

Abstract
Objective:
In this pilot study, we used untargeted metabolomics to identify biochemical mechanisms or biomarkers potentially underlying SLE-related fatigue.
Methods:
Metabolon conducted untargeted metabolomic plasma profiling using ultrahigh performance liquid chromatography/tandem mass spectrometry on plasma samples of 23 Black females with systemic lupus erythematosus (SLE) and 21 no SLE controls. Fatigue phenotypes of general fatigue, physical fatigue, mental fatigue, reduced activity, and reduced motivation were measured with the reliable and valid Multidimensional Fatigue Inventory (MFI).
Results:
A total of 290 metabolites were significantly different between the SLE and no SLE groups, encompassing metabolites related to glycolysis, TCA cycle activity, heme catabolism, branched chain amino acids, fatty acid metabolism, and steroids. Within the SLE group, controlling for age and co-morbidities, TCA cycle metabolites of alpha-ketoglutarate (AKG) and succinate were statistically significantly associated (p < 0.05) with physical and general fatigue.
Conclusion:
While pervasive perturbations in the entire TCA cycle have been implicated as a potential mechanism for fatigue, our results suggest individual metabolites of AKG and succinate may be potential biomarkers or targets of intervention for fatigue symptom management in SLE. Additionally, perturbations in heme metabolism in the SLE group provide additional insights into mechanisms that promote systemic inflammation.
Keywords: Systemic Lupus Erythematosus, metabolomics, patient-reported outcomes, fatigue, TCA cycle, bioenergetics
Systemic lupus erythematosus (SLE) occurs predominantly in women, and Black women have disproportionately poorer health outcomes across the trajectory of the disease, compared to women of other race/ethnicities.1 As evolving treatments have extended the lives of SLE patients, the focus of care has shifted to efforts to improve patient-reported outcomes and quality of life. Fatigue, defined as a sense of exhaustion that interferes with daily functioning,2 presents in 80% to 90% of SLE patients3, 4 with up to 50% of the patients reporting severe fatigue.5 Fatigue is a major driver of negative disease perception,6 poorer quality of life,7 discordant patient/provider views of health status,8 inability to maintain employment,9 difficulty caring for the household and children,10 and delays in discontinuing steroid therapy.11 However, evidence-based guidelines for clinician management of fatigue in SLE are lacking,12 in part because the underlying biochemical mechanisms for fatigue in SLE are largely unknown.
Clinically, SLE patients report physical fatigue as well as difficulty with concentration, a type of mental fatigue or “brain fog”.13 Inflammatory processes like those responsible for SLE can elicit fatigue, but studies have not demonstrated a clear link between fatigue and levels of systemic inflammation or the degree of lupus disease activity, indicating that multiple overlapping biological mechanisms for fatigue in SLE may present within the same individual.4 Metabolomics, the study of low molecular weight molecules within biological systems, has the potential to identify intermediate biomarkers or pathways for understanding the underlying molecular mechanisms of multiple disease processes,14 including fatigue, in SLE.
We conducted untargeted metabolomic plasma profiling in Black females with SLE and Black female no SLE controls to identify metabolite biomarkers and their associated metabolic pathways that distinguished between the two groups. We then examined whether these metabolites and metabolic pathways associated with fatigue and the different manifestations of fatigue, within the SLE group. In this exploratory study, our aim was to identify biochemical disturbances potentially underlying SLE-related fatigue to provide insights into novel targets for symptom management. Notably, we identified a significant association between the levels of the TCA cycle metabolites, succinate and AKG with physical and general fatigue in the plasma of SLE patients.
Methods
Patients, Samples, Clinical, and Symptom Data
Prior to initiating the study, the protocol was approved by the Emory University Institutional Review Board (IRB). All patients provided written informed consent. We analyzed blood plasma samples collected from 23 SLE patients in the context of a routine visit to their rheumatology physician in a single specialty outpatient clinic of a comprehensive health care system in Atlanta, Georgia. All patients had a diagnosis of SLE documented by rheumatologists in the electronic medical record (EMR) according to American College of Rheumatology/European Alliance of Associations of Rheumatology (ACR/EULAR) criteria.15 Blood specimens in the no SLE group were obtained as part of a pilot study of multiple chronic conditions in community-dwelling caregivers with a body mass index (BMI) of 30 or greater (Brewster, PI). Both SLE and no SLE controls met the following inclusion criteria: 1) self-reported African American or Black race (chosen from a fixed list of racial and ethnic categories) and 2) able to read, write, and understand English. The inclusion criterion for age was initially 30 to 64 years for SLE and no SLE control subjects. However, to increase recruitment, the inclusion criterion in SLE patients was broadened to include ages 18 to 64 years, as SLE tends to present in younger Black women,1 and 30 to 85 years for caregivers. Patients were excluded if they had an active major mental health disorder or uncontrolled hypertension. We restricted the study to those with female sex at birth to negate sex differences in the metabolome.16
Blood samples and questionnaire data related to sociodemographic, medical history, and fatigue were collected from both SLE patients and no SLE controls, and were subsequently processed and stored for analysis as part of harmonized research protocols. Blood was collected in EDTA tubes via venipuncture and kept on ice during transport to the laboratory for processing and storage. Time of data collection and fasting status were not controlled in the study as blood specimens were collected based on scheduled timing of clinical or study visits. Specimens were centrifuged at 2,000 × g for 10 minutes in a refrigerated centrifuge after which the plasma supernatant was pipetted into clean polypropylene tubes and stored at −80 degrees Celsius until defrosted in a batch for metabolomics analysis and measurement of cytokines interleukin 1 beta (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor (TNF), and C-reactive protein (CRP) via enzyme linked immunosorbent assay following standard laboratory procedures. Cytokines were measured using the Meso Scale Discovery V-Plex Custom Human Proinflammatory Panel. CRP was measured with the Human C-Reactive Protein/CRP Quantikine ELISA Kit from R & D Systems, Inc. (cat#DCRP00).
Demographic and clinical data were collected via completion of self-report questionnaires or a thorough review of the EMR. We calculated a co-morbidity score with the Charlson Co-morbidity Index (CMI).17 Using SELENA-SLEDAI score18 and current prescribed prednisone dosage, SLE participants’ disease activity was categorized by the rheumatology physician co-investigator into groups according to low (SLEDAI < 4 and prednisone less than or equal to 5 mg. daily), moderate (SLEDAI greater than 4 but less than 6 or SLEDAI of 5 with daily prednisone < 20 mg.), and high (SLEDAI ≥ 6 or daily prednisone 20 mg or greater) disease activity. Fatigue phenotypes were assessed with the 20 item reliable and valid patient-reported outcome measure, the Multi-dimensional Fatigue Inventory (MFI), measuring 5 dimensions of fatigue: general fatigue, physical fatigue, reduced activity, reduced motivation, and mental fatigue.19 Possible range of scores for all MFI subscales is 4 to 20 with higher scores indicating greater fatigue.
