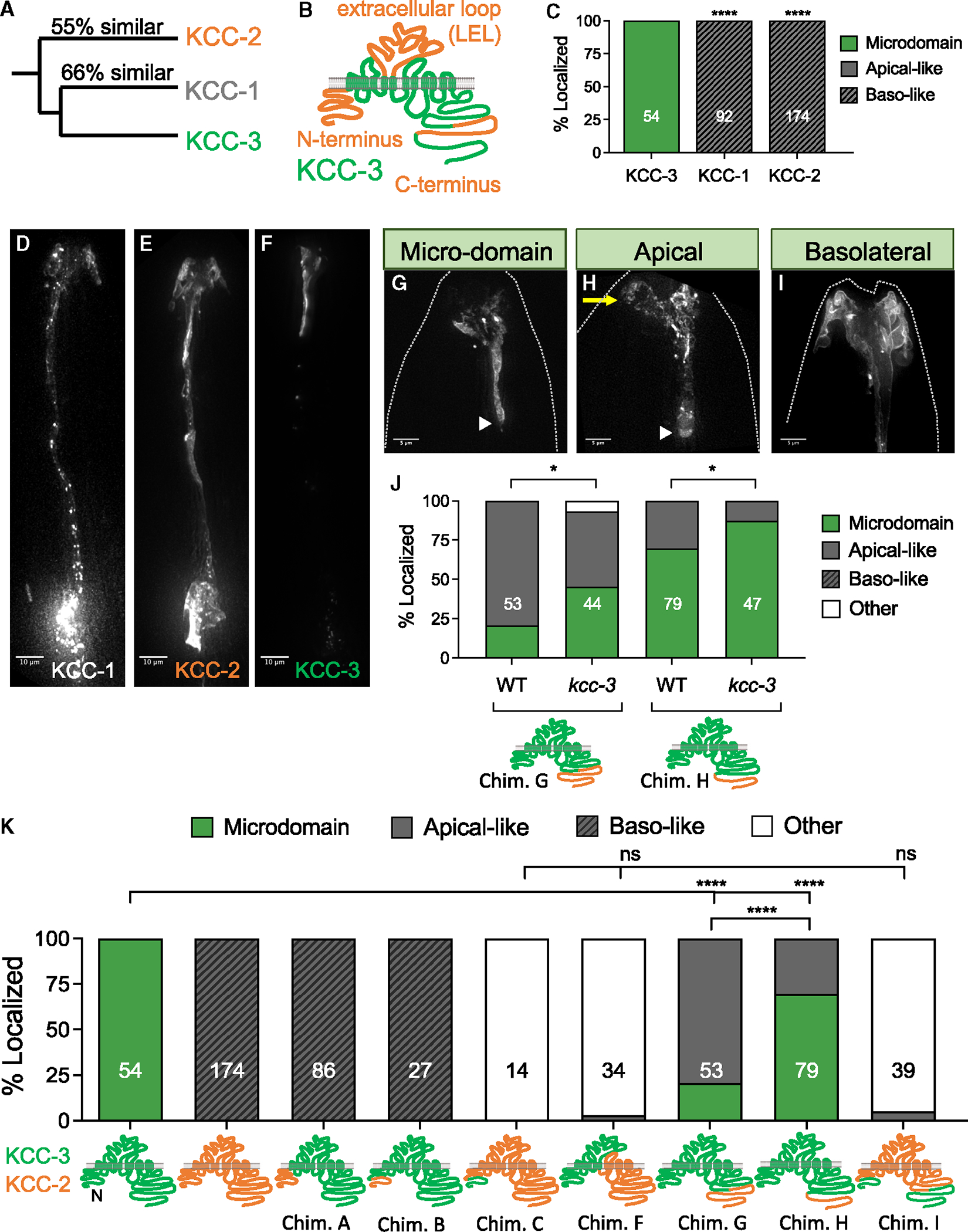
(A) Phylogenetic tree denoting the relationship and sequence similarity of the three C. elegans KCC proteins.
(B) Regions of high sequence dissimilarity between KCC-3 and KCC-1/2 from in silico sequence alignment studies, with orange denoting regions of high sequence dissimilarity.
(C) Quantification of KCC-1 and KCC-2 localization when expressed in AMsh glia compared with KCC-3. Data represent 3–5 biological replicates and 1–2 technical replicates.
(D–F) Fluorescence images of KCC-1 (D), KCC-2 (E), and KCC-3 (F). Scale bars: 10 μm.
(G–I) Fluorescence images of KCC localization patterns seen in KCC chimeras. A yellow arrow points to apical expression beyond the microdomain. A white arrowhead denotes the glial apical boundary (GAB). Dashed lines indicate the outline of the animal. Scale bar: 5 μm.
(J) Quantification of localization patterns seen in KCC chimeras in WT and kcc-3 mutant backgrounds. Data represent 2–3 biological replicates and 1–3 technical replicates.
(K) Quantification of localization patterns seen in KCC chimeras.
Worms were assessed over 2–5 biological replicates. *p < 0.05, ****p < 0.0001, Fisher’s exact test. n = number of animals on graph. All imaging data were collected over multiple days/biological replicates. See also Figure S14.
To identify protein regions driving the distinctive KCC-3 localization pattern, we generated protein chimeras between KCC-2 and KCC-3. First, we swapped the N-terminal 90-aa sequence of KCC-3 with the 84-aa equivalent aligned sequence of KCC-2 (Figure S4E; chimera A). This was sufficient to localize the chimera to basolateral membranes (Figures 4I and 4K). Narrowing this further, swapping the first 55-aa sequence of KCC-3 with the 41-aa equivalent aligned sequence of KCC-2 also drove the chimera to basolateral membranes (Figures 4K and S4E; chimera B). Swapping the first 10- or 20-aa sequence of KCC-3 with the equivalent aligned sequence of KCC-2 (Figure S4C; chimeras D and E), however, localized the chimera to the apical-like ad-neuronal microdomain, as wild-type KCC-3 (Figures 4G and S4D). Thus, sequences at 20–41 aa dictate KCC-2 basolateral localization. To test necessity, we swapped the first 41 aa of KCC-2 with the 55-aa equivalent aligned sequence of KCC-3 (Figure S4E; chimera C) and found that it no longer restricted KCC-2 to basolateral-like membranes (Figures 4K and S4B). Thus, the 19-aa N-terminal sequence (aa 20–41) of KCC-2 is necessary and sufficient to localize KCC proteins to ab-neuronal/basolateral membranes. We could not dissect this region further for a single sequence motif by either site-directed mutagenesis of predicted phosphorylation or dileucine sites or deletions (Figures S4C and S4D), which may be due to redundant targeting features within.
Through an analogous approach, we identified a distinct 87-aa microdomain-targeting sequence. A chimera that swapped the last 155 aa (aa 915–1,070) of KCC-3 with the equivalent 171 aa (aa 890–1,061) of KCC-2 (Figure S4G; chimera G) localized apically but not to a microdomain (Figures 5H and 5K). However, swapping the last 68 aa (aa 1,002–1,070) of KCC-3 with the equivalent 66 aa (aa 995–1,061) of KCC-2 (Figure S4G; chimera H) localized the chimera to a microdomain (Figure 4K). Thus, the 87-aa C-terminal sequence (aa 915–1,002) of KCC-3, highly dissimilar with KCC-1/2 (Figure S4G), directs KCC-3 to a microdomain.
Figure 5. Glial KCC-3 localization is regulated by distal non-AFD-NRE cilia.
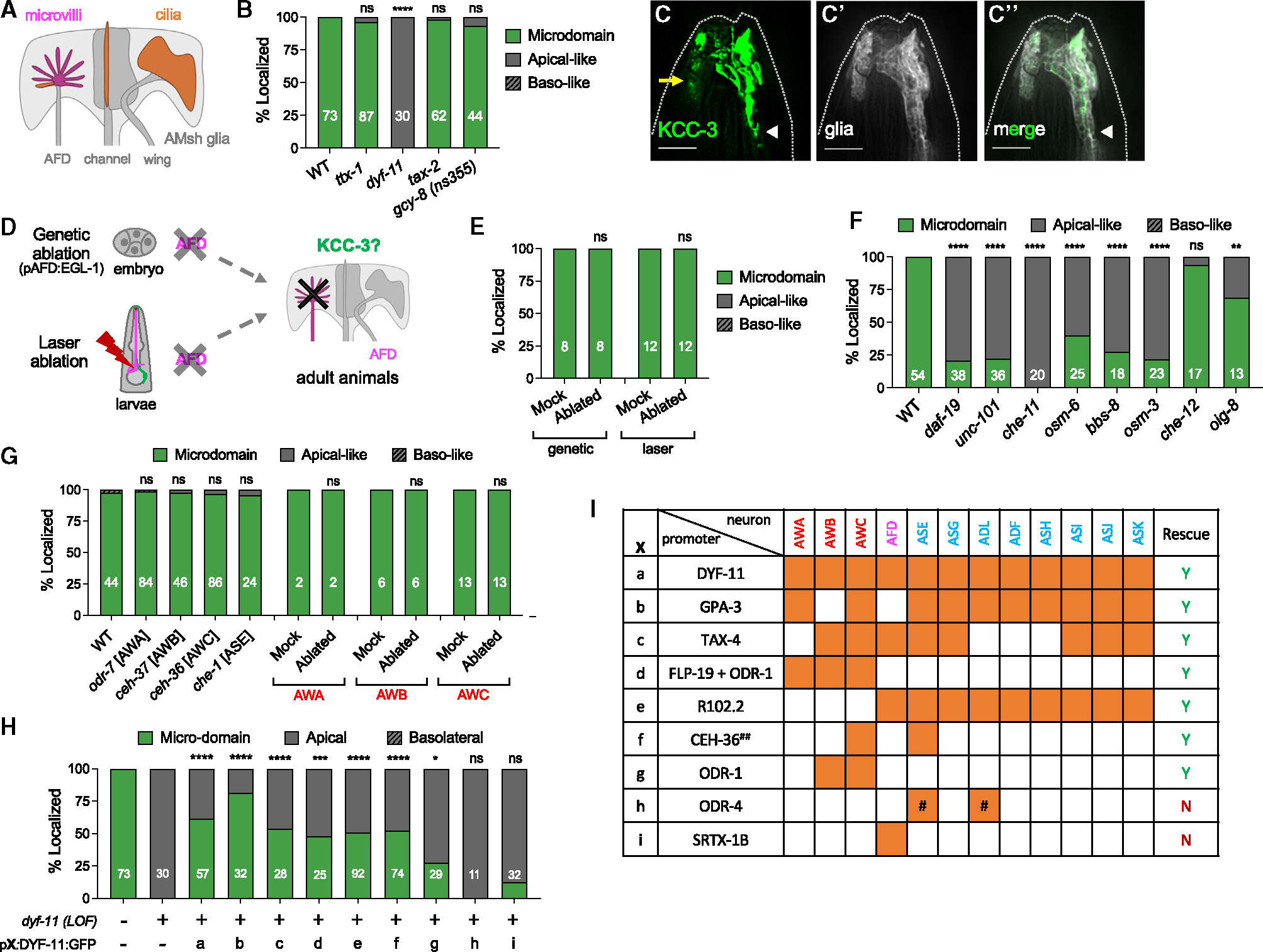
(A) Schematic showing distribution of microvilli and cilium structures in amphid NREs.
(B) Quantification of KCC-3 localization in ttx-1, dyf-11, tax-2, and gcy-8(ns355) mutants compared with the WT. Data represent 2–3 biological replicates.
(C–C″) Fluorescence images of KCC-3 (C, green), AMsh glia (C′, gray), and merge (C″) in dyf-11 cilium mutant animals. A yellow arrow points to apical expression beyond the microdomain. White arrowheads denote the GAB. Dashed lines indicate the outline of the animal. Scale bar: 5 mm.
(D) Schematic of genetic and laser ablation protocols to assess KCC-3 localization without AFD.
(E) Quantification of KCC-3 localization in adults after genetic and laser ablation compared with mock animals. Data represent 2 biological replicates.
(f) KCC-3 localization in cilium mutants. Data represent 1–3 biological replicates.
(G) Quantification of KCC-3 localization in amphid neuron identity mutants (odr-7, ceh-37, ceh-36, and che-1) and after wing neuron (AWA, AWB, and AWC) laser ablation. Data represent 1–4 biological replicates.
(H and I) Quantification of KCC-3 localization in DYF-11 rescue experiments (H). X refers to promoter(s) used for rescue experiments. The identity of X and the neurons in which the promoter(s) is (are) expressed are expanded in (I). Orange denotes expression in associated neurons. #, ODR-4 expresses in an ASX and an ADX neuron, but the exact identity of these neurons is unclear. ##, the CEH-36 rescue construct also has ODR-10 and R13H4.1, but only CEH-36 showed expression. Data represent 2–5 biological replicates and 1–3 technical replicates.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; Fisher’s exact test to WT. n = number of animals on graph. See also Figure S5.
