

Abstract
Neuropeptides represent a unique class of signaling molecules that have garnered much attention but require special consideration when gleaning identifications from mass spectra. With highly variable sequence lengths, neuropeptides must be analyzed in their endogenous state. Further, neuropeptides share great homology within families, differing by as little as a single amino acid residue, complicating even routine analyses and necessitating optimized computational strategies for confident and accurate identifications. We present EndoGenius, a database searching strategy designed specifically for elucidating neuropeptide identifications from mass spectra by leveraging optimized peptide-spectrum matching approaches, an expansive motif database, and a novel scoring algorithm to achieve broader representation of the neuropeptidome and minimize re-identification. This work describes an algorithm capable of reporting more neuropeptide identifications at 1% false discovery rate than alternative software in five Callinectes sapidus neuronal tissue types.
Keywords: Neuropeptide, EndoGenius, homology, peptide, mass spectrometry, database searching, FDR, peptidomics, endogenous, digest-free
Graphical Abstract
Introduction
Neuropeptides, essential signaling molecules originating from neurons or endocrine cells, serve as a resource for obtaining dynamic information of neuronal processes.1 To date, neuropeptides have provided valuable insight into biological disorders including Alzheimer’s disease2,3 and obesity.4–6 Mass spectrometry (MS) is revered for its high accuracy and sensitivity, and has been utilized in many neuropeptide studies to date.7–10 With regards to processing MS data, and subsequently obtaining peptide-spectrum matches (PSMs), database searching is a routine method, which compares the theoretical spectrum of a peptide with an experimental spectrum to make identifications. The area of database searching has seen tremendous growth in recent decades, with many software packages available for use including MSFragger,11 MaxQuant,12 PEAKS,13 Comet,14 and MetaMorpheus,15 each of which were designed for application in proteomics investigations.
While in principle these software packages can be used for neuropeptidomic analyses, an apparent discrepancy between the number of anticipated neuropeptide identifications, supported by the literature,1,16 and the much lower actual number of identified neuropeptides by MS is evident. This discrepancy can be explained by the unique nature of neuropeptides, which warrant atypical search considerations. In vivo, neuropeptides are cleaved from prohormones in a largely uncharacterized manner, producing fragments ranging from three to greater than 70 amino acids in length.1 Thus, typical sample preparation involving enzymatic digestion can be detrimental, and neuropeptides are thus analyzed in their endogenous form. Though many software packages offer the ability to search in a digest-free condition, neuropeptides share high levels of sequence similarity, sometimes sharing all but a single amino acid residue in common, further complicating search tasks.
Software optimized for neuropeptide identifications are sparse, though recently our group presented two packages that complement de novo sequencing analysis methods, which demonstrated the benefit of using optimized software packages.7,17 Herein we detail a novel platform, EndoGenius, which capitalizes on modifications of existing database searching principles to achieve optimized database searching of neuropeptide extracts. EndoGenius is the first stand-alone platform designed specifically for the identification of neuropeptides from data-dependent acquisition (DDA) mass spectra. Traditional database search strategies provided a nice framework to model our system after, however they make assumptions, such as identity of termini residues, that are not applicable to our neuropeptide analyses,18–20 thus providing an area of optimization for our neuropeptide applications, through which we optimized a scoring method to relay the confidence of an identification. The premise of our DDA searching algorithm lies in generating theoretical spectra for a given neuropeptide sequence, including any modifications, and then searching this against experimental spectra to identify any matches, scoring the matches to reflect the confidence of the match.21–23 Typical methods used to represent the quality of a spectral match employ one of the following two approaches, either examining a false-discovery rate (FDR), often including decoy database entries in the search to calculate the likelihood of identifying a false entry, or employing scoring methods, which rely on a number of factors to arrive at a value which represents the probability of the match to be true.18,24,25 Representing the accuracy of a neuropeptide match through an score alone is not intuitive, while calculation of FDR using a decoy database alone can be ineffective at determining the correct sequences, a unique challenge due to inherent neuropeptide homology.25 Algorithms where a score is calculated based on metrics rely on properties such as the number of missed cleavages or the identity of the end residues of a given peptide, features which are not applicable to neuropeptide searches.12,26 For the program described herein we developed a scoring system that is ideal for neuropeptides, free of any irrelevant terms. We then use these scores in-tandem with a target-decoy strategy, to determine a score threshold corresponding to a particular FDR value, where peptides with scores greater than the threshold are accepted as hits (Figure 1). Altogether, the novel platform described herein, EndoGenius, achieves improved identification of a broad selection of neuropeptides with respect to alternative software solutions.
Figure 1:
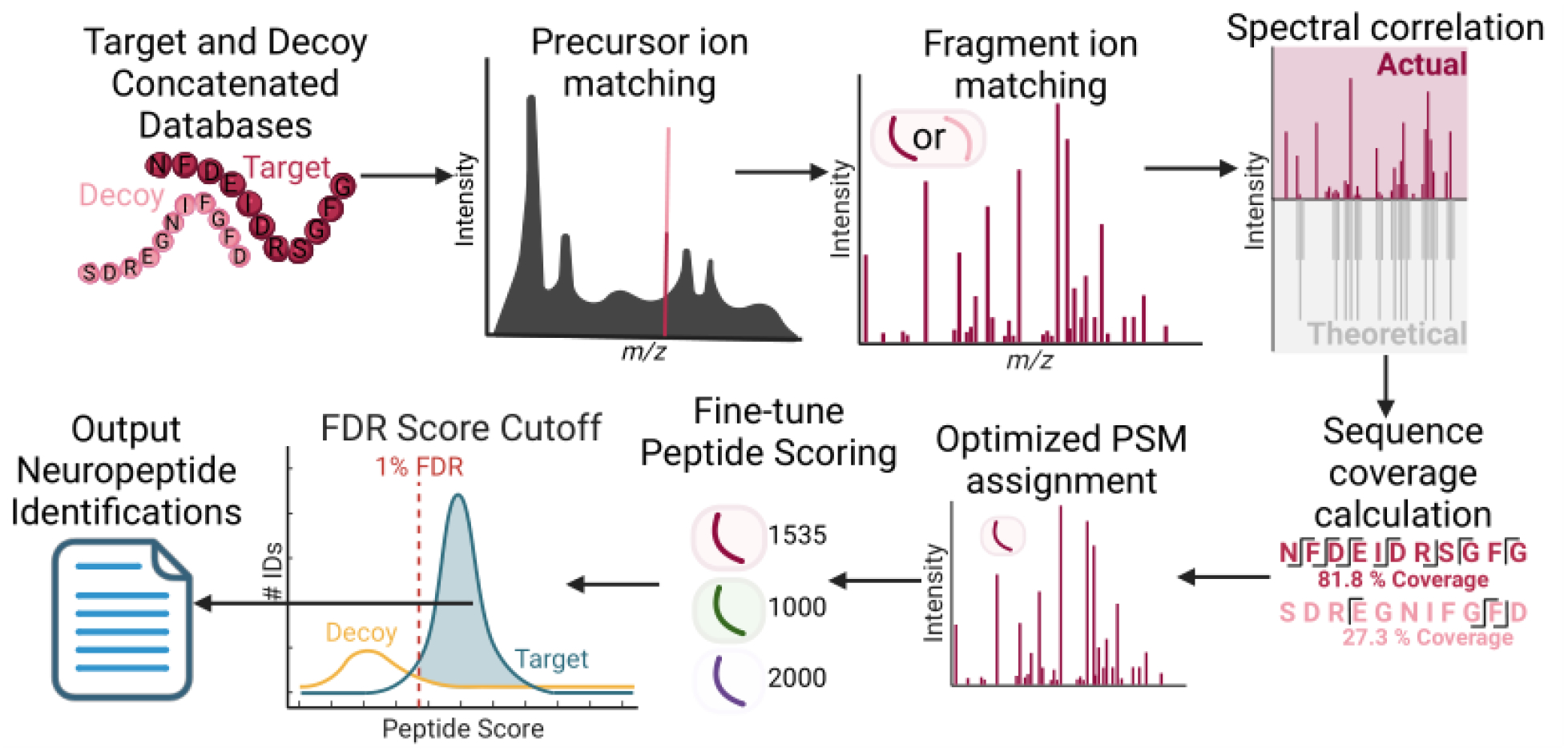
Generalized workflow of EndoGenius. From a target neuropeptide database, a shuffled decoy sequence is generated for each target. Target and decoy sequence databases are concatenated and shuffled to avoid biasing. Each peptide undergoes precursor and, if applicable, fragment ion matching, following which sequence coverage and spectral correlation calculations are completed. These data are input to the optimized peptide-spectrum match (PSM) assignment, which conducts a filtering to reward neuropeptide-typic attributes. Following a fine-tuning peptide scoring, a score is associated with a 1% false discovery rate (FDR) threshold, wherein peptides surpassing the threshold are exported as identifications.
Materials and Methods
Reagents and Materials
Crab saline components, as well as methanol (MeOH), acetonitrile (ACN), glacial acetic acid (GAA), ammonium bicarbonate, and formic acid (FA) were purchased from Fisher Scientific (Pittsburgh, PA). For this study, only HPLC grade or water (H2O) that was doubly distilled on a Millipore filtration system (Burlington, MA) were used. C18 Ziptips were purchased from Millipore (Burlington, MA). Optima Grade solvents were used for all LC (Fisher Scientific; Pittsburgh, PA).