Data Analysis
Descriptive statistics were used to characterize demographic, clinical, and fatigue measures. T tests were used to compare values of continuous variables between groups, and chi-squared tests were used to compare values of categorical variables between groups. Samples to measure cytokines and CRP were run in duplicate. Missing values that were below the detection limit were imputed as the lower limit of detection (LLOD) reported by the manufacturer. Values for technical replicates were averaged, and then the distributions of the data for each analyte were evaluated using Q-Q plots and Shapiro-Wilk tests of normality. Natural logarithm transformation improved the normality of most distributions, so the transformed values were used in subsequent analyses.
Liquid chromatography coupled to mass spectrometry (LC-MS) analysis
Untargeted metabolite identification and metabolomics analysis were conducted by Metabolon, Inc. (Durham, NC, USA). Samples were prepared using the automated MicroLab STAR system from Hamilton Company. Metabolon has published their approach to untargeted metabolomics analysis previously.20 Briefly, an integrated metabolomics platform was used for the identification and relative quantification, data reduction, and analysis of biochemicals that incorporated two separate ultrahigh performance liquid chromatography/tandem mass spectrometry (UHPLC/MS/MS2) injections focused on capturing different species of metabolites.
Multiple quality assurance and quality control measures were used in the workflow. A pooled matrix sample, generated by taking a small volume of each experimental sample, served as a technical replicate throughout the data set. Extracted water samples served as process blanks; and a mixture of QC standards that were carefully chosen not to interfere with the measurement of endogenous compounds were spiked into every analyzed sample, allowed instrument performance monitoring and aided chromatographic alignment. Experimental samples were randomized across the platform run with QC samples spaced evenly among the injections. All samples for the study were run in a single batch in random order using Metabolon’s in-house protocols.
Study metabolites were identified by comparison to Metabolon’s library that contains information on retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data) of more than 3,300 commercially authenticated purified standards as well as recurrent unknown entities repeatedly recognized on their platform. Biochemical identification was based on three criteria: retention index within a narrow RI window of the proposed identification, accurate mass match to the library +/− 10 ppm, and the MS/MS forward and reverse scores between the experimental data and authentic standards. Metabolon used proprietary visualization and interpretation software to confirm the consistency of peak identification among various samples. To quantify metabolites, peaks were quantified using area-under-the curve. Following log transformation and imputation of missing values with the minimum observed value for each compound, Welch’s two-sample t-test was used to identify biochemicals that differed significantly between the SLE and no SLE groups. To account for multiple comparisons errors, an estimate of the false discovery rate (q-value), or the proportion of metabolites falsely identified as differentially abundant between the SLE vs. no SLE groups, was calculated. Group differences noted to be statistically significant also had q values < 0.10. Differences between groups in metabolites were reported as fold change, with the ratio of the mean scaled intensity for a metabolite computed and displayed within a heatmap. Random Forest (RF) analysis was used to bin individual samples into groups based on their metabolite similarities and differences and define metabolites that contribute most strongly to group binning. Heatmaps were also created comparing the SLE vs. no SLE group, with green colors indicating a metabolite ratio < 1.0, indicating significantly lower metabolite intensity in the SLE group; whereas red heatmap color indicated the metabolite ratio was > 1.0, indicating significantly higher metabolite intensity in the SLE group. The small sample size within the SLE and no SLE groups prohibited formal metabolic pathway analysis; however, Metabolon’s identification platform helped identify the pathways to which metabolites of interest belonged.
We conducted univariate comparisons between the SLE vs. no SLE groups on fatigue severity, as well as compared groups on potential confounders for the metabolomics analysis including age, BMI, co-morbidities, smoking status, prednisone use, cytokines, and CRP. Two-tailed alpha was set at p < 0.05 for group comparisons. Spearman’s rho correlational analysis was conducted to determine if metabolites identified as significantly different between the SLE and no SLE groups were significantly associated with the 5 fatigue phenotypes in the SLE group. For figure 1, MetaboAnalyst 5.021 was used to analyse, perform statistics, and visualise the PCA plot and heatmap. Autoscaling of features (metabolites) was used for heatmap generation. For figure 2, Graphpad prism 9.0 software was used to generate bar plots and perform statistics.
Figure 1-.
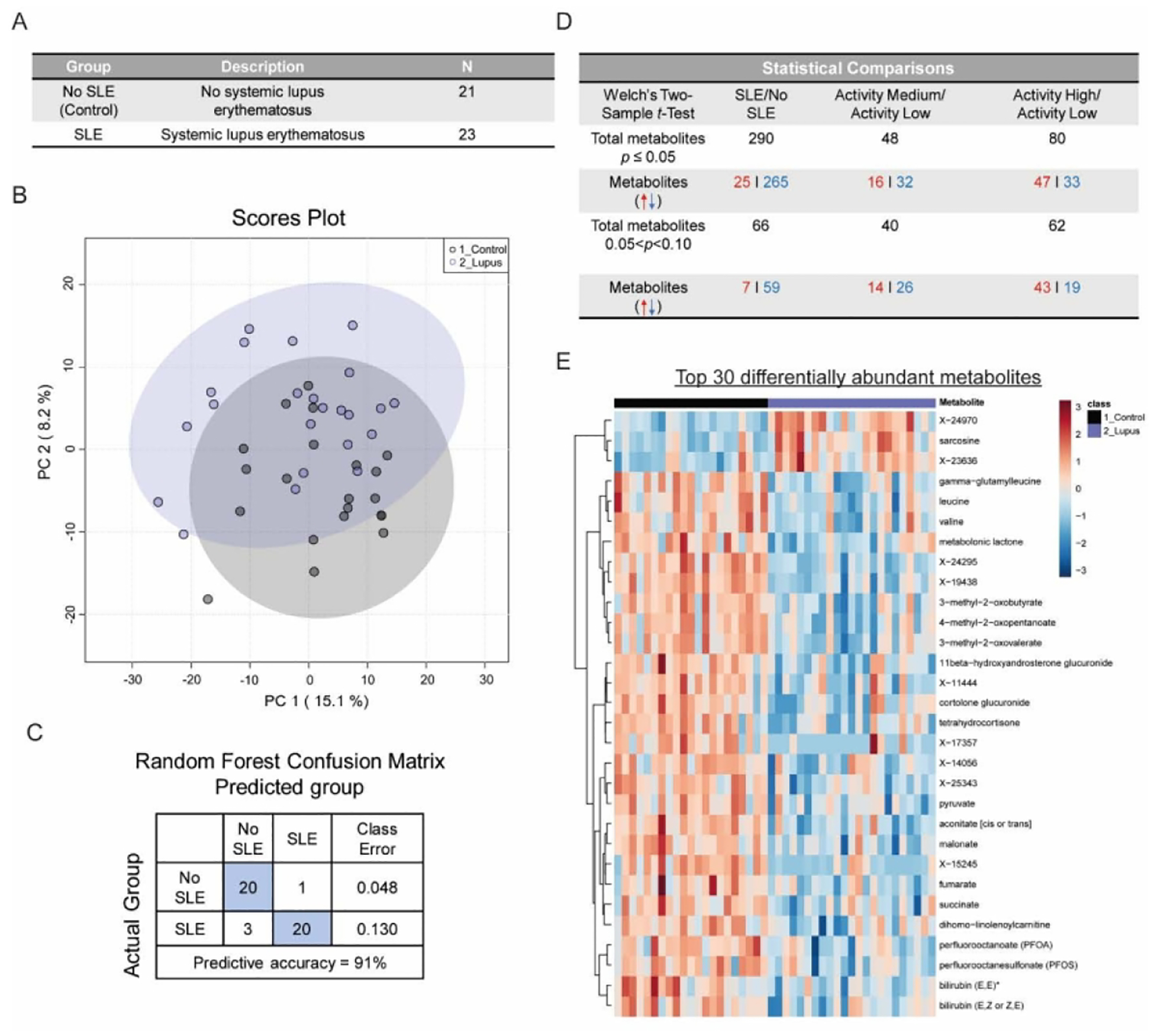
Overview of systemic lupus erythematosus metabolomics study experimental results
Figure 2-.
