

Abstract
The intestinal microbiota plays significant role in the physiology and functioning of host organisms. However, there is limited knowledge of the composition and evolution of microbiota-host relationships from wild ancestors to modern domesticated species. In this study, the 16S rRNA gene V3–V4 in the intestinal contents of different pig breeds was analyzed and was compared using high-throughput sequencing. This identified 18 323 amplicon sequence variants, of which the Firmicutes and Actinobacteria phyla and Bifidobacterium and Allobaculum genera were most prevalent in wild pigs (WP). In contrast, Proteobacteria and Firmicutes predominated in Chinese Shanxi Black pigs (CSB), while Firmicutes were the most prevalent phylum in Large White pigs (LW) and Iberian pigs (IB), followed by Bacteroidetes in IB and Proteobacteria in LW. At the genus level, Shigella and Lactobacillus were most prevalent in CSB and LW, while Actinobacillus and Sarcina predominated in IB. Differential gene expression together with phylogenetic and functional analyses indicated significant differences in the relative abundance of microbial taxa between different pig breeds. Although many microbial taxa were common to both wild and domestic pigs, significant diversification was observed in bacterial genes that potentially influence host phenotypic traits. Overall, these findings suggested that both the composition and functions of the microbiota were closely associated with domestication and the evolutionary changes in the host. The members of the microbial communities were vertically transmitted in pigs, with evidence of co-evolution of both the hosts and their intestinal microbial communities. These results enhance our understanding and appreciation of the complex interactions between intestinal microbes and hosts and highlight the importance of applying this knowledge in agricultural and microbiological research.
Supplementary information
The online version contains supplementary material available at 10.1007/s10123-023-00449-8.
Keywords: Pigs, Gut microbiota, Composition and function, Composition and evolution, Phylogeny
Introduction
The gut microbiota plays a major role in the overall health of mammals (Brestoff and Artis 2013). Domestic pigs (Sus scrofa) diverged from their wild ancestors in Eurasia approximately 10,000 years ago, and the selection of specific traits has resulted in significant phenotypic changes (Larson et al. 2005; Rubin et al. 2012). Pigs are used extensively as model animals in research on human diseases, development, and responses to infection (Lunney et al. 2021). The pig is also useful for investigating the evolution of the gut microbiota in a species as both the wild ancestor and a variety of domesticated breeds can be used for comparison (Ushida et al. 2016).
There have been a variety of recent investigations into the gut microbiota of the pig, many concerned with agricultural traits and applications (Crespo-Piazuelo et al. 2018; Gao et al. 2019; Kelly et al. 2017; Xiao et al. 2018; Yang et al. 2016; Zhao et al. 2015), such as weight gain (Mach et al. 2015; Ramayo-Caldas et al. 2016) and food intake and conversion (Camarinha-Silva et al. 2017; McCormack et al. 2017; Quan et al. 2018, 2019; Yang et al. 2017). Several studies have also reported on microbial composition in different intestinal regions. These studies have mostly been restricted to specific pig breeds, including the Large White (Zhao et al. 2015), Laiwu (Yang et al. 2016), Gloucestershire Old Spot (Kelly et al. 2017), Iberian pigs (Crespo-Piazuelo et al. 2018), Jinhua and Landrace (Xiao et al. 2018), and Shanxi Black breeds (Gao et al. 2019). Generally, vertebrate species show distinct variations in their gut microbiota that correlate with phylogenetic changes in the host (Brooks et al. 2016; Gaulke et al. 2017; Groussin et al. 2017; Ley et al. 2008). Evidence also suggests that specific microbial signatures are heritable (Koskella et al. 2017). These close relationships suggest the co-evolution of host and gut microbiota (Brooks et al. 2016; Gaulke et al. 2017; Groussin et al. 2017; Moeller et al. 2016). The compositions and evolution of the microbiota in the digestive tracts of wild pigs have been investigated (Yang et al. 2020). In addition, host genetics may be closely involved in structuring the gut microbial communities in different species, as shown by studies in humans (Wang et al. 2021), mice (Kemis et al. 2019; Suzuki et al. 2019), and pigs (Yang et al. 2022). However, relatively little is known about the relationship between the microbial compositions and functions in specific intestinal segments and their association with the genetics and evolutionary characteristics of pigs.
The present study investigated the microbial communities in four pig breeds (Crespo-Piazuelo et al. 2018; Gao et al. 2019; Yang et al. 2020). After collection of the contents of different intestinal regions, a high-throughput sequencing analysis of the 16S rRNA gene V3–V4 region was undertaken to determine gut microbial composition variations. The phylogenetic relationships between the hosts and gut microbiota were then analyzed and functional analysis of the microbiota was conducted to assess the differences in metabolic spatial structures and pathways in the different pig breeds and how they may have influenced the phenotype and adaptation of the hosts. These results will provide the theoretical basis for the composition and evolutionary of gut microbial communities in pigs.
Materials and methods
Animals and sample collection
The test animals were adult female (four years old) wild pigs (WP) (Yang et al. 2020), 150-day-old Large White pigs (LW, commercial pigs), Chinese Shanxi Black pigs (CSB, indigenous pigs) (Gao et al. 2019), and 120-day-old Iberian (IB, indigenous pigs) male pigs (Crespo-Piazuelo et al. 2018). In addition, we selected three unrelated wild pigs from populations in Xingyang County in Henan Province, China. These pigs were similar genetic backgrounds and had been reared under comparable conditions. Animals raised under controlled environmental conditions and on similar diets would be expected to have less variation in their microbiota (Yang et al. 2020). The wild pigs were fed twice daily with a controlled diet consisting of corn and soybean and supplemented with hay, which would be likely to reduce variability in the microbiota relative to pigs living in the wild. The animals had free access to water, and all were healthy and had not received any antibiotic treatment (Yang et al. 2020). The LW and CSB pigs were raised individually at the Datong Pig Breeding Farm (Shanxi Province, China) on standard diets based on the feeding standard of swine (NY/T 65-2004) issued by The Ministry of Agriculture of the People’s Republic of China (Gao et al. 2019). The Iberian pigs were fed ad libitum with a standard feed containing maize, wheat, barley, and soybean, with 3320 kcal of digestible energy and 15.6% of crude protein (Crespo-Piazuelo et al. 2018). Descriptions of the processes and instructions for animal slaughter, sample collection, sample preservation, DNA extraction, library construction, and sequencing are provided in the “References” section (Crespo-Piazuelo et al. 2018; Gao et al. 2019; Yang et al. 2020). Samples were taken from four to five regions of the intestine, namely, the duodenum (DU), jejunum (JE), ileum (IL), cecum CE), and colon (CO). The wild pig’s bacterial 16S rRNA V3–V4 region was amplified using the well-documented primer pair: 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Yang et al. 2020). The 341F-CCTAYGGGRBGCASCAG and 806R-GGACTACNNGGGTATCTAAT primers were used to amplify the hypervariable regions (V3 and V4) of 16S rRNA genes in Shanxi Black pigs and Large White pigs (Gao et al. 2019). The Iberian pig’s bacterial 16S rRNA V3–V4 region gene was amplified with two 16 S Amplicon PCR primers: Forward, 5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG, and Reverse, 5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG GACTACHVGGGTATCTAATC (Crespo-Piazuelo et al. 2018). The raw data have been uploaded to the NCBI Sequence Read Archive (SRA) database under the accession numbers PRJNA575288, SRP115844, and SRP136308. In additional, the study adhered to the guidelines on animal care of the Ministry of Science and Technology of China (Guidelines on Ethical Treatment of Experimental Animals (2006) No. 398) and the Ethics Committee of Shangqiu Normal University approved all the experiments (Shang (2022) No. 24).
Sequence analysis
Bioinformatic analysis of the microbiomes was conducted using QIIME2 (Bolyen et al. 2018) with several slight modifications as described in the official tutorials. Briefly, the raw sequence data were demultiplexed using the “demux” plugin and cut with primers using the “cutadapt” plugin (Martin. 2011). The sequences were filtered for quality, denoised, and merged, and chimeras were removed using the “DADA2” plugin (Callahan et al. 2016). The alpha-diversity metrics Chao1 (Chao 1984), Observed species, Shannon (Shannon 1948), Simpson (Simpson 1949), Faith’s PD (Faith 1992), Pielou’s evenness (Pielou 1966), Good’s coverage (Good 1953), and beta-diversity metrics were estimated with the “diversity” plugin, and samples were rarefied to 18 321 sequences per sample. The taxonomy of the ASVs was assigned with the “classify-sklearn” naïve Bayes taxonomy classifier in the “feature-classifier” plugin (Bokulich et al. 2018) against the Greengenes Database (13.8 version; DeSantis et al. 2006).
