

Abstract
Organisms exhibit extensive variation in ecological niche breadth, from very narrow (specialists) to very broad (generalists). Two general paradigms have been proposed to explain this variation: trade-offs between performance efficiency and breadth; and the joint influence of extrinsic (environmental) and intrinsic (genomic) factors. We assembled genomic, metabolic, and ecological data from nearly all known species of the ancient fungal subphylum Saccharomycotina (1,154 yeast strains from 1,051 species), grown in 24 different environmental conditions, to examine niche breadth evolution. We found that large differences in the breadth of carbon utilization traits between yeasts stem from intrinsic differences in genes encoding specific metabolic pathways, but limited evidence for trade-offs. These comprehensive data argue that intrinsic factors shape niche breadth variation in microbes.
One-Sentence Summary:
A nearly complete genomic and phenotypic catalog of the yeast subphylum illuminates the evolution of metabolic breadth.
Introduction
The ecological niche is a fundamental concept in ecology and evolutionary biology that explains the diversity and resource use of organisms through space and time. Species with broad niche breadths are defined as generalists, while those with narrow ones are specialists. There are many biotic and abiotic dimensions of the niche that can and do vary among organisms (1–3), begging the question: What factors contribute to niche breadth variation?
Two broad paradigms have been offered as answers across a variety of taxa. The first paradigm postulates that both niche generalism and specialism are governed by trade-offs between performance efficiency and niche breadth (4–9). In the context of metabolic niche breadth, selection for increased efficiency in utilizing a specific food source will be coupled to selection against utilizing other food sources and vice versa. Over the long-term, such selection produces generalists that utilize more substrates less efficiently and specialists that utilize fewer substrates more efficiently. Consistent with these expectations, selection for specialization in using a single food source in replicate populations of the bacterium Escherichia coli was coupled to a reduction in their ability to catabolize other food sources (10).
The second paradigm postulates that generalist and specialist phenotypes are the outcome of the joint influence of diverse extrinsic (environmental) and intrinsic (genomic) factors (11–16). Here, generalists and specialists are shaped by the environments in which they occur and the evolvability of their metabolic pathways, rather than by trade-offs. These specific conditions will result in a unifying set of extrinsic and intrinsic features that govern the evolution of generalist and specialist phenotypes.
Extrinsic factors are the environments in which species live. They can vary with respect to numerous abiotic and biotic factors, such as spatial and temporal heterogeneity, temperature, and carbon and nitrogen availability. For example, carbon sources have been shown to be limited within endothermic hosts (17, 18); temperatures and soil moisture can vary between woodland and meadow habitats due to canopy cover (19); and the availability of nitrogen sources (20, 21), carbon sources (22–24), and growth-inhibiting specialized metabolites can differ due to the activities of other organisms in the environment (25, 26). Variation in one or more of these extrinsic factors could exert selective pressure on traits, resulting in generalism and specialism (27).
Intrinsic factors that may influence niche breadth include the evolution of promiscuous enzymes responsible for the utilization of multiple resources (17, 28–31), as well as overlapping biochemical, developmental, and genetic pathways (15, 16). For example, yeast MAL and IMA genes are promiscuous enzymes associated with the utilization of multiple carbon sources in yeasts; that is, they can increase niche breadth by enabling broader consumption (17, 28). Conversely, gene loss due to drift or relaxed selection, which is likely in environments with lower nutrient diversity, could lead to narrower niche breadths (32). The diversity of traits and the genes that control them leads to the hypothesis that niche breadth variation may reflect the interplay between evolutionary and ecological forces acting on intrinsic factors.
The subphylum Saccharomycotina (phylum Ascomycota, kingdom Fungi), which includes the baker’s yeast Saccharomyces cerevisiae, the opportunistic pathogen Candida albicans, and the oleochemical cell factory Yarrowia lipolytica, exhibits extensive ecological, genomic, and metabolic diversity. Thus, it is a superb system for testing paradigms for the evolution of metabolic niche breadth (Fig. 1). The genomes of Saccharomycotina species, commonly referred to as yeasts, are highly diverse; levels of gene sequence divergence across yeasts are comparable to levels observed across plants and animals, and the subphylum also harbors considerable variation in gene content, including metabolic genes (28). In addition, extensive experimental work in model yeasts, such as S. cerevisiae (33) and C. albicans (34), provides validated functional genetic information.
Figure 1: Yeasts are morphologically, ecologically, and metabolically diverse.
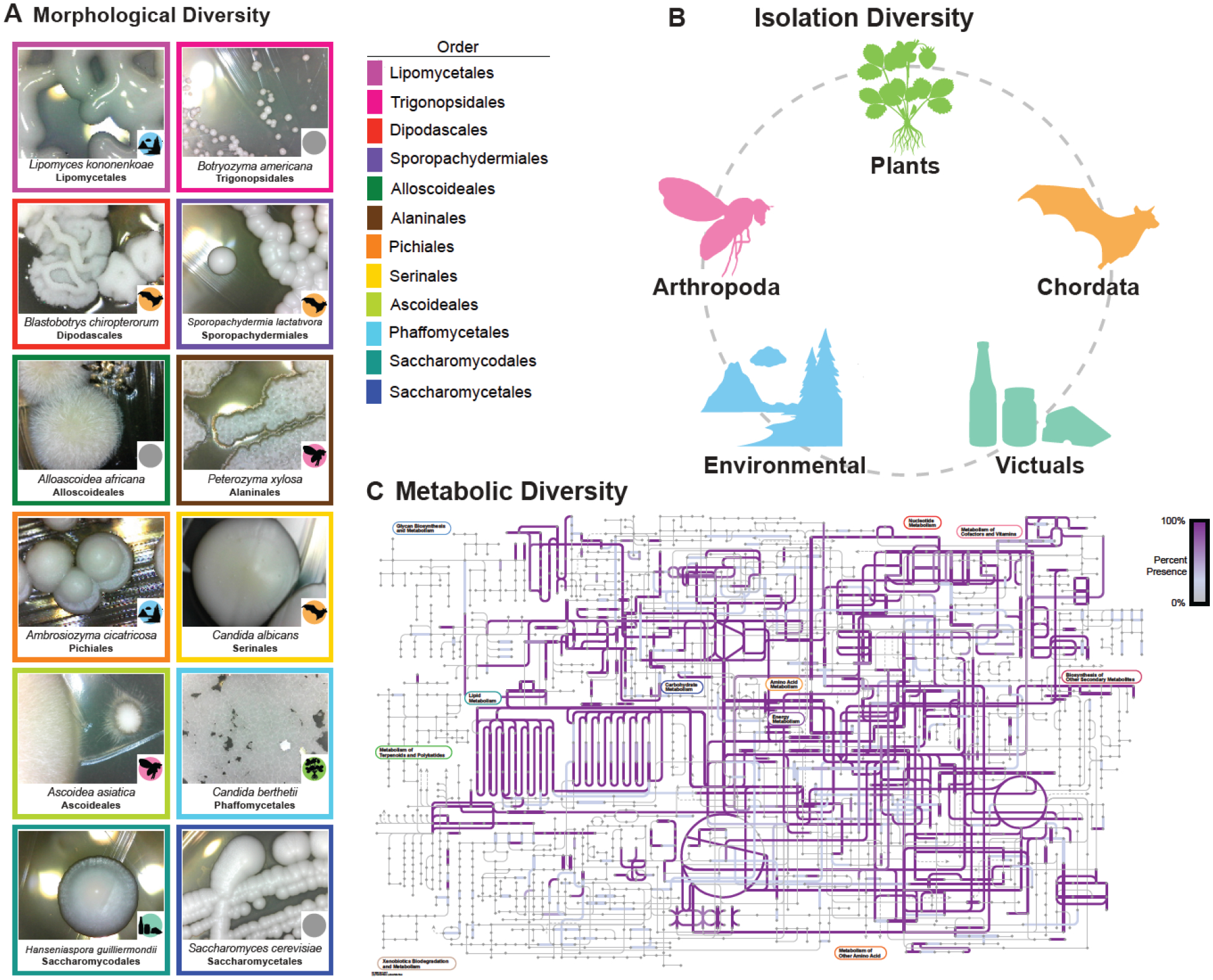
A. Images of yeasts from different orders. The color of the box surrounding the image indicates the species’ order. The color of the circle in the bottom right-hand corner of the image represents the isolation environment for the strain of the species sequenced and phenotyped during this study. Yeast colonies are morphologically diverse; they can vary in shape, color, size, dullness, etc.
B. Yeasts have been isolated from every biome and continent. Strains studied were found on plants, animals, in soil, and many other environments. Strain-level isolation data were placed into an ecological ontology to allow for identification of yeasts that shared higher-level ontological classes.
C. Yeasts are metabolically diverse. The image represents the KOs present across Saccharomycotina metabolic networks. Any pathway that is highlighted in purple is present across a subset of yeasts; the saturation of the purple represents the proportion of yeasts with the pathway.
Yeast growth profiles have been characterized across many carbon and nitrogen sources and environmental conditions (e.g., temperature), and they are highly variable across species (17, 28, 35). This phenotypic diversity is coupled to their ecological diversity. Yeasts are found in almost every biome on a wide array of substrates, and the isolation environments (defined as the specific environmental location a strain was originally isolated) of these yeasts are associated with specific phenotypic traits. For example, both glucose and sucrose fermentation are positively associated with living on fruits, fermented substrates, and juices (17), particularly among multiple yeast genera that have been linked to wine production and food spoilage (17, 36, 37). Opportunistic fungal pathogens have also evolved metabolic strategies that allow them to colonize the complex ecosystem of the human body, where carbon availability varies spatially and temporally (17, 38, 39). This treasure trove of genomic, metabolic, and environmental diversity across a subphylum makes Saccharomycotina an attractive and highly tractable system for studying niche breadth evolution.
To gain insight into the factors that contribute to metabolic niche breadth variation, we quantified variation in genome content, isolation environment, and carbon and nitrogen metabolism for 1,154 yeast strains, which represent nearly all known species in the subphylum Saccharomycotina. This dataset enabled us to evaluate the evidence for the two niche breadth evolution paradigms (trade-offs versus underlying intrinsic and extrinsic factors) across species with broad (generalists) and narrow (specialists) carbon niche breadths. Our evolutionary, machine learning, and network analyses uncovered a unifying set of intrinsic factors among generalists that were largely absent in specialists, and pinpointed specific genetic differences between generalists and specialists, including novel associations between carbon generalism and specific metabolic pathways. In contrast, we found limited evidence for trade-offs between carbon generalism and growth rate. Through ancestral trait reconstruction and coevolution analyses, we further demonstrated that generalists were more likely to have retained or gained traits, whereas specialists repeatedly arose through pervasive gene and trait loss. The genomic, metabolic, evolutionary, and ecological data for nearly all known species of the 400-million-year-old yeast subphylum Saccharomycotina provided here, coupled with the availability of multiple genetic models in the subphylum, present an inimitable resource and framework for linking genomic variation to phenotypic and ecological variation.
A genomic, evolutionary, and metabolic portrait of Saccharomycotina
We sequenced and assembled 953 new genomes in this study and combined them with 140 genomes previously sequenced by the Y1000+ Project (40) and 61 publicly available genomes (data S1A). Our dataset contained 1,154 genomes from 1,051 species, including 1,037 taxonomic type (i.e., ex-type) strains. Multiple strains were sequenced from 41 species, including a total of 19 recognized varieties distributed across nine species (i.e., two to three varieties per species). Sixty-one of the strains whose genomes were sequenced could not be assigned to any of the known species; thus, they are candidates for new species. The genomic dataset spans 96 yeast genera, which is about 90% of currently described genera (41). Excluded genera were typically those for which no living culture was available or those described after our last round of genome sequencing in February 2021. Our genome sequencing added between 1 and 336 species to each order, most notably expanding the order Serinales (previously major clade CUG-Ser1), which contains the human pathogens C. albicans and Candida auris, from 94 genomes to 430. All genome assemblies totaled ~15 billion base pairs. The assemblies had a mean N50 of 387.5 Kb, which was comparable to our previous smaller-scale dataset of 332 genomes (417.2 kb) (fig. S1A & data S1A) (28). All genomes were annotated to identify putative coding sequences. On average, 5,908 +/− 1069 (s.d.) protein-coding sequences were identified per genome with a range from 3,775 (Starmerella lactis-condensi) to 20,704 (Magnusiomyces magnusii) (fig. S1B) (42). Functional annotations were conducted using Kyoto Encyclopedia of Genes and Genomes (KEGG) and InterPro. GC content (subphylum mean = 41.1% +/− 6.61%) ranged from 23.9% (Candida bohioensis) to 66.8% (Candida pseudocylindracea), and genome size (subphylum mean = 13.2Mb +/− 3.5Mb) ranged from 7.2Mb (Starmerella lactis-condensi) to 41.3Mb (Magnusiomyces magnusii) (fig. S1C–D & data S1A). Of the 1,154 yeast genomes, 1,000 (~87%) had ≥ 90% of the 2,137 predefined single-copy orthologs defined by OrthoDB v10 (data S1A & data S1B) (43, 44).
At least three independent nuclear codon reassignments are known to have occurred during the evolution of the subphylum (45). Given the large number of newly added genomes, we inferred codon tables and tRNA genes to confirm the known reassignments and test for potential new reassignments (data S2). These results were consistent with the previously observed codon reassignments. Notably, genomes of the order Ascoideales had a diversity of tRNAs with CAG anticodons predicted to decode CUG codons, which is consistent with previous findings that these yeasts may stochastically decode CUG as both leucine and serine (46).
To infer the genome-scale phylogeny of the Saccharomycotina, we used 1,403 orthologous groups (OGs) from 1,154 Saccharomycotina genomes and 21 outgroups. Nearly all internodes in both concatenation-based (1,136/1,153; 99%) and coalescent-based (1,123/1,153; 97%) phylogenies received strong (≥ 95%) support (Fig. 2 and fig. S2 and fig. S3). The two phylogenies were highly congruent, with only 60/1,153 (5%) conflicting internodes (fig. S3). Moreover, relationships among the 12 recently circumscribed taxonomic orders (41) (previously major clades) were congruent with previous studies (28, 47, 48), including the placement of the Ascoideales (previously CUG-Ser2) and Alaninales (previously CUG-Ala).
Figure 2: Yeast traits are widely distributed across the phylogeny.