Two observations suggest that KCC-3 has a third ad-neuronal/apical targeting motif distinct from the two identified above. One, while deleting the basolateral targeting sequence (chimera C) in KCC-2 no longer drives it to ab-neuronal membranes, this protein still did not restrict to the ad-neuronal GAB, which was unlike KCC-3. We called this localization pattern “other” (neither clearly ad-neuronal/apical like nor ab-neuronal/basolateral like; Figures 4K and S4A). Two, a swap of the last 671 aa (aa 399–1,070) of KCC-3 with the equivalent 681 aa (aa 380–1,061) of KCC-2 also exhibited the “other” expression pattern (Figures 4K and S4F; chimera F). We found above that KCC-3 sequences until 915 aa localize to ad-neuronal/apical membranes (chimera G). Together, we inferred that regions between 390 and 915 aa on KCC-3 drive ad-neuronal/apical localization. To confirm this, we engineered a double-swap of both N-and C-terminal domains of KCC-2 to KCC-3 (leaving aa 390–915 as KCC-2) and again observed “other” localization (Figure 4K; chimera I). Thus, the aa 390–915 ad-neuronal/apical targeting motif enables subsequent microdomain enrichment.
Last, K/Cl proteins exist as oligomers through a C-terminal dimerization domain.45 We therefore considered whether ad-neuronal or microdomain localization of C-terminal chimeras F and G was, in fact, driven by endogenous wild-type KCC-3-dependent trafficking. To test this, we examined localization of these chimeras in kcc-3(ok228) null mutant animals and found that their localization profiles did not alter (Figure 4J). If at all, endogenous protein out-competed (not enabled) chimera localization. Thus, dimerization does not account for KCC-3’s ad-neuronal/apical and microdomain localization.
Altogether, these structure-function studies reveal that KCC-3 localization requires three sequence features: (1) lack of N-terminal basolateral targeting sequences, (2) presence of apical targeting sequences (aa 390–915), and (C) presence of microdomain-targeting sequences at the proximal C terminus (aa 910–1,002). Furthermore, apical targeting is a prerequisite for microdomain enrichment, indicating a two-step process of localization.
Glial KCC-3 localization is independent of AFD neuron shape or function
The motifs identified above did not suggest regulatory interactors, leaving open the question of whether KCC-3 localization is a cell-intrinsic feature or regulated by neuronal interactions. So, we undertook an orthogonal strategy to test this.
Since KCC-3 localizes around AFD-NRE, we first tested the model where AFD-NRE recruits glial KCC-3 to apposing membranes. AFD-NRE comprises ~40–50 microvilli and one pseudo-cilium (Figure 5A).19,24,46 ttx-1 mutants, which lack AFD-NRE microvilli, retained KCC-3 microdomain localization, suggesting that microvilli are not required (Figure 5B).47 We then examined KCC-3 localization in animals mutant for the ciliary protein DYF-11/IFTB/TRAF3IP1, whose loss leads to truncated cilia.19,48 Strikingly, in dyf-11 mutants, while the GAB is retained, glial KCC-3 was no longer constrained to an apical/ad-neuronal membrane microdomain but instead spread to the entirety of the AMsh glial anterior region (Figures 5B and 5C–5C″). This pattern was strikingly reminiscent to the ad-neuronal enriched chimera G. Thus, neuron cilia restrict KCC-3 to a microdomain.
AFD-NRE cilia house sensory transduction channels at their base.22,49,50 To test whether dyf-11mutant defects resulted from altered AFD-NRE activity, we examined KCC-3 localization in animals mutant for either the sole cyclic nucleotide-gated channel β subunit driving AFD activity, TAX-2,51 or animals with a gain-of-function gcy-8(ns335) mutation, which have constitutively activated levels of the transduction second messenger cGMP.24 Surprisingly, neither affected KCC-3 localization (Figure 5B). Further, cultivation temperature (AFD’s sensory input) also did not impact KCC-3 localization (Figure S5A). Thus, neuron cilium structure, but not activity, drives KCC-3’s microdomain localization.
To parse this further, we asked whether DYF-11 functions solely in AFD-NRE to localize KCC-3. To do so, we ablated the AFD neuron in two temporally distinct ways (Figure 5D). First, we genetically ablated AFD by expressing the pro-apoptotic factor EGL-1 under an AFD-specific promoter (Psrtx-1).24,52 The Psrtx-1 promoter is expressed shortly after AFD is born embryonically, indicating that it is competent to induce embryonic cell loss (Figures S5B–S5B″). Independently, we performed targeted laser microdissection to ablate AFD neurons in L1 larvae.53 With both protocols, we confirmed successful AFD ablation by disappearance of Psrtx-1:GFP (Figure S5D). Surprisingly, glial KCC-3 maintained localization to an ad-neuronal/apical microdomain in both scenarios (Figures 5E and S5C). In line with this, a rescuing DYF-11 cDNA expressed only in AFD neurons was unable to rescue KCC-3 localization defects of dyf-11 mutants (Figures 5H and 5I). Thus, while neuron cilia guide KCC-3 to a microdomain around AFD-NRE, the AFD itself is dispensable for this compartmentalization.
Ciliary transport drives glial KCC-3 microdomain localization
We next wondered whether other cilium mutants similarly affect KCC-3 localization. We performed a candidate screen of other regulators of cilium biogenesis and transport54,55 (Figure S5E). Briefly, mutations in the cilium biogenesis regulator DAF-19/RFX transcription factor, OSM-3/kinesin II, intraflagellar transport (IFT) CHE-11/IFT-A component, DYF-11/IFT-B component, OSM-6/IFT-B component, BBS-8/BBsome, or UNC-101/AP1 all led to aberrant expansion of glial KCC-3 to ad-neuronal regions beyond its microdomain (Figure 5F). Further, all mutants except unc-101 retained higher KCC-3 enrichment around AFD-NRE (Figure S5F). Finally, consistent with prior electron microscopy (EM) studies,10,19 we found that AMsh morphology in daf19 and dyf-11 mutant animals appeared grossly normal, although the AMsh glia anterior regions appeared smaller in daf-19 mutants (Figures S5G–S5I). Thus, these results indicated that KCC-3 localization defects in cilium mutants were not secondary to AMsh glia shape defects. Taken together, we concluded that ciliary IFT transport dictates loss of KCC-3 from non-AFD-NRE sites.
Cilia of two neurons drive glial KCC-3 microdomain localization
To determine which cilium NREs drive AMsh KCC-3 localization, we undertook both candidate screening and cell-specific rescue studies to pinpoint the DYF-11 site of action. Only sensory neurons in C. elegans are ciliated.56 Further, all non-AFD-NREs contacted by AMsh glia are cilium based and anatomically segregate into two classes: wing and channel NREs (Figure 5A). First, we examined KCC-3 localization in mutants for the OIG-8/Ig domain protein, which regulates elaboration of wing NRE cilia,57 and CHE-12/HEAT domain protein, which affects only channel neurons.58 oig-8 mutants exhibited KCC-3 localization defects similar to cilium mutants, although with significantly less penetrance, while che-12 mutants had no effect on KCC-3 localization (Figure 5F). Thus, wing neurons likely contribute to KCC-3 localization.
We then asked whether fate specification of wing neuron subtypes is relevant. We examined KCC-3 localization in animals bearing mutations in specific neuron identity determinants: ODR-7/nuclear hormone receptor (AWA), CEH-37/Otx homeo-domain (AWB), CEH-36/Otx (AWC), and CHE-1/GLASS zinc (Zn) finger (ASE).59–61 These genes act with DAF-19/RFX to specify the distinct NRE cilium shape for each neuron and, when mutated, result in intact but mis-specified NRE cilia of specific neurons (Figure S5J).62 We found that none of these mutants perturbed KCC-3 localization (Figure 5G). Consistent with this, ablation of each of the three wing neurons (AWA/B/C) individually also did not alter KCC-3 localization (Figure 5G). Thus, while cilia are required, the identity or presence of single wing neuron cilia does not regulate KCC-3 localization.
We hypothesized that this may reflect functional redundancy across multiple cilia. So, to identify the minimal set of cilia required, we performed cell-specific rescue experiments in dyf-11 mutant animals, which showed a complete loss of KCC-3 microdomain localization (Figures 5H and 5I). DYF-11 expressed under its native Pdyf-11, Pgpa-3, or Ptax-4 promoters, which express in all, 9, or 10 amphid neurons respectively, rescued KCC-3 localization (Figures 5H and 5I). Thus, DYF-11 rescuing cDNA is functional when expressed broadly. We then tested smaller, non-overlapping subsets of amphid neurons. PR102.2 (all channel neurons and AFD only) and Pflp-19 + Podr-1 (all wing neurons only) both exhibited equivalent rescue, suggesting that the localization cue(s) is (are) broadly expressed. Finally, expression under either combination of two neurons by Podr-1 (AWC and AWB) or Pceh-36 (AWC+ASE) was able to rescue. This was specific since expression in two other neurons by Podr-4 (2 channel neurons) did not rescue (Figures 5H and 5I). Thus, AWC+X is the minimal 2-neuron combination to guide KCC-3 localization, with X being another amphid neuron.
AFD-NRE shape requires AMsh glial KCC-3 microdomain enrichment
To address the functional consequence of this glial molecular microdomain, we examined the properties of both AFD and other NREs in kcc-3(ok228) mutant and mis-localized KCC-3 transgenic animals.
First, we asked whether KCC-3 localization impacts AFD-NRE shape by assessing whether mis-localized chimeras can rescue AFD-NRE shape of kcc-3 mutants (Figures 6A–A′).24 While expression of full-length KCC-3 in a kcc-3 background rescued AFD shape, the basolaterally localized chimera B failed to rescue AFD-NRE shape (Figure 6B). In contrast, the apically localized Chimera G, which also retains microdomain enrichment, could rescue kcc-3(ok228) mutant AFD-NRE defects (Figure 6B). In corollary, a mutation in unc-101 that completely loses AFD-NRE microdomain enrichment had defects in AFD-NRE shape, like kcc-3(ok228) mutant animals (Figure S6E). Thus, AFD-NRE shape and function require KCC-3 enrichment around AFD but are not impacted when KCC-3 expands to other ad-neuronal regions. In line with this, mutations in dyf-11 and osm-6, which had aberrantly expanded KCC-3 (Figure 3E), did not exhibit defects in AFD-NRE shape.24
Figure 6. Microdomain localization of KCC-3 regulates AFD shape.
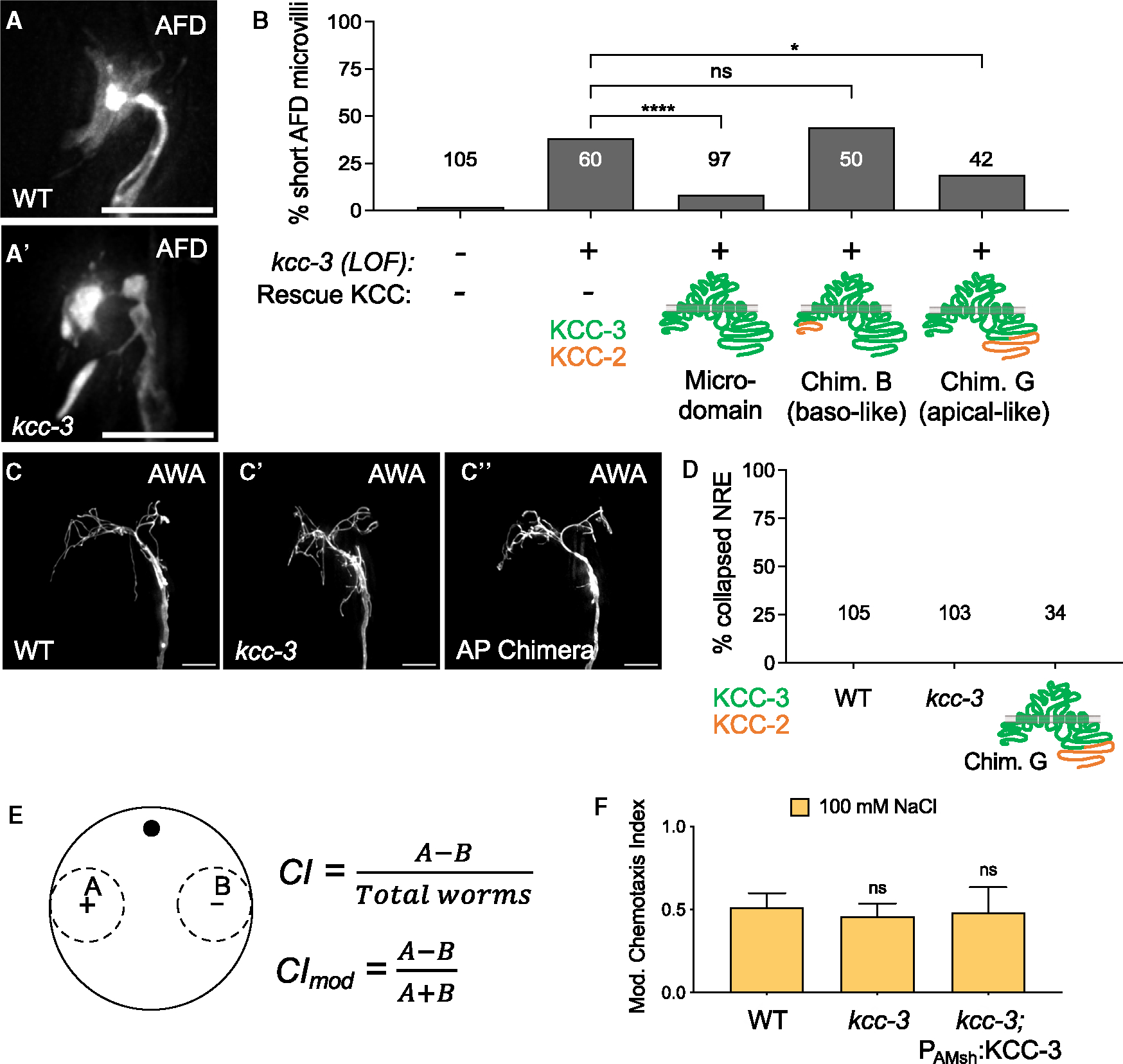
(A and A′) Fluorescence images of AFD-NRE in WT (A) and kcc-3(ok228) mutants (A′).
