

Abstract
Systems that can image in three dimensions at cellular resolution and across different locations within an organism may enable insights into complex biological processes, such as immune responses, for which a single location measurement may be insufficient. In this Letter, we describe an in vivo two-site imaging probe (TIP) that can simultaneously image two anatomic sites with a maximum separation of a few centimeters. The TIP consists of two identical bendable graded index (GRIN) lenses and is demonstrated by a two-photon two-color fluorescence imaging system. Each GRIN lens has a field of view of 162 × 162 × 170 μm3, a nominal numerical aperture of 0.5, a magnification of 0.7, and working distances of 0.2 mm in air for both ends. A blind linear unmixing algorithm is applied to suppress bleedthrough between channels. We use this system to successfully demonstrate two-site two-photon two-color imaging of two biomedically relevant samples, i.e., (1) a mixture of two autofluorescent anti-cancer drugs and (2) a live hybrid tumor consisting of two spectrally distinct fluorescent cell lines.
Fluorescence optical imaging (FOI) can provide single-cell tracking of biological processes and key insights into drug delivery in real time; e.g., imaging of fluorescently labeled immune cells has led to greater understanding of immune activation, trafficking, and immunotherapy responses [1]. FOI has also been critical in understanding the microscopic and macroscopic biodistribution and drug effects of these agents [2–4]; e.g., sunitinib and doxorubicin are two commonly used anti-cancer agents that are intrinsically detectable, and their in situ tumor distribution from localized delivery has been demonstrated by our group [5]. However, current FOI techniques are unable to simultaneously image multiple spatially discrete anatomic locations at the cellular resolution level. Such a multi-site FOI modality is desirable for many biomedical applications, including the establishment of highly kinetic pharmacokinetic and pharmacodynamic correlation between drug exposures in tumor and healthy organs (e.g., liver) following systemic or localized delivery [6,7].
To date, whole-body FOI systems have allowed multiple well separated locations to be imaged simultaneously by a far-field camera [8], but they lack the cellular resolution. High resolution in vivo FOI strategies, such as intravital imaging [9], are typically limited to a single superficial anatomic site. Multi-site or multifocus excitation using structured light has been implemented for selective illuminations in neuroscience [10], but the image is captured from the same field of view (FOV), thus essentially the same anatomic location. A mesoscope with a large FOV has been devised to simultaneously monitor neuron activities across different brain regions [11], but it cannot be used for microendoscopic imaging in deep tissues or organs. Therefore, there is an unmet need for a multi-location microendoscopic FOI modality with a three-dimensional (3D) cellular resolution.
In this work, we propose a two-site imaging probe (TIP) that can simultaneously image two anatomic locations, as conceptually shown in Fig. 1(a). In practice, each anatomic location may be within a distinct organ or diseased tissue for in vivo imaging. The TIP is enabled by bendable long graded index (GRIN) lenses recently proposed and demonstrated [12]. The TIP consists of two bendable GRIN lenses, where the proximal ends of all lenses are bundled and coupled to a single objective. The distal end of each GRIN lens is deflected and may be inserted into a separate anatomic site for microendoscopic imaging.
Fig. 1.
