

Abstract
The ability to introduce noncanonical amino acids as axial ligands in heme enzymes has provided a powerful experimental tool for studying the structure and reactivity of their FeIV=O (“ferryl”) intermediates. Here, we show that a similar approach can be used to perturb the conserved Fe coordination environment of 2-oxoglutarate (2OG) dependent oxygenases, a versatile class of enzymes that employ highly-reactive ferryl intermediates to mediate challenging C–H functionalizations. Replacement of one of the cis-disposed histidine ligands in the oxygenase VioC with a less electron donating Nδ-methyl-histidine (MeHis) preserves both catalytic function and reaction selectivity. Significantly, the key ferryl intermediate responsible for C–H activation can be accumulated in both the wildtype and the modified protein. In contrast to heme enzymes, where metal-oxo reactivity is extremely sensitive to the nature of the proximal ligand, the rates of C–H activation and the observed large kinetic isotope effects are only minimally affected by axial ligand replacement in VioC. This study showcases a powerful tool for modulating the coordination sphere of nonheme iron enzymes that will enhance our understanding of the factors governing their divergent activities.
Keywords: metal-oxo reactivity, C−H functionalization, genetic code expansion, noncanonical histidine analogue, 2OG-dependent hydroxylation
Introduction
IronII/2-oxoglutarate (2OG)-dependent dioxygenases are a versatile superfamily of enzymes that promote a wealth of selective C–H activation processes, including substrate hydroxylation, halogenation, C–C-forming cyclization, epoxidation, and desaturation.1−11 The catalytic capabilities of these enzymes have been expanded to include nonbiological processes, such as nitration and azidation.12,13 The catalytic FeII center is coordinated by a conserved His···Glu/Asp···His facial binding triad (Figure 1).14−17 In the case of halogenases and engineered azidation/nitration biocatalysts, the carboxylate is replaced by a noncoordinating residue (typically Ala/Gly), which leaves a vacant coordination site for anion binding.13,18−20 Reaction of the ferrous enzyme with molecular oxygen and 2OG generates the highly oxidizing ferryl (FeIV=O) intermediate, which effects selective H atom abstractions (HA) from bound substrates.21,22 Depending on the specific enzyme in question, the resulting FeIII–OH/R• intermediate can then undergo a variety of radical coupling processes, leading to distinct reaction outcomes.6,20
Figure 1.
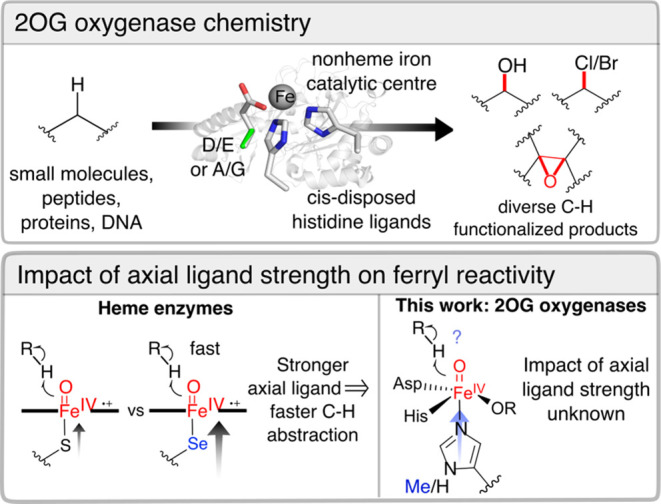
2OG oxygenases catalyze a wealth of oxidative chemistries. The effect of axial ligand substitutions is well-understood in heme enzymes, although the impact of axial ligand strength on nonheme iron 2OG oxygenases is unknown.
Given the broad array of valuable transformations catalyzed by 2OG oxygenases, there is considerable interest in understanding how their active site features govern the structure and reactivity of catalytic intermediates. To this end, a range of spectroscopic, kinetic, structural, and computational studies have advanced our understanding of the 2OG oxygenase catalytic cycles.23−37 Similarly, mutations of second and outer coordination sphere residues have advanced our understanding of 2OG oxygenase catalysis.38 However, performing structure–function relationships to probe the role of the conserved bis-histidine coordination environment has proven challenging, as mutation of either of the cis-disposed ligands abolishes catalytic function. If we were able to make functional ligand replacements, we could begin to address important questions such as the effect of ligand strength on FeIV=O reactivity or positioning (“in-line” vs “off-line”39,40) across family members. Previously, our group and others have shown that the axial ligands of heme enzymes can be replaced by noncanonical analogues of cysteine and histidine, creating a new way to probe structure–activity relationships directly in enzyme active sites.41−44,48 Here, we show that a similar approach can be used to modulate the primary coordination sphere of nonheme iron enzymes, providing new opportunities to probe and augment the catalytic mechanisms of this enzyme superfamily.
