

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by clinical symptoms of memory and cognitive deficiencies. Postmortem evaluation of AD brain tissue shows proteinopathy that closely associate with the progression of this dementing disorder, including the accumulation of extracellular beta amyloid (Aβ) and intracellular hyperphosphorylated tau (pTau) with neurofibrillary tangles (NFTs). Current therapies targeting Aβ have limited clinical efficacy and life-threatening side effects and highlights the need for alternative treatments targeting pTau and other pathophysiologic mechanisms driving AD pathogenesis. The brain’s extracellular matrices (ECM), particularly perineuronal nets (PNNs), play a crucial role in brain functioning and neurocircuit stability, and reorganization of these unique PNN matrices have been associated with the progression of AD and accumulation of pTau in humans. We hypothesize that AD-associated changes in PNNs may in part be driven the accumulation of pTau within the brain. In this work, we investigated whether the presence of pTau influenced PNN structural integrity and PNN chondroitin sulfate glycosaminoglycan (CS-GAG) compositional changes in two transgenic mouse models expressing tauopathy-related AD pathology, PS19 (P301S) and Tau4RTg2652 mice. We show that PS19 mice exhibit an age-dependent loss of hippocampal PNN CS-GAGs, but not the underlying aggrecan core protein structures, in association with pTau accumulation, gliosis, and neurodegeneration. The loss of PNN CS-GAGs were linked to shifts in CS-GAG sulfation patterns to favor the neuroregenerative isomer, 2S6S-CS. Conversely, Tau4RTg2652 mice exhibit stable PNN structures and normal CS-GAG isomer composition despite robust pTau accumulation, suggesting a critical interaction between neuronal PNN glycan integrity and neighboring glial cell activation. Overall, our findings provide insights into the complex relationship between PNN CS-GAGs, pTau pathology, gliosis, and neurodegeneration in mouse models of tauopathy, and offer new therapeutic insights and targets for AD treatment.
INTRODUCTION
AD is a progressive neurodegenerative disorder characterized by neuronal dysfunction and associated loss of memory and cognition (Terry et al. 1991; Busche & Konnerth 2015). The accumulation of Aβ and intracellular pTau are considered the hallmark neuropathologic criteria for diagnosing the AD progression (Braak & Braak 1991). Recent therapies targeting Aβ clearance using anti-Aβ monoclonal antibodies (mabs) during the early stages of AD provide minimal cognitive benefit, and about 40% of patients treated with the mab aducanumab experienced life-threatening side effects, including cerebral edemas (ARIA-E) and hemorrhages (ARIA-H) (Shi et al. 2022; van Dyck et al. 2023; Sims et al. 2023; Budd Haeberlein et al. 2022). As such, a dire need to explore treatment strategies targeting other neuropathological changes, including interactions between pTau and neurocircuit function, that may prove safer and more efficacious for treating a broader AD population.
Emerging concepts in the field of AD and other neurocognitive disorders suggest that neurocircuit functioning does not solely depend on neuron and glia cells but is also heavily influenced by the surrounding extracellular microenvironment. Constituting ~20% of the brain’s total volume in adults (Nicholson & Syková 1998), the highly understudied brain’s ECM may be one of the largest direct influencers of brain function. PNNs, which are a lattice-like brain matrix subtype that surround key GABAergic neurons involved in learning, memory, and cognition (Fawcett et al. 2022), are comprised of glycosylated chondroitin sulfate proteoglycans (CSPGs) with attached CS glycan chains.
The role for PNNs in underlying neurocognitive defects has gained recent attention in the field of AD research. Studies using Vicia villosa agglutinin (VVA) and Wisteria floribunda agglutinin (WFA) lectin staining of PNN CS-GAGs showed up to 70% reductions in the abundance of PNN CS glycan attachments in postmortem AD brain tissue (Kobayashi et al. 1989; Baig et al. 2005). Intriguingly, the loss of PNN CS-GAGs appears to occur independently to PNN core proteins (i.e., aggrecan and brevican), which remain stable in postmortem AD brain tissue (Brückner et al. 1999; Morawski et al. 2010a; Morawski et al. 2012; Lendvai et al. 2013; Howell et al. 2015). These reports imply that AD-associated ‘PNN loss’ is characterized by selective deglycosylation of the PNN CSPGs instead of core protein degradation and total loss of the PNN matrices themselves (Scarlett et al. 2022).
Notably, whereas there appears to be no significant overlap between extracellular Aβ deposits and PNN distribution in human AD brain tissue (Morawski et al. 2010a), the majority of neurons ensheathed by PNNs remain devoid of pTau accumulation (Brückner et al. 1999). These findings have given rise to the hypothesis that neurons surrounded by PNNs are ‘protected’ from NFT formation (Brückner et al. 1999; Morawski et al. 2012; Morawski et al. 2010a) and provide a strong rationale to support additional studies that further explore mechanisms driving the complex relationship between PNN CS-GAG loss and pTau accumulation in AD.
There have been multiple attempts to recapitulate the hallmark changes in PNNs observed in humans into a mouse model of AD, none of which have simultaneously shown reductions in PNN CS glycans while retain the underlying core proteins. The APP/PS1 mouse model of Aβ overexpression exhibits a significant increase in hippocampal WFA+ labeling of PNN CS-GAGs surrounding parvalbumin neurons (Végh et al. 2014), while the Tg2576/APPsw and APPNL-F mouse lines of amyloidosis showed no change in hippocampal PNN abundance or distribution compared to controls (Morawski et al. 2010b; Sos et al. 2020). The lack of PNN changes in mouse models of Aβ agree with human studies that show no significant overlap between extracellular Aβ deposits and PNN distribution in AD brain tissue (Morawski et al. 2010a). In contrast to these findings, 4-month-old 5xFAD mice that overexpress Aβ exhibit a significant decrease in WFA+ PNNs in the cortex and subiculum (Crapser et al. 2020), however, this loss occurred in parallel to the loss of PNN aggrecan core protein.
Studies investigating the relationship between PNNs and accumulation of pTau are much more limited. Although one study by Yang et al. showed no difference in the number of PNNs within the perirhinal cortex of 3-month-old TauP301S (PS19) mouse model of tauopathy compared to age-matched controls (Yang et al. 2015), this investigation was limited to both brain region (perirhinal cortex) and age (3 months). A recent study performed in 3-month-old and 6-month-old rTg4510 (P301L) mice, a mouse model of tauopathy associated with familial frontotemporal dementia, showed an age-associated decrease in cortical WFA+ PNNs only after the accumulation of pTau and gliosis (Kudo et al. 2023). Although this study implicates pTau and/or gliosis as a driver for PNN CS-GAG loss, core protein abundance was not examined.
To address the lack of appropriate mouse models capable of translating the changes in PNNs observed in human AD patients, we performed a thorough analysis between two distinct mouse models of tauopathy, including the mutant tau mouse line PS19 and the humanized tau mouse line Tau4RTg2652 (Tg2652). Whereas PS19 mice exhibit age-dependent accumulation of pTau, microgliosis, and neurodegeneration from 3 to 9 months of age, resulting in a shortened life span in this model (9–12 months of age) (Yoshiyama et al. 2007), Tg2652 mice exhibit early accumulation of pTau but live a normal life span without gliosis or neuronal loss (Wheeler et al. 2015). By investigating both mouse models of tauopathy, this body of work distinguishes age-associated changes in PNNs with pTau load and additional AD-associated pathologies, including gliosis and neurodegeneration.
MATERIALS AND METHODS
Study animals.
All animals were housed and handled in accordance with protocols approved by the University of Washington and the Veterans Affairs Puget Sound Health Care System’s Institutional Animal Care and Use Committee (IACUC) (Alonge UW protocol 2456–06/PROTO201600898 and Kraemer VA protocol 0708) and all experiments were conducted in an American Association for Accreditation of Laboratory Animal Care (AAALAC)-accredited animal research facility in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments reported follow the ARRIVE guidelines.
PS19 transgenic mice expressing human P301S mutant human tau was used in this study (RRID:IMSR_JAX:008169; Drs. Kraemer and Alonge’s in-house colonies). The PS19 mouse model is a well characterized mouse model of tauopathy and exhibits progressive gliosis and neurodegeneration with age (Yoshiyama et al. 2007). A second transgenic mouse model of tauopathy, the Tg2652 mouse line (Dr. Brian Kraemer’s in-house colony) that overexpresses wild type human tau, was also examined in this study. The Tg2652 mouse line exhibits early-stage tau pathology, including phosphorylated tau without neurofibrillary degeneration or gliosis (Wheeler et al. 2015). In this study, we used 3-month-old PS19 (3m-PS19) (n=9 WT, 6M/3F; n=8 PS19, 3M/5F), 6-month-old PS19 (6m-PS19) (n=8 WT, 4M/4F; n=10 PS19, 5M/5F), 9-month-old PS19 (9m-PS19) (n=10 WT, 5M/5F; n=10 PS19, 5M/5F), and 4-month-old Tg2652 (4m-Tg2652) (n=8 WT, 4M/4F; n=7 Tg2652, 3M/4F) mice. Both the PS19 and Tg2652 lines were backcrossed on a congenic C57BL/6J background, and all analyses were done in comparison to their respective wild type (wt), age-matched littermate controls. Mice were group-housed in a temperature-controlled room under 12 hour:12 hour light:dark cycle and ad-lib access to food and water. A power analysis for our main effect (loss of PNN CS glycans) was performed on the 9m-PS19 cohort; a mean difference of 58.5% was determined between wt controls vs 9m-PS19 mice, resulting in 13 for the (Z1-alpha/2+Z1-beta) squared value and a minimum sample size of n=2 to confirm alpha level of 0.05 at 95% power.