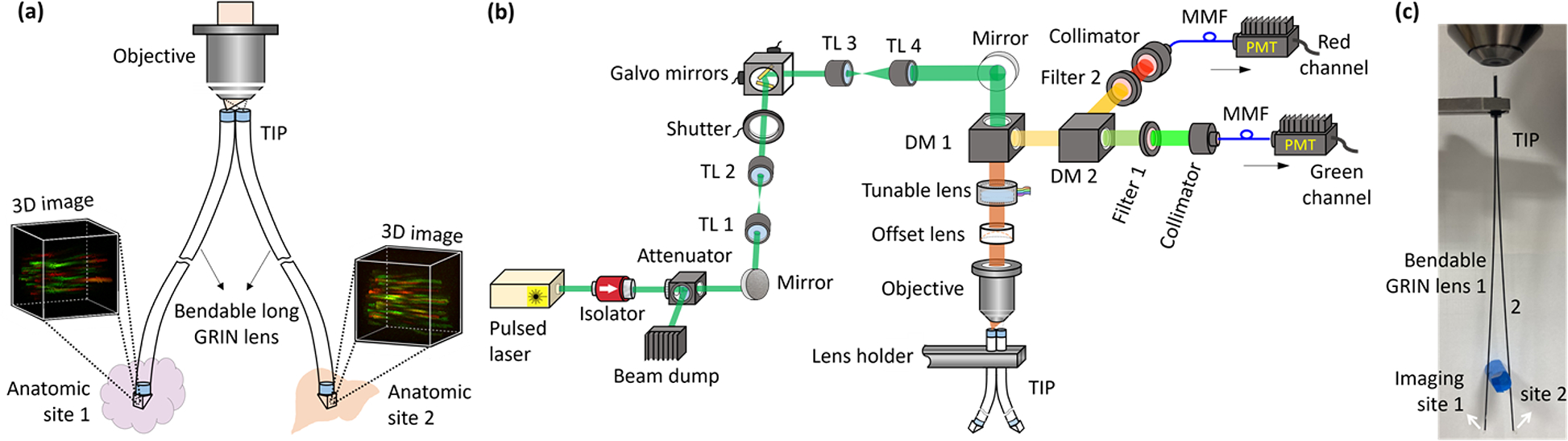
Illustration of the proposed TIP and the imaging system. (a) Conceptual demonstration of using a TIP for simultaneous imaging at two distinct locations, (b) schematic of the constructed two-photon two-color imaging system, and (c) an image of the demonstrated TIP made of two bendable GRIN lenses (diameter: 500 μm; length: ~110mm). TL, tube lens; DM, dichroic mirror; MMF, multimode optical fiber; PMT, photomultiplier tube.
A two-photon two-color fluorescence imaging system was constructed to demonstrate imaging through the TIP, see Fig. 1(b). A pulsed laser (FemtoTrain 1040–3, Spectra-Physics) fires excitation light centered around 1040 nm, which is followed by an optical isolator (ISO-FRDY-05-1030-W, Newport) used for blocking any reflected perturbations. A tunable attenuator (VA5-PBS253, Thorlabs) is applied to adjust the transmitted excitation power, and the unused beam is disposed of by a beam dump. The excitation beam passes through two tube lenses (TL 1 and TL 2) with the same focal length of 50 mm before reaching two galvanometer mirrors (GVS002, Thorlabs) for lateral scanning. A beam shutter (SHB1 T, Thorlabs) turns off the unnecessary illumination between acquisitions to minimize phototoxicity. The size of the beam is expanded by successively passing through two TLs with a focal length of 50 mm (TL 3) and 200 mm (TL 4), respectively. The size-expanded beam goes through a dichroic mirror (FF705-Di01, Semrock) before its divergence is regulated by a tunable lens (EL-10-30-TC-VIS-12D, Optotune) working in conjunction with an offset lens (LC4232, Thorlabs), which is then focused by a 20× microscope objective and coupled into the TIP. The objective numerical aperture (NA) is close to the coupling NA of the TIP to assure efficient coupling. In our experiments, the average power delivered to the sample was around 25 mW. The regulated divergence of the beam is converted into varying working distance (WD) of the objective, which in turn implements axial scanning for the TIP. The epifluorescence signals are directed to two channels (green and red in our case) after being separated by another dichroic mirror (FF556-SDi01, Semrock). A bandpass filter is used to clean up the collected fluorescence signal for each channel. The center wavelength and bandwidth of the filter are adjusted according to the tested sample. The cleaned fluorescence signals are collimated by two identical collimators (F950FC-A, Thorlabs) and subsequently sent to two identical photomultiplier tubes (H7421-40, Hamamatsu) for image acquisition through a multimode optical fiber (MMF). To increase the detection efficiency, we placed the collimators close to the objective and used a MMF with a large core diameter of 1 mm.
