

Abstract
Although a significant number of studies on vascular tissue engineering have been reported, the current availability of vessel substitutes in the clinic remains limited mainly due to the mismatch of their mechanical properties and biological functions with native vessels. In this study, a novel approach to fabricating a vessel graft for vascular tissue engineering was developed by promoting differentiation of human bone marrow mesenchymal stem cells (MSCs) into endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) on a native vascular extracellular matrix (ECM) scaffold in a rotary bioreactor. The expression levels of CD31 and vWF, and the LDL uptake capacity as well as the angiogenesis capability of the EC-like cells in the dynamic culture system were significantly enhanced compared to the static system. In addition, α-actin and smoothelin expression, and contractility of VSMC-like cells harvested from the dynamic model were much higher than those in a static culture system. The combination of on-site differentiation of stem cells towards vascular cells in the natural vessel ECM scaffold and maturation of the resulting vessel construct in a dynamic cell culture environment provides a promising approach to fabricating a clinically applicable vessel graft with similar mechanical properties and physiological functions to those of native blood vessels.
Introduction
Small-diameter vascular grafts are considered to be promising for the treatment of late stage vascular diseases, one of the largest causes of morbidity and mortality worldwide.1–3 However, current blood vessel substitutes are susceptible to failure mainly due to the mismatch of mechanical properties and biological functions with native vessels, and the rapid occlusion after implantation.4,5 Fabrication of vascular conduits with replicated mechanical properties and biological functions of native blood vessels remains a great challenge.6 Native blood vessels are composed of a single layer of endothelial cells (ECs) on the luminal surface and multiple layers of vascular smooth muscle cells (VSMCs) embedded in a fibrous extracellular matrix (ECM) of the blood vessel wall.7 Functional VSMCs and ECs, and a vessel scaffold that can replicate the mechanical properties and biocompatibility of the native blood vessel wall are the three major elements of tissue-engineered blood vessels (TEBVs). Compared to synthetic scaffolds that can provide desirable mechanical support to accommodate vascular cells and maintain durability in vivo,8 native decellularized ECM provides not only excellent mechanical strength and biocompatibility, but also beneficial bioactive components for cell growth and stem cell differentiation in tissue engineering and regenerative medicine.8–11
Limited proliferation capacity and rapid cell function attenuation during in vitro expansion of primary vascular cells have motivated the development of new approaches in which stem cells are induced to differentiate into ECs and VSMCs for vascular tissue engineering.12–16 However, the differentiation of stem cells into vascular cells was usually promoted by chemical stimuli in a planar culture system under static conditions,17,18 and the obtained cells have to be trypsinized and reseeded into the tubular scaffolds to constitute vessel grafts,8,14 which potentially impairs the phenotype and function of resulting cells. In addition, VSMCs lying in the media of blood vessel wall can withstand tensile stress as the vessel dilates in response to the pulsatile pressure exerted by the cardiac cycle, while ECs lining the lumen of the vessels experience shear stress caused by blood flow.19 These mechanical forces play a substantial role in the commitment of vascular cell functions and maintenance of vascular homeostasis.19 However, the impact of these mechanical cues on stem cell differentiation towards EC or VSMC phenotypes is not fully understood, despite the fact that some evidence suggested that substrate stiffness was able to alter the differentiation of stem cells towards VSMCs and ECs.20–23
In this context, this study aims to compare the differentiation efficiency of human bone marrow derived mesenchymal stem cells (MSCs) towards ECs and VSMCs on a native vessel ECM scaffold in a static culture system and in a dynamic cell culture system (rotary bioreactor). The mRNA and protein expression levels of the markers of ECs and VSMCs as well as EC- and VSMC-specific functions were compared for the two culture systems, where human aortic ECs and VSMCs were used as positive controls, respectively. In this study, we hypothesize that the soluble growth factor directed differentiation of human MSCs towards VSMCs and ECs would be enhanced by the bioactive components of the native arterial ECM and the mechanical stress generated in a rotary bioreactor.
Materials and methods
Preparation of decellularized ECM scaffolds and DNA quantification analysis
The decellularized pig carotid artery scaffolds (Fig. 1A) were prepared according to the procedures described in our previous studies.11 Briefly, carotid arteries were treated with 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) buffer (8 mM CHAPS, 1 M NaCl and 25 mM EDTA) for 22 h, followed by three brief washes with PBS. Next, carotid vessels were incubated with sodium dodecyl sulfate (SDS) buffer composed of 1.8 mM SDS, 1 M NaCl, and 25 mM EDTA for 24 h, followed by ten complete washes with PBS. Lastly, the carotid vessels were treated with 0.2 mg ml−1 DNase and 1 mg ml−1 RNase in PBS for 16 h at 37 °C, and then washed thoroughly with PBS ten times. All reagents were bought from Sigma Aldrich (Sigma, St. Louis, MO).
Fig. 1.

Schematic of the ex vivo bioreactor incorporated with a decellularized carotid artery scaffold. (A) Decellularized ECM scaffold. (B) MSCs were seeded on either the intimal or adventitial side of the decellularized carotid artery scaffold and mounted on the bioreactor insert. (C) The ECM-MSCs were cultured in a vascular bioreactor with VEGF medium inside the vessel scaffold and TGFβ1 medium outside the vessel scaffold. (D) The tissue engineered blood vessel (TEBV).
The decellularization efficiency was determined by DNA quantification analysis as described in our previous study.24 Briefly, the DNA content of fresh carotid arteries and decellularized vessel ECM scaffolds was measured using PicoGreen DNA assay following the protocol provided by the manufacturer (Invitrogen, Carlsbad, CA, USA). The fluorescence intensity for each sample was read at an excitation wavelength of 480 nm and an emission wavelength of 530 nm using a microplate reader (Infinite M200, Tecan, USA).
On-site differentiation of MSCs on decellularized vascular ECM scaffold in a rotary bioreactor
Human bone marrow-derived MSCs (P1–P5) were cultured in MSC culture medium supplemented with MSC growth kit. Human aortic SMCs (P1–P3) were grown in vascular cell basal medium supplemented with VSMC growth kit. Human aortic ECs (P1–P3) were cultured in vascular cell basal medium supplemented with EC growth kit. The three types of cells were subcultured when they reached approximately 90% confluence and the medium was refreshed every other day. All cell lines, culture media, and growth kits were purchased from ATCC (Manassas, VA, USA).
To evaluate the combined effects of a dynamic cell culture environment and the decellularized ECM scaffold on the differentiation of MSCs towards ECs, MSCs were grown under different culture conditions as follows: (1) MSCs cultured in complete MSC medium were defined as the MSC group; (2) MSCs seeded into the intimal surface of decellularized vessel ECM scaffolds (1 × 106 cells per scaffold) and cultivated in complete EC medium supplemented with 50 ng ml−1 vascular endothelial growth factor (VEGF, Invitrogen, Carlsbad, CA, USA) were defined as the static group; (3) the cells harvested from the vessel ECM scaffold in an ex vivo blood vessel bioreactor system (Synthecon, Houston, TX) were defined as the dynamic group, as schematically shown in Fig. 1.25 A laminar perfusion system throughout the whole circuit driven by a digital peristaltic pump not only exerted longitudinal shear stress on the cells seeded on the luminal side of the vessel scaffold, but also provided circumferential tensile stress for the cells grown on the adventitial side of the vessel scaffold. The decellularized pig carotid artery scaffold was mounted onto the insert of the bioreactor under sterile conditions (Fig. 1B). A circuit was constructed after placing the insert back into the bioreactor (Fig. 1C). Thereafter, 1 × 106 MSCs in 2 ml of EC medium supplemented with 50 ng ml−1 VEGF were injected into the lumen of the vessel scaffold from the port at the end of the rotary cylinder, and the rotary cylinder was rotated 90° every 30 min for 4 h to promote cell attachment to the vessel ECM scaffold. Finally, the perfusion through the whole circuit was started at a low flow rate of 0.5 ml min−1 for the first 24 h, and then increased to 1.5 ml min−1 for the next two weeks; (4) ECs cultured in complete EC medium were used as a positive control.
