

Abstract

Lithium iodide enables regioconvergent C–F bond functionalization of isomeric Morita–Baylis–Hillman fluorides with carbon, sulfur, and nitrogen nucleophiles. The defluorinative carbon–carbon and carbon–heteroatom bond formations give multifunctional compounds in excellent yields and with good to high diastereoselectivities at room temperature. The possibility of catalytic enantioselective allylation is also discussed.
Introduction
The general significance of fluorinated organic compounds in the life sciences has stimulated the introduction of many practical methods to make them.1,2 Organofluorines have become readily available starting materials that are rapidly growing in popularity among synthetic chemists. The usefulness of aryl fluorides in nucleophilic aromatic substitution and transition metal-catalyzed cross-coupling reactions is well documented. Csp2–F functionalization is a frequently employed venue to form carbon–carbon or carbon–heteroatom bonds, while applications of aliphatic substrates are less explored. Activation of a Csp3–F bond often requires strong Lewis acids and harsh conditions that may favor competing hydrodefluorination pathways and reduce functional group tolerance, although synthetically attractive protocols for carbon–carbon coupling,3−8 carbon–heteroatom bond formation,9−11 and halide exchange12,13 are known.14 Our laboratory has contributed to these efforts and introduced several methods that achieve C–F bond functionalization with a variety of alkyl fluorides under mild conditions.15−21
We have become increasingly interested in the development of synthetic methodologies that provide unique access to multifunctional compounds and exploit new reactivity patterns, in particular when these complement the outcome of existing reactions. To this end, we noticed that Shibata, Vilotijevic, and co-workers exploited silylated pronucleophiles that typically react at the allylic position in Morita–Baylis–Hillman (MBH) fluorides.22−31 By contrast, we envisioned that fluoride displacement might also be possible via attack at the vinylic carbon. Herein, we report that such a pathway by which the fluoride is replaced via formal SN2′ reaction can indeed be realized through activation with inexpensive lithium iodide at room temperature (Scheme 1). This protocol affords unprecedented regioselectivity control with carbon, sulfur, and nitrogen nucleophiles producing a variety of compounds in high yields and with good to excellent E/Z ratios. Moreover, this method allows regioconvergent substitution with isomeric MBH fluorides, which is attributed to the formation of a common (Z)-2-(iodomethyl)cinnamate intermediate that is readily consumed in the presence of a nucleophile.
Scheme 1. Allylic versus Vinylic Functionalization of MBH Fluorides.

In accordance with previous literature reports, we observed that the MBH fluoride 1 undergoes nucleophilic substitution at the allylic carbon when treated with silyl enol ethers 2 and 3 in the presence of catalytic amounts of DABCO,26−31 and we obtained 4 and 5 in 10% and 85% yields, respectively. The low yield of 4 was attributed to the low stability of silyl enol ether 2, which rapidly decomposed at room temperature. We discovered that employing enamines as nucleophiles switches the regioselectivity to the vinylic carbon resulting in SN2′ fluoride displacement (Scheme 2). We were pleased to find that both 6 and 7 afforded 8 and 9 in 74–83% yield and E/Z ratios of 16:1 and 20:1, respectively.
Scheme 2. Fluoride Substitution at the Allylic Carbon in the MBH Fluoride 1 with Silyl Enol Ethers versus SN2′ Displacement by the Corresponding Enamines.

When treating a 1:1 isomeric mixture of 10 and 11 with allylamine, 12, we observed that 10 reacts to the corresponding amine adduct 14 while 11 is not consumed. Further investigation revealed that the addition of LiI facilitates regioconvergent transformation of both 10 and 11 to a single iodide intermediate 13 which reacts quantitatively at room temperature with 12 to 14 exhibiting a high E/Z ratio of >20:1 (Scheme 3). We were able to isolate 13 to prove its central role in the regioconvergent defluorination pathway (see S1). However, we found that 10 can be directly transformed to 14 via SN2′ fluoride displacement in the absence of LiI, but consumption of 11 was not observed unless it was converted in situ to intermediate 13 which undergoes SN2 reaction with the amine nucleophile toward the same product. Alternatively, LiI can be replaced with TBAI, YI3, or YbI3, while TMSI proved less efficient, see SI. This generally points to negligible countercation effects at least when LiI, TBAI, etc., are used. According to Streitwieser,32 nucleophilic substitutions at allylic substrates by anionic nucleophiles are generally of the SN2 type, or when the SN2′ reaction prevails due to steric hindrance it proceeds with anti stereochemistry. The former is observed with the primary fluoride 11, while 10 is sterically hindered and therefore undergoes anti-SN2′ displacement. The diastereoselective conversion of 10 to the Z isomer of 13 is in agreement with an SN2′ transition state having the phenyl and the ester groups in a coplanar trans conformation according to a study of the reaction between phosphorus nucleophiles and MBH acetates by Georgiadis et al.33 This explains the regioconvergent generation of Z-13 from either allylic fluoride. Finally, SN2 displacement of the iodide in Z-13 with 12 gives E-14 in high yield and in excellent diastereomeric excess. This method is highly advantageous as it allows the use of both MBH fluoride isomers which are typically obtained as a mixture from their corresponding alcohols and are difficult to separate by column chromatography.