Mice Brain processing.
were anesthetized with urethane (4 g/kg; 0.2 mL; IP) and cardiac perfused first with 15 mLs of 0.1 M Phosphate Buffered Saline (PBS) followed by 10 mLs of 4% paraformaldehyde (PFA) in 0.1 M PBS using a 3-way stopcock. Brains were extracted, post-fixed for 24 hours in 4% PFA at 4°C, cryopreserved in 0.1 M PBS + 30% sucrose solution, frozen in optimal cutting temperature (OCT) compound on dry ice, and stored at −80°C degrees. Prior to sectioning, brains were acclimated overnight at −20°C and then sectioned with a Leica CM1950 cryostat at 30 μm-thick serial sections and stored in 0.1 M PBS + 0.02% sodium azide solution at 4°C as free-floating sections.
Immunofluorescent labeling, confocal microscopy, and mean fluorescence intensity quantifications.
30 μm-thick sections of free-floating mouse brain tissue sections were processed for immunofluorescence by first using antigen retrieval in 10 mM trisodium citrate (pH 8.5) and heated at 95°C for 20 min. Immunostaining was performed according to published methods (Alonge et al. 2020). Briefly, free-floating tissue sections were permeabilized for 25 min at room temperature (RT) in 0.1 M PBS + 0.2% Triton X-100 and blocked for 2 hours at 37°C in 0.1 M PBS + 0.05% Triton X-100 (PBS-T) + 10% normal donkey serum (NDS) (Jackson ImmunoResearch Labs Cat# 017–000-121, RRID:AB_2337258). Sections were then incubated overnight at 4°C using 1:1,000 dilution of biotin labeled Wisteria floribunda agglutinin (WFA; PNN CS-GAGs) (Sigma-Aldrich Cat# L-1516, RRID:AB_2620171), aggrecan (ACAN; PNN CSPG) (Millipore Cat# AB1031, RRID:AB_90460), glial fibrillary acidic protein (Aves Labs Cat# GFAP, RRID:AB_2313547), ionized calcium-binding adapter molecule1 (Iba1; microglia) (FUJIFILM Wako Shibayagi Cat# 019–19741, RRID:AB_839504), Phospho-Tau (Ser202, Thr205) (AT8) (Thermo Fisher Scientific Cat# MN1020B, RRID:AB_223648), and Cat-315 (Millipore Cat# MAB1581, RRID:AB_11214066) antibodies in PBS-T + 1% NDS. The following day, the sections were washed and incubated for 2 hours at RT in 1:1,000 secondary antibodies in PBS-T + 1% NDS. Sections were then counterstained for DAPI, mounted, and cover slipped using Floromount-G (ThermoFisher Cat# 4958–02).
Mean fluorescence intensity (MFI) of WFA, ACAN, GFAP, and Cat-315 were performed based on an established method for PNN quantification (Slaker et al. 2016) and applied using a stereological approach. We first subdivided the dorsal hippocampus into 5 Bregma positions (−1.4, −1.5, −1.6, −1.7, −1.8 mm from Bregma) and four subregions (CA1, CA2, CA3, DG) defined based on the anatomical location of hippocampal PNN populations of interest. From each defined region of interest, we used Fiji open-source imaging software to subtract a constant background and quantify the MFI (per mm2) for each target. We then computed the averaged normalized MFI for each target by first normalizing MFI to the controls for each of the 5 regions separately, and then averaging the normalized changes across all Bregma positions. Iba1+ microglia counts were performed on hippocampal subregions from 2 dorsal hippocampal sections (−1.5 and −1.6 mm from Bregma). From each section, Fiji was used to apply a standard threshold to each image and the number of Iba1+ cells (per mm2) were computed using the Analyze Particles function in Fiji using the following conditions: size (mm2) 0.4-Infinity pixel units and Circularity 0.3–1.00. Data is shown as an average of Iba1 counts between the two regions. Images taken for quantification were performed on the Keyence BZ-X800 fluorescent microscope (10X) and representative images were taken on the Nikon A1R HD confocal (10X).
TUNEL stain and cellular co-localization assays.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) stainings were performed on hippocampal subregions from 2 dorsal hippocampal sections (−1.5 and −1.6 mm from Bregma). For studies investigating cellular co-localization with TUNEL immunoreactivity, hippocampal sections were first labeled with primary antibodies again neurons (Aves Labs Cat# NUN, RRID:AB_2313556), oligodendrocytes (Aves Labs Cat# OLIG2–0100, RRID:AB_2924438), GFAP+ astrocytes, or Iba1+ microglia as described above. Tissues were then mounted to Superfrost histology slides, air-dried, washed 1×10 min with 0.1 M Tris Buffered Saline (TBS) in DNase-free water, followed by 1×10 min wash with DNase-free water. Sections were isolated by PapPen and 50 μL of TUNEL reaction mixture (Roche Cat# 11684795910), created by adding 19-parts labeling mix buffer: 1-part TdT enzyme, was applied to each section. Sections were then incubated at 37°C for 1.5 hours in a dark and humid chamber, followed by 1×10 min wash using 0.1 M TBS + 0.05% Triton X-100 (TBS-T), 1×10 min wash using 0.1 M TBS, dried, and coverslipped. To quantify total TUNEL abundance, Fiji was used to apply a standard threshold to each image and the % TUNEL area was quantified and averaged between the two hippocampal regions. To quantify co-localization, TUNEL immunoreactivity was manual identified in Fiji using the multi-point function and overlayed on neuronal, oligodendrocyte, astrocyte, or microglia labeling performed on the same tissues.
Hippocampal CS-GAG digestion and disaccharide isolation.
CS disaccharides were isolated and quantified according to our previously published methods (Alonge et al. 2019). Briefly, hippocampal sections were isolated from 30 μm PFA-fixed brain sections from the same tissues used in the immunofluorescent staining and TUNEL assays. The isolated hippocampal sections were washed 3x in Optima LC/MS-grade water and 1x in 50 mM ammonium bicarbonate at RT. ChondroitinaseABC (ChABC) digestion of CS-GAGs was performed using 500 mU/mL of ChABC (Sigma-Aldrich Cat# C3667) in 50 mM ammonium bicarbonate (pH 7.6) in a Thermo Scientific MaxQ4000 orbital shaker at 80 rpm, 37°C, for 24 hours. Supernatants were collected in sterile 1.7 mL microcentrifuges tubes and spun for 10 min at 14,000 × g to pellet any debris. The supernatant was then collected and dehydrated using a SpeedVac Concentrator and the lyophilized product was reconstituted in 30 μL of LC/MS-grade water.
LC-MS/MS + MRM quantification of hippocampal CS disaccharides.
CS samples were analyzed using a triple quadrupole mass spectrometer equipped with an electrospray ion source (Waters Xevo TQ-XS) operated in negative mode on a Waters Acquity I-class ultra-performance liquid chromatographic (UPLC) system coupled to the same Waters Xevo TQ-XS system. Disaccharides were resolved by a Hypercarb (2.1 × 50 mm, 3 μm) porous graphitic chromatography column (ThermoFisher Scientific Cat# 35003–052130) as described previously (Alonge et al. 2019). Assigned multiple reaction monitoring (MRM) channels: 4S-CS 458>300 m/z, 4S6S-CS 538>300 m/z, 0S-CS 378>175 m/z, 6S-C 458>282 m/z, and 2S6S-CS 268>282 m/z. MassLynx software version 4.1 (Waters) was used to acquire and quantify all data. Under the conditions described above, the ratios between peak areas produced from equimolar CS standard runs were normalized to the highest peak intensity and relative quantification of each CS isomer within a sample was achieved using a modified peak area normalization function (Alonge et al. 2019). Each CS isomer was expressed as a relative percent of the total CS isomer composition within each sample.
Statistical Analyses.
Statistical analyses were performed with GraphPad Prism® 10.0 (GraphPad Software, Inc., La Jolla, CA). Error bars represent the mean ± standard error of the mean (SEM). Data with two variables were first analyzed for normality using Shapiro-Wilk test and normal (Gaussian) distributed data were analyzed using parametric t-test and data that did not show normality were analyzed using non-parametric Mann Whitney test, and all comparison tests were two-tailed. All p-values, t-values (t), and degrees of freedom (df) are provided in the supplemental tables. Stereology data were compared using a 2-way ANOVA or mixed effects with matched regions and all p-values, degrees of freedom, and F-values are provided in the supplemental tables. Data used in ANOVA/Mixed Model and Linear Regression were not tested for normality. No pre-determined exclusion criteria were utilized in this study, all animals were included, outlier tests were not performed, and all viable data resulting from the analyses were included in this manuscript. Statistical analyses for Main Figure 1 are shown in Supplemental Table 1, statistical analyses for Main Figure 2 are shown in Supplemental Table 3, statistical analyses for Main Figure 3 are shown in Supplemental Table 4, statistical analyses for Main Figure 4 are shown in Supplemental Tables 5–6, and statistical analyses for Main Figure 5 are shown in Supplemental Tables 7–9. Investigators were blinded to study conditions during all quantitative analyses.