The differentiation of MSCs towards VSMCs in the bioreactor was conducted following a similar strategy to that used for the MSC–EC differentiation. (1) MSCs cultured in complete MSC medium were defined as the MSC group; (2) MSCs seeded onto the adventitial surface of decellularized vessel ECM scaffolds (1 × 106 cells per scaffold) and maintained in complete MSC medium supplemented with 10 ng ml−1 transforming growth factor-beta 1 (TGFβ1, Invitrogen, Carlsbad, CA, USA) were defined as the static group; (3) 1 × 106 MSCs were seeded manually onto the adventitial side of the vessel scaffold that was mounted on the insert of the bioreactor, and then the insert was placed back into the bioreactor and cultured overnight. Lastly, the perfusion was started at a lower flow rate of 0.5 ml min−1 for the first 24 h followed by 1.5 ml min−1 for the remainder two week culture period. The cells harvested from the ECM scaffold cultured in the rotary bioreactor system were defined as the dynamic group; and (4) VSMCs cultured in complete VSMC medium were used as a positive control. All the cells were incubated at 37 °C in a 5% CO2 and 95% humidity environment, and fed every other day. After a two-week incubation in the rotary bioreactor, the engineered blood vessel was harvested for the vascular cell functional characterization (Fig. 1D).
Mechanical force analysis for the bioreactor
The 2.5 cm long vessel scaffold (2.5 mm in diameter) in the bioreactor experiences forces derived from the pressure of the flowing media, the rotation of the scaffold, and the shear stress caused by the flowing media. The fluid velocity is calculated from the flow rate using eqn (1):
| (1) |
where is the volumetric flow rate and is the cross-sectional area. The fluid pressure is given by eqn (2):
| (2) |
where is the density of the media, in this case assumed to be water at 1000 kg m−3. Pressure within the vessel was calculated to be 1.25 × 10−2 Pa. Multiplying by the surface area of the vessel yields a force of 2.45 μN. The rotational stress is given by eqn (3):
| (3) |
where is the angular velocity, is the density of the tissue taken to be that of the muscle at 1059 kg m−3,26 and is the radius. Multiplying by the surface area gives the force over the whole scaffold at 3.39 μN. In the scaffold, the shear stress is given by eqn (4):
| (4) |
where is the viscosity assumed to be that of water at 10−3 Pa s, is the fluid velocity, and is the diameter of the scaffold. Multiplying the shear stress by the surface area of the scaffold yields a force of 0.39 μN.
Cell viability evaluation, histological analysis, and scanning electron microscopy examination
Cell viability evaluation.
To assess the effect of the dynamic culture system on MSC viability, the cells grown on decellularized ECM for two weeks were stained with calcein-AM/EthD-1 solution (Invitrogen, Carlsbad, CA, USA), as reported previously.24 Live cells were stained green because ubiquitous intracellular esterase activity is able to digest non-fluorescent calcein-AM into green fluorescent calcein, whereas dead cells were stained red since EthD-1 can pass through damaged membranes and emit red fluorescence upon binding to nucleic acids. The samples were observed under a confocal microscope (IX83 FV1200, Olympus Life Science, Center Valley, PA, USA).
Histological analysis.
Fresh carotid arteries, decellularized vessel ECM scaffolds, and the cell–ECM constructs after a 14-day cultivation under mechanical stimulation in the rotary bioreactor were fixed with 4% paraformaldehyde (Affymetrix, Santa Clara, CA, USA) at 4 °C overnight, and washed three times with PBS. The paraffin-embedded sections (5 μm thickness) were prepared for staining with hematoxylin and eosin dyes (Sigma, St. Louis, MO, USA) or Masson’s trichrome dyes (Sigma, St. Louis, MO, USA), respectively. Images were acquired using a phase contrast microscope (IX83, Olympus Life Science, Center Valley, PA, USA).
Scanning electron microscopy observation.
After 14-day cultivation under mechanical stimulation in the bioreactor, the cell–ECM constructs were fixed with 2.5% glutaraldehyde (Sigma, St. Louis, MO, USA) overnight at 4 °C, and dehydrated in a series of graded ethanol (25%, 50%, 75%, 95% and 100%) before lyophilization. The freeze-dried samples were then mounted onto stubs, sputtered with platinum, and observed with a scanning electron microscope (SEM, FEI Quanta 450, Beaverton, OR, USA).
Gene expression by real-time RT-PCR analysis.
To compare the mRNA expression levels of marker genes of ECs or VSMCs between different groups, total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol, and quantified by spectrophotometric analysis at 260 nm. 200 ng of RNA for each group was used for reverse transcription with a high capacity cDNA reverse transcription kit (Applied Biosystems, Förster City, CA, USA). Lastly, real-time PCR was performed with TaqMan gene expression assays (Applied Biosystems, Forster City, CA, USA) on a fast real-time PCR instrument (7500 Fast Real-Time PCR system, Applied Biosystems). The relative mRNA expression levels for CD31 (Hs01065279), von Willebrand factor (vWF, Hs01109446), fetal liver kinase-1 (flk-1, Hs00911700), α-actin (Hs00426835), calponin (Hs00206044), and smoothelin (Hs00199489) were estimated and normalized to β-actin (Hs01060665). Each sample was tested in triplicate.
Evaluation of protein expression with immunofluorescence staining
The difference in EC or VSMC specific protein expression level between different groups was determined by immunostaining. The samples from each experimental group were fixed in 4% paraformaldehyde at 4 °C overnight. The fixed vessel scaffolds seeded with cells in the static group and dynamic group were embedded in paraffin and sectioned at 5 μm thickness. After sequential antigen retrieval treatment with 10 mM sodium citrate solution (Invitrogen, Carlsbad, CA, USA), permeabilization with 0.5% Triton-X-100 solution (Sigma, St. Louis, MO, USA) and blocking with 1% bovine serum albumin (Sigma, St. Louis, MO, USA), the sections were stained with the following mouse anti-human antibodies (1 : 50): anti-CD31, anti-vWF, anti-flk-1, anti-smoothelin, anti-α-actin, and anti-calponin at 4 °C overnight. After complete rinsing with PBS, the samples were incubated with FITC-conjugated goat anti-mouse IgG (Invitrogen, Carlsbad, CA, USA) at 37 °C for 2 h, followed by staining with Hoechst 33342. The samples were observed under a confocal microscope. Except for the antibodies anti-μ-actin and anti-calponin that were purchased from Sigma (St. Louis, MO, USA), all other antibodies used in this experiment were purchased from Abcam (Cambridge, MA, USA).
EC-specific functional analysis
Acetylated low-density lipoprotein uptake assay was employed to perform the endothelial cell-specific functional analyses for the cells harvested from all four experimental groups. Briefly, the harvested cells from different groups were incubated with 10 μg ml−1 Dil-labeled acetylated low-density lipoprotein (Dil Ac-LDL, Invitrogen, Carlsbad, CA, USA) in serum-free medium at 37 °C for 4 h, followed by fixing with 4% paraformaldehyde. After that, the samples were stained with Hoechst 33342 to counterstain nuclei, and Dil Ac-LDL uptake was assessed quantitatively by using confocal microscopy.
Angiogenesis assay was carried out using Matrigel solution (Corning) at 5 mg ml−1. A 96-well plate was loaded with 50 μl of Matrigel in each well, and then incubated at 37 °C for 30 min before polymerization. The cells from different groups were harvested with trypsin and seeded on the Matrigel surface (8 × 103 cells per well). After 24 h of culture, five randomly selected fields for each sample were photographed using a phase contrast microscope. The number of junctions and segments as well as total segment length within each image was analyzed using Image J.
VSMC-specific functional analysis
Cell contractile capacity in different groups was evaluated by cell area reduction in response to vasoactive agonists, as described in our previous study.11 The cells from different groups were trypsinized and seeded in a 24-well plate with a final concentration of 1 × 104 cells per well. After overnight-culture, the cells were stimulated with 50 mM KCl or 1 mM carbachol (Sigma, St. Louis, MO, USA) for 15 min. Images were acquired using a phase contrast microscope (IX83, Olympus Life Science, Center Valley, PA, USA), and the cell area reduction was analyzed with Image J.
A collagen gel contraction experiment was performed using rat tail collagen I solution (Corning, New York, USA) at 2 mg ml−1. Briefly, the cells from different groups were trypsinized and mixed in a neutral collagen solution at 1.5 × 105 cells per ml cell density. Portions of 100 μl were added into a 12-well plate (Corning, New York, USA) and allowed to gelate for 1.5 h at 37 °C. After that, the collagen gels were detached with a spatula and allowed to contract for 24 h or 48 h. Images were captured using a stereo microscope (SZX10, Olympus Life Science, Center Valley, PA, USA) with a digital camera (DP22, Olympus Life Science, Center Valley, PA, USA), and the areas of contracted gels were calculated using Image J.