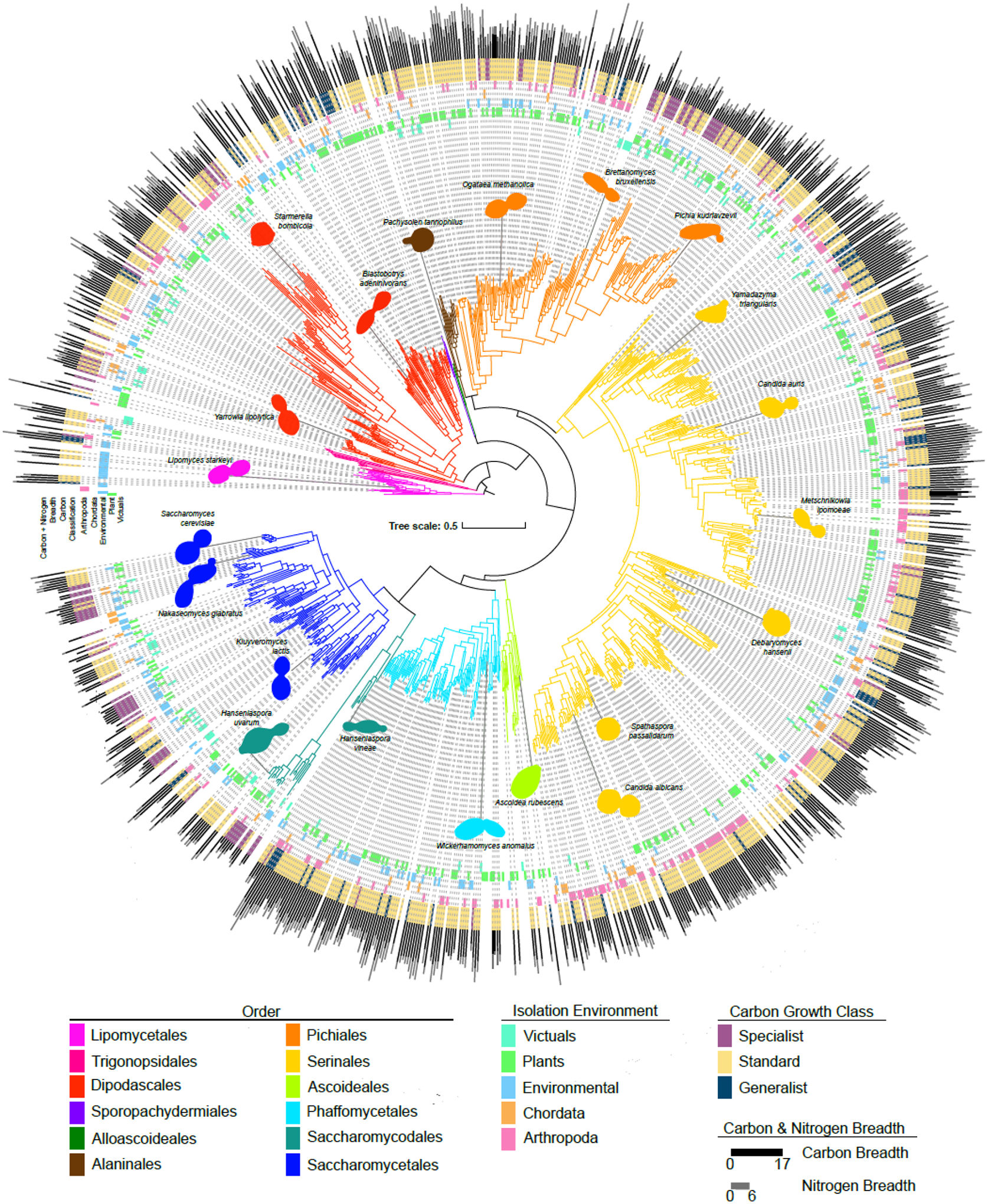
The phylogeny of 1,154 yeasts and fungal outgroups built from 1,403 orthologous groups of genes. Branches are colored according to their taxonomic assignment to an order of Saccharomycotina (41). The innermost rings are colored by the top-level type of isolation environment in which each specific strain was isolated. The purple, yellow, and blue ring identifies the carbon growth classification for each strain. This classification is based on the carbon niche breadth, which is represented by the bar graph on the exterior of the tree, along with nitrogen breadth. All traits illustrated (isolation environment, carbon growth class, nitrogen breadth, and carbon niche breadth) are widely distributed across the tree; no order has one trait exclusively.
To examine the evolution of metabolic niche breadth across Saccharomycotina, we quantified the growth rates of 853 yeast strains on 18 carbon sources, 6 nitrogen sources, and a no-carbon control (data S3). We found that yeasts displayed variation in growth rates across carbon (fig. S4A) and nitrogen sources (fig. S4B); on average, each yeast strain could metabolize eight carbon (Fig. 3A) and two nitrogen sources (fig. S5). Comparison of growth rates on different carbon sources revealed that 65.22% of yeasts (n = 557) grew fastest on glucose, while the remaining 34.78% (n = 297) grew faster on another carbon source (fig. S6). Mannose, an epimer of glucose not typically tested in yeast growth experiments, was the carbon source on which yeasts grew fastest, on average, after glucose (n = 112). We also found that 77 yeasts grew faster on fructose than glucose, including cases where their maximum growth rate was on a third carbon source. Several of these yeasts (n = 7) were in Dipodascales, which contains many known fructophilic yeasts (49). The ability to grow faster on fructose was independently verified in a second lab on a subset of yeasts (data S4).
Figure 3: Carbon specialists and generalists differ in nitrogen breadth, growth rate, and evolutionary history.
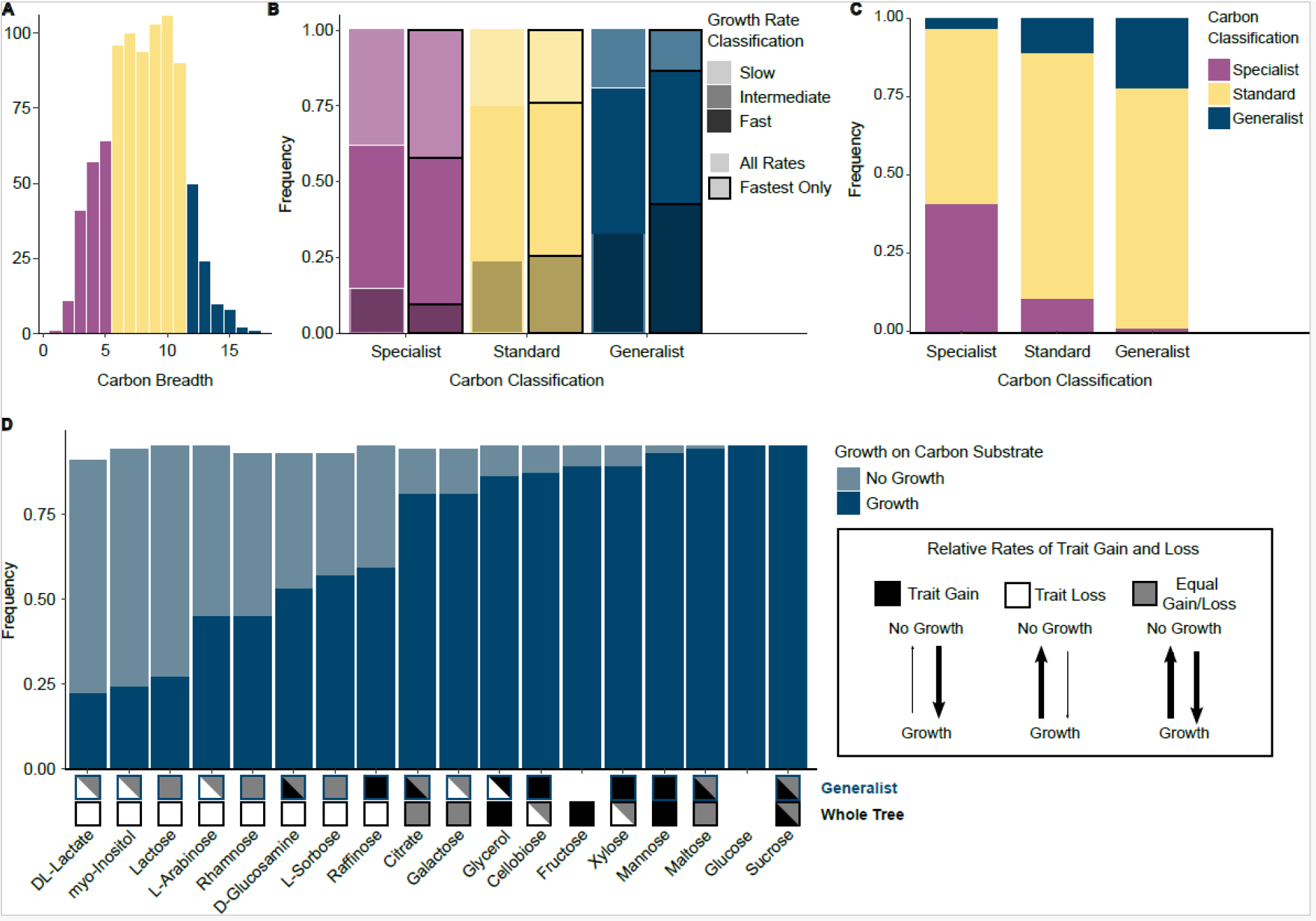
A. Histogram of carbon niche breadth across yeasts (n = 853). The colors of the bars represent the ranges for the different carbon classifications. Metabolic classifications were determined by permuting the binary carbon growth matrix (n = 1000 permutations). To determine the metabolic strategy of a yeast, we calculated the observed and expected (permuted) breadth for each yeast and calculated the binomial confidence intervals to determine significant differences in breadth. Generalists had a significantly larger carbon niche breadth than expected by chance, and specialists had a significantly smaller carbon niche breadth. If a yeast was not classified as either a generalist or a specialist, it was classified as standard.
B. The growth rates for each yeast on each of the 18 carbon sources were categorized as slow (bottom 25%), intermediate (median 50%), or fast (top 25%) using either all the rates per yeast (white outline) or only the highest rate per yeast (black outline). Carbon generalists had the highest proportion of fast growth rates (33% all rates, 43% fastest rates), while specialists had the smallest proportion (15% all rates, 9% fastest rates) The inverse was also true, with carbon generalists having the smallest proportion of slow growth rates (19% all rates, 14% fastest rates) and carbon specialists having the highest proportion of slow growth rates (38% all rates, 42% fastest rates).
C. Stacked bar graph of carbon metabolic strategies within each nitrogen metabolic strategy.
D. Carbon generalists shared many of the same growth traits: 10 out of 18 growth traits were found in more than 75% of generalists. Many of the carbon sources had different evolutionary trends in a generalist background as compared to across the whole tree. Three different evolutionary models are shown: trait gain (black), trait loss (white), and equal rates of trait gain and loss (gray). No box indicates that the trait was not co-evolving with background or across the tree. More than one evolutionary model is shown in cases where the reverse jump model spent 75% or less of the time on a single model. For example, the model testing correlated evolution between growth on D-glucosamine and generalist carbon classification reported a model string with a greater rate of gain in 55% of the run and a model string with equal rates of gain and loss in 29% of the run; therefore, we reported both the trait gain and equal gain/loss model in the generalist analysis.
A lack of evidence for trade-offs between carbon niche breadth and growth rates
We statistically classified yeasts into three categories for both carbon and nitrogen utilization niche breadths: specialist, standard, and generalist (data S3). We found that, for both carbon and nitrogen metabolism, most yeasts were classified as standard yeasts (i.e., yeasts that did not fall into the extremes for carbon niche breadth) (76.0%: 648/853 and 78.4%: 669/853, respectively) (data S3 & Fig. 3A). Of the remaining 24.0% (n = 205/853), 53.7% (n = 110/205) were specialists, and 46.3% (n = 95/205) were generalists for carbon sources (Fig. 3A). The median numbers of carbon sources used by specialist, standard, and generalist yeasts were four, eight, and twelve, respectively. Carbon generalists and specialists were widely distributed across the subphylum (Fig. 2), and all orders with more than 15 phenotyped strains (n = 8) featured both generalists and specialists. However, the relative proportion of generalists and specialists within orders varied greatly. For example, the order Saccharomycetales (n=82) had 3 generalists and 33 specialists, while the order Serinales (n= 347) had 53 generalists and 9 specialists. This result suggests that yeast orders exhibit distinct eco-evolutionary trajectories.
First, we tested for a trade-off between growth rate and carbon niche breadth by asking if specialists had a growth rate advantage over other yeasts in some conditions. We compared all growth rates within each carbon source by classifying growth into three categories: slow (growth rate in the lower quartile), intermediate, and fast (growth rate in the upper quartile.) We found a statistically significant interaction between carbon classification and growth rate (p-value < 2.2e-16); specialists were more often slow growers (38%: 146/381 growth rates) than fast growers (15%: 54/381), whereas generalists were more often fast growers (33%: 403/1,222 growth rates) than slow growers (20%: 238/1,222 growth rates) (Fig. 3B). Moreover, there were fewer specialists than generalists in the fast category across all tested carbon sources (data S5). We also examined linear phylogenetically corrected correlations between growth rates and carbon niche breadth. We found that growth rates on five carbon sources were positively correlated with carbon niche breadth when accounting for phylogeny and multiple-testing correction (glucose p = 0.0028, mannose p = 0.0056, myo-inositol p = 0.0083, galactose p=0.0024, and fructose p = 0.0111: all slopes between 0.001 and 0.002) (table S1 and fig. S7A). No significant negative correlations were identified, which would have indicated that specialists were faster growers.
Second, we repeated these analyses using only the fastest growth rate for each yeast because specialists might outperform other yeasts only in the environment in which they are specialized. We found that the proportion of fast-growing specialists was 9% (10/107), a decrease from the 15% of fast-growing specialists found when we compared all growth rates across all substrates, while the proportion of fast-growing generalists was 43% (38/89), an increase from 33% (Fig. 3B). Thus, the strong interaction between carbon classification and growth rates persisted when only the fastest rates were considered (p-value = 7.8 ×10−11). In this case, carbon niche breadth was significantly and positively correlated with growth rates on glucose (p-value = 0.0002, slope = 0.002), sucrose (p-value =0.0032, slope = 0.001), and fructose (p-value=0.0062, slope = 0.001) after accounting for multiple testing and phylogeny (table S1 and fig. S7B).
A third analysis using the fastest growth rate for each specialist compared to all other growth rates yielded similar results (table S1 and fig. S7C). In this analysis, the growth rate for a carbon source included only specialists whose growth rate was highest on that carbon source and any growth rates for standard and generalist yeasts. Moreover, specialists were not the fastest-growing yeast in any of the carbon sources tested, including glucose. Our findings suggest that generalists grow faster on more substrates than specialists, including under conditions preferred by specialists.