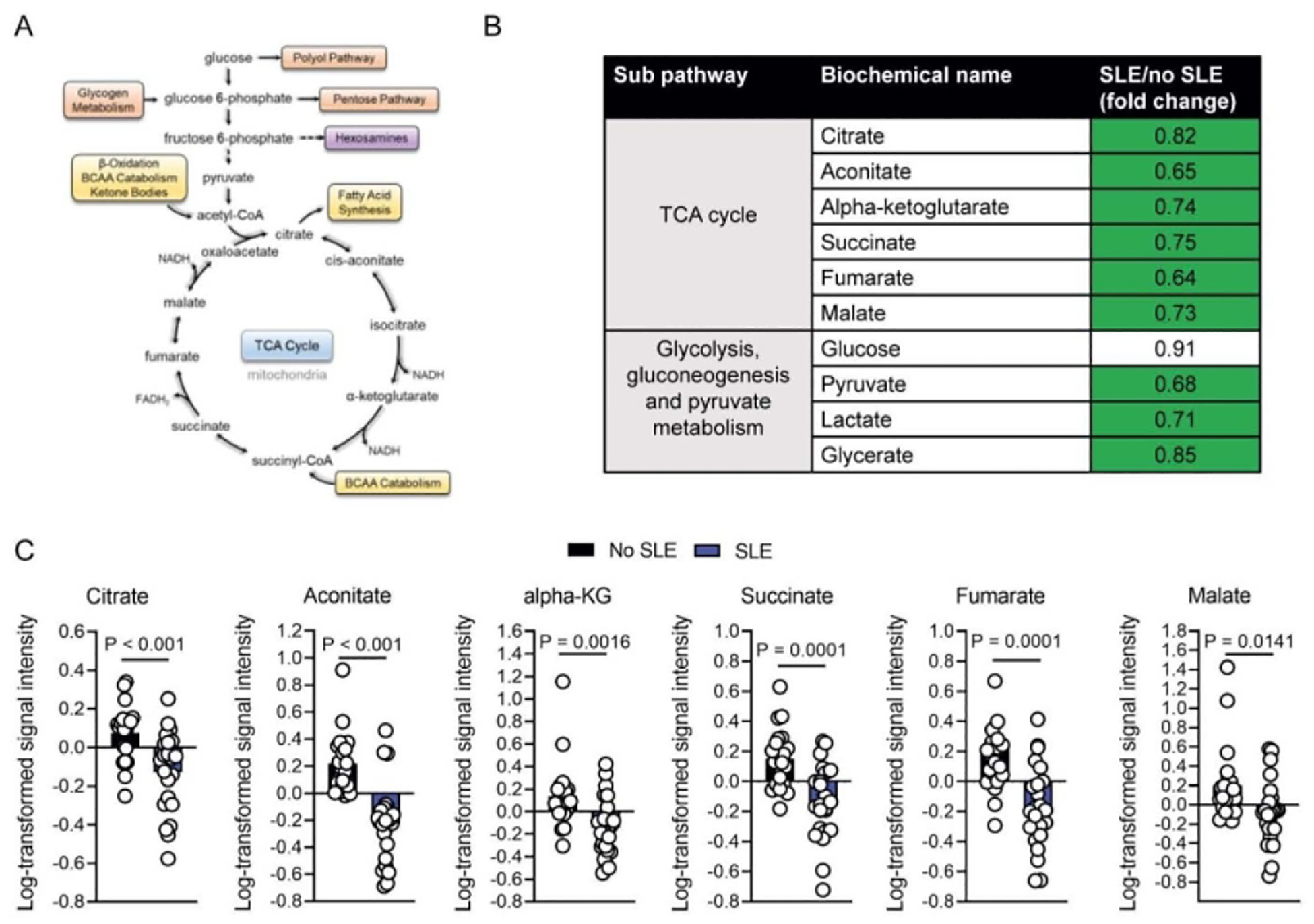
Altered glycolysis and TCA cycle metabolism in SLE patients
Results
The entire patient cohort self-identified as African American or Black (n=44); 23 participants with SLE and 21 controls (Figure 1A). SLE participants had been diagnosed with SLE for a mean of 11.6 years (SD 7.5), 39% (n=9) had history of lupus nephritis, and 70% (n=16) were taking hydroxychloroquine. Based on SLEDAI score and prednisone use, we categorized SLE participants’ disease activity as low in 61% (n=14), moderate in 26% (n=6) and high in 13% (n=3).
Compared to no SLE controls, SLE patients were significantly younger, had greater co-morbidities, and a greater proportion that were currently on prednisone or another oral form of steroid therapy. The groups did not differ by BMI score (Table 1). Only one SLE patient was a current smoker; data on the smoking status of the no SLE group were not collected. There were no statistically significant differences in levels of immune markers IL-1β, IL-6, TNF, or CRP between the SLE and no SLE group. Measured levels of IL-1β were low in all participants. The highest values of IL-6 and TNF were observed in SLE patients, but for most participants, these values were within the range considered normal. The majority of participants had CRP values less than 1 mg/dL, but a quarter of SLE patients and a third of participants with no SLE had moderately elevated CRP levels ranging from 1–3 mg/dL. Despite, on average, SLE participants being almost two decades younger compared to the controls; their average MFI fatigue scores were higher, but these differences were not statistically significant.
Table 1 -.