Bioinformatics and statistical analysis
Sequences were analyzed using QIIME2 (Bolyen et al. 2018) and several R packages (v3.2.0). The alpha diversity indices at the ASV level were determined using the ASV table in QIIME2 (Bolyen et al. 2018) and were visualized as box plots. ASV-level ranked abundance curves were created to assess richness and evenness. Jaccard (Jaccard 1908) and UniFrac (Lozupone and Knight 2005; Lozuponeet al. 2007) metrics were applied to assess structural variations in the microbiota, and the results were analyzed using PCoA and UPGMA hierarchical clustering (Ramette 2007); the significance of differentiation of microbiota structure among groups was assessed by PERMANOVA using R package “vegan.” Taxonomic differences were analyzed with MEGAN (Huson et al. 2011), GraPhlAn (Asnicar et al. 2015), and LEfSe (Segata et al. 2011). The LEfSe algorithm used a non-parametric factorial Kruskal-Wallis (KW) rank-sum test for the identification of significantly different ASVs, followed by Wilcoxon tests to examine between-group consistencies. LDA scores were used to estimate the effect sizes for differentially abundant taxa. Non-singleton ASVs were aligned using mafft (Katoh et al. 2002) and phylogenetic trees were constructed with fasttree2 (Price et al. 2009). Microbial functions were predicted by PICRUSt2 (Douglas et al. 2020) using the MetaCyc and KEGG databases.
Results
Data overview
Raw data analyzed was obtained from the NCBI SRA database (Crespo-Piazuelo et al. 2018; Gao et al. 2019; Yang et al. 2020). After quality control, the samples were processed using QIIME2 into 18 323 ASVs (Table S1). The presence of the ASVs was verified in the samples by species accumulation and rank-abundance curves. This showed similar patterns across the samples, indicating that the detectable microbial species were present in most samples (Fig. S1 A; Fig. S1 B).
Diversity analysis of the pig gut microbiota
To explore the microbial composition in specific intestinal regions in the different pig breeds, we performed an initial assessment of the alpha diversity of the microbiota for each region. Significant differences in the cecal samples between the pig breeds were observed. We calculated the Chao1, Faith_PD, Simpson, and Pielou_e indices for assessment of richness and evenness. This showed that the Chao1 and Faith_PD indices were significantly reduced in IBCE samples compared with those from LWCE (P < 0.05; Fig. 1), while the Good’s coverage diversity index was higher in IBCE in comparison with LWCE (P < 0.01). No differences were seen between WP or CSB CE samples and LWCE and IBCE (P > 0.05; Fig. 1). The Observed species and Shannon indices were also applied for the assessment of alpha diversity to analyze species numbers and their relative abundances in the different CE samples. Both indices were observed to be significantly higher in CSBCE and LWCE samples compared with IBCE samples (P < 0.05; Fig. 1). Furthermore, no significant differences were seen in the Shannon and Observed species indices in the DU, JE, IL, and CO gut segments, respectively (P > 0.05) (Fig. S2; Fig. S3; Fig. S4; Fig. S5).
Fig. 1.

The alpha-diversity comparisons for the Wild pig cecum (WPCE), Chinese Shanxi black pig cecum (CSBCE), Large White pig cecum (LWCE), Iberian pig cecum (IBCE)
We next investigated the differences and similarities within the microbial communities of the pig breeds, using analysis of the ASVs by PCoA and UPGMA. This showed significant alterations in the gut microbiota compositions in the different intestinal regions, with the microbial composition of the DU, JE, and IL regions differing from those in the CE and CO, which resembled each other (Fig. 2A). The PERMANOVA test based on the Bray-Curtis distance measures showed that the bacterial community structure was significantly (P < 0.01) different among these clusters grouped (Table S2). It was further confirmed that the change in bacterial community structure was significantly correlated with pig breeds. UniFrac distance metrics clustering showed that the gut microbes tended to cluster into four groups according to the pig breed, namely, WP, CSB, LW, and IB. Additional subgroups were observed within each of the main groups, with the CE (WPCE, CSBCE, LWCE, IBCE) and CO samples (WPCO, IBCO) clustering separately from those from the IL (WPIL, CSBIL, LWIL, IBIL), DU (WPDU, CSBDU, LWDU, IBDU), and JE (WPJE, CSBJE, LWJE, IBJE) regions (Fig. 2B; Fig. S6). These findings indicated that the microbiota compositions were not uniform along the intestine in the different pig breeds, with greater similarities seen between the CE and CO samples than between samples from the DU, JE, and IL regions. As seen in Figs. 2A and B, the gut microbiota from wild pigs clustered together separately from those of both commercial and domesticated indigenous pigs.
Fig. 2.

The beta-diversity comparisons for the different gut microbiota in pigs. A The principal coordinate analysis (PCoA) visualized via Bray-Curtis metrics and UniFrac distance metrics. Each symbol and color represents each gut location microbiota. B The hierarchical clustering analyses were performed by unweighted pair-group method with arithmetic means (UPGMA)
Microbial taxonomic composition analysis of the pig gut microbiota
The taxonomic distributions of the most abundant ASVs seen in the different intestinal regions and pig breeds were then investigated. Firstly, comparing the phyla present in specific intestinal regions across the pig breeds (Fig. 3A; Table S3), it was found that Firmicutes were most prevalent in WP, LW, and IB, followed by Actinobacteria in WP, Bacteroidetes in IB, and Proteobacteria in LW. In contrast, Proteobacteria predominated in CSB, followed by Firmicutes while Proteobacteria levels in both CSBCE and LWCE samples were low (3.57% and 3.10%, respectively). In terms of intestinal region Firmicutes and Bacteroidetes phyla predominated and accounted for more than 80% of the bacterial phyla in IB, with Firmicutes and Actinobacteria accounting for over 80% in WP and Firmicutes and Proteobacteria accounting for over 90% in CSB and LW. Comparison of the microbial communities in samples from the same intestinal regions showed that these differed significantly with the breed of pig. For instance, the IBJE microbiota (70.38%) contained the highest abundance of Firmicutes than the JE of the WP, CSB, and LW (33.86%, 22.15%, 61.88%, respectively). Overall, the microbial composition differed between WP, CSB, LW, and IB for the same intestinal regions. Furthermore, we observed dynamic changes in the proportions of Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria between the different breeds. These findings indicated that microbial communities differed between the different breeds for the same gut regions.
Fig. 3.

Community composition of the gut microbiota in different intestinal segments of Wild pigs, Chinese Shanxi black pigs, Large White pigs, and Iberian pigs at the phylum (A) and genus (B) levels, respectively
In terms of genera, Bifidobacterium and Allobaculum predominated in WP. In addition, Lactobacillus, Prevotella, and Clostridiaceae were found in DU samples from WPs, while Shigella and Lactobacillus were most abundant in CSB and LWP, and Actinobacillus, Sarcina, Streptococcus, Prevotella, Anaerovibrio, Anaerovibrio, and Lactobacillus predominated in IB (Fig. 3B; Table S4). In terms of distribution, it was found that only two genera (Sarcina and Actinobacillus) were absent from the five gut segments in WPs, with the lowest average distribution index of each genus seen in IL samples, suggesting that the IL microbiome is more even than that of other locations. Thus, although numerous genera were observed in the different regions of the intestine, there was a more uniform distribution in the IL (Fig. 3B; Table S4). Microbial community compositions in the same parts of the intestine also differed between wild and domesticated pigs as shown by PCoA where the domesticated pig samples were clustered and distinct from those of the wild pigs.
Differential analysis of bacterial taxa in pig gut microbiota
An LEfSe analysis was performed to determine the microbial species characteristics of different intestinal regions. Using LDA scores > 2, this showed differential abundances of 36, 47, 44, 89, and 79 ASVs in DU, JE, IL, CE, and CO samples, respectively, in the WP, CSB, LW, and IB groups (Fig. 4; Table S5). Differences were observed between the microbiomes of WPs and those of the LW, CBS, and IB pig breeds, seen at all taxonomic levels from phylum to genus (Fig. 4; Table S5). As seen in Fig. 4A, WPDU showed a significantly greater abundance in 17 taxa in Firmicutes, nine taxa in Actinobacteria, and five taxa each in Bacteroidetes and Proteobacteria. In comparison, IBDU samples contained greater abundances of 10 taxa in Proteobacteria, 11 in Firmicutes, and five taxa in Actinobacteria, while CSBDU showed had nine taxa in Proteobacteria, five taxa each in Bacteroidetes and Tenericutes, and four taxa in Actinobacteria. Thus, at the phylum level, Actinobacteria represented a significant biomarker for the various intestinal segments in WPs compared with IB, CSB, and LW. Similarly, Proteobacteria represented a biomarker for the DU and IL regions in CSB, in contrast to IB, WP, and LW, and was also a biomarker for IBCO compared with WPCO. At the genus level, it was found that Bifidobacterium and Allobaculum showed the greatest differences in abundance in the different intestinal regions of WPs compared with IB, WP, and LW. These differences were maintained in the order, class, and family levels (Fig. 4; Table S5). In addition, Sarcina and Streptococcus were more abundant throughout the gut of IB pigs, compared with CSB, WP, and LW, while Actinobacteria was more abundant in the DU and JE regions in CSB. WP samples showed enrichment of both Bifidobacterium and Allobaculum in comparison with domesticated pigs, while the latter showed a greater abundance of Shigella and Lactobacillus in CSB and LW and Actinobacillus and Sarcina in IB. These findings indicate that the domesticated pig microbiota has diverged from its ancestral state.