(B) Quantification of AFD-NRE shape rescue with WT KCC-3, a basolaterally localized KCC-2/KCC-3 chimera (chimera B), and an apically localized KCC-2/KCC-3 chimera (chimera G). Fisher’s exact test. *p < 0.05, ****p < 0.0001.
(C–C″) Fluorescence images of AWA in WT animals (C), kcc-3 mutant animals (C′), and animals that express the apically localized KCC-3 chimera (C″, AP chimera).
(D) Quantification of AWA shape in WT animals, kcc-3 mutant animals, and animals that express the apically localized KCC-3 chimera (chimera G). Fisher’s exact test.
(E) Schematic of chemotaxis assays, including the equations for the chemotaxis index (CI) and modified chemotaxis index (CImod).
(F) Behavioral quantifications for ASE-sensed tastant (100 mM NaCl). Data show mean + SD. One-way ANOVA with Tukey’s post hoc test.
Scale bars: 5 μm. n = number of animals on graph. Data represent at least 2 biological replicates. See also Figure S6.
AWC neuron activity requires the AMsh glial KCC-3 microdomain around AFD-NRE
To assess whether KCC-3 localization affected the shape and functions of non-AFD neurons, we first examined their NRE shape. We found that loss of kcc-3 did not impact the shape of any other NRE tested (wing, AWA/B/C; channel, ASE) (Figures 6C–C′, 6D, and S6A–S6D′). Further, overexpressing chimera G, which expresses in ad-neuronal/apical membranes outside of the KCC-3 microdomain, also did not affect the NRE shape of at least one of these neurons tested, AWA (Figures 6C″ and 6D). For completeness, we also tested whether neuron function was disrupted for channel and wing neurons by monitoring animal sensory behaviors. Loss of KCC-3 did not impact salt chemotaxis, mediated by the ASE channel neuron (Figures 6E and 6F). Unexpectedly, kcc-3 mutant animals showed impaired AWA and AWC neuron-driven animal behaviors. Specifically, kcc-3 mutant animals failed to chemotax toward attractive odorants sensed by AWA (methyl pyrazine and diacetyl) and AWC (isoamyl alcohol and benzaldehyde) (Figures 7A and S7A), with deficits comparable with impairments seen in either AMsh glia-ablated or dyf-11 sensory cilium-deficient animals (Figures S7B and S7C).10,58 ttx-1 mutants, which have defective AFD-NRE shape and function,47 have intact AWA and AWC behaviors, suggesting that the chemosensory defects of kcc-3 mutants are not secondary to impaired AFD-NRE or its downstream circuit neurons (Figure S7B). Further, expression of KCC-3 specifically in AMsh glia rescued the chemotaxis defects of kcc-3 mutants animals completely for AWC and significantly for AWA-driven behaviors, suggesting that these chemosensory defects were not due to a role of KCC-3 in other glia that associate with downstream circuit neurons, such as the CEPsh glia, which associates with the downstream interneurons63 (Figures 7A and S7A). Thus, AFD-localized AMsh glial KCC-3 modulates distal AWC and AWA chemosensory functions.
Figure 7. Schematic of KCC-3 localization in AMsh glia.
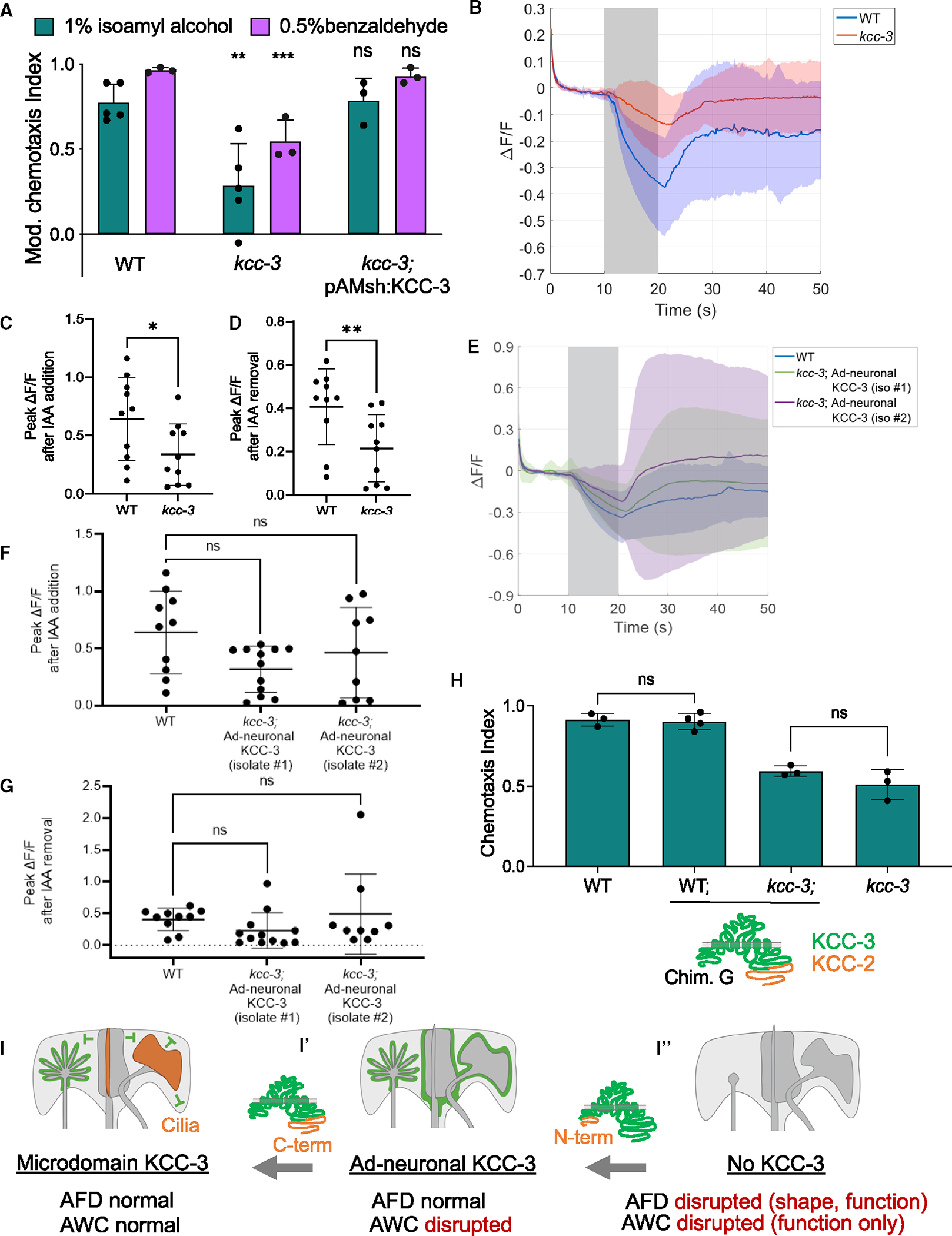
(A) Behavioral quantification of AWC-sensed odorants (1% isoamyl alcohol [IAA] and 0.5% benzaldehyde). Data show mean + SD. Significance compared with the WT. One-way ANOVA, Tukey’s post hoc test. **p < 0.01, ***p < 0.001.
(B) Averaged raw calcium transients in animals with the addition of 0.01% IAA in AWC neurons expressing GCaMP6s. Solid lines represent the average across 10–12 different animals in WT (blue) and kcc-3 (red) backgrounds.
(C) Peak calcium responses when the animal is presented with a stimulus (p = 0.043, Mann-Whitney test). (D) Peak calcium responses when IAA was removed (p = 0.0068, Mann-Whitney test).
(E) Averaged raw calcium transients in WT animals and two independently derived transgenic animal strains expressing the ad-neuronal KCC-3 in the kcc-3 mutant background.
(F and G) Peak calcium responses when IAA was added and removed, respectively (p = 0.15, p = 0.059, Kruskal-Wallis test).
(H) Behavioral quantification of AWC-sensed odorant (1% IAA) with animals expressing ad-neuronal KCC-3. Data show mean + SD. One-way ANOVA, Tukey’s post hoc test.
(I–I″) KCC-3 localization in a two-step process regulates multisensory processing. N-terminal sequences dictate ad-neuronal vs. basolateral localization. Neuron cilia as well as C-terminal KCC sequences determine microdomain localization.
Data represent at least 2 biological replicates. See also Figure S7.
To confirm this surprising result, we assessed AWC neuron function directly by monitoring intracellular Ca2+ dynamics via GCaMP imaging.63 We found that AWC neurons had significantly dampened responses to isoamyl alcohol (both presentation and withdrawal) in kcc-3 mutant animals (Figures 7B–7D; Videos S3 and S4). Furthermore, in animals expressing apical chimera F in the kcc-3(ok228) mutant background (localizes aberrantly to ad-neuronal regions beyond the microdomain), AWC exhibited aberrantly variable Ca2+ responses to isoamyl alcohol (Figures 7E, S7D, and S7E), although average response amplitudes across animals were not significantly impacted (Figures 7F and 7G). In line with this, chimera F was unable to rescue AWC-driven behavior deficits of kcc-3(ok228) mutant animals (Figure 7H) despite rescuing AFD-NRE shape defects (Figure 6B). Thus, distal AWC-NRE cilia localize AMsh glial KCC-3 to a microdomain around AFD-NRE to maintain its shape and function as well as the function of distal AWC-NRE. Altered KCC-3 localization has consequences for both thermosensory and chemosensory animal behaviors (Figures 7I–I″).
DISCUSSION
Our results provide single-molecule evidence that a single glial cell can differently regulate associated neurons. Study of the underlying biology suggests that neurons can interact with each other non-synaptically by regulating cues on shared glia. Using AMsh glia as a powerful single-glia experimental platform, we show that the glial apical-like ad-neuronal membrane is partitioned into multiple and distinct molecular microdomains around individual NRE contact sites. Further, apical-like ad-neuronal membranes extend into a boundary domain we term GAB. This is distinct from classic epithelial apical-basal polarity. Focusing on one microdomain cue, the K/Cl co-transporter KCC-3, as a molecular tool, we uncover a two-step model for microdomain localization. First, KCC-3 localizes to apical-like regions of the glia. Here, it is repelled by non AFD-NRE cilia, rendering it localized to AFD-NRE (Figures 7G–G″). Finally, we find that microdomain localization of KCC-3 to AFD-NRE not only regulates AFD-mediated thermosensory perception but also the fidelity of non-AFD chemosensory neuron functions. This implies that regulation of glial cue localization may be one mechanism by which glia-neuron units modulate and segregate information processing across circuits.
Neuron cilium regulation of glial cues
We found that non-AFD-NRE cilia localize a glial membrane cue to a microdomain through a signal transported by IFTA/B complex. Thus, a neuronal ciliary signal can modify localization of glial cues and thereby alter properties of other glia-associated neurons. Most mammalian cells have primary, non-motile cilia. In neurons and glia, their presence and functions are only recently being appreciated.64–66 To our knowledge, a role of neuron cilia in guiding glial properties has not yet been reported. While the molecular identity of this cue remains to be identified, possible suspects are ciliary extracellular vesicles.67,68
Glial cell polarity
We validate the prior observation27,28 that AMsh glia membrane are polarized, with ad-neuronal membranes localizing apical markers and ab-neuronal membranes localizing basolateral markers. We further report that membranes marked by one of three ad-neuronal molecular reporters (PH-PLCδ, KCC-3, or SAX-7) extend along the anterior-posterior axis up to a boundary we term the GAB. In contrast to the classic apical-basal epithelia polarity boundary, the GAB is not bound by the tight-junction proteins AJM-1/JAM or DLG-/DiscsLarge. Rather, it is a tube-within-tube configuration of apical and basolateral marked membranes. This distinguishing feature suggests that glial cell polarity is not a simple epithelial polarity. Instead, we propose that AMsh glia cell biology is conceptually analogous to the specialized polarity of astrocytes as well as neurons.69,70 Specifically, we posit that the GAB is a glial sorting center analogous to the neuronal axon initial segment (AIS) that delimits diffusion of membrane proteins across the cell’s polarized domains (neuronal axon/dendrite, glial ad-neuronal/ab-neuronal), a model we are currently testing.