SLE and No SLE Control Group Comparisons on Demographic, Clinical, and Fatigue Variables (n=44)
| Variable | SLE (n=23) |
No SLE Control (n=21) |
p value |
|---|---|---|---|
| n (%) or M ± SD | n (%) or M ± SD | ||
| Age | 42.5 ± 12.1 | 63.2 ± 6.4 | < 0.001 |
| BMI | 32.1 ± 10.3 | 34.9± 4.1 | 0.26 |
| CCI | 2.4 ±1.3 | 1.1± 1.3 | 0.002 |
| Current prednisone therapy | 11(47.8) | 1 (5) | 0.002 |
| Current smoking | 1(4) | Not collected | ----- |
| IL-1β (pg/mL) | 0.1 ± 0.06 | 0.1 ± 0.04 | 0.42 |
| IL-6 (pg/mL) | 1.9 ± 1.5 | 1.4 ± 0.7 | 0.34 |
| TNF (pg/mL) | 1.4 ± 0.6 | 1.1 ± 0.3 | 0.08 |
| CRP (mg/dL) | 0.6 ± 0.7 | 0.9 + 0.8 | 0.30 |
| MFI-General Fatigue | 14.6 ± 4.1 | 13.0 ± 4.0 | 0.23 |
| MFI-Physical Fatigue | 12.5 ± 4.6 | 11.1 ± 4.3 | 0.31 |
| MFI-Mental Fatigue | 12.1 ± 4.8 | 10.0 ± 4.4 | 0.14 |
| MFI-Reduced Activity | 11.8 ± 4.6 | 9.7 ± 4.6 | 0.16 |
| MFI-Reduced Motivation | 10.6 ± 2.9 | 9.7 ± 3.6 | 0.40 |
Note: SLE- systemic lupus erythematosus, BMI= Body mass index, CCI= Charlson Co-morbidity index, IL1β= interleukin 1 beta, IL-6= interleukin 6, TNF= tumor necrosis factor, CRP= C-reactive protein, MFI-Multidimensional Fatigue Inventory, M= mean, SD= standard deviation. n=21 for SLE symptom data and n=22 for SLE CRP data. Data were missing for menopausal status in both groups. P values derived from t test (continuous variables) or Χ2 test for categorical variables.
Within the SLE group, bivariate non-parametric correlational analyses revealed prednisone use was not significantly associated with any of the MFI subscales. Lower SLE disease activity was significantly associated with greater general fatigue (r = −.40, p = 0.03). There were no significant associations between SLE disease activity and the other MFI subscales of physical fatigue, mental fatigue, reduced activity, and reduced motivation. The immune markers measured were not significantly correlated with MFI scores of SLE patients with the exception of a positive association between CRP and MFI General Fatigue (r = 0.48, p = 0.03). There was also a trending correlation between TNF and MFI Mental Fatigue which did not reach the threshold for statistical significance (r = 0.42, p = 0.06).
Metabolomics Summary and Significantly Altered Biochemicals
The metabolomics data set in the study comprised a total of 1178 metabolites found in subjects’ plasma, 971 compounds of known identity (named metabolites) and 207 compounds of unknown structural identity (unnamed metabolites). Principal component analysis (PCA) revealed moderate separation between the SLE patient cohort and no SLE controls (Figure 1B). Random Forest (RF) comparison of SLE vs no SLE samples also resulted in a predictive accuracy of 91% in distinguishing the two groups, indicating that the model was successful in binning the samples to their appropriate groups based on biochemical profile (Figure 1C). Comparison of the SLE group with the no SLE controls revealed a total of 290 metabolites were significantly altered (25 increased, 265 decreased) at p ≤0.05. An additional 66 metabolites between p < 0.05 and p < 0.10 were also significantly altered (7 increased, 59 decreased) (Figure 1D). The top 30 significantly altered metabolites between SLE and no SLE groups was enriched for metabolites involved in glycolysis and TCA cycle activity, heme catabolism, branched chain amino acids (BCAAs), fatty acid metabolism, and steroids (Figure 1E and Table 2). Several unidentified metabolites also completed the top 30 differentially abundant metabolites. Except for rare exceptions, the SLE group demonstrated significantly decreased levels of these metabolites when compared to the no SLE group (Figure 1E and Table 2).
Table 2 -.
Plasma Metabolites with Statistically Significant Differences in SLE Subjects vs. No SLE Controls Grouped According to Major Metabolic and Sub Pathways (n=44)
| Major Pathway | Sub Pathway | Metabolite | Fold Change |
|---|---|---|---|
| Carbohydrate | Glycolysis, Gluconeogenesis, and Pyruvate Metabolism | pyruvate | 0.68 |
| lactate | 0.71 | ||
| glycerate | 0.85 | ||
| Energy | TCA Cycle | citrate | 0.82 |
| aconitate [cis or trans] | 0.65 | ||
| α-ketoglutarate | 0.74 | ||
| succinate | 0.75 | ||
| fumarate | 0.64 | ||
| malate | 0.73 | ||
| Co-Factors | Hemoglobin and Porphyrin Metabolism | heme | 1.92 |
| bilirubin (Z,Z) | 0.69 | ||
| bilirubin (E,E)* | 0.53 | ||
| bilirubin (E,Z or Z,E)* | 0.59 | ||
| Amino Acids | Leucine, Isoleucine and Valine Metabolism | leucine | 0.76 |
| N-acetylleucine | 0.73 | ||
| 1-carboxyethylleucine | 0.58 | ||
| 4-methyl-2-oxopentanoate | 0.63 | ||
| alpha-hydroxyisocaproate | 0.69 | ||
| isoleucine | 0.81 | ||
| N-acetylisoleucine | 0.72 | ||
| 1-carboxyethylisoleucine | 0.62 | ||
| 3-methyl-2-oxovalerate | 0.65 | ||
| 3-hydroxy-2-ethylpropionate | 0.70 | ||
| ethylmalonate | 0.84 | ||
| valine | 0.78 | ||
| 1-carboxyethylvaline | 0.62 | ||
| 3-methyl-2-oxobutyrate | 0.71 | ||
| 3-hydroxyisobutyrate | 0.74 | ||
| Glutathione Metabolism | cysteinylglycine | 0.73 | |
| cysteinylglycine disulfide* | 0.83 | ||
| cys-gly, oxidized | 0.74 | ||
| 5-oxoproline | 0.82 | ||
| 2-hydroxybutyrate/2-hydroxyisobutyrate | 0.65 | ||