Animals
Female blue crabs, C. sapidus, were obtained from Midway Asian Foods and subsequently stored in artificial seawater at 35 parts per thousand (ppt) salinity, 13–16 °C, and 8–10 ppm O2. Prior to sacrifice, crabs were anesthetized on ice for 20 minutes. Brain, sinus glands (SG), pericardial organs (PO), thoracic ganglia (TG), and commissural ganglia (CoG) were collected as previously described.27 Dissections were conducted in chilled (10 °C) physiological saline, composed of 440 mM NaCl, 11 mM KCl, 13 mM CaCl2, 26 mM MgCl2, and 10 mM Trizma acid. Saline was adjusted to pH 7.4 with NaOH.
Neuropeptide Sample Preparation and Data Acquisition
For each tissue type, three tissue samples were pooled. Neuropeptides were extracted from tissue using 90/9/1 (v/v/v) MeOH/H2O/GAA, and subsequently desalted via Millipore Ziptips. A solution of 0.1% FA in water was used to reconstitute the neuropeptide extracts and was then loaded onto a 15 cm capillary (75 μm internal diameter) which was packed with 1.7 μm diameter Ethylene Bridged Hybrid C18 material. The integrated emitter tip was confirmed to be in line with the instrument inlet.
Untargeted profiling of neuropeptides was conducted via LC-MS/MS with a Thermo Q-Exactive HF mass spectrometer coupled to a Dionex Ultimate 3000 LC system. Mobile phase A, 0.1% FA in H2O, and mobile phase B, 0.1% FA in ACN, were used to separate peptides with a gradient elution of 10 to 20% B over 70 min, and 20 to 95% B over 20 min at a flow rate of 300 nL/min. Profile mode was used to acquire full MS scans ranging from 200 to 2000 m/z at a resolution of 60 K. The automatic gain control (AGC) target was set to 1 × 106 with a maximum injection time of 250 ms. Tandem mass spectra were acquired in centroid mode. The top 10 most abundant precursor ions were selected for higher-energy collisional dissociation (HCD) fragmentation with a dynamic exclusion window of 30 s. A resolution power of 15 K, isolation window of 2.0 Th, normalized collision energy (NCE) of 30, maximum injection time of 120 ms, AGC target of 2×105, and fixed first mass of m/z 100 were set as parameters for data-dependent acquisition (DDA). Each sample was injected in technical triplicate.
Software Notes
The EndoGenius algorithm and graphical user-interface (GUI) were written in Python. The program is compatible with Python 3 and was validated with Python v.3.10 in an Anaconda environment. The program is open-source and freely available at https://github.com/lingjunli-research/EndoGenius, with a user manual and tutorial included. A schematic of the program workflow is shown in Figure 1. The GUI was built in Figma and converted to Python using Tkinter Designer program.
All data files were processed on a Dell Inc. Precision Tower 5810 computer using Windows 10 with a 64-Bit processor and 4 cores operating at 3.10 GHz and 128 GB installed RAM.
Target Decoy Database
A target-decoy database was built using a decoy-shuffle strategy, with a previously reported crustacean neuropeptide database (891 unique neuropeptides).7 A Biopython package was used to parse the neuropeptide database from .FASTA format.28 Using the Python Itertools package, decoy-shuffle sequences were generated and concatenated to the original database. The sequence order of all entries within the concatenated database were scrambled to minimize biasing.
Motif Database
The motif database was built upon the previously reported crustacean neuropeptide motif database, using the same methodology.17 In brief, the neuropeptide database, described previously,7 was divided into families, derived from their structural homology.29 All peptides within a family were aligned through the UniProt Align platform.30 The alignment produced a series of peptides adjusted to the same length, which were input to the WebLogo platform.31 A full motif was defined as a series of unambiguous, continuous amino acids within all familial sequences. A partial motif was designed as an extension to a full motif, where one variable or ambiguous amino acid position was allowed, provided it was flanked by two unambiguous amino acid residues. A complete depiction of the motif database generation workflow is shown in Figure 2a.
Figure 2:
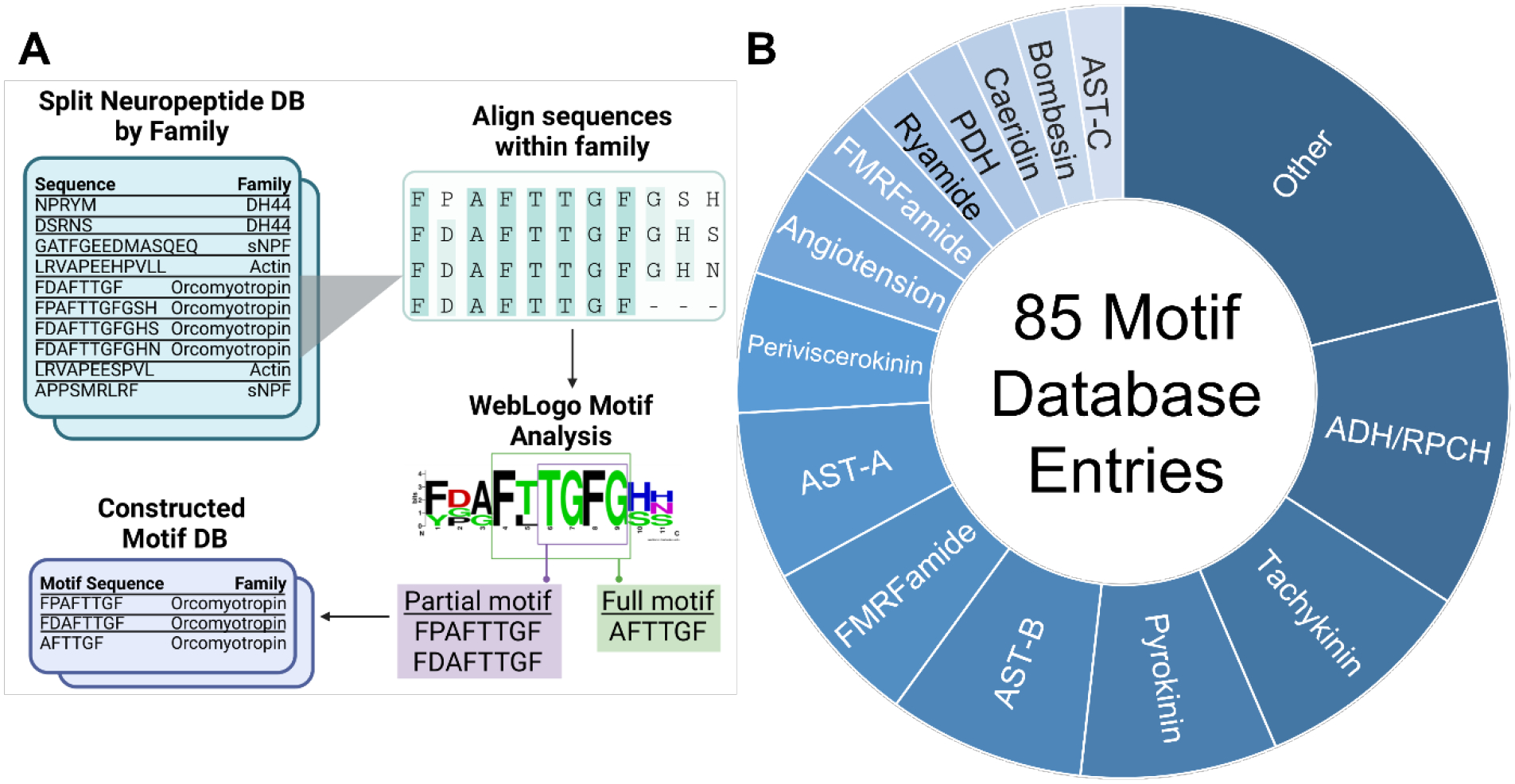
(A) A motif database generation workflow was constructed, by which a list of target neuropeptides was divided into sublists based on familial association. All peptides within a given neuropeptide family were aligned, from which full motifs, or regions of complete alignment, or partial motifs, wherein two regions of complete alignment were separated by a single variable residue, were assigned. (B) Using the motif algorithm, a selection of 85 motif entries were assigned, corresponding to 23 families.