The TIP demonstrated in this work was composed of two 500 μm thick and 110 mm long bendable GRIN lenses with nominal NA of 0.5 for both object and image sides, see Fig. 1(c). The design WD was 0.2 mm in air for both ends, and the magnification was 0.7. The split distal ends were used to image at two different tissue sites, with the location at lenses 1 and 2 referred to as sites 1 and 2, respectively. The bundled proximal ends are aligned with the coupling objective. Thin GRIN lenses are used to both guarantee flexibility and make use the proximal ends fit into the objective FOV, which, however, leads to reduced FOV for each GRIN lens. Each lens was subject to a cantilever-beamlike deflection, which has been described in detail in our previous work [12]. This deflection may cause a slight lateral shift to the FOV, but no changes to the resolution were detected. Using the simulation method previously established [12], the maximum FOV shift is calculated to be around 9.5 μm when the distal ends are separated by about 2 cm. This shift is on the order of the diameter of a typical mammalian cell. Since the lens separation will be fixed during testing, the FOV shift may not be present in one imaging round. In addition, the lateral FOV shift would be even further reduced for TIPs made of longer GRIN lenses whose feasibility has been theoretically investigated [13]. While the separation of a few centimeters is sufficient for many applications in laboratory mice, the separation may be increased further if a biocompatible coating (e.g., polyimide) is used to mechanically protect the GRIN lenses. Protected bendable GRIN lenses may undergo a tighter bend without breaking, providing more flexibility for applications requiring asymmetric deformations to the lenses (thus different axial offsets at the distal end). Furthermore, note that each GRIN lens is not rotatable during testing, which, however, would not be an issue for bendable GRIN lenses without a prism for the side-view.
Because the TIP was composed of more than one bendable GRIN lens, each of them was subject to an offset relative to the objective axis, see Fig. 2(a), in which the FOV orientation is defined as well. The same FOV orientation is used throughout this work. We first investigated the influence of this offset on the resolution using one single lens. A piece of polydimethylsiloxane (PDMS) with embedded 1-μm-diameter fluorescent beads was attached to the prism surface for testing. Figure 2(b) shows a fine-scanned image of one 1-μm-diameter fluorescent bead, which was used to calculate both the lateral (X and Y) and axial (Z) resolution. The obtained resolution, defined as the full width at half maximum (FWHM), as a function of the offset is summarized in Fig. 2(c). We did not observe an appreciable degradation for both the lateral (~2.7 μm) and axial (~24.5 