Statistical analysis
All data were expressed as mean ± standard error of mean (SEM). Statistical analysis was performed by a two-sample t test assuming equal variances, and P < 0.05 was considered statistically significant.
Results
Characterization of decellularized vessel scaffolds
To determine the decellularization efficiency, hematoxylin and eosin (H&E) staining of both native carotid and decellularized ECM scaffolds was performed and the results (Fig. S1, ESI†) clearly showed that cellular and nucleic components were completely removed in the decellularized ECM (Fig. S1C and S1D, ESI†) compared to the native carotid (Fig. S1A and S1B, ESI†). Furthermore, decellularized ECM scaffolds showed an average residual DNA of 0.018 ± 0.005 μg mg−1, which was significantly lower than 0.213 ± 0.026 μg mg−1 contained in the native carotid (Fig. S1E, ESI†). The significantly decreased residual DNA content in the decellularized scaffolds supported the histological staining observations.
Cell viability and morphology of EC-like cells grown on the luminal side of the decellularized ECM scaffold in the bioreactor
Cell viability and morphological characteristics of the EC-like cells grown on the luminal side were examined to evaluate the effect of the dynamic microenvironment provided by both the ECM and bioreactor on MSC differentiation. The cell viability was evaluated by live-dead staining after a two-week culture in the dynamic model. As shown in Fig. 2A, MSCs reseeded on the intimal side of the decellularized ECM scaffold in the dynamic cell culture system displayed high cell viability after two weeks, indicating that a beneficial microenvironment was provided by the dynamic cell culture model. In addition, the intimal surface of the ECM scaffolds recellularized with MSCs was observed by SEM. After two weeks of cultivation, an intact cell layer covered with secreted matrix was observed on the intimal surface of the decellularized scaffold (Fig. 2B). Moreover, both HE staining and Masson’s trichrome staining were applied to visualize the morphological characteristics of MSCs loaded into the dynamic cell culture system. A dense EC-like cell layer was formed on the intimal side of the decellularized ECM scaffold (Fig. 2C–F). All these observations supported the fact that the combination of decellularized ECM and the mechanical force generated by the rotary bioreactor was favorable for MSC attachment, growth, and differentiation.
Fig. 2.
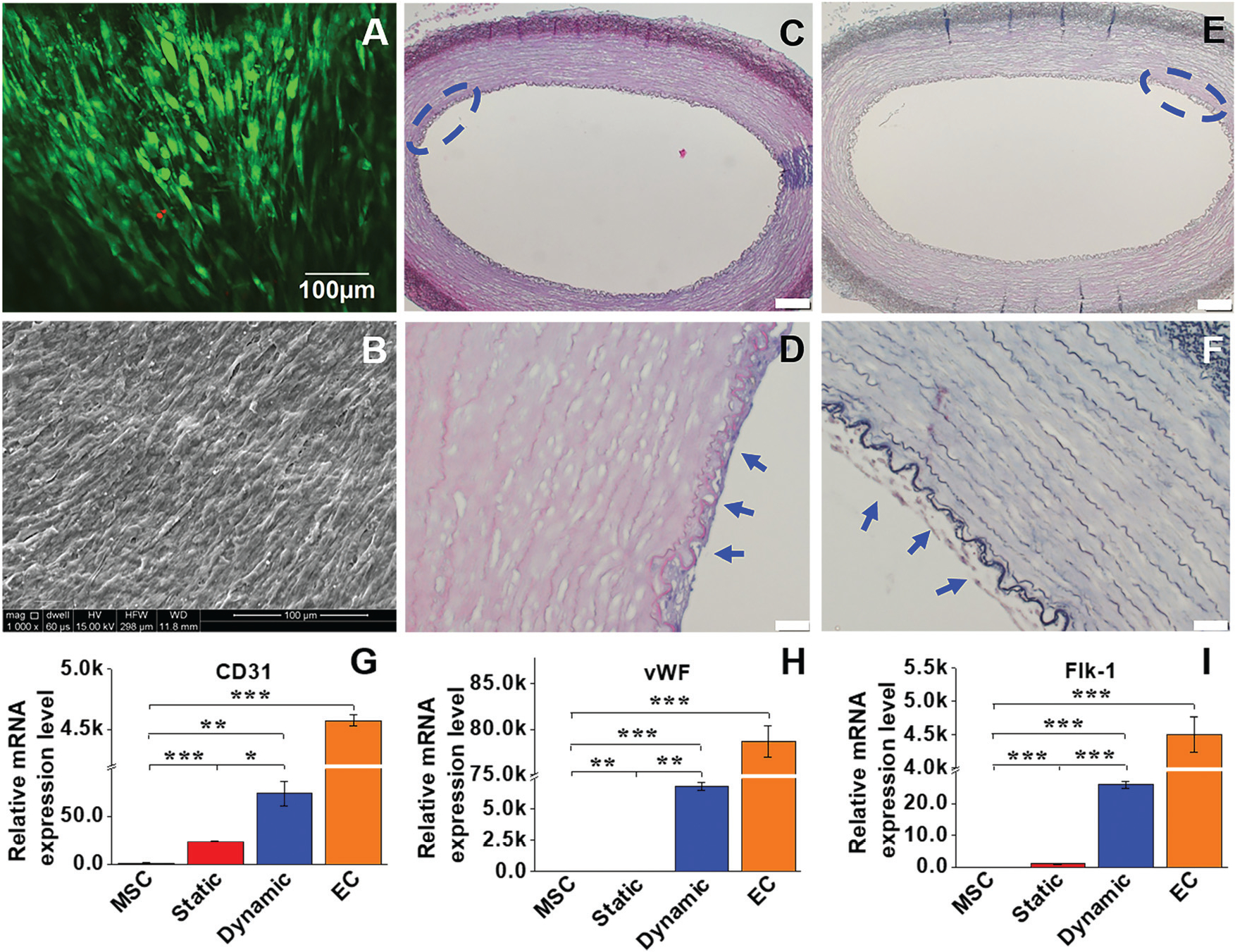
Cell viability and morphological characteristics, and real time RT-PCR analysis of the EC marker gene expression. (A) MSCs reseeded on the intimal side of decellularized ECM in the dynamic model displayed high cell viability after two weeks. (B) An intact cell layer covered with secreted ECM was observed on the intimal surface of the decellularized scaffolds. (C and D) H&E staining and (E and F) Masson’s trichrome staining confirmed that a dense layer of EC-like cells was formed on the intimal side of the decellularized ECM (blue arrows). Each marker gene was significantly upregulated for both static and dynamic groups compared to the negative control (MSCs), and the transcript expression level of each marker gene in the dynamic culture system was significantly higher than that in the static culture system (G–I). Data are expressed as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Gene and protein expression of EC-specific markers
To compare the differentiation efficiency of MSCs towards EC-like cells in the static culture system and in the dynamic bioreactor system, both the mRNA expression and protein expression levels of three EC-specific markers were determined by real time RT-PCR and immunofluorescence staining, respectively. As shown in Fig. 2G–I, each marker gene, including CD31, vWF and flk-1, was significantly upregulated in both the static and dynamic groups compared to MSCs (negative control), but lower than that of the ECs (positive control). More importantly, the transcript expression level of each marker gene in the dynamic culture system was significantly higher than that of the static culture system, suggesting the substantial roles played by mechanical forces in facilitating the VEGF-directed differentiation of MSCs towards the EC-like phonotype. Consistent with the real-time PCR results, immunofluorescence staining for CD31 and vWF showed that the protein expression levels in the dynamic group were much higher than those in the static group and MSC control (Fig. 3), although they were still lower than those in the ECs (positive control). These results provide robust evidence for the fact that the dynamic culture system incorporated with native vascular ECM played an important regulatory role in MSC–EC differentiation.
Fig. 3.