Figure 1. 9m-PS19 mice exhibit loss of WFA+ PNNs in the presence of pTau accumulation and gliosis.
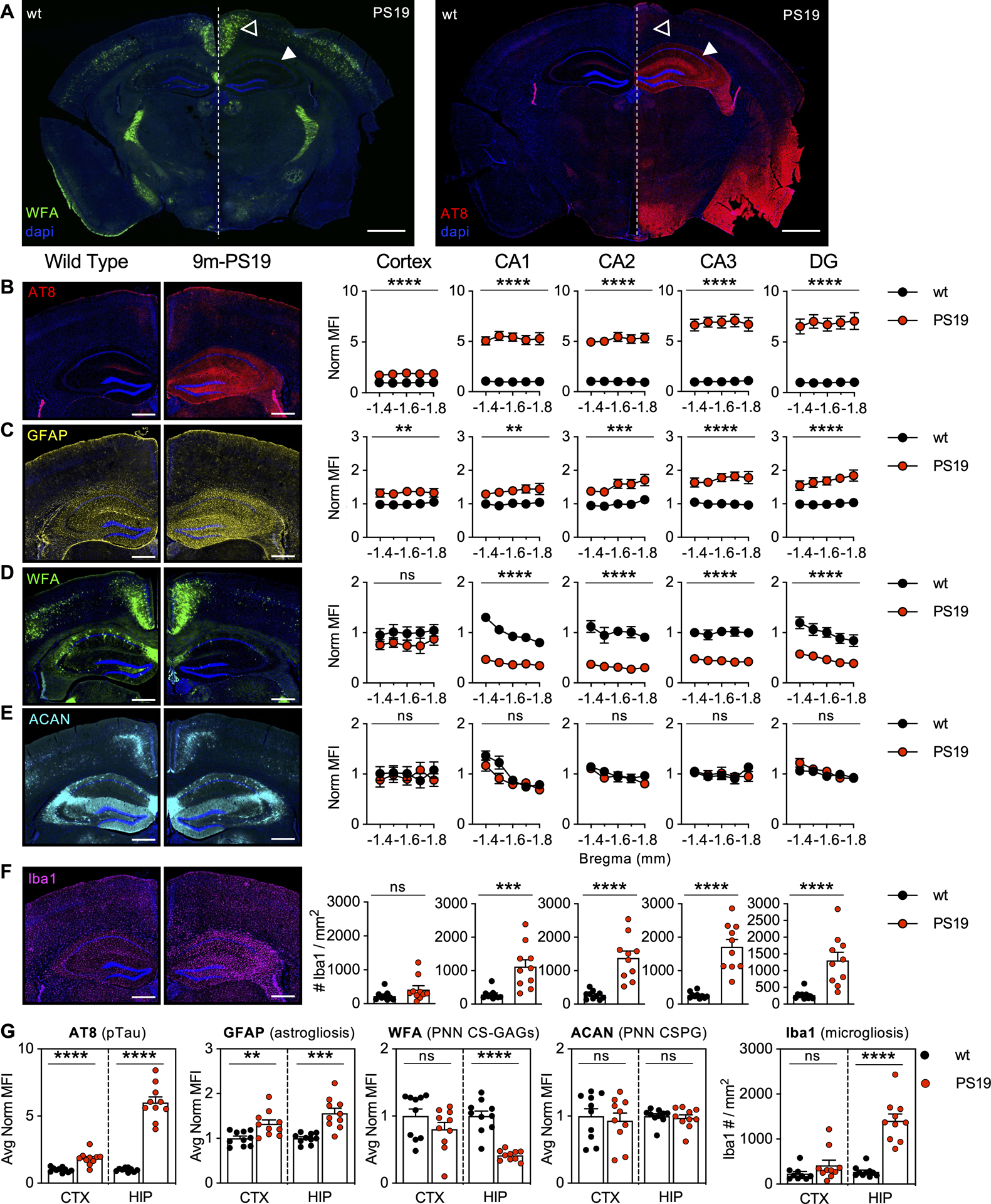
9-month-old PS19 mice exhibit A) striking loss of hippocampal WFA+ PNNs in association with regional pTau accumulation. 5-region stereology analysis (Bregma −1.4 to −1.8 mm) of B) AT8 (pTau), C) GFAP (astrogliosis), D) WFA (PNN CS-GAGs), E) ACAN (PNN CSPG), and F) Iba1 (microgliosis) were performed and then G) averaged and quantified. Solid arrows: dorsal hippocampus; Open arrow: retrosplenial cortex. Scale bar: A) 1 mm, B-F) 0.5 mm, representative images from male mice, dapi included in all images. Statistics reported in Supplemental Table 1.
Figure 2. 6m-PS19 mice exhibit stable PNNs in the presence of pTau accumulation without gliosis.
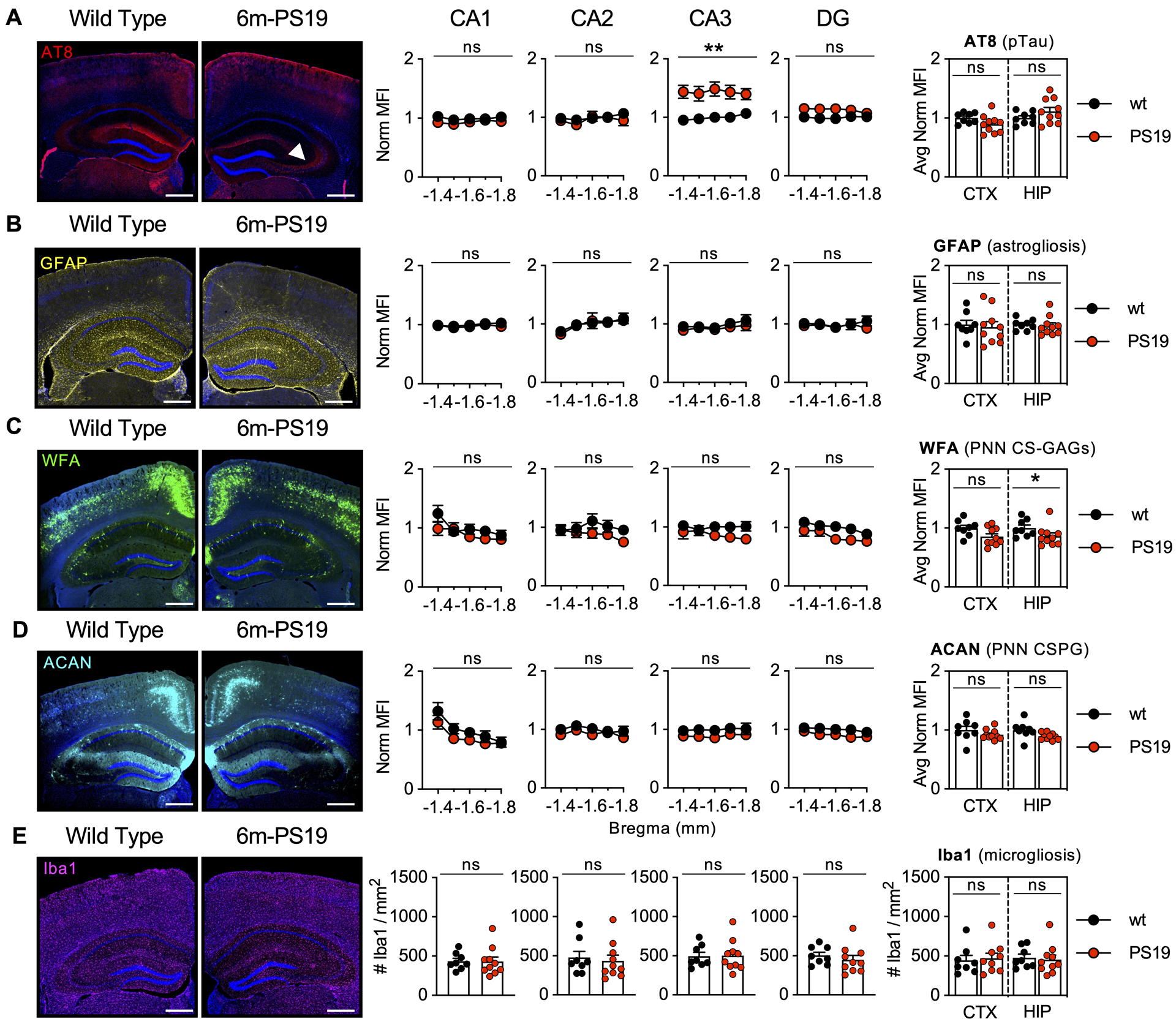
5-region stereology analysis (Bregma −1.4 to −1.8 mm) of 6-month-old PS19 mice exhibit regional accumulation of A) pTau (AT8) in the CA3 (white arrow) without changes in B) astrogliosis (GFAP), C) WFA (PNN CS-GAGs), D) ACAN (PNN CSPG), or E) microgliosis (Iba1). Scale bar: 0.5 mm, representative images from male mice, dapi included in all images. Statistics reported in Supplemental Table 3.