μm) resolutions as the lens offset increased. Furthermore, we also tested the signal efficiency and observed that the signal dropped as the lens offset increased. We found that this signal drop was due to the misaligned coupling between the slightly off-axis epifluorescence signal and the MMF. If the coupling efficiency is too low, it may be alleviated by translating the MMF (or proximal end of the TIP) or using an objective with a lower magnification (but with a high NA for better resolution).
Fig. 2.
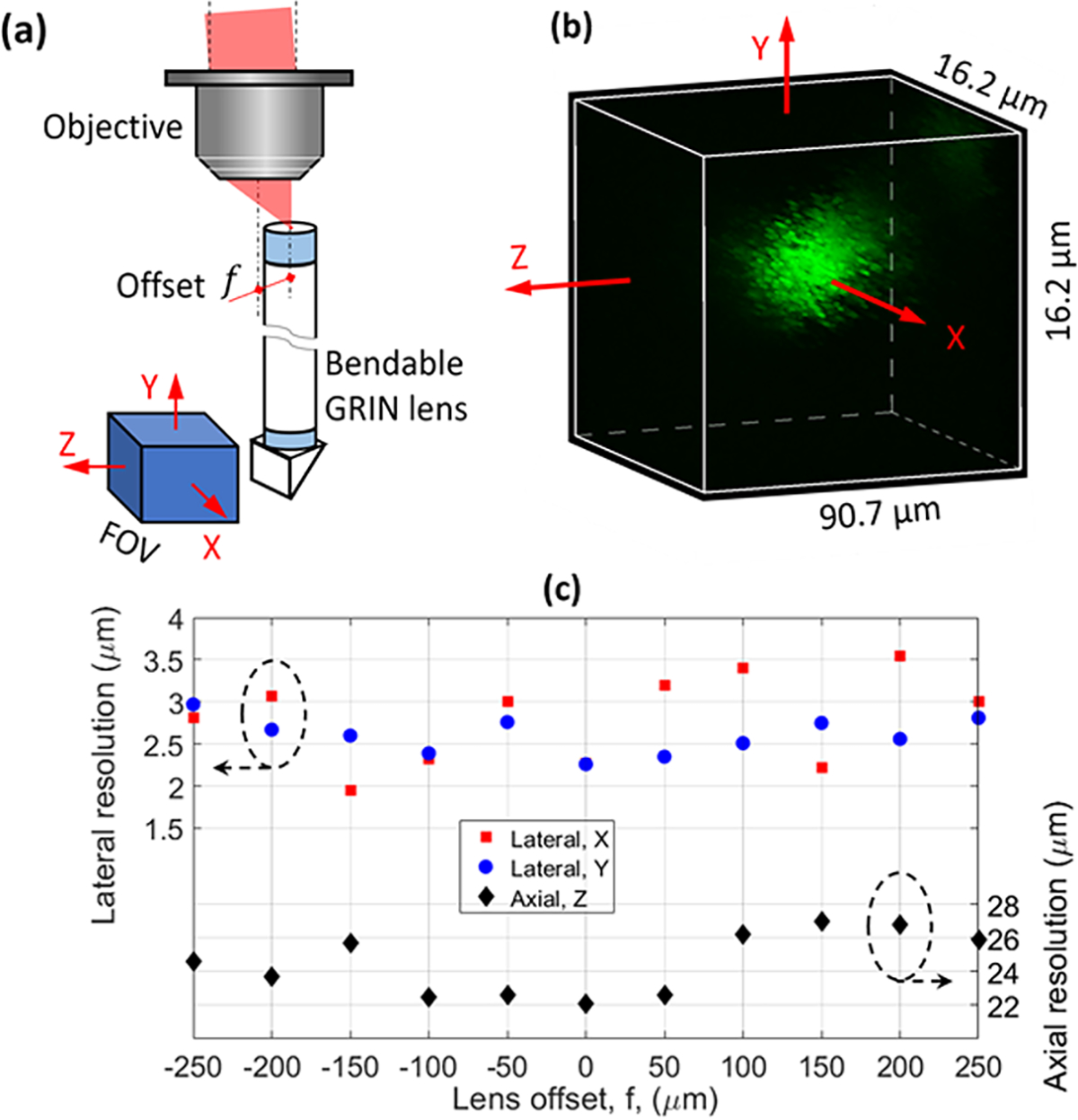
Resolution characterization. (a) Schematic showing the lens offset, (b) typical fine-scanned image of a 1-μm-diameter bead used for resolution calculation, and (c) measured resolution versus lens offset.
We then testified two-site two-photon two-color imaging through the TIP using fluorescent beads. To implement two-color imaging with minimum cross talk, in addition to selecting appropriate emission filters, one typically needs to develop different excitation wavelengths to minimize bleedthrough between channels, which is, however, highly costly for two-photon imaging systems. Besides hardware development, cost-effective image processing methods have also reduced spectral bleedthrough [14–17]. In this work, we implement a recently proposed PICASSO unmixing method [17] to suppress this bleedthrough. This blind linear unmixing method does not require prior knowledge of emission spectra of the fluorophores, which is a desirable advantage for practical applications. PICASSO assumes that the ratio of fluorescence signals between different channels for each individual fluorophore is fixed; thus a linear correlation is established. The goal of PICASSO is to minimize this correlation by iteratively subtracting significantly scaled down (in intensity) images acquired from other channels.
To ensure that PICASSO works properly for our application, site 1 (lens 1) of the TIP was arranged to image one type of beads (red in this case) as a control. The original image with a maximum tested FOV of 162 μm × 170 μm × 162 μm at each site is shown in the left part of Fig. 3(a), which suggests that the beads had strong bleedthrough in both channels (the overlay of green and red turns yellow). The image after the PICASSO unmixing is shown in the left part of Fig. 3(b), which indicates that only the red channel survived the unmixing, while the green channel was significantly suppressed. To visualize the change in correlation after unmixing, the bivariate histogram of the original image is shown in Fig. 3(c). Bleedthrough-related linear correlation is clearly visible, as evidenced by the pattern denoted by the red dashed arrow. The bleedthrough diminished after unmixing, leaving only a vertical (i.e., uncorrelated) pattern in the histogram designated by the red dashed arrow in Fig. 3(d).
Fig. 3.