Fig. 4.

Bacterial taxa differentially represented in duodenum (A), ileum (B), jejunum (C), cecum (D), and colon (E) gut locations in Wild pigs, Chinese Shanxi black pigs, Large White pigs, and Iberian pigs identified by LEFSe using a LDA score threshold of > 2.0
Phylogenetic analysis of the pig gut microbiota
A phylogenetic tree of the microbial ASVs was created to investigate the evolutionary relationships between the gut microbiota from the different pig breeds, using distance matrices calculated from the sequencing analysis. The tree was generated using the most abundant 50 bacterial genomes represented in the pig microbiome (Fig. 5; Fig. S7; Fig. S8; Fig. S9; Fig. S10; Fig. S11). This showed that the genomes spanned many microbiota-associated phyla. Each branch of the tree represents one gut microbiota. Phylogenetic trees for [Prevotella] (ASV39), Prevotella (ASV11), Lactobacillus (ASV3), Shigella (ASV2), Bifidobacterium (ASV9), Actinobacillus (ASV22), Clostridium (ASV21), Sarcina (ASV1), Streptococcus (ASV5), and Campylobacter (ASV14) showed that their microbiomes represent “core” microbial genera that do not cluster together. Interestingly, the top 50 most abundant microbiota were located on different branches according to their intestinal location with related organisms clustered together. Several clusters of co-existing bacteria were identified by visual inspection, with the 10 most abundant genera indicated in Fig. 5. The different clusters tended to be associated with pig breeds, for example, clusters 1 (ASV_32, ASV_7, ASV_350, ASV_240, ASV_95, ASV_50, ASV_47, ASV_56, ASV_15, ASV_164), 2 (ASV_136, ASV_39, ASV_131, ASV_373, ASV_126, ASV_132, ASV_11), and 3 (ASV_2, ASV_120, ASV_69, ASV_155, ASV_30) are associated with different phyla, Firmicutes, Bacteroidetes, and Proteobacteria in different pig breeds. This relationship with the host type is suggestive of specific interactions between the microbiota and the host. Cluster 1, for example, while consisting largely of Firmicutes, also includes other phyla, while in cluster 3, the constituents are present in all the breeds but belong to the same microbial group. Thus, phylogeny is able to demonstrate the evolution of microbiota during the domestication of pig.
Fig. 5.

Phylogenetic tree with ASV abundance distribution. Species abundance distribution was aligned to the tree and visualized as boxplots. The phylum information was used to color symbolic points on the tree and also species abundance distributions
Functional analysis of the pig gut microbiota
PICRUSt2 was used for metagenomic assessment of the identified bacteria, as this may provide insight into metabolic differences in the various intestinal regions of the host. The ASVs were assigned to genes using available genetic annotations, and the functions of the genes were analyzed by KEGG pathway enrichment. This yielded 7746 genes (Table S6) associated with 393 pathways (Table S7). Seven pathways were observed to differ between the different intestinal regions. The most enriched functional modules in all samples were “human diseases,” “organismal systems,” and “metabolism,” while the most significant pathways were also similar in the different samples. Six of these pathways were related to metabolism (amino acid, biosynthesis, carbohydrate, energy, cofactors and vitamins, and lipid), with one pathway involved in the processing of environmental information (member transport) and two pathways associated with the processing of genetic information (translation and replication, and repair) which were common to all samples (Table S7). The associations of these pathways with the same intestinal samples from different pig breeds were then examined. As seen in Fig. 6, the pathway enrichment differed according to the intestinal region in the different breeds with 12 pathways showing significant enrichment in the DU, five in the JE, seven in the IL, 26 in the CE, and 13 in the CO (P < 0.05; Fig. 6; Table S8). The enriched pathways in the DU were “fatty acid degradation” (ko00071) in LWDU and “fatty acid biosynthesis” (ko00061) in IBDU. The DU also had a greater abundance of microbial genes in the “methane metabolism” (ko00680) and “starch and sucrose metabolism” (ko00500) in WPDU. The pathways enriched in JE and IL samples were “lysine degradation” (ko00310), “ubiquinone and other terpenoid quinone biosynthesis” (ko00130) in CSBIL, “fatty acid biosynthesis” (ko00061), “folate biosynthesis” (ko00790), and “amino sugar and nucleotide sugar metabolism” (ko00520) in IBJE and IBIL, “nicotinate and nicotinamide metabolism” (ko00760), “seleno-compound metabolism” (ko00450), “ribosome biogenesis in eukaryotes” (ko03008), “fatty acid degradation” (ko00071), “valine, leucine, and isoleucine biosynthesis” (ko00290), and “tryptophan metabolism” (ko00380) in WPJE and WPIL. Furthermore, it was found that CSBCE samples had highest abundance of microbial genes in “zeatin biosynthesis” (ko00908), “drug metabolism other enzymes” (ko00983), “other glycan degradation” (ko00511), “glycosaminoglycan degradation” (ko00531), “citrate cycle TCA cycle” (ko00020), “RNA degradation” (ko03018), “beta alanine metabolism” (ko00410), “folate biosynthesis” (ko00790), “RNA polymerase” (ko03020), and “methane metabolism” (ko00680) in CSB compared with WPCE (starch and sucrose metabolism (ko00500), biosynthesis of ansamycins (ko01051), streptomycin biosynthesis (ko00521), histidine metabolism (ko00340), biosynthesis of vancomycin group antibiotics (ko01055), ABC transporters (ko02010) and IBCE (phosphonate and phosphinate metabolism (ko00440), toluene degradation (ko00623), glycerophospholipid metabolism (ko00564), and butanoate metabolism (ko00650) in the WP and IB breeds. The IBCO microbial genes were related to “fructose and mannose metabolism” (ko00051) and “toluene degradation” (ko00623) which had low abundance in the IB samples. In contract, WPCO samples showed greater enrichment in “nicotinate and nicotinamide metabolism” (ko00760), “seleno-compound metabolism” (ko00450), “pentose and glucuronate interconversions” (ko00040), “RNA degradation” (ko03018), “histidine metabolism” (ko00340), and “lysine biosynthesis” (ko00300). The clustered heatmap showed a clear distinction between the WP and IB samples and the CSB and LW samples (Fig. 7; Table S9). Notably, more microbial genes associated with metabolism were found in wild pigs. However, the relative abundance of microbial genes related to the metabolism of amino acids and carbohydrates, as well as resistance, was greater in WPs in comparison with the LW, CSB, and IB domesticated pigs, while genes associated with lipid metabolism were more abundant in IB pigs compared with LW, CSB, and WP pigs. These results suggest that the metabolic functions of the microbiota differ according to pig breeds, with specific pathways predominating in certain breeds.
Fig. 6.

Predicted functional differentially of the bacterial genus represented in the duodenum (A), jejunum (B), ileum (C), cecum (D), and colon (E) gut locations in different pig breeds identified by LEFSe using a LDA score threshold of > 2.0
Fig. 7.

Heatmap clustered by KEGG pathway showing different enrichments in the different gastrointestinal sites and different pig breeds. The vertical columns represent groups, and the horizontal rows depict metabolic pathways. The color coding is based on row z-scores
Discussion
The literature contains relatively few studies on the microbial composition and potential functionality of the fecal microbiota in wild pigs, domestic indigenous pigs, and commercial pigs (Ushida et al. 2016; Huang et al. 2020). The relationships between specific microbial composition and evolution have been characterized in wild pigs (Yang et al. 2020). An analysis of the gut microbiota in the gastrointestinal tracts of wild boar in the karst region of Southwest China also demonstrated the importance of environmental adaption (Cao et al. 2022). To date, few studies have specifically focused on the effect of domestication on the evolutionary of the gut microbiota in pigs, and it has been assumed that domestication has resulted in only minor changes in community richness. Furthermore, the key to understanding the role of disrupted microbiota in pigs is to specifically consider the composition and evolution of the microbiota themselves and whether inter-group microbial differences can be linked to domestication. We, therefore, systematically characterized changes in the microbial composition, distribution, phylogeny, and potential functionality in specific sections of the digestive tract in different pig breeds. This showed vast differences in microbial composition along the length of the gut, and we, furthermore, provide additional insights into the composition and evolutionary of the gut microbiota in each section and their potential functional consequences in pigs.
