

Abstract
To clarify the binding properties of hepatitis C virus (HCV) core protein and its viral RNA for the encapsidation, morphogenesis, and replication of HCV, the specific interaction of HCV core protein with its genomic RNA synthesized in vitro was examined in an in vivo system. The positive-sense RNA from the 5′ end to nucleotide (nt) 2327, which covers the 5′ untranslated region (5′UTR) and a part of the coding region of HCV structural proteins, interacted with HCV core protein, while no interaction was observed in the same region of negative-sense RNA and in other regions of viral and antiviral sense RNAs. The internal ribosome entry site (IRES) exists around the 5′UTR of HCV; therefore, the interaction of the core protein with this region of HCV RNA suggests that there is some effect on its cap-independent translation. Cells expressing HCV core protein were transfected with reporter RNAs consisting of nt 1 to 709 of HCV RNA (the 5′UTR of HCV and about two-thirds of the core protein coding regions) followed by a firefly luciferase gene (HCV07Luc RNA). The translation of HCV07Luc RNA was suppressed in cells expressing the core protein, whereas no significant suppression was observed in the case of a reporter RNA possessing the IRES of encephalomyocarditis virus followed by a firefly luciferase. This suppression by the core protein occurred in a dose-dependent manner. The expression of the E1 envelope protein of HCV or β-galactosidase did not suppress the translation of both HCV and EMCV reporter RNAs. We then examined the regions that are important for suppression of translation by the core protein and found that the region from nt 1 to 344 was enough to exert this suppression. These results suggest that the HCV core protein interacts with viral genomic RNA at a specific region to form nucleocapsids and regulates the expression of HCV by interacting with the 5′UTR.
Hepatitis C virus (HCV) is the main cause of posttransfusion and sporadic non-A, non-B hepatitis (10, 18, 20, 26, 56). HCV persists for a long period and frequently leads to liver cirrhosis and hepatocellular carcinoma (4, 45). Although some cell lines have been shown to support partial replication of HCV (20, 23, 47), there are currently no efficient in vitro systems that will grow HCV. It was recently reported that a full-length HCV RNA transcribed from a cDNA clone of HCV was infectious in a chimpanzee by direct injection into the liver (13, 25, 60). However, the presence of infectious RNA in cell culture has not been reported. The lack of a conventional cell culture system for HCV hampers study of the replication mechanism of HCV.
HCV has approximately 9.5 kb of positive-strand RNA that possesses one open reading frame encoding one polyprotein (12). After translation, a capsid protein (core), envelope glycoproteins (E1 and E2), and nonstructural proteins (NS2, NS3a, NS3b, NS4A, NS4B, NS5A, and NS5B) are processed from the polyprotein by cellular and viral proteases (8, 11, 16, 17, 50, 55). HCV RNA has a long untranslated region at the 5′ end (5′UTR) whose sequence is highly conserved among different HCV isolates (8). The region around the 5′UTR contains the internal ribosome entry site (IRES) which is important for the initiation of cap-independent translation (57). Reynolds et al. (43) have shown that the functional region of the HCV IRES mapped between 40 and 370 nucleotides (nt) from the 5′ end includes a part of the core protein coding region.
The HCV core protein has many biological properties. It has four basic amino acid clusters, and the second cluster from the N terminus has been shown to be a nuclear localization signal (54). The C-terminal region of the core protein contains many hydrophobic amino acid residues and is an anchor that binds with the endoplasmic reticulum (34, 46). It has been suggested that it is also important for binding with the E1 protein (29). On the other hand, the N-terminal region of the core protein is involved not only in multimerization (32) but also in the protein’s interaction with the cytoplasmic tail of the lymphotoxin-β receptor (31) and modulation of its signal pathway (9). The core protein activates human c-myc, the Rous sarcoma virus long terminal repeat (LTR), and simian virus 40 early promoters, while it suppresses c-fos, p21, and human immunodeficiency virus LTR promoters (24, 40, 42) and the expression of coinfecting genomes of hepatitis B virus (48). The core protein modulates sensitivity to apoptosis (41, 44, 62) and, with H-ras, transforms primary rat embryo fibroblasts to a tumorigenic phenotype (39). Furthermore, the core protein induces liver steatosis (36), which eventually develops into hepatocellular carcinoma in transgenic mice (35). It has recently been reported that the HCV core protein suppresses the host immune response, and this result illustrates the persistence of HCV (27). In addition to the biological properties described above, HCV core protein may play an important role as a structural protein in the formation of viral nucleocapsids. In positive-strand RNA viruses, specific interactions between the nucleocapsid protein and its viral sense RNA have been demonstrated (for example, in Sindbis virus [15, 58, 59], rubella virus [28], and coronavirus [30, 53]).