| Gamma-glutamyl Amino Acid | gamma-glutamylglutamate | 0.65 | |
| gamma-glutamylisoleucine* | 0.70 | ||
| gamma-glutamylleucine | 0.63 | ||
| gamma-glutamyl-alpha-lysine | 0.69 | ||
| gamma-glutamylmethionine | 0.69 | ||
| gamma-glutamylphenylalanine | 0.73 | ||
| gamma-glutamyltryptophan | 0.70 | ||
| gamma-glutamyltyrosine | 0.68 | ||
| gamma-glutamylvaline | 0.72 | ||
| Lipid | Fatty acid synthesis | malonate | 0.69 |
| Short Chain Fatty Acid | butyrate/isobutyrate (4:0) | 0.55 | |
| Medium Chain Fatty Acid | (2 or 3)-decenoate (10:1n7 or n8) | 0.58 | |
| 10-undecenoate (11:1n1) | 0.71 | ||
| laurate (12:0) | 0.78 | ||
| Long Chain Saturated Fatty Acid | myristate (14:0) | 0.63 | |
| pentadecanoate (15:0) | 0.66 | ||
| palmitate (16:0) | 0.70 | ||
| margarate (17:0) | 0.61 | ||
| stearate (18:0) | 0.70 | ||
| nonadecanoate (19:0) | 0.72 | ||
| Long Chain Monounsaturated Fatty Acid | oleate/vaccenate (18:1) | 0.62 | |
| 10-nonadecenoate (19:1n9) | 0.61 | ||
| eicosenoate (20:1) | 0.65 | ||
| nervonate (24:1n9)* | 0.81 | ||
| Long Chain Polyunsaturated Fatty Acid (n3 and n6) | stearidonate (18:4n3) | 0.52 | |
| eicosapentaenoate (EPA; 20:5n3) | 0.58 | ||
| docosapentaenoate (n3 DPA; 22:5n3) | 0.42 | ||
| docosahexaenoate (DHA; 22:6n3) | 0.46 | ||
| linoleate (18:2n6) | 0.57 | ||
| linolenate [alpha or gamma; (18:3n3 or 6)] | 0.54 | ||
| dihomo-linoleate (20:2n6) | 0.52 | ||
| dihomo-linolenate (20:3n3 or n6) | 0.55 | ||
| arachidonate (20:4n6) | 0.58 | ||
| adrenate (22:4n6) | 0.62 | ||
| docosadienoate (22:2n6) | 0.62 | ||
| Fatty Acid Metabolism (also Branch Chain Amino Acid Metabolism | butyrylcarnitine (C4) | 0.80 | |
| propionylglycine | 1.75 | ||
| 2-methylmalonylcarnitine (C4-DC) | 0.69 | ||
| Fatty Acid Metabolism (Acyl Glycine) | picolinoylglycine | 0.75 | |
| Fatty Acid Metabolism (Acyl Carnitine, Short Chain) | acetylcarnitine (C2) | 0.79 | |
| Fatty Acid Metabolism (Acyl Carnitine, Medium Chain | hexanoylcarnitine (C6) | 0.75 | |
| octanoylcarnitine (C8) | 0.65 | ||
| decanoylcarnitine (C10) | 0.74 | ||
| laurylcarnitine (C12) | 0.85 | ||
| Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | myristoylcarnitine (C14) | 0.79 | |
| palmitoylcarnitine (C16) | 0.81 | ||
| stearoylcarnitine (C18) | 0.76 | ||
| Fatty Acid Metabolism (Acyl Carnitine, Monounsaturated) | cis-4-decenoylcarnitine (C10:1) | 0.68 | |
| palmitoleoylcarnitine (C16:1)* | 0.80 | ||
| oleoylcarnitine (C18:1) | 0.79 | ||
| eicosenoylcarnitine (C20:1)* | 0.80 | ||
| Fatty Acid Metabolism (Acyl Carnitine, Polyunsaturated) | linoleoylcarnitine (C18:2)* | 0.82 | |
| dihomo-linoleoylcarnitine (C20:2)* | 0.71 | ||
| arachidonoylcarnitine (C20:4) | 0.64 | ||
| dihomo-linolenoylcarnitine (C20:3n3 or 6)* | 0.66 | ||
| adrenoylcarnitine (C22:4)* | 0.63 | ||
| docosapentaenoylcarnitine (C22:5n3)* | 0.68 | ||
| Fatty Acid Metabolism (Acyl Carnitine, Dicarboxylate) | octadecanedioylcarnitine (C18-DC)* | 0.74 | |
| Fatty Acid Metabolism (Acyl Carnitine, Hydroxy) | (R)-3-hydroxybutyrylcarnitine | 0.48 | |
| (S)-3-hydroxybutyrylcarnitine | 0.58 | ||
| 3-hydroxyoctanoylcarnitine (2) | 0.82 | ||
| Carnitine Metabolism | deoxycarnitine | 0.89 | |
| Ketone bodies | 3-hydroxybutyrate (BHBA) | 0.59 | |
| Fatty acid metabolism (Acyl Choline) | palmitoylcholine | 0.65 | |
| oleoylcholine | 0.68 | ||
| dihomo-linolenoyl-choline | 0.64 | ||
| linoleoylcholine* | 0.69 | ||
| stearoylcholine* | 0.56 | ||
| docosahexaenoylcholine | 0.45 | ||
| arachidonoylcholine | 0.61 | ||
| Fatty Acid, Monohydroxy | 2-hydroxypalmitate | 0.71 | |
| 2-hydroxystearate | 0.68 | ||
| 3-hydroxyhexanoate | 0.68 | ||
| 3-hydroxydecanoate | 0.69 | ||
| 3-hydroxylaurate | 0.62 | ||
| 3-hydroxymyristate | 0.76 | ||
| 3-hydroxyoleate* | 0.62 | ||
| Lipid | Pregnenolone | pregnenolone sulfate | 0.63 |
| Steroids | 21-hydroxypregnenolone disulfate | 0.64 | |
| pregnenediol sulfate (C21H3405S)* | 0.66 | ||
| pregnenediol disulfate (C21H34O8S2)* | 0.65 | ||
| pregnenetriol sulfate* | 0.51 | ||
| pregnenetriol disulfate* | 0.67 | ||
| Corticosteroids | tetrahydrocortisol glucuronide | 0.60 | |
| tetrahydrocortisone glucuronide (5) | 0.45 | ||
| cortolone glucuronide (1) | 0.53 | ||
| Androgenic Steroids | dehydroepiandrosterone sulfate (DHEA-S) | 0.62 | |
| 16a-hydroxy DHEA 3-sulfate | 0.71 | ||
| androsterone glucuronide | 0.65 | ||
| epiandrosterone sulfate | 0.55 | ||
| androsterone sulfate | 0.51 | ||
| 11beta-hydroxyetiocholanolone glucuronide* | 0.40 | ||
| androstenediol (3beta,17beta) monosulfate (1) | 0.70 | ||
| androstenediol (3beta,17beta) monosulfate (2) | 0.74 | ||
| androstenediol (3beta,17beta) disulfate (1) | 0.71 | ||
| androstenediol (3beta,17beta) disulfate (2) | 0.69 | ||
| androstenediol (3alpha, 17alpha) monosulfate (3) | 0.53 | ||
| 5alpha-androstan-3alpha,17beta-diol monosulfate (1) | 0.54 | ||
| 5alpha-androstan-3beta,17beta-diol disulfate | 0.56 | ||
| andro steroid monosulfate C19H28O6S (1)* | 0.91 | ||
| 11beta-hydroxyandrosterone glucuronide | 0.42 |
Note: In the heat map, green (p ≤ .05) boxes represent significantly decreased concentrations of metabolites. Red (p ≤ .05) boxes represent significantly increased concentrations of metabolites.