Figure 3. 4m-Tg2652 mice exhibit stable PNNs in the presence of pTau accumulation without gliosis.
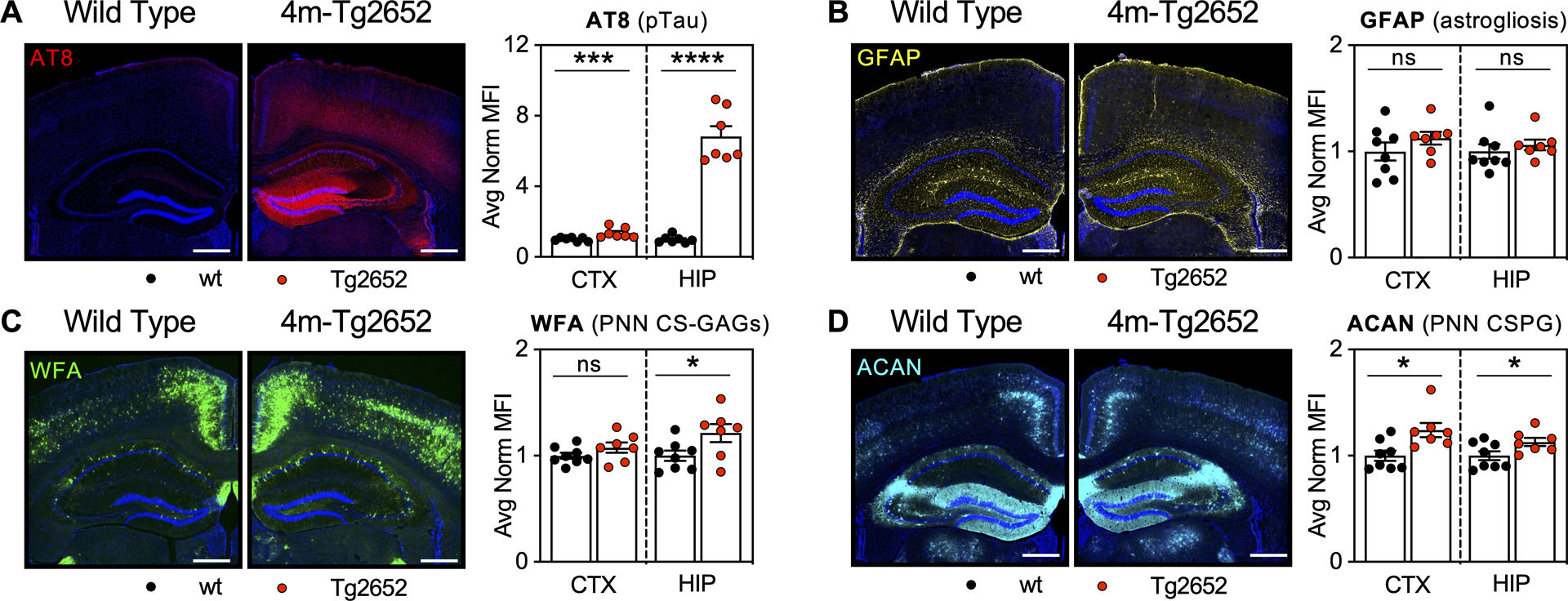
5-region stereology analysis (Bregma −1.4 to −1.8 mm) of 4-month-old Tg2652 mice exhibit robust accumulation of A) pTau (AT8) without B) gliosis (GFAP, astrogliosis) in association with increased C) WFA+ PNNs (PNN CS-GAGs) and D) ACAN (PNN CSPG). Scale bar: 0.5 mm, representative images from male mice, dapi included in all images. Statistics reported in Supplemental Table 4.
Figure 4. 9m-PS19 mice exhibit altered hippocampal CS isomer composition in association with PNN glycan loss, pTau accumulation, and neuroinflammation.
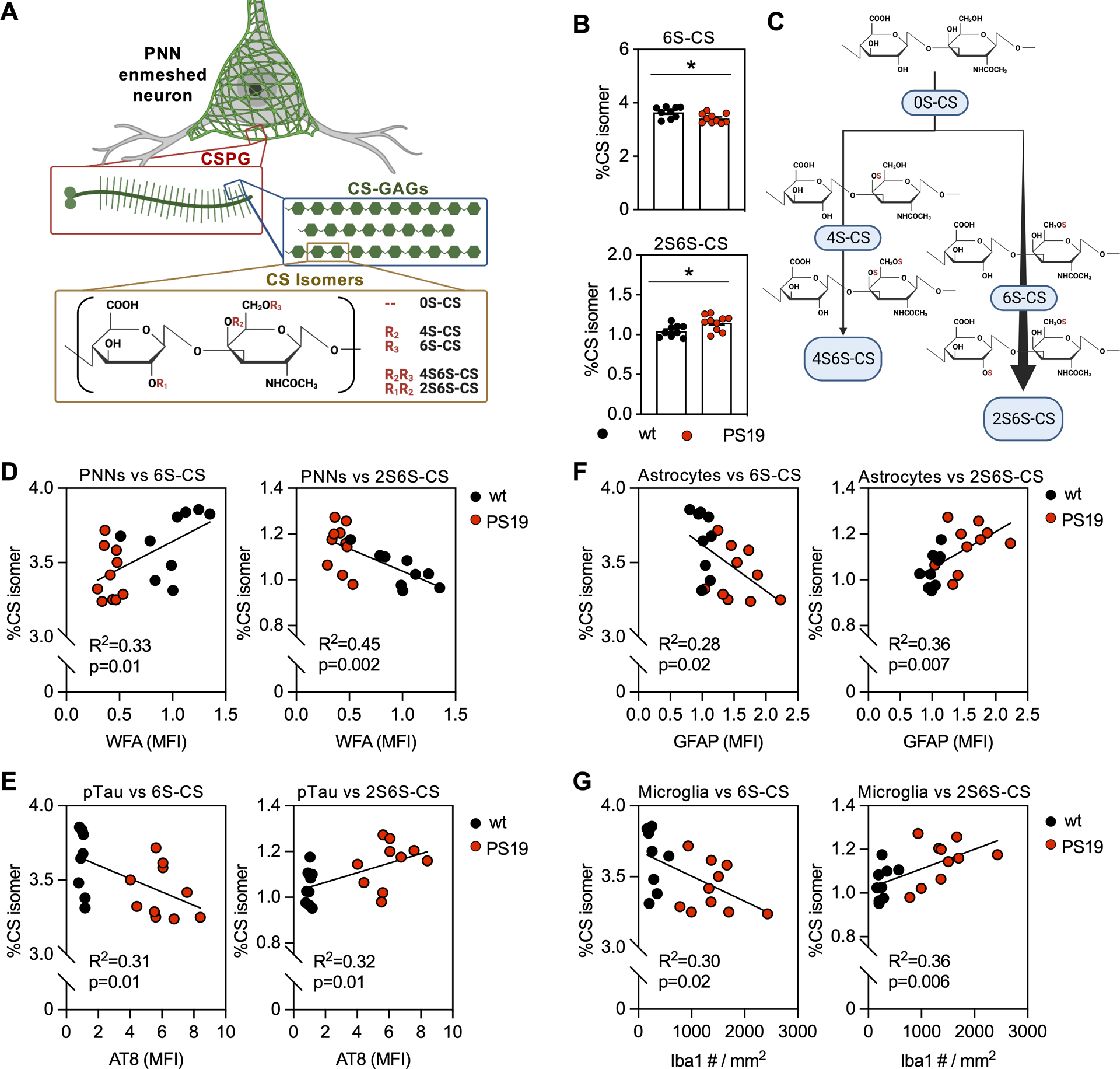
PNN matrices are comprised of A) non-sulfated (0S-CS), mono-sulfated (4S-CS, 6S-CS) and di-sulfated (2S6S-, 4S6S-CS) isomers. 9-month-old PS19 mice exhibit B) decreased mono-sulfated 6S-CS isomer and increased 2S6S-CS isomer compared to controls, which is visualized in C) the illustration representing flux through the 2S6S-CS isomer biosynthesis branch. These isomer changes occur in association with D) loss of WFA (PNN CS-GAGs) and gain of E) AT8 (pTau), F) GFAP (astrogliosis), and G) Iba1 (microgliosis). Statistics reported in Supplemental Tables 5 and 6.
Figure 5. 9m-PS19 mice exhibit increased neurodegeneration in association with pTau accumulation, gliosis, and altered CS-GAG sulfation patterning.