Glial regulation of K/Cl transporters
In C. elegans, KCC-3 acts in AMsh glia to regulate AFD thermosensory neuron shape and function.24 We report here that its localization is restricted to AFD’s contact site on AMsh glia. This localization is independent of the canonical K/Cl regulators, which were identified primarily in KCC-2 studies, and depends on motifs that remain to be characterized. Thus, glial regulation of KCC-3 is mechanistically distinct from how other cell types, including neurons, regulate KCC-1/2 (Figures 3A–3D), highlighting the relevance of cell-specific molecular studies.
KCC-3 is a SLC12A6 electroneutral K/Cl co-transporter implicated in neurological diseases, including autism, epilepsy, and schizophrenia.71–74 Multiple C. elegans glia, as well as other glia across species, localize KCC-3 to molecular microdomains. In rodents, Schwann cell peripheral glia localize KCC-3 to apical microvilli around nodes.75 In mammals, inner ear Deiter cells (glia-like support cells) localize KCC-3 to basal poles of hair cells.71,76 CNS astrocytes and microglia also express KCC-3 at ad-neuronal membranes.77 Defining how glia regulate KCC-3 sub-cellular localization and function, then, will be broadly relevant to understanding neural functions and KCC-3-associated neurological diseases.
Glial microdomains and cross-modal information processing
Prior work hinted that AMsh glia may regulate different neurons unequally.10,23,24,78 Our identifying that it makes at least three distinct molecular microdomains of regulatory cues (KCC-3, LIT-1, and neither) around different NREs offers a mechanistic basis for this glia-neuron specificity at single-cell resolution. Furthermore, an intriguing finding of this study is that microdomain-localized KCC-3 also affects distal NRE functions without altering their shape. How might this happen? Prior studies have shown that many glia-associated amphid neurons express receptor guanylyl cyclases (rGCs) at glia-interfacing ciliary membranes, and many of these rGCs in silico have motifs reminiscent of the canonical chloride-binding pocket, like AFD’s GCY-8/rGC24,79,80 (data not shown). Further, many of these neurons, including AWCON wing neurons, respond to K+, Cl−, or Na+.81–83 Thus, aberrant expression of glial KCC-3 at these NRE contact sites may change the regional ionic balance and thus alter sensitivity to cues (Figures 6 and 7). An alternate mechanism is based on the role of K+-Cl− co-transporters in regulating cell volume.84 In this case, aberrant KCC-3 activity around nonAFD-NREs may alter glial cell volume and, thereby, glia-NRE intermembrane spacing. If so, then this could alter the concentration of other regulators at glia:NRE intercellular microenvironments, leading to aberrant neuronal responses. While direct assessment of K+ or Cl− levels or dynamic cell volume at these glia-NRE subcompartments is currently technically limited, these models will be exciting to test in future studies.
Independent functional studies have previously reported glial roles in tuning synapses and animal behavior through neurotransmitter release85 and glia exhibiting functional Ca2+ microdomain responses to sensory cues and neuron activity.9,86–89 While the correlation between functional and molecular microdomains remains to be determined, these insights together lead us to speculate that glial cue microdomains may mediate cross-circuit information processing by enabling non-synaptic neuron crosstalk.
Limitations of the study
To determine where DYF-11 acts, we overexpressed DYF-11:GFP under different promoters as extrachromosomal arrays. While multiple arrays were examined per conclusion, and expression was tracked via bicistronic fluorescent tags, there may also be weak promoter expression in other cells not detected by fluorescent reporters. Similarly, due to technical limitations, we utilized multicopy extrachromosomal arrays of chimera proteins to study aberrant KCC-3 localization.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aakanksha Singhvi (asinghvi@fredhutch.org).
Materials availability
Plasmids and stable strains generated in this study are available without restrictions by contacting the lead contact.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Caenorhabditis elegans
C. elegans were cultured as previously described.98,99 Bristol N2 strain was used as wild type. Animals were raised at 20°C (unless noted) for at least one week without starvation. L4 larval animals were picked to fresh plates and assayed 24 h later, unless otherwise noted. See key resources table for a full list of mutant and transgenic strains used in this study.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E. coli OP50 | CGC | OP50; RRID:WB-STRAIN: WBStrain00041969 |
| E. coli HT115 | CGC | HT115: RRID:WB-STRAIN: WBStrain00041079 |
| E. coli DH5α competent cells | Fisher Scientific | Cat#: 18-265-017 |
| E. coli XL10-Gold ultracompetent cells | Agilent Technologies | Cat#: 200314 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Sodium Azide | Sigma-Aldrich | Cat#: S-2002 |
| Taq DNA Polymerase | Sibgene | Cat#: SG-1-500 |
| Q5 High-Fidelity DNA polymerase | New England Biolabs (NEB) | Cat#: M0491L |
| ExTaq DNA polymerase | Takara | Cat#: RR01a.m. |
| PfU Ultra High-Fidelity DNA polymerase | Agilent Technologies | Cat#: 600380 |
| T4 DNA ligase | New England Biolabs (NEB) | Cat#: M0202L |
| AgeI-HF | New England Biolabs (NEB) | Cat#: R3552S |
| EcoRI-HF | New England Biolabs (NEB) | Cat#: R3101L |
| ApaI | New England Biolabs (NEB) | Cat#: R0114S |
| BamHI-HF | New England Biolabs (NEB) | Cat#: R3136L |
| SalI-HF | New England Biolabs (NEB) | Cat#: R3138L |
| SphI-HF | New England Biolabs (NEB) | Cat#: R3182L |
| Ethanol, ACS Reagent Grade | Sigma-Aldrich | Cat#: 459844-4L |
| Isoamyl Alcohol | Sigma-Aldrich | Cat#: W205702 |
| Benzaldehyde | Sigma-Aldrich | Cat#: 418099 |
| 2-Methylpyrazine | Sigma-Aldrich | Cat#: W330906 |
| 2,3-Dutanedione | Sigma-Aldrich | Cat#: 11038 |
| NaCl | Fisher Scientific | Cat#: S271-500 |
| Chloroform | Fisher Scientific | Cat#: C298-500 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Gibson Assembly Master Mix | New England Biolabs (NEB) | Cat#: E2611S |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs (NEB) | Cat#: E0552S |
| QIAprep Spin Miniprep Kit | Qiagen | Cat#: 27106 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Wildtype | CGC | N2; RRID:WB-STRAIN: WBStrain00000001 |
| kcc-3(ok228) II | Singhvi et al. (2016)24 | LX1024 |
| unc-23(e25) V | CGC | CB25 |
| che-14 (ok193) I | CGC | ML514 |
| daf-6(e1377) X | CGC | CB1377 |
| lit-1(ns132) III | Oikonomou et al. (2011)34 | OS3402 |
| snx-1(ns133) X | Oikonomou et al. (2012)35 | OS4343 |
| wnk-1(tm487)/mec-3(e1338) IV | NBRP | FX00487 |
| R08C7.2(ok1681) IV/nT1 [qIs51] (IV; V) | CGC | VC1258 |
| argk-1(ok2973) V | CGC | MAH172 |
| ttx-1(p767) V | CGC | PR767 |
| dyf-11 (mn392) X | CGC | SP1713 |
| tax-2(p691) I | CGC | PR691 |
| gcy-8(ns335) IV | Singhvi et al. (2016)24 | OS8595 |
| daf-19(m86) II; daf-12(sa204) X | CGC | JT6924 |
| unc-101(m1) I | CGC | DR1 |
| che-11(e1810) V | CGC | CB3330 |
| osm-6(p811) V | CGC | PR811 |
| bbs-8(nx77) V | CGC | MX52 |
| osm-3(p802) IV | CGC | PR802 |
| che-12(e1812) V | CGC | CB3332 |
| oig-8(ot818) II | CGC | OH13813 |
| che-1(p679) I | CGC | PR679 |
| ceh-37(ok642) X | CGC | RB823 |
| odr-7(ky4) X | CGC | CX4 |
| ceh-36(ky646) X | CGC | CX5893 |
| nsIs228 [Psrtx-1:GFP + Punc-122:RFP] I | Singhvi et al. (2016)24 | OS4565 |
| nsIs373 [Psrtx-1b:GFP + Punc-122:RFP] X | Singhvi et al. (2016)24 | OS7270 |
| kyIs37 [Podr-10:GFP + lin-15(+)] II | CGC | CX3260 |
| kyIs104 [Pstr-1:GFP] lin-15B&lin-15A(n765) X | CGC | CX3553 |
| kyIs140 [Pstr-2:GFP + lin-15(+)] I | CGC | CX3695 |
| kyIs136 [Pstr-2:GFP + lin-15(+)] X | Troemel et al. (1999)90 | CX3261 |
| ntIs1 [Pgcy-5:GFP + lin-15(+)] V | CGC | OH3192 |
| nsEx5658 [kcc-3:GFP fosmid + Pmig-24:venus] | Singhvi et al. (2016)24 | OS9978 |
| dnaEx117 [pCM11{PF53F4.13:SAX-7:mApple} + Pmig-24:venus] | Martin et al. (2022)28 | ASJ382 |
| dnaEx95 [pCM7{PF53F4.13:SAX-7deltacyt:sfGFP} + Pmig-24:venus] | Martin et al. (2022)28 | ASJ345 |
| dnaIs19 [pCM7{PF53F4.13:SAX-7deltacyt:sfGFP} + Pmig-24:venus]? | Martin et al. (2022)28 | |
| hmnIs30 [PF16F9.3:AJM-1:YFP]? | Lowetal. (2019)27 | CHB2522 |
| xnIs17 [PDLG-1:DLG-1:GFP + rol-6(su1006)]? | CGC | FT63 |
| nsEx2606 [pG038{PTO2B11.3:GFP:UT-1}] | Oikonomou et al. (2011)34 | OS4546 |
| nsEx4131 [Pvap-1:VAP-1:sfGFP + Pmig-24:venus] | This study | OS7635 |
| nsIs105 [Phlh-17:GFP] I | Katzetal. (2019)91 | OS1914 |
| oyIs44 [Podr-1-RFP] V | CGC | PY2417 |
| oyIs87[Pga4Δ6:myr-GFP] III | Maurya and Sengupta (2021)92 | PY10421 |
| nsEx4394 [pJF29{Psrtx-1B:DYF-11:GFP} + Pelt-2:mCherry] | Raiders etal. (2021)93 | OS8259 |
| nsIs228 [Psrlx-1:GFP + Punc-122:RFP] I; kcc-3(ok228) II | Singhvi et al. (2016)24 | OS9081 |
| nsIs109 [PF16F9.3:DTA(G53E) + Punc-122:GFP]? | Bacaj et al. (2008)10 | OS1932 |
| pekIs123 [Podr-1:GCaMP6s:SL2:mCherry + PrpI-28S:neoR:rpl-28UTR + Punc-122:mCherry] | Gift from Jihong Bai | BJH878 |
| dnaEx6 [pSR7{PF53F4.13:KCC-3:mScarlet} (5 ng/μL) + pAS326{PVAP-1:VAP-1:sfGFP} (20 ng/μL) + Pelt-2:mCherry (10 ng/μL) + pBluescript (65 ng/μL)] | This study | ASJ30 |
| dnaEx28 [pSR7{PF53F4.13:KCC-3:mScarlet} (5 ng/μL) + Pmig-24:venus (30 ng/μL) + pBluescript (65 ng/μL)] | This study | ASJ134 |
| dnaEx54 [pSR11{PF53F4.13:CFP:SL2:KCC-3:mScarlet} (5 ng/μL) + Pmig-24:venus (530 ng/μL) + pBluescript (65 ng/μL)] | This study | ASJ221 |
| dnaIs10 [integration of dnaEx28] V 2x OUT | This study | ASJ286 |
| dnaIs15 [integration of dnaEx54] IV 4x OUT | This study | ASJ463 |
| kcc-3(ok228) II; kyIs136 X | This paper | OS3909 |
| kcc-3(ok228) II; kyIs104 X | This study | ASJ237 |
| kcc-3(ok228) II; ntIs1 V | This study | ASJ229 |
| kcc-3(ok228) II; nsEx4131 | This study | ASJ1195 |
| kcc-3(ok228) II; nsEx2606 | This study | ASJ1225 |
| kcc-3(ok228) II; oyIs87 III | This study | ASJ628 |
| dnaEx6; ttx-1(p767) IV | This study | ASJ128 |