We selected the 2OG-dependent hydroxylase VioC as a model enzyme for active site reengineering. VioC catalyzes a regio- and stereoselective hydroxylation of its amino acid substrate, l-arginine, to form 3(S)-hydroxy-l-arginine, a vital step in the biosynthesis of the nonribosomal peptide tuberactinomycin antibiotic, viomycin.45,46 The catalytic iron is coordinated by His168, Asp170, and His316, with His316 trans to the site at which oxygen is proposed to bind.45,47
Results
To probe the effect of axial ligand electron donation, we replaced His316 by a noncanonical Nδ-methyl-histidine (MeHis) ligand using an engineered pyrrolysyl-tRNA synthetase/pyrrolysyl-tRNA pair (PylRS_MeHis/tRNAPyl), which selectively encodes MeHis in response to the amber (UAG) stop codon.49 Stoichiometric replacement of histidine by MeHis was confirmed by mass spectrometry (Supporting Table S1). A 1.6 Å X-ray crystal structure of VioC MeHis316 (Supporting Table S2) superimposes well with a published wildtype structure (secondary structure superposed RMSD of 0.2 Å),47 with no significant changes in the location of the iron cofactor or the imidazole planes of the axial His/MeHis ligands (Figures 2 and S1). Introduction of the additional methyl substituent on the axial ligand displaces an ordered water, which, in wildtype VioC, forms hydrogen bonding interactions with the Nδ-H of His316, the Asn311 side chain, and the backbone carbonyl of Ala314. The substrate l-arginine (occupancy 0.8) and succinate (occupancy 0.8) adopt similar conformations to those observed in the wildtype structure, with the l-Arg pro-3S hydrogen apparently well-positioned for selective removal by the ferryl complex.
Figure 2.
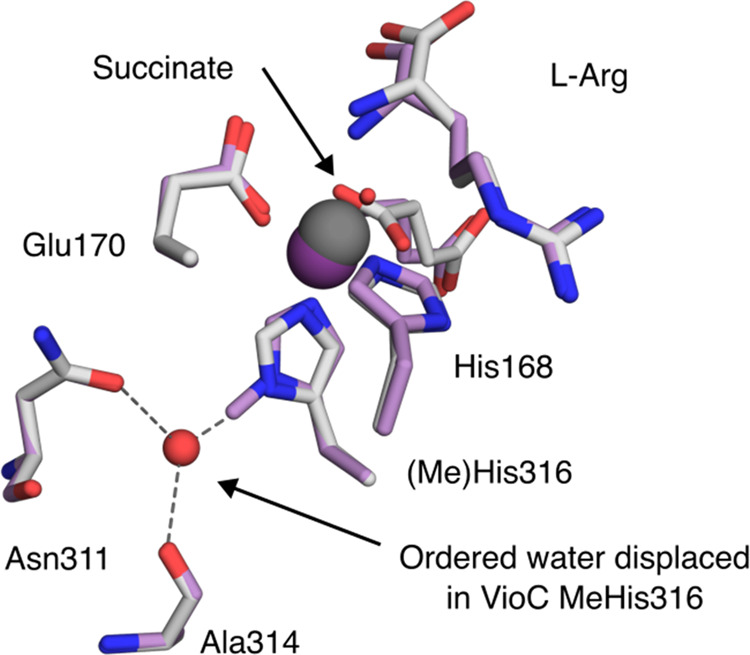
Structural characterization of VioC MeHis316. The active sites of VioC (PDB: 6ALQ) and VioC MeHis316 (PDB: 9EQF). Active site residues, succinate, and bound substrate l-Arg are shown as atom-colored sticks (gray and purple carbons for VioC and VioC MeHis316, respectively). Iron is shown as dark gray or dark purple spheres in VioC (occupancy 1) and VioC MeHis316 (occupancy 0.4), respectively. An ordered water molecule that coordinates the Nδ of His316 (shown as a red sphere) is only present in the structure of VioC.
We next explored the impact of ligand substitution on l-arginine hydroxylase activity. Assays were performed under standard conditions (1 mM FeII, 1 mM l-Arg, 5 mM 2OG, and 1 mM sodium ascorbate, 25 °C) using either wildtype VioC (1 μM) or VioC MeHis316 (1 μM) as a biocatalyst. The conversion of l-arginine to 3(S)–OH-l-arginine was monitored by reverse-phase high-performance liquid chromatography (HPLC) following derivatization with N-(9-fluorenylmethoxycarbonyloxy)succinimide (Fmoc-OSu). The wildtype enzyme achieved ∼600 turnovers under these conditions, a value in line with previous reports.50−52 Remarkably, despite mutation of the primary iron coordination sphere, VioC MeHis316 was able to perform >400 turnovers, with no observed change in regioselectivity (Figure 3). Product retention times in biotransformations with VioC MeHis316 are indistinguishable from those observed with the wildtype enzyme, suggesting that the high degree of site selectivity achieved by wildtype VioC is retained upon axial ligand substitution, as anticipated, given the similar l-arginine conformations observed in the wildtype and VioC MeHis316 crystal structures.
Figure 3.