We next tested whether there was a trade-off between carbon and nitrogen breadth. We found significantly fewer carbon generalists that were also nitrogen specialists (n = 1) and carbon specialists that were also nitrogen generalists (n = 2) than expected by chance (p-value = 3.26×10−14) (Fig. 3C). Moreover, trait-trait co-evolutionary analysis found that carbon generalists tended to also be nitrogen generalists (Bayes factor >2). Furthermore, our analyses of co-evolution between carbon and nitrogen generalism showed that nitrogen generalism arises almost exclusively in a genetic background of carbon generalism (i.e. in carbon generalism lineages; table S2). In other words, carbon generalism mainly arises before and may facilitate nitrogen generalism. Additionally, phylogenetic regression analysis showed a strong positive correlation between carbon and nitrogen niche breadth (reported p-value of 0.000, slope of correlation = 0.92; table S2). These results suggest that there is an evolutionarily conserved functional connection between carbon and nitrogen metabolism in yeasts. Consistent with our finding, it is well known that certain amino acids can serve as both a carbon and nitrogen source and, as such, are dually regulated by both carbon and nitrogen signaling systems (50, 51). Additionally, many metabolic pathways are known to be controlled by signals from other compounds or nutrients. In bacteria, nitrogen, sulfur, phosphorus, and iron metabolism can even be controlled by carbon metabolism (50, 52).
Our previous analysis of 332 yeasts identified a pervasive pattern of trait loss (28), which suggests that generalists have either retained carbon-acquisition traits over long evolutionary timescales or gained traits, unlike their non-generalist relatives. To test these hypotheses, we compared the relative rates of carbon trait gain or loss, either across all yeasts or specifically within generalist lineages, while taking phylogeny into account (Fig. 3D, table S3). For the eight carbon traits found in less than 75% of generalists, we identified a strong trend of trait loss across the entire phylogeny but some evidence of trait gain in the generalist background. Therefore, carbon generalists appear to have both gained and retained carbon traits that were otherwise lost broadly across the rest of the subphylum.
Intrinsic factors shape carbon niche breadth variation in yeasts
Given the extreme carbon niche breadths of generalists and specialists, we next tested whether these two groups have independent factors favoring generalist and specialist phenotypes. Extrinsic factors, such as carbon availability in an isolation environment, could shape variation in metabolic niche breadth. Similar environments, which are likely to share extrinsic factors, may favor the evolution of generalists or specialists. To explore the possibility that some environments contain extrinsic factors that shape carbon niche breadth, we identified the precise isolation environment for each possible yeast strain (1,088 total). We then grouped strains by similar environments using a formal hierarchical ontology of isolation environments. This ontology contained 1,597 classes (specific environments) (fig. S8, data S6). Environment classifications at the highest level of our ontology generally contained similar numbers of generalists and specialists: Arthropoda (24 generalists and 16 specialists), Chordata (7 and 8), plants (25 and 31), and food or drink (5 and 16). Furthermore, generalists and specialists shared environments. For example, Hyphopichia homilentoma (generalist) and Wickerhamomyces sydowiorum (specialist) were both isolated from tunnels of the wood-boring beetle Sinoxylon ruficorne in the red bushwillow Combretum apiculatus. Given the limited number of generalists and specialists within an environment and the fact that we only had a single environment per strain, we were unable to rigorously test for extrinsic factors that favor generalists or specialists. We anticipate that incorporation of improved characterizations of yeast habitats and the addition of isolation environment data into our formal ontology will enable future investigations of the environmental factors shaping carbon niche breadth evolution.
We next hypothesized that the genomes of generalists may contain a larger number of metabolic genes, which are intrinsic factors, than those of specialists. We found that both the total number of genes and the number of KEGG ortholog groups (KOs) were both positively and significantly associated with carbon niche breadth (Fig. 4A & fig. S10A–B). Strikingly, we found that, for every additional carbon source a yeast could metabolize, its genome contained, on average, an additional 36 genes and 2 KOs.
Figure 4: Generalist and specialist metabolism differs in expected and unexpected ways.
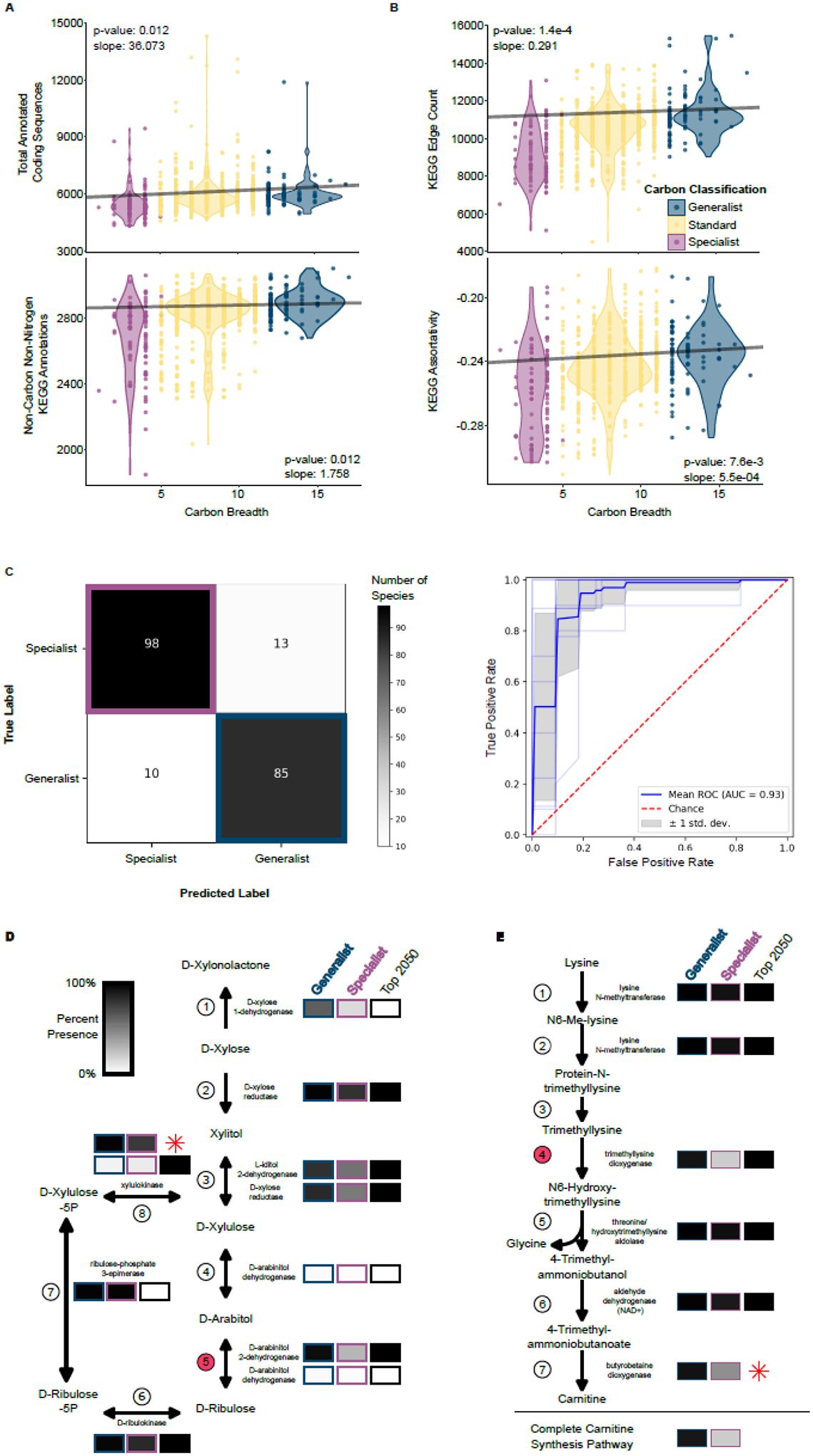
A. Total annotated coding sequences (top) and total number of annotated KEGG ortholog groups (KOs; bottom) are both positively and significantly correlated with carbon niche breadth using a Phylogenetic Generalized Least Squares (PGLS) analysis. One outlier with a predicted number of coding sequences is not visualized but was included in the analysis (Magnusiomyces magnusii, number of protein-coding genes = 20,704, carbon niche breadth = 9).
B. Two KEGG network statistics were significantly and positively correlated with carbon niche breadth when taking into account phylogenetic relatedness (PGLS). KEGG Edge Count (top) and KEGG Assortativity (bottom) were both elevated in carbon generalists.
C. Yeasts were classified into generalists and specialists using a machine learning algorithm trained on the KOs. The correct classification occurred in 88% of specialists and 89% of generalists. The ROC analysis suggests that both the sensitivity and specificity of our model is excellent (AUC=0.93).
D. Multiple reactions in the pentose and glucuronate interconversions pathway were important in classifying yeasts into generalists and specialists as determined by the leave-out analysis, which identified 2,050 informative KOs (black boxes.) Boxes are shaded as the percent of each carbon classification with at least one enzyme in that step of the reaction. The reaction with the third highest relative importance in the machine learning analysis is shown in Step 5 and is facilitated by D-arabinitol 2-dehydrogenase. Interestingly, experimental studies suggest that yeast D-arabinitol 2-dehydrogenase is also capable of completing the reaction in Step 4 (93). Step 8 was among the top features used in the machine learning analysis, despite the fact that KEGG only partially annotated this gene. The xylulokinase encoded by yeast XYL3 is well studied (58). Therefore, we re-annotated the XYL3 gene and have shown its relative abundance (red star).
E. The carnitine biosynthesis pathway includes multiple reactions that are important for classifying carbon generalists and specialists. The reaction in Step 4 had the fourth highest relative importance in the machine learning classification of carbon classification. Step 7 was not annotated by KEGG in any of our yeasts, but this step had been previously characterized in Candida albicans as being facilitated by the trimethyllysine dioxygenase enzyme encoded by BBH2 (64). We re-annotated BBH2 using this reference sequence and calculated the relative abundance in each carbon classification (red star). Finally, we determined the number of yeasts that could hypothetically complete the lysine to carnitine biosynthesis pathway.
Metabolic networks, including the carbon metabolism network, are more complex than just the total number of genes because they are highly interconnected due to shared enzymes and pathways. To examine whether metabolic network structure varied between generalists and specialists, we used KOs to build metabolic networks for all yeasts and tested for a correlation between carbon niche breadth and six common network properties that reflect biological complexity (Fig. 4B and fig. S10C–F, data S7) (53, 54). Relative to carbon specialists, carbon generalists had a higher edge-count, or more connections between nodes of the network (Fig. 4B) (55). Both carbon generalists and specialists had disassortative networks, or networks with high levels of connection between nodes with dissimilar properties, a property of all biological networks (56). However, relative to specialists, the generalist networks were less disassortative, or had more highly interconnected nodes (Fig. 4B). There were no significant correlations between carbon niche breadth and the other network properties (fig. S10C–F). Despite the extreme difference in carbon metabolism capabilities, carbon generalists and specialists had only slight differences in the size and shape of their global KEGG metabolic networks. These results suggest that generalist and specialist networks are overall similar in size and shape but differ in how they are wired.
We next investigated differences in the composition of generalist and specialist networks. Generalists and specialists largely showed similar compositions across KOs, but a small set of KOs was depleted (presence < 20%) in specialists and enriched (presence >85%) in generalists (table S4). Generalist-enriched KOs were related to nitrogen, fructose, mannose, and galactose metabolisms. Enrichment of these terms suggests that differences in gene content contribute to the overall carbon metabolism trait differences observed between generalists and specialists.
Unifying genetic features of carbon niche breadth generalists
To gain further insight into the genes and pathways contributing to the observed carbon niche breadth variation across the yeast subphylum, we employed machine learning. Specifically, we trained a supervised random forest classifier to use KO presence and absence as predictive features for carbon niche breadth classification. Niche breadth classification of generalists and specialists was used instead of the actual number of carbon sources because there were insufficient numbers of yeasts for some values to adequately train our model (e.g., there was only one yeast that grew on 17/18 carbon sources, but there were 64 yeasts that grew on five carbon sources). The resulting classifier was both highly sensitive and specific, correctly classifying 88% of specialists and 89% of generalists (AUC=0.93; Fig. 4C). The high accuracy suggests that generalist and specialist KEGG networks differ in ways that were not detected in the KO enrichment analysis.
Examination of the features on which the classifier relied using dropout analysis identified 2,050 KOs that significantly contributed to classification accuracy. Approximately 5,000 unique yeast KOs were used to train the algorithm, suggesting that many KOs contributed some information to niche breadth classification. We further examined the top four features because the fifth feature had only half the relative importance score of each of the fourth. Two of the top four features had direct links to the catabolism of specific carbon substrates, demonstrating the power and precision of our algorithm. The KO for manB (K01192), which encodes a β-mannosidase, had the second highest relative importance (relative importance 0.048). This KO was identified in 7% of specialists (8/111) and 80% of generalists (76/95). β-mannosidases are known to have a role in microbial utilization of N-glycans as a carbon source (57). Almost all the carbon generalists (93/95) can utilize mannose, which leads to the hypothesis that generalists likely use the mannose moieties present in N-glycans as a carbon and energy source.
The KO with the third highest importance was K17738 (relative importance 0.043), which is the ARD gene encoding D-arabinitol 2-dehydrogenase, an important component of the pentose and glucuronate interconversions pathway (Fig. 4D, step 5). This KO was more frequently present in the genomes of generalists (96%, 91/95) than in the genomes of specialists (71%, 79/111). Indeed, in a portion of this pathway, 5 of the 8 reactions were among the 2,050 KOs (with two falling in the top 100 KOs) that contributed to the classification of carbon generalists and specialists (black boxes in Fig. 4D). Importantly, growth on xylose was included in our carbon classification, and the xylose metabolism genes XYL1 (Step 2 in Fig. 4D), XYL2 (Step 3), and XYL3 (Step 8) were all identified as important features (with XYL1 falling within the top 100), suggesting that xylose metabolism genes may be promiscuous and have multiple metabolic capabilities (58). This result also supports the hypothesis that intrinsic genetic factors contribute to niche breadth by connecting pathways.