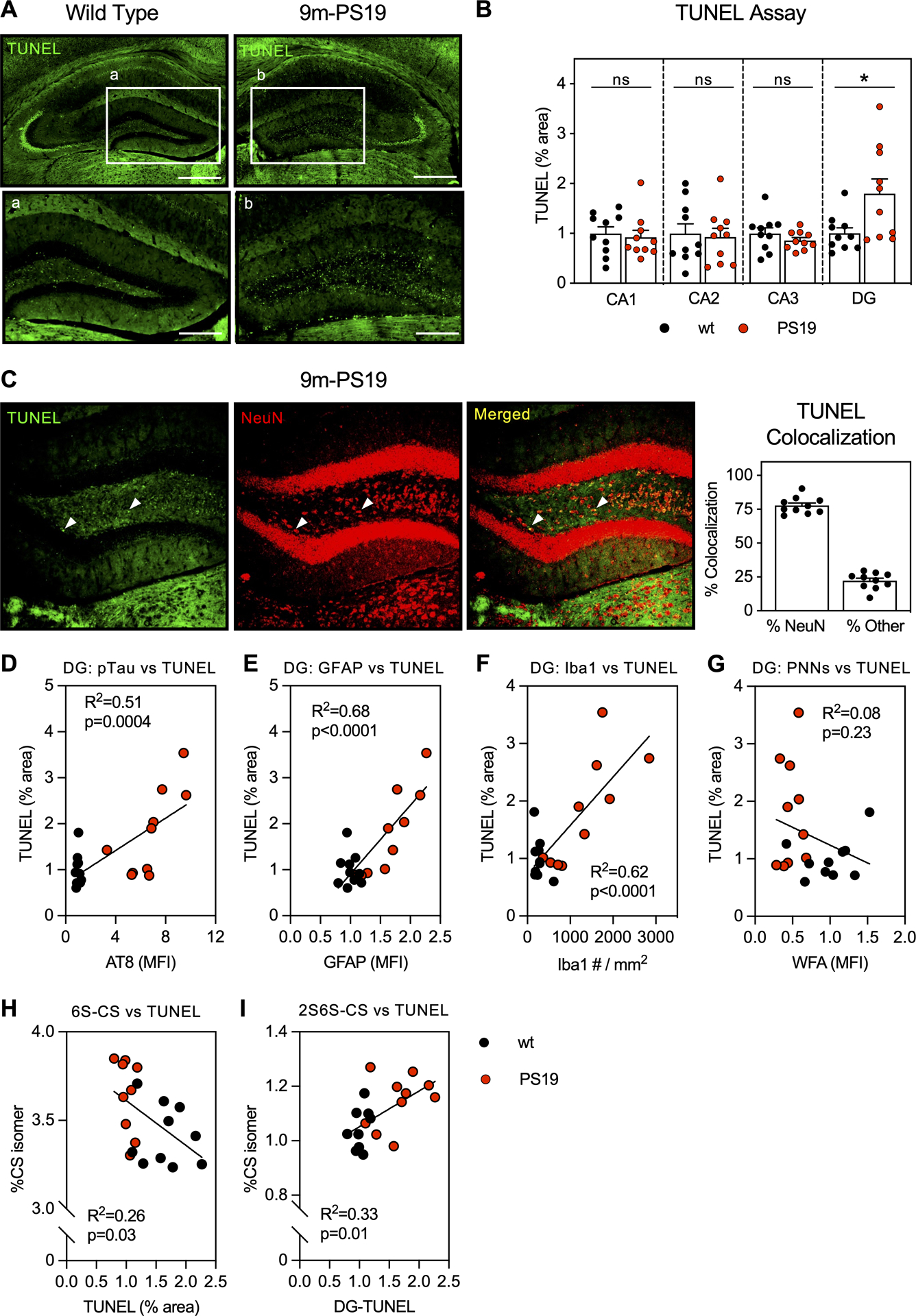
TUNEL staining in 9-month-old A) wt and age-matched PS19 mice showed selective increase in apoptosis within the B) DG of the dorsal hippocampus that positively colocalized with C) neurons labeled with NeuN. The increase in DG-TUNEL associated with D) pTau (AT8), E) astrogliosis (GFAP), and F) microgliosis (Iba1), but not G) WFA (PNN CS-GAG) loss. DG-TUNEL staining also associated with H) decreased 6S-CS isomer and I) increased 2S6S-CS isomer hippocampal content. Scale bar: A,B) 0.5 mm, a,b) 0.250 mm, representative images from male mice, dapi included in all images. Statistics reported in Supplemental Tables 7–9.
RESULTS
PS19 mice exhibit age-dependent loss of glycosylated PNNs in association with progressive pTau accumulation and gliosis.
We first set out to characterize the spatial profile of PNN changes in PS19 mice by performing stereological mapping of PNN CS-GAGs (WFA) and the PNN core protein aggrecan (ACAN) in relation to both the accumulation of pTau (AT8) and the induction of neuroinflammation (GFAP, astrogliosis; Iba1, microgliosis) throughout the hippocampus and adjacent retrosplenial cortex of 9-month-old PS19 mice (Fig. 1A). Compared to age-matched, wt littermate controls, 9m-PS19 mice displayed a robust accumulation of hippocampal pTau (p<0.0001) (Fig. 1B), astrogliosis (p<0.0001) (Fig. 1C), and microgliosis (p<0.0001) (Fig. 1F). These changes were also apparent to a lesser extent in the adjacent retrosplenial cortex (Fig. 1G; Supplemental Table 1). The increase in pTau and gliosis occurred in parallel to a striking (58.5%) decrease of hippocampal WFA+ PNN CS-GAGs (p<0.0001) (Fig. 1D), independent to significant changes in cortical WFA+ PNNs (p=0.19) (Fig. 1G), which suggests the extent of WFA+ PNN loss may relate the abundance of pTau and gliosis unique to these brain regions. Surprisingly, we did not observe distinguishable changes in hippocampal ACAN+ PNNs using an antibody against the PNN core protein (p=0.63) (Fig. 1E). The finding that PNN core proteins remain intact in 9m-PS19 mice was confirmed after staining with a second antibody against aggrecan, Cat-315 (p=0.75) (Supplemental Fig. 1) (Matthews et al. 2002), implying ACAN+ PNNs remain intact in these mice. Overall, these results show 9m-PS19 mice exhibit regional PNN deglycosylation without changes in ACAN core protein abundance, like that observed in postmortem brains of human AD patients.
To determine whether the loss of PNN CS-GAGs occurrs prior to pathogenic accumulation of pTau and resulting gliosis, we performed a similar analysis in 3-month-old and 6-month-old PS19 mice. Complementing the original study by Yang et al., which showed no difference in the number of WFA+ PNNs in the perirhinal cortex of 3m-PS19 mice (Yang et al. 2015), we also observed no changes in hippocampal or cortical PNNs in the absence of gliosis in 3m-PS19 mice (Supplemental Fig. 2, Supplemental Table 2). Meanwhile, stereology analysis of 6m-PS19 mice showed intraregional differences in hippocampal pTau accumulation (Fig. 2A), with the CA3 region exhibiting the greatest accumulation of pTau at this age (p=0.003). However, unlike 9m-PS19 mice, we failed to detect increased hippocampal astrogliosis (p=0.72) or microgliosis (p=0.77) in 6m-PS19 compared to wt controls (Fig. 2B,E). Although gliosis appeared unaltered, we did observe a slight (13.7%) decrease in PNN CS-GAGs (Fig. 2C) independent to changes in ACAN (Fig. 2D) at this age (p=0.04) (Supplemental Table 3). The differences in PNN integrity between 6m- and 9m-PS19 mice suggest that the presence of low abundant pTau accumulation alone is insufficient to result in complete loss of WFA+ PNNs in this mouse model of tauopathy.
Tg2652 mice exhibit robust pTau accumulation in the absence of gliosis and changes in PNNs.
Given the progressive loss of glycosylated PNNs from 6m- to 9m-PS19 mice, we wanted to next determine whether this effect might be driven by robust accumulation of pTau in the absence of neuroinflammation. Therefore, we evaluated PNN changes in second mouse line, the Tg2652 mouse model of tauopathy, which exhibits abundant pTau accumulation without gliosis. We first observed robust hippocampal pTau accumulation (p<0.0001) (Fig. 3A) without changes in astrogliosis (p=0.50) (Fig. 3B) in 4-month-old Tg2652 mice compared to age-matched, wt littermate controls (Supplemental Fig. 3, Supplemental Table 4), at an age where pTau accumulation peaks in this mouse model of tauopathy (Wheeler et al. 2015). Unexpectedly, 4m-Tg2652 mice exhibited a significant increase in the immunoreactivity of both hippocampal WFA+ PNN CS-GAGs (p=0.004) (Fig. 3C) and aggrecan+ PNN core protein expression (p<0.0001) (Fig. 3D). Taken together, results obtained from 4m-Tg2652 mice describe a mouse model in which robust pTau accumulation in the absence of gliosis does not result in PNN loss.
9m-PS19 mice exhibit changes in hippocampal CS-GAG sulfation patterns in association with pTau and gliosis.