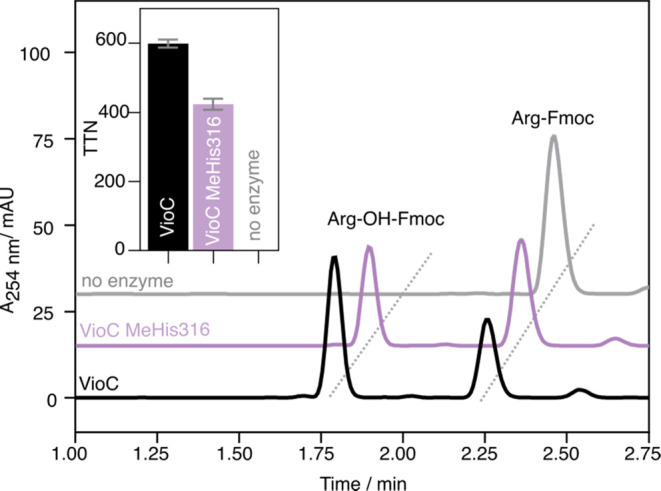
HPLC analysis of VioC-catalyzed hydroxylation. HPLC trace at 254 nm showing Fmoc-derivatized substrate Arg-Fmoc and product Arg-OH-Fmoc. Reaction conditions: 1 μM enzyme, 1 mM Arg, 5 mM 2OG, 1 mM (NH4)2Fe(SO4)2(H2O)6, 1 mM Asc in 100 μL 100 mM Tris pH 7.5, 18 h shaking (300 rpm) at 25 °C. Fmoc derivatization was achieved by the addition of 100 μL of 5 mM Fmoc-OSu in ACN. (inset) The total turnover numbers achieved by VioC (black) and VioC MeHis316 (purple) after 18 h shaking. Measurements are given as the average of four replicates with the standard deviation presented as an error bar.
With a functional modified enzyme in hand, we were well placed to investigate the effect of electron donation by the axial ligand on the VioC catalytic mechanism. To this end, we created cluster models of the ferryl intermediates of VioC and VioC MeHis316 that include first and second coordination sphere of the metal and substrate (Supporting Figure S2). Replacing His316 and its coordinating water with an axial MeHis ligand has little effect on ferryl structure, Fe–N bond length, electronic configuration, and spin state ordering (Supporting Tables S3 and S4). Introduction of the MeHis ligand increases the electron affinity of the ferryl intermediate slightly (ΔΔE = 1.3 kcal mol–1, Supporting Table S5). In both systems, in silico reduction results in electron transfer into the virtual σ*z2 orbital along the Fe-His316 bond. These calculations suggest that MeHis is a less electron donating ligand than the native histidine, which is polarized by hydrogen bonding to an ordered water (Supporting Table S5). Analogous calculations performed on the wildtype enzyme in the absence of this ordered water molecule confirm that hydrogen bonding interactions with His316 increase its electron donating properties, as evidenced by an increase in ferryl electron affinity (Supporting Table S5). These trends observed with VioC and VioC MeHis316 are in agreement with our previous observations in heme proteins, where polarized His ligands have been shown to be more electron-donating than MeHis in both peroxidases and myoglobin.41,44
To further explore the impact of ligand substitution on ferryl reactivity, we calculated the energy landscapes for C3-hydroxylation of l-arginine by VioC and VioC MeHis316. In this case we generated a larger cluster model, to include a greater number of residues interacting with the arginine substrate (Figure 4). Large cluster models of this type were previously shown to reproduce experimental product distributions and selectivities well.53,54 Recent computational studies on nonheme iron dioxygenases showed that experimentally determined free energies of activation and regioselectivities can only be reproduced with either large QM cluster models or QM/MM with a large QM region, hence the former approach was used here.55 Density functional theory (DFT) calculations were performed on the active-site models using unrestricted hybrid density functional UB3LYP in combination with a LANL2DZ basis set on iron (with core potential) and 6-31G* on the rest of the atoms (basis set BS1) (see Supporting Methods for further details).56−60 In agreement with previous calculations and experimental EPR/Mössbauer studies on the ferryl species of VioC and related nonheme iron dioxygenases the ground state is the quintet spin state.61−67 The hydrogen atom abstraction step is rate-determining with a free energy of activation of ΔG = 15.2 kcal mol–1 for VioC and ΔG = 15.8 kcal mol–1 for VioC MeHis316. The subsequent OH rebound leads to hydroxyarginine products with large exothermicity. These calculations show that the energy barrier for C–H cleavage is minimally affected by the His316MeHis modification (ΔΔE = 0.6 kcal mol–1, Figure 4 and Supporting Table S6). Both HA transition-state structures have large imaginary frequencies and indeed, replacing the transferring hydrogen atom by deuterium gives a large predicted kinetic isotope effect (KIE), suggesting a large quantum chemical tunnelling contribution to the C–H abstraction step (Supporting Table S7 and Figure S5).
Figure 4.

Computational analysis of VioC MeHis316. (A) Energy profile for hydrogen abstraction and rebound steps in the l-Arg hydroxylation reaction by VioC (black) and VioC MeHis316 (purple). UB3LYP/BS2 energies with zero-point energies included in kcal mol–1. (B) The QM cluster model B of the ferryl intermediate of wildtype VioC, generated to probe the energy landscapes for l-Arg hydroxylation. (C) The optimized geometries of model B in VioC (left) and VioC MeHis316 (right) in the 5Re (reactant) state.