Associations Between TCA Cycle Metabolites and Fatigue Phenotypes Within the SLE Group
Reduced glycolytic and TCA cycle metabolite abundance was the most striking difference between SLE and no SLE patients (Figure 2A–C). Reduced TCA cycle activity as a bioenergetic pathway has previously been implicated in the excessive fatigue experienced by SLE patients22, 23. Therefore, we conducted correlational analyses of relationships between fatigue phenotypes and individual TCA cycle metabolites (citrate, aconitate, AKG, succinate, fumarate, and malate) within the SLE group. The metabolites of interest were those that were identified as significantly different between the SLE and no SLE groups in the metabolomics analysis. To control for variables that were different between the SLE and no SLE groups which could serve as potential confounders in the within SLE group analysis of relationships between metabolites and fatigue, we conducted univariate correlational analyses to examine relationships between age, co-morbidity index (CMI), prednisone use, and the TCA cycle metabolites of interest. Both age and CMI, but not prednisone use, were significantly associated (p < 0.05) with metabolites; with age associated with citrate, AKG, and fumarate and CMI associated with AKG, fumarate, and malate. Therefore, age and CMI were controlled in subsequent analyses of relationships between fatigue phenotypes and TCA cycle metabolites within the SLE group. Greater concentrations of the individual metabolites of AKG and succinate were significantly associated with greater general and physical fatigue when age and CMI were controlled (Table 3).
Table 3 -.
Partial Correlations Among TCA Cycle Metabolites and Multidimensional Fatigue Inventory Phenotypes Within the SLE Group Controlling for Age and Co-Morbidities
| Multi-Dimensional Fatigue Inventory Subscales | ||||||
|---|---|---|---|---|---|---|
| General Fatigue | Physical Fatigue | Reduced Activity | Reduced Motivation | Mental Fatigue | ||
| TCA Cycle Metabolites | Citrate | −.06 | .24 | −.10 | −.42 | −.24 |
| Aconitate | .24 | .31 | −.01 | −.39 | −.02 | |
| Alpha-Ketoglutarate | .51 * | .60 ** | .42 | .10 | .31 | |
| Succinate | .49 * | .61 ** | .04 | −.02 | .12 | |
| Fumarate | −.03 | .29 | −.25 | −.14 | −.19 | |
| Malate | −.07 | .33 | −.25 | −.11 | −.16 | |
Note: df =17 for partial correlation.
p < 0.05,
p < 0.01 two tailed.
Significant correlations bolded.
Discussion
Corroborating prior metabolomics investigations of SLE patients,23 we found pervasively altered, and for the most part, reduced abundance of circulating metabolites associated with the major metabolic pathways of glycolysis, TCA cycle, fatty acid metabolism, and BCAA metabolism in participants with SLE compared to those with no SLE. Many of the observed differences between groups related to energy metabolism. Glucose metabolism is a major source of energy in tissues and organs. Glycolysis, which occurs in the cytosol of the cell, involves the oxidation of glucose into pyruvate, with the net production of two energy rich adenosine triphosphate (ATP) molecules and two NADH reducing equivalents. Pyruvate can subsequently be reduced to lactate by lactate dehydrogenase (LDH) to regenerate NAD+ for the maintenance of glycolytic flux or it can enter the TCA cycle where it is completely oxidized to CO2 and generates GTP/ATP, NADH and FADH2. NADH and FADH2 generated here can then be used to transfer electrons to the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS).24 Circulating lactate generated by glycolysis has been found to feed the TCA cycle in most tissues except for the brain.25 The reduced pyruvate and lactate in the context of no significant alterations in glucose suggested decreased glycolytic activity in the SLE participants. Moreover, the multiple TCA cycle metabolites with significantly lower concentrations in the SLE group suggested reduced TCA cycle activity which has been observed in multiple metabolomics studies in SLE.22, 23
Fatty acids (FAs) regulate cell membrane structure and function, intracellular signaling, and transcription and expression of genes,26 as well as provide carbon units for oxidative TCA cycle activity. We observed reduced concentrations of short, medium, and long chain fatty acids within the SLE group compared to the no SLE group, particularly free fatty acids (FFA). Reduced FFA have been observed in SLE; however one metabolomics SLE study found both elevated and decreased concentrations of FFA compared to healthy controls.27 In addition to FAs, citrate levels were lower in SLE patients (Table 2). Citrate generated by the TCA cycle can be exported from mitochondria to the cytosol, where it is converted to acetyl-CoA by ATP-citrate lyase (ACLY).28 Acetyl-CoA can undergo additional modification to generate malonyl-CoA, an essential precursor in the synthesis of FAs. Levels of acyl-carnitines, which transport acyl-CoA species into mitochondria as part of β-oxidation,29 were also significantly depleted in SLE patients. Together these data suggest an impairment in both lipid synthesis and catabolism in SLE patients.
The BCAAs, leucine, isoleucine, and valine, are essential amino acids for humans and must be acquired through dietary means.24 BCAAs can undergo transamination to generate alpha-ketoacids which then enter the mitochondria where they can subsequently be further catabolized to act as fuel for oxidative TCA cycle metabolism or as an anaplerotic source of TCA cycle intermediates. In this study, significantly reduced levels of leucine, isoleucine, valine as well as alpha-ketoacids were observed in SLE participants compared to no SLE controls. These results are suggestive of either decreased absorption of BCAAs from diet, an impairment in BCAA catabolism, and/or the result of depletion of stores from increased utilization as an energy source.
Group differences were also noted in steroids, with the SLE group having lower concentrations of circulating pregnenolone and androgenic steroids and corticosteroids. Many of these metabolites were sulfated forms of the steroids. Sulfated steroids are actively transported into the cell where they are desulfated and rendered active through steroid sulfatase (STS) enzyme activity.30 Inflammation has been speculated to influence STS activity; which could, in turn affect levels of sulfated steroids.30 The reduced concentrations of steroids could also be related to adrenal gland suppression secondary to prednisone therapy.31
The focus of our work is on the molecular basis of fatigue in females with SLE. Researchers have implicated disruptions in the TCA cycle metabolic pathway as potential molecular mechanisms for clinical fatigue in SLE.22 Our study advances molecular understanding of fatigue, as to our knowledge, no prior studies have associated individual TCA cycle metabolites with a reliable and valid measure of fatigue within the same study. Within the SLE group, we identified statistically significant correlations between general fatigue, physical fatigue and the individual TCA cycle metabolites of AKG and succinate, controlling for potential confounders of age and co-morbidities. Individual metabolites are bioactive, and particularly under inflammatory conditions, influence cellular metabolism to support cell energy needs, biosynthesis of substrates needed for critical metabolic processes, and control of cellular redox activity. While we did not observe significantly higher levels of the four inflammatory markers measured in this study in SLE patients compared to participants without SLE, some of the values measured were elevated, and we did identify a significant association between levels of CRP and MFI General Fatigue in SLE patients, supporting the idea of a low-grade inflammatory state which could impact metabolism. AKG, a key intermediate in the TCA cycle, supports cellular energy metabolism,32 but also has multiple other physiological functions, including modulating collagen synthesis and bone development, and extending the life span of mice by suppressing chronic inflammation and inducing production of IL-10.33 Consequently, decreased availability of AKG may impact fatigue through its effect on inflammatory pathways. AKG is a source of glutamine and glutamate, and enhances immune system response by increasing activity of neutrophils and phagocytosis.32 AKG and succinate are epigenetic modifiers that may contribute to DNA and histone remodeling by acting in conjunction with the chromatin-modifying enzymes.34 Dietary interventions that optimize AKG and other metabolites could be an important therapeutic target to support more normal immunologic function.34
