

Abstract
Protein-protein interactions (PPIs) are fundamental to understanding biological systems as protein complexes are the active molecular modules critical for carrying out cellular functions. Dysfunctional PPIs have been associated with various diseases including cancer. Systems-wide PPI analysis not only sheds light on pathological mechanisms, but also represents a paradigm in identifying potential therapeutic targets. In recent years, cross-linking mass spectrometry (XL-MS) has emerged as a powerful tool for defining endogenous PPIs of cellular networks. While proteome-wide studies have been performed in cell lysates, intact cells and tissues, applications of XL-MS in clinical samples have not been reported. In this study, we adopted a DSBSO-based in vivo XL-MS platform to map interaction landscapes from two breast cancer patient-derived xenograft (PDX) models. As a result, we have generated a PDX interaction network comprising 2,557 human proteins and identified interactions unique to breast cancer subtypes. Interestingly, most of the observed differences in PPIs correlated well with protein abundance changes determined by TMT-based proteome quantitation. Collectively, this work has demonstrated the feasibility of XL-MS analysis in clinical samples, and established an analytical workflow for tissue cross-linking that can be generalized for mapping PPIs from patient samples in the future to dissect disease-relevant cellular networks.
Keywords: XL-MS, breast cancer, PDX, PPI, Alkyne-A-DSBSO
Graphical Abstract
Introduction
Protein-protein interactions (PPIs) constitute the fundamental basis of complex biological networks that govern cellular functionality and physiological states1,2. It is well-known that altered and dysregulated PPIs have been intimately linked to a variety of human pathologies3–5, prominently including cancer6. As such, there is a paramount importance to develop methodologies to unravel the molecular mechanisms behind these interactions and identify clinical biomarkers for developing therapeutic treatments7. Within the last several decades, affinity purification mass spectrometry (AP-MS) has proven powerful and been utilized to profile PPIs at the global scale from many organisms due to its sensitivity, versatility, speed and scope8,9. The interaction data generated by large-scale AP-MS studies have contributed to the expansion of current protein interaction databases9,10. However, the potential reorganization of PPIs upon cell lysis and difficulty in maintaining weak and transient interactions prevent the full characterization of native PPIs. In recent years, AP-MS has been coupled with proximity labeling (e.g. APEX (ascorbate peroxidase), BioID) to improve the detection of dynamic and transient interactions11,12. However, these approaches only provide a list of putative interactors and can result in high background due to label diffusion and/or nonspecific binding13. In addition, affinity purification-based strategies require cell engineering to express tagged or fusion proteins in host cells, and often involve iterative experiments with a large number of bait proteins to unravel PPI networks at the systems-level, limiting their applicability to clinical samples.
Cross-linking mass spectrometry (XL-MS) has emerged as a powerful tool for studying PPIs both in vitro and in vivo, offering a unique capability to capture native PPIs from cellular environments14–20. This approach reveals endogenous PPI identities and interaction contacts concurrently to enable the differentiation between direct and indirect interactors. The cross-links formed between proximal residues of interacting proteins can further serve as distance constraints to assist in refining existing structures and elucidating architectures of native protein complexes14–17,21–23. To advance XL-MS studies, we have previously developed a series of sulfoxide-containing MS-cleavable cross-linkers to facilitate the detection and identification of cross-linked peptides24,25, which have proven effective for global PPI mapping25–27. While various cross-linking platforms have been utilized for whole-proteome PPI mapping of cell lysates25,27–29, intact cells26,30–32, organelles33–35, and tissues18,33,36, XL-MS analysis of clinical samples has not been explored. To identify disease-specific PPIs and biomarkers from clinical samples, we have taken our previously developed XL-MS platform featuring enrichable and MS-cleavable cross-linkers i.e. Alkyne/Azide-A-DSBSO (aka DSBSO)37,38, for in-cell cross-linking26 and adapted it for XL-MS at the tissue level.
Patient-derived xenografts (PDXs) have become indispensable models in cancer research due to their capacity to faithfully reproduce the characteristics and behavioral attributes of human tumors39. Thus far, these models have been extensively employed to study cancer biology through genomic sequencing40, transcriptional profiling41, immunohistochemical staining42, and proteomics43,44. While comprehensive PPI characterization of PDX and primary tumors is still lacking, recent studies have identified similar genomic signatures and protein profiles between breast cancer PDX and its original tumors45,46. These studies collectively suggest that PDX models are a valuable preclinical system for disease PPI identification. Therefore, we performed XL-MS analysis of two major breast cancer subtypes: basal and luminal PDX models. The luminal subtype is the most prevalent (~45% of cases) and considered as low grade, which expresses hormone receptors and responds well to hormone therapy, but poorly to chemotherapy. The basal subtype constitutes around 15% of cases and is characterized by high-grade, proliferative, and hormone receptor-negative tumors with no targeted therapy and poor prognosis. Characterization of these cancer subtypes at the PPI level is crucial for understanding their distinct clinical features and identifying potential targets for diagnosis, prognosis, and therapy. To this end, we have adapted the DSBSO-based in vivo XL-MS platform to analyze the two types of breast cancer PDX tissues and obtained a comprehensive XL-PPI network composed of 2,557 proteins. To the best of our knowledge, this study represents the first application of XL-MS to PDX samples, highlighting its effectiveness for defining interaction networks in clinical samples. The analytical strategy established here can be generalized to study protein interactions in other disease models to help us delineate molecular networks underlying human pathologies.
EXPERIMENTAL PROCEDURES
Cross-Linking, Protein Extraction and Trypsin Digestion of Mouse Xenograft Tissue
Patient-derived xenograft (PDX) tumor tissues used in this study were from NCI Patient-Derived Models Repository (PDMR), including basal (P96) and luminal (P97) subtypes of breast cancers. Tumor pieces for each subtype were dissected from mouse as previously described44. Thirty mg of breast cancer patient-derived xenograft tissues (basal and luminal) were cryopulverized prior to suspension in cross-linking buffer (20 mM HEPES (4-(2-Hydroxyethyl)piperazine-1-ethane-sulfonic acid), 150 mM NaCl, 1.5 mM MgCl2, pH 7.8) containing 2 mM Alkyne-A-DSBSO. The tissue powders were then incubated for 1 h with rotation at 37 °C. Following quenching of remaining cross-linker with 50 mM ammonium bicarbonate for 15 min, the samples were homogenized by 22G needle. The supernatant was clarified by centrifugation at 14,000 g and protein concentration was measured by Bradford assay. 1 mg protein from each PDX sample was digested using FASP (filter aided sample preparation) procedure as previously described26 (see Supplemental Methods). The digested peptides were desalted and stored in −80 °C prior to subsequent “click”-based labeling and enrichment.
Click Chemistry-Based Cross-Link Enrichment.
Click labeling of BPA (biotin picolyl azide) to Alkyne-A-DSBSO cross-linked peptides was performed as previously described26. Briefly, BPA, BTTAA, CuSO4, and sodium ascorbate were added to peptide digests (40 uL) to a final concentration of 1.2 mM, 1 mM, 500 μM, and 5 mM, respectively. The mixture was then rotated in the dark at room temperature for 2 h. After labeling, binding buffer (25 mM sodium phosphate, 150 mM NaCl, pH 7.5) was added to a final volume of 200 μL and rotated with 250 μL of streptavidin beads for 2 h at room temperature. After washing with binding buffer twice, the DSBSO cross-linked peptides were eluted from the beads through acid cleavage using 10% formic acid at 37 °C overnight.
Cross-link Identification and Data Analysis
The eluted peptides were first separated by SEC (size exclusion chromatography); the two primary fractions with cross-linked peptides (F23–25min, F25–27min) were collected and injected for LC-MSn analysis using an UltiMate 3000 RSLC coupled in-line to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher, San Jose, CA). MSn data were extracted using PAVA and subjected to database searching using Batch-Tag within Protein Prospector (v.6.3.5). Cross-linked peptides were identified by the integration of MSn data with database search results using the in-house software xl-Tools as described26 (Supplemental Methods). PPI analysis and structural mapping were derived based on DSBSO XL-MS data. Gene Ontology enrichment was performed using the R package “ClusterProfiler”47. BioPlex (https://bioplex.hms.harvard.edu/)9, BioGrid (https://thebiogrid.org/)48 and CORUM (http://mips.helmholtz-muenchen.de/corum)49 were used to annotate PPIs and map protein complexes. Mapping of cross-links to high-resolution structures of protein complexes was performed using PyMOL as previously described26.
TMT Quantification of Mouse PDX Proteomes
100 μg of peptides from each sample were labeled with TMT-16-plex in the presence of 30% acetonitrile. Samples were labeled for 60 min at room temperature. After labeling, the reaction was quenched by adding 5% hydroxylamine followed by incubation at RT for 15 min. Then, the labeled peptides were pooled and desalted for subsequent LC-MS/MS analysis. Mass spectrometry analysis and MS data searching were performed as previously described44 (see Supplemental Methods).