At the phylum level, the primary findings centered on the structural composition and distribution of the intestinal microbiota in WP, LW, CSB, and IB pig breeds (Fig. 3A; Table S3). Earlier work has observed the core microbiota of domestic pigs to be dominated by Firmicutes and Bacteroidetes (Crespo-Piazuelo et al. 2018; Gao et al. 2019; Xiao et al. 2018; Yang et al. 2016; Mach et al. 2015; Ramayo-Caldas et al. 2016; Quan et al. 2018, 2019; He et al. 2016; Ivarsson et al. 2014; Kraler et al. 2016; Liu et al. 2012; Slifierz et al. 2015). Here, we observed that the four most abundant phyla were Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria. The Bifidobacterium and Allobaculum genera predominated in WPs, while Shigella and Lactobacillus genera were most abundant in CSB and LWP and Actinobacillus and Sarcina in IB pigs. In contrast, neither Sarcina and Actinobacillus were found in any of the gut regions of WPs (Fig. 3B; Table S4). These findings indicate that many bacterial species are uniformly distributed throughout the intestine in same pig breeds. Previous studies have reported marked differences in pig microbiota at the genus level. For instance, Lactobacillus, Prevotella, and Treponema were predominant in Duroc pigs (Yang et al. 2016), while Xiao et al. (2017) observed a greater abundance of Prevotella, Clostridium, SMB53, and Streptococcus in Landrace, and Yorkshire and Hampshire pigs. These differences may result from differences in feed composition and environment, as well as from inherent breed differences. However, it is difficult to make detailed comparisons of the specific effects of diet and environment in the studied populations.
Multiple factors are known to affect the gut microbiota, including genetics. Human genetic variation is documented to influence the gut microbiota by both environmental and host-associated factors (Bonder et al. 2016; Qin et al. 2022; Lopera-Maya et al. 2022). Genetic variations result in phenotypic differences, which are also known to influence the gut microbiota (Kemis et al. 2019; Suzuki et al. 2019; Chen et al. 2020; Wang et al. 2021; Yang et al. 2022). Chen et al. (2021a) determined the stability and variation in human microbiota in response to physiological changes in the host. Yang et al. (2022) have shown that, under conditions of significant genetic diversity and environmental uniformity, the composition of the microbiota is heritable. These results provide strong evidence for the influence of host genotype on the abundance of specific bacteria in the intestine. Thus, the pig genotype can affect the composition of the gut microbiota. Differences at the levels of phylum, class, order, family, and genus were apparent in samples from the same intestinal region in different pig breeds (Fig. 4; Table S5). Within-group stability was observed for both microbial composition and abundance. We also found that the genetic stability of the microbial communities varied significantly according to pig breed, with Bifidobacterium species showing relatively high within-group stability over extended periods in WP populations. Notably, it has been found that some Bifidobacterium species colonize wild pig populations and subsequently show a high degree of genetic stability (Ushida et al. 2016). This supports the observation that the microbiota of both indigenous and commercial pigs have diversified from their state in ancestral pig populations. Martínez and colleagues (2018) reported that each person has a unique and stable community of gut microbes that is as personal as a “fingerprint.” Studies have shown that an individual’s genetics, diet, environment, lifestyle, and physiological state all make small contributions to variations in the gut microbiome among individuals. However, less than 30% of this variation can be explained, and even identical twins, who share the same genotype and often diets and lifestyle, have distinct gut microbiomes. It is interesting that, despite living in varied habitats, wild pigs show remarkable similarities in their microbiota. More importantly, the microbiota of domesticated pigs differed significantly from those of their wild ancestors. These findings also suggest that different pig breeds may have specific microbial compositions that differ from others and may persist for long periods of time. The genetic profile of the gut microbiota may thus represent a host “marker” that distinguishes the specific host from others. Overall, these results demonstrate that both the composition and functional influence of the microbiota may be closely linked with the genotype of the host. Gut microbes are vertically transmitted in pigs, and thus, there is a symbiotic relationship between the hosts and their associated microbial communities. Therefore, it is not surprising that the effects of these factors were greater in modern pig breeds. The aim of the study was not to demonstrate that domestication and the development of the modern breeds are the most critical factors affecting the pig gut microbiota. Instead, we investigated and confirmed that pig domestication resulted in a specific shift in the composition of the pig microbiome. Moreover, we argue that the transition from ancestral wild pigs to modern pig breeds is masked to some extent by the influence of genotype, which may independently drive changes in the microbiome.
Phylogenetic analysis showed that pig genotypes were associated with a variety of microbiota phyla (Fig. 5). Each branch on the tree represents one gut microbiota. Phylogenetic trees for Prevotella (ASV11), Lactobacillus (ASV3), Shigella (ASV2), Bifidobacterium (ASV9), Sarcina (ASV1), and Streptococcus (ASV5) showed the presence of “core” microbial species in different groups. Interestingly, the phylogenetic trees showed that the top 50 most abundant gut microbes from different intestinal locations were located on different branches in different pig populations and were clustered according to the taxonomy of the microorganisms. Several clusters of bacteria were visible after clustering, as shown in Fig. 5. These bacteria belonged to a variety of species, usually belonging to the same order. PCoA and UPGMA analyses showed a lack of uniformity in the gut microbiota along the intestine among the different pig breeds, while within the same breed, there tended to be clustering of the microbiota species. Specifically, the gut microbial clusters from WPs were distinct from those of the domesticated CSB, IB, and LW (Fig. 3A and B). Sharpton has described mechanisms through which gut microbes in vertebrates affect the physiology and fitness of the hosts (Sharpton 2018). Wibowo et al. (2021) performed a large-scale de novo assembly of microbial genomes from palaeofeces, establishing that palaeofeces with well-preserved DNA are abundant sources of microbial genomes, including previously undescribed microbial species, which may elucidate the evolutionary histories of human microbiomes. The phylogenetic analysis showed significant correspondences between the gut microbes and their hosts, strongly suggestive of co-evolution. This also suggests that the adaptation is to the host genotype, as this had a significant effect on the composition of the microbial communities. Thus, phylogenetic analysis can be used to investigate the evolution of microbiota during the change from wild to domesticated conditions. In all, the findings suggest that the composition and functional influence of the microbiota are closely linked with the evolutionary adaptation of the host, suggestive of the co-evolution of pigs and their microbiota.
In the final section, we performed a metagenomic analysis of the functional influence of the microbiota in the different pig breeds. A total of 7746 genes (Table S6) associated with 393 pathways (Table S7) were identified by KEGG. As shown in Fig. 6, the five intestinal regions differed in the functions of their microbiota among the four pig populations (P < 0.05; Fig. 6; Table S8). The clustered heatmap shows a clear distinction between WP and IB samples and CSB and LW samples (Fig. 7; Table S8). Specifically, the microbiota from WPs contained greater numbers of metabolism-related genes. Different abundances were seen in approximately half of the bacterial species and pathways, together with within-group differences in microbial genotypes. Genes associated with amino acid and carbohydrate metabolism, as well as resistance, were more abundant in WPs compared with LW, CSB, and IB, while genes associated with lipid metabolism were more common in IB pigs than in LW, CSB, and WP pigs. These four types of pigs differ in their lipid content: CSB and IB pigs have higher fat deposition with lower proportions of meat compared with the leaner LW and WP pigs. Gao et al. (2019) observed that Prevotella was more abundant in CSB pigs compared with their LW counterparts, suggesting that CSB pigs may be more efficient in their absorption of nutrients than LW pigs. Crespo-Piazuelo et al. (2018) reported that energy pathways associated with the gut microbiota varied along the intestine in IB pigs, suggesting an explanation for the influence of the microbiota on lipid metabolism. Anaerobic bacteria compete for the degradation of plant polysaccharides and fatty acid production in the colon. Prevotella is usually abundant in pig feces (Yang et al. 2016; Kim et al. 2017) and is known to be involved in polysaccharide degradation and amino acid metabolism to influence the level of intramuscular fat in pigs (Fang et al. 2017). Chen et al. (2021b) suggested that Prevotella copri in the intestines of pigs fed on commercial diets influences chronic inflammatory responses in the host, mediated by TLR4 and mTOR signaling to increase fat deposition in the host. Thus, differences in the lipid content of the host may be caused by microbes such as Prevotella spp. Further investigation is necessary to verify these possible relationships. Furthermore, we observed that Actinobacteria predominated in the intestines of WPs, whereas Bacteroidetes were more abundant in domesticated pigs. Bacteroidetes are able to degrade bacterial exopolysaccharides in the animal gut (Lammerts van Bueren et al. 2015), while Actinobacteria produce various compounds that influence immunity and metabolism and are vital to host health (Matsui et al. 2012). The change from dominance by Actinobacteria in wild pigs to Bacteroidetes in domesticated pigs may be the result of a variety of influences, including genetics, evolution, environmental change, and the type of food. Actinobacteria are also associated with resistance against infection and higher levels of this phylum in wild pigs may be associated with increased infection resistance in contrast to domesticated pigs. Thus, it is apparent that domestication has resulted in a shift in the composition of the microbiota, and although there are clear similarities between wild and domesticated animals in terms of microbial dominance, there are clear differences between the two populations.