Because of its amino acid residues and the similarity of its gene organization to other positive-strand RNA viruses, especially flaviviruses, the HCV core protein is also thought to bind to genomic RNA so that nucleocapsids form in the virus particles (33). However, there has been no unambiguous report of a study reproducing a specific interaction of the core protein with the genomic RNA either in vitro or in vivo, an interaction which is likely to occur in virion formation. Therefore, the binding properties of the core protein and viral RNA must be clarified to understand the mechanisms of viral encapsidation, morphogenesis, and replication.
In this study, we established an in vivo system to analyze the specific interactions of transiently expressed HCV core protein with transfected RNA synthesized in vitro. By using this system, we demonstrated that viral sense RNAs containing the 5′UTR to the E2 protein coding region are responsible for the specific interaction with HCV core protein. Furthermore, we found that expression of the HCV core protein specifically suppresses the translation of RNA possessing the 5′UTR of HCV. These findings suggest that HCV core protein may be implicated not only in encapsidating but also in modulating the expression of viral proteins leading to the establishment of a persistent HCV infection.
MATERIALS AND METHODS
Cells.
A human hepatocellular carcinoma cell line, HepG2, was obtained from the American Type Culture Collection. Cells were maintained in Dulbecco’s modified Eagle’s medium (GIBCO Laboratories, Grand Island, N.Y.) containing 2 mM l-glutamine, penicillin (50 IU/ml), and streptomycin (50 μg/ml) and supplemented with 10% fetal calf serum.
Recombinant baculoviruses.
Recombinant baculoviruses were constructed for expression of the proteins in mammalian cells (49). AcCA39, AcCA816, and AcCAlacZ possess cDNAs for HCV core (amino acids [aa] 1 to 191), envelope protein E1 (aa 192 to 383; E1), and β-galactosidase (β-Gal) under the CAG promoter (38), respectively. AcCAG has no insert and was used as a negative control.
Plasmids.
The cDNA clones of HCV genotype 1b, NIHJ1, used in this study were originally isolated from a blood sample of an HCV carrier that was infectious for both humans and chimpanzees (1). Each portion of the cDNAs was cloned under the T3 and T7 promoters in order to synthesize both strands of RNAs in vitro (Fig. 1). pBlue094, which contains nt 62 to 9402 of the HCV genome, was constructed by inserting the HindIII fragment of the HCV cDNA into the same site of pBluescriptII SK(−) (Stratagene, La Jolla, Calif.). pBlue014 which, covers nt 62 to 1358, was constructed by the ligation of the BamHI fragment from pBR094′ (see below) into pBluescriptII KS(−) (pBlue; Stratagene). pBR 094′ was constructed by insertion of 697 bp of the HindIII-ClaI (nt 14 to 710) fragment of the HCV cDNA and 8,692 bp of the ClaI-HindIII (nt 711 to 9402) fragment of HCV cDNA of pBR394 (1) into the HindIII site of pBR322 vector. pBlue1123, pBlue2333, and pBlue3247 were constructed by ligation of SalI-SacI (nt 1124 to 2327), XhoI-SacII (nt 2282 to 3313), and PstI-BamHI (nt 3212 to 4739) fragments of the HCV cDNA into the same site of pBlue, respectively. pBlue4763 was constructed by insertion of the BamHI-SacII (nt 4740 to 6345) fragment of pBlue4775 (see below) into the same site of pBlue. pBlue4775 was constructed by insertion of the BamHI (nt 4740 to 7476) fragment of pBlue094 into the same site of pBlue. pBlue6275 was constructed by insertion of the PvuII-BamHI (nt 6204 to 7476) fragment of pBlue4775 into the SmaI site of pBlue after blunting with T4 polymerase. pBlue7495 was constructed by ligation of the SalI-HindIII (nt 7420 to 9548) fragment of pT73′X possessing an entire HCV cDNA under the T7 promoter (1) into pBlue. pBlue7486, which included nt 4720 to 8648, was constructed by digestion of pBlue7495 with SmaI and self-ligation. pBlue8395, which contains nt 8280 to 9548, was constructed by self-ligation of about 4.2 kbp of the XhoI-BstEII fragment of pBlue7495 after blunting with T4 polymerase.