RESULTS AND DISCUSSION
DSBSO-Based XL-MS Analysis of Breast Cancer PDX Tissues
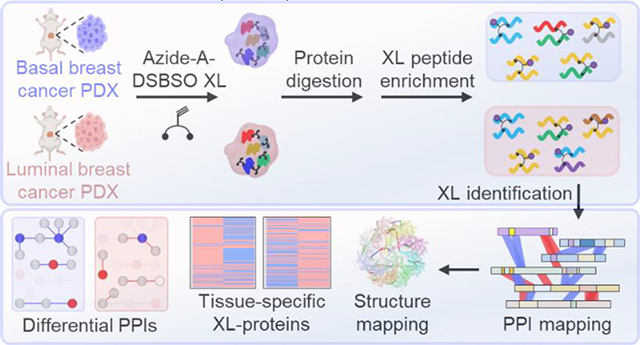
To expand the application of XL-MS to clinical samples and assess the feasibility to identify disease-relevant PPIs, we have adapted the DSBSO XL-MS strategy and applied it to two (basal and luminal) breast cancer PDX models (Figure 1). To enable cross-linking of frozen tissues, PDX samples were first cryopulverized before cross-linking with Alkyne-A-DSBSO. Next, the cross-linked tissues were lysed in urea-containing buffer, and the extracted proteins were enzymatically digested (Figure 1). The resulting cross-linked peptides were attached to a biotin tag through click chemistry-based conjugation, followed by binding to streptavidin resin26. After extensive washing and acid elution, cross-linked peptides were isolated from non-cross-linked peptides. To enhance the detection and identification of the most structurally informative inter-linked peptides, the enriched cross-linked peptides were further separated by peptide SEC and the fractions containing interlinked peptides were then subjected to LC MSn for unambiguous identification50.
Figure 1.
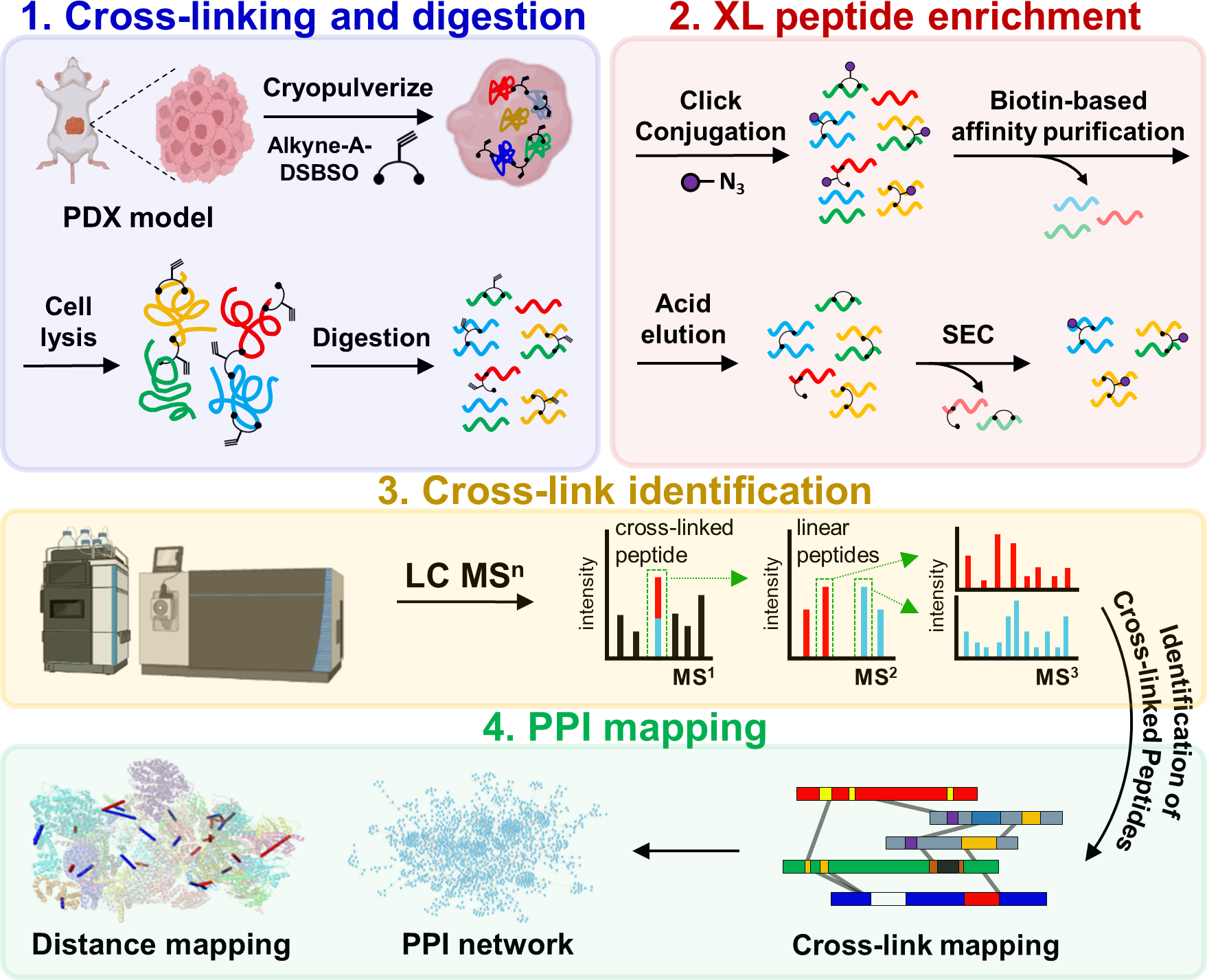
The DSBSO-based XL-MS platform for global PPI mapping in breast cancer PDX models. Alkyne-A-DSBSO was used for tissue cross-linking in this study.
Here, we performed XL-MS analyses on two biological replicates of basal and luminal breast cancer PDX samples. In total, 15,952 CSMs (cross-link peptide spectrum matches) were identified at an FDR of 1.8%, generating an XL-proteome composed of 2,557 human proteins (Table S1). The overall results are comparable to our previous proteome-wide XL-data of HEK 293 cell lysates27 and cells26, demonstrating the effectiveness of the DSBSO-based workflow for PDX tissues.
XL-Proteomes of Breast Cancer PDX Tissues
From basal breast cancer PDX samples, a total of 8,707 CSMs were obtained from 1,908 cross-linked proteins. Similarly, a total of 7,245 CSMs were obtained for luminal breast cancer PDX samples from 1,720 cross-linked proteins (Table S1). While the total number of cross-linked proteins identified from basal and luminal breast cancer are similar, 836 and 649 proteins were unique to basal breast cancer and luminal breast cancer respectively, with 41.9% overlap between the two compared samples (Figure 2A). In addition, 34.6% of K-K linkages were found in both types of the samples, suggesting subtle differences in cancer interactomes. To better assess the cross-linking data, we have performed tandem mass tag (TMT)-based quantitative analysis on both PDX models to determine the relative protein abundances within their proteomes. As a result, we identified and quantified a total of 8,855 human proteins from basal and luminal PDX models, referred to as the TMT-proteome from this point onward (Table S2).
Figure 2.