The Bifidobacterium and Allobaculum genera predominated in most of the WP samples (Fig. 3B; Table S4). Bifidobacteria are found in the digestive tracts of both mammals and insects (Ventura et al. 2012) and, together with Allobaculum, are classified as probiotic bacteria as they protect the mucosal barrier of the intestine (Furusawa et al. 2013). These bacteria contain numerous genes encoding enzymes responsible for the breakdown of complex carbohydrates (Milani et al. 2014). Bifidobacterium degrade the hexose sugars through a specific pathway, the “bifid shunt.” dependent on the fructose-6-phosphate phosphoketolase enzyme (Pokusaeva et al. 2011). The end-products of this pathway are ATP and short-chain fatty acids that protect the mucosal barrier against pathogenic infection (Sánchez et al. 2010); an example is acetate produced by Bifidobacterium which has been shown to protect epithelial cells from infection (Fukuda et al. 2011). Bifidobacterium has also been found to protect against rotaviral enteritis (Rigo-Adrover et al. 2017) and necrotizing enterocolitis in newborn rats (Satoh et al. 2016; Wu et al. 2013) and to enhance inflammation and the immune response in colitis-affected rats during weaning (Izumi et al. 2015). Similarly, one of the most abundant genera observed in wild pigs was Allobaculum, a member of the Erysipelotrichaceae family and about which relatively little is known. The first reported member of the genus is Allobaculum stercoricanis from dog feces (Greetham et al. 2004). Allobaculum is also negatively associated with adiposity in mice (Baldwin et al. 2016) and has been found to utilize glucose and produce both lactate and butyrate (Herrmann et al. 2017). Allobaculum species have also been linked to inflammation in mice (Cox et al. 2014), while Miyauchi et al. (2020) observed a causative association between an Allobaculum strain and a predisposition to autoimmune encephalitis. Van Muijlwijk et al. (2021) observed that A. mucolyticum secreted numerous O-glycan carbohydrate-degrading enzymes, resulting in an ability to degrade intestinal mucins, resulting in effective colonization and degradation of the intestinal mucus layer. Thus, it is possible that the high proportion of Bifidobacterium and Allobaculum in the microbiome may enhance both nutrient absorption and resistance to disease in wild pigs. However, Tsuchida et al. (2017) observed that intestinal bacteria from wild pigs contained fiber-degrading enzymes while domestic pigs expressed genes associated with tetracycline resistance. It is possible that the latter may be linked to the routine use of antibiotics during the rearing of domestic pigs. Overall, these reports suggest that throughout porcine evolution and domestication, alterations in both the environment and nutrient source, as well as artificial selection, have led to divergence in the composition of the intestinal microbiome, with modern domestic pigs having microbiota associated with fast growth but reduced disease resistance compared with wild pigs (Yang et al. 2020). The metagenomic results suggest that the microbiota have distinct spatial and functional attributes that enhance the degradation and utilization of nutrients as well as maintaining gut homeostasis. We have also demonstrated a possible causal association between microbial, metabolites, and the host phenotype. There is the potential to expand knowledge of how physical damage to the gastrointestinal tract alters microbial content. An environmental application of the pig gastrointestinal model could evaluate the effects of climate change on the microbiome and gut health (Lunney et al. 2021). Our future studies will focus on the microbial strain, investigating genetics and evolution by metagenomic sequencing or whole genome sequencing, as well as the potential therapeutic effects of diet on the gut microbiome.
Conclusion
Significant differences in microbial compositions and functions were found in the same intestinal regions of different pig breeds. The results suggest that the evolution of these aspects of the microbiota is strongly linked to both the domestication and genetics of the pig. Microbiota were found to be genetically stable and specific, with vertical transmission in the host, suggesting co-evolution of the host and its microbiome. The gut microbiota influences the host, with signaling pathways affecting the phenotype of the host. Nevertheless, the influence of the highly variable microbial genomes on the metabolism of the host remains to be explored. These findings provide insight into the complexity of microbial-host relationships and highlight the significance of applying this knowledge in agricultural practice.
Supplementary information
Below is the link to the electronic supplementary material.
{kind=link}
Supplementary file1 (PNG 418 KB) Fig. S1 Species accumulation (A) and rank-abundance (B) curves analysis of the different gut intestinal tract samples at 97% sequences identity. If the curves reach or nearly reach a plateau, it indicates that most of the species present in all samples have been observed.
{kind=link}
Supplementary file2 (PNG 78 KB) Fig. S2 The alpha-diversity comparisons for the duodenum (DU) in different pig populations.
{kind=link}
Supplementary file3 (PNG 71 KB) Fig. S3 The alpha-diversity comparisons for the jejunum (JE) in different pig populations.
{kind=link}
Supplementary file4 (PNG 70 KB) Fig. S4 The alpha-diversity comparisons for the ileum (IL) in different pig populations.
{kind=link}
Supplementary file5 (PNG 57 KB) Fig. S5 The alpha-diversity comparisons for the colon (CO) in different pig populations.
{kind=link}
Supplementary file6 (PNG 1499 KB) Fig. S6 The hierarchical clustering analyses were performed by unweighted pair-group method with arithmetic means (UPGMA).
{kind=link}
Supplementary file7 (PNG 115 KB) Fig. S7 Phylogenetic tree with ASV abundance distribution in duodenum (DU) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
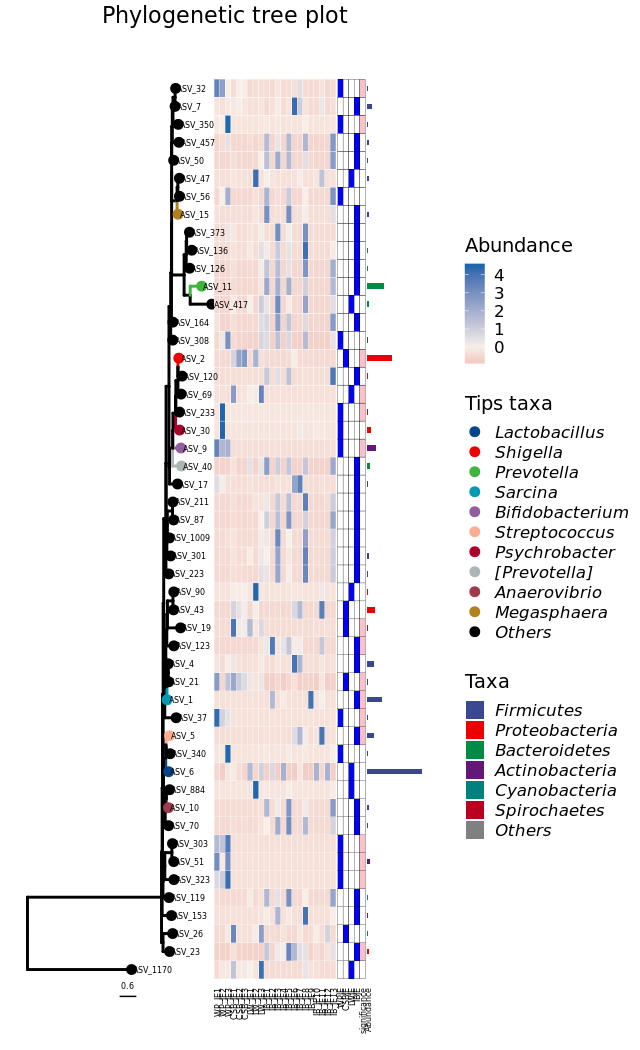{kind=link}
Supplementary file8 (PNG 110 KB) Fig. S8 Phylogenetic tree with ASV abundance distribution in jejunum (JE) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
{kind=link}
Supplementary file9 (PNG 118 KB) Fig. S9 Phylogenetic tree with ASV abundance distribution in ileum (IL) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
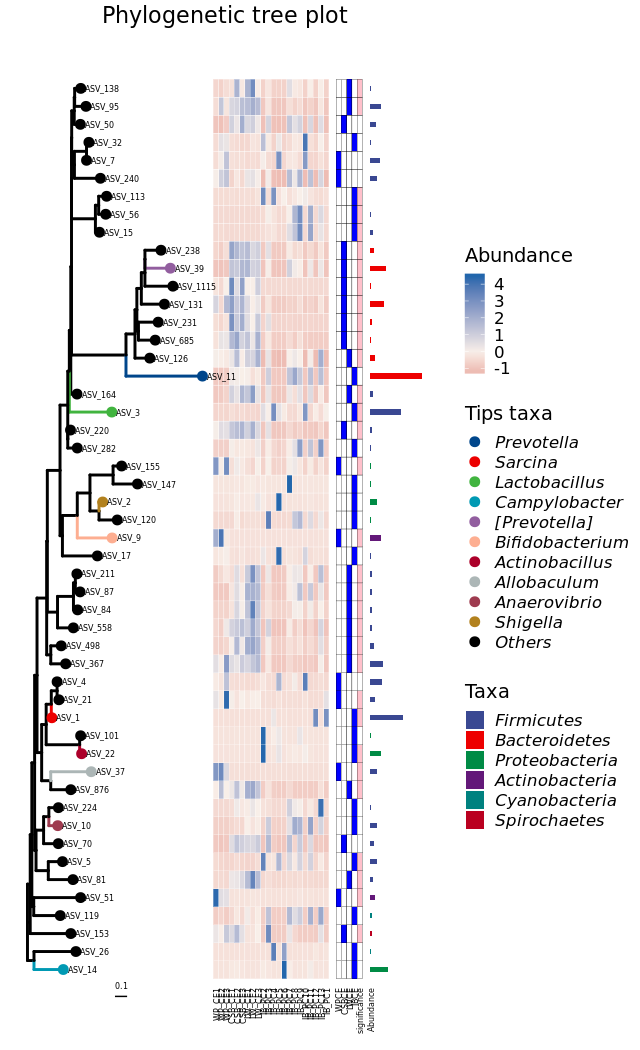{kind=link}
Supplementary file10 (PNG 116 KB) Fig. S10 Phylogenetic tree with ASV abundance distribution in cecum (CE) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
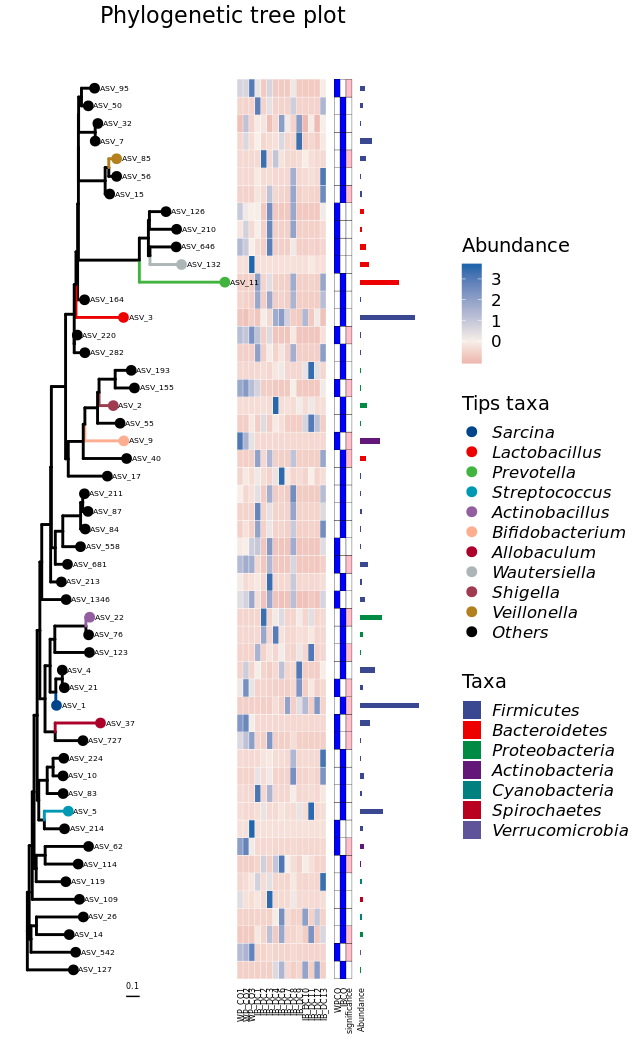{kind=link}
Supplementary file11 (PNG 108 KB) Fig. S11 Phylogenetic tree with ASV abundance distribution in colon (CO) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
Author contribution
GY and ML designed the research. ZW, ZL, CS, FG, and YC contributed to experimental work. SZ, HZ, and CZ performed the data analysis and wrote the manuscript. GY and ML revised the manuscript. All authors reviewed and approved the manuscript.
Funding
This study was supported by the Foundation of Industry University Research Cooperation Project of He’nan Science and Technology Committee of China (182107000041) and the Key R&D and Promotion Program in Henan Province of China (092102110088, 212102110001, 22210320010).