Scheme 3. Regioconvergent Addition of Amine 12 to an Isomeric Mixture of the MBH Fluorides 10 and 11 via Iodide Intermediate 13.

Intrigued by the regioconvergence and high diastereoselectivity of this reaction, we began screening various conditions including base additives, stoichiometry of reactants, and solvents (see SI). We determined that optimal results are obtained with two equivalents of nucleophile, diisopropylethylamine, and LiI in dichloromethane at room temperature. It is noteworthy, however, that only slightly lower yields were obtained with one equivalent of lithium iodide, and the reaction was found to proceed with catalytic amounts, generating 14 in 52% yield as well as 25% of a dialkylation byproduct, see SI. Next, we evaluated the substrate scope under optimized conditions. As shown in Scheme 4, a diverse array of nucleophiles undergoes the desired regioconvergent allylic substitution with isomeric mixtures of MBH fluorides in high yields and good to excellent diastereoselectivity. MBH fluorides with different ester groups gave the corresponding products 14–17 in almost quantitative yields and 20:1 dr when treated with allylamine. Overall, amine nucleophiles are well tolerated and give yields ranging from 83% to 99%. The reaction with primary, secondary, and heterocyclic amines all afford the desired products 18 and 20–25 in >20:1 dr. A moderate decrease in dr (10:1) was observed when aniline was used in the synthesis of 19, while yields were not affected. Interestingly, thiols are also tolerated, and we obtained 26 in 99% yield and 10:1 dr. A noticeable drop in the diastereoselectivity was observed with carbon nucleophiles that can, however, be generated in situ with Hünig’s base, thus eliminating the need to prepare enamines. The use of dimethyl malonate, 1-pyrrolidino-1-cyclohexene, and 2-carbethoxycyclopentanone afforded the desired products 27–29 with yields ranging from 81% to 95% and dr’s between 4:1 and 10:1. The reaction outcome proved sensitive to the presence of electron-withdrawing and electron-donating groups in the phenyl ring of the MBH fluoride. The 4-cyanophenyl and 4-nitrophenyl derivatives quantitatively converted to the intermediate 13, but subsequent amination with 12 was not observed even after heating to 50 °C overnight. By contrast, overalkylation to the tertiary amine byproduct could not be controlled with the 3-methoxyphenyl MBH fluoride despite the use of two equivalents of 12.
Scheme 4. Scope of the Regioconvergent Allylic Substitution Reaction.