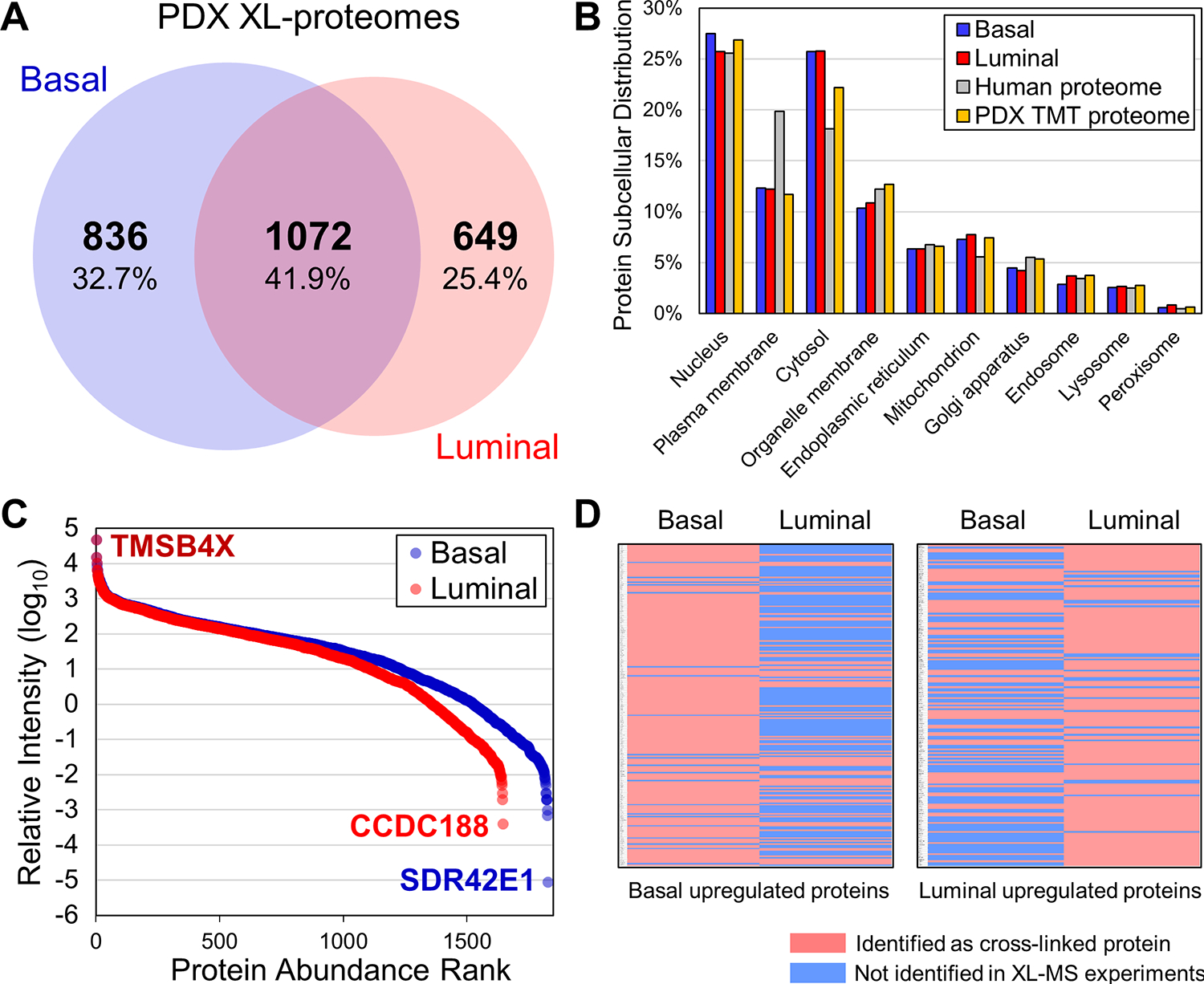
Annotation of the PDX XL proteomes. (A) Venn diagram depicting overlap of the basal and luminal XL-proteomes. (B) Comparison of subcellular compartmental distribution between basal and luminal XL-proteomes, as well as the human proteome and the PDX TMT-proteome. (C) Protein abundance distributions of basal and luminal XL-proteomes. (D) Heatmaps showing the identification of cross-linked proteins that were upregulated in basal and luminal samples respectively, illustrating good correlations between the cross-link detectability and protein relative abundance in the two compared samples.
To understand the breadth of the XL-proteomes, we first compared their subcellular compartment distributions to those of the human51 and TMT-proteomes. As shown in Figure 2B, the PDX XL- and human proteomes shared similar subcellular distributions that differed only slightly in the representation of cytosolic and plasma membrane proteins, similar to results from previous XL-MS data of HEK293 cells26 and lysates25,27. To examine the dynamic range of proteins comprising the PDX XL-proteomes, we plotted the abundance distribution of cross-linked proteins against that of the integrated human proteome obtained through PAXDB52 (Figure 2C). As illustrated, the overall relative intensities of cross-linked proteins spanned a range over 7 orders of magnitude, comparable to previously published data26,27. While the overall content of the XL-proteome coincides well with the TMT-proteome, 581 proteins were only identified by XL-MS analyses. This includes the highest abundant protein (i.e. TMSB4X, ~5 kDa) and the two lowest abundant proteins (i.e. SDR42E1 and CCDC188) found in our XL-MS data (Figure 2C), suggesting that XL-MS analysis can offer complementary protein identification to shotgun proteomics and enhance proteome coverage. The observed differences can be attributed to the enrichment of cross-links and the dependence of cross-link identification on the abundance of protein-protein interactions rather than solely on the abundance of individual proteins. In addition, cross-linkability at the interaction interfaces and the resulting cross-linked peptides can influence their identification.
Within the PDX TMT-proteome, 937 proteins exhibited downregulation in luminal breast cancer (luminal/basal ≤ 0.5), and 801 proteins displayed upregulation in luminal breast cancer (luminal/ basal ≥ 2) (Table S2). Of the 937 downregulated proteins, 212 were identified as cross-linked proteins. Of these, 56 were found in both samples, while 135 and 21 were unique to basal and luminal, respectively. Similarly, of the 801 upregulated proteins, 175 were identified by XL-MS with 25 unique to basal, 93 to luminal, and 57 shared in both samples (Figure 2D). Notably, the observed variations in protein abundance between the two breast cancer subtypes determined by TMT-based quantitation exhibited a high degree of accordance with differential identification of cross-linked proteins by XL-MS (Figure 2D and Table S2). This consistency reaffirms the reliability of our XL-data and the differences observed in the two PDX models.
Gene Ontology Analysis of the PDX XL-Proteomes
To examine the context of the XL-proteomes, we first performed Gene Ontology (GO) analysis of cellular components. Distinct enrichment patterns were unveiled between basal and luminal breast cancer proteomes, as well as between breast cancer53 and normal breast tissue54. Both basal and luminal breast cancer showed common enrichments in cellular components, such as cell-substrate junction, focal adhesion, actin cytoskeleton, and ribosome. In comparison, luminal breast cancer exhibited a unique focus on ficolin-1-rich granules, suggesting differences in vesicular trafficking and immune-related processes (Figure 3B). Interestingly, cellular component GO analyses on normal breast tissue and breast cancer proteomes (Figures 3C, D) also revealed pronounced disparities, with the cancer proteome displaying enrichments in cell-substrate junction and focal adhesion—the two most enriched cellular components in both basal and luminal XL-proteomes.
Figure 3.
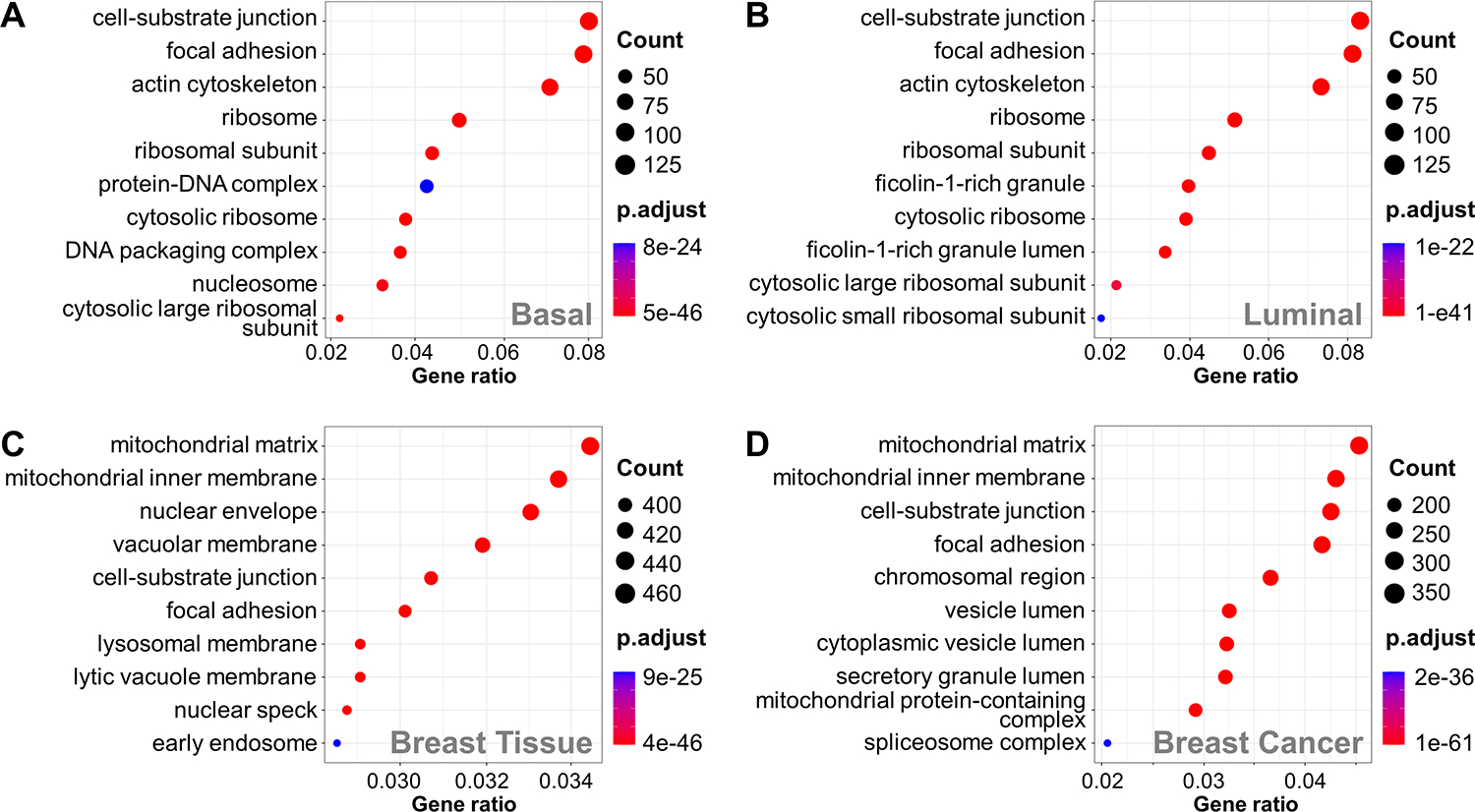
GO enrichment analysis of PDX XL-proteomes and normal/cancer breast tissue proteomes. Cell component enrichment analysis of (A) basal XL-proteome, (B) luminal XL-proteome, (C) normal breast proteome, and (D) breast cancer proteome
Next, molecular function GO analysis revealed that both basal breast cancer and luminal breast cancer highlighted cadherin binding, actin binding, and ATP hydrolysis activity (Figure S1A, B). While normal breast tissue also showed enrichments in cadherin binding, actin binding and ATP hydrolysis activity were only enriched in the breast cancer proteome (Figure S1C, D), illustrating an expected similarity between breast cancer proteins and basal or luminal cross-linked proteins. Finally, the XL-proteomes of basal and luminal breast cancer were found to be enriched in biological processes associated with increased cellular activity, such as cytoplasmic translation, nucleosome assembly, chromatin remodeling, and protein folding (Figures S1E, F). These results suggest an augmented demand for protein synthesis, genomic organization, and cellular machinery—hallmarks of the proliferative nature of cancer cells, that correlate well with the enriched biological processes observed in the breast cancer but not normal breast tissue proteome (Figure S1G, H). Taken together, the PDX XL-proteomes contain molecular information that provides insights into the subtle differences between basal and luminal breast cancer and their divergence from normal breast tissue.