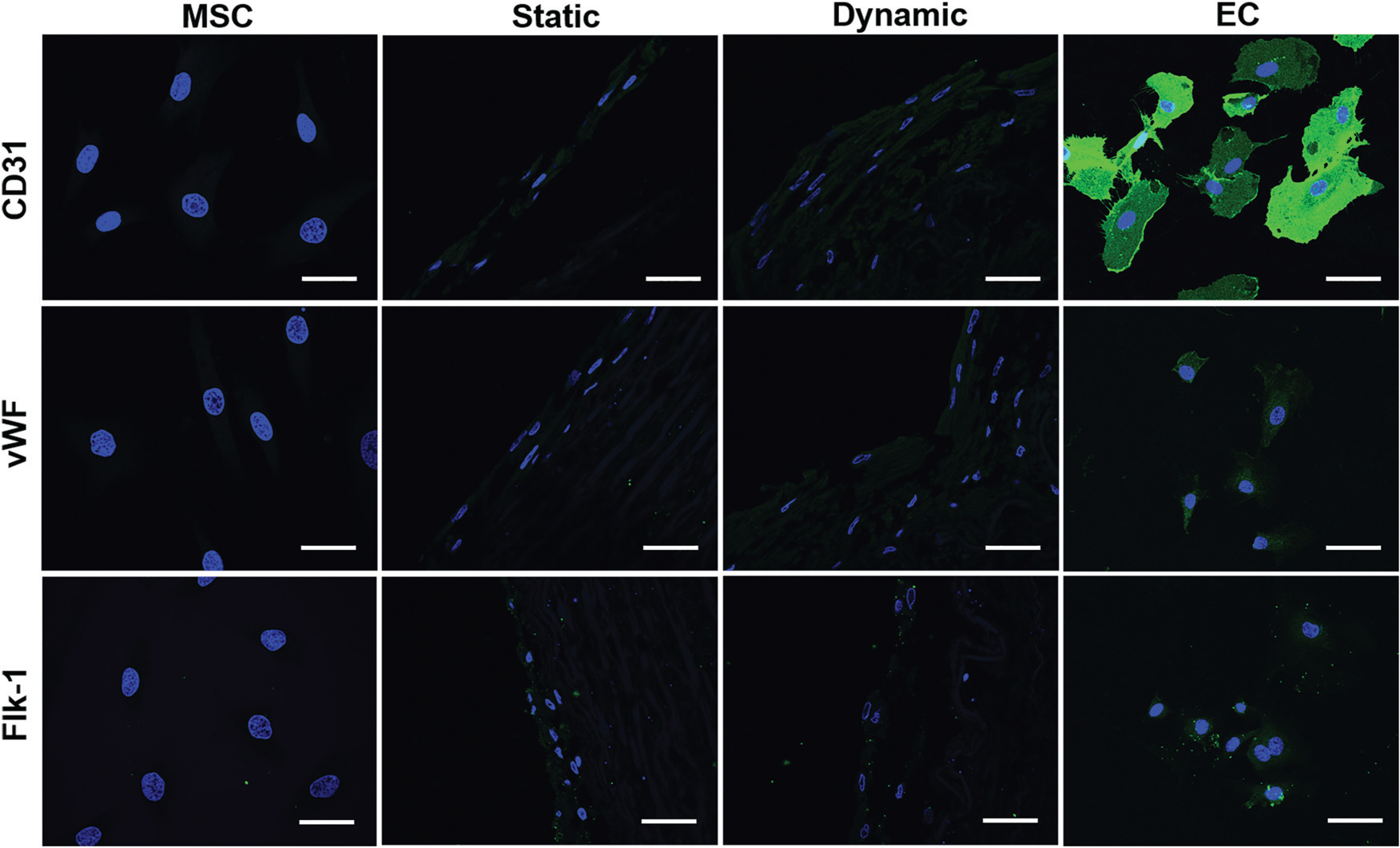
Immunofluorescent staining for EC-specific proteins CD31, vWF, and flk-1. Each marker protein expression level in both the static and dynamic groups was much higher than that in the MSC control group. Both CD31 and vWF expression levels in the dynamic group were stronger than those in the static group. The images represent at least three independent experiments. Scale bars = 50 μm.
Functional evaluation of the MSC-derived EC-like cells
LDL uptake assay and Matrigel angiogenesis were employed to detect the functional activity of EC-like cells differentiated from MSCs in the different culture systems. As shown in Fig. 4A–D, MSCs (negative control) were unable to uptake Dil Ac-LDL, whereas positive control ECs exhibited a high Dil Ac-LDL uptake capacity. Compared to the cells in the static group with a low capability to uptake Dil Ac-LDL, the cells differentiated from the dynamic group showed much higher Dil Ac-LDL uptake capacity, indicating the high MSC–EC differentiation efficiency under shear stress stimulation. The capabilities of four different cell groups to form capillary networks was tested on Matrigel (Fig. 4E–H). The negative control MSCs exhibited a low capacity to form capillary networks (Fig. 4E), whereas a dense capillary network was generated by control ECs on Matrigel after 24 h (Fig. 4H). The cells differentiated in the static group (Fig. 4F) evidently formed more networks than the MSC group, but much less than the dynamic group (Fig. 4G). Quantitative analysis of junction number, segment number, and total segment length for each group was further performed to compare their network formation capabilities. All quantitative parameters of the dynamic group were significantly higher than those of the static group (Fig. 4I–K). Taken together, consistent with genetic and protein assays, all the functional evaluation results confirmed that the microenvironment provided by the native carotid ECM and mechanical cues generated in the bioreactor was able to enhance MSC–EC differentiation.
Fig. 4.
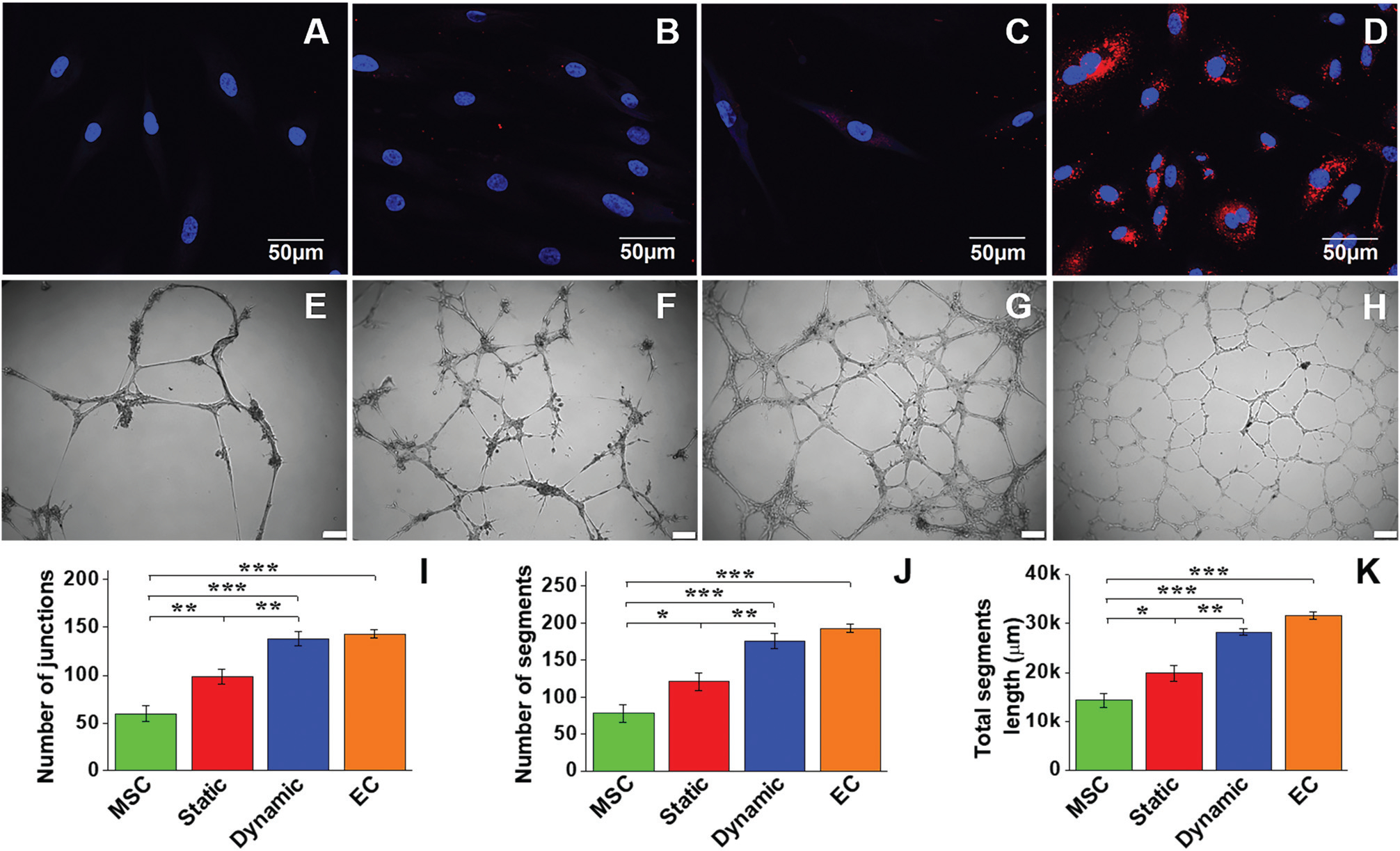
LDL-uptake assay and Matrigel angiogenesis evaluation of MSC-derived EC-like cells. Negative control MSCs were unable to uptake Dil Ac-LDL (A), whereas positive control ECs exhibited a high capability for Dil Ac-LDL uptake (D). Compared to the static culture group (B), the dynamic culture group showed a much higher Dil Ac-LDL uptake capability (C). Negative control MSCs showed a small amount of capillary network formed on the Matrigel after 24 h (E). In contrast, a dense capillary network was generated by positive control ECs on the Matrigel after 24 h (H). The cells of the static group (F) apparently formed more networks than the MSC group, but much less than the dynamic group (G). Scale bars = 100 μm. Quantitative analysis of the parameters of the capillary network showed that significant differences existed among the four different cell groups (I–K). Data are expressed as mean ± SEM of five randomly selected images (48 cm × 36 cm). *P < 0.05, **P < 0.01, ***P < 0.001.