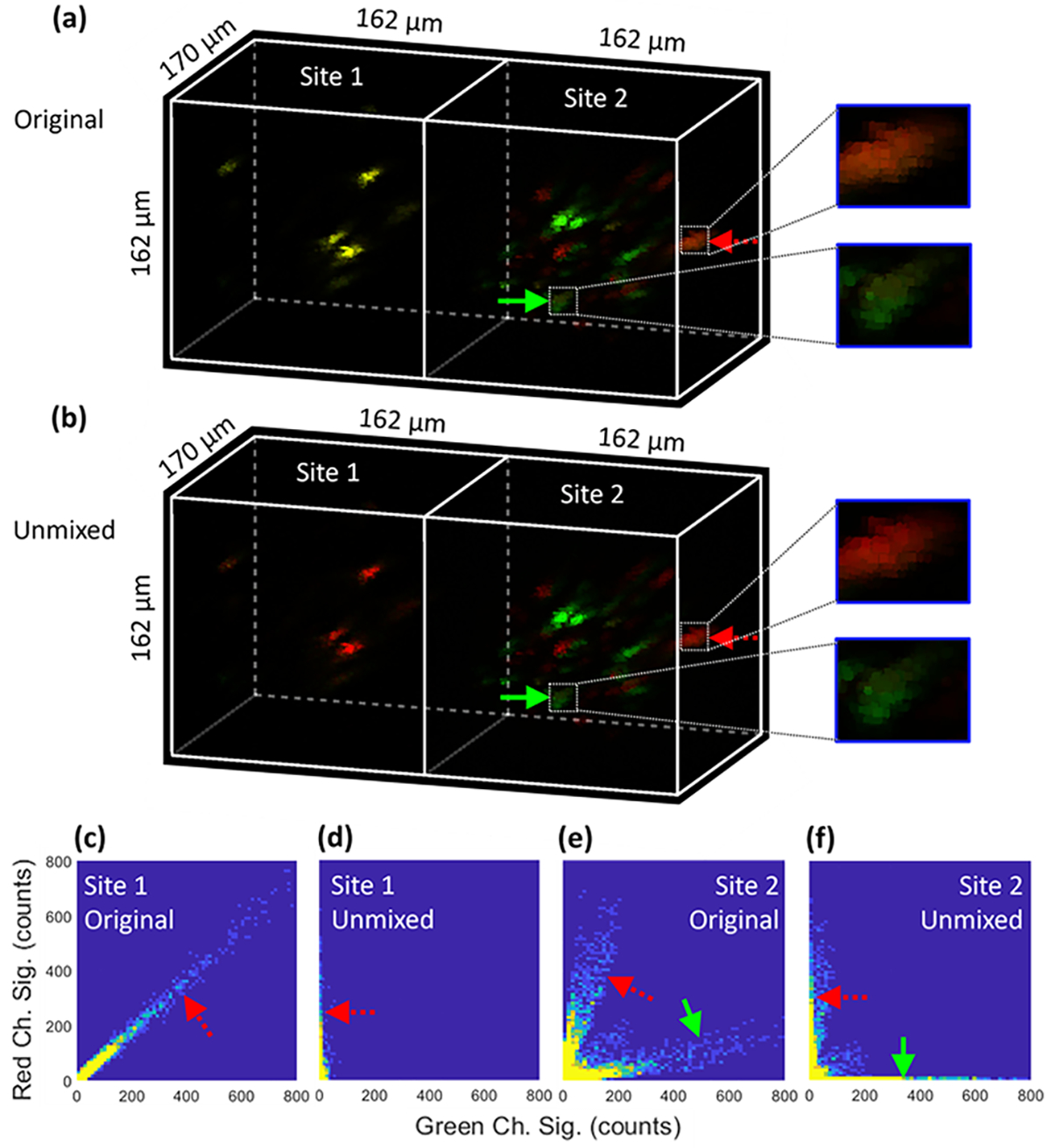
Two-site two-color imaging demonstrated and verified using discrete fluorescent beads and PICASSO image unmixing. (a) Original and (b) unmixed images, and bivariate histogram for the (c) original and (d) unmixed images at site 1 and (e) original and (f) unmixed images at site 2.
With the effectiveness of PICASSO verified by the one-bead-type imaging at site 1, we then applied PICASSO to unmix images of two types of fluorescent beads (green and red) at site 2 (lens 2) of the TIP. The original overlay image is shown in the right part of Fig. 3(a), in which the green solid arrow and red dashed arrow point toward a green bead and red bead, respectively. The two beads are visualized as slightly yellow due to bleedthrough, see the close-up views in Fig. 3(a). The bleedthrough is more clearly visible in the bivariate histogram of the original image in Fig. 3(e). After the PICASSO unmixing, the image is displayed in the right part of Fig. 3(b), in which the yellow component in the two designated beads is essentially eliminated, see the close-up views in Fig. 3(b). This finding is confirmed by the histogram shown in Fig. 3(f), where the resulting horizontal and vertical patterns indicate uncorrelation.