Validation of Cross-links by 3-D Structural Mapping
To assess the validity of the identified cross-links, we first compared our XL-proteome against the CORUM protein complex database. As a result, 1,908 cross-linked proteins from basal breast cancer were distributed into 560 CORUM protein complexes, in which 356 (63.6%) and 79 (14.11%) were identified with ≥ 50% and ≥ 90% protein composition, respectively (Table S3). In comparison, 1,720 cross-linked proteins from the XL-proteome of luminal breast cancer PDX comprised 426 CORUM protein complexes. Similarly, 258 (60.1%) of the protein complexes were identified with ≥ 50% protein compositions and 64 protein complexes (15.2%) were determined with subunit recovery exceeding 90% (Table S3). These results illustrate the capability of XL-MS analysis to uncover endogenous protein complexes from breast cancer tissues.
Based on the number of identified K-K linkages, the CCT, cytoplasmic ribosome, and Nop56p-associated pre-rRNA complexes were best represented in our XL-data. The CCT complex was identified with 8 subunits (100% composition) in both samples. To examine cross-link validity, we mapped the identified linkages to a high-resolution structure of the CCT complex (PDB: 7LUP) (Figure 4A). 84.6% (22/26) and 87.9% (29/33) of the linkages from basal and luminal PDX samples, respectively, corresponded to Cα-Cα distances ≤ 35 Å, thereby satisfying the maximum DSBSO distance threshold (Figure 4C). While 3 out of the 4 non-satisfactory cross-links spanned distances ranging from 43–64 Å, one linkage (CCT8:K466-TCP1:K494) was found to span 129.3 Å when mapped to a single CCT ring. However, when mapped between two adjacent rings, its distance was reduced to 22.2 Å, supporting the presence of a multimer consisting of dimerized CCT rings55 (Figure S2). Another major complex identified here was the cytoplasmic ribosome complex, which consists of 79 subunits and was identified with 110 linkages from 65 subunits in basal and 93 linkages from 58 subunits in luminal cancer. When mapped to the available structure (PDB entry: 6OLG) (Figure 4B), more than 95% cross-links were ≤ 35 Å, demonstrating excellent cross-link distance satisfaction (Figure 4C). Interestingly, this satisfaction rate is comparable to that of previously reported in-cell XL-data (94%) 26, but much better than that of cell lysate-derived XL-data (72~76%)27. It is known that the ribosomal complex is dynamic, and its interactions are prone to change during cell lysis. As tissue cross-linking preserves ribosomal interactions similarly to in-cell cross-linking, our results suggest that the DSBSO XL-MS workflow is applicable for capturing endogenous protein interactions from frozen PDX tissues. When all cross-links were mapped to available high-resolution structures of 254 protein complexes, more than 95% of them were satisfactory, further reaffirming cross-link identification reliability (Figure 4D, Table S4).
Figure 4.
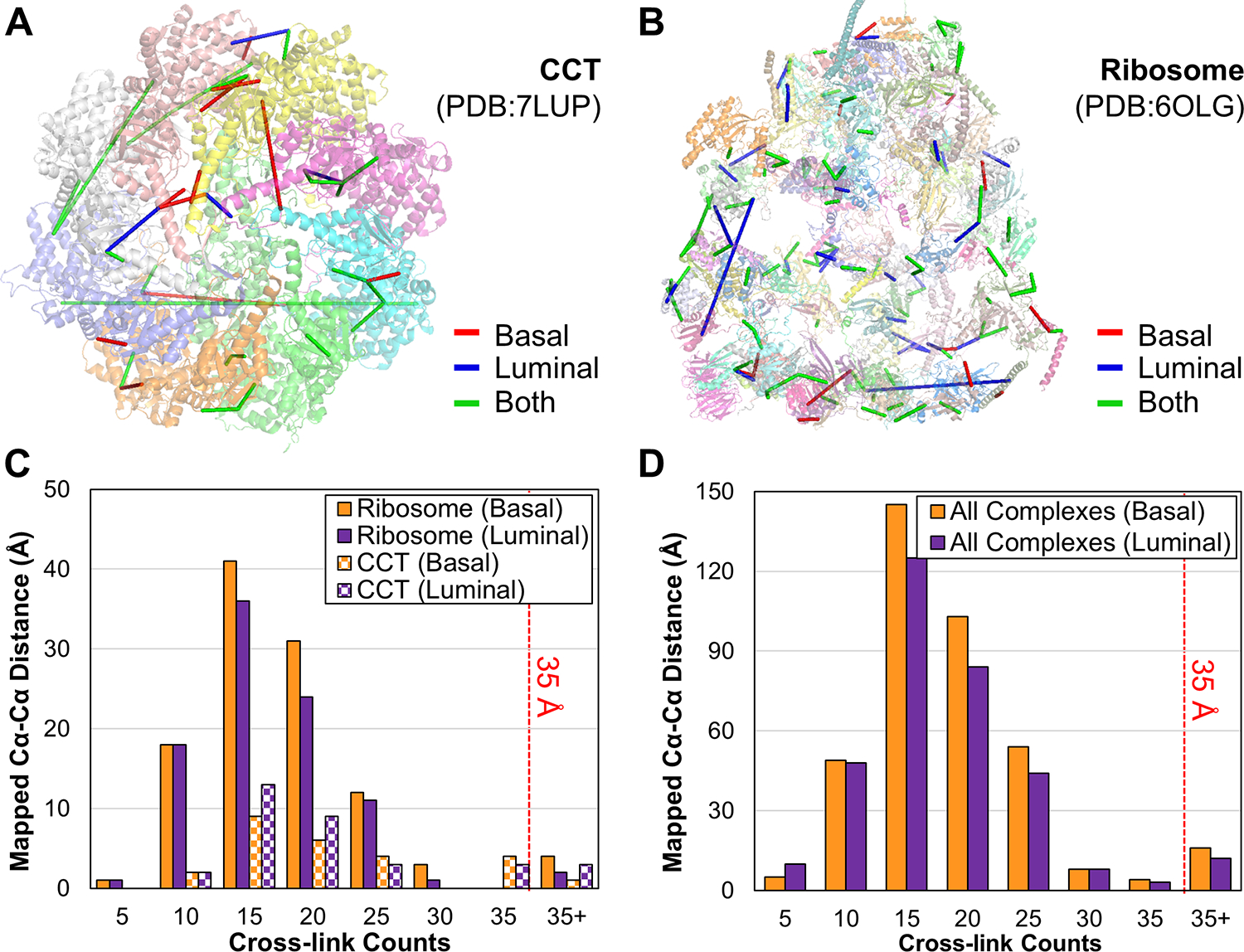
Evaluation of cross-link validity by structural mapping. Mapping of cross-links onto high-resolution structures of (A) CCT (PDB: 7LUP) and (B) ribosome (PDB: 6OLG) complexes. Cross-links unique to basal PDX shown in red, unique to luminal PDX shown in blue, and identified in both samples depicted in green. (C) Distance distributions of the mapped cross-links from ribosome and CCT complexes. (D) Distance distributions of mapped cross-links from all available high-resolution structures.