The feature with the highest relative importance was K03940 (relative importance 0.062), which encodes an NADH ubiquinone oxidoreductase core subunit (NDUFS7 in humans) of Complex I of the mitochondrial electron transport chain. This KO was identified in 29% of specialists (32/111) and 95% of generalists (90/95). Interestingly, Complex I is known to vary widely, in presence and makeup, including the presence of an alternative pathway in some yeasts (59). For example, in S. cerevisiae, the NADH oxidoreductase function of Complex I is conducted by three single-subunit enzymes (Ndi1p, Nde1p, or Nde2p) (60). Conversely, in Y. lipolytica, Complex I is composed of 42 subunits, including the NADH ubiquinone oxidoreductase NUKM (K03940) (61). Thirty additional Complex I enzymes were within the top 2,050 KOs, and two fell within the top 10%: K03941 and K03966, which are both NADH ubiquinone oxidoreductases in the β subcomplex (KEGG map00190). The Saccharomycetales and Saccharomycodales have both completely lost the canonical Complex I and contain many specialist yeasts (59). The relatively high importance of K03940, however, is not solely due to these orders, as the effect is widespread. For example, within the Pichiales, 100% (5/5) of generalist genomes encode K03940, in contrast to only 18% (6/33) of specialists. Complex I has been implicated in C. albicans growth and virulence (62), as a global regulator of fungal secondary metabolism in Aspergillus (63), and results in a higher proton motive force compared to the alternative pathway in S. cerevisiae. The presence of Complex I in generalists, therefore, may support increased carbon niche breadth and elevated growth rates.
The last KO we investigated was K00474 (relative importance 0.043), which encodes a trimethyllysine dioxygenase involved in lysine degradation. Every step in the pathway that degrades lysine to carnitine, except the last step, was identified as important in the machine learning classification. The last step (Fig. 4E, Step 7) was not annotated by KEGG in any of our yeasts. Therefore, we annotated the BBH2 gene, which encodes the trimethyllysine dioxygenase, directly from our predicted coding sequences using previously published reference sequences (64). After manual annotation of BBH2, we found that most carbon generalists were predicted to be able to complete the carnitine biosynthesis pathway (91%: 86/95), while relatively few carbon specialists were predicted to do so (20%: 22/111). Carnitine plays an important role in the transport of acetyl coenzyme A (acetyl-CoA), which in turn is a major metabolite that contributes to many metabolic pathways, including the production of ATP in the mitochondrial tricarboxylic acid (TCA) cycle. Acetyl-CoA can be produced within the mitochondria when glucose is available or, when glucose is unavailable, it can be transported into the mitochondria using the carnitine shuttle (65). Some yeasts, including C. albicans, rely solely on the carnitine shuttle for this transport (64), while other yeasts, such as S. cerevisiae, can use a carnitine-independent method for acetyl-CoA transport (66). Similarly, some yeasts, such as C. albicans, can synthesize carnitine; others, such as S. cerevisiae, cannot and rely on exogenous sources. A complete carnitine synthesis pathway may ensure acetyl-CoA transport when glucose is unavailable, especially in species that rely solely on the carnitine shuttle.
Additionally, carnitine and carnitine acetyltransferases can be essential for growth on some nonfermentable carbon sources. These include ethanol, as well as glycerol in certain S. cerevisiae mutants with disrupted citrate metabolism (67). We found that 90.5% (86/95) of generalists can grow on glycerol compared to only 24.5% (27/110) of specialists (table S2). Moreover, specialists that could grow on glycerol were more likely to have the complete carnitine synthesis pathway than those that did not (z-test, χ2= 10.425, p-value = 0.0186). These results suggest that carnitine production affords metabolic flexibility and carbon niche breadth.
Human yeast pathogens include both carbon generalists and specialists
This comprehensive dataset and analytical framework provide the opportunity to study how the observed genomic, metabolic, and environmental variation across the subphylum is associated with any complex trait of interest (68–70). To illustrate this potential, we examined the metabolic niche breadths of yeast pathogens of humans compared to those of their non-pathogenic close relatives (using a specific phylogenetic distance cutoff to standardize the clades) (Fig. 5). The World Health Organization (WHO) recently released its first-ever fungal priority pathogens list, which included six Saccharomycotina species (71). We defined 11 yeasts as opportunistic human pathogens because they are known to cause human infections and generally require biosafety level 2 (BSL-2) precautions in research laboratories.
Figure 5: Carbon generalism and specialism are not associated with yeast pathogenicity.
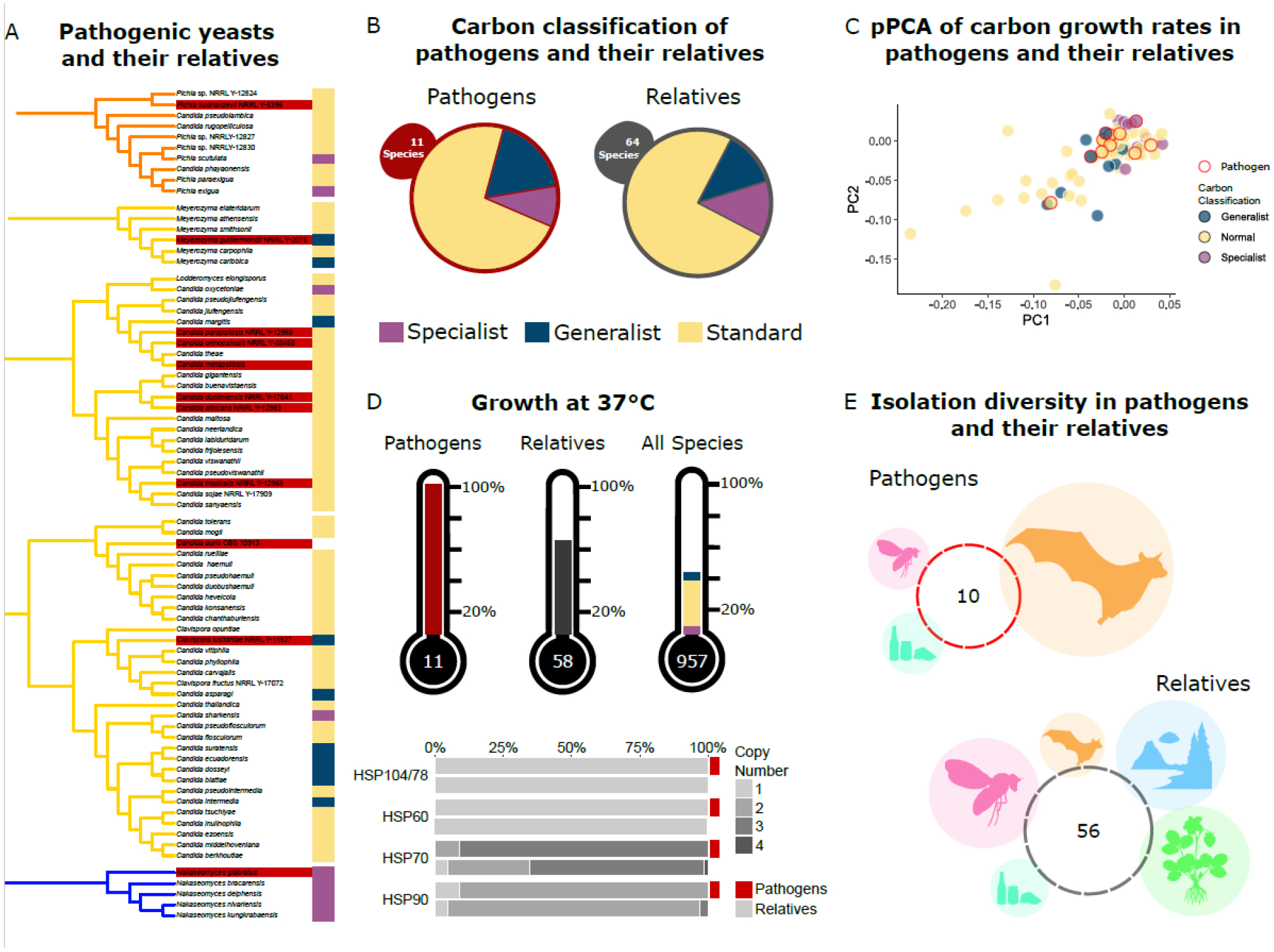
A. The phylogenetic clades containing human fungal pathogens. Clades reflect all species within a specific phylogenetic distance from the identified pathogen. Pathogens are found in three different orders, and at least one pathogen is classified in the generalist, specialist, and standard categories.
B. Pathogens and their relatives had nearly identical proportions of generalist, specialist, and standard yeasts. This result suggests that carbon niche breadth is not a defining or predictive factor for the potential of a species to gain the ability to infect humans.
C. Pathogens and their relatives did not differ substantially in their growth rates on carbon substrates. The phylogenetically corrected principal component analysis (pPCA) was constructed using growth rates on carbon substrates and projected onto the first two components (totaling 80% of the total variance.) Pathogens did not cluster together, while generalists and specialists appeared further apart. This result suggests that pathogens do not have shared growth rate characteristics.
D. Proportion of yeasts that can grow at 37°C in pathogens, their relatives, and all sampled yeasts. All yeasts identified as pathogens can grow at 37°C. Pathogenic yeasts were significantly more likely to grow at 37°C than their non-pathogenic relatives (χ2, p = 0.042). Heat shock protein (HSP) gene copy number was determined using InterPro and KEGG orthologs. HSP gene copy number was not significantly associated with pathogenicity.
E. Isolation environment for the specific strains of pathogens and their relatives. Circles are proportional to the percent of yeasts isolated from Chordata (orange), Arthropoda (pink), Victuals (teal), Environmental (blue), and Plants (green).
Carbon sources and availability vary in vivo in humans, suggesting that carbon niche breadth may play an important role in promoting or preventing fungal pathogenesis (72). Yeasts are subject to diverse micro-environments characterized by varying nutrients within a host (39, 72, 73). Their capacity to survive under fluctuating carbon conditions has been closely associated with virulence. For example, lactate assimilation across the C. albicans clade and, in Nakaseomyces glabratus (syn. Candida glabrata), is associated with increased antifungal and osmotic stress resistance and has been shown to reduce phagocytosis within the host (73). Interestingly, these pathogens exhibit reduced resistance to the antifungal drug amphotericin B when grown in culture media containing lactate relative to culture media containing glucose (73). We found that pathogens spanned the range of carbon niche breadth classifications and included specialist, standard, and generalist yeasts. Carbon niche breadths within pathogenic yeasts ranged from 15 in Meyerozyma guilliermondii to only 2 in N. glabratus (74). Furthermore, the proportion of pathogenic yeasts classified as standard, generalist, and specialist was similar to that of their non-pathogenic relatives (Fig. 5A–B). Collectively, these results suggest that yeast pathogenicity is not associated with carbon niche breadth.
Previous work in C. albicans linked its pathogenicity to its high growth rate (75). To examine whether this link holds across yeast pathogens, we visualized all pathogenic yeasts and their relatives on a phylogenetically corrected principal component analysis using all our growth rate data (Fig. 5C). We observed no clustering of pathogenic yeasts using carbon growth rates. Moreover, yeast pathogens within the same clade varied in their growth rate on glucose by almost 3-fold: Candida parapsilosis had a growth rate of 0.042, while Candida tropicalis had a growth rate of 0.124. Our growth rate data, however, were collected at a specific temperature in defined media and may not reflect growth rates in human infections.
We also examined the role of temperature, gene content, and environment in yeast pathogenicity. One feature known to be necessary, but insufficient, for pathogenicity is growth at human body temperature or 37°C (Fig. 5D) (39). We observed that relatives of human pathogens had an elevated rate of growth at 37°C (~64%) compared to all yeasts for which growth at this temperature was measured (~41%). This result likely reflects the necessity of growth at 37°C to evolve prior to pathogenicity. Heat shock proteins (HSPs) are also known to impact temperature tolerance (76). Examination of copy number variation in the genes encoding HSPs in the pathogenic species and their relatives identified a slight increase in HSP70 gene copy number among pathogenic yeasts (Fig. 5D). Finally, we found that pathogenic yeasts and their relatives had been isolated from all examined environments (Fig 5E). The analyses shown here suggest that pathogenicity can emerge in species across the spectrum of carbon metabolic breadth. Moreover, the lack of notable differences between yeast pathogens and their non-pathogenic relatives supports the hypothesis that the traits and genetic elements contributing to pathogenicity are not broadly shared across pathogens but unique to each (77). The data and analyses presented here provide a model for the investigation of other complex traits across Saccharomycotina using our ensemble of genomic, metabolic, and environmental data.
Conclusions
Here we focused on two predominant paradigms proposed to underlie the evolution of yeast carbon niche breadth. The first paradigm, where trade-offs dominate, was not supported when we analyzed over 10,000 growth rates measured across 853 yeasts. We found that generalists typically grew faster on carbon sources than specialists, even on those carbon sources for which specialists had their maximum growth rates. Thus, the ability to metabolize additional carbon sources does not come at the cost of reduced growth rates on other carbon sources. Carbon metabolism traits found within generalists were either maintained across evolutionary time or gained, even though there was a strong overall trend for trait loss across the subphylum. Of course, trade-offs between carbon metabolism traits likely exist in natural habitats. Future experiments along gradients of different environmental conditions, such as temperature, competition, or oxygen availability may shed additional light on condition-specific trade-offs in carbon niche breadth evolution.
In contrast, we found strong support for the second paradigm in the form of intrinsic factors that underlie the generalist phenotype. Machine learning allowed us to identify specific genes, complexes, and pathways shared by generalists but largely absent from specialists. These genes were directly involved in carbon and energy metabolism, often by enhancing metabolic flexibility and robustness. This finding supports the second paradigm because we identify a shared set of intrinsic genomic features across the generalist phenotype, even though generalists vary in the specific carbon sources they can metabolize. This finding does not support the hypothesis of trade-offs for two reasons. First, the pathways enriched in generalists are hypothesized to increase metabolic efficiency, which is contrary to the proposed trade-off between carbon niche breadth and efficiency. Second, under the trade-off paradigm, specialists and generalists would both have unique traits that provide them with a selective advantage. However, we found that generalists, as compared to specialists, have more genes in their genomes, including those not directly associated with carbon metabolism.
Given the advantages of wide carbon niche breadth and the absence of detectable efficiency costs, the question remains: what forces are shaping specialist yeasts? In some cases, carbon specialism could be associated with rapid gene loss. For example, in the genus Hanseniaspora (10/14 or 71.4% specialists), there were widespread gene losses, including of genes involved in DNA repair and carbon metabolism (78). Another hypothesis is that each specialist is subject to unique evolutionary pressures that would obviate unifying features. Finally, it is also possible that there are growth-associated trade-offs that we are unable to measure. Features, such as enhanced carbon sequestration, killer yeast toxins, pathogenicity, and microbial community composition, could provide specialists with advantages in highly specific environments. For example, Hanseniaspora species have a growth advantage over other species, including S. cerevisiae, on grapes at harvest and in the early stages of alcoholic fermentation (79). Further investigations into the evolution of yeast generalism and specialism will likely be fruitful, but a plethora of additional questions could be addressed with these data including: quantifying correlations among genes, traits, and/or ecologies; investigations of gene family evolution; research into the origins of pathogenesis; and genome-informed bioprospecting of yeasts and their genes for the sustainable production of cellulosic biofuels and bioproducts. More broadly, by coupling a comprehensive dataset with a robust analytical framework for studying macroevolutionary processes, the Y1000+ Project provides a roadmap that connects DNA to diversity.