Data availability
All data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The animal care and use guidelines put forth by the Ministry of Science and Technology of China (Guidelines on Ethical Treatment of Experimental Animals (2006) No. 398) were followed for this study, with the Ethics Committee of Shangqiu Normal University having approved all experiments herein (Shang (2022) No. 24).
Consent to participate
For this type of study, consent is not required.
Consent for publication
Consent for publication is not required in this study.
Competing interest
The authors declare no competing interests.
Footnotes
Shuhong Zhang, Huan Zhang, and Cheng Zhang are as co-first authors.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ming Li, Email: liming@henau.edu.cn.
Guangli Yang, Email: guangliyang@163.com.
References
- Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N (2015) Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin J, Collins B, Wolf PG, Martinez K, Shen W, Chuang CC, Zhong W, Cooney P, Cockrell C, Chang E, Gaskins HR, McIntosh MK (2016) Table grape consumption reduces adiposity and markers of hepatic lipogenesis and alters gut microbiota in butter fat-fed mice. J Nutr Biochem 27:123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with Qiime 2´s q2-feature-classifier plugin. Microbiome 6:90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Caporaso JG (2018) QIIME 2: reproducible, interactive, scalable, and extensible microbiome data science. Peer J Preprints 6:e27295v2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, Zhernakova DV, Jankipersadsing SA, Jaeger M, Oosting M, Cenit MC, Masclee AAM, Swertz MA, Li Y, Kumar V, Joosten L, Harmsen H, Weersma RK, Franke L, Hofker MH, Xavier RJ, Jonkers D, Netea MG, Wijmenga C, Fu J, Zhernakova A (2016) The effect of host genetics on the gut microbiome. Nat Genet 48(11):1407–1412 [DOI] [PubMed] [Google Scholar]
- Brestoff JR, Artis D (2013) Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 14(7):676–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR (2016) Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol 14(11):e2000225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, Mcmurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) Dada2: high-resolution sample inference from illumina amplicon data. Nat Methods 13(17):581–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarinha-Silva A, Maushammer M, Wellmann R, Vital M, Preuss S, Bennewitz J (2017) Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs. Genetics 206(3):1637–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Yang X, Peng C, Wang Y, Guo Q, Su H (2022) Gut microbiota reveals the environmental adaption in gastro-intestinal tract of wild boar in karst region of Southwest China. Ann Microbiol 72(1):1–15 [Google Scholar]
- Chao A (1984) Nonparametric estimation of the number of classes in a population. Scand J Stat 11:265–270 [Google Scholar]
- Chen L, van den Munckhof ICL, Schraa K, ter Horst R, Koehorst M, van Faassen M, der Ley C, Doestzada M, Zhernakova DV, Kurilshikov A, Bloks VW, Groen AK, Riksen NP, Rutten JHW, Joosten LAB, Wijmenga C, Alexandra Z, Netea MG, Fu J, Kuipers F (2020) Genetic and microbial associations to plasma and fecal bile acids in obesity relate to plasma lipids and liver fat content. Cell Rep 33(1):108212 [DOI] [PubMed] [Google Scholar]
- Chen C, Fang S, Wei H, He M, Huang L (2021a) Prevotella copri increases fat accumulation in pigs fed with formula diets. Microbiome 9(1):175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang D, Garmaeva S, Kurilshikov A, Vila AV, Gacesa R, Sinha T, Segal E, Weersma RK, Wijmenga C (2021b) The long-term genetic stability and individual specificity of the human gut microbiome. Cell 184(9):2302–2315 [DOI] [PubMed] [Google Scholar]
- Cox LM, Yamanishi S, Sohn J, Alekseyenko VA, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, Zárate Rodriguez JG, Rogers AB, NicolasRobine LP, Blaser MJ (2014) Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158(4):705–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Piazuelo D, Estellé J, Revilla M, Criado-Mesas L, Ramayo-Caldas Y, Óvilo C, Fernández AI, Ballester M, Folch JM (2018) Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci Rep 8(1):12727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Micro 72(7):5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas GM, Maffei VJ, Zaneveld J, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI (2020) PICRUSt2: an improved and customizable approach for metagenome inference. BioRxiv 672295. 10.1101/672295
- Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Conserv 61(1):1–10 [Google Scholar]
- Fang S, Xiong X, Su Y, Huang L, Chen C (2017) 16S rRNA gene-based association study identified microbial taxa associated with pork intramuscular fat content in feces and cecum lumen. BMC Microbiol 17(1):162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T (2011) Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469(7331):543–547 [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504(7480):446–450 [DOI] [PubMed] [Google Scholar]
- Gao P, Liu Y, Le B, Qin B, Liu M, Zhao Y, Guo X, Cao G, Liu J, Li B, Duan Z (2019) A comparison of dynamic distributions of intestinal microbiota between Large White and Chinese Shanxi Black pigs. Arch Microbiol 201(3):357–367 [DOI] [PubMed] [Google Scholar]
- Gaulke CA, Arnold HK, Kembel SW, O’Dwyer JP, Sharpton TJ (2017) Ecophylogenetics reveals the evolutionary associations between mammals and their gut microbiota. BioRxiv 2017:182212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good IJ (1953) The population frequency of species and the estimation of the population parameters. Biometrika 40(3–4):237–264 [Google Scholar]
- Greetham HL, Gibson GR, Giffard C, Hippe H, Merkhoffer B, Steiner U, Falsen E, Collins MD (2004) Allobaculum stercoricanis gen. nov., sp. nov., isolated from canine feces. Anaerobe 10(5):301–307 [DOI] [PubMed] [Google Scholar]
- Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, Alm EJ (2017) Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat Commun 8:14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Fang S, Huang X, Zhao Y, Ke S, Yang H, Li Z, Gao J, Chen C, Huang L (2016) Evaluating the contribution of gut microbiota to the variation of porcine fatness with the cecum and fecal samples. Front Microbiol 7:2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann E, Wayne Y, Douglas R, Verena RG, Riedel CU, Ralf C, Markus E (2017) RNA-based stable isotope probing suggests Allobaculum spp. as particularly active glucose assimilators in a complex murine microbiota cultured in vitro. Biomed Res Int 2017:1829685 [DOI] [PMC free article] [PubMed]
- Huang J, Zhang W, Fan R, Liu Z, Huang T, Li J, Du T, Xiong T (2020) Composition and functional diversity of fecal bacterial community of wild boar, commercial pig and domestic native pig as revealed by 16S rRNA gene sequencing. Arch Microbiol 202(4):843–857 [DOI] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21(9):1552–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivarsson E, Roos S, Liu HY, Lindberg JE (2014) Fermentable non-starch polysaccharides increases the abundance of Bacteroides-Prevotella-Porphyromonas in ileal microbial community of growing pigs. Animal 8:1777–1787 [DOI] [PubMed] [Google Scholar]