Cell viability and morphology of VSMC-like cells differentiated on the adventitial side of the ECM scaffold in the bioreactor
To evaluate the effect of the microenvironment provided by the adventitial side of the ECM scaffold on cell viability, live-dead staining was also conducted for the dynamic model after two weeks of culture. As shown in Fig. 5A, MSCs seeded on the outside surface of the ECM scaffold exhibited high viability on day 14, supporting the fact that the dynamic culture system with a decellularized ECM is favorable for MSC survival on both the intimal and adventitial sides. SEM imaging further demonstrated that MSCs seeded on the adventitial side of the ECM scaffold were able to secrete ECM proteins and form an intact cell layer with a uniform topology after two weeks of cultivation (Fig. 5B). The histological morphology of the dynamic cell group was characterized by both H&E and Masson’s trichrome staining (Fig. 5C–F), where a number of VSMC-like cell layers were formed on the adventitial surface of the vessel scaffolds. Therefore, the decellularized carotid ECM is an ideal scaffold to support MSC growth and differentiation on both sides for fabricating vessel grafts for vascular tissue engineering.
Fig. 5.
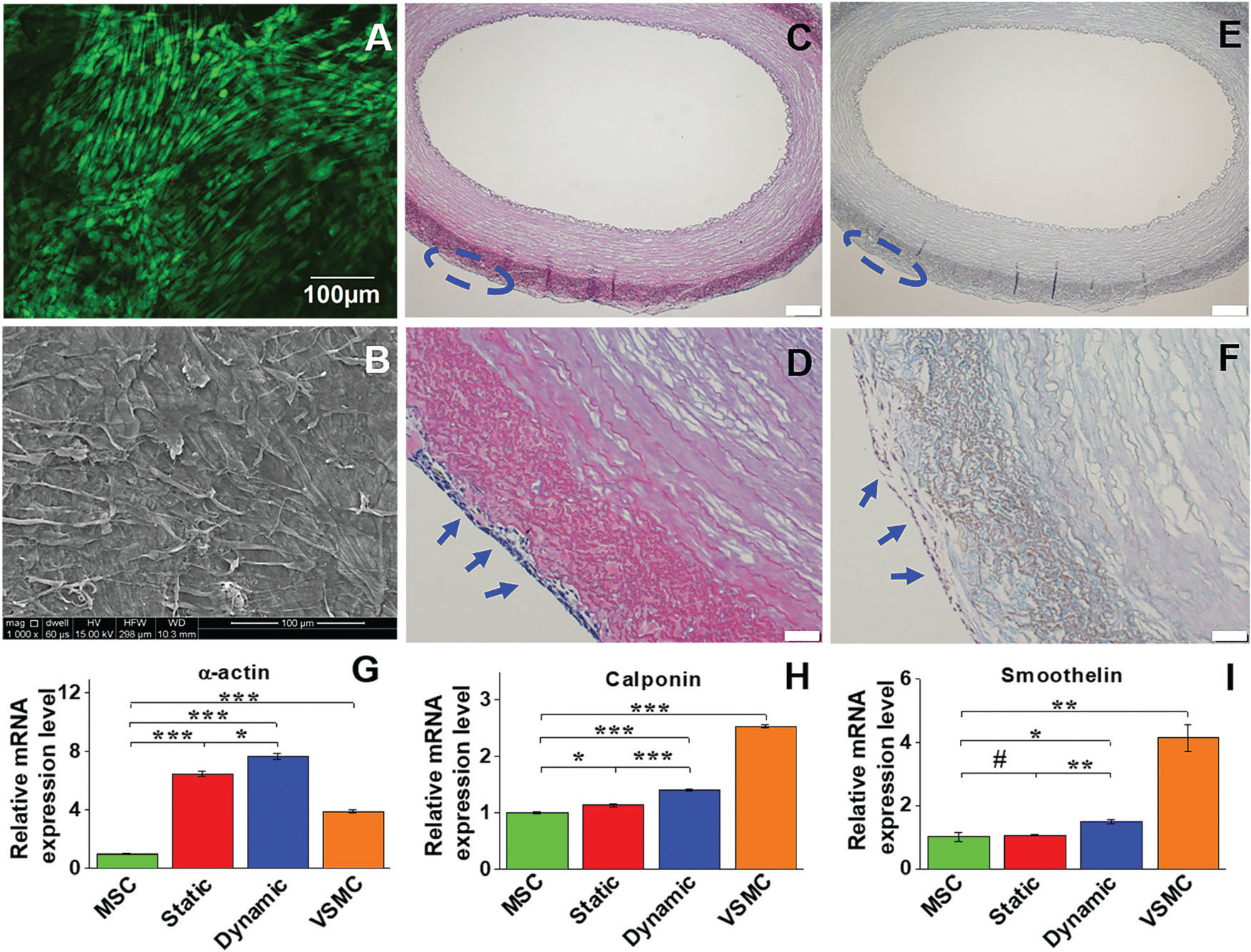
Cell viability and morphology, and mRNA expression of VSMC marker proteins in different experimental groups. Live-dead staining showed that cells cultured on the adventitial side of the vessel scaffold exhibited high viability on day 14 (A). SEM imaging also demonstrated that the cells cultured in the dynamic system were able to form an intact cell layer with uniform topology after two weeks (B). H&E staining (C and D) and Masson’s trichrome staining (E and F) showed multiple layers of VSMC-like cells grown on the adventitial side of the vessel scaffolds (blue arrows). Real time RT-PCR revealed that gene expression of α-actin and calponin was significantly upregulated in each differentiation group compared to the control MSCs (G). The mRNA expression level of each marker protein in the dynamic group was significantly higher than that in the static group, and α-actin expression levels in both the dynamic and static groups were even higher than that in the positive control VSMC (H). Data are expressed as mean ± SEM (n = 3). #P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Evaluation of mRNA and protein expression of VSMC markers
The expression levels of the three VSMC marker proteins, α-actin, calponin, and smoothelin, were quantitatively analyzed at both transcript and protein expression levels by real time RT-PCR and immunostaining. As shown in Fig. 5G–I, α-actin and calponin were significantly upregulated in each experimental group compared to the negative control (MSCs). The mRNA expression levels of all three VSMC marker proteins in the dynamic culture group were statistically higher than those in the static group, and α-actin expression levels in both dynamic and static groups were even higher than that of the positive control group (VSMCs). Consistent with the real time RT-PCR results, immunostaining for different cell groups also demonstrated that the fluorescence intensity of α-actin in the dynamic group was higher than that in the static group, and smoothelin was slightly upregulated in the dynamic group compared to the static group (Fig. 6). These results indicated that the combination of a decellularized vessel ECM and the circumferential tensile stress generated in a rotary bioreactor benefited TGFβ1-induced MSC–VSMC differentiation.
Fig. 6.