Motif Scoring
To assign a score to a motif-sequence match, a ratio score (SA) was first assigned, determined by dividing the length of the motif (LM) by the length of the neuropeptide (LN; Equation 1). As this first step can be biased against a singular, short motif present in a lengthy neuropeptide, we applied a square root normalization procedure (Equation 2).32,33 The normalized ratio score was then multiplied by the neuropeptide’s sequence coverage (CS), or the number of expected fragment ions present, to reward motif-peptide matches with strong experimental evidence. The sequence coverage calculation was produced by dividing the number of experimental fragment ions present (NE) by the number of theoretical fragment ions (NT) expected (Equation 3). The final motif score (SF) is reported as the normalized ratio score (SB) multiplied by the sequence coverage (CS; Equation 4).
| Equation 1 |
| Equation 2 |
| Equation 3 |
| Equation 4 |
Database Search Methods
In preparation of database searching, Thermo .RAW files were converted to .MS2 files via RawConverter.34 Raw files were also converted to .MZML format for compatibility with the PyOpenMS.35 Conversion was done using MSconvert under default settings.36 All search settings selected are designed to be adjustable by the user for best performance. For the purposes of this manuscript, the following parameters were used: precursor error cutoff, 20 ppm; fragment error cutoff, 0.02 Da; minimum m/z value, 50; minimum intensity value, 1000; maximum number of modifications per peptide, 3. Modifications considered in search were C-terminal amidation, oxidation of methionine, N-termini cyclization of glutamic acid and glutamine, and sulfation of tyrosine, common modifications documented within neuropeptides.37,38 Database searching was completed first at the precursor ion level, followed by the fragment ion level. Following this analysis, two metrics were calculated: percent sequence coverage (Cseq) and correlation values. The percent sequence coverage (Equation 5) reflects the quantity of fragment ions identified (Ni) with respect to the length of the peptide (Lp). The correlation value reflects the level of similarity of a peptide’s theoretical spectrum to its actual spectrum. Correlation values were calculated using the Hyperscore algorithm via the PyOpenMS python package.35
| Equation 5 |
PSM Assignment
A PSM algorithm was carefully designed to reward the hallmark attributes of neuropeptides when determining if a spectrum corresponds to any number of candidate matched peptides. As chimeric spectra are frequently associated with neuropeptides, this module also includes the ability to assess if a spectrum is indeed chimeric. The principle of chimeric spectra refers to when two or more peptides have similar mass and retention time, resulting in coelution and co-fragmentation at the MS/MS level, often culminating in fewer identifications overall.39 Thus, the highly homologous nature of neuropeptides makes them further pre-dispositioned to producing chimeric spectra, warranting careful consideration in PSM filtering, as outlined in Figure S1. After the filtering of putative PSMs, two of peptides remain, and these share adjacent, swapped residues (e.g. PNFLRF and PFNLRF), these peptides will be assumed to be chimeric, and both will be subjected to scoring and subsequent FDR filtering. While many of the metrics outlined in Figure S1 are rather intuitive, such as average fragment ion error, other metrics were specifically generated for this task. For example, we first search for if a motif is present in a peptide. If multiple peptide candidates have a motif, we then look to identify what percentage of the peptide is comprised of that motif. When some motif sequences are as short as three residues, the likelihood of a decoy possessing this motif by pure chance can be quite high. Thus, including a metric of this nature helps to parse this out. In addition, a metric of confidence of sequence coverage is included as a downstream metric. This is included as there is literature stating that it is reasonable to assume a spectrum-peptide match is correct above a certain percent sequence coverage threshold,40 thus this was applied throughout the PSM assignment decision tree. For the purpose of this manuscript, PSM assignment metric filtering values were adjusted to be: confidence sequence coverage threshold, 70%; maximum number of adjacent swapped amino acids, 2; minimum motif length, 3; number of substituted amino acids, 1.
Definitive Screening Design and Fine-tune Scoring
A definitive screening design (DSD) was generated and interpreted with JMP Pro 15.0.0, as outlined previously.41 A series of continuous attributes were selected to represent levels of spectrum-peptide match quality. These terms included average fragment error, precursor error, number of consecutive b-ions, number of consecutive y-ions, average number of annotations per fragment ion, average number of annotations per fragment ion that were not associated with a neutral loss of water or ammonia, hyperscore,35 and motif score. Here, number of annotations describes the number of fragment ions identified for a theoretical fragment ion type, for example, fragment ion y5 could potentially be identified at multiple charge states, as well as in water and ammonia neutral loss forms, each representing an annotation. For each metric, the corresponding value was extracted, and normalized across the whole of peptide identifications to a value of 1, where the maximum value was equal to 1, and all other values were scaled accordingly. The DSD was applied to adequately determine the necessary magnitude of these factors in building a final calculation to reflect the confidence of the PSM. These continuous metrics were assigned a value of 0, 5, or 10, representing the integer for the normalized metric value, from above, to be multiplied by. Each metric was also assigned a categorial value of “multiply” or “divide” to determine whether the factor should be rewarded or penalized in the final score. The design was assessed on the basis of 15 responses, reflecting the number of unique IDs found at 1% FDR across three technical replicates of five crustacean tissues: brain, commissural ganglia (CoG), pericardial organs (PO), sinus glands (SG), thoracic ganglia (TG). A response goal of unique ID maximization was indicated within the design. Using the JMP Fit Definitive Screening tool, significant factors were revealed. FDR was calculated as the ratio of the number of decoy identifications (Ndecoy) to the number of target identifications (Ntarget), as reported elsewhere (Eq. 6).7,13 The DSD and accompanying responses across the 50 prescribed combination of runs is outlined in Supplemental File 1.
Upon determining the significant factors across all tissues, discrepancies in factor values were parsed through application of a full-factorial design for just those factors (Supplemental File 2). The factors assessed through the full-factorial design were precursor error, number of consecutive b-fragment ions, motif score, average number of non-neutral fragment ions per amino acid, percent sequence coverage, average number of annotations per amino acid, and average number of fragment ions per amino acid. These factors were assigned a continuous component from 0 through 10, and a categorial component of multiple and divide, as above. The design was generated in JMP Pro 15.0.0. After obtaining responses for each prescribed combination of factor values, given the exhaustive nature of this design, the optimized factor values were determined to be those that provided the greatest number of identifications at 1% FDR.
The fine-tune scoring was assessed for biasing by searching against an entrapment database alone, comprised of sequences from non-crustacean neuropeptides. The database used was previously described.7 Search settings were held consistent to those described above.
| Equation 6: |
Alternative Software Evaluation
PEAKS analysis of neuropeptides was performed as described previously.7 All analyses were conducted in PEAKS Studio version 10.6.13,42 Analysis parameters were set to the equivalent of all EndoGenius parameters: parent mass error tolerance, 20 ppm; fragment mass error tolerance, 0.02 Da precursor mass search type, monoisotopic; enzyme, none; max missed cleavages, 100; digest mode, unspecific; max variable PTM per peptide; 3. Variable PTMs included: C-termini amidation; Oxidation of M; Pyro-glu from Q; Pyro-glu from E; Sulfation. Results were exported by adjusting the peptide −10lgP value to be greater than or equal to the equivalent of 1% FDR. Results were filtered to include only significant peptides. The output peptide report was referenced for all results described herein.
MetaMorpheus evaluation was conducted using version 1.0.5.43 Error tolerance and variable PTMs were the same as above. The protease selected was “top-down”. Search for truncated proteins and proteolysis products was selected as false. The maximum number of missed cleavages was set to 2. All other MetaMorpheus parameters were left as default.
MSFragger version 4.0 was operated via FragPipe version 18.0.44 Once more, precursor mass tolerance was 20 ppm and fragment mass tolerance was 0.02 Da. The cleavage type was set to non-specific with up to 12 missed cleavages. PTMs were set as above, with up to three variable PTMs tolerated. All other MSFragger parameters were left as default. Validation tools were run, specifically Percolator, with a minimum probability value of 0.5 (default).
EndoGenius Validation with Alternative Dataset
Brain, SG, and PO tissue from 3 bioreplicates of blue crab, C. sapidus, were collected and processed as with the first dataset. Tissues were analyzed once more on a LC-MS/MS with a Thermo Q-Exactive HF mass spectrometer coupled to a Dionex Ultimate 3000 LC system, using the same acquisition parameters. These spectra were processed through EndoGenius using the same parameters as before. Data were filtered to an EndoGenius score of 1000.
Results and Discussion
EndoGenius Outperforms Alternative Software Strategies
As most software implemented in neuropeptide workflows are typically designed for proteomics-based inquiries, the subsequent search results often lack optimization, illustrated by few PSMs and unique identifications. PEAKS Studio is a program routinely used in neuropeptide investigations, with notably increased performance compared to alternative software, attributed primarily to its hybrid de novo sequencing and database searching approach, which helps parse through highly homologous neuropeptides.45 In particular, its de novo sequencing algorithm is exhaustive, searching all possible combinations of amino acids, enabling a more sensitive algorithm compared with more common spectral graph-based algorithms.17 Our group has recently released two software packages designed to redirect PEAKS results through pre- and post-processing strategies aimed to increase the frequency of neuropeptide identification.7,17 The work described herein presents the first fully independent, open-source database searching strategy for neuropeptides. Through careful optimization of each step of traditional database searching workflows (Figure 1),14,23 including decoy database generation, precursor and fragment ion matching, post-translational modification (PTM) identification and localization, spectral correlation evaluations, and scoring algorithms, as well as strategic inclusion of a motif database (Figure 2a), we have successfully developed a platform capable of effectively and efficiently identifying a wide breadth of neuropeptides.
In the current application, results were sought from crustacean neuropeptide extracts. As the crustacean neuropeptide database only contains 891 peptide entries,7 specific filtering strategies are necessary to enable statistical leverage to achieve a reasonable number of identifications. For example, in previous work, the FDR threshold was required to be extended from the typical 1% value to 5% to glean a reasonable number of identifications, following by manual inspection, given that only a few hundred neuropeptides are expected to be identified, and thus a single decoy identification can rapidly decrease the number of results.41 Thus, this work leverages statistical power by implementing strategic filtering steps to gradually increase precision of identification as the workflow progresses.