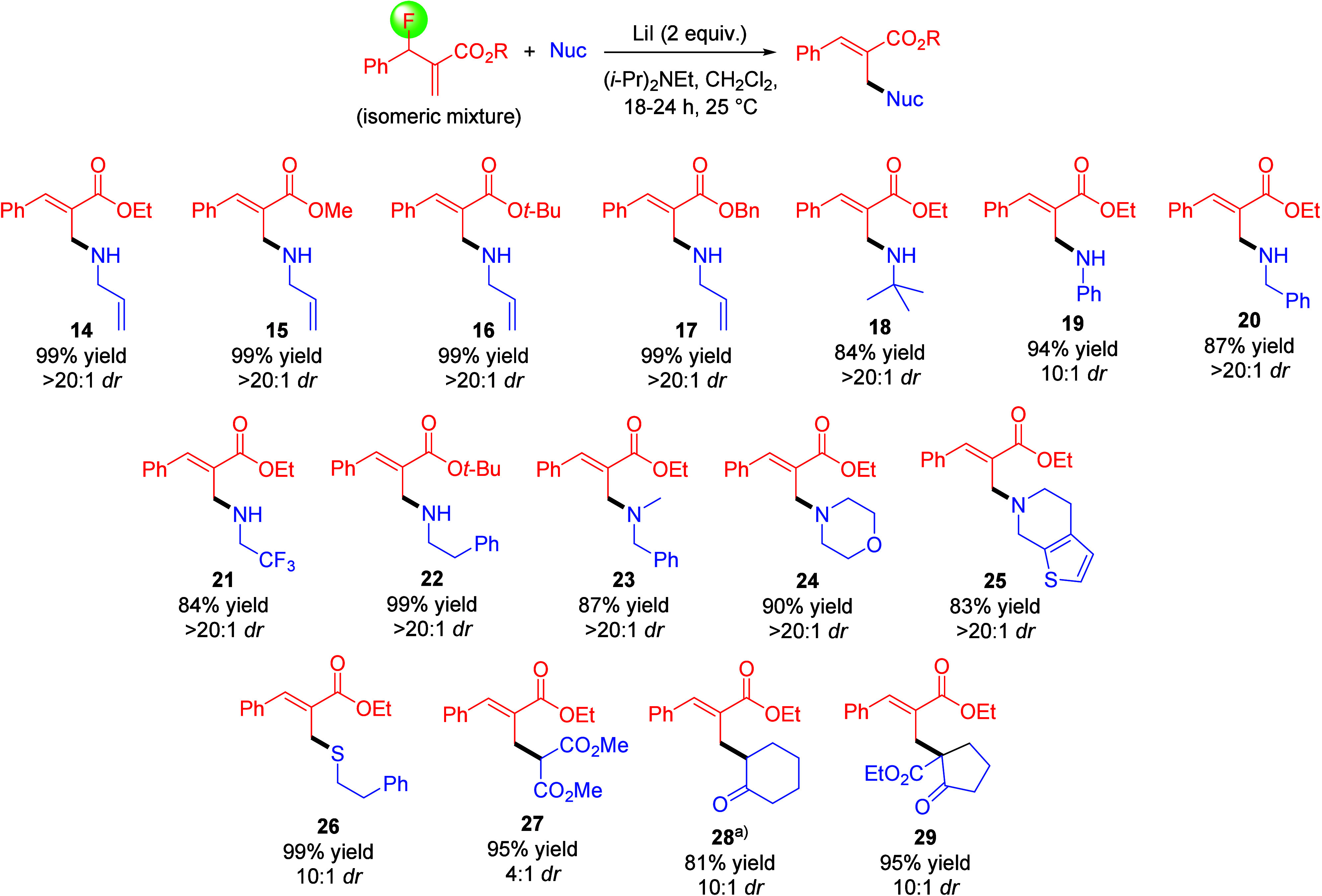
Conditions: MBH fluoride (0.1 mmol), nucleophile (0.2 mmol), LiI (0.2 mmol), DIPEA (0.2 mmol) in anhydrous dichloromethane (0.5 mL). aPrepared from 1-pyrrolidino-1-cyclohexene.
We discovered that HFIP-assisted palladium-catalyzed asymmetric alkylation is also possible and proceeds exclusively at the same carbon atom. Similar to our LiI protocol, we discovered that isomeric mixtures of MBH fluorides 10 and 11 react in a regioconvergent mechanism with the palladium catalyst following fluoride abstraction with HFIP. The comprehensive screening of palladium complexes and reaction conditions revealed that 29 can be obtained in 95% yield, 65% ee, and 10:1 diastereomeric ratio, Scheme 5 and SI.
Scheme 5. Palladium-Catalyzed Asymmetric Allylic Alkylation Using Ketoester 30 and a Mixture of the Fluorides 10 and 11.

In conclusion, we have introduced a practical method that allows smooth substitution at the vinylic carbon of MBH fluorides with excellent regioconvergence, yield, and E/Z diastereoselectivity with carbon, sulfur, and nitrogen nucleophiles. The use of LiI increases the practicality of this chemistry by enabling regioconvergent substitution of isomeric fluoride mixtures via a common intermediate. This affords multifunctional MBH derivatives in 81–99% yields and high dr’s in most cases. Asymmetric catalytic substitution at the vinylic carbon is also possible, albeit with only moderate enantioselectivity. This method complements previously reported regioselective substitutions of MBH fluorides with silylated pronucleophiles that react at the allylic carbon center. In addition, the necessity to separate E/Z isomers of the MBH fluoride starting materials is overcome with inexpensive lithium iodide which produces a common allylic iodide intermediate and thus significantly increases overall yields.