To probe the effect of ligand substitution on ferryl reactivity experimentally, we next used stopped-flow absorption measurements to define the kinetics of C–H cleavage in VioC and VioC MeHis316. Spectral changes were monitored following exposure of an anoxic reactant complex (enzyme:FeII:2OG:Arg) to molecular oxygen within air-saturated buffer. Consistent with previous reports, VioC exhibits accumulation of a transient ferryl intermediate, as evidenced by the emergence of a broad absorption feature centered at 320 nm that reaches maximum intensity at ∼50 ms (Figure 5).61 Time-dependent absorbance changes were fitted to a sequential a → b → c model (See Supporting Information) to derive rate constants for the formation and decay of the ferryl intermediate in wildtype VioC (kform,obs = 41.2 ± 0.7 s–1 and kH = 11.9 ± 0.1 s–1, respectively; Tables 1 and S8). Experiments performed with per-d7-l-Arg lead to an extended lifetime of the ferryl intermediate (Figure 5 and Table 1), which was found to decay with a rate constant (kD) of 0.28 ± 0.002 s–1 to give a kinetic isotope effect (KIE = kD/kH) of 43.2 ± 0.7 for C–H/C–D abstraction (cf. a KIE of 43 reported previously, Supporting Table S9). Analogous experiments with VioC MeHis316 also reveal the formation of a ferryl intermediate with spectral features that are almost indistinguishable from those seen with the wildtype enzyme (Figure 5 inset). Remarkably, introduction of the less electron donating MeHis ligand has minimal effect on the rate constants of ferryl formation (kform,obvs = 46.8 ± 1.0 s–1), the rate of C–H abstraction (kH = 8.3 ± 0.3 s–1) and the observed KIE (28.9 ± 0.9). The modest reduction in KIE observed with VioC MeHis316 could plausibly arise from changes in the electronic structure of the ferryl intermediate and/or from minor adjustments in substrate positioning or active site dynamics. This result is in stark contrast to the situation encountered in heme enzymes, where there is a strong correlation between axial ligand strength and ferryl reactivity.43,44 Plausibly, the differing response to ligand substitution originates in the large contribution of quantum tunnelling to C–H abstraction by 2OG oxygenases, as evidenced by the high KIE.
Figure 5.
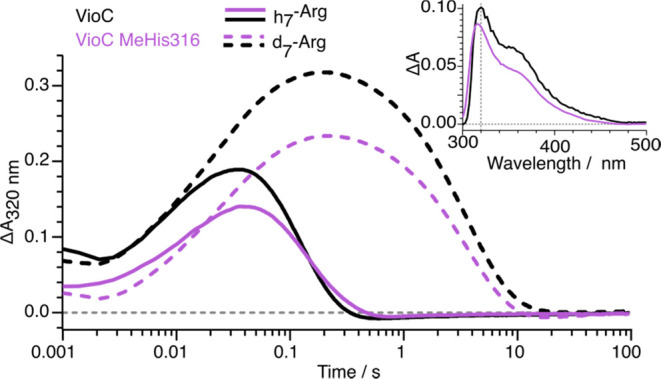
Stopped-flow UV–visible absorption analysis. Kinetics of formation and decay of the ferryl intermediates in the reactions of VioC (black) and VioC MeHis316 (purple) with l-Arg (solid lines) and per-d7-l-Arg (dashed lines) monitored by the absorbance changes at λ = 320 nm. Absorbance changes shown are the average of triplicate measurements. An anoxic solution of enzyme in complex with (per-d7-)-l-Arg and 2OG was mixed at 5 °C with an equal volume of 5 °C air-saturated buffer, to give a final concentration of 500 μM enzyme, 3 mM 2OG, 3 mM l-Arg or per-d7-l-Arg, and 500 μM (NH4)2Fe(SO4)2(H2O)6. (inset) The difference spectra of ferryl intermediates observed with VioC (black) and VioC MeHis316 (purple) 100 ms after rapid mixing a substrate complex (enzyme:FeII:d7-l-Arg:2OG) with air-saturated buffer.
Table 1. Stopped-flow Kinetic Analysis of VioC and VioC MeHis316a.
| VioC |
VioC
MeHis316 |
|||
|---|---|---|---|---|
| h7-l-Arg | d7-l-Arg | h7-l-Arg | d7-l-Arg | |
| kH or kD, s–1 | 11.9 ± 0.1 | 0.28 ± 0.01 | 8.3 ± 0.1 | 0.29 ± 0.01 |
| KIE | 43 ± 1 | 29 ± 1 | ||
The rates of C–H abstraction given with protonated and deuterated l-Arg as the substrate are the average of triplicate measurements.
Conclusions
In summary, we have demonstrated that an expanded genetic code enables perturbation of the primary coordination sphere of nonheme iron enzymes while maintaining catalytic function. This approach enables new probes of structure–function relationships directly in enzyme active sites that were previously not possible within the constraints of the genetic code. Against our expectations, modulation of axial ligand strength has minimal effect on the kinetics of C–H/D abstraction, with large kinetic isotope effects (30–40) observed in both wildtype VioC and VioC MeHis316, consistent with a substantial tunnelling contribution in both proteins. Moving forward, there are clear opportunities to expand our methodology to explore the impact of equatorial ligand substitutions on 2OG oxygenase catalysis. Moreover, we anticipate that our ability to desymmetrize the bis-histidine coordination sphere of nonheme iron enzymes will open new avenues to characterizing key metal-oxo intermediates (e.g., through selective isotopic labeling) or perhaps even unlock alternative modes of reactivity in this versatile superfamily of enzymes.