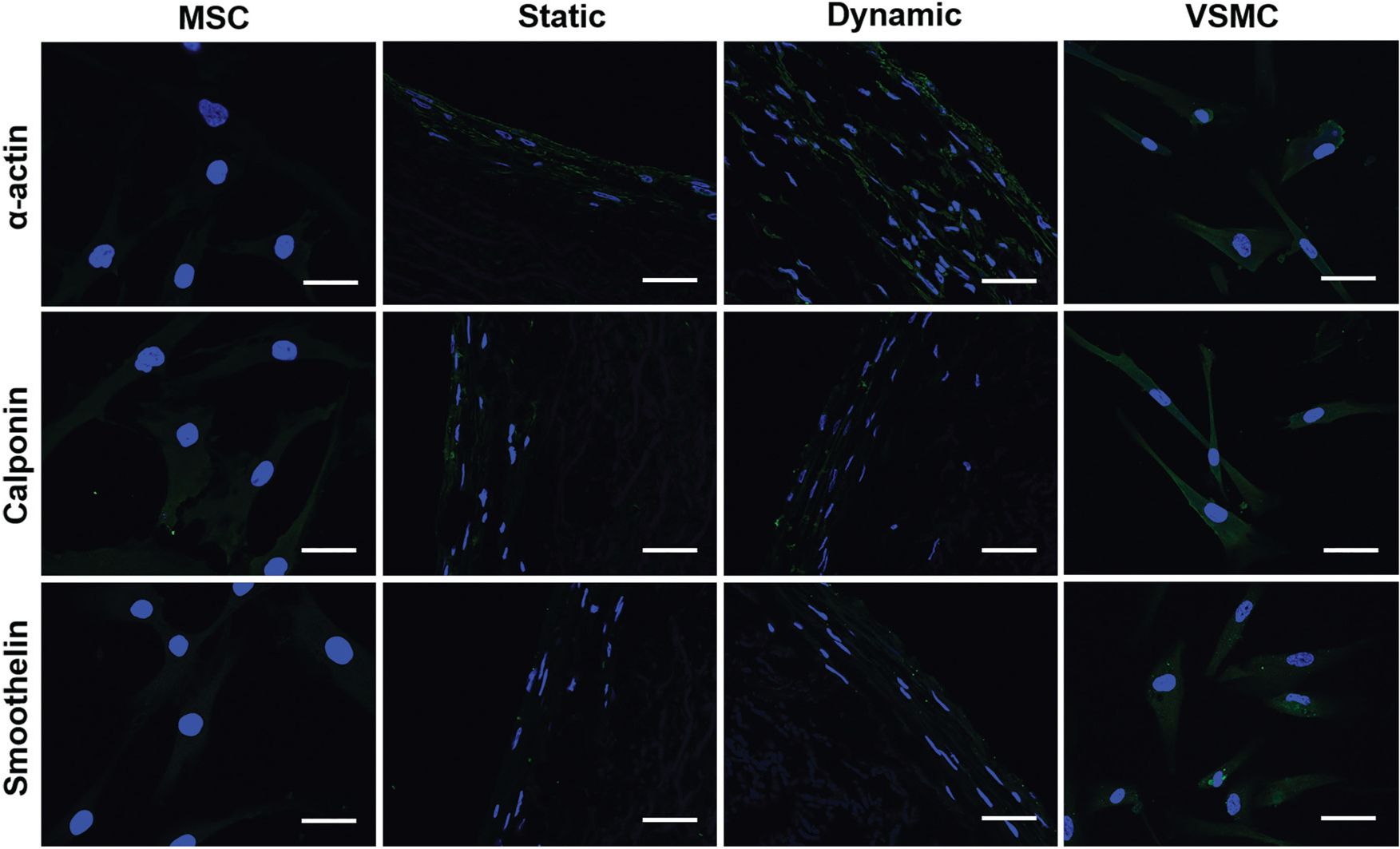
Immunostaining of VSMC marker proteins for different cell groups. The fluorescence intensity of α-actin in the dynamic group was much higher than that in the static group, and smoothelin in the dynamic group was slightly upregulated compared to the static group. The images represent at least three independent experiments. Scale bars = 50 μm.
Functional analysis of the MSC-derived VSMC-like cells
To further investigate the roles played by the dynamic culture system in MSC–VSMC differentiation, cell area reduction upon treatment with vasoactive agonists and collagen gel contraction capability were evaluated for the different cell groups. As shown in Fig. S2 and S3 (ESI†), most of the cells in each group appeared to contract in response to both KCl and carbachol. Negative control MSCs exhibited the lowest contractile capability with 16.54 ± 2.21% and 13.58 ± 1.42% cell area reduction upon exposure to KCl (Fig. 7A) and carbachol (Fig. 7B), respectively. Both KCl and carbachol induced a significantly higher reduction in cell area for the static and dynamic groups compared to MSCs, and the reduction percentage of the cell area for the dynamic group was higher than that of the static group.
Fig. 7.
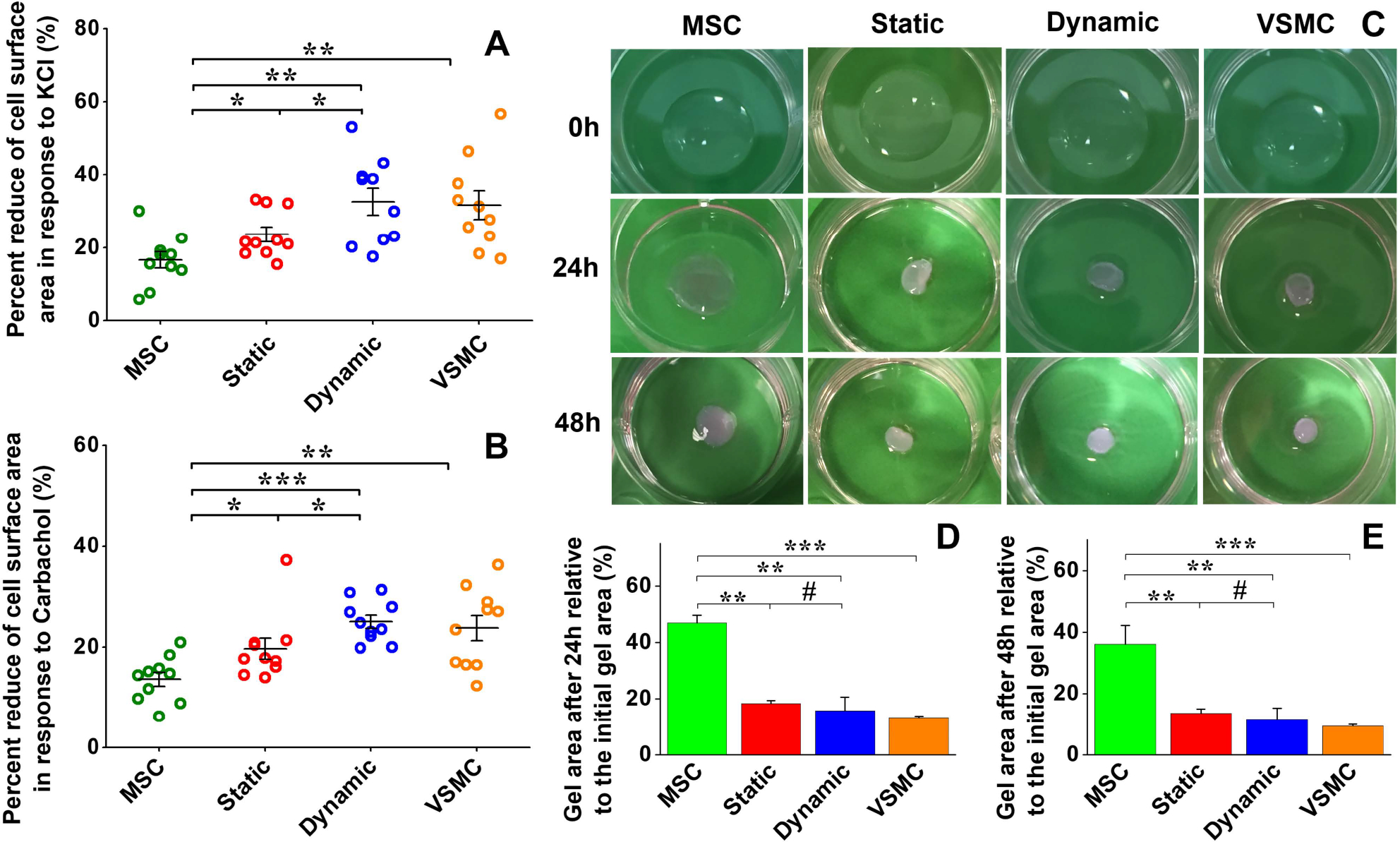
Functional analysis of MSC-derived VSMC-like cells. Cell area reduction assay showed that MSCs have the lowest contractile capability upon exposure to KCl (A) and carbachol (B), respectively. Both KCl and carbachol induced significant decreases in cell area for the static and dynamic groups compared to the negative control MSCs, and the cell area for the dynamic group was reduced more than that for the static group (A and B). Collagen lattice contraction assay showed that four groups of cells embedded in collagen gel could induce apparent contraction of the collagen gel, and the positive VSMC group exhibited the highest contraction capability among them (C). Quantitative analysis demonstrated that there was no significant difference between the collagen gel areas for the static and dynamic groups (D and E). Data are expressed as mean ± SEM (n = 3). #P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Collagen lattice contraction assay was conducted to further evaluate cell contractility. As shown in Fig. 7C, all four cell groups embedded in collagen gel could induce apparent contraction of the gel as the culture time increased, and the positive VSMC group displayed the highest contractile capability. Quantitative analysis demonstrated that both the static and dynamic groups had a significantly higher level of contraction of collagen lattices compared to the MSC group, and the dynamic group exhibited a slightly higher capacity for gel contraction compared to the static group (p > 0.05) (Fig. 7D and E), which is consistent with the results of real time RT-PCR and immunostaining assay.