Finally, with the two-site two-photon two-color imaging verified by fluorescent beads, we moved on to demonstrate the imaging of two biomedically relevant samples. The first sample was a nonuniform mixture of two fluorescent anti-cancer drugs, sunitinib (green fluorescence) and doxorubicin (red fluorescence), which are widely used clinically [18,19]. Each drug was formulated and dried separately and then coarsely blended to form the mixture. It is critical that the mixture is nonuniform for the unmixing algorithm to function properly (a uniform mixture would be regarded as a single new drug). A small piece of this mixture was attached to each site of the TIP, see the photos in Fig. 4(a). An acquired image after unmixing is shown in Fig. 4(b), where both locations show a blend of the two types of drug grains. For more detail, one lateral 2D slice from both site 1 (slice 1) and site 2 (slice 2) is shown in Figs. 4(c) and 4(d), respectively, with the original version displayed in the left and the unmixed version displayed in the right. The green solid (or red dashed) arrows point to locations where bleedthrough of sunitinib (or doxorubicin) in the red (or green) channel was originally present but suppressed after unmixing.
Fig. 4.
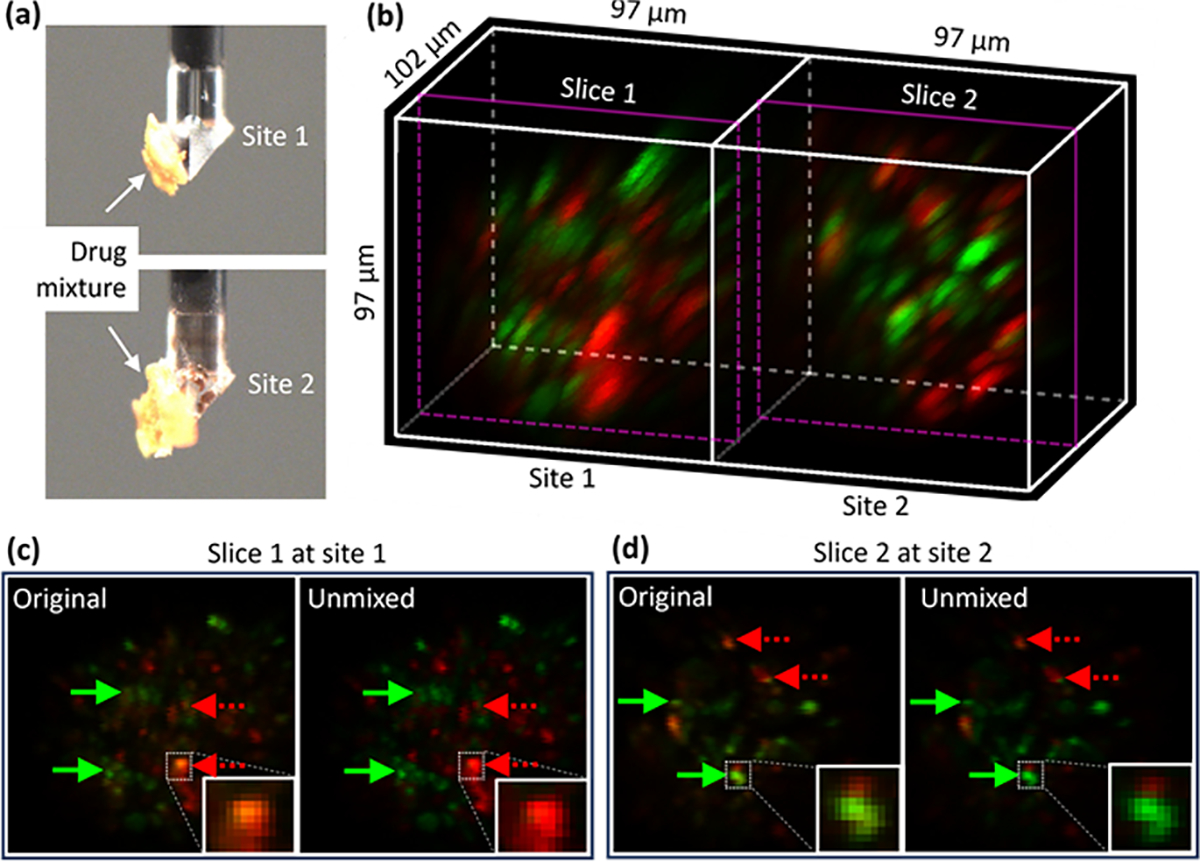
Two-site imaging of anti-cancer drug mixtures. (a) Photos of the drug mixtures, (b) acquired image after unmixing, and 2D sectioning at (c) slice 1 and (d) slice 2 without (left) and with (right) unmixing.