| dnaEx6; tax-2(p691) I | This study | ASJ129 |
| dnaEx6; gcy-8(ns335) IV | This study | ASJ136 |
| dnaEx28; kyIs37 II | This study | ASJ202 |
| dnaEx28; kyIs104 X | This study | ASJ203 |
| dnaEx28; ntIs1 V | This study | ASJ188 |
| dnaEx28; che-1(p679) I | This study | ASJ277 |
| dnaEx28; ceh-37(ok642) X | This study | ASJ285 |
| dnaEx28; odr-7(ky4) X | This study | ASJ307 |
| dnaEx28; ceh-36(ky646) X | This study | ASJ308 |
| dnaIs10 V; osm-3(p802) IV | This study | ASJ1105 |
| dnaIs10 V; kyIs104 X | This study | ASJ787 |
| dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ583 |
| dnaIs10 V; kyIs140 I | This study | ASJ304 |
| dnaIs10 V; nsIs228 I | This study | ASJ306 |
| dnaIs10 V; che-14 (ok193) I | This study | ASJ467 |
| dnaIs10 V; nsIs228 I; daf-6(e1377) X | This study | ASJ424 |
| dnaIs10 V; nsIs228 I; lit-1(ns132) III | This study | ASJ468 |
| dnaIs10 V; nsIs228 I; snx-1(ns133) X | This study | ASJ373 |
| dnaIs10 V; wnk-1 (tm487)/nTI (IV; V) | This study | ASJ1214 |
| dnaIs10 V; xnIs17? | This study | ASJ1160 |
| dnaIs10 V; kyIs140 I; unc-23(e25) V | This study | ASJ316 |
| dnaIs10 V; hmnIs30? | This study | ASJ305 |
| dnaIs10 V; nsIs228 I; kcc-3(ok228) II | This study | ASJ341 |
| dnaIs15 IV; argk-1(ok2973) V | This study | ASJ511 |
| dnaIs15 IV; daf-19(m86) II; daf-12(sa204) X | This study | ASJ1074 |
| dnaIs15 IV; unc-101(m1) I | This study | ASJ1075 |
| dnaIs15 IV; che-11(e1810) V | This study | ASJ1098 |
| dnaIs15 IV; osm-6(p811) V | This study | ASJ676 |
| dnaIs15 IV; bbs-8(nx77) V | This study | ASJ1111 |
| dnaIs15 IV; che-12(e1812) V | This study | ASJ1094 |
| dnaIs15 IV; oig-8(ot818) II | This study | ASJ1102 |
| dnaIs15 IV; oyIs87 III | This study | ASJ824 |
| dnaIs15 IV; nsIs228 I; dyf-11(mn392) X | This study | ASJ536 |
| nsIs373 X; unc-101(m1) I | This study | ASJ1189 |
| oyIs44 V; nsEx4131 | This study | ASJ55 |
| dnaEx515 [pOO1{PF53F4.13:PH-PLCdelta:GFP} (5 nq/μL) + Punc-122:RFP (20 nq/μL) + pBluescript (65 nq/μL)] | This study | ASJ1141 |
| dnaEx312 [pSR7{PF53F4.13:KCC-3:mScarlet} (5 nq/μL) + Punc-122:RFP (25 nq/μL) + pBluescript (70 nq/μL)]; dnaIs19? | This study | ASJ790 |
| dnaEx256 [kcc-3:GFP fosmid (50 nq/μL) + pCM11{PF53F4.13:SAX-7:mAppfe} (5 nq/μL) + Punc-122:RFP (15 nq/μL) + pBluescript (30 nq/μL)] | This study | ASJ753 |
| dnaIs19?; dnaEx224 [pCM11{PF53F4.13:SAX-7.mAppfe} (2.5 nq/μL) + Punc-122:RFP (30 nq/μL) + pBluescript (67.5 nq/μL)] | This study | ASJ694 |
| dnaEx514 [pPG6 {Phlh-17:KCC-3:mScarlet} (20 ng/μL) + coel:RFP (20 ng/μL) + pBluescript (60 ng/μL)]; nsIs105 I | This study | ASJ1140 |
| dnaEx84 [pSR17{PF53F4.13:KCC-1a:mScarlet} (5 ng/μL) + Pmig-24:venus (30 ng/μL) + pBluescript (60 ng/μL)] | This study | ASJ317 |
| dnaEx75, 76 [pSR15{PF53F4.13:KCC-2a:mScarlet} (5 ng/μL) + Pmig-24:venus (30 ng/μL) + pBluescript (60 ng/μL)] | This study | ASJ278, 279 |
| dnaEx153 [pSR24{PF53F4.13:ChimeraA:mScarlet} (5 ng/μL) + Pmig-24:venus (30 ng/μL) + pBluescript (65 ng/μL)] | This study | ASJ469 |
| dnaEx193 [pSR27{PF53F4.13:ChimeraB:mScarlet} (5 ng/μL) + Pmig-24:venus (30 ng/μL) + pBluescript (65 ng/μL)] | This study | ASJ632 |
| dnaEx206 [pSR33{PF53F4i3:ChimeraC:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ671 |
| dnaEx314, 315, 316 [pSR43{PF53F4.13:ChimeraD:mScarlet} (5 ng/μL) + Punc-122:RFP (25 ng/μL) + pBluescript (70 ng/μL)] | This study | ASJ792, 793, 794 |
| dnaEx325, 351, 352 [pSR45{PF53F4.13:chimeras:mScarlet} (5 ng/μL) + Punc-122:RFP (25 ng/μL) + pBluescript (70 ng/μL)] | This study | ASJ803, 834, 835 |
| dnaEx322 [pSR49{PF53F4.13:ChimeraF:mScarlet} (5 ng/μL) + Punc-122:RFP (25 ng/μL) + pBluescript (70 ng/μL)] | This study | ASJ800 |
| dnaEx454 [pSR25{PF53F4.13:ChimeraG:mScarlet} (5 ng/μL) + Punc-122:GFP (15 ng/μL) + pBluescript (60 ng/μL)]; nsIs228 I | This study | ASJ1133 |
| dnaEx410 [pSR83{PF53F4.13:ChimeraH:mScarlet} (5 ng/μL) + Punc-122:GFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ931 |
| dnaEx446 [pSR87{PF53F4.13:ChimeraI:mScarlet} (5 ng/μL) + Punc-122:GFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ1015 |
| dnaEx410; nsIs228 I; kcc-3(ok228) II | This study | ASJ1185 |
| dnaEx454; nsIs228 I; kcc-3(ok228) II | This study | ASJ1024 |
| dnaEx454; oyIs87 III | This study | ASJ1177 |
| dnaEx454; oyIs87 III; kcc-3(ok228) II | This study | ASJ1218 |
| dnaEx386 [pSR69{PF53F4.13:KCC-2deletion1:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ878 |
| dnaEx413 [pSR71{PF53F4.13:KCC-2deletion2:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ934 |
| dnaEx430 [pSR66{PF53F4.13:KCC-3deletion3:mScarlet} (5 ng/μL) + Punc-i22:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ970 |
| dnaEx407 [pSR73{PF53F4.13:KCC-3deletion4:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ928 |
| dnaEx387 [pSR67{PF53F4.13:KCC-3deletion5:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ879 |
| dnaEx425 [pSR75{PF53F4.13:KCC-3deletion6:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ960 |
| dnaEx252, 257, 302 [pSR39{PF53F4.13:KCC-2LL>AA:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ744, 754, 764 |
| dnaEx253, 254 [pSR41{PF53F4.13:KCC-3ST>AA:mScarlet} (5 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (80 ng/μL)] | This study | ASJ745, 746 |
| dnaEx339 [pSR59 {PF53F4.13-KCC-3TVGE>AVGE:mSc3rlet} (5 ng/μL) + Punc-122:RFP (25 ng/μL) + pBluescript (70 ng/μL)] | This study | ASJ820 |
| dnaEx332 [pSR61{PF53F4.13:KCC-3TTS>AAA:mScarlet} (5 ng/μL) + Punc-122:RFP (25 ng/μL) + pBluescript (70 ng/μL)] | This study | ASJ813 |
| dnaEx79 [pJF48 {Psrtx-1:EGL-1} (50 ng/μL)+ Pelt-2:mCherry (10 ng/μL) + pBluescript (60 ng/μL)]; nsIs228 I | This study | ASJ289 |
| dnaEx79; dnaIs10 V; nsIs228 I | This study | ASJ309 |
| dnaEx221 [pAN1{Pdyf-11:DYF-11:GFP} (70 ng/μL) + Punc-122:RFP (30 ng/μL)]; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ691 |
| dnaEx450, 453, 488 [pSR85{PR102.2:DYF-11:GFP} (50 ng/μL) + Punc-122:RFP (15 ng/μL) + pBluescript (35 ng/μL)]; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ1019, 1023, 1076 |
| dnaEx337 [pSR57{Pgpa-3:DYF-11:GFP} (50 ng/μL) + Punc-122:RFP (30 ng/μL) + pBluescript (20 ng/μL)]; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ818 |
| dnaEx317 [pSR53{Ptax-4:DYF-11:GFP} (10 ng/μL) + Punc-122:RFP (30 ng/μL) + pBluescript (60 ng/μL)]; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ795 |
| dnaEx334 [pSR35{Podr-1:DYF-11:GFP} (10 ng/μL) + pSR56 {Pflp-19:DYF-11:GFP} (10 ng/μL) + Punc-122:RFP (30 ng/μL) + pBluescript (50 ng/μL)]; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ815 |
| dnaEx418 [pSR77{PR13H4.1:DYF-11:GFP} (30 ng/μL) + pSR79{Pceh-36:DYF-11:GFP} (30 ng/μL) + pSR82{Podr-10:DYF-11:GFP} (30 ng/μL) + Punc-122:RFP (15 ng/μL)]; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ943 |
| dnaEx207 [pSR35{Podr-1:DYF-11:GFP (5 ng/μL)+ Punc-122:RFP (30 ng/μL)} + pBluescript (65 ng/μL); dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ672 |
| dnaEx326 [pSR52{Podr-4:DYF-11:GFP (10 ng/μL) + Punc-122:RFP (30 ng/μL)} + pBluescript (40 ng/μL)}; dnaIs10 V; dyf-11 (mn392) X) | This study | ASJ804 |
| nsEx4394; dyf-11 (mn392) X) | This study | ASJ701 |
| dnaEx444 [pSR27{PF53F4.13:ChimeraB:mScarlet} (5 ng/μL) + Punc-122:GFP] (15 ng/μL) + pBluescript (80 ng/μL); nsIs228 I; kcc-3(ok228) II | This study | ASJ1013 |
| dnaEx449 [pSR27{PF53F4.13:ChimeraB:mScarlet} (5 ng/μL) + Punc-122:GFP] (15 ng/μL) + pBluescript (80 ng/μL); nsIs228 I; kcc-3(ok228) II | This study | ASJ1018 |
| dnaEx462 [pSR25{PF53F4.13:ChimeraG:mScarlet} (5 ng/μL) + Punc-122:GFP] (15 ng/μL) + pBluescript (80 ng/μL); nsIs228 I; kcc-3(ok228) II | This study | ASJ1034 |
| pekIs123 4x OUT | This study | ASJ560 |
| pekIs123; kcc-3(ok228) II | This study | ASJ592 |
| pekIs123; kcc-3(ok228) II; dnaEx599 [pSR25 {PF53F4.13:ChimeraG:mScarlet} (5 ng/μL) + Punc-122:GFP (20 ng/μL) + pBluescript (75 ng/μL)] | This study | ASJ1282 |
| pekIs123; kcc-3(ok228) II; dnaEx600 [pSR25 {PF53F4.13:ChimeraG:mScarlet} (5 ng/μL) + Punc-122 GFP (20 ng/μL) + pBluescript (75 ng/μL)] | This study | ASJ1283 |
|
| ||
| Oligonucleotides | ||
|
| ||
| mScarlet F mutagenesis primer: CGACTCTAAGTCGACGGTACCGGTtatggtatcgaagggagaggc | This study | N/A |
| mScarlet R mutagenesis primer: GCCTCTCCCTTCGATACCATAACCGGTACCGTCGACTTAGAGTCG | This study | N/A |
| Fse-CFP F primer: aaGGCCGGCCATGAGTAAAG | This study | N/A |
| Xba-CFP R primer: cttctagaCTATTTGTATAGTTCATCCATGCC | This study | N/A |
| Asc-SL2 R primer: ttGGCGCGCCacagcagttt | This study | N/A |
| KCC-1 a segA F primer: aaggatccATGACAACCGGCA | This study | N/A |
| KCC-1 a segA R primer: ACGAATTCCGTCTCCCCATTCT | This study | N/A |
| KCC-1 a segB F primer: CGGAATTCGTGGACTTGCTCTC | This study | N/A |
| KCC-1 a segB F primer: ttGTCGACatCGAGCTCTCCG | This study | N/A |
| See Table S1 for additional oligonucleotides | N/A | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSR1 {PF53F4.13:mScarlet:unc-54 3’UTR} | This study | pASJ1/pSR1 |
| pSR5 {PF53F4.13:KCC-3:mScarlet:unc-54 3’UTR (out of frame)} | This study | pASJ5/pSR5 |
| pSR7 {PF53F4.13:KCC-3:mScarlet:unc-54 3’UTR (in frame)} | This study | pASJ7/pSR7 |
| pSR9 {PF53F4.13:CFP:SL2:mKate2:unc-54 3’UTR} | This study | pASJ32/pSR9 |
| pSR11 {PF53F4.13:CFP:SL2:KCC-3:mScarlet:unc-54 3’UTR} | This study | pASJ35/pSR11 |
| pSR15 {PF53F4.13:KCC-2a:mScarlet:unc-54 3’UTR} | This study | pASJ61/pSR15 |
| pSR17 {PF53F4.13:KCC-1a:mScarlet:unc-54 3’UTR} | This study | pASJ94/pSR17 |
| pSR24 {PF53F4.13:ChimeraA:mScarlet:unc-54 3’UTR} | This study | pASJ113/pSR24 |
| pSR25 {PF53F4.13:ChimeraG:mScarlet:unc-54 3’UTR} | This study | pASJ117/pSR25 |
| pSR27 {PF53F4.13:ChimeraB:mScarlet:unc-54 3’UTR} | This study | pASJ132/pSR27 |
| pSR33 {PF53F4.13:ChimeraC:mScarlet:unc-54 3’UTR} | This study | pASJ146/pSR33 |
| pSR35 {Podr-1:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ150/pSR35 |
| pSR39 {PF53F4.13:KCC-2LL>AA:mScarlet:unc-54 3’UTR} | This study | pASJ201/pSR39 |
| pSR41 {PF53F4.13:KCC-3ST>AA:mScarlet:unc-54 3’UTR} | This study | pASJ203/pSR41 |
| pSR43 {PF53F4.13:ChimeraD:mScarlet:unc-54 3’UTR} | This study | pASJ227/pSR43 |