- Izumi H, Minegishi M, Sato Y, Shimizu T, Sekine K, Takase M (2015) Bifidobacterium breve alters immune function and ameliorates DSS-induced inflammation in weanling rats. Pediatr Res 78(4):407–416 [DOI] [PubMed] [Google Scholar]
- Jaccard P (1908) Nouvelles recherches sur la distribution florale. Bull Soc Vaud Sci Nat 44:223–270 [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J, Daly K, Moran AW, Ryan S, Bravo D, Shirazi-Beechey SP (2017) Composition and diversity of mucosa-associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences. Environ Microbiol 19(4):1425–1438 [DOI] [PubMed] [Google Scholar]
- Kemis JH, Linke V, Barrett KL, Boehm FJ, Traeger LL, Keller MP, Rabaglia ME, Schueler KL, Stapleton DS, Gatti DM, Churchill GA, Amador-Noguez D, Russell JD, Yandell BS, Broman KW, Coon JJ, Attie AD, Rey FE (2019) Genetic determinants of gut microbiota composition and bile acid profiles in mice. PLoS Genet 15:e1008073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Park SH, Sin HS, Jang SH, Lee SW, Kim SY, Kwon B, Yu KY, Kim SY, Yang DK (2017) Hypocholesterolemic effects of probiotic mixture on diet-induced hypercholesterolemic rats. Nutrients 9(3):E293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B, Hall LJ, Metcalf CJE (2017) The microbiome beyond the horizon of ecological and evolutionary theory. Nat Ecol Evol 1(11):1606–1615 [DOI] [PubMed] [Google Scholar]
- Kraler M, Ghanbari M, Domig KJ, Schedle K, Kneifel W (2016) The intestinal microbiota of piglets fed with wheat bran variants as characterised by 16S rRNA next-generation amplicon sequencing. Arch Anim Nutr 70(3):173–189 [DOI] [PubMed] [Google Scholar]
- Lammerts van Bueren A, Saraf A, Martens EC, Dijkhuizen L (2015) Differential metabolism of exopolysaccharides from probiotic Lactobacilli by the human gut symbiont Bacteroides thetaiotaomicron. Appl Environ Microbiol 81(12):3973–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson G, Dobne K, Albarella U, Fang M, Matisoo-Smith E, Robins J, Lowden S, Finlayson H, Brand T, Willerslev E, Rowley-Conwy P, Andersson L, Cooper A (2005) World-wide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307(5715):1618–1621 [DOI] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R (2008) Evolution of mammals and their gut microbes. Science 320(5883):1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Ivarsson E, Dicksved J, Lundh T, Lindberg JE (2012) Inclusion of Chicory (Cichorium intybus L) in pigs’ diets affects the intestinal microenvironment and the gut microbiota. Appl Environ Microbiol 78(12):4102–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopera-Maya EA, Kurilshikov A, van der Graaf A, Hu S, Andreu-Sánchez S, Chen L, Vila AV, Gacesa R, Sinha T, Collij V, Klaassen MAY, Bolte LA, Brandao Gois MF, Neerincx PBT, Swertz MA, Harmsen HJM, Wijmenga C, Fu J, Weersma RK, Zhernakova A, Sanna S (2022) Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat Genet 54(2):143–151 [DOI] [PubMed] [Google Scholar]
- Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71(12):8228–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73(3):1576–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunney JK, Van Goor A, Walker KE, Hailstock T, Franklin J, Dai C (2021) Importance of the pig as a human biomedical model. Sci Transl Med 13(621):eabd5758 [DOI] [PubMed] [Google Scholar]
- Mach N, Berri M, Estellé J, Levenez F, Lemonnier G, Denis C, Leplat JJ, Chevaleyre C, Billon Y, Doré J, Rogel-Gaillard C, Lepage P (2015) Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ Microbiol Rep 7:554–569 [DOI] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17(1):10–12 [Google Scholar]
- Martínez I, Maldonado-Gomez MX, Gomes-Neto JC, Kittana H, Ding H, Schmaltz R, Joglekar P, Cardona RJ, Marsteller NL, Kembel SW, Benson AK, Peterson DA, Ramer-Tait AE, Walter J (2018) Experimental evaluation of the importance of colonization history in early-life gut microbiota assembly. Elife 7:e36521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Tanaka J, Namihira T, Shinzato N (2012) Antibiotics production by an actinomycete isolated from the termite gut. J Basic Microbiol 52(6):731–735 [DOI] [PubMed] [Google Scholar]
- McCormack UM, Curião T, Buzoianu SG, Prieto ML, Ryan T, Varley P, Crispie F, Magowan E, Metzler-Zebeli BU, Berry D, O’Sullivan O, Cotter PD, Gardiner GE, Lawlor PG (2017) Exploring a possible link between the intestinal microbiota and feed efficiency in pigs. Appl Environ Microb 83(15):e00380-e417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C, Lugli GA, Duranti S, Turroni F, Bottacini F, Mangifesta M, Sanchez B, Viappiani A, Mancabelli L, Taminiau B, Delcenserie V, Barrangou R, Margolles A, van Sinderen D, Ventura M (2014) Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl Environ Microb 80(20):6290–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi E, Kim S, Suda W, Kawasumi M, Onawa S, Taguchi-Atarashi N, Morita H, Taylor TD, Hattori M, Ohno H (2020) Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature 585(7823):102–106 [DOI] [PubMed] [Google Scholar]
- Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, Ochman H (2016) Cospeciation of gut microbiota with hominids. Science 3353(6297):380–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielou EC (1966) The measurement of diversity in different types of biological collections. J Theor Biol 13:131–144 [Google Scholar]
- Pokusaeva K, Fitzgerald GF, Van Sinderen D (2011) Carbohydrate metabolism in Bifidobacteria. Genes Nutr 6(3):285–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26(7):1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Havulinna AS, Liu Y, Jousilahti P, Ritchie SC, Tokolyi A, Sanders JG, Valsta L, Brożyńska M, Zhu Q, Tripathi A, Vázquez-Baeza Y, Loomba R, Cheng S, Jain M, Niiranen T, Lahti L, Knight R, Salomaa V, Inouye M, Méric G (2022) Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat Genet 54(2):134–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan J, Cai G, Ye J, Yang M, Ding R, Wang X, Zheng E, Fu D, Li S, Zhou S, Liu D, Yang J, Wu Z (2018) A global comparison of the microbiome compositions of three gut locations in commercial pigs with extreme feed conversion ratios. Sci Rep 8(1):4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan J, Cai G, Yang M, Zeng Z, Ding R, Wang X, Zhuang Z, Zhou S, Li S, Yang H, Li Z, Zheng E, Huang W, Yang J, Wu Z (2019) Exploring the fecal microbial composition and metagenomic functional capacities associated with feed efficiency in commercial DLY pigs. Front Microbiol 10:52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramayo-Caldas Y, Mach N, Lepage P, Levenez F, Denis C, Lemonnier G, Leplat JJ, Billon Y, Berri M, Doré J, Rogel-Gaillard C, Estellé J (2016) Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J 10(12):2973–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramette A (2007) Multivariate analyses in microbial ecology. FEMS Microbiol Ecol 62(2):142–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo-Adrover M, Saldaña-Ruíz S, van Limpt K, Knipping K, Garssen J, Knol J, Franch A, Castell M, Pérez-Cano FJ (2017) A combination of scGOS/lcFOS with Bifidobacterium breve M-16V protects suckling rats from rotavirus gastroenteritis. Eur J Nutr 56(4):1657–1670 [DOI] [PubMed] [Google Scholar]