Acknowledgments
The authors gratefully acknowledge the European Research Council (ERC Starter Grant, grant number 757991, to A.P.G.), the Biotechnology and Biological Sciences Research Council (David Phillips Fellowship BB/M027023/1, to A.P.G.), and the UK Catalysis Hub funded by the EPSRC (grants EP/R026815/1, EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1, and EP/M013219/1, to A.P.G.). F.J.H was supported by an EPSRC Doctoral Prize Fellowship EP/W524347/1. The MCC is funded by EPSRC (EP/F067496). We thank the Diamond Light Source for access to beamlines (proposal number MX31850). The authors acknowledge the use of the Protein Structure Facility and Ultrafast Biophysics Facility at Manchester Institute of Biotechnology. Mass spectrometry data were acquired by R. Spiess, Manchester Institute of Biotechnology. The authors would like to acknowledge the assistance given by Research IT and the use of the Computational Shared Facility at The University of Manchester. Additional computing facilities for this work were provided by ARCCA at Cardiff University, HPC Wales.
Glossary
Abbreviations
- HPLC
high performance liquid chromatography
- DFT
density functional theory
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.4c02365.
Additional experimental details, materials, and methods, additional references, fits of stopped-flow data to kinetic models, protein mass spectrometry, data collection and refinement statistics for structure determination of VioC MeHis316, and the Cartesian coordinates of the DFT models A and B (PDF)
Author Contributions
F.J.H. and C.J.T. carried out molecular biology and protein production. F.J.H. carried out stopped-flow spectroscopy, protein crystallization, and hydroxylation assays. F.J.H. and C.L. interpreted, analyzed, and presented structural data. F.J.H., M.O., D.J.H. and J.Z. contributed to experimental design and data analysis. F.J.H., M.O., D.J.H. and A.P.G. discussed the results and participated in writing the manuscript. All authors provided input throughout project progression. A.P.G. initiated and directed the research. The DFT calculations and analysis were performed by M.Q., E.G., H.S.A., and S.P.d.V.
The authors declare no competing financial interest.
Supplementary Material
References
- Schofield C. J.; Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struct. Biol. 1999, 9 (6), 722–731. 10.1016/S0959-440X(99)00036-6. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S.-K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y.-S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100 (1), 235–350. 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104 (2), 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Abu-Omar M. M.; Loaiza A.; Hontzeas N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105 (6), 2227–2252. 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- De Visser S. P.; Kumar D.. Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; The Royal Society of Chemistry, 2011. [Google Scholar]
- Martinez S.; Hausinger R. P. Catalytic mechanisms of Fe(II)- and 2-Oxoglutarate-dependent oxygenases. J. Biol. Chem. 2015, 290 (34), 20702–20711. 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M. D.; Flashman E. Catalytic strategies of the non-heme iron dependent oxygenases and their roles in plant biology. Curr. Opin. Chem. Biol. 2016, 31, 126–135. 10.1016/j.cbpa.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser S. P.; Mukherjee G.; Ali H. S.; Sastri C. V. Local Charge Distributions, Electric Dipole Moments, and Local Electric Fields Influence Reactivity Patterns and Guide Regioselectivities in α-Ketoglutarate-Dependent Non-heme Iron Dioxygenases. Acc. Chem. Res. 2022, 55 (1), 65–74. 10.1021/acs.accounts.1c00538. [DOI] [PubMed] [Google Scholar]
- Wojdyla Z.; Borowski T. Properties of the Reactants and Their Interactions within and with the Enzyme Binding Cavity Determine Reaction Selectivities. The Case of Fe(II)/2-Oxoglutarate Dependent Enzymes. Chem. - Eur. J. 2022, 28 (18), e202104106 10.1002/chem.202104106. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Fujimori D. G.; Walsh C. T.; Bollinger J. M. Non-Heme Fe(IV)–Oxo Intermediates. Acc. Chem. Res. 2007, 40 (7), 484–492. 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausinger R. P.Biochemical Diversity of 2-Oxoglutarate-Dependent Oxygenases. In 2-Oxoglutarate-Dependent Oxygenases; Schofield C., Hausinger R., Eds.; The Royal Society of Chemistry, 2015; pp 1–58. [Google Scholar]