Summary of Methods
Detailed materials and methods can be found in the supplementary materials (80). All data generated as a part of the project have been deposited in a FigShare repository (42).
Genome sequencing, annotation, and phylogenomics
Strains were obtained primarily from the NRRL (USA) and CBS (Netherlands) culture collections (USA). We sequenced pair-end libraries using the Illumina HiSeq 2500 platform and assembled genomes using the meta-assembler pipeline iWGS (81). We assessed assembly quality using Benchmarking Universal Single-Copy Orthologs (BUSCO) (44) and filtered the assemblies to remove mitochondrial and bacterial DNA contaminants. Genomes were functionally annotated using KEGG (55) and InterPro (82, 83) databases. We constructed a phylogenomic data matrix from 1,403 orthologous groups (taxon occupancy for each group ≥ 50%; 719,591 amino acid sites); we inferred the phylogeny of the subphylum using both concatenation and coalescence under maximum likelihood using IQ-Tree (84) and ASTRAL-III (85) respectively, and estimated the yeast time tree using the RelTime method (86).
Phenotyping, niche breadth classification, and testing for trade-offs and trait co-evolution
We generated quantitative growth data on 18 carbon and 6 nitrogen sources for 853 yeasts, measuring optical density every two hours for a week on the BMG Omega SpectroStar Plate Reader. We conducted all experiments in triplicate, and a new yeast colony was picked for each yeast across replicates. We calculated growth rates using a logistic model using the R package grofit (87). We classified yeasts as specialist, standard, or generalist for both carbon and nitrogen metabolism by calculating the binomial confidence intervals of carbon and nitrogen breadth relative to randomized growth data. We measured the correlation between carbon and nitrogen breadth and tested for trade-offs between carbon niche breadth and efficiency (by measuring the correlation between growth rates and carbon niche breadth classifications) using phylogenetic generalized least squares analyses with PGLScaper (88). Finally, we inferred the co-evolution of carbon traits and carbon generalism/specialism using BayesTraits (http://www.evolution.reading.ac.uk).
Underlying factors driving generalist and specialist phenotypes
We identified strain-specific isolation environments for 1,088 yeasts and standardized them by creating an ontology of environments and their hierarchical network using Web Protégé (https://github.com/protegeproject/webprotege). To identify underlying genomic features contributing to generalists and specialist phenotypes, we used genome annotations to build metabolic networks and quantify network variation among generalists and specialists while accounting for phylogeny. We also identified KEGG ontologies enriched in generalists and specialists using a KEGG enrichment analysis (89). Finally, we constructed a machine learning algorithm using the XGBoost random forest classifier (90), which we trained using 90% of the genomic data and using the remaining 10% for cross validation, to identify genes whose presence/absence was most strongly associated with carbon generalism and specialism.
Supplementary Material
Acknowledgments:
We thank Linda C. Horianopoulos, Kaitlin J. Fisher, Liang Sun, David J. Krause, Kyle T. David, Christina M. Chavez, David C. Rinker, Trey K. Sato, and Hittinger Lab and Rokas Lab members for helpful discussions; Barbara Robbertse and Conrad Schoch for coordinating GenBank taxonomy; the yeast community for publicly depositing taxonomic type strains; the University of Wisconsin Biotechnology Center DNA Sequencing Facility (Research Resource Identifier – RRID:SCR_017759) for providing DNA sequencing facilities and services; Wisconsin Energy Institute staff for computational support; and the Center for High-Throughput Computing at the University of Wisconsin-Madison (https://doi.org/10.21231/GNT1-HW21). This work was performed in part using resources contained within the Advanced Computing Center for research and Education at Vanderbilt University in Nashville, TN. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding:
National Science Foundation Grant DEB-1442148 (CTH)
National Science Foundation Grant DEB-2110403 (CTH)
National Science Foundation Grant DEB-1442113 (AR)
National Science Foundation Grant DEB-2110404 (AR)
DOE Great Lakes Bioenergy Research Center, funded by BER Office of Science Grant DE-SC0018409 (CTH)
USDA National Institute of Food and Agriculture Hatch Project 1020204 (CTH)
USDA National Institute of Food and Agriculture Hatch Project 7005101 (CTH)
H. I. Romnes Faculty Fellow, supported by the Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (CTH)
National Institutes of Health/National Institute of Allergy and Infectious Diseases Grant R56 AI146096 (AR)
National Institutes of Health/National Institute of Allergy and Infectious Diseases Grant R01 AI153356 (AR)
Burroughs Wellcome Fund (AR)
Research supported by the National Key R&D Program of China Grant 2022YFD1401600 (XXS)
National Science Foundation for Distinguished Young Scholars of Zhejiang Province Grant LR23C140001 (XXS)
Fundamental Research Funds for the Central Universities Grant 226-2023-00021 (XXS)
National Institutes of Health Grant T32 HG002760-16 (JFW)
National Science Foundation Grant Postdoctoral Research Fellowship in Biology 1907278 (JFW)
Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program (JLS, AR)
National Science Foundation Graduate Research Fellowship Grant DGE-1256259 (QLK)
Predoctoral Training Program in Genetics, funded by the National Institutes of Health Grant 5T32GM007133 (QLK)
Slovenian Research Agency Grant P4–0116 (NC)
Slovenian Research Agency Grant MRIC-UL ZIM, IP-0510 (NC)
Fundação para a Ciência e a Tecnologia Grant UIDB/04378/2020 (CG, PG, JPS)
Fundação para a Ciência e a Tecnologia Grant LA/P/0140/2020 (CG, PG, JPS)
Fundação para a Ciência e a Tecnologia Grant PTDC/BIA-EVL/0604/2021 (CG)
Fundação para a Ciência e a Tecnologia Grant PTDC/BIA-EVL/1100/2020 (PG)
Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brazil CNPq Grant 408733/2021 (CAR)
Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brazil CNPq Grant 406564/2022-1 - “INCT Yeasts: Biodiversity, preservation and biotechnological innovation” (CAR)
MINCyT Grant PICT-2020-SERIE A-00226 (DL)
CONICET Grant PIP 11220200102948CO (DL)
UNComahue Grant 04/B247 (DL)
JLS is a Howard Hughes Medical Institute Awardee of the Life Sciences Research Foundation
Footnotes
Competing interests: JLS was a scientific adviser for WittGen Biotechnologies and is an adviser for ForensisGroup, Inc. AR is a scientific consultant for LifeMine Therapeutics, Inc. The other authors declare no other competing interests.
Supplementary Materials
Data and materials availability:
All genome sequence assemblies and raw sequencing data have been deposited in GenBank under the accessions noted in Data S1. All other data, including data on growth on different carbon and nitrogen sources and isolation environment data, have been deposited in Figshare at https://doi.org/10.25452/figshare.plus.c.6714042 (42). All code has been deposited in GitHub at https://zenodo.org/records/10709452 (91) and https://zenodo.org/doi/10.5281/zenodo.10711058 (92) and is available in Figshare at https://doi.org/10.25452/figshare.plus.c.6714042 (42). Nearly all strains came from globally recognized yeast culture collections and may be ordered from the United States Department of Agriculture (https://nrrl.ncaur.usda.gov for NRRL strains) or Westerdijk Fungal Biodiversity Institute (https://wi.knaw.nl for CBS strains) under their respective Material Transfer Agreements (MTAs) for publicly deposited strains; currently, NRRL only requires an MTA for strains requiring BSL-2 precautions. Strains from the Hittinger Lab that represent candidates for novel species that have not yet been formally described or deposited at CBS or NRRL may be obtained from cthittinger@wisc.edu under the Uniform Biological MTA or other mutually acceptable MTA.
References
- 1.Bruns EL, Antonovics J, Hood ME, From generalist to specialists: Variation in the host range and performance of anther-smut pathogens on Dianthus. Evolution (N Y) 75, 2494–2508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preston DB, Johnson SG, Generalist grasshoppers from thermally variable sites do not have higher thermal tolerance than grasshoppers from thermally stable sites-A study of five populations. J Therm Biol 88, 102527 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Wenger JW, Piotrowski J, Nagarajan S, Chiotti K, Sherlock G, Rosenzweig F, Hunger Artists: Yeast Adapted to Carbon Limitation Show Trade-Offs under Carbon Sufficiency. PLoS Genet 7, e1002202 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacArthur RH, Geographical Ecology: Patterns in the Distribution of Species (Princeton University Press, 1984; https://books.google.com/books?id=3NAYEKc--WAC)Biology / Princeton University Press. [Google Scholar]
- 5.Wilson DS, Yoshimura J, On the coexistence of specialists and generalists. Am Nat 144, 692–707 (1994). [Google Scholar]
- 6.Burmeister AR, Fortier A, Roush C, Lessing AJ, Bender RG, Barahman R, Grant R, Chan BK, Turner PE, Pleiotropy complicates a trade-off between phage resistance and antibiotic resistance. Proceedings of the National Academy of Sciences 117, 11207–11216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Z, Liu J, Zhao P, Jia T, Li C, Chai B, Biogeographic patterns and assembly mechanisms of bacterial communities differ between habitat generalists and specialists across elevational gradients. Front Microbiol 10, 169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seebacher F, Ducret V, Little AG, Adriaenssens B, Generalist–specialist trade-off during thermal acclimation. R Soc Open Sci 2, 140251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Napier JD, Grabowski PP, Lovell JT, Bonnette J, Mamidi S, Gomez-Hughes MJ, VanWallendael A, Weng X, Handley LH, Kim MK, A generalist–specialist trade-off between switchgrass cytotypes impacts climate adaptation and geographic range. Proceedings of the National Academy of Sciences 119, e2118879119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostrowski EA, Woods RJ, Lenski RE, The genetic basis of parallel and divergent phenotypic responses in evolving populations of Escherichia coli. Proceedings of the Royal Society B: Biological Sciences 275, 277–284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Q, Luo G, Guo J, Xiao Y, Zhang F, Guo S, Ling N, Shen Q, Microbial generalist or specialist: Intraspecific variation and dormancy potential matter. Mol Ecol 31 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Kassen R, The experimental evolution of specialists, generalists, and the maintenance of diversity. J Evol Biol 15, 173–190 (2002). [Google Scholar]
- 13.Hesse E, Best A, Boots M, Hall AR, Buckling A, Spatial heterogeneity lowers rather than increases host–parasite specialization. J Evol Biol 28, 1682–1690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Büchi L, Vuilleumier S, Coexistence of specialist and generalist species is shaped by dispersal and environmental factors. Am Nat 183, 612–624 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Bleuven C, Landry CR, Molecular and cellular bases of adaptation to a changing environment in microorganisms. Proceedings of the Royal Society B: Biological Sciences 283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caspeta L, Nielsen J, Thermotolerant yeast strains adapted by laboratory evolution show trade-off at ancestral temperatures and preadaptation to other stresses. mBio 6, e00431–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Opulente DA, Rollinson EJ, Bernick-Roehr C, Hulfachor AB, Rokas A, Kurtzman CP, Hittinger CT, Factors driving metabolic diversity in the budding yeast subphylum. BMC Biol 16, 26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown AJP, Brown GD, Netea MG, Gow NAR, Metabolism impacts upon Candida immunogenicity and pathogenicity at multiple levels. Trends Microbiol 22, 614–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanneste T, Govaert S, Spicher F, Brunet J, Cousins SAO, Decocq G, Diekmann M, Graae BJ, Hedwall P-O, Kapás RE, Contrasting microclimates among hedgerows and woodlands across temperate Europe. Agric For Meteorol 281, 107818 (2020). [Google Scholar]
- 20.Grzyb A, Wolna-Maruwka A, Niewiadomska A, The significance of microbial transformation of nitrogen compounds in the light of integrated crop management. Agronomy 11, 1415 (2021). [Google Scholar]
- 21.Manuel E-U, Juan-Luis R, Expression of a Pseudomonas putidaAminotransferase Involved in Lysine Catabolism Is Induced in the Rhizosphere. Appl Environ Microbiol 67, 5219–5224 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schädel C, Blöchl A, Richter A, Hoch G, Quantification and monosaccharide composition of hemicelluloses from different plant functional types. Plant physiology and Biochemistry 48, 1–8 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Ebringerová A, Hromádková Z, Heinze T, Hemicellulose. Polysaccharides I: Structure, characterization and use, 1–67 (2005). [Google Scholar]
- 24.Jaeger Iii CH, Lindow SE, Miller W, Clark E, Firestone M, Mapping of sugar and amino acid availability in soil around roots with bacterial sensors of sucrose and tryptophan. Appl Environ Microbiol 65, 2685–2690 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdel-Mawgoud AM, Aboulwafa MM, Hassouna NA-H, Characterization of surfactin produced by Bacillus subtilis isolate BS5. Appl Biochem Biotechnol 150, 289–303 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Demain AL, From natural products discovery to commercialization: a success story. J Ind Microbiol Biotechnol 33, 486–495 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Tate EG, Pitt AL, Little MD, Tavano JJ, Nickerson MA, Factors contributing to the range expansion and population increase of a native generalist species. Amphibia-Reptilia 43, 299–311 (2022). [Google Scholar]