XL-PPI Networks of the PDX Models
To define and compare the interactomes of basal and luminal breast cancer, we generated a combined XL-PPI network of both PDX models comprising 2,557 nodes and 3,643 edges denoting 2,516 inter-protein and 1,127 intra-protein interactions (Figure S3). Among them, about 31% of the total XL-PPIs were found in both samples. While the number of inter-protein XL-PPIs captured from basal and luminal breast cancer were comparable (1,600 vs. 1,411), their overlap was only 19.7% (Figure S4A), suggesting noticeable differences in their PPI networks. To evaluate the authenticity of identified XL-PPIs, we cross-referenced the interactions within the STRING database56. Similar to our previous studies26,27, 85% of the XL-PPIs present in STRING (211 from basal and 189 from luminal) corresponded to STRING scores above 0.9, suggesting high confidence of the identified interactions (Figure S4B). Next, we compared our XL-PPIs against the BioPlex and BioGrid PPI databases, as well as other published proteome-wide XL-MS data. Out of the 1,600 XL-PPIs identified from basal breast cancer, 373 were found in the selected PPI databases and 200 have been reported by previous proteome-wide XL-MS studies25–29,57,58, with the remaining 1,027 XL-PPIs representing novel interactions (Table S5A). For the 1,411 XL-PPIs identified from luminal breast cancer, 313 were found in the PPI databases and 160 were reported in the proteome-wide XL-MS data, leaving 938 XL-PPIs as novel interactions (Table S5B).
Interestingly, the 155 novel XL-PPIs shared by both PDX models involve 234 proteins including cancer-specific proteins. For instance, a known candidate biomarker for head and neck squamous cell carcinoma59, TMSB4X, has been found to interact with two breast cancer-related proteins, UACA and RAI14 (Figure 5). UACA has been identified as a modulator of chemoresistance and survival in breast cancer60, while down-regulation of RAI14 can inhibit the proliferation and invasion of breast cancer cells61. Another protein known to be important in regulating tumorigenesis and metastasis in breast cancer is TMSB1062, which has been found to interact with actin. As actins hold central importance in cell motility63, their interaction suggests a potential connection to cancer cell metastasis and invasion. Moreover, high expression of GRHL2 is strongly correlated with lower relapse-free survival in all four subtypes of breast cancer64. GSN was found to have higher expression in breast cancer tissue than histologically normal epithelium65. The novel interaction of GRHL2 and GSN identified here presents a link connecting their functions in tumorigenesis. In addition to the novel PPIs found in both breast cancer samples, our study has unveiled novel PPIs specific to basal and luminal breast cancer subtypes (Table S5A–B). For example, an interaction between CCDC188 and ATP5A1 was only detected in luminal breast cancer samples (Figure 5). While the role of CCDC188 in cancer is unclear, ATP5A1, a subunit of the ATP synthase complex, has been reported to be abnormally upregulated in breast cancer and used as a prognostic indicator and a therapeutic target for breast7 and lung cancer66, suggesting potential importance of their interaction in breast cancer.
Figure 5.
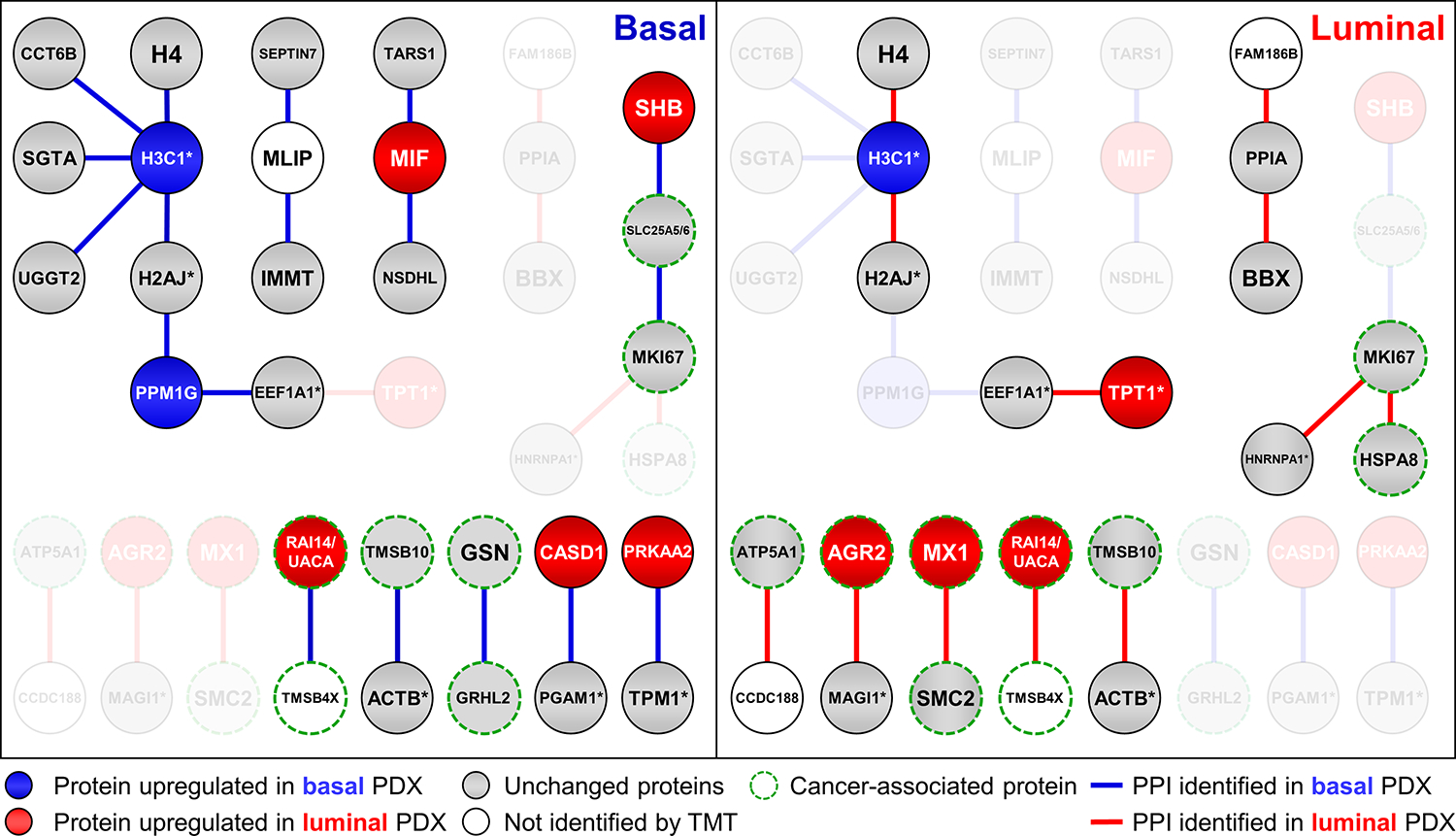
Selected XL-PPIs from base and luminal PDX models. Node colors represent relative TMT quantitation in basal and luminal PDX samples. Blue: protein upregulated in basal PDX; red: protein upregulated in luminal PDX; gray: protein unchanged in basal vs. luminal PDX; no color: protein not identified in TMT-proteome. Blue edges denote XL-PPIs identified in basal PDX; red edges correspond to those identified in luminal PDX. Edges and corresponding missing nodes from each PDX sample grayed out. Note: “*” signifies a protein group containing multiple homologous proteins.