| pSR45 {PF53F4.13:chimeras:mScarlet:unc-54 3’UTR} | This study | pASJ228/pSR45 |
| pSR49 {PF53F4.13:ChimeraF:mScarlet:unc-54 3’UTR} | This study | pASJ230/pSR49 |
| pSR52 {Podr-4:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ233/pSR52 |
| pSR53 {Ptax-4:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ234/pSR53 |
| pSR56 {PfIp-19:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ252/pSR56 |
| pSR57 {Pgpa-3:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ253/pSR57 |
| pSR59 {PF53F4.13:KCC-3TVGE>AVGE:mScarlet:unc-54 3’UTR} | This study | pASJ254/pSR59 |
| pSR61 {PF53F4.13:KCC-3TTS>AAA:mScarlet:unc-54 3’UTR} | This study | pASJ256/pSR61 |
| pSR66 {PF53F4.13:KCC-3deletlon3:mScarlet:unc-54 3’UTR} | This study | pASJ275/pSR66 |
| pSR67 {PF53F4.13:KCC-3deletion5:mScarlet:unc-54 3’UTR} | This study | pASJ276/pSR67 |
| pSR69 {PF53F4.13:KCC-2deletion1:mScarlet:unc-54 3’UTR} | This study | pASJ277/pSR69 |
| pSR71 {PF53F4.13:KCC-2deletion2:mScarlet:unc-54 3’UTR} | This study | pASJ290/pSR71 |
| pSR73 {PF53F4.13:KCC-3deletion4:mScarlet:unc-54 3’UTR} | This study | pASJ291/pSR73 |
| pSR75 {PF53F4.13:KCC-3deletion6:mScarlet:unc-54 3’UTR} | This study | pASJ292/pSR75 |
| pSR77 {PR13H4.1:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ293/pSR77 |
| pSR79 {Pceh-36:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ295/pSR79 |
| pSR82 {Podr-10:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ297/pSR82 |
| pSR83 {PF53F4.13:ChimeraH:mScarlet:unc-54 3’UTR} | This study | pASJ328/pSR83 |
| pSR85 {PR102.2:DYF-11:GFP:unc-54 3’UTR} | This study | pASJ381/pSR85 |
| pSR87 {PF53F4.13:ChimeraI:mScarlet:unc-54 3’UTR} | This study | pASJ383/pSR87 |
| pCM7 {PF53F4.13:SAX-7deltacyt:sfGFP:unc-54 3’UTR} | Martin et al. (2022)28 | pASJ74/pCM7 |
| pCM11 {PF53F4.13:SAX-7:mApple:unc-54 3’UTR} | Martin et al. (2022)28 | pASJ82/pCM11 |
| pAS272 {PF53F4.13:KCC-3:mCherry:unc-54 3’UTR} | This study | pAS272 |
| pCF27 {PVAP-1:VAP-1:sfGFP:unc-54 3’UTR} | This study | pAS326/pCF27 |
| pAB47 {PF53F4.13:SL2:mKate2:unc-54 3’UTR} | This study | pAS548/pAB47 |
| pJF48 {Psrtx-1:EGL-1:unc-54 3’UTR} | Raiders etal. (2021)93 | pAS447/pJF48 |
| pAB44 {PF53F4.13:mKate2:unc-54 3’UTR} | This study | pAS545/pAB44 |
| pOO1 {PF53F4.13:PH-PLCdelta:GFP} | This study | pASJ350/pOO1 |
| pAN1 {PDYF-11:DYF-11:GFP} | This study | pASJ9/pAN 1 |
| pPG6 {Phlh-17:KCC-3:mScarlet} | This study | pASJ410/pPG6 |
| Pmig-24:venus | Abraham et al. (2007)94 | N/A |
| Punc-122:GFP | Miyabayashi et al. (1999)95 | N/A |
| Punc-122:RFP | Miyabayashi et al. (1999)95 | N/A |
| Pelt-2:mCherry | Armenti et al. (2014)96 | N/A |
| pBluescript | Melloetal. (1991)97 | N/A |
| L4440 empty vector | Ahringer RNAi library | N/A |
| pros-1 RNAi | Ahringer RNAi library | WBGene00000448 |
| wnk-1 RNAi | Ahringer RNAi library | WBGene00006941 |
| kcc-3:GFP fosmid | TransgeneOme | WBGene00019205 Clone ID:9914866399944241 H10 |
|
| ||
| Software and algorithms | ||
|
| ||
| FIJI | ImageJ | https://fiji.sc/; RRID:SCR_002285 |
| Prism 10 | GraphPad | https://www.graphpad.com/; RRID:SCR_002798 |
| ApE | Wayne Davis | https://jorgensen.biology.utah.edu/wayned/ape/; RRID:SCR_014266 |
METHOD DETAILS
Germline transformation and integration
Germ-line transformations by micro-injection to generate unstable extra-chromosomal array transgenes were carried out using standard protocols.97 Integration of extra-chromosomal arrays was performed using UV irratdiation. All transgenic arrays were generated with Pmig-24:Venus, Punc-122:GFP, Pelt-2:mCherry, or Punc-122:RFP as co-injection markers.94–96
Plasmids
All plasmids transformed into either DH5-alpha or XL-10 Gold competent cells.
PAMsh:KCC-3:worm-mScarlet and PAMsh:CFP:SL2:KCC-3:worm-mScarlet
Codon-optimized worm mScarlet was synthesized by Genewiz with flanking AgeI/EcoRI digestion sites and cloned into pAB44 using AgeI/EcoRI to make pSR1 (pF53:worm-mScarlet). mScarlet and the unc-54 3′UTR was cloned into pAS272 (PAMsh:KCC-3:mcherry) using Age1/Apa1 from pSR1 to make pSR5 (PAMsh:KCC-3:mScarlet [out of frame]). Site-directed mutagenesis was used to add TATG in front of mScarlet in pSR5 to make pSR7 (PAMsh:KCC-3:mScarlet [in frame]).
CFP was PCR amplified from pIL43 (gift from Max Heiman) with flanking FseI/XbaI sites and inserted into pAS548 (pSM:SL2:mCherry) to make pSR9 (pSM:CFP:SL2:mCherry). CFP-SL2 was then PCR amplified from pSR9 and inserted into pSR7 (PAMsh:KCC-3:mScarlet) using FseI/AscI to make pSR11 (PAMsh:CFP:SL2:KCC-3:mScarlet).
KCC-1/KCC-2
cDNA for C. elegans KCC-1 (WB-GENE: WBGene00006504) and KCC-2 (WB-GENE: WBGene00019205) were PCR amplified in one (KCC-2) or two (KCC-1) segments from a mixed stage cDNA library and cloned into pSR7 using BamH1/EcoRI (KCC-1) or BamH1/SalI (KCC-2) to generate pSR17 (PAMsh:KCC-1:mScarlet) and pSR61/62 (PAMsh:KCC-2:mScarlet).
KCC-2/KCC-3 chimeras
We used a PCR fusion based approach to create KCC-2/KCC-3 chimera proteins.10 Briefly, 2 or more segments of KCC-2 or KCC-3 were PCR amplified with nested primers. The 3′ primer for every segment but the last included a 24bp overhang to the following segment. In the first PCR, all KCC segments are amplified independently. In the second PCR, nested primers fuse the independent segments to create one contiguous KCC chimeric protein. This product is then inserted into pSR7 using BamH1/Sal1 sites to create the final plasmid.
KCC-2/KCC-3 deletions and mutations
Small (<12 bp) deletions in KCC-3 or KCC-2 were induced using the Q5 site-directed mutagenesis kit. Base pair changes were induced using standard site-directed mutagenesis protocols.101
DYF-11 rescue constructs
All promoters were PCR amplified from gDNA of mixed stage animals and inserted into pAN1 (Pdyf-11:DYF-11:GFP) using either SphI/SalI or Gibson assembly. Promoters cloned: Pgpa-3 (5.4 kb upstream start codon), Ptax-4 (3.1 kb upstream start codon), Pflp-19 (3.6 kb upstream start codon), Podr-1 (2.4 kb upstream start codon), PR102.2 (602 bp upstream start codon), Pceh-36 (3 kb upstream start codon), and Podr-4 (2.3 kb upstream start codon). PR13H4.1 (5.7 kb upstream start codon) and Podr-10 (1 kb upstream start codon) were also cloned but failed to show expression.
RNAi
Plasmids expressing double-stranded RNA were obtained from the Ahringer Library.102 The L4440 empty vector was used as a negative control and pros-1 RNAi used as positive control. Synchronized L1 animals were fed RNAi bacteria.103 L4 animals were moved to a fresh plate of RNAi bacteria and scored 24 h later for KCC-3 localization. RNAi experiments were done on ASJ306 (dnaIs10 V; nsIs228 I).
Microscopy, image processing and analysis
Worms were immobilized with 40mM sodium azide and placed on 2% SEAKEM agarose pads. Images were collected on a Deltavision Elite RoHS wide-field deconvolution system, 40x/1.3 NA oil-immersion or OLY 100×/1.40 NA oil-immersion objective and a DV Elite CMOS Camera. Some images were also captured on a VisiTech iSIM super resolution microscope. Image processing was done in FIJI ImageJ.
Laser ablation
Laser ablation was done on the photoactivation system on the Deltavision Elite RoHS wide-field microscope. L1 animals were picked and mounted on an agar pad on a glass microscope slide. One AFD neuron was focused with a 100× objective plus digital zoom. The neuron was blasted with a 405 laser at 100% for 2s to kill the neuron. Animals were rescued off the agar pad and allowed to recover on an NGM plate. AFD cell death was assessed the following day for lack of GFP fluorescence in AFD. Animals were scored for KCC-3 localization one day later, in day 1 adults.
Dyf-11 rescue experiments
All constructs were injected into ASJ583 (dyf-11(mn392); dnaIs10 [PAMsh:KCC-3:mScarlet]). For blinded scoring, both extra-chromosomal array-positive and negative animals were assessed for KCC-3 localization first and then for presence of the rescue construct.
Chimera neuron shape rescue experiments
For blinded scoring, both extra-chromosomal array-positive and negative animals were assessed for neuron (AFD/AWA) shape first and then for presence of the rescue construct.
Behavioral assays
All chemotaxis behavioral assays as previously described.82 Briefly, animals are placed at the black dot, 1 μL odorant diluted in ethanol is placed at +, and 1 μL ethanol is placed at − (Figure 5G). 1 μL of 1M sodium azide is also placed at both + and − points to anesthetize animals. Animals are allowed to explore on the plate for 1hr. All assays performed on day 1 adult animals. The Chemotaxis Index (CI) was calculated as and Modified Chemotaxis Index (CImod) was calculated as ; to account for the larger population of “non-deciding” animals. Statistical analysis was performed with unpaired t test (Graphpad).
Calcium imaging
Calcium imaging was conducted in microfluidic chambers.104 Imaging was performed at 7 Hz on a Leica DMi8 inverted microscope with a 63x/1.40 NA oil immersion objective and an Andor iXon Life 888 EMCCD camera, using Leica LAS-X software. L4 animals were picked 24 h prior to imaging and left in 20°C overnight. Animals were immobilized using 1 mM levamisole prior to being loaded into the imaging chamber. Animals were presented with alternating S-Basal buffer and 0.01% isoamyl alcohol diluted in S-basal buffer. Image analysis was performed using ImageJ. ΔF/F was calculated by (F-F0)/F0, where F0 was designated by the average fluorescent intensity prior to odor presentation (t = 1s - t = 10s). All data was collected as “nascent” response, wherein animals were not pre-exposed to IAA prior to assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data and statistics were graphed and analyzed using Prism9. Proportional data is presented as the proportional sum across multiple days of data collection ±95% confidence interval. Two-Sided Fisher’s Exact tests were performed to compare across genotypes. One-way ANOVAs with Bonferroni’s multiple comparisons were performed to assess peak responses for the calcium imaging dataset.
Supplementary Material
Highlights.