- Rubin CJ, Megens HJ, Barrio AM, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg Ö, Jern P, Jørgensen C, Archibald A, Fredholm M, Groenen MAM, Andersson L (2012) Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci USA 109(48):19529–19536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez B, Urdaci MC, Margolles A (2010) Extracellular proteins secreted by probiotic bacteria as mediators of effects that promote mucosa-bacteria interactions. Microbiology 156(11):3232–3242 [DOI] [PubMed] [Google Scholar]
- Satoh T, Izumi H, Iwabuchi N, Odamaki T, Namba K, Abe F, Xiao JZ (2016) Bifidobacterium breve prevents necrotising enterocolitis by suppressing inflammatory responses in a preterm rat model. Benef Microbes 7(1):75–82 [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423 [Google Scholar]
- Sharpton TJ (2018) Role of the gut microbiome in vertebrate evolution. mSystems 3: e00174–17. [DOI] [PMC free article] [PubMed]
- Simpson EH (1949) Measurement of species diversity. Nature 163:688 [Google Scholar]
- Slifierz MJ, Friendship RM, Weese JS (2015) Longitudinal study of the early-life fecal and nasal microbiotas of the domestic pig. BMC Microbiol 15(1):184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki TA, Phifer-Rixey M, Mack KL, Sheehan MJ, Lin D, Bi K, Nachman MW (2019) Host genetic determinants of the gut microbiota of wild mice. Mol Ecol 28(13):3197–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida S, Maruyama F, Ogura Y, Toyoda A, Hayashi T, Okuma M, Ushida K (2017) Genomic characteristics of Bifidobacterium thermacidophilum pig isolates and wild boar isolates reveal the unique presence of a putative mobile genetic element with tetW for pig farm isolates. Front Microbiol 8:1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushida K, Tsuchida S, Ogura Y, Toyoda A, Maruyama F (2016) Domestication and cereal feeding developed domestic pig-type intestinal microbiota in animals of suidae. Anim Sci J 87(6):835–841 [DOI] [PubMed] [Google Scholar]
- Van Muijlwijk GH, van Mierlo G, Jansen PW, Vermeulen M, Bleumink-Pluym NM, Palm NW, van Putten JPM, de Zoete MR (2021) Identification of Allobaculum mucolyticum as a novel human intestinal mucin degrader. Gut Microbes 13:1966278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Turroni F, van Sinderen D (2012) Probiogenomics as a tool to obtain genetic insights into adaptation of probiotic bacteria to the human gut. Bioengineered 3:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Doestzada M, Chen L, Andreu-Sánchez S, van den Munckhof ICL, Augustijn HE, Koehorst M, Ruiz-Moreno AJ, Bloks VW, Riksen NP, Rutten JHW, Joosten LAB, Netea MG, Wijmenga C, Zhernakova A, Kuipers F, Fu J (2021) Characterization of gut microbial structural variations as determinants of human bile acid metabolism. Cell Host Microbe 29(12):1802–1814 [DOI] [PubMed] [Google Scholar]
- Wibowo MC, Yang Z, Borry M, Hübner A, Huang KD, Tierney BT, Zimmerman S, Barajas-Olmos F, Contreras-Cubas C, García-Ortiz H, Martínez-Hernández A, Luber JM, Kirstahler P, Blohm T, Smiley FE, Arnold R, Ballal SA, Pamp SJ, Russ J, Maixner F, Rota-Stabelli O, Segata N, Reinhard K, Orozco L, Warinner C, Snow M, LeBlanc S, Kostic AD (2021) Reconstruction of ancient microbial genomes from the human gut. Nature 594(7862):234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SF, Chiu HY, Chen AC, Lin HY, Lin HC, Caplan M (2013) Efficacy of different probiotic combinations on death and necrotizing enterocolitis in a premature rat model. J Pediatr Gastr Nutr 57(1):23–28 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Li K, Xiang Y, Zhou W, Gui G, Yang H (2017) The fecal microbiota composition of boar Duroc, Yorkshire, Landrace and Hampshire pigs. Asian-Australas J Anim Sci 30:1456–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Kong F, Xiang Y, Zhou W, Wang J, Yang H, Zhang G, Zhao J (2018) Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci Rep 8(1):5985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Huang X, Fang S, Xin W, Huang L, Chen C (2016) Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci Rep 6:27427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Huang X, Fang S, He M, Zhao Y, Wu Z, Yang M, Zhang Z, Chen C, Huang L (2017) Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficency in pigs. Front Microbiol 8:1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Shi C, Zhang S, Liu Y, Li Z, Gao F, Cui Y, Yan Y, Li M (2020) Characterization of the bacterial microbiota composition and evolution at different intestinal tract in wild pigs (Sus scrofa ussuricus). PeerJ 8:e9124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wu J, Huang X, Zhou Y, Zhang Y, Liu M, Liu Q, Ke S, He M, Fu H, Fang S, Xiong X, Jiang H, Chen Z, Wu Z, Gong H, Tong X, Huang Y, Ma J, Gao J, Charlier C, Coppieters W, Shagam L, Zhang Z, Ai H, Yang B, Georges M, Chen C, Huang L (2022) ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs. Nature 606(7913):358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wang Y, Liu S, Huang J, Zhai Z, He C, Ding J, Wang J, Wang H, Fan W, Zhao J, Meng H (2015) The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 10(20):e0117441 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 (PNG 418 KB) Fig. S1 Species accumulation (A) and rank-abundance (B) curves analysis of the different gut intestinal tract samples at 97% sequences identity. If the curves reach or nearly reach a plateau, it indicates that most of the species present in all samples have been observed.
Supplementary file2 (PNG 78 KB) Fig. S2 The alpha-diversity comparisons for the duodenum (DU) in different pig populations.
Supplementary file3 (PNG 71 KB) Fig. S3 The alpha-diversity comparisons for the jejunum (JE) in different pig populations.
Supplementary file4 (PNG 70 KB) Fig. S4 The alpha-diversity comparisons for the ileum (IL) in different pig populations.
Supplementary file5 (PNG 57 KB) Fig. S5 The alpha-diversity comparisons for the colon (CO) in different pig populations.
Supplementary file6 (PNG 1499 KB) Fig. S6 The hierarchical clustering analyses were performed by unweighted pair-group method with arithmetic means (UPGMA).
Supplementary file7 (PNG 115 KB) Fig. S7 Phylogenetic tree with ASV abundance distribution in duodenum (DU) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
Supplementary file8 (PNG 110 KB) Fig. S8 Phylogenetic tree with ASV abundance distribution in jejunum (JE) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
Supplementary file9 (PNG 118 KB) Fig. S9 Phylogenetic tree with ASV abundance distribution in ileum (IL) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
Supplementary file10 (PNG 116 KB) Fig. S10 Phylogenetic tree with ASV abundance distribution in cecum (CE) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
Supplementary file11 (PNG 108 KB) Fig. S11 Phylogenetic tree with ASV abundance distribution in colon (CO) of pigs. Species abundance distribution was aligned to the tree and visualized as boxplots. The Phylum information was used to color symbolic points on the tree and also species abundance distributions.
Data Availability Statement
All data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.