Correlation of XL-PPIs with Protein Abundance in PDX Samples
To better understand the correlation of XL-PPIs to protein abundance, we constructed an XL-PPI subnetwork containing 821 differentially expressed proteins with 835 edges (Figure S5), with 145 interactions present in both cancer subtypes, 403 exclusively detected in basal breast cancer and 287 unique to luminal breast cancer. The majority of these PPIs displayed a strong correlation with their relative abundance in the two compared samples (Table S2). For example, a downregulated protein in luminal breast cancer, H3C1, was found to interact with CCT6B, SGTA, UGGT2 in basal breast cancer (Figure 5). PPM1G, another downregulated protein in luminal breast cancer, was engaged in interactions with two protein groups that were exclusively identified in basal breast cancer. Similarly, TPT1, an upregulated protein in luminal breast cancer, was observed to interact with 1 protein group in the luminal breast cancer context (Figure 5). AGR2 (cancer-relevant protein, luminal/basal, 20.4) and MX1(luminal/basal 18.1) are two highly upregulated proteins in luminal breast cancer (Table S2), and their interactions with others were only detected in luminal breast cancer (Figure 5).
While most of the differentially detected XL-PPIs correlate well with relative protein abundance in the two compared samples, some do not follow the same trend. For instance, for four upregulated proteins in luminal breast cancer, MIF, SHB, CASD1 and PRKAA2 (Table S5), their pair-wise interactions (MIF-NSDHL, MIF-TARS, SHB-SLC25A5/6, CASD1-PGAM1/2/4 and PRKAA2-TPM1/2/3/4) were only identified in basal breast cancer. Notably, PRKAA2 is a catalytic subunit of AMP-activated protein kinase and tropomyosin proteins (TPM1/2/3/4) are known phosphorylated proteins. The PRKAA2-TPM interaction may be involved in modulating the phosphorylation states of TPM proteins during tumorigenesis.
Apart from the PPIs involving differentially expressed proteins, our constructed XL-PPI interactome also contains a subnetwork of unchanged proteins (luminal/basal, 0.5–2) in the TMT-proteome that were reproducibly and differentially detected in our XL-data. This subnetwork contains 586 nodes and 662 edges, in which 490 PPIs were identified in both cancer subtypes, 123 unique to basal, and 49 to luminal breast cancer. Interestingly, despite no apparent abundance differences of these proteins between the two cancer subtypes, some PPIs were exclusively detected in one or the other. For instance, the interactions of MLIP with two cancer related proteins, IMMT and SEPTIN7, were only found in basal breast cancer. SEPTIN7 has been reported to have an inhibitory effect on cancer cells migration and invasion67, while IMMT serves as an independent diagnostic biomarker for advanced clinical status and poor relapse-free survival rate in breast cancer patients68. Similarly, the two pair-wise interactions (PPIA-BBX and PPIA-FAM186B) were only detected in luminal breast cancer. PPIA has been shown to play a crucial role in the development of various human cancers, such as gastric cancer69, colon cancer70, hepatocellular carcinoma71, lung adenocarcinoma72, and multiple myeloma73. Notably, the same cross-link between PPIA and FAM186B was found in previous XL-MS data of the HEK293 cells26, reaffirming their interaction.
Another unchanged protein is MKI67, a known biomarker for breast cancer proliferation, which exhibited different interactions in the two cancer subtypes. As shown in Figure 5, MKI67 was found to interact with SLC25A5/6 in basal, whereas its interaction with HSPA8 and HNRNPA protein group were identified in luminal. Remarkably, all of the MKI67-interacting proteins identified in our XL-proteomes have been shown to be associated with cancer. HSPA8 has a similar function to MKI67 for cell proliferation in breast cancer74, and their interaction is further supported by our previous XL-data of HEK 293 cells26. Furthermore, increased expression of SLC25A5/6 has been reported in cancer cells75, and members of the heterogeneous nuclear ribonucleoprotein family (i.e. HNRNPD, HNRNPK) have been implicated as having oncogenic potential or being capable of modulating oncogenic pathways. The identification of these cancer-specific PPIs suggests that MKI67 may coordinate with different proteins during breast cancer development. Collectively, our XL-MS analysis was able to identify tissue-specific PPIs that involve cancer-relevant proteins associated with tumorigenesis. Our results suggest that the XL-PPI networks of the PDX models are modulated by multiple factors including protein abundance and disease states, and the network rewiring is associated with breast cancer subtypes.
Conclusion
In this study, we have adapted the DSBSO-based in vivo XL-MS platform to define protein interactions in basal and luminal breast cancer PDX models. The scope and depth of the resulting XL-proteomes are comparable to systems-wide XL-MS analyses of intact cells and cell lysates, demonstrating the effectiveness of tissue cross-linking. While the PDX XL-proteome only comprises a portion of the proteome, its current content contains molecular information that exhibits the enrichment of cellular components and molecular functions specific to breast cancer. In comparison with the PDX proteome determined by TMT quantitation, differential detection of cross-linked proteins in the two compared samples corresponded very well with their relative abundance, supporting the reliability of the XL-MS data. The PPI network analysis revealed that a diverse group of protein interactions have been identified with a number of interactions involving cancer-relevant proteins. Importantly, tissue-specific interaction profiles have been observed, suggesting potential network rewiring in the two selected cancer subtypes. Interestingly, XL-PPIs only identified in a single cancer subtype did not necessarily correlate with their relative abundances. This observation supports the notion that alterations in PPIs are not limited to protein abundance changes, but also affected by cellular states, uncovering new insights on molecular differences between cancer subtypes beyond conventional proteome profiling. Notably, apart from molecular differences in the two selected cancer subtypes, other factors such as genetic and environmental alterations could contribute to the observed PPI differences. Therefore, multiple sample groups within a given subtype would be needed to better understand the attributes of network differences in future studies. Additionally, the integration of isotope-labeling based quantitative XL-MS strategies will allow accurate determination of condition-specific PPIs for revealing cancer-specific interactomes and their association with disease progression76. Moreover, coupling cross-linking with co-Fractionation can facilitate the elucidation of functional protein modules77. Overall, the analytical workflow established here offers a promising avenue for comprehending PPIs in disease contexts to advance our understanding of human pathologies and uncover new molecular targets for improved therapeutic strategies.
Supplementary Material
Acknowledgments
We wish to thank Prof. A.L. Burlingame, Drs. Peter Baker and Robert Chalkley for their support of Protein Prospector. This work was supported by National Institutes of Health grants R35GM145249 to L.H. and U24CA271079 to H.Z. and D.C..
ABBREVIATIONS
- PPIs
protein-protein interactions
- XL-MS
cross-linking mass spectrometry
- Azide/Alkyne-A-DSBSO
Azide/Alkene-tagged, acid-cleavable disuccinimidyl bis-sulfoxide
- DSBSO
Azide/Alkyne-A-DSBSO
- PDX
patient-derived xenografts
- MSn
multi-stage mass spectrometry
- CID
collisional induced dissociation
- LC-MSn
liquid chromatography multistage mass spectrometry
- CSM
cross-link peptide spectrum match
- BPA
biotin picolyl azide
- BTTAA
2-(4-((bis((1-(tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)acetic acid
- TMT
tandem mass tag
- FASP
filter aided sample preparation
- HEPES
4-(2-Hydroxyethyl)piperazine-1-ethane-sulfonic acid
- SEC
size exclusion chromatography
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
The following supporting information is available free of charge at:
https://pubs.acs.org/doi/10.1021/acs.jproteome.3c00832.
Supplemental Methods and GO analysis of XL-proteomes from basal and luminal breast PDX samples, normal breast tissue proteome and breast cancer proteome (Figure S1), Distance mapping of CCT8:K466-TCP1:K494 (Figure S2), XL-PPI network of DSBSO cross-linked basal and luminal breast cancer PDX samples (Figure S3), Comparison of PPIs identified from basal and luminal breast cancer PDX samples and their STRING score distribution (Figure S4), and Comparison of XL-PPIs from differential regulated proteins (Figure S5). Identified cross-linked peptides (Table S1.xlsx), TMT quantitation results (Table S2.xlsx), CORUM protein complex analysis (Table S3.xlsx), distance mapping of cross-links (Table S4.xlsx), and comparison of PPI with databases and literature (Table S5.xlsx).