Succinate is a bioactive pro-inflammatory molecule that acts through multiple mechanisms, including generating mitochondrial reactive oxygen species (mROS), activation of hypoxia-inducible factor-1 alpha (HIF-1α), and intra and extra-cellular signaling in regulating inflammation35 as a ligand for the SUCNR1 receptor.36 SUCNR1 receptors are located in multiple cells and tissues throughout the body. Consequently, the bioactivity of succinate is vast. One potential mechanism by which succinate may affect fatigue in SLE is through increased inflammation caused by succinate signaling in macrophages, a key cell within the innate immune system and a major producer of inflammatory cytokines.37 Succinate accumulates in immunologically activated macrophages and this accumulation has a pro-inflammatory effect by inhibiting prolyl hydroxylase domain (PHD) activity. When PHD activity is inhibited in the cell, the transcription of HIF- 1α is stabilized, supporting encoding of genes producing pro-inflammatory cytokines (IL- 1β)35 and prolonging inflammation. Elevated succinate in normoxic cells has a similar stabilizing effect on HIF-1α perpetuating inflammation similar to what would occur when cells were in hypoxic conditions.35
In the current study, circulating heme was significantly elevated in SLE patients compared to no SLE controls, while intermediate bilirubin catabolites were significantly reduced, suggesting a disruption of the heme catabolic pathway. The heme catabolism pathway regulates multiple physiologic processes including apoptosis, inflammation, and redox homeostasis.38 Excessive levels of free heme promote oxidative stress and inflammation.38 Heme oxygenase (HO) is an immunomodulatory enzyme that catalyzes the degradation of heme to generate biliverdin/bilirubin, carbon monoxide, and iron; together protecting cells from heme-induced injury.38 Reduced activity of HO, especially its isoform HO-1, and resultant build-up of free heme may potentially exacerbate inflammation in SLE.39
Limitations and Conclusions
This investigation was a pilot study. We controlled for biological sex by including only females; however, we did not collect data about menopausal status and exercise behavior; smoking data were missing for the no SLE control group. Additionally, we did not control for dietary intake, fasting status, or time of day the blood specimens were collected. All of these factors could potentially have influenced our results40, 41. While we statistically controlled co-morbidities and prednisone use, the excessive co-morbidities in the SLE group as well as differences between groups with respect to other medications may have impacted the findings beyond the impact of SLE and its treatment, particularly cardiovascular co-morbidities where, in animal models with hypertension42, accumulating succinate is released into the circulation.
In interpreting our results, we hypothesized about both intracellular and extracellular mechanisms by which AKG and/or succinate could affect fatigue; particularly mechanisms involving the immune cell compartment. However, the mechanisms by which these metabolites might be detected in the plasma are largely unknown in SLE. One potential mechanism may be through tissue damage; for example, kidney damage in the form of lupus nephritis is a common clinical feature in SLE that develops in more than 50% of patients.43 The succinate receptor SUCRN1 is expressed in the kidney with elevated succinate levels associated with local tissue stress and damage.44 Researchers have emphasized the need for more precise understanding of blood biomarkers including the cellular origin as well as potential pathways by which biomarkers reach the circulation.45 Moreover, the intestinal microbiome, especially among obese individuals, is a major source of succinate within the circulation46 and future multi-omic studies that account for the impact of the intestinal microbiome on metabolites are indicated.
Individuals with SLE are often on powerful treatment regimens to control their disease including glucocorticoids, immunosuppressives, and biologic therapies, and, while these would be expected to influence levels of immune markers measured in this study, to date, there are limited controlled studies about how these pharmacologic agents influence the metabolome with respect to the drug itself, the dose, and the length of treatment.23 Glucocorticoids are especially concerning as a confounder of metabolomics studies, as an investigation in males receiving dexamethasone demonstrated time dependent changes in 150 metabolites;47 however, relevant to this investigation, few changes were noted in TCA cycle metabolites. A critical next step in future metabolomics studies in the SLE population will be to design robust metabolomics studies to evaluate the impact of pharmacologic impact of treatment over time.23
We found that lower disease activity was associated with greater fatigue. Our research is consistent with prior studies that found that even when clinical data indicate that patients with SLE are having low disease activity, they can experience debilitating fatigue leading to discordance between patient and physicians regarding disease control.48 Some investigators have even argued that psychological issues are to blame for low disease activity-SLE patients’ complaints of fatigue, recommending psychological assessment and counseling in these cases.49 However, our findings suggest a potential biological mechanism for underlying fatigue symptoms for SLE patients with noticeable differences in TCA cycle metabolite levels in an apparently low disease activity state. Individuals with SLE with low disease activity while simultaneously debilitated by fatigue are an important population for whom interventions are greatly needed.50
Acknowledgments
This work was supported by National Institutes of Health Funding for the P30 Center for the Study of Symptom Science, Metabolomics, and Multiple Chronic Conditions P30NR018090-02S1. Dr. Kimble was funded with a research re-entry supplement under this parent award.
Contributor Information
Laura P. Kimble, Emory University, Atlanta, GA.
Arezou Khosroshahi, School of Medicine, Emory University, Atlanta, GA.
Glenna S. Brewster, Emory University, Atlanta, GA.
Sandra B. Dunbar, Emory University, Atlanta, GA.
Dylan Ryan, University of Cambridge, Cambridge, Great Britain.
Nicole Carlson, Emory University, Atlanta, GA.
Ron Eldridge, Emory University, Atlanta, GA.
Madelyn Houser, Emory University, Atlanta, GA.
Elizabeth Corwin, Columbia University, New York, NY.