Target-Decoy Database Approach
A neuropeptide database for crustacea has been described previously,7 and was employed in this work. Use of a target-decoy search strategy is routine in database searching software, particularly useful in assigning a false-discovery rate (FDR), or the ratio of the number of decoy identifications to the number of target identifications. The key to applying this approach is to develop decoy sequences that share similar characteristics so that a decoy sequence is reasonably similar to its corresponding target sequence.46 Recently, we reported the advantage of using a decoy-shuffle database for neuropeptide application, as opposed to other strategies such as decoy-reverse, decoy-random, and decoy-hybrid.7 Thus, the decoy-shuffle strategy was also employed here, where all target sequences from the neuropeptide database file were shuffled and concatenated with the target database, and all sequences were shuffled to minimize bias toward a particular database.
PSM Assignment
Database searching begins by calculating the theoretical m/z values for all target and decoy peptides, identifying precursor m/z values that align with these within an indicated error threshold. Given that a target peptide and its corresponding decoy peptide have the same precursor mass, it is expected that at least two peptides will match to a single precursor peak. Thus, we generate a shortlist of candidate peptides for a particular spectrum on the basis of precursor m/z alone. Following this, we impart filtering strategies to delineate the strongest peptide match within the shortlist for the spectrum. This is conducted through use of a decision tree, outlined in Figure S1. Here, we incorporate additional criteria for putative PSM filtering, such as the score of spectrum correlation with theoretical spectra and percent sequence coverage of fragment ions. As shown previously, neuropeptides often share conserved sequence motif with high degree of homology,7 particularly within neuropeptide families, with two distinct neuropeptides differing by as little as a single amino acid residue.1 This homology can lead to plenty of putative peptide matches for a single spectrum, producing the need for a more sophisticated assignment algorithm. In our method of PSM assignment, we placed much emphasis on a motif database, which describes expected and conserved neuropeptide sequences, to aid in the assignment process. The crustacean neuropeptide motif database (Supplemental File 3) utilized in this study is an elaborated version of the one previously described by our lab,17 with motifs representing 23 neuropeptide families (Figure 2b). This elaborated version contained both new, full motifs, as well as a newer concept, partial motifs. Partial motifs are generated when two conserved motif regions are joined by a singular, variable amino acid. To retain the knowledge of this motif, while accommodating the interior, variating amino acid, we incorporated multiple entries within the motif database to account for these subtle changes. Further, to address the differences between partial- and full-motifs, we generated a novel motif scoring algorithm. While previous work scored a motif through the ratio of the motif length to the length of the peptide (Equation 1),17 we noted that this could inadvertently impart biasing against neuropeptides of longer lengths, given that some peptide prohormone families can extend beyond 200 amino acid residues in length. To overcome this, we applied a square-root normalization procedure,33 wherein the original motif score was normalized by multiplying by the square root of the length of the neuropeptide (Equation 2), reducing this biasing. We then reward this score for the presence of corresponding fragment ions, multiplying by sequence coverage (Equation 3). These key advances upon previous neuropeptide motif analysis strategies were imperative to improved identification of neuropeptides. Indeed, we found that inclusion of the motif database in PSM assignment substantially improved the number of unique neuropeptide identifications at 1% FDR (Figure S2).
We wanted to assess any biasing of our model toward targets over decoys, ensuring that the identifications produced by EndoGenius were real. To do this, we utilized an entrapment database described previously.7 We wanted to ensure that, in the presence of no relevant peptides, EndoGenius did not provide identifications. Indeed, we found only less than 5 identifications for each of the 15 raw spectra files searched (Figure S3).
The improvement of this intricate PSM assignment algorithm was evident when comparing results from EndoGenius to the results of PEAKS, routinely used for neuropeptide identification. As shown in Figure 3, across 15 samples (encompassing 5 tissues), with our algorithm there were 86 times that a neuropeptide backbone was identified just one time, whereas in PEAKS this was only present 16 times. More notably, there was a single neuropeptide backbone that was identified 757 times in PEAKS across these 15 samples, underscoring the need for an optimized software program to minimize routine re-identification and re-assignment of highly homologous neuropeptides. Alternatively, the most frequently a single neuropeptide backbone was identified in EndoGenius 170 times. These findings carry through to the final results of the program, where we find a substantial increase in the number of unique neuropeptides identified from a single experiment (Figure 4).
Figure 3:
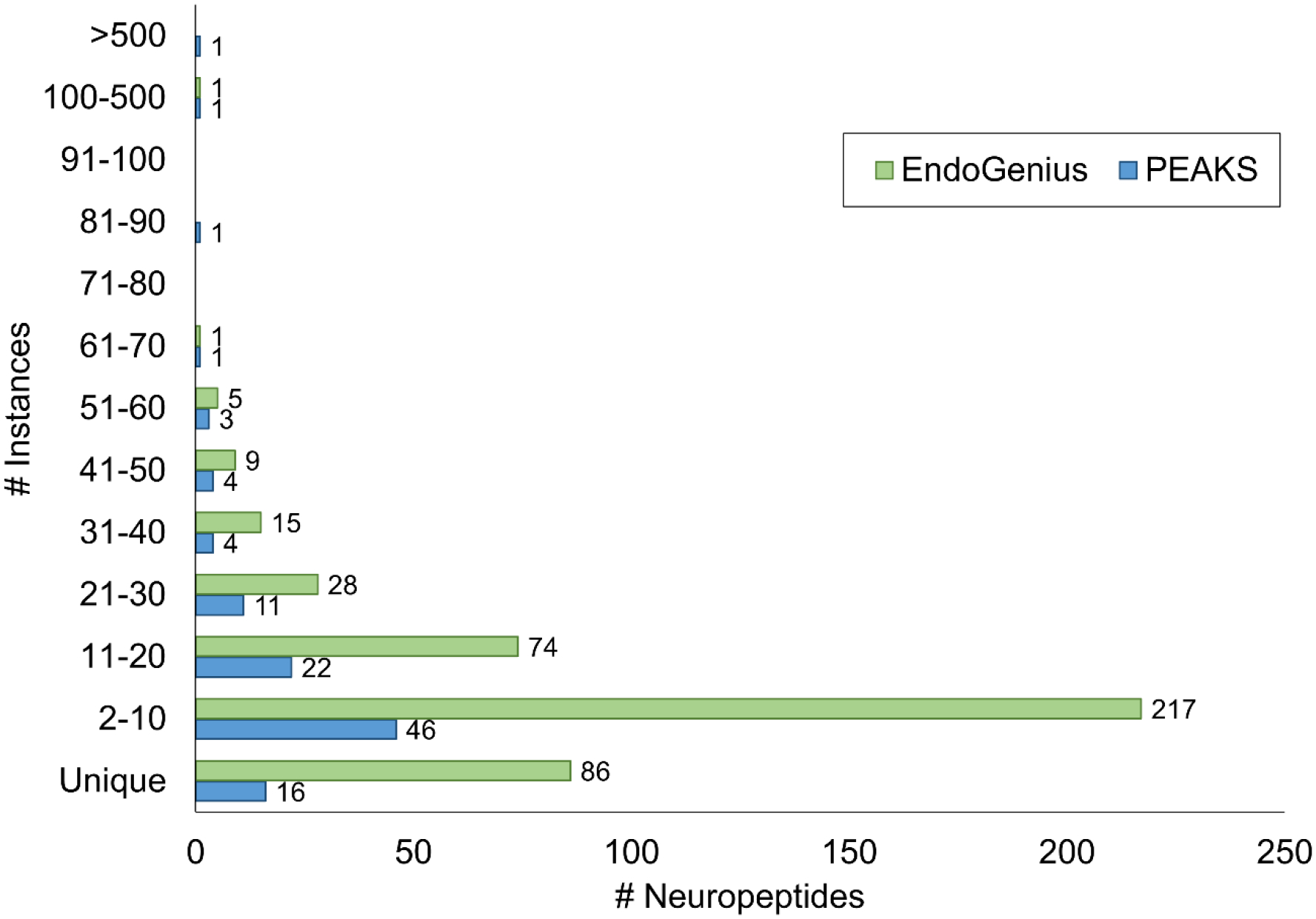
Comparison of the frequency of neuropeptide backbone re-identification in PEAKS compared to EndoGenius across 15 samples, corresponding to 3 technical replicates of 5 crustacean tissue samples.