- Matthews M. L.; Chang W.-c.; Layne A. P.; Miles L. A.; Krebs C.; Bollinger J. M. Jr. Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat. Chem. Biol. 2014, 10 (3), 209–215. 10.1038/nchembio.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui J.; Zhao Q.; Huls A. J.; Soler J.; Paris J. C.; Chen Z.; Reshetnikov V.; Yang Y.; Guo Y.; Garcia-Borràs M.; Huang X. Directed evolution of nonheme iron enzymes to access abiological radical-relay C(sp3)-H azidation. Science 2022, 376 (6595), 869–874. 10.1126/science.abj2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshlyakov D. A.; Henshaw T. F.; Monterosso G. R.; Ryle M. J.; Hausinger R. P. Direct Detection of Oxygen Intermediates in the Non-Heme Fe Enzyme Taurine/α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc. 2004, 126 (4), 1022–1023. 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- McDonough M. A.; Loenarz C.; Chowdhury R.; Clifton I. J.; Schofield C. J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20 (6), 659–672. 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Islam M. S.; Leissing T. M.; Chowdhury R.; Hopkinson R. J.; Schofield C. J. 2-Oxoglutarate-Dependent Oxygenases. Annu. Rev. Biochem. 2018, 87, 585–620. 10.1146/annurev-biochem-061516-044724. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer M. E.; Sumida K. H.; Pelton J. G.; McMurry J. L.; Marchand J. A.; Chang M. C. Y. A family of radical halogenases for the engineering of amino acid-based products. Nat. Chem. Biol. 2019, 15, 1009–1016. 10.1038/s41589-019-0355-x. [DOI] [PubMed] [Google Scholar]
- Mitchell A. J.; Dunham N. P.; Bergman J. A.; Wang B.; Zhu Q.; Chang W. C.; Liu X.; Boal A. K. Structure-Guided Reprogramming of a Hydroxylase to Halogenate Its Small Molecule Substrate. Biochemistry 2017, 56 (3), 441–444. 10.1021/acs.biochem.6b01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. D.; Srnec M.; Matthews M. L.; Liu L. V.; Kwak Y.; Park K.; Bell C. B.; Alp E. E.; Zhao J.; Yoda Y.; et al. Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 2013, 499 (7458), 320–323. 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Tirupati B.; Bollinger J. M.; Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42 (24), 7497–7508. 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Glass T. E.; Krebs C.; Bollinger J. M. Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α -Ketoglutarate Dioxygenase (TauD). J. Am. Chem. Soc. 2003, 125 (43), 13008–13009. 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- Chang W.-c.; Guo Y.; Wang C.; Butch S. E.; Rosenzweig A. C.; Boal A. K.; Krebs C.; Bollinger J. M. Jr. Mechanism of the C5 stereoinversion reaction in the biosynthesis of carbapenem antibiotics. Science 2014, 343 (6175), 1140–1144. 10.1126/science.1248000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham N. P.; Chang W. C.; Mitchell A. J.; Martinie R. J.; Zhang B.; Bergman J. A.; Rajakovich L. J.; Wang B.; Silakov A.; Krebs C.; et al. Two Distinct Mechanisms for C-C Desaturation by Iron(II)- and 2-(Oxo)glutarate-Dependent Oxygenases: Importance of α-Heteroatom Assistance. J. Am. Chem. Soc. 2018, 140 (23), 7116–7126. 10.1021/jacs.8b01933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho R. Y. N.; Mehn M. P.; Hegg E. L.; Liu A.; Ryle M. J.; Hausinger R. P.; Que L. Jr. Resonance Raman studies of the iron(II)-α-keto acid chromophore in model and enzyme complexes. J. Am. Chem. Soc. 2001, 123 (21), 5022–5029. 10.1021/ja0041775. [DOI] [PubMed] [Google Scholar]
- Elkins J. M.; Ryle M. J.; Clifton I. J.; Hotopp J. C. D.; Lloyd J. S.; Burzlaff N. I.; Baldwin J. E.; Hausinger R. P.; Roach P. L. X-ray crystal structure of Escherichia coli taurine/α-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry 2002, 41 (16), 5185–5192. 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- Bollinger J. M.; Krebs C. Stalking intermediates in oxygen activation by iron enzymes: Motivation and method. J. Inorg. Biochem. 2006, 100 (4), 568–605. 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Martinie R. J.; Pollock C. J.; Matthews M. L.; Bollinger J. M.; Krebs C.; Silakov A. Vanadyl as a Stable Structural Mimic of Reactive Ferryl Intermediates in Mononuclear Nonheme-Iron Enzymes. Inorg. Chem. 2017, 56 (21), 13382–13389. 10.1021/acs.inorgchem.7b02113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Wang B.; Shaik S.; Lai W. QM/MM Calculations Reveal the Important Role of α-Heteroatom Substituents in Controlling Selectivity of Mononuclear Nonheme HppE-Catalyzed Reactions. ACS Catal. 2020, 10 (16), 9521–9532. 10.1021/acscatal.0c01803. [DOI] [Google Scholar]
- Cho K.-B.; Hirao H.; Shaik S.; Nam W. To rebound or dissociate? This is the mechanistic question in C–H hydroxylation by heme and nonheme metal–oxo complexes. Chem. Soc. Rev. 2016, 45 (5), 1197–1210. 10.1039/C5CS00566C. [DOI] [PubMed] [Google Scholar]