- 28.Shen X-X, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MABB, Wisecaver JH, Wang M, Doering DT, Boudouris JT, Schneider RM, Langdon QK, Ohkuma M, Endoh R, Takashima M, Manabe R, Čadež N, Libkind D, Rosa CA, DeVirgilio J, Hulfachor AB, Groenewald M, Kurtzman CP, Hittinger CT, Rokas A, Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersson DI, Jerlström-Hultqvist J, Näsvall J, Evolution of New functions De novo and from preexisting genes. Cold Spring Harb Perspect Biol 7, a017996 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voordeckers K, Brown CA, Vanneste K, van der Zande E, Voet A, Maere S, Verstrepen KJ, Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLoS Biol 10, e1001446 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patrick WM, Quandt EM, Swartzlander DB, Matsumura I, Multicopy suppression underpins metabolic evolvability. Mol Biol Evol 24, 2716–2722 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McBride CS, Arguello JR, O’Meara BC, Five Drosophila genomes reveal nonneutral evolution and the signature of host specialization in the chemoreceptor superfamily. Genetics 177, 1395–416 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christie KR, Weng S, Balakrishnan R, Costanzo MC, Dolinski K, Dwight SS, Engel SR, Feierbach B, Fisk DG, Hirschman JE, Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res 32, D311–D314 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnaud MB, Costanzo MC, Skrzypek MS, Binkley G, Lane C, Miyasato SR, Sherlock G, The Candida Genome Database (CGD), a community resource for Candida albicans gene and protein information. Nucleic Acids Res 33, D358–D363 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurtzman C, Fell JW, Boekhout T, The Yeasts: A Taxonomic Study (Elsevier, 2011). [Google Scholar]
- 36.Rozpędowska E, Hellborg L, Ishchuk OP, Orhan F, Galafassi S, Merico A, Woolfit M, Compagno C, Piškur J, Parallel evolution of the make–accumulate–consume strategy in Saccharomyces and Dekkera yeasts. Nat Commun 2, 302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naumov GI, Naumova ES, Sniegowski PD, Saccharomyces paradoxus and Saccharomyces cerevisiae are associated with exudates of North American oaks. Can J Microbiol 44, 1045–1050 (1998). [PubMed] [Google Scholar]
- 38.Hewitt SK, Foster DS, Dyer PS, Avery SV, Phenotypic heterogeneity in fungi: Importance and methodology. Fungal Biol Rev 30, 176–184 (2016). [Google Scholar]
- 39.V Ene I, Brunke S, Brown AJP, Hube B, Metabolism in fungal pathogenesis. Cold Spring Harb Perspect Med 4, a019695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hittinger CT, Rokas A, Bai F-Y, Boekhout T, Gonçalves P, Jeffries TW, Kominek J, Lachance M-A, Libkind D, Rosa CA, Sampaio JP, Kurtzman CP, Genomics and the making of yeast biodiversity. Curr Opin Genet Dev 35, 100–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groenewald M, Hittinger CT, Bensch K, Opulente DA, Shen X-X, Li Y, Liu C, LaBella AL, Zhou X, Limtong S, A genome-informed higher rank classification of the biotechnologically important fungal subphylum Saccharomycotina. Stud Mycol (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Data is available at: 10.25452/figshare.plus.c.6714042. [DOI] [Google Scholar]
- 43.V Kriventseva E, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA, Zdobnov EM, OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47, D807–D811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, V Kriventseva E, Zdobnov EM, BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35, 543–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krassowski T, Kominek J, Shen XX, Opulente DA, Zhou X, Rokas A, Hittinger CT, Wolfe KH, Multiple Reinventions of Mating-type Switching during Budding Yeast Evolution. Current Biology 29, 2555–2562.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mühlhausen S, Schmitt HD, Pan K-T, Plessmann U, Urlaub H, Hurst LD, Kollmar M, Endogenous stochastic decoding of the CUG codon by competing Ser-and Leu-tRNAs in Ascoidea asiatica. Current Biology 28, 2046–2057 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Steenwyk JL, Chang Y, Wang Y, James TY, Stajich JE, Spatafora JW, Groenewald M, Dunn CW, Hittinger CT, A genome-scale phylogeny of the kingdom Fungi. Current Biology 31, 1653–1665 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen X-X, Steenwyk JL, LaBella AL, Opulente DA, Zhou X, Kominek J, Li Y, Groenewald M, Hittinger CT, Rokas A, Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci Adv 6, eabd0079 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonçalves C, Wisecaver JH, Kominek J, Oom MS, Leandro MJ, Shen X-X, Opulente DA, Zhou X, Peris D, Kurtzman CP, Evidence for loss and reacquisition of alcoholic fermentation in a fructophilic yeast lineage. Elife 7, e33034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Commichau FM, Forchhammer K, Stülke J, Regulatory links between carbon and nitrogen metabolism. 9, 167–172 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Parada JL, Magasanik B, Expression of the hut operons of Salmonella typhimurium in Klebsiella aerogenes and in Escherichia coli. J Bacteriol 124, 1263–1268 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noda-Garcia L, Romero Romero ML, Longo LM, Kolodkin-Gal I, Tawfik DS, Bacilli glutamate dehydrogenases diverged via coevolution of transcription and enzyme regulation. EMBO Rep 18, 1139–1149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner A, Metabolic networks and their evolution. Adv Exp Med Biol 751 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Proulx SR, Promislow DEL, Phillips PC, Network thinking in ecology and evolution. Trends Ecol Evol 20, 345–353 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Kanehisa M, Goto S, KEGG: Kyoto Encyclopedia of Genes and Genomes. [Preprint] (2000). 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barabási A-L, Oltvai ZN, Network biology: understanding the cell’s functional organization. Nat Rev Genet 5, 101–113 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Cordeiro RL, Santos CR, Domingues MN, Lima TB, Pirolla RAS, Morais MAB, Colombari FM, Miyamoto RY, Persinoti GF, Borges AC, Mechanism of high-mannose N-glycan breakdown and metabolism by Bifidobacterium longum. Nat Chem Biol 19, 218–229 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nalabothu RL, Fisher KJ, LaBella AL, Meyer TA, Opulente DA, Wolters JF, Rokas A, Hittinger CT, Codon optimization improves the prediction of xylose metabolism from gene content in budding yeasts. Mol Biol Evol, msad111 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolters JF, LaBella AL, Opulente DA, Rokas A, Hittinger CT, Mitochondrial Genome Diversity across the Subphylum Saccharomycotina. bioRxiv (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luttik MAH, Overkamp KM, Kötter P, De Vries S, Van Dijken JP, Pronk JT, The Saccharomyces cerevisiae NDE1 and NDE2 genes encode separate mitochondrial NADH dehydrogenases catalyzing the oxidation of cytosolic NADH. Journal of Biological Chemistry 273, 24529–24534 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Wirth C, Brandt U, Hunte C, Zickermann V, Structure and function of mitochondrial complex I. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857, 902–914 (2016). [DOI] [PubMed] [Google Scholar]
- 62.She R, Jarosz DF, Mapping causal variants with single-nucleotide resolution reveals biochemical drivers of phenotypic change. Cell 172, 478–490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bromley M, Johns A, Davies E, Fraczek M, Mabey Gilsenan J, Kurbatova N, Keays M, Kapushesky M, Gut M, Gut I, Mitochondrial complex I is a global regulator of secondary metabolism, virulence and azole sensitivity in fungi. PLoS One 11, e0158724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strijbis K, van Roermund CWT, Visser WF, Mol EC, van den Burg J, MacCallum DM, Odds FC, Paramonova E, Krom BP, Distel B, Carnitine-dependent transport of acetyl coenzyme A in Candida albicans is essential for growth on nonfermentable carbon sources and contributes to biofilm formation. Eukaryot Cell 7, 610–618 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strijbis K, Distel B, Intracellular acetyl unit transport in fungal carbon metabolism. [Preprint] (2010). 10.1128/EC.00172-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Roermund CWT, Hettema EH, van den Berg M, Tabak HF, Wanders RJA, Molecular characterization of carnitine-dependent transport of acetyl-CoA from peroxisomes to mitochondria in Saccharomyces cerevisiae and identification of a plasma membrane carnitine transporter, Agp2p. EMBO J 18, 5843–5852 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swiegers JH, Dippenaar N, Pretorius IS, Bauer FF, Carnitine-dependent metabolic activities in Saccharomyces cerevisiae: three carnitine acetyltransferases are essential in a carnitine-dependent strain. Yeast 18, 585–595 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Harrison M-C, Ubbelohde EJ, LaBella AL, Opulente DA, Wolters JF, Zhou X, Shen X-X, Groenewald M, Hittinger CT, Rokas A, Machine learning illuminates how diet influences the evolution of yeast galactose metabolism. bioRxiv, 2023.07.20.549758 (2023). [Google Scholar]
- 69.Gonçalves C, Harrison M-C, Steenwyk JL, Opulente DA, LaBella AL, Wolters JF, Zhou X, Shen X-X, Groenewald M, Hittinger CT, Rokas A, Diverse signatures of convergent evolution in cacti-associated yeasts. bioRxiv, 2023.09.14.557833 (2023). [Google Scholar]
- 70.David KT, Harrison M-C, Opulente DA, LaBella AL, Wolters JF, Zhou X, Shen X-X, Groenewald M, Pennell M, Hittinger CT, Rokas A, Saccharomycotina yeasts defy longstanding macroecological patterns. bioRxiv, 2023.08.29.555417 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.World Health Organization, “WHO fungal priority pathogen list to guide research, development and public health action” (2022); https://www.who.int/publications/i/item/9789240060241.
- 72.Ries LNA, Beattie S, Cramer RA, Goldman GH, Overview of carbon and nitrogen catabolite metabolism in the virulence of human pathogenic fungi. Mol Microbiol 107, 277–297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ene IV, Adya AK, Wehmeier S, Brand AC, Maccallum DM, Gow NAR, Brown AJP, Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell Microbiol 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Masako T, Takashi S, Taxonomy of Pathogenic Yeasts Candida, Cryptococcus, Malassezia, and Trichosporon. Med Mycol J 63, 119–132 (2022). [DOI] [PubMed] [Google Scholar]
- 75.Magee PT, Fungal pathogenicity and morphological switches. Nat Genet 42, 560–561 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Mayer FL, Wilson D, Hube B, Candida albicans pathogenicity mechanisms. Virulence 4, 119–128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rokas A, Mead ME, Steenwyk JL, Oberlies NH, Goldman GH, Evolving moldy murderers: Aspergillus section Fumigati as a model for studying the repeated evolution of fungal pathogenicity. PLoS Pathog 16, e1008315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steenwyk JL, Opulente DA, Kominek J, Shen XX, Zhou X, Labella AL, Bradley NP, Eichman BF, Čadež N, Libkind D, De Virgilio J, Hulfachor AB, Kurtzman CP, Hittinger CT, Rokas A, Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol 17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fugelsang KC, Edwards CG, Wine Microbiology: Practical Applications and Procedures (2007).
- 80.Supplementary Materials: Genomic factors shape carbon and nitrogen metabolic niche breadth across Saccharomycotina yeasts. Science (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou X, Peris D, Kominek J, Kurtzman CP, Hittinger CT, Rokas A, In silico Whole Genome Sequencer and Analyzer (iWGS): a computational pipeline to guide the design and analysis of de novo genome sequencing studies. G3: Genes, Genomes, Genetics 6, 3655–3662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S, InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blum M, Chang HY, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, Richardson L, Salazar GA, Williams L, Bork P, Bridge A, Gough J, Haft DH, Letunic I, Marchler-Bauer A, Mi H, Natale DA, Necci M, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A, Finn RD, The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49, D344–D354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R, Teeling E, IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang C, Rabiee M, Sayyari E, Mirarab S, ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kumar S, Stecher G, Tamura K, MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33, 1870–1874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kahm M, Hasenbrink G, Lichtenberg-Fraté H, Ludwig J, Kschischo M, grofit: fitting biological growth curves with R. J Stat Softw 33, 1–21 (2010).20808728 [Google Scholar]
- 88.Orme D, The caper package: Comparative analysis of phylogenetics and evolution in R. R package version 5, 1–36 (2013). [Google Scholar]
- 89.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G, clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen T, Guestrin C, “XGBoost: A scalable tree boosting system” in Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (2016)vols. 13–17-August-2016. [Google Scholar]
- 91.Code available for growth and enrichment analyses at. https://zenodo.org/records/10709452.
- 92.Code available for machine learning analysis at. https://zenodo.org/doi/10.5281/zenodo.10711058.