Discussion
In this study, we developed a promising approach to fabricating an autologous vessel graft for vascular tissue engineering by the combination of on-site differentiation of human bone marrow derived MSCs towards vascular cells on a natural blood vessel ECM scaffold and maturation of the vessel construct in a dynamic cell culture system. The powerful combinatorial promotive effect of the native vascular ECM scaffold and the mechanical cues generated by a rotary bioreactor on MSC differentiation was evaluated by comparing the differentiation efficiency and the physiological functions of the resulting cells with those of the static cell culture system. Growth factors VEGF and TGFβ1 were respectively used as chemical cues to induce the differentiation of MSCs towards ECs and VSMCs in both dynamic and static culture systems.27–30 Our results demonstrated that the cells cultured on the native vessel ECM scaffold in the dynamic system significantly increased the efficiency of MSC differentiation towards both EC- and VSMC-like cells compared to the static culture system, as evidenced by the upregulated expression of marker proteins for ECs (Fig. 2 and 3) and VSMCs (Fig. 5 and 6), as well as elevated EC- (Fig. 4) and VSMC- (Fig. 7) specific physiological functions.
The endothelial layer provides multiple anti-thrombogenic functions for native blood vessels, thus a lack of EC lining in the autologous vessel graft lumen is considered as one of the major problems that may potentially cause a poor patency of engineered vessel constructs.6,31–33 In this study, the EC-like cells obtained by on-site differentiation of MSCs on a decellularized vessel scaffold could potentially overcome this drawback caused by the EC deficiency in the engineered vessel graft. EC-like cells differentiated from MSCs in a static culture system incorporated with decellularized carotid scaffold exhibited a much higher EC-specific function compared to the negative control MSCs (Fig. 4). This is consistent with a recent report that found that decellularized ovine artery matrix could serve as an efficient culture system for promoting differentiation of adipose-derived MSCs towards ECs.34 The capability of the decellularized carotid vessel scaffold to facilitate MSC–VSMC differentiation has been reported in our previous study.11 In addition, decellularized ECM was also reported to direct MSC differentiation in other directions that facilitate tissue regeneration, such as osteogenic and chondrogenic differentiation.35,36 The underlying mechanism by which the decellularized ECM scaffold promotes MSC-vascular cell differentiation may lie in the bioactive component proteins in the ECM, such as laminin, fibronectin, vitronectin, collagen, and gelatin.37–39 For example, laminin plays a critical role in enhancing the expression of EndoTF FOXC2 and its downstream gene, thereby inducing the endothelial differentiation of MSCs.37 Gluck et al. found that laminin and vitronectin enhanced vascular EC differentiation in a traditional 2D cell culture system, while fibronectin enhanced VSMC differentiation in a 3D system.38 More importantly, optimal combinatorial ECMs were able to enhance endothelial differentiation, compared to many single-factor ECMs, in part through an integrin β3-mediated pathway.39 Furthermore, ideal mechanical support provided by each side of the native vascular ECM might be another reasonable explanation for the inductive effect on the MSC differentiation exerted by the ECM. A similar finding was reported by Wingate et al. whereby lineage commitment of MSCs towards specific vascular cells could be controlled by carefully designing the substrate modulus.40 Additionally, MSCs cultured on poly(e-caprolactone) (PCL) films with uniaxial stretching have been demonstrated to obtain a contractile SMC-like phenotype, with ordered organization of cellular stress filaments and upregulated expression of the contractile makers, including SM-α-actin, calponin, and SM-MHC.41 Floren et al. also reported the ability to augment MSC differentiation into mature VSMCs by combining appropriate silk fibroin hydrogel stiffness (33 kPa) with growth factor (TGFβ1).20
In addition to the native vascular ECM, mechanical cues generated by a rotary bioreactor are a critical contributor to the dynamic cell culture system developed in this study for the differentiation of MSCs towards vascular cells and integration with the ECM scaffold to form a physiologically functional vascular graft. Actually, it is well recognized that perfusion bioreactor systems are beneficial for vascular tissue engineering because they expose the vascular cells to flow and shear stresses, which affect various cellular responses, arterial formation, and maintenance.42 Therefore, chemical factor treatments may not be sufficient to develop tissue engineered vascular grafts and co-administration of biomechanical forces may be needed to improve stem cell differentiation.43 Our findings are consistent with previous studies performed by Hasanzadeh et al. that found that shear stress exerted by blood flow alone could be an effective promoter for the differentiation of adipose-derived MSCs towards ECs.44 Growing evidence supports the hypothesis that biomechanical cues play important roles in vascular cell derivation from stem cells.43,45,46 Sivarapatna et al. reported that a biomimetic flow bioreactor was an effective means for human induced pluripotent stem cell–EC differentiation towards an arterial-like phenotype,43 and shear force was also documented to exert a synergistic effect with VEGF in promoting the differentiation of adipose-derived stem cells into ECs.45,46 Additionally, similar to our studies, Park et al. compared the effects of biaxial and uniaxial strain on the differentiation of bone marrow MSCs, and reported that uniaxial strain, a better mimicked mechanical environment experienced by VSMCs in vivo, could successfully promote MSC differentiation towards VSMCs.47 More specifically, mechanical stress causes VSMCs to have proper cellular orientation, increased synthesis of collagen fiber bundles, and alters the phenotype from synthetic to contractile.42 The study by Lee et al. was able to confirm that cell spreading, stress fiber formation, and apparent microvascular formation by ECs and MSCs in collagen gels are affected by fluid shear stress.48 In terms of the underlying mechanism relating to how mechanical cues regulate the differentiation of MSCs into vascular cells, Kurpinski et al. found that cells adjust their orientation along the direction of mechanical forces to minimize the stress applied on the cytoskeleton and other cellular components, and this adaptation in cell orientation may in turn influence MSC differentiation.47,49,50 Besides, growing evidence suggests that genes responsive to shear stress are connected to cell proliferation and differentiation, vascular tone, cellular adhesion, and immune responses.42
Conclusions
Our findings demonstrate that the growth factors VEGF and TGFβ1 directed differentiation of human MSCs towards VSMCs and ECs can be significantly enhanced by the synergistic combination of bioactive components of native arterial ECM and mechanical forces in a dynamic cell culture system. These on-site differentiated vascular cells are able to integrate into a native vascular ECM scaffold and the vessel construct can be further matured under mechanical stimulation in a rotary bioreactor, thus providing a promising approach to fabricating a clinically available vessel graft with mechanical properties and biological functions similar to those of native blood vessels. Future work of this study will be to implant the engineered vessel graft into a model host to study its application in vascular tissue engineering.
Supplementary Material
Funding
This work was supported in part by the American Heart Association (15SDG25420001), South Dakota Board of Regents (UP1600205) and National Science Foundation/EPSCoR Cooperative Agreement (IIA-1355423).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb03348j
Data availability
All experimental data required to reproduce the findings from this study will be made available upon request.