- Usharani D.; Janardanan D.; Shaik S. Does the TauD Enzyme Always Hydroxylate Alkanes, While an Analogous Synthetic Non-Heme Reagent Always Desaturates Them?. J. Am. Chem. Soc. 2011, 133 (2), 176–179. 10.1021/ja107339h. [DOI] [PubMed] [Google Scholar]
- Berger M. B.; Walker A. R.; Vázquez-Montelongo E. A.; Cisneros G. A. Computational investigations of selected enzymes from two iron and α-ketoglutarate-dependent families. Phys. Chem. Chem. Phys. 2021, 23 (39), 22227–22240. 10.1039/D1CP03800A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; Lord R. L.; Cisneros G. A. Ab Initio QM/MM Calculations Show an Intersystem Crossing in the Hydrogen Abstraction Step in Dealkylation Catalyzed by AlkB. J. Phys. Chem. B 2013, 117 (21), 6410–6420. 10.1021/jp403116e. [DOI] [PubMed] [Google Scholar]
- Chaturvedi S. S.; Rifayee S. B. J. S.; Waheed S. O.; Wildey J.; Warner C.; Schofield C. J.; Karabencheva-Christova T. G.; Christov C. Z. Can Second Coordination Sphere and Long-Range Interactions Modulate Hydrogen Atom Transfer in a Non-Heme Fe(II)-Dependent Histone Demethylase?. JACS Au 2022, 2 (9), 2169–2186. 10.1021/jacsau.2c00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed S. O.; Ramanan R.; Chaturvedi S. S.; Ainsley J.; Evison M.; Ames J. M.; Schofield C. J.; Christov C. Z.; Karabencheva-Christova T. G. Conformational flexibility influences structure–function relationships in nucleic acid N-methyl demethylases. Org. Biomol. Chem. 2019, 17 (8), 2223–2231. 10.1039/C9OB00162J. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. Differences in and Comparison of the Catalytic Properties of Heme and Non-Heme Enzymes with a Central Oxo–Iron Group. Angew. Chem., Int. Ed. 2006, 45 (11), 1790–1793. 10.1002/anie.200503841. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Hay S.; de Visser S. P. An Active Site Tyr Residue Guides the Regioselectivity of Lysine Hydroxylation by Nonheme Iron Lysine-4-hydroxylase Enzymes through Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2024, 146 (17), 11726–11739. 10.1021/jacs.3c14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A.; Waheed S. O.; Varghese A.; Cherilakkudy F. H.; Schofield C. J.; Karabencheva-Christova T. G. Unusual catalytic strategy by non-heme Fe(ii)/2-oxoglutarate-dependent aspartyl hydroxylase AspH. Chem. Sci. 2024, 15 (10), 3466–3484. 10.1039/D3SC05974J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K. M.; Altmyer M.; Martinie R. J.; Schaperdoth I.; Krebs C.; Bollinger J. M.; Boal A. K. Structure of a Ferryl Mimic in the Archetypal Iron(II)- And 2-(Oxo)-glutarate-Dependent Dioxygenase, TauD. Biochemistry 2019, 58 (41), 4218–4223. 10.1021/acs.biochem.9b00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Li Y.; Yuan W.; Zhang X.; Si Y.; Wang B. Conformational Isomerization of the Fe(III)–OH Species Enables Selective Halogenation in Carrier-Protein-Independent Halogenase BesD and Hydroxylase-Evolved Halogenase. ACS Catal. 2024, 14, 9342–9353. 10.1021/acscatal.4c02704. [DOI] [Google Scholar]
- Green A. P.; Hayashi T.; Mittl P. R. E.; Hilvert D. A Chemically Programmed Proximal Ligand Enhances the Catalytic Properties of a Heme Enzyme. J. Am. Chem. Soc. 2016, 138 (35), 11344–11352. 10.1021/jacs.6b07029. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Hilvert D.; Green A. P. Engineered Metalloenzymes with Non-Canonical Coordination Environments. Chem. - Eur. J. 2018, 24, 11821–11830. 10.1002/chem.201800975. [DOI] [PubMed] [Google Scholar]
- Onderko E. L.; Silakov A.; Yosca T. H.; Green M. T. Characterization of a selenocysteine-ligated P450 compound I reveals direct link between electron donation and reactivity. Nat. Chem. 2017, 9 (7), 623–628. 10.1038/nchem.2781. [DOI] [PubMed] [Google Scholar]
- Ortmayer M.; Fisher K.; Basran J.; Wolde-Michael E. M.; Heyes D. J.; Levy C.; Lovelock S. L.; Anderson J. L. R.; Raven E. L.; Hay S.; et al. Rewiring the “ Push-Pull” Catalytic Machinery of a Heme Enzyme Using an Expanded Genetic Code. ACS Catal. 2020, 10, 2735–2746. 10.1021/acscatal.9b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmetag V.; Samel S. A.; Thomas M. G.; Marahiel M. A.; Essen L. O. Structural basis for the erythro-stereospecificity of the L-arginine oxygenase VioC in viomycin biosynthesis. FEBS J. 2009, 276 (13), 3669–3682. 10.1111/j.1742-4658.2009.07085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.; Zabriskie T. M. VioC is a Non-Heme Iron, a -Ketoglutarate-Dependent Oxygenase that Catalyzes the Formation of 3 S -Hydroxy-L-Arginine during Viomycin Biosynthesis. ChemBioChem 2004, 5, 1274–1277. 10.1002/cbic.200400082. [DOI] [PubMed] [Google Scholar]