Data Availability
Data are available via ProteomeXchange with identifier PXD046966
References
- (1).Alberts B The cell as a collection of protein machines: preparing the next generation of molecular biologists cell 1998, 92, 291–294. [DOI] [PubMed] [Google Scholar]
- (2).Leitner A; Faini M; Stengel F; Aebersold R Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines Trends in biochemical sciences 2016, 41, 20–32. [DOI] [PubMed] [Google Scholar]
- (3).Rual JF; Venkatesan K; Hao T; Hirozane-Kishikawa T; Dricot A; Li N; Berriz GF; Gibbons FD; Dreze M; Ayivi-Guedehoussou N Towards a proteome-scale map of the human protein–protein interaction network Nature 2005, 437, 1173–1178. [DOI] [PubMed] [Google Scholar]
- (4).Cheng F; Zhao J; Wang Y; Lu W; Liu Z; Zhou Y; Martin WR; Wang R; Huang J; Hao T Comprehensive characterization of protein–protein interactions perturbed by disease mutations Nature genetics 2021, 53, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lim J; Hao T; Shaw C; Patel AJ; Szabó G; Rual J-F; Fisk CJ; Li N; Smolyar A; Hill DE A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration Cell 2006, 125, 801–814. [DOI] [PubMed] [Google Scholar]
- (6).Gulati S; Cheng TM; Bates PA In Seminars in cancer biology; Elsevier, 2013, pp 219–226. [DOI] [PubMed] [Google Scholar]
- (7).Li Z; Ivanov AA; Su R; Gonzalez-Pecchi V; Qi Q; Liu S; Webber P; McMillan E; Rusnak L; Pham C The OncoPPi network of cancer-focused protein–protein interactions to inform biological insights and therapeutic strategies Nature communications 2017, 8, 14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Huttlin EL; Ting L; Bruckner RJ; Gebreab F; Gygi MP; Szpyt J; Tam S; Zarraga G; Colby G; Baltier K The BioPlex network: a systematic exploration of the human interactome Cell 2015, 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Huttlin EL; Bruckner RJ; Navarrete-Perea J; Cannon JR; Baltier K; Gebreab F; Gygi MP; Thornock A; Zarraga G; Tam S Dual proteome-scale networks reveal cell-specific remodeling of the human interactome Cell 2021, 184, 3022–3040. e3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Michaelis AC; Brunner AD; Zwiebel M; Meier F; Strauss MT; Bludau I; Mann M The social and structural architecture of the yeast protein interactome Nature 2023, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Qin W; Cho KF; Cavanagh PE; Ting AY Deciphering molecular interactions by proximity labeling Nature methods 2021, 18, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu X; Salokas K; Weldatsadik RG; Gawriyski L; Varjosalo M Combined proximity labeling and affinity purification– mass spectrometry workflow for mapping and visualizing protein interaction networks Nature protocols 2020, 15, 3182–3211. [DOI] [PubMed] [Google Scholar]
- (13).May DG; Scott KL; Campos AR; Roux KJ Comparative application of BioID and TurboID for protein-proximity biotinylation Cells 2020, 9, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yu C; Huang L Cross-linking mass spectrometry (XL-MS): An emerging technology for interactomics and structural biology Analytical chemistry 2018, 90, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tang X; Wippel HH; Chavez JD; Bruce JE Crosslinking mass spectrometry: a link between structural biology and systems biology Protein Science 2021, 30, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Piersimoni L; Kastritis PL; Arlt C; Sinz A Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein–Protein Interactions– A Method for All Seasons Chemical Reviews 2021, 122, 7500–7531. [DOI] [PubMed] [Google Scholar]
- (17).Lee K; O’Reilly FJ Cross-linking mass spectrometry for mapping protein complex topologies in situ Essays in Biochemistry 2023, 67, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chavez JD; Mohr JP; Mathay M; Zhong X; Keller A; Bruce JE Systems structural biology measurements by in vivo cross-linking with mass spectrometry Nature protocols 2019, 14, 2318–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sinz A Cross-linking/mass spectrometry for studying protein structures and protein–protein interactions: where are we now and where should we go from here? Angewandte Chemie International Edition 2018, 57, 6390–6396. [DOI] [PubMed] [Google Scholar]
- (20).Matzinger M; Mechtler K Cleavable cross-linkers and mass spectrometry for the ultimate task of profiling protein–protein interaction networks in vivo Journal of Proteome Research 2020, 20, 78–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).O’Reilly FJ; Rappsilber J Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology Nature structural & molecular biology 2018, 25, 1000–1008. [DOI] [PubMed] [Google Scholar]
- (22).Britt HM; Cragnolini T; Thalassinos K Integration of mass spectrometry data for structural biology Chemical Reviews 2021, 122, 7952–7986. [DOI] [PubMed] [Google Scholar]
- (23).Graziadei A; Rappsilber J Leveraging crosslinking mass spectrometry in structural and cell biology Structure 2022, 30, 37–54. [DOI] [PubMed] [Google Scholar]
- (24).Gutierrez CB; Block SA; Yu C; Soohoo SM; Huszagh AS; Rychnovsky SD; Huang L Development of a novel sulfoxide-containing MS-cleavable homobifunctional cysteine-reactive cross-linker for studying protein–protein interactions Analytical chemistry 2018, 90, 7600–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jiao F; Salituro LJ; Yu C; Gutierrez CB; Rychnovsky SD; Huang L Exploring an alternative cysteine-reactive chemistry to enable proteome-wide PPI analysis by cross-linking mass spectrometry Analytical chemistry 2023, 95, 2532–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wheat A; Yu C; Wang X; Burke AM; Chemmama IE; Kaake RM; Baker P; Rychnovsky SD; Yang J; Huang L Protein interaction landscapes revealed by advanced in vivo cross-linking–mass spectrometry Proceedings of the National Academy of Sciences 2021, 118, e2023360118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Jiao F; Yu C; Wheat A; Wang X; Rychnovsky SD; Huang L Two-dimensional fractionation method for proteome-wide cross-linking mass spectrometry analysis Analytical chemistry 2022, 94, 4236–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bartolec TK; Vázquez-Campos X; Norman A; Luong C; Johnson M; Payne RJ; Wilkins MR; Mackay JP; Low JK Cross-linking mass spectrometry discovers, evaluates, and corroborates structures and protein–protein interactions in the human cell Proceedings of the National Academy of Sciences 2023, 120, e2219418120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu F; Rijkers DT; Post H; Heck AJ Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry Nature methods 2015, 12, 1179–1184. [DOI] [PubMed] [Google Scholar]
- (30).Jiang PL; Wang C; Diehl A; Viner R; Etienne C; Nandhikonda P; Foster L; Bomgarden RD; Liu F A Membrane-Permeable and Immobilized Metal Affinity Chromatography (IMAC) Enrichable Cross-Linking Reagent to Advance In Vivo Cross-Linking Mass Spectrometry Angewandte Chemie International Edition 2022, 61, e202113937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gao H; Zhao L; Zhong B; Zhang B; Gong Z; Zhao B; Liu Y; Zhao Q; Zhang L; Zhang Y In-depth in vivo crosslinking in minutes by a compact, membrane-permeable, and alkynyl-enrichable crosslinker Analytical Chemistry 2022, 94, 7551–7558. [DOI] [PubMed] [Google Scholar]
- (32).Zhao L; Zhong B; An Y; Zhang W; Gao H; Zhang X; Liang Z; Zhang Y; Zhao Q; Zhang L Enhanced protein–protein interaction network construction promoted by in vivo cross-linking with acid-cleavable click-chemistry enrichment Frontiers in Chemistry 2022, 10, 994572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chavez JD; Lee CF; Caudal A; Keller A; Tian R; Bruce JE Chemical crosslinking mass spectrometry analysis of protein conformations and supercomplexes in heart tissue Cell systems 2018, 6, 136–141. e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liu F; Lössl P; Rabbitts BM; Balaban RS; Heck AJ The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes Molecular & Cellular Proteomics 2018, 17, 216–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ryl PS; Bohlke-Schneider M; Lenz S; Fischer L; Budzinski L; Stuiver M; Mendes MM; Sinn L; O’reilly FJ; Rappsilber J In situ structural restraints from cross-linking mass spectrometry in human mitochondria Journal of Proteome Research 2019, 19, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Caudal A; Tang X; Chavez JD; Keller A; Mohr JP; Bakhtina AA; Villet O; Chen H; Zhou B; Walker MA Mitochondrial interactome quantitation reveals structural changes in metabolic machinery in the failing murine heart Nature cardiovascular research 2022, 1, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kaake RM; Wang X; Burke A; Yu C; Kandur W; Yang Y; Novtisky EJ; Second T; Duan J; Kao A A new in vivo cross-linking mass spectrometry platform to define protein–protein interactions in living cells Molecular & Cellular Proteomics 2014, 13, 3533–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Burke AM; Kandur W; Novitsky EJ; Kaake RM; Yu C; Kao A; Vellucci D; Huang L; Rychnovsky SD Synthesis of two new enrichable and MS-cleavable cross-linkers to define protein–protein interactions by mass spectrometry Organic & biomolecular chemistry 2015, 13, 5030–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hidalgo M; Amant F; Biankin AV; Budinská E; Byrne AT; Caldas C; Clarke RB; de Jong S; Jonkers J; Mælandsmo