The biological functions of PNNs are dictated by the composition of their attached CS-GAGs. These CS-GAGs are comprised of repeating units of glucuronic acid and N-acetylgalatosamine isomers that are uniquely modified by the addition of sulfates to the disaccharide unit, including non-sulfated (0S-CS), mono-sulfated (4S-CS and 6S-CS), and di-sulfated (2S6S-CS and 4S6S-CS) variants (Fig. 4A) (Djerbal et al. 2017). The relative abundance of CS isomers incorporated into the CS-GAG chains has been linked to key biological functions within the brain including charged molecule diffusion (0S-CS (Nicholson & Hrabětová 2017)), neurocircuit plasticity (4S-CS and 6S-CS (Miyata et al. 2012; Yang et al. 2021)), glial scarring (4S6S-CS (Brown et al. 2012)), and neurite outgrowth (2S6S-CS (Shida et al. 2019; Clement et al. 1999)).
We previously optimized a liquid chromatography–tandem mass spectrometry (LC-MS/MS) technique capable of quantifying the relative abundance of each non-, mono-, and di-sulfated CS isomer extracted from fixed mouse and human brain tissues using ChABC enzymatic digestion (Alonge et al. 2019). To determine if the loss of WFA+ PNN CS-GAG staining in 9m-PS19 mice (Fig. 1) associates with changes in their CS-GAG sulfation patterns, we performed LC-MS/MS on CS isomers extracted from fixed hippocampal tissue sections from the same mouse cohort. Compared to age-matched, wt littermate controls, we observed a clear shift in 9m-PS19 hippocampal CS sulfation patterning, including decreased monosulfated 6S-CS isomer (p=0.02) and increased disulfated 2S6S-CS isomer (p=0.02) abundance (Fig. 4B), suggesting apparent flux of disaccharides through the 2S6S-CS branch of the CS biosynthesis pathway (Fig. 4C). Moreover, these effects were not seen in the hippocampus of 6m-PS19 or 4m-Tg2652 mice (Supplemental Table 5). To further explore the relationship between these hippocampal CS isomer changes and our histopathological findings, we associated changes in hippocampal CS sulfation with loss of WFA+ PNNs, pTau accumulation, and gliosis in the 9m-PS19 cohort. Linear regression analyses showed changes in 6S-CS and 2S6S-CS isomers significantly correlated with the decrease in WFA+ PNNs (p=0.01, p=0.002) (Fig. 4D), increase in pTau accumulation (p=0.01, p=0.01) (Fig. 4E), and elevation in both astrogliosis (p=0.02, p=0.007) (Fig. 4F) and microgliosis (p=0.02, p=0.006) (Fig. 4G) in these mice (Supplemental Table 6). Although changes both 6S-CS and 2S6S-CS isomers correlated with the histological findings in 9m-PS19 mouse brain tissue, we note that the strongest correlation occurred between 2S6S-CS and the loss of WFA+ PNNs (R2=0.45) followed by 2S6S-CS and the induction of microgliosis (R2=0.42) and astrogliosis (R2=0.36).
9m-PS19 mice exhibit regional neurodegeneration in association with histopathological changes in pTau, gliosis, and PNN deglycosylation.
Neurodegeneration is a hallmark of AD in both demented patients and aged PS19 mice (Yoshiyama et al. 2007; Terry et al. 1991). TUNEL staining, which detects DNA breaks formed during cellular apoptosis, is a widely applied histological method used to measure neurodegeneration (Linsley et al. 2019). In this regard, we sought to determine the regional extent of neurodegeneration in 9m-PS19 mice and correlate these changes to cellular markers, accumulation of AD pathology, and changes in CS glycan sulfation patterns specific to the 9m-PS19 cohort (Fig. 5A). Compared to age-matched, wt littermate controls, 9m-PS19 mice exhibited a significant increase in TUNEL immunoreactivity within the dentate gyrus (DG) (p=0.02) subregion of the hippocampus (Fig. 5B; Supplemental Table 7). Colocalization of TUNEL staining with NeuN showed 77.8% of TUNEL immunoreactivity positively colocalized with neurons (Fig. 5C). Meanwhile, colocalization with glial markers for astrocytes (GFAP), microglia (Iba1), and oligodendrocytes (Olig2) showed minimal overlap with TUNEL immunoreactivity (Supplemental Figure 4), indicating neuronal loss as the primary source of apoptosis within the DG of PS19 mice. We observed a moderate correlation between DG-TUNEL staining and the accumulation of pTau (p=0.004) (Fig. 5D), and a strong correlation between DG-TUNEL staining and GFAP astrogliosis (p<0.0001) (Fig. 5E) and Iba1 microgliosis (p<0.0001) (Fig. 5F) (Supplemental Table 8). These data suggest that both pTau accumulation and increased gliosis observed in 9m-PS19 mice may play a role in neurodegeneration within the DG, potentially contributing to the cognitive deficits often observed in this AD mouse model (Yoshiyama et al. 2007).
Lastly, we sought to correlate the induction of neurodegeneration (TUNEL) within the DG with changes in PNN CS-GAG abundance and composition in the 9m-PS19 cohort (Supplemental Table 9). Although we did not observe a significant association between TUNEL staining and WFA+ PNN CS-GAG loss within the DG (Fig. 5G), we did discover that DG-TUNEL stain correlated with decreased 6S-CS (p=0.03) (Fig. 5H) and increased 2S6S-CS (p=0.01) isomer abundance (Fig. 5I), implying neuronal loss itself does not drive the loss of WFA+ PNNs but may directly influence their CS-GAG compositions. Overall, we conclude that 9m-PS19 mice exhibit increased neurodegeneration within the DG subregion of the hippocampus in association with accumulated pTau, elevated gliosis, and shifts in CS-GAG sulfation patterning.
DISCUSSION
Here, the results of our study show an age-dependent loss in PNN CS-GAGs without changes in PNN core protein abundance in the PS19 mouse model of tauopathy, which to our knowledge, is the first mouse model of AD to recapitulate PNN deglycosylation without core protein loss first observed in postmortem AD brain tissue. By investigating two different mouse models of tauopathy, this body of work distinguishes age-associated changes in PNNs with pTau load and additional AD-associated pathologies, including gliosis and neurodegeneration. Whereas 3-month-old PS19 mice do not exhibit pTau accumulation (Yoshiyama et al. 2007), gliosis, or changes in PNN matrices (Supplemental Fig. 1), 9-month-old PS19 mice exhibit robust pTau accumulation in the presence of astrogliosis, microgliosis, and neurodegeneration (Fig. 1 and Fig. 5). At this age, PS19 mice also exhibit a striking loss of hippocampal PNN-associated CS-GAGs, but not the underlying PNN aggrecan core protein, within regions of high pTau burden and gliosis (Fig. 1). Changes in PNN CS-GAG abundance occurred in parallel to changes in CS-GAG composition, which included increased expression of the 2S6S-CS neuroregenerative isomer (Fig. 4), suggesting an age-dependent glycosylation defect in this model.
In contrast to the PS19 mouse model of tauopathy, 4-month-old Tg2652 mice show stable PNN matrices in the presence of high pTau burden without gliosis (Fig. 3), implicating that the presence of pTau alone does not influence hippocampal PNNs. Instead, a complex relationship between pTau, gliosis, and neurodegeneration appears to drive PNN CS-GAG changes. The inflection point for PNN CS-GAG changes in PS19 mice appears to emerge at 6 months of age, where both the appearance of increased pTau accumulation specific to the CA3 hippocampal subregion and loss of WFA+ PNNs are first observed (Fig. 2).
Since these PNN CS-GAG changes occur just prior to, or in parallel with, the induction of gliosis in 6-month-old PS19 mice, this pivotal pathophysiologic window could be exploited for therapeutic intervention targeting PNN preservation in AD by preventing the induction of gliosis even before the detection of pTau. Recent work suggests that microgliosis represents a potential mechanism driving PNN loss in models of neurodegeneration (Miyata et al. 2012; Miyata & Kitagawa 2016; Crapser et al. 2021), and here we present additional evidence that astrogliosis may also exacerbate this effect. Although epidemiological evidence suggest that non-steroidal anti-inflammatory drugs may reduce the risk of AD, the majority of NSAIDs fail to reduce the prevalence of AD or influence the progression of AD-associated cognitive decline (Rivers-Auty et al. 2020). However, the anti-inflammatory drug diclofenac was recently found to associate with the slowing of cognitive decline (Stuve et al. 2020). Unlike the other pain-relievers, diclofenac represses the release of the inflammatory cytokine interleukin-1β and reduces activation of microglia (Stopschinski et al. 2023). Considering that other NSAIDs tested do not slow the progression of cognitive decline in AD, the beneficial effects of diclofenac are likely not through traditional COX-2 inhibition. Instead, diclofenac targeting of glia-associated mechanisms of neuroinflammation provides a strong rational for future studies to investigate the effects of diclofenac on the preservation PNN glycosylation and neurocircuit functioning in AD.