- 93.Zhang G, Lin Y, He P, Li L, Wang Q, Ma Y, Characterization of the sugar alcohol-producing yeast Pichia anomala. J Ind Microbiol Biotechnol 41 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Sylvester K, Wang Q-M, James B, Mendez R, Hulfachor AB, Hittinger CT, Temperature and host preferences drive the diversification of Saccharomyces and other yeasts: A survey and the discovery of eight new yeast species. FEMS Yeast Res 15, fov002 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Spurley WJ, Fisher KJ, Langdon QK, V Buh K, Jarzyna M, Haase MAB, Sylvester K, V Moriarty R, Rodriguez D, Sheddan A, Wright S, Sorlie L, Hulfachor AB, Opulente DA, Hittinger CT, Substrate, temperature, and geographical patterns among nearly 2000 natural yeast isolates. Yeast n/a, 55–68 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kominek J, Doering DT, Opulente DA, Shen X-X, Zhou X, DeVirgilio J, Hulfachor AB, Groenewald M, Mcgee MA, Karlen SD, Eukaryotic acquisition of a bacterial operon. Cell 176, 1356–1366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song L, Florea L, Langmead B, Lighter: fast and memory-efficient sequencing error correction without counting. Genome Biol 15, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chikhi R, Medvedev P, Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I, ABySS: A parallel assembler for short read sequence data. Genome Res 19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA, The MaSuRCA genome assembler. Bioinformatics 29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zerbino DR, Birney E, Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weib CL, Pais M, Cano LM, Kamoun S, Burbano HA, nQuire: A statistical framework for ploidy estimation using next generation sequencing. BMC Bioinformatics 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang S, Kang M, Xu A, HaploMerger2: Rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gurevich A, Saveliev V, Vyahhi N, Tesler G, QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM, BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kriventseva EV, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA, Zdobnov EM, OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, V Kriventseva E, Zdobnov EM, BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35, 543–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M, BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stanke M, Diekhans M, Baertsch R, Haussler D, Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24 (2008). [DOI] [PubMed] [Google Scholar]
- 112.Brůna T, Lomsadze A, Borodovsky M, GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform 2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, Ogata H, KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shulgina Y, Eddy SR, A computational screen for alternative genetic codes in over 250,000 genomes. Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Steenwyk JL, Rokas A, orthofisher: A broadly applicable tool for automated gene identification and retrieval. G3: Genes, Genomes, Genetics 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Price MN, Dehal PS, Arkin AP, FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One 5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Le SQ, Gascuel O, An improved general amino acid replacement matrix. Mol Biol Evol 25 (2008). [DOI] [PubMed] [Google Scholar]
- 118.Katoh K, Standley DM, MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T, trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Letunic P Bork, Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krassowski T, Coughlan AY, Shen XX, Zhou X, Kominek J, Opulente DA, Riley R, Grigoriev IV, Maheshwari N, Shields DC, Kurtzman CP, Hittinger CT, Rokas A, Wolfe KH, Evolutionary instability of CUG-Leu in the genetic code of budding yeasts. Nat Commun 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.LaBella AL, Opulente DA, Steenwyk JL, Hittinger CT, Rokas A, Signatures of optimal codon usage in metabolic genes inform budding yeast ecology. PLoS Biol 19, e3001185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO, Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464 (2010). [DOI] [PubMed] [Google Scholar]
- 124.Harrell FE Jr, Dupont MC, The Hmisc package. R package version 3, 0–12 (2006). [Google Scholar]
- 125.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res 13 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Csardi G, Nepusz T, Csárdi G, Nepusz T, Csardi G, Nepusz T, The igraph software package for complex network research. InterJournal Complex Systems 1695, 1–9 (2006). [Google Scholar]
- 127.Ofir A, Hofmann K, Weindling E, Gildor T, Barker KS, Rogers PD, Kornitzer D, Role of a Candida albicans Nrm1/Whi5 homologue in cell cycle gene expression and DNA replication stress response. Mol Microbiol 84, 778–794 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Al-Reedy RM, Malireddy R, Dillman CB, Kennell JC, Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genetics and Biology 49 (2012). [DOI] [PubMed] [Google Scholar]
- 129.Baker CR, Tuch BB, Johnson AD, Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc Natl Acad Sci U S A 108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baker EC, Wang B, Bellora N, Peris D, Hulfachor AB, Koshalek JA, Adams M, Libkind D, Hittinger CT, The genome sequence of Saccharomyces eubayanus and the domestication of lager-brewing yeasts. Mol Biol Evol 32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Batista TM, Moreira RG, Hilário HO, Morais CG, Franco GR, Rosa LH, Rosa CA, Draft genome sequence of Sugiyamaella xylanicola UFMG-CM-Y1884T, a xylan-degrading yeast species isolated from rotting wood samples in Brazil. Genom Data 11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Borelli G, José J, Teixeir PJPL, dos Santos LV, Pereira GAG, De novo assembly of Candida sojae and Candida boidinii genomes, unexplored xylose-consuming yeasts with potential for renewable biochemical production. Genome Announc 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brejová B, Lichancová H, Brázdovič F, Cillingová A, Neboháčová M, Tomáška L, Vinař T, Nosek J, Draft genome sequence of an obligate psychrophilic yeast, Candida psychrophila NRRL Y-17665T. Genome Announc 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brejová B, Lichancová H, Hodorová V, Neboháčová M, Tomáška Ľ, Vinař T, Nosek J, Genome Sequence of an Arthroconidial Yeast, Saprochaete fungicola CBS 625.85. Microbiol Resour Announc 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Brejová B, Lichancová H, Brázdovič F, Hegedűsová E, Forgáčová Jakúbková M, Hodorová V, Džugasová V, Baláž A, Zeiselová L, Cillingová A, Neboháčová M, Raclavský V, Tomáška Ľ, Lang BF, Vinař T, Nosek J, Genome sequence of the opportunistic human pathogen Magnusiomyces capitatus. Curr Genet 65 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Butler G, Rasmussen MD, Lin MF, Santos MAS, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJP, Brunke S, Costanzo MC, Fitzpatrick DA, De Groot PWJ, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, Santos MC, Schmitzberger FF, Sherlock G, Shah P, Silverstein KAT, Skrzypek MS, Soll D, Staggs R, Stansfield I, Stumpf MPH, Sudbery PE, Srikantha T, Zeng Q, Berman J, Berriman M, Heitman J, Gow NAR, Lorenz MC, Birren BW, Kellis M, Cuomo CA, Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Čadež N, Bellora N, Ulloa R, Hittinger CT, Libkind D, Genomic content of a novel yeast species Hanseniaspora gamundiae sp. nov. from fungal stromata (Cyttaria) associated with a unique fermented beverage in Andean Patagonia, Argentina. PLoS One 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Čadež N, Bellora N, Ulloa R, Tome M, Petković H, Groenewald M, Hittinger CT, Libkind D, Hanseniaspora smithiae sp. nov., a Novel Apiculate Yeast Species From Patagonian Forests That Lacks the Typical Genomic Domestication Signatures for Fermentative Environments. Front Microbiol 12, 679894 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chatterjee S, Alampalli SV, Nageshan RK, Chettiar ST, Joshi S, Tatu US, Draft genome of a commonly misdiagnosed multidrug resistant pathogen Candida auris. BMC Genomics 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dietrich FS, Voegeli S, Kuo S, Philippsen P, Genomes of Ashbya fungi isolated from insects reveal four mating-type loci, numerous translocations, lack of transposons, and distinct gene duplications. G3 (Bethesda) 3, 1225–1239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, Mohr C, Pöhlmann R, Luedi P, Choi S, Wing RA, Flavier A, Gaffney TD, Philippsen P, The Ashbya gossypii Genome as a Tool for Mapping the Ancient Saccharomyces cerevisiae Genome. Science (1979) 304 (2004). [DOI] [PubMed] [Google Scholar]
- 142.Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuvéglise C, Talla E, Genome evolution in yeasts. Nature 430, 35–44 (2004). [DOI] [PubMed] [Google Scholar]
- 143.Duplessis S, Cuomo CA, Lin YC, Aerts A, Tisserant E, Veneault-Fourrey C, Joly DL, Hacquard S, Amselem J, Cantarel BL, Chiu R, Coutinho PM, Feaue N, Field M, Frey P, Gelhaye E, Goldberg J, Grabherr MG, Kodira CD, Kohler A, Kües U, Lindquist EA, Lucas SM, Mago R, Mauceli E, Morin E, Murat C, Pangilinan JL, Park R, Pearson M, Quesneville H, Rouhier N, Sakthikumar S, Salamov AA, Schmutz J, Selles B, Shapiro H, Tanguay P, Tuskan GA, Henrissat B, Van De Peer Y, Rouzé P, Ellis JG, Dodds PN, Schein JE, Zhong S, Hamelin RC, Grigoriev IV, Szabo LJ, Martin F, Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci U S A 108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Freel KC, Friedrich A, Sarilar V, Devillers H, Neuvéglise C, Schacherer J, Whole-genome sequencing and intraspecific analysis of the yeast species Lachancea quebecensis. Genome Biol Evol 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gabaldón T, Martin T, Marcet-Houben M, Durrens P, Bolotin-Fukuhara M, Lespinet O, Arnaise S, Boisnard S, Aguileta G, Atanasova R, Bouchier C, Couloux A, Creno S, Almeida Cruz J, Devillers H, Enache-Angoulvant A, Guitard J, Jaouen L, Ma L, Marck C, Neuvéglise C, Pelletier E, Pinard A, Poulain J, Recoquillay J, Westhof E, Wincker P, Dujon B, Hennequin C, Fairhead C, Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Galeote V, Bigey F, Devillers H, Neuvéglise C, Dequin S, Genome sequence of the food spoilage yeast Zygosaccharomyces bailii CLIB 213T. Genome Announc 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gazis R, Kuo A, Riley R, LaButti K, Lipzen A, Lin J, Amirebrahimi M, Hesse CN, Spatafora JW, Henrissat B, Hainaut M, Grigoriev IV, Hibbett DS, The genome of Xylona heveae provides a window into fungal endophytism. Fungal Biol 120 (2016). [DOI] [PubMed] [Google Scholar]
- 148.Souciet JL, Dujon B, Gaillardin C, Johnston M, Baret PV, Cliften P, Sherman DJ, Weissenbach J, Westhof E, Wincker P, Jubin C, Poulain J, Barbe V, Ségurens B, Artiguenave F, Anthouard V, Vacherie B, Val ME, Fulton RS, Minx P, Wilson R, Durrens P, Jean G, Marck C, Martin T, Nikolski M, Rolland T, Seret ML, Casarégola S, Despons L, Fairhead C, Fischer G, Lafontaine I, Leh V, Lemaire M, De Montigny J, Neuvéglise C, Thierry A, Blanc-Lenfle I, Bleykasten C, Diffels J, Fritsch E, Frangeul L, Goëffon A, Jauniaux N, Kachouri-Lafond R, Payen C, Potier S, Pribylova L, Ozanne C, Richard GF, Sacerdot C, Straub ML, Talla E, Comparative genomics of protoploid Saccharomycetaceae. Genome Res 19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Goffeau A, Barrell G, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG, Life with 6000 genes. Science (1979) 274 (1996). [DOI] [PubMed] [Google Scholar]
- 150.Gordon JL, Armisen D, Proux-Weŕa E, ÓhEígeartaigh SS, Byrne KP, Wolfe KH, Evolutionary erosion of yeast sex chromosomes by mating-type switching accidents. Proc Natl Acad Sci U S A 108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Haase MAB, Kominek J, Opulente DA, Shen XX, LaBella AL, Zhou X, DeVirgilio J, Hulfachor AB, Kurtzman CP, Rokas A, Hittinger CT, Repeated horizontal gene transfer of GALactose metabolism genes violates Dollo’s law of irreversible loss. Genetics 217 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hane JK, Lowe RGT, Solomon PS, Tan KC, Schoch CL, Spatafora JW, Crous PW, Kodira C, Birren BW, Galagan JE, Torriani SFF, McDonald BA, Oliver RP, Dothideomycete-plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 19 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hartmann FE, Rodríguez RC De La Vega, Gladieux P, Ma WJ, Hood ME, Giraud T, Falush D, Higher Gene Flow in Sex-Related Chromosomes than in Autosomes during Fungal Divergence. Mol Biol Evol 37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hodorová V, Lichancová H, Zubenko S, Sienkiewicz K, Penir SMU, Afanasyev P, Boceck D, Bonnin S, Hakobyan S, Smyczynska U, Zhivkoplias E, Zlatohurska M, Tralle E, Frolova A, Pryszcz LP, Brejová B, Vinař T, Nosek J, Genome Sequence of the Yeast Saprochaete ingens CBS 517.90. Microbiol Resour Announc 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jackson AP, Gamble JA, Yeomans T, Moran GP, Saunders D, Harris D, Aslett M, Barrell JF, Butler G, Citiulo F, Coleman DC, De Groot PWJ, Goodwin TJ, Quail MA, McQuillan J, Munro CA, Pain A, Poulter RT, Rajandream MA, Renauld H, Spiering MJ, Tivey A, Gow NAR, Barrell B, Sullivan DJ, Berriman M, Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans. Genome Res 19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, Schmutz J, Lindquist E, Dehal P, Shapiro H, Jin YS, Passoth V, Richardson PM, Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol 25 (2007). [DOI] [PubMed] [Google Scholar]
- 157.Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, Davis RW, Scherer S, The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A 101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kämper J, Kahmann R, Bölker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Müller O, Perlin MH, Wösten HAB, De Vries R, Ruiz-Herrera J, Reynaga-Peña CG, Snetselaar K, McCann M, Pérez-Martín J, Feldbrügge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, González-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Münch K, Rössel N, Scherer M, Vraněs M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho ECH, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li W, Sanchez-Alonso P, Schreier PH, Häuser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schlüter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Güldener U, Münsterkötter M, Haase D, Oesterheld M, Mewes HW, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW, Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444 (2006). [DOI] [PubMed] [Google Scholar]
- 159.Kellis M, Birren BW, Lander ES, Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428 (2004). [DOI] [PubMed] [Google Scholar]
- 160.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäck B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Costa MD, Doré J, Floudas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Högberg N, Johansson T, Khouja HR, Labutti K, Lahrmann U, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, Tunlid A, Grigoriev IV, Hibbett DS, Martin F, Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet 47 (2015). [DOI] [PubMed] [Google Scholar]
- 161.Krause DJ, Kominek J, Opulente DA, Shen XX, Zhou X, Langdon QK, DeVirgilio J, Hulfachor AB, Kurtzman CP, Rokas A, Hittinger CT, Functional and evolutionary characterization of a secondary metabolite gene cluster in budding yeasts. Proc Natl Acad Sci U S A 115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lachance MA, Hurtado E, Hsiang T, A stable phylogeny of the large-spored Metschnikowia clade. Yeast 33 (2016). [DOI] [PubMed] [Google Scholar]