Figure 4:
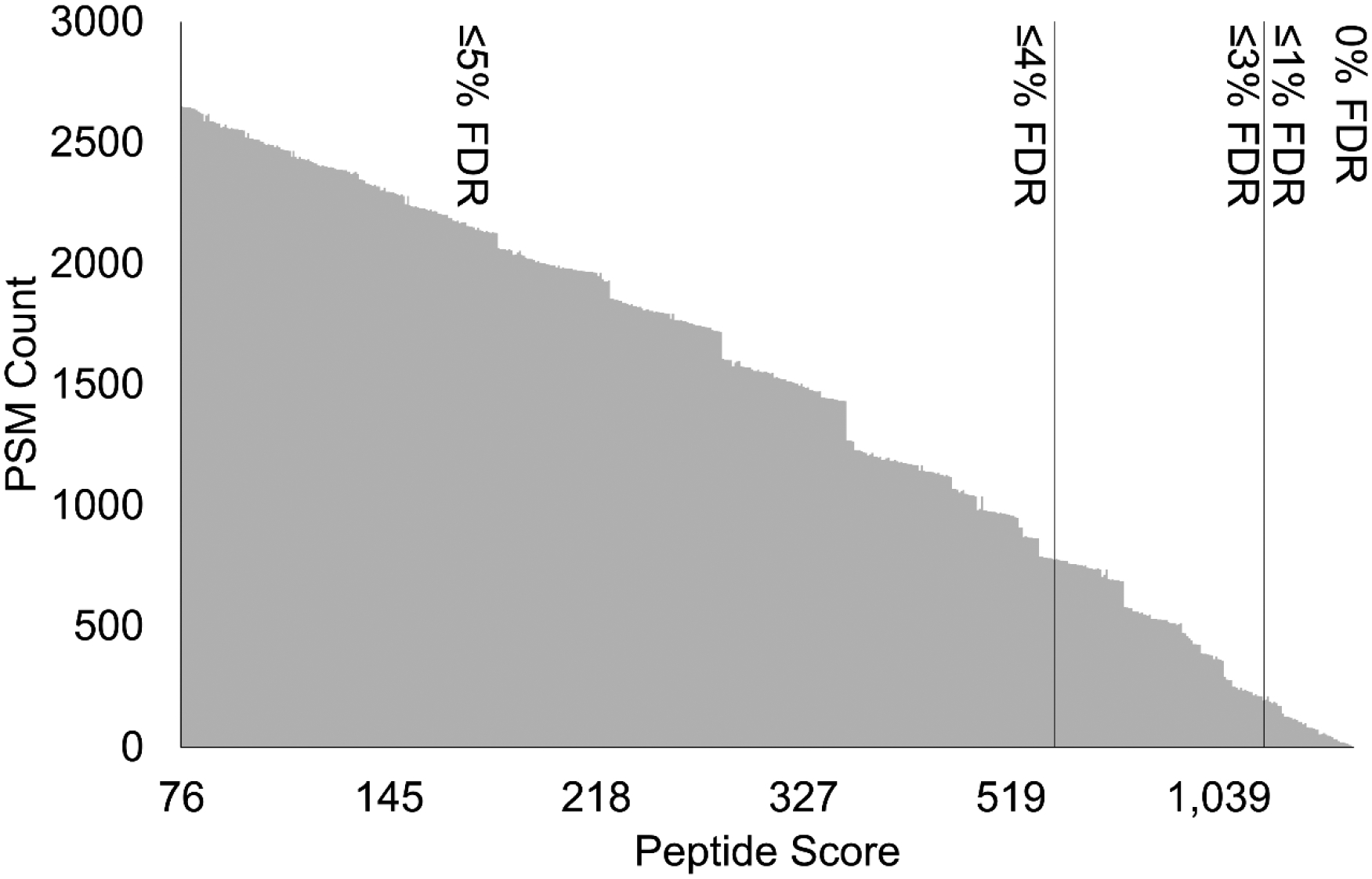
Representative plots describing peptide score versus the number of PSMs, with FDR thresholds of 0–5% indicated. Plots here describe sinus gland (SG) sample results.
Fine-tune Scoring
Many established programs, particularly PEAKS, have adopted a scoring algorithm to reflect the confidence of a score, used to establish score cutoffs to accompany FDR values. In PEAKS, this proprietary algorithm is termed the -logP value. While the details of this calculation are not publicly available, previous reports have shown that factors contributing to the score include precursor mass error, charge state, and maximum length of the consecutively matched fragment ion series.13 With this inspiration, we sought to apply this same theory to develop our own score calculation algorithm to be used to effectively represent and filter PSM matches. To effectively craft a scoring algorithm in an unbiased fashion, we employed use of a DSD, a statistical practice routinely used in engineering fields, that has recently been adopted as application in neuropeptidomic applications, albeit for data acquisition purposes.41,47 DSDs can be used to leverage statistical power to optimize conditions whilst minimizing the number of assessment experiments necessary.48 For example, in the work described herein, we evaluated 20 different factors through conducting 50 experimental runs prescribed by the DSD. Alternatively, probing the effects of each factor manipulation in a full-factorial manner would require 8,000 experiments. In this context, an experiment refers to the evaluation of a given combination of factor values. Each combination from the DSD was evaluated and assessed based on the number of unique identifications resulting at 1% FDR (Supplemental File 1). We started with 10 components we hypothesized could contribute to a score that effectively described the likelihood of a strong match: average fragment error (in Da), precursor error (in ppm), number of consecutive b-fragment ions identified, number of consecutive y-fragment ions identified, average number of annotations per fragment ion (including neutral loss fragments and multiple charge states), percent sequence coverage (Equation 5), average number of fragment ions per amino acid (all corresponding b- and y-ions), average number of fragment ions not corresponding to a neutral-loss per amino acid, spectral correlation (Hyperscore),35 and motif score (Equation 4). Each of these factors were treated as continuous, to determine the optimal integer-based weighting of this factor. Each of these components were also designated a categorial factor, equal to +1 or −1, which would determine if the component should contribute to the final score (multiply) or if detract from the final score (divide). Following fitting the DSD with its responses using the JMP Fit Definitive Screening function, the statistical response revealed any significant and insignificant metrics that resulted in the number of IDs, the response selected for which to maximize desirability. This analysis revealed that each of the samples had different factors that were influential in the high number of identifications. Associated response residuals are located in Figure S4. It should be noted that some components, seemingly complementary, such as the number of consecutive b- and y-fragment ions, revealed differences in significance, where the b-fragment ions were significant and y-fragment ions were not. While the underlying mechanistic reason for this significance was not probed, it can be speculated that this is due to the discrepancy in ionization efficiency, kinetic stability, and subsequent fragment ion abundance, between b- and y-type fragment ions. In fact, with non-tryptic, doubly charged peptides, a higher abundance of b-type fragment ions has been noted previously,49 and is perhaps a reasonable hypothesis given the high propensity for neuropeptide precursors to appear at a +2 charge state.50 Interestingly, a handful of significant factors had widely diverging optimal values across tissue types (Figure S4). Thus, a second, full-factorial design was generated for these remaining, significant factors: precursor error, precursor error operation, # consecutive b-ions, motif score, and average number of non-neutral-loss fragment ions per amino acid. This second design was necessary to identify the true optimal factor combination that was fitting for all tissue types (Supplemental File 2). Significant factors, as well as their final, optimal value are illustrated in Table 1. Upon determination of the optimal combination of factors, a FDR evaluation script was written to determine the fine tune score associated with a selected FDR % threshold. This script simply probed score thresholds iteratively, localizing on the lowest score that produced an FDR threshold less than or equal to the specified value, based on Equation 6. Herein, the selected FDR was 1%. Figure 4 describes the number of IDs and the corresponding fine-tune score at FDR cutoff intervals.
Table 1.
Final DSD factors. Each factor and values were proven significant by definitive screening design (DSD) and subsequent analysis. Factor refers to the value that the normalized result is multiplied by, and the operation refers to how the value is included in the final calculation, either having a positive role, or multiplication, or a negative role, by division. Other factors searched, but deemed insignificant were average fragment error, # consecutive y-ions, average number of annotations per fragment, average number of fragment ions per amino acid, hyperscore, and motif score.
| Factor | Value | Operation |
|---|---|---|
| Precursor error | 10 | Divide |
| # Consecutive b-ions | 10 | Multiply |
| % Sequence coverage | 10 | Multiply |
| # Fragment ions not from neutral-loss per AA | 10 | Divide |
Benchmarking of EndoGenius with Other software
As PEAKS software has routinely been used for neuropeptide analyses, it was imperative to benchmark the presented results of EndoGenius against the results generated by PEAKS. Additionally, as PEAKS is a commercial software, we sought to benchmark EndoGenius also against MSFragger44 and MetaMorpheus,43 both popular tools that are freely available and open source. It was immediately apparent that there were unique profiles of neuropeptides, signified by the differing quantities of unique peptide IDs, reported in each method (Figure 5a). When further investigating identifications, it was largely apparent that a statistically significant (p-value<0.05) increase in number of peptide backbones, defined as a peptide sequence alone, were consistently identified by EndoGenius (Figure 5b). These results are in line with the aforementioned finding, in which it was evident that PEAKS has a higher frequency of re-identifying the same peptides, likely a result of the high level of homology between individual neuropeptides. These results were underscored in the number of unique IDs achieved by EndoGenius that were not identified with PEAKS, while still largely corroborating the identifications from PEAKS (Figure 5c).