C. elegans AMsh glia form distinct molecular microdomains of cues around different neurons
Glial KCC-3 localizes only to membranes contacting AFD temperature-sensing neurons
Distal non-AFD neuron cilia repel glial KCC-3 and constrain it to a microdomain around AFD
Glial KCC-3 localization regulates AFD and non-AFD neuron function and associated behaviors
ACKNOWLEDGMENTS
We thank the Singhvi lab and Jihong Bai for discussions and comments on the manuscript. We thank lab members Cecilia Martin, James Bent, Alex Neitz, Violet Sorrentino, and Olivia Okamoto; Piali Sengupta, Shai Shaham, Max Heiman, and Jihong Bai for generously sharing reagents; and the Bai lab including Aaradhya Pant for help with calcium imaging studies. S.R. was funded by a T32 training grant (5T32NS099578-03). This work was funded by a Simons Foundation/SFARI grant (488574), the Esther A. & Joseph Klingenstein Fund and the Simons Foundation Award in Neuroscience (227823), a Brain Research Foundation seed grant (BRFSG-2023-10), and NIH/NINDS funding (NS114222) (to A.S.). This work was performed while A.S. was a Glenn Foundation for Medical Research and AFAR Junior Faculty Grant Awardee. A.S. is sincerely thankful for philanthropic support to her laboratory, including from the Stephanus, George Brown, and Van Sloun Foundations. This research was supported by the Cellular Imaging and Genomics Shared Resources of the Fred Hutch/University of Washington/Seattle Children’s Cancer Consortium (P30 CA015704). Some strains were sourced from the CGC (funding: NIH Office of Research Infrastructure Programs, P40 OD010440), the International C. elegans Gene Knockout Consortium (funding: NIH), the C. elegans Reverse Genetics Core Facility at the University of British Columbia (funding: Canadian Institute for Health Research, Genome Canada, Genome BC, the Michael Smith Foundation, and NIH), and the National BioResource Project (NBRP), Japan.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113844.
REFERENCES
- 1.von Bartheld CS, Bahney J, and Herculano-Houzel S (2016). The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 524, 3865. 10.1002/CNE.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barres BA (2008). Perspective The Mystery and Magic of Glia : A Perspective on Their Roles in Health and Disease. Neuron 60, 430–440. 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Allen NJ, and Eroglu C (2017). Cell Biology of Astrocyte-Synapse Interactions. Neuron 96, 697–708. 10.1016/j.neuron.2017.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung WS, Welsh CA, Barres BA, and Stevens B (2015). Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 18, 1539–1545. 10.1038/nn.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eroglu Ç, Allen NJ, Susman MW, O’Rourke NA, Park CY, Özkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, et al. (2009). Gabapentin Receptor α2δ−1 Is a Neuronal Thrombospondin Receptor Responsible for Excitatory CNS Synaptogenesis. Cell 139, 380–392. 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji R-R, and Eroglu C (2017). Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551, 192–197. 10.1038/nature24638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sparrrow R,J, Hicks DP, Hamel C, Sparrow JR, Hicks D, Hamel CP, Sparrrow R,J, Hicks D, and Hamel CP (2010). The Retinal Pigment Epithelium in Health and Disease. Curr. Mol. Med. 10, 802–823. 10.2174/156652410793937813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez YA, Roebber JK, Dvoryanchikov G, Makhoul V, Roper SD, and Chaudhari N (2021). “Tripartite Synapses” in Taste Buds: A Role for Type I Glial-like Taste Cells. J. Neurosci. 41, 9860–9871. 10.1523/JNEUROSCI.1444-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khakh BS, and Sofroniew MV (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952. 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bacaj T, Tevlin M, Lu Y, and Shaham S (2008). Glia are essential for sensory organ function in C. elegans. Science 322, 744–747. 10.1126/science.1163074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang SM, Michel K, Jokhi V, Nedivi E, and Arlotta P (2020). Neuron-class specific responses govern adaptive myelin remodeling in the neocortex. Science 370. 10.1126/SCIENCE.ABD2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belin S, Zuloaga KL, and Poitelon Y (2017). Influence of mechanical stimuli on schwann cell biology. Front. Cell. Neurosci. 11, 347. 10.3389/FNCEL.2017.00347/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pogodalla N, Kranenburg H, Rey S, Rodrigues S, Cardona A, and Klämbt C (2021). Drosophila ßHeavy-Spectrin is required in polarized ensheathing glia that form a diffusion-barrier around the neuropil. Nat. Commun. 12. 10.1038/S41467-021-26462-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L, and Oliet SHR (2015). Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 18, 219–226. 10.1038/NN.3901. [DOI] [PubMed] [Google Scholar]
- 15.Singhvi A, and Shaham S (2019). Glia-Neuron Interactions in Caenorhabditis elegans. Annu. Rev. Neurosci. 42, 149–168. 10.1146/annurev-neuro-070918-050314. [DOI] [PubMed] [Google Scholar]
- 16.Singhvi A, Shaham S, and Rapti G (2023). Glia development and function in the nervous system of Caenorhabditis elegans. Cold Spring Harbor Perspect. Biol. 10.1101/cshperspect.a041346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oikonomou G, and Shaham S (2011). The glia of caenorhabditis elegans. Glia 59, 1253–1263. 10.1002/glia.21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward S, Thomson N, White JG, and Brenner S (1975). Electron microscopical reconstruction of the anterior sensory anatomy of the nematode caenorhabditis elegans. J. Comp. Neurol. 160, 313–337. 10.1002/cne.901600305. [DOI] [PubMed] [Google Scholar]
- 19.Perkins LA, Hedgecock EM, Thomson JN, and Culotti JG (1986). Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev. Biol. 117, 456–487. 10.1016/0012-1606(86)90314-3. [DOI] [PubMed] [Google Scholar]
- 20.Perens EA, and Shaham S (2005). C. elegans daf-6 encodes a patched-related protein required for lumen formation. Dev. Cell 8, 893–906. 10.1016/j.devcel.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 21.Kimura KD, Miyawaki A, Matsumoto K, and Mori I (2004). The C. elegans Thermosensory Neuron AFD Responds to Warming. Curr. Biol. 14, 1291–1295. 10.1016/J.CUB.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 22.Goodman MB, and Sengupta P (2018). The extraordinary AFD thermosensor of C. elegans. Pflügers Archiv 470, 839. 10.1007/S00424-017-2089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace SW, Singhvi A, Liang Y, Lu Y, Shaham S, Wallace SW, Singhvi A, Liang Y, Lu Y, and Shaham S (2016). PROS-1/Prospero Is a Major Regulator of the Glia- Specific Secretome Controlling Sensory-Neuron Shape and Function in C. elegans Article PROS-1/Prospero Is a Major Regulator of the Glia-Specific Secretome Controlling Sensory-Neuron Shape and Function. Cell Rep. 15, 550–562. 10.1016/j.celrep.2016.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singhvi A, Liu B, Friedman CJ, Fong J, Lu Y, Huang XY, and Shaham S (2016). A Glial K/Cl Transporter Controls Neuronal Receptive Ending Shape by Chloride Inhibition of an rGC. Cell 165, 936–948. 10.1016/j.cell.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raiders S, Han T, Scott-Hewitt N, Kucenas S, Lew D, Logan MA, and Singhvi A (2021). Engulfed by Glia: Glial Pruning in Development, Function, and Injury across Species. J. Neurosci. 10.1523/JNEUROSCI.1660-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida A, Nakano S, Suzuki T, Ihara K, Higashiyama T, and Mori I (2016). A glial K+/Cl− cotransporter modifies temperature-evoked dynamics in Caenorhabditis elegans sensory neurons. Gene Brain Behav. 15, 429–440. 10.1111/gbb.12260. [DOI] [PubMed] [Google Scholar]
- 27.Low IIC, Williams CR, Chong MK, Mclachlan IG, Wierbowski BM, Kolotuev I, and Heiman MG (2019). Morphogenesis of Neurons and Glia within an Epithelium. Development 146. 10.1242/dev.171124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin CG, Bent JS, and Singhvi A (2022). Epithelia delimits glial apical polarity against mechanical shear to maintain glia-neuron - architecture. Preprint at bioRxiv. 10.1101/2022.12.26.521704. [DOI] [Google Scholar]
- 29.Mahon MJ (2011). Apical membrane segregation of phosphatidylinositol-4,5-bisphosphate influences parathyroid hormone 1 receptor compartmental signaling and localization via direct regulation of ezrin in LLC-PK1 cells. Cell. Signal. 23, 1659. 10.1016/J.CELL-SIG.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin K, Fogg VC, and Margolis B (2006). Tight Junctions and Cell Polarity. Annu. Rev. Cell Dev. Biol. 22, 207–235. 10.1146/ANNUREV.CELLBIO.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 31.McMahon L, Legouis R, Vonesch JL, and Labouesse M (2001). Assembly of C. elegans apical junctions involves positioning and compaction by LET-413 and protein aggregation by the MAGUK protein DLG-1. J. Cell Sci. 114, 2265–2277. 10.1242/JCS.114.12.2265. [DOI] [PubMed] [Google Scholar]
- 32.Segbert C, Johnson K, Theres C, Van Fürden D, and Bossinger O (2004). Molecular and functional analysis of apical junction formation in the gut epithelium of Caenorhabditis elegans. Dev. Biol. 266, 17–26. 10.1016/j.ydbio.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 33.Kö ppen M, Simske JS, Sims PA, Firestein BL, Hall DH, Radice AD, Rongo C, and Hardin JD (2001). Cooperative regulation of AJM-1 controls junctional integrity in Caenorhabditis elegans epithelia. Nat. Cell Biol. 3, 983–991. 10.1038/NCB1101-983. [DOI] [PubMed] [Google Scholar]
- 34.Oikonomou G, Perens EA, Lu Y, Watanabe S, Jorgensen EM, and Shaham S (2011). Opposing Activities of LIT-1/NLK and DAF-6/Patched-Related Direct Sensory Compartment Morphogenesis in C. elegans. PLoS Biol. 9, e1001121. 10.1371/journal.pbio.11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oikonomou G, Perens EA, Lu Y, and Shaham S (2012). Some, but not all, retromer components promote morphogenesis of C. elegans sensory compartments. Dev. Biol. 362, 42–49. 10.1016/j.yd-bio.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purice MD, Quitevis EJA, Manning RS, Severs LJ, Tran N-T, Sorrentino V, Setty M, and Singhvi A (2023). Molecular heterogeneity of C. elegans glia across sexes. Preprint at bioRxiv. 10.1101/2023.03.21.533668. [DOI] [Google Scholar]
- 37.Tanis JE, Bellemer A, Moresco JJ, Forbush B, and Koelle MR (2009). The Potassium Chloride Cotransporter KCC-2 Coordinates Development of Inhibitory Neurotransmission and Synapse Structure in Caenorhabditis elegans. J. Neurosci. 29, 9943–9954. 10.1523/JNEUROSCI.1989-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hisamoto N, Moriguchi T, Urushiyama S, Mitani S, Shibuya H, and Matsumoto K (2008). Caenorhabditis elegans WNK-STE20 pathway regulates tube formation by modulating ClC channel activity. EMBO Rep. 9, 70–75. 10.1038/sj.embor.7401128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payne JA, Rivera C, Voipio J, and Kaila K (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206. 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 40.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, and Kahle KT (2014). The WNK-SPAK/OSR1 pathway: Master regulator of cationchloride cotransporters. Sci. Signal. 7, 1–11. 10.1126/sci-signal.2005365. [DOI] [PubMed] [Google Scholar]
- 41.Kaila K, Price TJ, Payne JA, Puskarjov M, and Voipio J (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blaesse P, Airaksinen MS, Rivera C, and Kaila K (2009). CationChloride Cotransporters and Neuronal Function. Neuron 61, 820–838. 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Salin-Cantegrel A, Rivière JB, Shekarabi M, Rasheed S, DaCal S, Laganière J, Gaudet R, Rochefort D, Lesca G, Gaspar C, et al. (2011). Transit defect of potassium-chloride co-transporter 3 is a major pathogenic mechanism in hereditary motor and sensory neuropathy with agenesis of the corpus callosum. J. Biol. Chem. 286, 28456–28465. 10.1074/jbc.M111.226894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burgess A, Shah K, Hough O, and Hynynen K (2016). C. elegans S6K Mutants Require a Creatine Kinase-Like Effector for Lifespan Extension. Cell Rep. 15, 477–491. 10.1586/14737175.2015.1028369.Focused. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simard CF, Bergeron MJ, Frenette-Cotton R, Carpentier GA, Pelchat ME, Caron L, and Isenring P (2007). Homooligomeric and Heterooligomeric Associations between K+-Cl− Cotransporter Isoforms and between K+-Cl− and Na+-K+-Cl− Cotransporters. J. Biol. Chem. 282, 18083–18093. 10.1074/JBC.M607811200. [DOI] [PubMed] [Google Scholar]