The finding that PNN CS-GAGs are altered in 9m-PS19 mice agree with a recent report that identified gene changes in chondroitin sulfate metabolism underlying the vulnerability of pTau (but not Aβ) accumulation and neurodegeneration in human AD brain tissue (Grothe et al. 2018). Our lab also reported striking differences in CS-GAG sulfation patterns from the middle frontal gyrus of human postmortem AD brain tissue, changes that positively correlated to the degree of pTau accumulation (Logsdon et al. 2022). Although there are similarities between pTau-associated CS-GAG compositional changes in human AD brain and 9m-PS19 mice, we note some critical differences between these two datasets. In our human cohort, we observed a stepwise increase in cortical 6S-CS isomer expression from non-demented Braak 0-II to non-demented Braak III/IV subjects, as well as a second increase from non-demented Braak III/IV to demented Braak V/VI subjects (Logsdon et al. 2022). This two-step increase suggests that the elevation in brain 6S-CS isomer occurs prior to the development of AD clinicopathology in humans, which is then further elevated in the demented brain. However, we did not observe the same changes in PS19 mice. Specifically, we report no discernable differences in 6S-CS isomer expression in the presence of low pTau (6m-PS19 mice), or robust pTau accumulation (4m-Tg2652 mice) (Supplemental Table 5). We interpret these results to suggest that whereas increased 6S-CS may drive the appearance of pTau in humans, it does not appear necessary for pTau expression in PS19 or Tg2652 mice.
Meanwhile, we did observe an increase in cortical 2S6S-CS isomer expression unique to the demented Braak V/VI patient population (Logsdon et al. 2022), implying this isomer is upregulated only after the appearance of pTau in AD brain tissue. This finding mirrors the increase in 2S6S-CS abundance observed in 9m-PS19 mice (Fig. 4), which also increases after considerable pTau accumulation and gliosis at this age. We also recently reported a significant increase in the 2S6S-CS isomer after gliosis-associated traumatic brain injury in mice (Alonge et al. 2021), further support a novel link between gliosis and elevated 2S6S-CS isomer expression. The increase in hippocampal 2S6S-CS in 9m-PS19 mice tightly correlated to increased TUNEL immunoreactivity and neurodegeneration in the DG (Fig. 5). This finding is of notable importance, since adult neurogenesis within the DG is well-established in rodents (Altman & Das 1965). Future studies are thus warranted to determine whether the increase in TUNEL immunoreactivity associates with the loss of established neurons or newly regenerated neurons, potentially using doublecortin (DCX) immunostaining (Brown et al. 2003), as a first step towards understanding the influence of PNN glycan changes on neurodegeneration and neurogenesis in the DG.
Since the 2S6S-CS isomer is a critical variant shown to exhibit neuroregenerative properties, including increasing neurite bearing neurons (Clement et al. 1999), process length (Clement et al. 1999), and outgrowth in hippocampal cell cultures (Shida et al. 2019), we predict that the increase in 2S6S-CS isomer in demented Braak V/VI patients and 9m-PS19 mice may represent a biological response to pTau-associated neurodegeneration. Drug treatments that increase the 2S6S-CS isomer may thus be of therapeutic interest for the treatment of neurodegeneration in AD, and fibroblast growth factor-1 (FGF-1) is once such therapy shown to influence both PNN abundance and composition. We recently showed that central administration of FGF-1 into rats 1) induces >2x increase in WFA+ PNNs after 24h, and 2) significantly increases the relative abundance of the 2S6S-CS isomer (Alonge et al. 2020). Studies that investigate the influence of FGF-1 treatment on PNN stability and composition in mouse models of tauopathy are currently ongoing.
There are several limitations of the interpretation of this study worth discussing. We acknowledge that our CS-GAG analyses from whole hippocampal brain tissue may not reflect intraregional and cell-specific changes within this brain region. If neurodegeneration is a key driver for the CS biosynthesis flux from 6S-CS to 2S6S-CS isomer expression (illustrated in Fig. 4C), then these changes may be limited to specific hippocampal subregions or cellular populations (e.g., DG). Indeed, we recently reported striking regional differences in brain CS-GAG composition throughout wt mouse brain tissue (Scarlett et al. 2022), thus providing strong evidence that CS-GAG patterning is not universal throughout the brain. Follow-up studies using laser capture microdissection (LMD) and/or matrix assisted laser desorption ionization (MALDI) imaging mass spectrometry may be useful in determining spatial differences in CS isomer expression throughout the hippocampus. A second limitation to this study is that our bulk LC-MS/MS technique may also be detecting changes in CS sulfation patterns of non-PNN matrices in the brain, including the interstitial matrix and so-called ‘glial-scarring’ deposits. Regardless of submatrix expression, we predict that the increase in 2S6S-CS isomer may for pericellular matrix coats and interact similarly with enmeshed neurons, thus providing important clues as to how the brain may attempt to regenerate neural circuitry in response to neurodegeneration.
Finally, we would like to comment on the unequal binding specificity of WFA to CS-GAGs. A recent study published by Nadanaka et al. showed WFA lectin preferentially recognizes and binds to non-sulfated 0S-CS tetrasaccharide units compared to sulfated isomer variants (Nadanaka et al. 2020). WFA binding affinity is highly influenced by the abundance of 0S-CS isomer units incorporated into the PNN CS-GAGs, and as such, should not be thought of as universal, non-specific histological label for PNN CS-GAGs (Scarlett et al. 2022; Härtig et al. 2022). Since we did not observe significant differences in the 0S-CS isomer in 9m-PS19 mice compared to wt controls (Supplemental Table 5), we do not predict WFA binding affinity differences will contribute to the histochemical loss of WFA+ PNNs (Fig. 1) in these mice.
Overall, this study investigates the influence of pTau on hippocampal PNNs in mouse models of tauopathy. We revealed that PS19 mice exhibit an age-associated loss of WFA+ PNNs, in the absent of changes in PNN core protein abundance, and that this effect was exacerbated in the presence of gliosis and neurodegeneration. Our findings also identify 6-month-old PS19 mice as a critical inflection point for the loss of WFA+ PNNs and suggest treatment prior to, or in parallel with, the induction of neuroinflammation may represent a beneficial therapeutic window for the treatment of AD. Future and ongoing studies investigating the effects of anti-inflammatory drug therapies (e.g., diclofenac) and PNN stabilizers (e.g., FGF-1) on PNN glycosylation are of intense interest to the AD community. In conclusion, this research provides novel insights into the dynamics between PNNs, pTau, and gliosis, and presents a rationale to test potential AD therapies through mechanisms involving PNN glycosylation and CS-GAG associated neuroregeneration.
Supplementary Material
FUNDING AND ACKNOWLEDGEMENTS
The authors are grateful for Linda Robinson for outstanding mouse colony management and Kathryn Cui and Nicole Richardson for histology technical assistance. Histochemical work was performed by the National Institute of Diabetes and Digestive and Kidney Diseases (NIH-NIDDK) sponsored Diabetes Research Center (DRC) Cellular and Molecular Imaging Core P30 DK017047. Mass spectrometry work was supported by University of Washington School of Pharmacy’s Mass Spectrometry Center (MSC). This work was also supported by the National Institute on Aging (NIH-NIA) grants P30 AG066509 (KMA), R21 AG074152 (KMA), R01 AG066211 (BCK), and T32 AG052354 (AFL). Additional funding includes the National Institute of Allergy and Infectious Diseases (NIAID) grant DP2 AI171150 (KMA) and the Department of Defense (DoD) grant AZ210089 (KMA).