- 163.Lichancová H, Hodorová V, Sienkiewicz K, Penir SMU, Afanasyev P, Boceck D, Bonnin S, Hakobyan S, Krawczyk PS, Smyczynska U, Zhivkoplias E, Zlatohurska M, Odrzywolski A, Tralle E, Frolova A, Pryszcz LP, Brejová B, Vinař T, Nosek J, Genome Sequence of Flavor-Producing Yeast Saprochaete suaveolens NRRL Y-17571. Microbiol Resour Announc 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liti G, Nguyen Ba AN, Blythe M, Müller CA, Bergström A, Cubillos FA, Dafhnis-Calas F, Khoshraftar S, Malla S, Mehta N, Siow CC, Warringer J, Moses AM, Louis EJ, Nieduszynski CA, High quality de novo sequencing and assembly of the Saccharomyces arboricolus genome. BMC Genomics 14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, Tsai IJ, Bergman CM, Bensasson D, O’Kelly MJT, Van Oudenaarden A, Barton DBH, Bailes E, Nguyen AN, Jones M, Quail MA, Goodhead I, Sims S, Smith F, Blomberg A, Durbin R, Louis EJ, Population genomics of domestic and wild yeasts. Nature 458 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lobo FP, Gonçalves D, Alves SL, Gerber AL, de V. ATR, Basso LC, Franco GR, Soares MA, Cadete RM, Rosa CA, Stambuk BU, Gonçalves DL, Alves SL, Gerber AL, de V. ATR, Basso LC, Franco GR, Soares MA, Cadete RM, Rosa CA, Stambuk BU, Draft genome sequence of the D-xylose-fermenting yeast Spathaspora arborariae UFMG-HM19.1AT. Genome Announcments 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, Vamathevan J, Miranda M, Anderson IJ, Fraser JA, Allen JE, Bosdet IE, Brent MR, Chiu R, Doering TL, Donlin MJ, D’Souza CA, Fox DS, Grinberg V, Fu J, Fukushima M, Haas BJ, Huang JC, Janbon G, Jones SJM, Koo HL, Krzywinski MI, Kwon-Chung JK, Lengeler KB, Maiti R, Marra MA, Marra RE, Mathewson CA, Mitchell TG, Pertea M, Riggs FR, Salzberg SL, Schein JE, Shvartsbeyn A, Shin H, Shumway M, Specht CA, Suh BB, Tenney A, Utterback TR, Wickes BL, Wortman JR, Wye NH, Kronstad JW, Lodge JK, Heitman J, Davis RW, Fraser CM, Hyman RW, The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science (1979) 307 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lopes DD, Cibulski SP, Mayer FQ, Siqueira FM, Rosa CA, Hector RE, Ayub MAZ, Draft genome sequence of the D-xylose-fermenting yeast Spathaspora xylofermentans UFMG-HMD23.3. Genome Announc 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lopes MR, Morais CG, Kominek J, Cadete RM, Soares MA, Uetanabaro APT, Fonseca C, Lachance M-A, Hittinger CT, Rosa CA, Genomic analysis and D-xylose fermentation of three novel Spathaspora species: Spathaspora girioi sp. nov., Spathaspora hagerdaliae f. a., sp. nov. and Spathaspora gorwiae f. a., sp. nov. FEMS Yeast Res 16 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Lopes MR, Batista TM, Franco GR, Ribeiro LR, Santos ARO, Furtado C, Moreira RG, Goes-Neto A, Vital MJS, Rosa LH, Lachance MA, Rosa CA, Scheffersomyces stambukii f.A., sp. Nov., a D-xylose-fermenting species isolated from rotting wood. Int J Syst Evol Microbiol 68 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Love KR, Shah KA, Whittaker CA, Wu J, Bartlett MC, Ma D, Leeson RL, Priest M, Borowsky J, Young SK, Love JC, Comparative genomics and transcriptomics of Pichia pastoris. BMC Genomics 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.MacCallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, Malek J, McKernan K, Ranade S, Shea TP, Williams L, Young S, Nusbaum C, Jaffe DB, ALLPATHS 2: Small genomes assembled accurately and with high continuity from short paired reads. Genome Biol 10 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Martinez D, Challacombe J, Morgenstern I, Hibbett D, Schmoll M, Kubicek CP, Ferreira P, Ruiz-Duenas FJ, Martinez AT, Kersten P, Hammel KE, Vanden Wymelenberg A, Gaskell J, Lindquist E, Sabat G, BonDurant SS, Larrondo LF, Canessa P, Vicuna R, Yadav J, Doddapaneni H, Subramanian V, Pisabarro AG, Lavín JL, Oguiza JA, Master E, Henrissat B, Coutinho PM, Harris P, Magnuson JK, Baker SE, Bruno K, Kenealy W, Hoegger PJ, Kües U, Ramaiya P, Lucas S, Salamov A, Shapiro H, Tu H, Chee CL, Misra M, Xie G, Teter S, Yaver D, James T, Mokrejs M, Pospisek M, Grigoriev IV, Brettin T, Rokhsar D, Berka R, Cullen D, Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci U S A 106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Martinez D, Larrondo LF, Putnam N, Sollewijn Gelpke MD, Huang K, Chapman J, Helfenbein KG, Ramaiya P, Detter JC, Larimer F, Coutinho PM, Henrissat B, Berka R, Cullen D, Rokhsar D, Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol 22 (2004). [DOI] [PubMed] [Google Scholar]
- 175.Morais CG, Batista TM, Kominek J, Borelli BM, Furtado C, Moreira RG, Franco GR, Rosa LH, Fonseca C, Hittinger CT, Lachance MA, Rosa CA, Spathaspora boniae sp. nov., a D-xylose-fermenting species in the Candida albicans/lodderomyces clade. Int J Syst Evol Microbiol 67 (2017). [DOI] [PubMed] [Google Scholar]
- 176.Morales L, Noel B, Porcel B, Marcet-Houben M, Hullo MF, Sacerdot C, Tekaia F, Leh-Louis V, Despons L, Khanna V, Aury JM, Barbe V, Couloux A, Labadie K, Pelletier E, Souciet JL, Boekhout T, Gabaldon T, Wincker P, Dujon B, Complete DNA sequence of Kuraishia capsulata illustrates novel genomic features among budding yeasts (saccharomycotina). Genome Biol Evol 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Morel G, Sterck L, Swennen D, Marcet-Houben M, Onesime D, Levasseur A, Jacques N, Mallet S, Couloux A, Labadie K, Amselem J, Beckerich JM, Henrissat B, Van De Peer Y, Wincker P, Souciet JL, Gabaldón T, Tinsley CR, Casaregola S, Differential gene retention as an evolutionary mechanism to generate biodiversity and adaptation in yeasts. Sci Rep 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Naseeb S, James SA, Alsammar H, Michaels CJ, Gini B, Nueno-Palop C, Bond CJ, McGhie H, Roberts IN, Delneri D, Saccharomyces jurei sp. Nov., isolation and genetic identification of a novel yeast species from Quercus robur. Int J Syst Evol Microbiol 67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Neafsey DE, Barker BM, Sharpton TJ, Stajich JE, Park DJ, Whiston E, Hung CY, McMahan C, White J, Sykes S, Heiman D, Young S, Zeng Q, Abouelleil A, Aftuck L, Bessette D, Brown A, FitzGerald M, Lui A, Macdonald JP, Priest M, Orbach MJ, Galgiani JN, Kirkland TN, Cole GT, Birren BW, Henn MR, Taylor JW, Rounsley SD, Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res 20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Riccombeni A, Vidanes G, Proux-Wéra E, Wolfe KH, Butler G, Sequence and analysis of the genome of the pathogenic yeast Candida orthopsilosis. PLoS One 7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Riley R, Haridas S, Wolfe KH, Lopes MR, Hittinger CT, Göker M, Salamov AA, Wisecaver JH, Long TM, Calvey CH, Aerts AL, Barry KW, Choi C, Clum A, Coughlan AY, Deshpande S, Douglass AP, Hanson SJ, Klenk H-P, LaButti KM, Lapidus A, Lindquist EA, Lipzen AM, Meier-Kolthoff JP, Ohm RA, Otillar RP, Pangilinan JL, Peng Y, Rokas A, Rosa CA, Scheuner C, Sibirny AA, Slot JC, Stielow JB, Sun H, Kurtzman CP, Blackwell M, V Grigoriev I, Jeffries TW, Comparative genomics of biotechnologically important yeasts. Proceedings of the National Academy of Sciences 113, 9882–9887 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sarilar V, Devillers H, Freel KC, Schacherer J, Neuvéglise C, Draft genome sequence of Lachancea lanzarotensis CBS 12615T, an ascomycetous yeast isolated from grapes. Genome Announc 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Scannell DR, Frank AC, Conant GC, Byrne KP, Woolfit M, Wolfe KH, Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc Natl Acad Sci U S A 104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Scannell DR, Zill OA, Rokas A, Payen C, Dunham MJ, Eisen MB, Rine J, Johnston M, Hittinger CT, The awesome power of yeast evolutionary genetics: New genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3: Genes, Genomes, Genetics 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schäfer B, Hansen M, Lang BF, Transcription and RNA-processing in fission yeast mitochondria. RNA 11 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sonnenberg ASM, Gao W, Lavrijssen B, Hendrickx P, Sedaghat-Tellgerd N, Foulongne-Oriol M, Kong WS, Schijlen EGWM, Baars JJP, Visser RGF, A detailed analysis of the recombination landscape of the button mushroom Agaricus bisporus var. bisporus. Fungal Genetics and Biology 93 (2016). [DOI] [PubMed] [Google Scholar]
- 187.Suzuki T, Hoshino T, Matsushika A, Draft genome sequence of Kluyveromyces marxianus strain DMB1, isolated from sugarcane bagasse hydrolysate. Genome Announc 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Toome M, Ohm RA, Riley RW, James TY, Lazarus KL, Henrissat B, Albu S, Boyd A, Chow J, Clum A, Heller G, Lipzen A, Nolan M, Sandor L, Zvenigorodsky N, Grigoriev IV, Spatafora JW, Aime MC, Genome sequencing provides insight into the reproductive biology, nutritional mode and ploidy of the fern pathogen Mixia osmundae. New Phytologist 202 (2014). [DOI] [PubMed] [Google Scholar]
- 189.Vakirlis N, Sarilar V, Drillon G, Fleiss A, Agier N, Meyniel JP, Blanpain L, Carbone A, Devillers H, Dubois K, Gillet-Markowska A, Graziani S, Huu-Vang N, Poirel M, Reisser C, Schott J, Schacherer J, Lafontaine I, Llorente B, Neuvéglise C, Fischer G, Reconstruction of ancestral chromosome architecture and gene repertoire reveals principles of genome evolution in a model yeast genus. Genome Res 26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Van Kan JAL, Stassen JHM, Mosbach A, Van Der Lee TAJ, Faino L, Farmer AD, Papasotiriou DG, Zhou S, Seidl MF, Cottam E, Edel D, Hahn M, Schwartz DC, Dietrich RA, Widdison S, Scalliet G, A gapless genome sequence of the fungus Botrytis cinerea. Mol Plant Pathol 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vaux S, Criscuolo A, Desnos-Ollivier M, Diancourt L, Tarnaud C, Vandenbogaert M, Brisse S, Coignard B, Dromer F, Multicenter outbreak of infections by Saprochaete clavata, an unrecognized opportunistic fungal pathogen. mBio 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Vega-Alvarado L, Gómez-Angulo J, Escalante-García Z, Grande R, Gschaedler-Mathis A, Amaya-Delgado L, Sanchez-Flores A, Arrizon J, High-quality draft genome sequence of Candida apicola NRRL Y-50540. Genome Announc 3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Vervoort Y, Herrera-Malaver B, Mertens S, Guadalupe Medina V, Duitama J, Michiels L, Derdelinckx G, Voordeckers K, Verstrepen KJ, Characterization of the recombinant Brettanomyces anomalus β-glucosidase and its potential for bioflavouring. J Appl Microbiol 121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wendland J, Walther A, Chromosome number reduction in eremothecium coryli by two telomere-to-telomere fusions. Genome Biol Evol 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wohlbach DJ, Kuo A, Sato TK, Potts KM, Salamov AA, LaButti KM, Sun H, Clum A, Pangilinan JL, Lindquist EA, Lucas S, Lapidus A, Jin M, Gunawan C, Balan V, Dale BE, Jeffries TW, Zinkel R, Barry KW, Grigoriev IV, Gasch AP, Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. Proc Natl Acad Sci U S A 108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wortman JR, Gilsenan JM, Joardar V, Deegan J, Clutterbuck J, Andersen MR, Archer D, Bencina M, Braus G, Coutinho P, von Döhren H, Doonan J, Driessen AJM, Durek P, Espeso E, Fekete E, Flipphi M, Estrada CG, Geysens S, Goldman G, de Groot PWJ, Hansen K, Harris SD, Heinekamp T, Helmstaedt K, Henrissat B, Hofmann G, Homan T, Horio T, Horiuchi H, James S, Jones M, Karaffa L, Karányi Z, Kato M, Keller N, Kelly DE, Kiel JAKW, Kim JM, van der Klei IJ, Klis FM, Kovalchuk A, Krasevec N, Kubicek CP, Liu B, Maccabe A, Meyer V, Mirabito P, Miskei M, Mos M, Mullins J, Nelson DR, Nielsen J, Oakley BR, Osmani SA, Pakula T, Paszewski A, Paulsen I, Pilsyk S, Pócsi I, Punt PJ, Ram AFJ, Ren Q, Robellet X, Robson G, Seiboth B, van Solingen P, Specht T, Sun J, Taheri-Talesh N, Takeshita N, Ussery D, vanKuyk PA, Visser H, van de Vondervoort PJI, de Vries RP, Walton J, Xiang X, Xiong Y, Zeng AP, Brandt BW, Cornell MJ, van den Hondel CAMJJ, Visser J, Oliver SG, Turner G, The 2008 update of the Aspergillus nidulans genome annotation: a community effort. Fungal Genet Biol 46 Suppl 1 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xiao H, Shao Z, Jiang Y, Dole S, Zhao H, Exploiting Issatchenkia orientalis SD108 for succinic acid production. [Preprint] (2014). 10.1186/s12934-014-0121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Xu J, Saunders CW, Hu P, Grant RA, Boekhout T, Kuramae EE, Kronstad JW, DeAngelis YM, Reeder NL, Johnstone KR, Leland M, Fieno AM, Begley WM, Sun Y, Lacey MP, Chaudhary T, Keough T, Chu L, Sears R, Yuan B, Dawson TL, Dandruff-associated Malassezia genomes reveal convergent and divergent virulence traits shared with plant and human fungal pathogens. Proc Natl Acad Sci U S A 104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yang J, Wang L, Ji X, Feng Y, Li X, Zou C, Xu J, Ren Y, Mi Q, Wu J, Liu S, Liu Y, Huang X, Wang H, Niu X, Li J, Liang L, Luo Y, Ji K, Zhou W, Yu Z, Li G, Liu Y, Li L, Qiao M, Feng L, Zhang KQ, Genomic and proteomic analyses of the fungus arthrobotrys oligospora provide insights into nematode-trap formation. PLoS Pathog 7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhang X, Cheng X, Liu L, Liu S, Genome Sequence Resource for the Plant Pathogen Sclerotinia sclerotiorum WH6 Isolated in China. Plant Dis 105 (2021). [DOI] [PubMed] [Google Scholar]
- 201.Kunze G, Gaillardin C, Czernicka M, Durrens P, Martin T, Böer E, Gabaldón T, Cruz JA, Talla E, Marck C, Goffeau A, Barbe V, Baret P, Baronian K, Beier S, Bleykasten C, Bode R, Casaregola S, Despons L, Fairhead C, Giersberg M, Gierski PP, Hähnel U, Hartmann A, Jankowska D, Jubin C, Jung P, Lafontaine I, Leh-Louis V, Lemaire M, Marcet-Houben M, Mascher M, Morel G, Richard GF, Riechen J, Sacerdot C, Sarkar A, Savel G, Schacherer J, Sherman DJ, Stein N, Straub ML, Thierry A, Trautwein-Schult A, Vacherie B, Westhof E, Worch S, Dujon B, Souciet JL, Wincker P, Scholz U, Neuvéglise C, The complete genome of Blastobotrys (Arxula) adeninivorans LS3 - A yeast of biotechnological interest. Biotechnol Biofuels 7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Opulente DA, Langdon QK, Jarzyna M, Buh KV, Haase MAB, Groenewald M, Hittinger CT. Taxogenomic analysis of a novel yeast species isolated from soil, Pichia galeolata sp. nov. Yeast 40 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genome sequence assemblies and raw sequencing data have been deposited in GenBank under the accessions noted in Data S1. All other data, including data on growth on different carbon and nitrogen sources and isolation environment data, have been deposited in Figshare at https://doi.org/10.25452/figshare.plus.c.6714042 (42). All code has been deposited in GitHub at https://zenodo.org/records/10709452 (91) and https://zenodo.org/doi/10.5281/zenodo.10711058 (92) and is available in Figshare at https://doi.org/10.25452/figshare.plus.c.6714042 (42). Nearly all strains came from globally recognized yeast culture collections and may be ordered from the United States Department of Agriculture (https://nrrl.ncaur.usda.gov for NRRL strains) or Westerdijk Fungal Biodiversity Institute (https://wi.knaw.nl for CBS strains) under their respective Material Transfer Agreements (MTAs) for publicly deposited strains; currently, NRRL only requires an MTA for strains requiring BSL-2 precautions. Strains from the Hittinger Lab that represent candidates for novel species that have not yet been formally described or deposited at CBS or NRRL may be obtained from cthittinger@wisc.edu under the Uniform Biological MTA or other mutually acceptable MTA.