Figure 5:
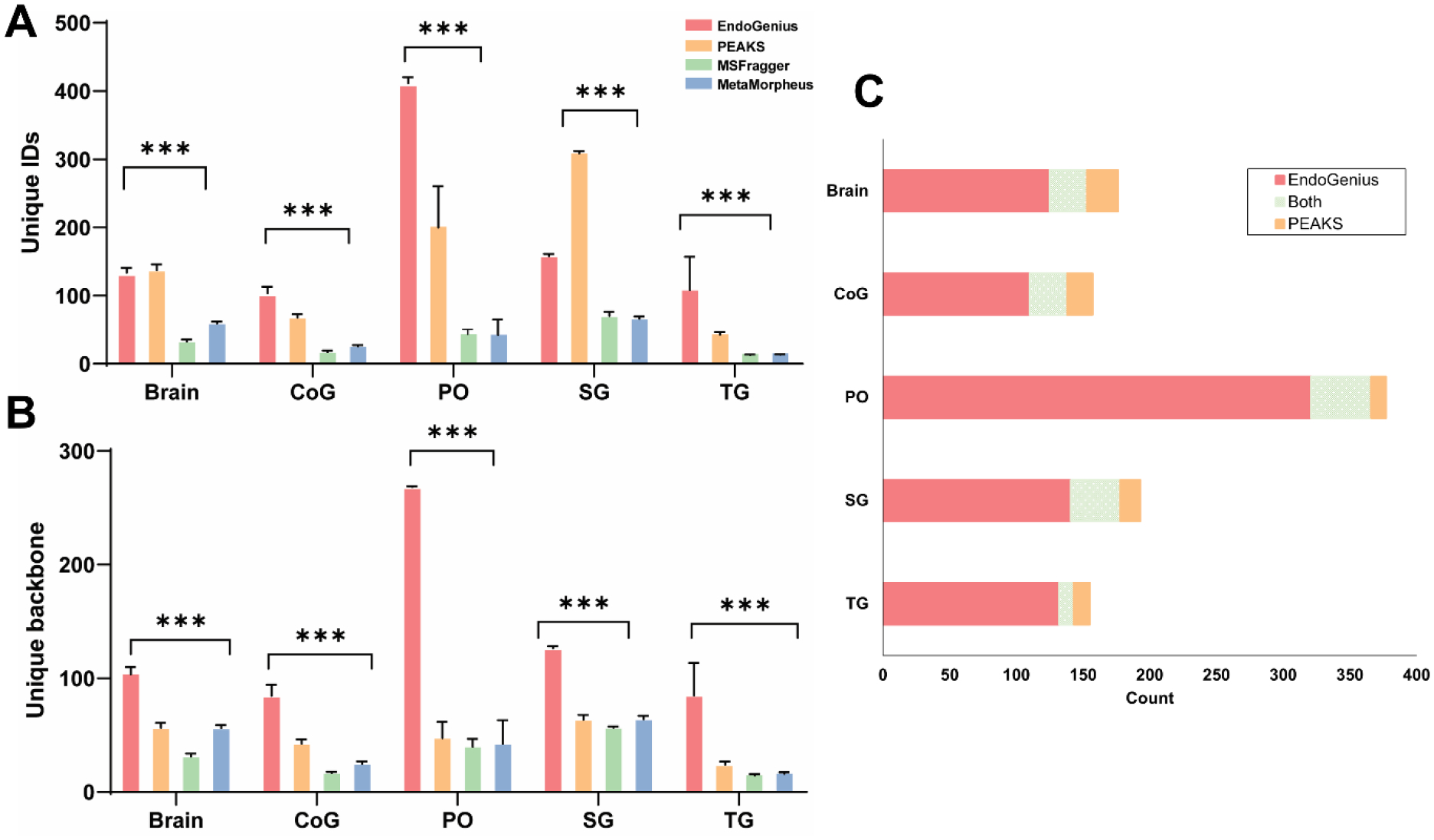
Analysis of results from EndoGenius, PEAKS, MSFragger, and MetaMorpheus, of brain, commissural ganglion (CoG), pericardial organs (PO), sinus glands (SG) and thoracic ganglion (TG) tissue types. (A) Unique peptide IDs identified all software. Unique IDs are defined as peptides including PTMs. (B) Unique backbones identified in all software. Unique backbones are defined as the peptide sequence only. For A & B, bar graph shows the ANOVA test result, error bars, mean ± s.d. (* p-value <0.05, ** p-value <0.001, ***p-value<0.0001). (C) Overlap of neuropeptide backbone identifications from EndoGenius and PEAKS across 5 tissue types.
Application of EndoGenius to an Unknown Dataset
While using a 1% FDR threshold for comparison of identifications across platforms provided a relatively translatable metric for comparison, we hypothesized that for our own method, defining a threshold using our “EndoGenius Score” may be more fruitful for generating reproducible identifications. The logic here is simple. The FDR threshold was initially generated for proteomics, in which thousands of proteins are expected to be identified. With samples in which the analyte of interest is sparce, such as neuropeptides, an FDR can be detrimental to producing accurate results. In these experiments, in which sometimes less than 100 neuropeptides are anticipated, just one false positive (decoy) identification can quickly raise the FDR. Thus, in many instances the reported number of peptides at 1% FDR typically include no decoy identifications, and the results are actually more equal to 0% FDR. This well-documented challenge has been addressed in a few different ways. Perhaps the simplest method has been to increase the FDR threshold to 5% and exercise more caution to manually verify results.41 Alternatively, non-FDR thresholds have been suggested, such as in PEAKS, where it is recommended to use a −10logP, their proprietary scoring method, of 20.13 We noted in our work that this value routinely corresponds to approximately 3–5% FDR. To address this FDR concern within our own dataset, to enable the ability to search very neuropeptide-sparse samples, we sought to establish a confident scoring value with respect to our own EndoGenius score, that routinely reported a low number of false identifications. As we compared the EndoGenius score to FDR across 15 samples, we noted consistency in the EndoGenius score, with an increased score agreeing with a reduced FDR (Figure S5). From this, we began to speculate if an EndoGenius score threshold application, rather than an FDR, could more consistently separate true from false peptides, while ensuring adequate representation of neuropeptides, not lost to the statistical shortcomings of the FDR value.
We used this knowledge to apply our optimized EndoGenius platform for the identification of neuropeptides from an unknown dataset. We analyzed brain, PO, and SG samples obtained from blue crabs (C. sapidus). In doing so, we found results comparable to our initial work, when examining both unique identifications (Figure 6a) and unique backbones (Figure 6b). Here, a unique identification represents a peptide and any PTMs, while the unique backbone represents the amino acid sequence alone.
Figure 6:
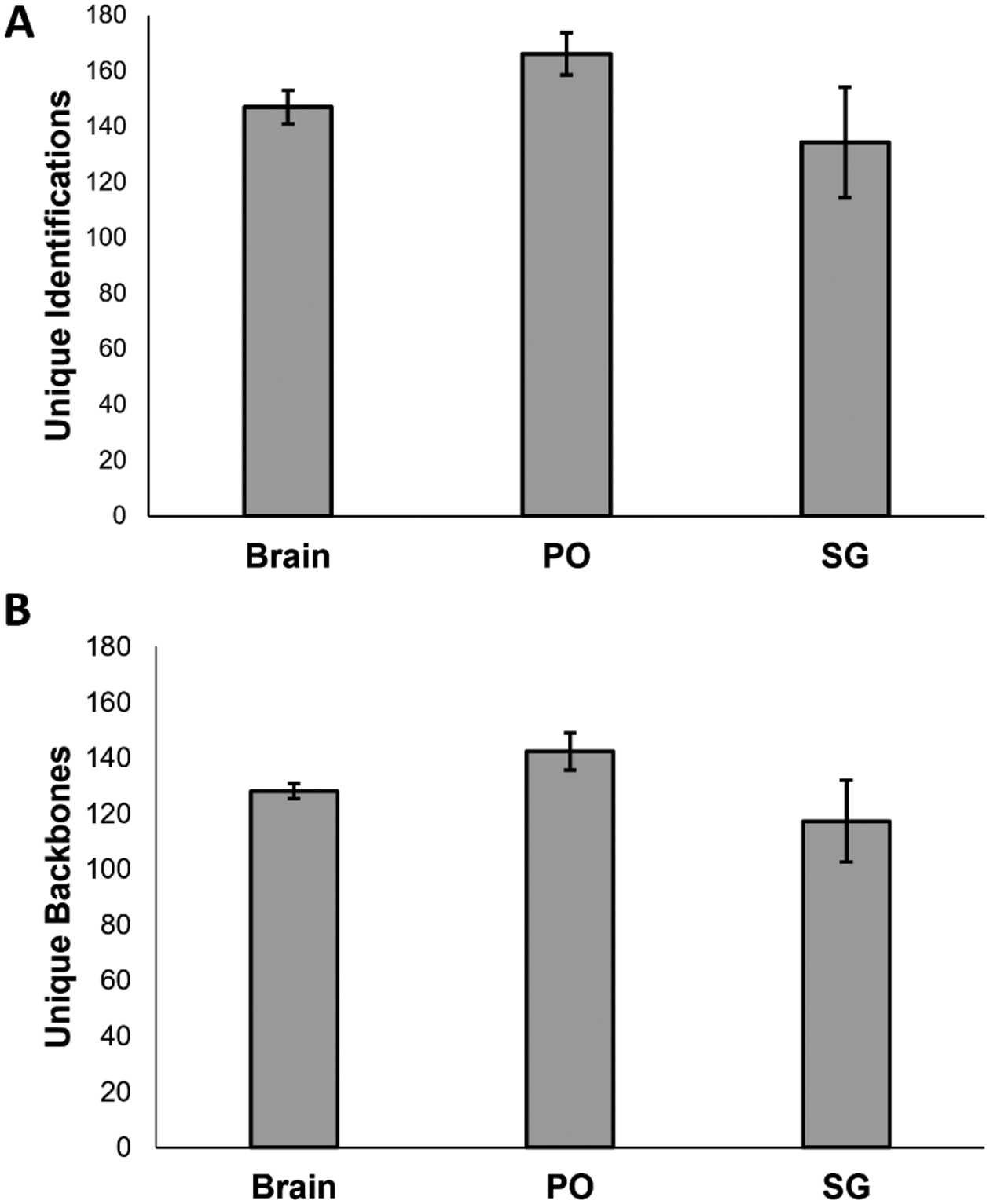
Results obtained through use of EndoGenius on an unknown dataset (A) average number of unique identifications, including modifications (B) average number of unique backbones. Error bars represent standard deviation.