Experimental Section
General Information
All chemicals and solvents were used as purchased without further purification. The MBH fluorides were synthesized following literature procedures.22−24,26−29 NMR spectra were obtained at 400 MHz (1H NMR) and 100 MHz (13C NMR) in deuterated chloroform or methanol. Chemical shifts are reported in ppm relative to the solvent peak. Reaction products were purified by column chromatography on silica gel (particle size 40–63 μm) as described below.
General Procedure of the Regioconvergent Substitution of MBH Fluorides with C-, N-, and S-Nucleophiles
A vial was charged with LiI (0.2 mmol), MBH fluoride (0.1 mmol), nucleophile (0.2 mmol), diisopropylethylamine (0.2 mmol), and anhydrous dichloromethane (0.5 mL). The mixture was stirred at room temperature under N2 atmosphere for 24 h. The residue of the crude reaction mixture was directly dry loaded onto silica gel and purified by flash chromatography using hexanes–ethyl acetate mixtures as mobile phase as described below.
General Procedure of the Regioconvergent Substitution of MBH Fluorides with Enamines
A vial was charged with the MBH fluoride (0.1 mmol), enamine (0.1 mmol), and THF (0.5 mL) under nitrogen. The reaction was stirred at room temperature for 18 h. The mixture was quenched with saturated ammonium chloride and extracted with CH2Cl2, followed by purification of the residue by flash chromatography as described below.
General Procedure of the Regioconvergent Substitution of MBH Fluorides with Silyl Enol Ethers
A vial was charged with the silyl enol ether (0.1 mmol), DABCO (0.01 mmol), MBH fluoride (0.1 mmol), and anhydrous CH2Cl2 (0.5 mL) under nitrogen. The reaction was stirred for 18 h. The mixture was quenched with saturated ammonium chloride and extracted with CH2Cl2, followed by purification of the residue by flash chromatography as described below.
Asymmetric Allylic Alkylation Procedure
A vial was charged with (S)-(+)-(3,5-dioxa-4-phospha-cyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)bis[(1R)-1-phenylethyl]amine (0.024 mmol, 24 mol %) and [η3-C3H5ClPd]2 (0.01 mmol, 5.0 mol %) in anhydrous dichloromethane (0.5 mL). The mixture was stirred at room temperature under N2 atmosphere for 1 h. HFIP (0.2 mmol) was added followed by diisopropylethylamine (0.2 mmol), ketoester 30 (0.2 mmol), and the MBH fluoride 10 (0.1 mmol). The resulting mixture was stirred at room temperature for 2 days. The residue of the crude reaction mixture was directly dry loaded onto silica gel and purified by flash chromatography as described below.
Representative Examples
Ethyl 2-(Naphthalen-2-yl(2-oxocyclohexyl)methyl)acrylate (5)
Structure 5 was produced as a colorless oil in 85% yield (28.6 mg, 0.09 mmol) from ethyl 2-(fluoro(naphthalen-2-yl)methyl)acrylate (25.8 mg, 0.1 mmol) and (cyclohex-1-en-1yloxy)trimethylsilane (17.0 mg, 0.1 mmol) after 18 h at 25 °C using the protocol provided above and hexanes/EtOAc (95:5) as the mobile phase. The dr was determined as >20:1 by 1H NMR analysis. 1H NMR (400 MHz, chloroform-d) δ 7.79–7.71 (m, 3H), 7.66 (m, 1H), 7.48–7.38 (m, 2H), 7.33 (m, 1H), 6.26 (s, 1H), 5.66 (s, 1H), 4.40 (d, J = 11.2 Hz, 1H), 4.14–3.97 (m, 2H), 3.17 (m, 1H), 2.55–2.31 (m, 2H), 2.02 (m, 1H), 1.82–1.48 (m, 4H), 1.39–1.25 (m, 1H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, chloroform-d) δ 212.0, 166.7, 143.2, 138.1, 133.4, 132.4, 128.1, 127.8, 127.7, 127.6, 126.5, 126.0, 125.6, 60.8, 54.6, 45.6, 42.5, 33.4, 29.1, 24.6, 14.0. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C22H24O3Na 359.1619, found 359.1616.