ABBREVIATIONS
- AD
Alzheimer’s disease
- Aβ
beta amyloid
- ACAN
aggrecan
- ARIA-E
amyloid-related imaging abnormalities (cerebral edemas)
- ARIA-H
amyloid-related imaging abnormalities (hemorrhages)
- ChABC
chondroitinaseABC
- CS
chondroitin sulfate
- CS-GAG
chondroitin sulfate-glycosaminoglycan
- CSPG
chondroitin sulfate proteoglycan
- DG
dentate gyrus
- ECM
extracellular matrix
- FGF-1
fibroblast growth factor-1
- GFAP
glial fibrillary acidic protein
- Iba1
ionized calcium-binding adapter molecule1
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LMD
laser capture microdissection
- mabs
monoclonal antibodies
- MALDI
matrix-assisted laser desorption ionization
- MFI
mean fluorescence intensity
- MRM
multiple reaction monitoring
- NDS
normal donkey serum
- NFTs
neurofibrillary tangles
- OCT
optimal cutting temperature
- PFA
paraformaldehyde
- PBS
phosphate buffered saline
- PBS-T
0.1 M PBS + 0.05% Triton X-100
- PNNs
perineuronal nets
- PS19
tau P301S
- pTau
phosphorylated tau
- RT
room temperature
- SEM
standard error of the mean
- Tg2652
tau4RTg2652
- TBS
tris buffered saline
- TBS-T
0.1 M TBS + 0.05% Triton X-100
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- UPLC
ultra-performance liquid chromatographic
- VVA
vicia villosa agglutinin
- WFA
wisteria floribunda agglutinin
- WT
wild type
Footnotes
CONFLICT OF INTEREST
AFL reports affiliation with the University of Washington and is also the Scientific Director for Psycheceutical, Inc. The research presented in this manuscript was conducted at the University of Washington independently and prior to affiliating with Psycheceutical, Inc., and there is no overlap between the activities of Psycheceutical, Inc and the work performed in this study. All other authors declare no conflict of interest.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Alonge KM, Herbert MJ, Yagi M, Cook DG, Banks WA and Logsdon AF (2021) Changes in Brain Matrix Glycan Sulfation Associate With Reactive Gliosis and Motor Coordination in Mice With Head Trauma. Front Behav Neurosci 15, 745288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge KM, Logsdon AF, Murphree TA, Banks WA, Keene CD, Edgar JS, Whittington D, Schwartz MW and Guttman M (2019) Quantitative analysis of chondroitin sulfate disaccharides from human and rodent fixed brain tissue by electrospray ionization-tandem mass spectrometry. Glycobiology 29, 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge KM, Mirzadeh Z, Scarlett JM et al. (2020) Hypothalamic perineuronal net assembly is required for sustained diabetes remission induced by fibroblast growth factor 1 in rats. Nat Metab 2, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J and Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124, 319–335. [DOI] [PubMed] [Google Scholar]
- Baig S, Wilcock GK and Love S (2005) Loss of perineuronal net N-acetylgalactosamine in Alzheimer’s disease. Acta Neuropathol 110, 393–401. [DOI] [PubMed] [Google Scholar]
- Braak H and Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Brown JM, Xia J, Zhuang B et al. (2012) A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc Natl Acad Sci U S A 109, 4768–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L and Kuhn HG (2003) Transient expression of doublecortin during adult neurogenesis. J Comp Neurol 467, 1–10. [DOI] [PubMed] [Google Scholar]
- Brückner G, Hausen D, Härtig W, Drlicek M, Arendt T and Brauer K (1999) Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer’s disease. Neuroscience 92, 791–805. [DOI] [PubMed] [Google Scholar]
- Budd Haeberlein S, Aisen PS, Barkhof F et al. (2022) Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J Prev Alzheimers Dis 9, 197–210. [DOI] [PubMed] [Google Scholar]
- Busche MA and Konnerth A (2015) Neuronal hyperactivity--A key defect in Alzheimer’s disease? Bioessays 37, 624–632. [DOI] [PubMed] [Google Scholar]
- Clement AM, Sugahara K and Faissner A (1999) Chondroitin sulfate E promotes neurite outgrowth of rat embryonic day 18 hippocampal neurons. Neuroscience Letters 269, 125–128. [DOI] [PubMed] [Google Scholar]
- Crapser JD, Arreola MA, Tsourmas KI and Green KN (2021) Microglia as hackers of the matrix: sculpting synapses and the extracellular space. Cellular & Molecular Immunology 18, 2472–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapser JD, Spangenberg EE, Barahona RA, Arreola MA, Hohsfield LA and Green KN (2020) Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. eBioMedicine 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djerbal L, Lortat-Jacob H and Kwok J (2017) Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj J 34, 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Fyhn M, Jendelova P, Kwok JCF, Ruzicka J and Sorg BA (2022) The extracellular matrix and perineuronal nets in memory. Molecular Psychiatry 27, 3192–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Sepulcre J, Gonzalez-Escamilla G, Jelistratova I, Schöll M, Hansson O and Teipel SJ (2018) Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 141, 2755–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtig W, Meinicke A, Michalski D, Schob S and Jäger C (2022) Update on Perineuronal Net Staining With Wisteria floribunda Agglutinin (WFA). Front Integr Neurosci 16, 851988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell MD, Bailey LA, Cozart MA, Gannon BM and Gottschall PE (2015) Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol Commun 3, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Emson PC and Mountjoy CQ (1989) Vicia villosa lectin-positive neurones in human cerebral cortex. Loss in Alzheimer-type dementia. Brain research 498, 170–174. [DOI] [PubMed] [Google Scholar]
- Kudo T, Takuwa H, Takahashi M et al. (2023) Selective dysfunction of fast-spiking inhibitory interneurons and disruption of perineuronal nets in a tauopathy mouse model. iScience 26, 106342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai D, Morawski M, Négyessy L et al. (2013) Neurochemical mapping of the human hippocampus reveals perisynaptic matrix around functional synapses in Alzheimer’s disease. Acta Neuropathol 125, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley JW, Reisine T and Finkbeiner S (2019) Cell death assays for neurodegenerative disease drug discovery. Expert Opin Drug Discov 14, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Francis KL, Richardson NE et al. (2022) Decoding perineuronal net glycan sulfation patterns in the Alzheimer’s disease brain. Alzheimers Dement 18, 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Kelly GM, Zerillo CA, Gray G, Tiemeyer M and Hockfield S (2002) Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J Neurosci 22, 7536–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S and Kitagawa H (2016) Chondroitin 6-Sulfation Regulates Perineuronal Net Formation by Controlling the Stability of Aggrecan. Neural Plast 2016, 1305801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S, Komatsu Y, Yoshimura Y, Taya C and Kitagawa H (2012) Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat Neurosci 15, 414–422, S411–412. [DOI] [PubMed] [Google Scholar]
- Morawski M, Brückner G, Jäger C, Seeger G and Arendt T (2010a) Neurons associated with aggrecan-based perineuronal nets are protected against tau pathology in subcortical regions in Alzheimer’s disease. Neuroscience 169, 1347–1363. [DOI] [PubMed] [Google Scholar]
- Morawski M, Bruckner G, Jager C, Seeger G, Matthews RT and Arendt T (2012) Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain pathology (Zurich, Switzerland) 22, 547–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski M, Pavlica S, Seeger G, Grosche J, Kouznetsova E, Schliebs R, Brückner G and Arendt T (2010b) Perineuronal nets are largely unaffected in Alzheimer model Tg2576 mice. Neurobiology of Aging 31, 1254–1256. [DOI] [PubMed] [Google Scholar]
- Nadanaka S, Miyata S, Yaqiang B, Tamura J-I, Habuchi O and Kitagawa H (2020) Reconsideration of the Semaphorin-3A Binding Motif Found in Chondroitin Sulfate Using Galnac4s-6st-Knockout Mice. Biomolecules 10, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C and Hrabětová S (2017) Brain Extracellular Space: The Final Frontier of Neuroscience. Biophys J 113, 2133–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C and Syková E (1998) Extracellular space structure revealed by diffusion analysis. Trends in Neurosciences 21, 207–215. [DOI] [PubMed] [Google Scholar]
- Rivers-Auty J, Mather AE, Peters R, Lawrence CB and Brough D (2020) Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun 2, fcaa109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett JM, Hu SJ and Alonge KM (2022) The “Loss” of Perineuronal Nets in Alzheimer’s Disease: Missing or Hiding in Plain Sight? Frontiers in Integrative Neuroscience 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Chu F, Zhu F and Zhu J (2022) Impact of Anti-amyloid-β Monoclonal Antibodies on the Pathology and Clinical Profile of Alzheimer’s Disease: A Focus on Aducanumab and Lecanemab. Front Aging Neurosci 14, 870517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shida M, Mikami T, Tamura JI and Kitagawa H (2019) Chondroitin sulfate-D promotes neurite outgrowth by acting as an extracellular ligand for neuronal integrin αVβ3. Biochim Biophys Acta Gen Subj 1863, 1319–1331. [DOI] [PubMed] [Google Scholar]
- Sims JR, Zimmer JA, Evans CD et al. (2023) Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaker ML, Harkness JH and Sorg BA (2016) A standardized and automated method of perineuronal net analysis using Wisteria floribunda agglutinin staining intensity. IBRO Reports 1, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos KE, Mayer MI, Takács VT et al. (2020) Amyloid β induces interneuron-specific changes in the hippocampus of APPNL-F mice. PloS one 15, e0233700–e0233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopschinski BE, Weideman RA, McMahan D, Jacob DA, Little BB, Chiang HS, Saez Calveras N and Stuve O (2023) Microglia as a cellular target of diclofenac therapy in Alzheimer’s disease. Ther Adv Neurol Disord 16, 17562864231156674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuve O, Weideman RA, McMahan DM, Jacob DA and Little BB (2020) Diclofenac reduces the risk of Alzheimer’s disease: a pilot analysis of NSAIDs in two US veteran populations. Ther Adv Neurol Disord 13, 1756286420935676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA and Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580. [DOI] [PubMed] [Google Scholar]
- van Dyck CH, Swanson CJ, Aisen P et al. (2023) Lecanemab in Early Alzheimer’s Disease. N Engl J Med 388, 9–21. [DOI] [PubMed] [Google Scholar]
- Végh MJ, Heldring CM, Kamphuis W et al. (2014) Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun 2, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JM, McMillan PJ, Hawk M et al. (2015) High copy wildtype human 1N4R tau expression promotes early pathological tauopathy accompanied by cognitive deficits without progressive neurofibrillary degeneration. Acta Neuropathol Commun 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Cacquevel M, Saksida LM et al. (2015) Perineuronal net digestion with chondroitinase restores memory in mice with tau pathology. Exp Neurol 265, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Gigout S, Molinaro A et al. (2021) Chondroitin 6-sulphate is required for neuroplasticity and memory in ageing. Mol Psychiatry 26, 5658–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B et al. (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.