Conclusion
The work described herein presents an optimized database searching program, ideal for analysis of neuropeptides. This program utilizes a strategic PSM assignment algorithm in conjuncture with a fine-tune scoring calculation to achieve a substantial increase in the number of neuropeptide PSMs, unique peptides, and unique peptide backbones. This finding is in-part achieved through the referencing of a motif database, capable of parsing highly-homologous neuropeptide sequences, to greatly increase the diversity of neuropeptide identifications, where other programs may repeatedly re-identify a single neuropeptide. Future work in this area will include integration of an automated motif library-building program. Altogether, EndoGenius represents the first standalone, open-source program optimized for identification of neuropeptides in their endogenous state from mass spectrum. Source code, in addition to usage instructions (Supplemental File 4) can be found online at https://github.com/lingjunli-research/EndoGenius).
Supplementary Material
Supplemental File 1: Definitive screening design
Supplemental File 2: Full-factorial design
Supplemental File 3: Crustacean neuropeptide motif database
Supplemental File 4: EndoGenius user manual
Supplemental Figures: PSM assignment decision tree (Figure S1), evaluation of motif database perturbations (Figure S2), target-decoy entrapment search results (Figure S3), DSD results (Figure S4), FDR versus EndoGenius score evaluation (Figure S5).
Acknowledgements
This work was supported in part by National Science Foundation (CHE-2108223) and National Institutes of Health (NIH) through grant R01DK071801 and R01NS029436. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. L.F. was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM008505 (Chemistry–Biology Interface Training Program). N.Q.V. was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM008349 (Biotechnology Training Program) and by the National Heart Lung and Blood Institute of the National Institutes of Health under Award Number T32HL007936 (Training Program in Translational Cardiovascular Science). T.C.D. was supported in part by the SciMed Graduate Research Scholars Fellowship through the University of Wisconsin-Madison. Support for this fellowship is provided by the Graduate School, part of the Office of Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison, with funding from the Wisconsin Alumni Research Foundation and the UW-Madison. L.L. would like to acknowledge NIH grants R01AG052324, R01AG078794, R21AG065728, S10OD028473, and S10OD025084, as well as funding support from a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation. We would also like to thank Jimmy Eng for guidance in the development of the peptide fragmentation portion of the program. TOC and Figure 1 were generated with the use of BioRender.
Abbreviations:
- MS
mass spectrometry
- PSM
peptide-spectrum match
- DDA
data-dependent acquisition
- FDR
false-discovery rate
- MeOH
methanol
- ACN
acetonitrile
- GAA
glacial acetic acid
- FA
formic acid
- H2O
water
- SG
sinus glands
- PO
pericardial organs
- TG
thoracic ganglia
- CoG
commissural ganglia
- ACG
automatic gain control
- HCD
higher-energy collisional dissociation
- NCE
normalized collision energy
- GUI
graphical user-interface
- SA
ratio score
- LM
motif length
- LN
neuropeptide length
- CS
motif sequence coverage
- NE
experimental fragment ions present
- NT
theoretical fragment ions present
- SF
motif score
- SB
normalized ratio score
- Cseq
percent sequence coverage
- Ni
fragment ions identified
- LP
peptide length
- DSD
definitive screening design
- PTM
post-translational modification
Footnotes
Data deposition:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the MassIVE partner repository with the dataset identifier MSV000092654.
References
- (1).Christie AE; Stemmler EA; Dickinson PS Crustacean Neuropeptides. Cellular and Molecular Life Sciences 2010, 67 (24), 4135–4169. 10.1007/s00018-010-0482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Roeske LC; Auchus AP Neuropeptide Changes in Cortical and Deep Gray Structures in Alzheimer’s Disease. Rev Neurosci 1995, 6, 317–328. [DOI] [PubMed] [Google Scholar]
- (3).Chen XY; Du YF; Chen L Neuropeptides Exert Neuroprotective Effects in Alzheimer’s Disease. Front Mol Neurosci 2019, 11. 10.3389/fnmol.2018.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Beck B; Burlet A; Bazin R; Nicolas J-P; Burlet C Early Modification of Neuropeptide Y but Not of Neurotensin in the Suprachiasmatic Nucleus of the Obese Zucker Rat; 1992; Vol. 136. [DOI] [PubMed] [Google Scholar]
- (5).Hillebrand JJG; de Wied D; Adan RAH Neuropeptides, Food Intake and Body Weight Regulation: A Hypothalamic Focus. Peptides (N.Y.) 2002, 23 (12), 2283–2306. 10.1016/S0196-9781(02)00269-3. [DOI] [PubMed] [Google Scholar]
- (6).Zhang Y; DeLaney K; Hui L; Wang J; Sturm RM; Li L A Multifaceted Mass Spectrometric Method to Probe Feeding Related Neuropeptide Changes in Callinectes Sapidus and Carcinus Maenas. J Am Soc Mass Spectrom 2018, 29 (5), 948–960. 10.1007/s13361-017-1888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Vu NQ; Yen H-C; Fields L; Cao W; Li L HyPep: An Open-Source Software for Identification and Discovery of Neuropeptides Using Sequence Homology Search. J Proteome Res 2023, 22 (2), 420–431. 10.1021/acs.jproteome.2c00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fields L; Ma M; DeLaney K; Phetsanthad A; Li L A Crustacean Neuropeptide Spectral Library for Data-Independent Mass Spectrometry Applications. Proteomics- manuscript accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ye H; Wang J; Tian Z; Ma F; Dowell J; Bremer Q; Lu G; Baldo B; Li L Quantitative Mass Spectrometry Reveals Food Intake-Induced Neuropeptide Level Changes in Rat Brain: Functional Assessment of Selected Neuropeptides as Feeding Regulators. Molecular and Cellular Proteomics 2017, 16 (11), 1922–1937. 10.1074/mcp.RA117.000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Buchberger A; Yu Q; Li L Advances in Mass Spectrometric Tools for Probing Neuropeptides. Annual Review of Analytical Chemistry 2015, 8, 485–509. 10.1146/annurev-anchem-071114-040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kong AT; Leprevost F. v.; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cox J; Mann M MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat Biotechnol 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (13).Zhang J; Xin L; Shan B; Chen W; Xie M; Yuen D; Zhang W; Zhang Z; Lajoie GA; Ma B PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Molecular & Cellular Proteomics 2012, 11 (4), M111.010587. 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Eng JK; Jahan TA; Hoopmann MR Comet: An Open-Source MS/MS Sequence Database Search Tool. Proteomics 2013, 13 (1), 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- (15).Shortreed MR; Wenger CD; Frey BL; Sheynkman GM; Scalf M; Keller MP; Attie AD; Smith LM Global Identification of Protein Post-Translational Modifications in a Single-Pass Database Search. J Proteome Res 2015, 14 (11), 4714–4720. 10.1021/acs.jproteome.5b00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ma M; Gard AL; Xiang F; Wang J; Davoodian N; Lenz PH; Malecha SR; Christie AE; Li L Combining in Silico Transcriptome Mining and Biological Mass Spectrometry for Neuropeptide Discovery in the Pacific White Shrimp Litopenaeus Vannamei. Peptides (N.Y.) 2010, 31 (1), 27–43. 10.1016/j.peptides.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).DeLaney K; Cao W; Ma Y; Ma M; Zhang Y; Li L PRESnovo: Prescreening Prior to de Novo Sequencing to Improve Accuracy and Sensitivity of Neuropeptide Identification. J Am Soc Mass Spectrom 2020, 31 (7), 1358–1371. 10.1021/jasms.0c00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ivanov M. v.; Levitsky LI; Lobas AA; Panic T; Laskay ÜA; Mitulovic G; Schmid R; Pridatchenko ML; Tsybin YO; Gorshkov M. v. Empirical Multidimensional Space for Scoring Peptide Spectrum Matches in Shotgun Proteomics. J Proteome Res 2014, 13 (4), 1911–1920. 10.1021/pr401026y. [DOI] [PubMed] [Google Scholar]
- (19).Carvalho PC; Fischer JSG; Xu T; Cociorva D; Balbuena TS; Valente RH; Perales J; Yates JR; Barbosa VC Search Engine Processor: Filtering and Organizing Peptide Spectrum Matches. Proteomics 2012, 12 (7), 944–949. 10.1002/pmic.201100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Carvalho PC; Lima DB; Leprevost F. v.; Santos MDM; Fischer JSG; Aquino PF; Moresco JJ; Yates JR; Barbosa VC Integrated Analysis of Shotgun Proteomic Data with PatternLab for Proteomics 4.0. Nat Protoc 2015, 11 (1), 102–117. 10.1038/nprot.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tabb DL; Narasimhan C; Strader MB; Hettich RL DBDigger: Reorganized Proteomic Database Identification That Improves Flexibility and Speed. Anal Chem 2005, 77 (8), 2464–2474. 10.1021/ac0487000. [DOI] [PubMed] [Google Scholar]