Ethyl (E)-3-(Naphthalen-2-yl)-2-((2-oxocyclopentyl)methyl)acrylate (8)
Compound 8 was formed as a colorless oil in 83% yield (26.8 mg, 0.08 mmol) from ethyl 2-(fluoro(naphthalen-2-yl)methyl)acrylate (25.8 mg, 0.1 mmol) and 1-(cyclopent-1-en-1-yl)pyrrolidine (13.7 mg, 0.1 mmol) after 18 h at 25 °C using the general protocol provided above and hexanes/EtOAc (95:5) as the mobile phase. The dr was determined as 16:1 by 1H NMR analysis. 1H NMR (400 MHz, chloroform-d) δ (m, 5H), 7.52–7.39 (m, 3H), 4.29 (q, J = 7.1 Hz, 2H), 3.16 (m, 1H), 2.66 (m, 1H), 2.41 (m, 1H), 2.26 (m, 1H), 2.08 (m, 1H), 1.89 (m, 1H), 1.62 (m, 1H), 1.45 (m, 1H), 1.36 (t, J = 7.1 Hz, 3H), 0.83 (m, 1H). 13C NMR (100 MHz, chloroform-d) δ 219.8, 168.2, 140.0, 133.1, 133.0, 132.9, 131.7, 129.1, 128.4, 128.2, 127.6, 126.8, 126.7, 126.5, 61.0, 48.7, 37.7, 29.7, 27.1, 20.5, 14.3. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C21H22O3Na 345.1461, found 345.1459.
Ethyl (E)-2-(Allylamino)methyl)-3-phenyl acrylate (14)
Structure 14 was obtained as a colorless oil in 99% yield (24.5 mg, 0.1 mmol) from ethyl 2-(fluoro(phenyl)methyl)acrylate (20.0 mg, 0.1 mmol) and allyl amine (11.0 mg, 0.2 mmol) after 18 h at 25 °C using the general protocol provided above and hexanes/EtOAc (92:8) as the mobile phase. The dr was determined as >20:1 by 1H NMR analysis. 1H NMR (400 MHz, methanol-d4) δ 7.83 (s, 1H), 7.44–7.31 (m, 5H), 5.82 (m, 1H), 5.11–5.00 (m, 2H), 4.28 (q, J = 7.1 Hz, 2H), 3.59 (s, 2H), 3.17 (ddd, J = 6.3, 1.4, 1.4 Hz, 2H), 1.33 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, methanol-d4) δ 167.8, 141.9, 141.8, 135.8, 134.8, 130.0, 128.9, 127.8, 116.4, 59.8, 50.5, 43.9, 12.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C15H19NO2Na 268.1313, found 268.1308.
This reaction was repeated on a larger scale, and 14 was obtained as a colorless oil in 94% yield (230.4 mg, 0.94 mmol) from ethyl 2-(fluoro(phenyl)methyl)acrylate (208.2 mg, 1.0 mmol) and allyl amine (114.0 mg, 2.0 mmol) after 18 h at 25 °C using the procedure provided above and hexanes/EtOAc (92:8) as the mobile phase. The dr was determined as >20:1 by 1H NMR analysis.
Ethyl (Z)-2-((Phenethylthio)methyl)-3-phenyl acrylate (26)
Structure 26 was produced as a colorless oil in 99% yield (32.6 mg, 0.1 mmol) from ethyl 2-(fluoro(phenyl)methyl)acrylate (20.0 mg, 0.1 mmol) and 2-phenylethane-1-thiol (27.6 mg, 0.2 mmol) after 18 h at 25 °C using the general protocol provided above and hexanes/EtOAc (96:4) as the mobile phase. Rf = 0.66 (hexanes/EtOAc, 8:2). The dr was determined as 10:1 by 1H NMR analysis. 1H NMR (400 MHz, methanol-d4) δ 7.80 (s, 1H), 7.43–7.30 (m, 5H), 7.28–7.09 (m, 5H), 4.16 (q, J = 7.1 Hz, 2H), 3.60 (s, 2H), 2.80–2.67 (m, 4H), 1.23 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, methanol-d4) δ 167.6, 141.8, 141.6, 139.4, 134.5, 130.6, 129.1, 128.8, 128.4, 128.2, 125.4, 60.72, 49.7, 44.5, 35.0, 13.0. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C20H22NO2SNa 349.1238, found 349.1235.