GM Patient-derived xenograft models: an emerging platform for translational cancer research Cancer discovery 2014, 4, 998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sun H; Cao S; Mashl RJ; Mo CK; Zaccaria S; Wendl MC; Davies SR; Bailey MH; Primeau TM; Hoog J Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment Nature communications 2021, 12, 5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Caeser R; Egger JV; Chavan S; Socci ND; Jones CB; Kombak FE; Asher M; Roehrl MH; Shah NS; Allaj V Genomic and transcriptomic analysis of a library of small cell lung cancer patient-derived xenografts Nature Communications 2022, 13, 2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Guillen KP; Fujita M; Butterfield AJ; Scherer SD; Bailey MH; Chu Z; DeRose YS; Zhao L; Cortes-Sanchez E; Yang CH A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology Nature cancer 2022, 3, 232–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mirhadi S; Tam S; Li Q; Moghal N; Pham NA; Tong J; Golbourn BJ; Krieger JR; Taylor P; Li M Integrative analysis of non-small cell lung cancer patient-derived xenografts identifies distinct proteotypes associated with patient outcomes Nature communications 2022, 13, 1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mertins P; Tang LC; Krug K; Clark DJ; Gritsenko MA; Chen L; Clauser KR; Clauss TR; Shah P; Gillette MA Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry Nature protocols 2018, 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ding L; Ellis MJ; Li S; Larson DE; Chen K; Wallis JW; Harris CC; McLellan MD; Fulton RS; Fulton LL Genome remodelling in a basal-like breast cancer metastasis and xenograft Nature 2010, 464, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Huang KL; Li S; Mertins P; Cao S; Gunawardena HP; Ruggles KV; Mani D; Clauser KR; Tanioka M; Usary J Proteogenomic integration reveals therapeutic targets in breast cancer xenografts Nature communications 2017, 8, 14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yu G; Wang LG; Han Y; He QY clusterProfiler: an R package for comparing biological themes among gene clusters Omics: a journal of integrative biology 2012, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stark C; Breitkreutz BJ; Reguly T; Boucher L; Breitkreutz A; Tyers M BioGRID: a general repository for interaction datasets Nucleic acids research 2006, 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tsitsiridis G; Steinkamp R; Giurgiu M; Brauner B; Fobo G; Frishman G; Montrone C; Ruepp A CORUM: the comprehensive resource of mammalian protein complexes–2022 Nucleic acids research 2023, 51, D539–D545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kao A; Chiu CL; Vellucci D; Yang Y; Patel VR; Guan S; Randall A; Baldi P; Rychnovsky SD; Huang L Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes Molecular & Cellular Proteomics 2011, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Aleksander SA; Balhoff J; Carbon S; Cherry JM; Drabkin HJ; Ebert D; Feuermann M; Gaudet P; Harris NL The Gene Ontology knowledgebase in 2023 Genetics 2023, 224, iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wang M; Herrmann CJ; Simonovic M; Szklarczyk D; von Mering C Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines Proteomics 2015, 15, 3163–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Tyanova S; Albrechtsen R; Kronqvist P; Cox J; Mann M; Geiger T Proteomic maps of breast cancer subtypes Nature communications 2016, 7, 10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Fagerberg L; Hallström BM; Oksvold P; Kampf C; Djureinovic D; Odeberg J; Habuka M; Tahmasebpoor S; Danielsson A; Edlund K Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics Molecular & cellular proteomics 2014, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cuéllar J; Ludlam WG; Tensmeyer NC; Aoba T; Dhavale M; Santiago C; Bueno-Carrasco MT; Mann MJ; Plimpton RL; Makaju A Structural and functional analysis of the role of the chaperonin CCT in mTOR complex assembly Nature communications 2019, 10, 2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Mering CV; Huynen M; Jaeggi D; Schmidt S; Bork P; Snel B STRING: a database of predicted functional associations between proteins Nucleic acids research 2003, 31, 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yugandhar K; Wang TY; Leung AKY; Lanz MC; Motorykin I; Liang J; Shayhidin EE; Smolka MB; Zhang S; Yu H MaXLinker: proteome-wide cross-link identifications with high specificity and sensitivity Molecular & Cellular Proteomics 2020, 19, 554–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Liu F; Lössl P; Scheltema R; Viner R; Heck AJ Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification Nature communications 2017, 8, 15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chi LH; Chang WM; Chang YC; Chan YC; Tai CC; Leung KW; Chen CL; Wu AT; Lai TC; Li YC Global proteomics-based identification and validation of thymosin Beta-4 X-linked as a prognostic marker for head and neck squamous cell carcinoma Scientific reports 2017, 7, 9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhu Q; Schultz E; Long J; Roh JM; Valice E; Laurent CA; Radimer KH; Yan L; Ergas IJ; Davis W UACA locus is associated with breast cancer chemoresistance and survival NPJ breast cancer 2022, 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Gu M; Zheng W; Zhang M; Dong X; Zhao Y; Wang S; Jiang H; Liu L; Zheng X Downregulation of RAI14 inhibits the proliferation and invasion of breast cancer cells Journal of Cancer 2019, 10, 6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhang X; Ren D; Guo L; Wang L; Wu S; Lin C; Ye L; Zhu J; Li J; Song L Thymosin beta 10 is a key regulator of tumorigenesis and metastasis and a novel serum marker in breast cancer Breast Cancer Research 2017, 19, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Dominguez R; Holmes KC Actin structure and function Annual review of biophysics 2011, 40, 169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Mooney SM; Talebian V; Jolly MK; Jia D; Gromala M; Levine H; McConkey BJ The GRHL2/ZEB feedback loop—a key axis in the regulation of EMT in breast cancer Journal of cellular biochemistry 2017, 118, 2559–2570. [DOI] [PubMed] [Google Scholar]
- (65).Chen ZY; Wang PW; Shieh DB; Chiu KY; Liou YM Involvement of gelsolin in TGF-beta 1 induced epithelial to mesenchymal transition in breast cancer cells Journal of biomedical science 2015, 22, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Lu Z; Song Q; Yang J; Zhao X; Zhang X; Yang P; Kang J Comparative proteomic analysis of anti-cancer mechanism by periplocin treatment in lung cancer cells Cellular Physiology and Biochemistry 2014, 33, 859–868. [DOI] [PubMed] [Google Scholar]
- (67).Jia ZF; Huang Q; Kang CS; Yang WD; Wang GX; Yu SZ; Jiang H; Pu PY Overexpression of septin 7 suppresses glioma cell growth Journal of neuro-oncology 2010, 98, 329–340. [DOI] [PubMed] [Google Scholar]
- (68).Lin HY; Wu HJ; Chu PY Multi-omics and experimental analysis unveil theragnostic value and immunological roles of inner membrane mitochondrial protein (IMMT) in breast cancer Journal of Translational Medicine 2023, 21, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Bai Z; Ye Y; Liang B; Xu F; Zhang H; Zhang Y; Peng J; Shen D; Cui Z; Zhang Z Proteomics-based identification of a group of apoptosis-related proteins and biomarkers in gastric cancer International journal of oncology 2011, 38, 375–383. [DOI] [PubMed] [Google Scholar]
- (70).Rosen AW; Gögenur M; Paulsen IW; Olsen J; Eiholm S; Kirkeby LT; Pedersen OB; Pallisgaard N; Gögenur I Perioperative changes in cell-free DNA for patients undergoing surgery for colon cancer BMC gastroenterology 2022, 22, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Mou L; Jia C; Wu Z; Xin B; Liang Zhen CA; Wang B; Ni Y; Pu Z Clinical and Prognostic Value of PPIA, SQSTM1, and CCL20 in Hepatocellular Carcinoma Patients by Single-Cell Transcriptome Analysis Cells 2022, 11, 3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Rho JH; Roehrl MH; Wang JY Tissue proteomics reveals differential and compartment-specific expression of the homologs transgelin and transgelin-2 in lung adenocarcinoma and its stroma Journal of proteome research 2009, 8, 5610–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Cohen YC; Zada M; Wang S-Y; Bornstein C; David E; Moshe A; Li B; Shlomi-Loubaton S; Gatt ME; Gur C Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing Nature medicine 2021, 27, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ying B; Xu W; Nie Y; Li Y HSPA8 is a new biomarker of triple negative breast cancer related to prognosis and immune infiltration Disease Markers 2022, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Liu AR; Liu YN; Shen SX; Yan LR; Lv Z; Ding HX; Wang A; Yuan Y; Xu Q Comprehensive analysis and validation of solute carrier family 25 (SLC25) in pan-cancer 2021. [DOI] [PMC free article] [PubMed]
- (76).Yu C; Huang L New advances in cross-linking mass spectrometry toward structural systems biology Current Opinion in Chemical Biology 2023, 76, 102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Wang Y; Hu Y; Hoti N; Huang L; Zhang H Characterization of In Vivo Protein Complexes via Chemical Cross-Linking and Mass Spectrometry Analytical chemistry 2022, 94, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available via ProteomeXchange with identifier PXD046966