References
- 1.Lim SS, Drenkard C. Epidemiology of lupus: an update. Curr Opin Rheumatol. 2015;27(5):427–432. [DOI] [PubMed] [Google Scholar]
- 2.Zielinski MR, Systrom DM, Rose NR. Fatigue, Sleep, and Autoimmune and Related Disorders. Front Immunol. 2019;10:1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fonseca R, Bernardes M, Terroso G, et al. Silent Burdens in Disease: Fatigue and Depression in SLE. Autoimmune Dis. 2014;2014:790724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tench CM, McCurdie I, White PD, et al. The prevalence and associations of fatigue in systemic lupus erythematosus. Rheumatology (Oxford). 2000;39(11):1249–1254. [DOI] [PubMed] [Google Scholar]
- 5.Overman CL, Kool MB, Da Silva JA, et al. The prevalence of severe fatigue in rheumatic diseases: an international study. Clin Rheumatol. 2016;35(2):409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dima A, Caraiola S, Delcea C, et al. Self-reported disease severity in women with systemic lupus erythematosus. Rheumatol Int. 2019;39(3):533–539. [DOI] [PubMed] [Google Scholar]
- 7.Sutanto B, Singh-Grewal D, McNeil HP, et al. Experiences and perspectives of adults living with systemic lupus erythematosus: thematic synthesis of qualitative studies. Arthritis Care Res (Hoboken). 2013;65(11):1752–1765. [DOI] [PubMed] [Google Scholar]
- 8.Golder V, Ooi JJY, Antony AS, et al. Discordance of patient and physician health status concerns in systemic lupus erythematosus. Lupus. 2018;27(3):501–506. [DOI] [PubMed] [Google Scholar]
- 9.Booth S, Price E, Walker E. Fluctuation, invisibility, fatigue - the barriers to maintaining employment with systemic lupus erythematosus: results of an online survey. Lupus. 2018;27(14):2284–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon C, Isenberg D, Lerstrom K, et al. The substantial burden of systemic lupus erythematosus on the productivity and careers of patients: a European patient-driven online survey. Rheumatology (Oxford). 2013;52(12):2292–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tani C, Elefante E, Signorini V, et al. Glucocorticoid withdrawal in systemic lupus erythematosus: are remission and low disease activity reliable starting points for stopping treatment? A real-life experience. RMD Open. 2019;5(2):e000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider M Guidelines for the management of systemic lupus erythematosus: great synthesis of evidence and eminence with limited focus on patient’s needs. Rheumatology (Oxford). 2018;57(1):12–13. [DOI] [PubMed] [Google Scholar]
- 13.Mackay M Lupus brain fog: a biologic perspective on cognitive impairment, depression, and fatigue in systemic lupus erythematosus. Immunol Res. 2015;63(1–3):26–37. [DOI] [PubMed] [Google Scholar]
- 14.Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17(7):451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78(9):1151–1159. [DOI] [PubMed] [Google Scholar]
- 16.Krumsiek J, Mittelstrass K, Do KT, et al. Gender-specific pathway differences in the human serum metabolome. Metabolomics. 2015;11(6):1815–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charlson ME, Pompei P, Ales KL, et al. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373–383. [DOI] [PubMed] [Google Scholar]
- 18.Castrejon I, Tani C, Jolly M, et al. Indices to assess patients with systemic lupus erythematosus in clinical trials, long-term observational studies, and clinical care. Clin Exp Rheumatol. 2014;32(5 Suppl 85):S-85–95. [PubMed] [Google Scholar]
- 19.Smets EM, Garssen B, Bonke B, et al. The Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995;39(3):315–325. [DOI] [PubMed] [Google Scholar]
- 20.Evans AM, DeHaven CD, Barrett T, et al. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81(16):6656–6667. [DOI] [PubMed] [Google Scholar]
- 21.Pang Z, Chong J, Zhou G, et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49(W1):W388–W396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu T, Xie C, Han J, et al. Metabolic disturbances associated with systemic lupus erythematosus. PLoS One. 2012;7(6):e37210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang T, Mohan C. Caution in studying and interpreting the lupus metabolome. Arthritis Res Ther. 2020;22(1):172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson D, Cox MM. Lehninger Principles of Biochemistry. New York, NY: W. H. Freeman Macmillan Learning; 2017. [Google Scholar]
- 25.Hui S, Ghergurovich JM, Morscher RJ, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calder PC. Functional Roles of Fatty Acids and Their Effects on Human Health. JPEN J Parenter Enteral Nutr. 2015;39(1 Suppl):18S–32S. [DOI] [PubMed] [Google Scholar]
- 27.Shin TH, Kim HA, Jung JY, et al. Analysis of the free fatty acid metabolome in the plasma of patients with systemic lupus erythematosus and fever. Metabolomics. 2017;14(1):14. [DOI] [PubMed] [Google Scholar]
- 28.Wellen KE, Hatzivassiliou G, Sachdeva UM, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33(5):469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster PA, Mueller JW. SULFATION PATHWAYS: Insights into steroid sulfation and desulfation pathways. J Mol Endocrinol. 2018;61(2):T271–T283. [DOI] [PubMed] [Google Scholar]
- 31.Oray M, Abu Samra K, Ebrahimiadib N, et al. Long-term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457–465. [DOI] [PubMed] [Google Scholar]
- 32.Wu N, Yang M, Gaur U, et al. Alpha-Ketoglutarate: Physiological Functions and Applications. Biomol Ther (Seoul). 2016;24(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asadi Shahmirzadi A, Edgar D, Liao CY, et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020;32(3):447–456 e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Long H, Chang C, et al. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: a comprehensive overview. Cell Mol Life Sci. 2018;75(18):3353–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills E, O’Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 2014;24(5):313–320. [DOI] [PubMed] [Google Scholar]
- 36.Ryan DG, Murphy MP, Frezza C, et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab. 2019;1:16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Veledo S, Ceperuelo-Mallafre V, Vendrell J. Rethinking succinate: an unexpected hormone-like metabolite in energy homeostasis. Trends Endocrinol Metab. 2021;32(9):680–692. [DOI] [PubMed] [Google Scholar]
- 38.Wu B, Wu Y, Tang W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front Pharmacol. 2019;10:825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrada AA, Llanos C, Mackern-Oberti JP, et al. Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology. 2012;136(4):414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu T, Holzapfel C, Dong X, et al. Effects of smoking and smoking cessation on human serum metabolite profile: results from the KORA cohort study. BMC Med. 2013;11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rangel-Huerta OD, Pastor-Villaescusa B, Gil A. Are we close to defining a metabolomic signature of human obesity? A systematic review of metabolomics studies. Metabolomics. 2019;15(6):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sadagopan N, Li W, Roberds SL, et al. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens. 2007;20(11):1209–1215. [DOI] [PubMed] [Google Scholar]
- 43.Oku K, Atsumi T. Systemic lupus erythematosus: nothing stale her infinite variety. Mod Rheumatol. 2018;28(5):758–765. [DOI] [PubMed] [Google Scholar]
- 44.Deen PM, Robben JH. Succinate receptors in the kidney. J Am Soc Nephrol. 2011;22(8):1416–1422. [DOI] [PubMed] [Google Scholar]
- 45.Kawata K, Liu CY, Merkel SF, et al. Blood biomarkers for brain injury: What are we measuring? Neurosci Biobehav Rev. 2016;68:460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serena C, Ceperuelo-Mallafre V, Keiran N, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018;12(7):1642–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bordag N, Klie S, Jurchott K, et al. Glucocorticoid (dexamethasone)-induced metabolome changes in healthy males suggest prediction of response and side effects. Sci Rep. 2015;5:15954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elera-Fitzcarrald C, Vega K, Gamboa-Cardenas RV, et al. Discrepant Perception of Lupus Disease Activity: A Comparison Between Patients’ and Physicians’ Disease Activity Scores. J Clin Rheumatol. 2020;26(7S Suppl 2):S165–S169. [DOI] [PubMed] [Google Scholar]
- 49.Arnaud L, Gavand PE, Voll R, et al. Predictors of fatigue and severe fatigue in a large international cohort of patients with systemic lupus erythematosus and a systematic review of the literature. Rheumatology (Oxford). 2019;58(6):987–996. [DOI] [PubMed] [Google Scholar]
- 50.Felten R, Sagez F, Gavand PE, et al. 10 most important contemporary challenges in the management of SLE. Lupus Sci Med. 2019;6(1):e000303. [DOI] [PMC free article] [PubMed] [Google Scholar]