3-Ethyl 1,1-Dimethyl (E)-4-phenylbut-3-ene-1,1,3-tricarboxylate (27)
Structure 27 was produced as a colorless oil in 95% yield (30.2 mg, 0.1 mmol) from ethyl 2-(fluoro(phenyl)methyl)acrylate (20.0 mg, 0.1 mmol) and dimethyl malonate (26.4 mg, 0.2 mmol) after 18 h at at 25 °C using the procedure provided above and hexanes/EtOAc (95:5) as the mobile phase. The dr was determined as 4:1 by 1H NMR (400 MHz, methanol-d4) δ 7.76 (s, 1H), 7.44–7.29 (m, 5H), 4.25 (q, J = 7.1 Hz, 2H), 3.74 (t, J = 7.8 Hz, 1H), 3.57 (s, 6H), 3.14 (d, J = 7.9 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, methanol-d4) δ 169.2, 167.5, 141.6, 141.4, 137.4.0, 134.9, 128.8, 128.4, 60.8, 51.4, 50.3, 25.9, 13.1. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H20O6Na 343.1158, found 343.1152.
Ethyl (E)-1-(2-(Ethoxycarbonyl)-3-phenylallyl)-2-oxocyclopentane-1-carboxylate ((E)-29)
Compound (E)-29 (32.8 mg, 0.95 mmol) was isolated as a colorless oil in 95% yield from ethyl 2-(fluoro(phenyl)methyl)acrylate (20.8 mg, 0.1 mmol) and ethyl 2-oxocyclopentanecarboxylate (31.2 mg, 0.2 mmol) after 48 h at 25 °C using the general protocol provided above and hexanes/EtOAc (94:6) as the mobile phase. The dr was determined as 10:1 by 1H NMR analysis. The ee was determined by HPLC (Chiracel OJ-H, hexanes/i-PrOH 98:2, flow rate 1 mL/min, λ = 254 nm) as 65% ee, tR (minor) = 43.8 min, tR (major) = 59.7 min. 1H NMR (400 MHz, methanol-d4) δ 7.73 (s, 1H), 7.42–7.30 (m, 5H), 4.20 (q, J = 7.1 Hz, 2H), 4.06–3.86 (m, 2H), 3.40 (d, J = 14.4 Hz, 1H), 2.96 (d, J = 14.4 Hz, 1H), 2.41–2.22 (m, 2H), 2.13 (m, 1H), 1.85–1.72 (m, 3H), 1.31 (t, J = 7.1 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, methanol-d4) δ 214.3, 170.8, 168.3, 141.5, 135.2, 129.4, 129.1, 128.8, 128.5, 128.1, 61.3, 60.8, 59.5, 36.6, 32.8, 29.8, 18.9, 13.0, 12.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C20H24O5Na 367.1521, found 367.1517.
Acknowledgments
We gratefully acknowledge financial support from the US National Institutes of Health, GM106260. E.N. thanks the Henry Luce Foundation for a Clare Boothe Luce Graduate Fellowship.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.4c00660.
Optimization, mechanistic studies, NMR spectra, chiral HPLC results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kirk L. K. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Process Res. Dev. 2008, 12, 305–321. 10.1021/op700134j. [DOI] [Google Scholar]
- Champagne P. A.; Desroches J.; Hamel J.-D.; Vandamme M.; Paquin J.-F. Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev. 2015, 115, 9073–9174. 10.1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- Gu W.; Haneline M. R.; Douvris C.; Ozerov O. V. Carbon–Carbon Coupling of C(sp3)–F Bonds Using Alumenium Catalysis. J. Am. Chem. Soc. 2009, 131, 11203–11212. 10.1021/ja903927c. [DOI] [PubMed] [Google Scholar]
- Champagne P. A.; Benhassine Y.; Desroches J.; Paquin J.-F. Friedel-Crafts Reaction of Benzyl Fluorides: Selective Activation of C-F Bonds as Enabled by Hydrogen Bonding. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. 10.1002/anie.201406088. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Perez M.; Caputo C. B.; Stephan D. W. Use of Trifluoromethyl Groups for Catalytic Benzylation and Alkylation with Subsequent Hydrodefluorination. Angew. Chem., Int. Ed. 2016, 55, 1417–1421. 10.1002/anie.201510494. [DOI] [PubMed] [Google Scholar]