- 46.Doroquez DB, Berciu C, Anderson JR, Sengupta P, and Nicastro D (2014). A high-resolution morphological and ultrastructural map of anterior sensory cilia and glia in Caenorhabditis elegans. Elife 2014, 1–35. 10.7554/eLife.01948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satterlee JS, Sasakura H, Kuhara A, Berkeley M, Mori I, and Sengupta P (2001). Specification of thermosensory neuron fate in C. elegans requires ttx-1, a homolog of otd/Otx. Neuron 31, 943–956. 10.1016/S0896-6273(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 48.Swoboda P, Adler HT, and Thomas JH (2000). The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. elegans. Mol. Cell 5, 411–421. 10.1016/S1097-2765(00)80436-0. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen PAT, Liou W, Hall DH, and Leroux MR (2014). Ciliopathy proteins establish a bipartite signaling compartment in a C. elegans thermosensory neuron. J. Cell Sci. 127, 5317–5330. 10.1242/JCS.157610/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kunitomo H, Uesugi H, Kohara Y, and Iino Y (2005). Identification of ciliated sensory neuron-expressed genes in Caenorhabditis elegans using targeted pull-down of poly(A) tails. Genome Biol. 6, R17. 10.1186/gb-2005-6-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coburn CM, and Bargmann CI (1996). A putative cyclic nucleotide-gated channel is required for sensory development and function in C. elegans. Neuron 17, 695–706. 10.1016/S0896-6273(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 52.Nehme R, and Conradt B (2009). egl-1: a key activator of apoptotic cell death in C. elegans. Oncogene 27, S30–S40. 10.1038/onc.2009.41. [DOI] [PubMed] [Google Scholar]
- 53.Sulston JE (1983). Neuronal cell lineages in the nematode Caenorhabditis elegans. Cold Spring Harbor Symp. Quant. Biol. 48 (Pt 2), 443–452. 10.1101/sqb.1983.048.01.049. [DOI] [PubMed] [Google Scholar]
- 54.Dwyer ND, Adler CE, Crump JG, L’Etoile ND, and Bargmann CI (2001). Polarized Dendritic Transport and the AP-1 μ1 Clathrin Adaptor UNC-101 Localize Odorant Receptors to Olfactory Cilia. Neuron 31, 277–287. 10.1016/S0896-6273(01)00361-0. [DOI] [PubMed] [Google Scholar]
- 55.Lechtreck KF (2015). IFT-cargo interactions and protein transport in cilia. Trends Biochem. Sci. 40, 765. 10.1016/J.TIBS.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bae YK, and Barr MM (2008). Sensory roles of neuronal cilia: Cilia development, morphogenesis, and function in C. elegans. Front. Biosci. 13, 5959. 10.2741/3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Howell K, and Hobert O (2017). Morphological Diversity of C. elegans Sensory Cilia Instructed by the Differential Expression of an Immunoglobulin Domain Protein. Curr. Biol. 27, 1782–1790. 10.1016/J.CUB.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 58.Bacaj T, Lu Y, and Shaham S (2008). The Conserved Proteins CHE12 and DYF-11 Are Required for Sensory Cilium Function in Caenorhabditis elegans. Genetics 178, 989. 10.1534/GENETICS.107.082453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lanjuin A, VanHoven MK, Bargmann CI, Thompson JK, and Sengupta P (2003). Otx/otd homeobox genes specify distinct sensory neuron identities in C. elegans. Dev. Cell 5, 621–633. 10.1016/s1534-5807(03)00293-4. [DOI] [PubMed] [Google Scholar]
- 60.Uchida O, Nakano H, Koga M, and Ohshima Y (2003). The C. elegans che-1 gene encodes a zinc finger transcription factor required for specification of the ASE chemosensory neurons. [DOI] [PubMed] [Google Scholar]
- 61.Sengupta P, Colbert HA, and Bargmann CI (1994). The C. elegans gene odr-7 encodes an olfactory-specific member of the nuclear receptor superfamily. Cell 79, 971–980. 10.1016/0092-8674(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 62.Lanjuin A, and Sengupta P (2004). Specification of chemosensory neuron subtype identities in Caenorhabditis elegans. Curr. Opin. Neurobiol. 14, 22–30. 10.1016/J.CONB.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 63.Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB, and Bargmann CI (2007). Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature 450, 63–70. 10.1038/nature06292. [DOI] [PubMed] [Google Scholar]
- 64.Sengupta P (2017). Cilia and sensory signaling: The journey from “animalcules” to human disease. PLoS Biol. 15, e2002240. 10.1371/JOURNAL.PBIO.2002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Green JA, and Mykytyn K (2014). Neuronal Primary Cilia: An Underappreciated Signaling and Sensory Organelle in the Brain. Neuropsycho-pharmacology 39, 244. 10.1038/NPP.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ki SM, Jeong HS, and Lee JE (2021). Primary Cilia in Glial Cells: An Oasis in the Journey to Overcoming Neurodegenerative Diseases. Front. Neurosci. 15, 1227. 10.3389/FNINS.2021.736888/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Razzauti A, and Laurent P (2021). Ectocytosis prevents accumulation of ciliary cargo in c. Elegans sensory neurons. Elife 10. 10.7554/ELIFE.67670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang J, and Barr MM (2018). Cell-cell communication via ciliary extracellular vesicles: clues from model systems. Essays Biochem. 62, 205–213. 10.1042/EBC20170085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leterrier C (2018). The Axon Initial Segment: An Updated Viewpoint. J. Neurosci. 38, 2135–2145. 10.1523/JNEUROSCI.1922-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mazare N, Oudart M, and Cohen-Salmon M (2021). Local translation in perisynaptic and perivascular astrocytic processes - a means to ensure astrocyte molecular and functional polarity? J. Cell Sci. 134. 10.1242/JCS.251629. [DOI] [PubMed] [Google Scholar]
- 71.Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, et al. (2003). Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 22, 5422–5434. 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Delpire E, and Kahle KT (2017). The KCC3 cotransporter as a therapeutic target for peripheral neuropathy. Expert Opin. Ther. Targets 21, 113–116. 10.1080/14728222.2017.1275569. [DOI] [PubMed] [Google Scholar]
- 73.Garneau AP, Marcoux A-A, Frenette-Cotton R, Mac-Way F, Lavoie JL, and Isenring P (2017). Molecular insights into the normal operation, regulation and multisystemic roles of K + -Cl − cotransporter 3 (KCC3). Am. J. Physiol. Cell Physiol. 3, 00106. 10.1152/ajpcell.00106.2017. [DOI] [PubMed] [Google Scholar]
- 74.Shekarabi M, Moldrich RX, Rasheed S, Salin-Cantegrel A, Laganiere J, Rochefort D, Hince P, Huot K, Gaudet R, Kurniawan N, et al. (2012). Loss of Neuronal Potassium/Chloride Cotransporter 3 (KCC3) Is Responsible for the Degenerative Phenotype in a Conditional Mouse Model of Hereditary Motor and Sensory Neuropathy Associated with Agenesis of the Corpus Callosum. J. Neurosci. 32, 3865–3876. 10.1523/JNEUROSCI.3679-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun YT, Lin TS, Tzeng SF, Delpire E, and Shen MR (2010). Deficiency of electroneutral K+-Cl− cotransporter 3 causes a disruption in impulse propagation along peripheral nerves. Glia 58, 1544–1552. 10.1002/glia.21028. [DOI] [PubMed] [Google Scholar]
- 76.Ray S, and Singhvi A (2021). Charging Up the Periphery: Glial Ionic Regulation in Sensory Perception. Front. Cell Dev. Biol. 9, 1989. 10.3389/FCELL.2021.687732/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Annunziato L, Boscia F, and Pignataro G (2013). Ionic transporter activity in astrocytes, microglia, and oligodendrocytes during brain ischemia. J. Cerebr. Blood Flow Metabol. 33, 969. 10.1038/JCBFM.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Y, D’Urso G, and Bianchi L (2012). Knockout of glial channel ACD-1 exacerbates sensory deficits in a C. elegans mutant by regulating calcium levels of sensory neurons. J. Neurophysiol. 107, 148–158. 10.1152/jn.00299.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van Den Akker F, Zhang X, Miyagl M, Huo X, Misono KS, and Yee VC (2000). Structure of the dimerized hormone-binding domain of a guanylyl-cyclase-coupled receptor. Nature 406, 101–104. 10.1038/35017602. [DOI] [PubMed] [Google Scholar]
- 80.Dutzler R, Campbell EB, and MacKinnon R (2003). Gating the selectivity filter in ClC chloride channels. Science 300, 108–112. 10.1126/SCIENCE.1082708. [DOI] [PubMed] [Google Scholar]
- 81.Bargmann CI, Thomas JH, and Horvitz HR (1990). Chemosensory cell function in the behavior and development of Caenorhabditis elegans. Cold Spring Harbor Symp. Quant. Biol. 55, 529–538. 10.1101/SQB.1990.055.01.051. [DOI] [PubMed] [Google Scholar]
- 82.Bargmann CI, Hartwieg E, and Horvitz HR (1993). Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell 74, 515–527. 10.1016/0092-8674(93)80053-H. [DOI] [PubMed] [Google Scholar]
- 83.Zaslaver A, Liani I, Shtangel O, Ginzburg S, Yee L, and Sternberg PW (2015). Hierarchical sparse coding in the sensory system of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 112, 1185–1189. 10.1073/PNAS.1423656112/SUPPL_FILE/PNAS.201423656SI.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kahle KT, Khanna AR, Alper SL, Adragna NC, Lauf PK, Sun D, and Delpire E (2015). K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 21, 513–523. 10.1016/j.molmed.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang X, Liu Y, Hong X, Li X, Meshul CK, Moore C, Yang Y, Han Y, Li WG, Qi X, et al. (2021). NG2 glia-derived GABA release tunes inhibitory synapses and contributes to stress-induced anxiety. Nat. Commun. 12, 1–18. 10.1038/s41467-021-25956-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agarwal A, Wu PH, Hughes EG, Fukaya M, Tischfield MA, Langseth AJ, Wirtz D, and Bergles DE (2017). Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 93, 587–605. 10.1016/j.neuron.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ding G, Zou W, Zhang H, Xue Y, Cai Y, Huang G, Chen L, Duan S, and Kang L (2015). In Vivo tactile stimulation-evoked responses in Caenorhabditis elegans amphid sheath glia. PLoS One 10. 10.1371/journal.pone.0117114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Duan D, Zhang H, Yue X, Fan Y, Xue Y, Shao J, Ding G, Chen D, Li S, Cheng H, et al. (2020). Sensory Glia Detect Repulsive Odorants and Drive Olfactory Adaptation. Neuron 108, 707–721.e8. 10.1016/j.neuron.2020.08.026. [DOI] [PubMed] [Google Scholar]
- 89.Wang Y, Apicella A, Lee SK, Ezcurra M, Slone RD, Goldmit M, Schafer WR, Shaham S, Driscoll M, and Bianchi L (2008). A glial DEG/ENaC channel functions with neuronal channel DEG-1 to mediate specific sensory functions in C. elegans. EMBO J. 27, 2388–2399. 10.1038/emboj.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Troemel ER, Sagasti A, and Bargmann CI (1999). Lateral signaling mediated by axon contact and calcium entry regulates asymmetric odorant receptor expression in C. elegans. Cell 99, 387–398. 10.1016/S0092-8674(00)81525-1. [DOI] [PubMed] [Google Scholar]
- 91.Katz M, Corson F, Keil W, Singhal A, Bae A, Lu Y, Liang Y, and Shaham S (2019). Glutamate spillover in C. elegans triggers repetitive behavior through presynaptic activation of MGL-2/mGluR5. Nat. Commun. 10, 1–13. 10.1038/s41467-019-09581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maurya AK, and Sengupta P (2021). xbx-4, a homolog of the Joubert syndrome gene FAM149B1, acts via the CCRK and RCK kinase cascade to regulate cilia morphology. Curr. Biol. 31, 5642. 10.1016/J.CUB.2021.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raiders S, Black EC, Bae A, MacFarlane S, Klein M, Shaham S, and Singhvi A (2021). Glia actively sculpt sensory neurons by controlled phagocytosis to tune animal behavior. Elife 10. 10.7554/eLife.63532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abraham MC, Lu Y, and Shaham S (2007). A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev. Cell 12, 73–86. 10.1016/J.DEV-CEL.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 95.Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, and Sengupta P (1999). Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans. Dev. Biol. 215, 314–331. 10.1006/DBIO.1999.9470. [DOI] [PubMed] [Google Scholar]
- 96.Armenti ST, Lohmer LL, Sherwood DR, and Nance J (2014). Repurposing an endogenous degradation system for rapid and targeted depletion of C. elegans proteins. Development 141, 4640–4647. 10.1242/DEV.115048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mello CC, Kramer JM, Stinchcomb D, and Ambros V (1991). Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970. 10.1002/J.1460-2075.1991.TB04966.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. 10.1093/GENETICS/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stiernagle T (2006). Maintenance of C. elegans. WormBook 11, 1–11. 10.1895/WORMBOOK.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hobert O (2018). PCR Fusion-Based Approach to Create Reporter Gene Constructs for Expression Analysis in Transgenic C. elegans. Bio-techniques 32, 728–730. 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
- 101.Liu H, and Naismith JH (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91. 10.1186/1472-6750-8-91/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kamath RS, and Ahringer J (2003). Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321. 10.1016/S1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 103.Timmons L (2004). Endogenous inhibitors of RNA interference in Caenorhabditis elegans. Bioessays 26, 715–718. 10.1002/BIES.20078. [DOI] [PubMed] [Google Scholar]
- 104.Chronis N, Zimmer M, and Bargmann CI (2007). Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat. Methods 4, 727–731. 10.1038/NMETH1075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.