References
- 1.Unal B, Critchley JA and Capewell S, J. Epidemiol. Community Health, 2003, 57, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laslett LJ, Alagona P, Clark BA, Drozda JP, Saldivar F, Wilson SR, Poe C and Hart M, J. Am. Coll. Cardiol, 2012, 60, S1–S49. [DOI] [PubMed] [Google Scholar]
- 3.Isenberg BC, Williams C and Tranquillo RT, Circ. Res, 2006, 98, 25–35. [DOI] [PubMed] [Google Scholar]
- 4.Greco Song HH, Rumma RT, Ozaki CK, Edelman ER and Chen CS, Cell Stem Cell, 2018, 22, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurobe H, Maxfield MW, Breuer CK and Shinoka T, Stem Cells Transl. Med., 2012, 1, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meiring M, Khemisi M, Laker L, Dohmen PM and Smit FE, Med. Sci. Monit. Basic Res., 2017, 23, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, Zhou JY, Lu QP, Wei YJ and Hu SS, Biotechnol. Bioeng, 2008, 99, 1007–1015. [DOI] [PubMed] [Google Scholar]
- 8.Zhao YL, Zhang S, Zhou JY, Wang JL, Zhen MC, Liu Y, Chen JB and Qi ZQ, Biomaterials, 2010, 31, 296–307. [DOI] [PubMed] [Google Scholar]
- 9.Negishi J, Hashimoto Y, Yamashita A, Zhang YW, Kimura T, Kishida A and Funamoto S, J. Biomed. Mater. Res., Part A, 2017, 105, 1293–1298. [DOI] [PubMed] [Google Scholar]
- 10.Syazwani N, Azhim A, Morimoto Y, Furukawa KS and Ushida T, J. Med. Biol. Eng, 2015, 35, 258–269. [Google Scholar]
- 11.Li N, Sanyour H, Remund T, Kelly P and Hong ZK, Mater. Sci. Eng., C, 2018, 93, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho SW, Park HJ, Ryu JH, Kim SH, Kim YH, Choi CY, Lee MJ, Kim JS, Jang IS, Kim DI and Kim BS, Biomaterials, 2005, 26, 1915–1924. [DOI] [PubMed] [Google Scholar]
- 13.Song YS, Lee HJ, Park IH, Kim WK, Ku JH and Kim SU, Int. J. Impotence Res, 2007, 19, 378–385. [DOI] [PubMed] [Google Scholar]
- 14.Dash BC, Levi K, Schwan J, Luo J, Bartulos O, Wu HW, Qiu CH, Yi T, Ren YM, Campbell S, Rolle MW and Qyang YB, Stem Cell Rep, 2016, 7, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundaram S, Echter A, Sivarapatna A, Qiu CH and Niklason L, Tissue Eng., Part A, 2014, 20, 740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang YY, Hu J, Jiao J, Liu ZN, Zhou Z, Zhao C, Chang LJ, Chen YE, Ma PX and Yang B, Biomaterials, 2014, 35, 8960–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang CG, Li Y, Yang M, Zou YH, Liu HH, Liang ZY, Yin Y, Niu GC, Yan ZG and Zhang BH, Eur. J. Vasc. Endovasc. Surg, 2018, 55, 257–265. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Yin S, Cen L, Liu QH, Liu W, Cao YL and Cui L, Tissue Eng., Part A, 2010, 16, 1201–1213. [DOI] [PubMed] [Google Scholar]
- 19.Henderson K, Sligar AD, Le VP, Lee J and Baker AB, Adv. Healthcare Mater, 2017, 6, 1700556. [DOI] [PubMed] [Google Scholar]
- 20.Floren M, Bonani W, Dharmarajan A, Motta A, Migliaresi C and Tan W, Acta Biomater, 2016, 31, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Handorf AM, Zhou YX, Halanski MA and Li WJ, Organogenesis, 2015, 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subbiah R, Hwang MP, Du P, Suhaeri M, Hwang JH, Hong JH and Park K, Macromol. Biosci, 2016, 16, 1723–1734. [DOI] [PubMed] [Google Scholar]
- 23.Wingate K, Floren M, Tan Y, Tseng PON and Tan W, Tissue Eng., Part A, 2014, 20, 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li N, Wang DD, Sui ZG, Qi XY, Ji LY, Wang XL and Yang L, Tissue Eng., Part C, 2013, 19, 708–719. [DOI] [PubMed] [Google Scholar]
- 25.Shi Q, Hodara V, Simerly CR, Schatten GP and VandeBerg JL, Stem Cells Dev., 2013, 22, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward SR and Lieber RL, J. Biomech, 2005, 38, 2317–2320. [DOI] [PubMed] [Google Scholar]
- 27.Seruya M, Shah A, Pedrotty D, du Laney T, Melgiri R, McKee JA, Young HE and Niklason LE, Cell Transplantation, 2004, 13, 93–101. [DOI] [PubMed] [Google Scholar]
- 28.Kinner B, Zaleskas JM and Spector M, Exp. Cell Res, 2002, 278, 72–83. [DOI] [PubMed] [Google Scholar]
- 29.Chen MY, Lie PC, Li ZL and Wei X, Exp. Hematol, 2009, 37, 629–640. [DOI] [PubMed] [Google Scholar]
- 30.Nourse MB, Halpin DE, Scatena M, Mortisen DJ, Tulloch NL, Hauch KD, Torok-Storb B, Ratner BD, Pabon L and Murry CE, Arterioscler., Thromb., Vasc. Biol, 2010, 30, U80–U189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchell SL and Niklason LE, Cardiovasc. Pathol, 2003, 12, 59–64. [DOI] [PubMed] [Google Scholar]
- 32.Barron V, Lyons E, Stenson-Cox C, McHugh PE and Pandit A, Ann. Biomed. Eng, 2003, 31, 1017–1030. [DOI] [PubMed] [Google Scholar]
- 33.Heyligers JMM, Arts CHP, Verhagen HJM, de Groot PG and Moll FL, Ann. Vasc. Surg, 2005, 19, 448–456. [DOI] [PubMed] [Google Scholar]
- 34.Zhang WB, Huo YH, Wang XL, Jia YM, Su L, Wang CX, Li Y, Yang YH and Liu YY, Heart Vessels, 2016, 31, 1874–1881. [DOI] [PubMed] [Google Scholar]
- 35.Gao CY, Huang ZH, Jing W, Wei PF, Jin L, Zhang XH, Cai Q, Deng XL and Yang XP, J. Mater. Chem. B, 2018, 6, 7471–7485. [DOI] [PubMed] [Google Scholar]
- 36.Liang Y, Idrees E, Szojka ARA, Andrews SHJ, Kunze M, Mulet-Sierra A, Jomha NM and Adesida AB, Acta Biomater, 2018, 80, 131–143. [DOI] [PubMed] [Google Scholar]
- 37.Wang CH, Wang TM, Young TH, Lai YK and Yen ML, Biomaterials, 2013, 34, 4223–4234. [DOI] [PubMed] [Google Scholar]
- 38.Gluck JM, Delman C, Chyu J, MacLellan WR, Shemin RJ and Heydarkhan-Hagvall S, J. Biomed. Mater. Res., Part B, 2014, 102, 1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou LQ, Kim JJ, Wanjare M, Patlolla B, Coller J, Natu V, Hastie TJ and Huang NF, Sci. Rep, 2017, 7, 6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wingate K, Bonani W, Tan Y, Bryant SJ and Tan W, Acta Biomater, 2012, 8, 1440–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang ZY, Teoh SH, Johana NB, Chong MSK, Teo EY, Hong MH, Chan JKY and Thian ES, J. Mater. Chem. B, 2014, 2, 5898–5909. [DOI] [PubMed] [Google Scholar]
- 42.Elliott MB and Gerecht S, J. Mater. Chem. B, 2016, 4, 3443–3453. [DOI] [PubMed] [Google Scholar]
- 43.Sivarapatna A, Ghaedi M, Le AV, Mendez JJ, Qyang YB and Niklason LE, Biomaterials, 2015, 53, 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasanzadeh E, Amoabediny G, Haghighipour N, Gholami N, Mohammadnejad J, Shojaei S and Salehi-Nik N, In Vitro Cell. Dev. Biol.: Anim, 2017, 53, 818–826. [DOI] [PubMed] [Google Scholar]
- 45.Fischer LJ, McIlhenny S, Tulenko T, Tulenko T, Golesorkhi N, Zhang P, Larson R, Lombardi J, Shapiro I and DiMuzio PJ, J. Surg. Res, 2009, 152, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colazzo F, Alrashed F, Saratchandra P, Carubelli I, Chester AH, Yacoub MH, Taylor PM and Somers P, Growth Factors, 2014, 32, 139–149. [DOI] [PubMed] [Google Scholar]
- 47.Park JS, Chu JSF, Cheng C, Chen FQ, Chen D and Li S, Biotechnol. Bioeng, 2004, 88, 359–368. [DOI] [PubMed] [Google Scholar]
- 48.Lee EJ and Niklason LE, Tissue Eng., Part C, 2010, 16, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurpinski K, Chu J, Hashi C and Li S, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 16095–16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurpinski K, Chu J, Wang DJ and Li S, Cell. Mol. Bioeng, 2009, 2, 606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data required to reproduce the findings from this study will be made available upon request.