- (22).Taylor JA; Johnson RS Sequence Database Searches via de Novo Peptide Sequencing by Tandem Mass Spectrometry. COMMUNICATIONS IN MASS SPECTROMETRY 1997, 11, 1067–1075. [DOI] [PubMed] [Google Scholar]
- (23).Eng JK; McCormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom 1994, 5 (11), 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- (24).Elias JE; Gygi SP Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry. Nat Methods 2007, 4 (3), 207–214. 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- (25).Moosa JM; Guan S; Moran MF; Ma B Repeat-Preserving Decoy Database for False Discovery Rate Estimation in Peptide Identification. J Proteome Res 2020, 19 (3), 1029–1036. 10.1021/acs.jproteome.9b00555. [DOI] [PubMed] [Google Scholar]
- (26).Craig R; Beavis RC TANDEM: Matching Proteins with Tandem Mass Spectra. Bioinformatics 2004, 20 (9), 1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- (27).Gutierrez GJ; Grashow RG Cancer Borealis Stomatogastric Nervous System Dissection. J Vis Exp 2009, 25 (25), 1–5. 10.3791/1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cock PJA; Antao T; Chang JT; Chapman BA; Cox CJ; Dalke A; Friedberg I; Hamelryck T; Kauff F; Wilczynski B; de Hoon MJL Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25 (11), 1422–1423. 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Russo AF Overview of Neuropeptides: Awakening the Senses? Headache: The Journal of Head and Face Pain 2017, 57 (S2), 37–46. 10.1111/head.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bateman A; Martin M-J; Orchard S; Magrane M; Ahmad S; Alpi E; Bowler-Barnett EH; Britto R; Bye-A-Jee H; Cukura A; Denny P; Dogan T; Ebenezer T; Fan J; Garmiri P; da Costa Gonzales LJ; Hatton-Ellis E; Hussein A; Ignatchenko A; Insana G; Ishtiaq R; Joshi V; Jyothi D; Kandasaamy S; Lock A; Luciani A; Lugaric M; Luo J; Lussi Y; MacDougall A; Madeira F; Mahmoudy M; Mishra A; Moulang K; Nightingale A; Pundir S; Qi G; Raj S; Raposo P; Rice DL; Saidi R; Santos R; Speretta E; Stephenson J; Totoo P; Turner E; Tyagi N; Vasudev P; Warner K; Watkins X; Zaru R; Zellner H; Bridge AJ; Aimo L; Argoud-Puy G; Auchincloss AH; Axelsen KB; Bansal P; Baratin D; Batista Neto TM; Blatter M-C; Bolleman JT; Boutet E; Breuza L; Gil BC; Casals-Casas C; Echioukh KC; Coudert E; Cuche B; de Castro E; Estreicher A; Famiglietti ML; Feuermann M; Gasteiger E; Gaudet P; Gehant S; Gerritsen V; Gos A; Gruaz N; Hulo C; Hyka-Nouspikel N; Jungo F; Kerhornou A; Le Mercier P; Lieberherr D; Masson P; Morgat A; Muthukrishnan V; Paesano S; Pedruzzi I; Pilbout S; Pourcel L; Poux S; Pozzato M; Pruess M; Redaschi N; Rivoire C; Sigrist CJA; Sonesson K; Sundaram S; Wu CH; Arighi CN; Arminski L; Chen C; Chen Y; Huang H; Laiho K; McGarvey P; Natale DA; Ross K; Vinayaka CR; Wang Q; Wang Y; Zhang J UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51 (D1), D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Crooks GE; Hon G; Chandonia J-M; Brenner SE WebLogo: A Sequence Logo Generator: Figure 1. Genome Res 2004, 14 (6), 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hancock AA; Bush EN; Stanisic D; Kyncl JJ; Lin CT Data Normalization before Statistical Analysis: Keeping the Horse before the Cart. Trends Pharmacol Sci 1988, 9 (1), 29–32. 10.1016/0165-6147(88)90239-8. [DOI] [PubMed] [Google Scholar]
- (33).Deininger S-O; Cornett DS; Paape R; Becker M; Pineau C; Rauser S; Walch A; Wolski E Normalization in MALDI-TOF Imaging Datasets of Proteins: Practical Considerations. Anal Bioanal Chem 2011, 401 (1), 167–181. 10.1007/s00216-011-4929-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).He L; Diedrich J; Chu YY; Yates JR Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal Chem 2015, 87 (22), 11361–11367. 10.1021/acs.analchem.5b02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Röst HL; Sachsenberg T; Aiche S; Bielow C; Weisser H; Aicheler F; Andreotti S; Ehrlich H-C; Gutenbrunner P; Kenar E; Liang X; Nahnsen S; Nilse L; Pfeuffer J; Rosenberger G; Rurik M; Schmitt U; Veit J; Walzer M; Wojnar D; Wolski WE; Schilling O; Choudhary JS; Malmström L; Aebersold R; Reinert K; Kohlbacher O OpenMS: A Flexible Open-Source Software Platform for Mass Spectrometry Data Analysis. Nat Methods 2016, 13 (9), 741–748. 10.1038/nmeth.3959. [DOI] [PubMed] [Google Scholar]
- (36).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak M-Y; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat Biotechnol 2012, 30 (10), 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Anapindi KDB; Romanova EV; Checco JW; Sweedler JV Mass Spectrometry Approaches Empowering Neuropeptide Discovery and Therapeutics. Pharmacol Rev 2022, 74 (3), 662–679. 10.1124/pharmrev.121.000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Akhtar MN; Southey BR; Andrén PE; Sweedler JV; Rodriguez-Zas SL Evaluation of Database Search Programs for Accurate Detection of Neuropeptides in Tandem Mass Spectrometry Experiments. J Proteome Res 2012, 11 (12), 6044–6055. 10.1021/pr3007123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Houel S; Abernathy R; Renganathan K; Meyer-Arendt K; Ahn NG; Old WM Quantifying the Impact of Chimera MS/MS Spectra on Peptide Identification in Large-Scale Proteomics Studies. J Proteome Res 2010, 9 (8), 4152–4160. 10.1021/pr1003856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fort KL; Cramer CN; Voinov VG; Vasil’Ev YV; Lopez NI; Beckman JS; Heck AJR Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J Proteome Res 2018, 17 (2), 926–933. 10.1021/acs.jproteome.7b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Phetsanthad A; Carr AV; Fields L; Li L Definitive Screening Designs to Optimize Library-Free DIA-MS Identification and Quantification of Neuropeptides. J Proteome Res 2023, 22 (5), 1510–1519. 10.1021/acs.jproteome.3c00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ma B; Zhang K; Hendrie C; Liang C; Li M; Doherty-Kirby A; Lajoie G PEAKS: Powerful Software for Peptide de Novo Sequencing by Tandem Mass Spectrometry. Rapid Communications in Mass Spectrometry 2003, 17 (20), 2337–2342. 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- (43).Solntsev SK; Shortreed MR; Frey BL; Smith LM Enhanced Global Post-Translational Modification Discovery with MetaMorpheus. J Proteome Res 2018, 17 (5), 1844–1851. 10.1021/acs.jproteome.7b00873. [DOI] [PubMed] [Google Scholar]
- (44).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry–Based Proteomics. Nat Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).De La Toba EA; Anapindi KDB; Sweedler JV Assessment and Comparison of Database Search Engines for Peptidomic Applications. J Proteome Res 2023. 10.1021/acs.jproteome.2c00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Elias JE; Gygi SP Target-Decoy Search Strategy for Mass Spectrometry-Based Proteomics. In Proteome Bioinformatics; Hubbard SJ, Jones AR, Eds.; Humana Press: Totowa, NJ, 2010; pp 55–71. 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sauer CS; Li L Mass Spectrometric Profiling of Neuropeptides in Response to Copper Toxicity via Isobaric Tagging. Chem Res Toxicol 2021, 34 (5), 1329–1336. 10.1021/acs.chemrestox.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Jones B; Nachtsheim CJ A Class of Three-Level Designs for Definitive Screening in the Presence of Second-Order Effects. Journal of Quality Technology 2011, 43 (1), 1–15. 10.1080/00224065.2011.11917841. [DOI] [Google Scholar]
- (49).Bensadek D; Monigatti F; Steen JAJ; Steen H Why b, y’s? Sodiation-Induced Tryptic Peptide-like Fragmentation of Non-Tryptic Peptides. Int J Mass Spectrom 2007, 268 (2–3), 181–189. 10.1016/j.ijms.2007.06.014. [DOI] [Google Scholar]
- (50).Fields L; Ma M; DeLaney K; Phetsanthad A; Li L A Crustacean Neuropeptide Spectral Library for Data‐independent Acquisition (DIA) Mass Spectrometry Applications. Proteomics 2024. 10.1002/pmic.202300285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1: Definitive screening design
Supplemental File 2: Full-factorial design
Supplemental File 3: Crustacean neuropeptide motif database
Supplemental File 4: EndoGenius user manual
Supplemental Figures: PSM assignment decision tree (Figure S1), evaluation of motif database perturbations (Figure S2), target-decoy entrapment search results (Figure S3), DSD results (Figure S4), FDR versus EndoGenius score evaluation (Figure S5).