- Dryzhakov M.; Moran J. Autocatalytic Friedel–Crafts Reactions of Tertiary Aliphatic Fluorides Initiated by B(C6F5)3·H2O. ACS Catal. 2016, 6, 3670–3673. 10.1021/acscatal.6b00866. [DOI] [Google Scholar]
- Yoshida S.; Shimomori K.; Kim Y.; Hosoya T. Single C–F Bond Cleavage of Trifluoromethylarenes with an ortho-Silyl Group. Angew. Chem., Int. Ed. 2016, 55, 10406–10409. 10.1002/anie.201604776. [DOI] [PubMed] [Google Scholar]
- Hamel J. D.; Paquin J.-F. Activation of C–F Bonds α to C–C Multiple Bonds. Chem. Commun. 2018, 54, 10224–10239. 10.1039/C8CC05108A. [DOI] [PubMed] [Google Scholar]
- Traff A. M.; Janjetovic M.; Hilmersson G. C–F Bond Substitution via Aziridinium Ion Intermediates. Chem. Commun. 2015, 51, 13260–13263. 10.1039/C5CC04723D. [DOI] [PubMed] [Google Scholar]
- Liu X.-W.; Echavarren J.; Zarate C.; Martin R. Ni-Catalyzed Borylation of Aryl Fluorides via C–F Cleavage. J. Am. Chem. Soc. 2015, 137, 12470–12473. 10.1021/jacs.5b08103. [DOI] [PubMed] [Google Scholar]
- Jaiswal A. K.; Prasad P. K.; Young R. D. Nucleophilic Substitution of Aliphatic Fluorides via Pseudohalide Intermediates. Chem. Eur. J. 2019, 25, 6290–6294. 10.1002/chem.201806272. [DOI] [PubMed] [Google Scholar]
- Traff A. M.; Janjetovic M.; Ta L.; Hilmersson G. Selective C-F Bond Activation: Substitution of Unactivated Alkyl Fluorides Using YbI3. Angew. Chem., Int. Ed. 2013, 52, 12073–12076. 10.1002/anie.201306104. [DOI] [PubMed] [Google Scholar]
- Janjetovic M.; Ekebergh A.; Traff A. M.; Hilmersson G. Catalytic Iodination of the Aliphatic C–F Bond by YbI3(THF)3: Mechanistic Insight and Synthetic Utility. Org. Lett. 2016, 18, 2804–2807. 10.1021/acs.orglett.6b01022. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Huang Y.-G.; Liu C.; Xiao J.-C.; Chen Q.-Y.; Guo Y. Review of Recent Advances in C-F Bond Activation of Aliphatic Fluorides. J. Fluorine Chem. 2015, 179, 14–22. 10.1016/j.jfluchem.2015.07.007. [DOI] [Google Scholar]
- Balaraman K.; Wolf C. Catalytic Enantioselective and Diastereoselective Allylic Alkylation with Fluoroenolates: Efficient Access to C3-Fluorinated and All-Carbon Quaternary Oxindoles. Angew. Chem., Int. Ed. 2017, 56, 1390–1395. 10.1002/anie.201608752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding R.; Bakhshi P. R.; Wolf C. Organocatalytic Insertion of Isatins into Aryl Difluoronitromethyl Ketones. J. Org. Chem. 2017, 82, 1273–1278. 10.1021/acs.joc.6b02704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman K.; Wolf C. Chemodivergent Csp3-F Bond Functionalization and Cross-Electrophile Alkyl-Alkyl Coupling with Alkyl Fluorides. Sci. Adv. 2022, 8, eabn7819 10.1126/sciadv.abn7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman K.; Kyriazakos S.; Palmer R.; Thanzeel F. Y.; Wolf C. Selective Csp3-F Bond Functionalization with Lithium Iodide. Synthesis 2022, 54, 4320–4328. 10.1055/s-0041-1738383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman K.; Wolf C. Palladium and Nickel Catalyzed Suzuki Cross-Coupling with Alkyl Fluorides. Org. Lett. 2021, 23, 8994–8999. 10.1021/acs.orglett.1c03515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figula B.; Kane D. L.; Balaraman K.; Wolf C. Organocuprate Cross–Coupling Reactions with Alkyl Fluorides. Org. Lett. 2022, 24, 8719–8723. 10.1021/acs.orglett.2c03775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane D. L.; Figula B. C.; Balaraman K.; Bertke J. A.; Wolf C. General Alkyl Fluoride Functionalization via Short-lived Carbocation-organozincate Ion Pairs. Nat. Commun. 2024, 15, 1866. 10.1038/s41467-024-45756-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimine T.; Fukushi K.; Shibata N.; Taira H.; Tokunaga E.; Yamano A.; Shiro M.; Shibata N. Kinetic Resolution of Allyl Fluorides by Enantioselective Allylic Trifluoromethylation Based on Silicon-Assisted C-F Bond Cleavage. Angew. Chem., Int. Ed. 2014, 53, 517–520. 10.1002/anie.201308071. [DOI] [PubMed] [Google Scholar]