- Mitchell A. J.; Dunham N. P.; Martinie R. J.; Bergman J. A.; Pollock C. J.; Hu K.; Allen B. D.; Chang W. C.; Silakov A.; Bollinger J. M.; et al. Visualizing the reaction cycle in an Iron(II)- and 2-(Oxo)-glutarate-dependent hydroxylase. J. Am. Chem. Soc. 2017, 139 (39), 13830–13836. 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 2013, 342 (6160), 825–829. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H.; Peters F. B.; Yang P.-Y.; Reed S.; Chittuluru J. R.; Schultz P. G. Genetic incorporation of histidine derivatives using an engineered pyrrolysyl-tRNA synthetase. ACS Chem. Biol. 2014, 9 (5), 1092–1096. 10.1021/cb500032c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson M.; McNamee M.; Fan R.; Guo Y.; Chang W.-c. C. Repurposing Nonheme Iron Hydroxylases To Enable Catalytic Nitrile Installation through an Azido Group Assistance. J. Am. Chem. Soc. 2019, 141 (8), 3419–3423. 10.1021/jacs.8b13906. [DOI] [PubMed] [Google Scholar]
- Voss M.; Malca S. H.; Buller R. Exploring the Biocatalytic Potentialof Fe/a-Ketoglutarate-Dependent Halogenases. Chem. - Eur. J. 2020, 26, 7336–7345. 10.1002/chem.201905752. [DOI] [PubMed] [Google Scholar]
- Zhou S.; Pan J.; Davis K. M.; Schaperdoth I.; Wang B.; Boal A. K.; Krebs C.; Bollinger J. M. Steric Enforcement of cis-Epoxide Formation in the Radical C–O-Coupling Reaction by Which (S)-2-Hydroxypropylphosphonate Epoxidase (HppE) Produces Fosfomycin. J. Am. Chem. Soc. 2019, 141 (51), 20397–20406. 10.1021/jacs.9b10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H. S.; Henchman R. H.; Warwicker J.; de Visser S. P. How Do Electrostatic Perturbations of the Protein Affect the Bifurcation Pathways of Substrate Hydroxylation versus Desaturation in the Nonheme Iron-Dependent Viomycin Biosynthesis Enzyme?. J. Phys. Chem. A 2021, 125 (8), 1720–1737. 10.1021/acs.jpca.1c00141. [DOI] [PubMed] [Google Scholar]
- Mokkawes T.; de Visser S. P. Caffeine Biodegradation by Cytochrome P450 1A2. What Determines the Product Distributions?. Chem. - Eur. J. 2023, 29 (32), e202203875 10.1002/chem.202203875. [DOI] [PubMed] [Google Scholar]
- Ali H. S.; de Visser S. P. Electrostatic Perturbations in the Substrate-Binding Pocket of Taurine/α-Ketoglutarate Dioxygenase Determine its Selectivity. Chem. - Eur. J. 2022, 28 (9), e202104167 10.1002/chem.202104167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37 (2), 785 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82 (1), 270–283. 10.1063/1.448799. [DOI] [Google Scholar]
- Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54 (2), 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]
- Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77 (7), 3654–3665. 10.1063/1.444267. [DOI] [Google Scholar]
- Dunham N. P.; Mitchell A. J.; Del Río Pantoja J. M.; Krebs C.; Bollinger J. M.; Boal A. K. α-Amine Desaturation of d -Arginine by the Iron(II)- and 2-(Oxo)glutarate-Dependent l -Arginine 3-Hydroxylase, VioC. Biochemistry 2018, 57 (46), 6479–6488. 10.1021/acs.biochem.8b00901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Price J. C.; Hoffart L. M.; Barr E. W.; Krebs C. Mechanism of Taurine: α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem. 2005, 2005, 4245–4254. 10.1002/ejic.200500476. [DOI] [PubMed] [Google Scholar]
- Chen H.; Lai W.; Yao J.; Shaik S. Perferryl FeV–Oxo Nonheme Complexes: Do They Have High-Spin or Low-Spin Ground States?. J. Chem. Theory Comput. 2011, 7 (10), 3049–3053. 10.1021/ct200614g. [DOI] [PubMed] [Google Scholar]
- Kulik H. J.; Drennan C. L. Substrate Placement Influences Reactivity in Non-heme Fe(II) Halogenases and Hydroxylases. J. Biol. Chem. 2013, 288 (16), 11233–11241. 10.1074/jbc.M112.415570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójcik A.; Radoń M.; Borowski T. Mechanism of O2 Activation by α-Ketoglutarate Dependent Oxygenases Revisited. A Quantum Chemical Study. J. Phys. Chem. A 2016, 120 (8), 1261–1274. 10.1021/acs.jpca.5b12311. [DOI] [PubMed] [Google Scholar]
- Ghafoor S.; Mansha A.; de Visser S. P. Selective Hydrogen Atom Abstraction from Dihydroflavonol by a Nonheme Iron Center Is the Key Step in the Enzymatic Flavonol Synthesis and Avoids Byproducts. J. Am. Chem. Soc. 2019, 141 (51), 20278–20292. 10.1021/jacs.9b10526. [DOI] [PubMed] [Google Scholar]
- Waheed S. O.; Varghese A.; Chaturvedi S. S.; Karabencheva-Christova T. G.; Christov C. Z. How Human TET2 Enzyme Catalyzes the Oxidation of Unnatural Cytosine Modifications in Double-Stranded DNA. ACS Catal. 2022, 12 (9), 5327–5344. 10.1021/acscatal.2c00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.