The second biomedical sample tested was a hybrid live murine MC38 tumor consisting of eYFP (green) and mCherry (red)-labeled cells. Both cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM), and a homogenous mixture of the two cell lines (1:1 ratio) with a total of 2 million cells in 200 μl of Matrigel was injected into each of the two flank regions of an athymic nude mouse. The tumors reached around 10 mm after about 10 days, and then in vivo imaging in one tumor was performed as the mouse was anesthetized, see Fig. 5(a). A shallow incision was introduced to the tumor to facilitate insertion of the TIP by a few millimeters. The motion artifact was minimized by displacing and fixing the tumor with a tape and by the flexibility of our bendable GRIN lenses.
Fig. 5.
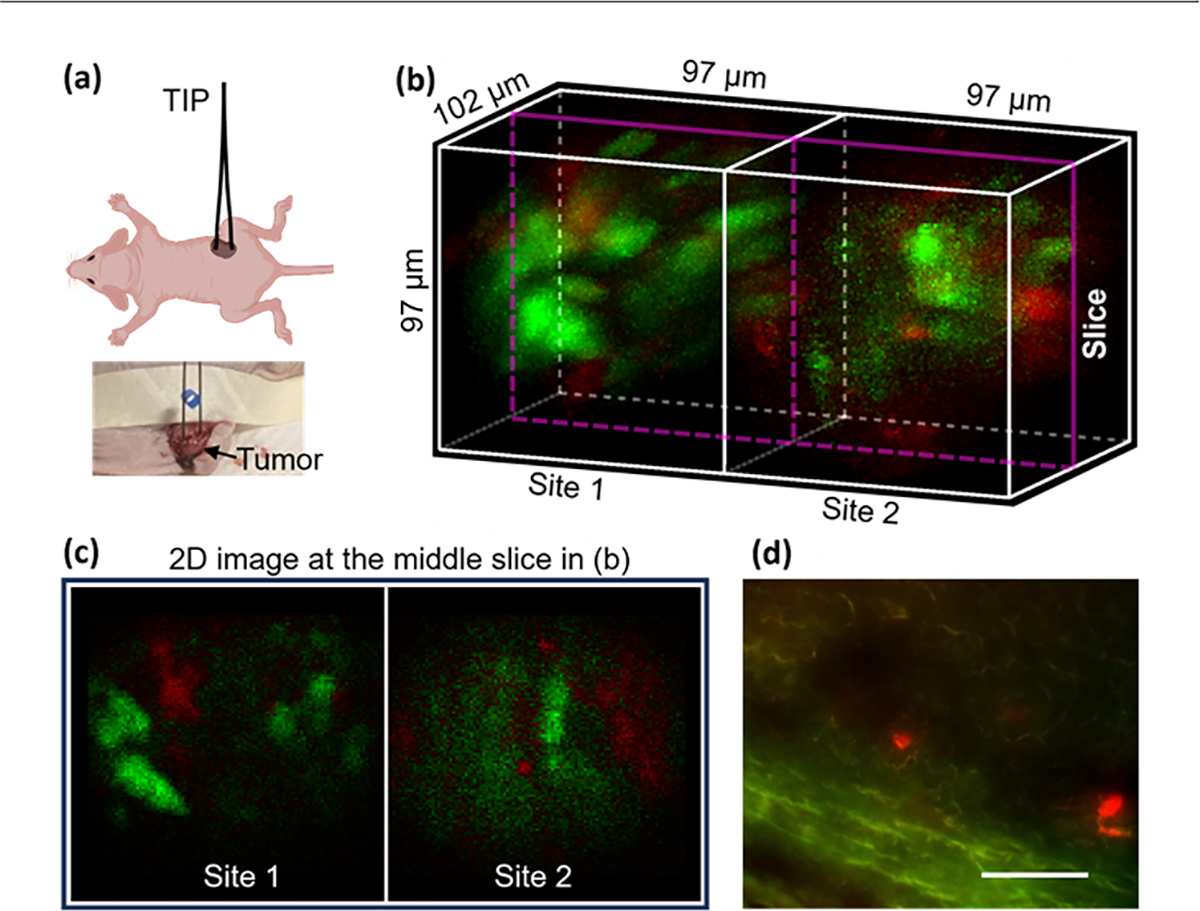
Two-site in vivo imaging of a hybrid tumor in a living mouse. (a) Schematic and photo of the testing in the animal, (b) an acquired image (green: eYFP; red: mCherry), (c) 2D image at the middle slice in (b), and (d) image of a cryosection of the tumor, scale bar: 60 μm.