- Nishimine T.; Taira H.; Tokunaga E.; Shiro M.; Shibata N. Enantioselective Trichloromethylation of MBH-Fluorides with Chloroform Based on Silicon-assisted C–F Activation and Carbanion Exchange Induced by a Ruppert–Prakash Reagent. Angew. Chem., Int. Ed. 2016, 55, 359–363. 10.1002/anie.201508574. [DOI] [PubMed] [Google Scholar]
- Okusu S.; Okazaki H.; Tokunaga E.; Soloshonok V. A.; Shibata N. Organocatalytic Enantioselective Nucleophilic Alkynylation of Allyl Fluorides Affording Chiral Skipped Ene-ynes. Angew. Chem., Int. Ed. 2016, 55, 6744–6748. 10.1002/anie.201601928. [DOI] [PubMed] [Google Scholar]
- Nishimine T.; Taira H.; Mori S.; Matsubara O.; Tokunaga E.; Akiyama H.; Soloshonok V. A.; Shibata N. Synthesis of Chiral (Tetrazolyl)methyl-containing Acrylates via Silicon-induced Organocatalytic Kinetic Resolution of Morita–Baylis–Hillman Fluorides. Chem. Commun. 2017, 53, 1128–1131. 10.1039/C6CC08830A. [DOI] [PubMed] [Google Scholar]
- Zi Y.; Lange M.; Schultz C.; Vilotijevic I. Latent Nucleophiles in Lewis Base Catalyzed Enantioselective N-Allylations of N-Heterocycles. Angew. Chem., Int. Ed. 2019, 58, 10727–10731. 10.1002/anie.201903392. [DOI] [PubMed] [Google Scholar]
- Lange M.; Zi Y.; Vilotijevic I. Enantioselective Synthesis of Pyrrolizin-1-ones via Lewis Base Catalyzed N-Allylation of N-Silyl Pyrrole Latent Nucleophiles. J. Org. Chem. 2020, 85, 1259–1269. 10.1021/acs.joc.9b02819. [DOI] [PubMed] [Google Scholar]
- Sumii Y.; Nagasaka T.; Wang J.; Uno H.; Shibata N. Synthesis of Chiral gem-Difluoromethylene Compounds by Enantioselective Ethoxycarbonyldifluoromethylation of MBH Fluorides via Silicon-Assisted C–F Bond Activation. J. Org. Chem. 2020, 85, 15699–15707. 10.1021/acs.joc.0c02201. [DOI] [PubMed] [Google Scholar]
- Zi Y.; Lange M.; Vilotijevic I. Enantioselective Lewis Base Catalyzed Phosphonyldifluoromethylation of Allylic Fluorides Using a C-silyl Latent Pronucleophile. Chem. Commun. 2020, 56, 5689–5692. 10.1039/D0CC01815E. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Lange M.; Zi Y.; Görls H.; Vilotijevic I. Latent Pronucleophiles in Lewis Base Catalysis: Enantioselective Allylation of Silyl Enol Ethers with Allylic Fluorides. Chem. Eur. J. 2023, 29, e202300641 10.1002/chem.202300641. [DOI] [PubMed] [Google Scholar]
- Duran J.; Mateos J.; Moyano A.; Companyó X. Catalytic Asymmetric Defluorinative Allylation of Silyl Enol Ethers. Chem. Sci. 2023, 14, 7147–7153. 10.1039/D3SC01498C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streitwieser A.; Jayasree E. G.; Hasanayn F.; Leung S. S.-H. A Theoretical Study of SN2′ Reactions of Allylic Halides: Role of Ion Pairs. J. Org. Chem. 2008, 73, 9426–9434. 10.1021/jo8020743. [DOI] [PubMed] [Google Scholar]
- Kalyva M.; Zografos A. L.; Kapourani E.; Giambazolias E.; Devel L.; Papakyriakou A.; Dive V.; Lazarou Y. G.; Georgiadis D. Probing the Mechanism of Allylic Substitution of Morita–Baylis–Hillman Acetates (MBHAs) by using the Silyl Phosphonite Paradigm: Scope and Applications of a Versatile Transformation. Chem. Eur. J. 2015, 21, 3278–3289. 10.1002/chem.201405626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.