An acquired and unmixed two-site image of the tumor is shown in Fig. 5(b). Both types of cells were successfully captured at both sites (green and red blobs). The hybrid tumor is observed to have distinct spatial architecture of the two cell types as well as nonuniform fluorescence intensity across locations, which is due to the well-documented tumor heterogeneity [4]. A 2D slice image is shown in Fig. 5(c), in which the cells measured around 10 μm in diameter. As a comparison, an image acquired from one tumor cryosection is displayed in Fig. 5(d). The bright red cells also measured 10 μm, which is consistent with the two-site image.
In summary, we have proposed and demonstrated a TIP capable of simultaneous microendoscopic 3D imaging at two anatomic locations. By integrating with a two-photon two-color imaging system, two-site imaging of anti-cancer drug mixtures and an in vivo murine hybrid tumor was successfully implemented.
Funding.
National Institutes of Health (5P41EB015871, 5P41EB028741, 5R37CA224144, K25EB032900).
Footnotes
Disclosures. O.J. is a consultant to Kibur Medical, Inc. His interest was reviewed and is managed by BWH and MGB Healthcare in accordance with their outside interest policies.
Data availability.
Data underlying the results presented in this paper are not publicly available at this time but may be obtained from the authors upon reasonable request.
REFERENCES
- 1.Pereira M. d. S. F., Thakkar A, and Lee DA, Methods Cell Biol. 167, 163 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Shah S, Chandra A, Kaur A, et al. , J. Photochem. Photobiol., B 170, 65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Etrych T, Lucas H, Chytil OJP, et al. , J. Controlled Release 226, 168 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Minchinton AI and Tannock IF, Nat. Rev. Cancer 6, 583 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Peruzzi P, Dominas C, Fell G, et al. , Sci. Transl. Med. 15, eadi0069 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonas O, Landry HM, Fuller JE, et al. , Sci. Transl. Med. 7, 284ra257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatarova Z, Blumberg DC, Korkola JE, et al. , Nat. Biotechnol. 40, 1823 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leblond F, Davis SC, Valdés PA, et al. , J. Photochem. Photobiol., B 98, 77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pittet MJ and Weissleder R, Cell 147, 983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castanares ML, Gautam V, Drury J, et al. , Biomed. Opt. Express 7, 5325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sofroniew NJ, Flickinger D, King J, et al. , eLife 5, e14472 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu G, Kang JW, Bhagavatula S, et al. , Opt. Express 30, 36651 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G, Kang JW, and Jonas O, Materials 14, 3392 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiménez-Sánchez D, Ariz M, Morgado JM, et al. , Bioinformatics 36, 1590 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Neher RA, Mitkovski M, Kirchhoff F, et al. , Biophys. J. 96, 3791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McRae TD, Oleksyn D, Miller J, et al. , PLoS One 14, e0225410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seo J, Sim Y, Kim J, et al. , Nat. Commun. 13, 2475 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow LQM and Eckhardt SG, J. Clin. Oncol. 25, 884 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Rivankar S, J. Can. Res. Ther. 10, 853 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data underlying the results presented in this paper are not publicly available at this time but may be obtained from the authors upon reasonable request.