FIG. 1.

HCV RNAs used in this study. The gene organization of HCV is shown at the top. The gray and white bars indicate the RNAs used in the experiments. Numbers on the bars indicate the positions of both ends of the RNAs. Gray bars indicate regions of viral sense RNA exhibiting a specific interaction with the core protein, as shown in Fig. 2 to 4.
pBlue03 was constructed by cloning a HindIII-AccI (nt 1 to 334) fragment of pUCAB (see below) into the same site of pBlue. pUCAB was constructed by cloning the HindIII-NcoI fragment (nt 1 to 83) of pUCA into the same site of pUCB (1). pBlue37 containing nt 329 to 716 was constructed by self-ligation of the fragment derived from pBlue352 (nt 329 to 5231) digested with ClaI. pBlue714 was constructed by cloning a BamHI-ClaI fragment (nt 708 to 1357) of pBlue014 into the BamHI-ClaI site of pBlue. pBlue1423 was constructed by deletion of the SalI and BamHI (nt 1358 to 2327) fragment from pBlue1123.
pT7HCVLuc, which has been described previously (5), carries cDNA for the 5′UTR (nt 1 to 341) of HCV, a firefly luciferase gene (luciferase gene), cDNA for the coding region of the C terminus of NS5B and the 3′UTR (nt 9354 to 9523) of HCV, a ribozyme of hepatitis D virus, and a T7 terminator. pT7HCV07Luc was constructed as follows. pUC007 carrying nt 1 to 730 of the HCV cDNA under the T7 promoter (1) was digested with ClaI, blunt ended with Klenow enzyme, ligated with BamHI linker [d(CGCGGATCCGCG); New England Biolabs, Inc., Beverly, Mass.], and digested with BamHI (about 0.8-kbp fragment). This BamHI fragment was cloned into the same site of the PicaGene vector (this plasmid contains a luciferase gene; Toyo Ink Co. Ltd., Tokyo, Japan). In pT7HCV07Luc, the luciferase gene was connected in frame to the coding region of two-thirds of the core protein. pHCV09Luc, which has been described previously (61), has nt 1 to 924 of the HCV cDNA, the luciferase gene, cDNA for the coding region of the C terminus of NS5B and the 3′UTR of HCV, a ribozyme of hepatitis D virus, and a T7 terminator under the T7 promoter. In this plasmid, the luciferase gene was connected in frame to the E1 coding gene. pRL-null (Promega, Madison, Wis.) carrying the Renilla luciferase (RLuc) gene under the T7 promoter and pT7EMCVLuc possessing the IRES (nt 271 to 831) of encephalomyocarditis virus (EMCV), which is essential for the function of the IRES, and a firefly luciferase gene under the T7 promoter (5, 37) were used as templates for the synthesis of RNAs.
Preparation of RNAs.
The plasmids were linearized by digestion with appropriate restriction enzymes, and these DNA fragments were used as templates for the synthesis of RNAs. pT7HCVLuc, pT7HCV07Luc, and pT7HCV09Luc were linearized by digestion with XhoI for in vitro RNA synthesis. By XhoI digestion, the coding region of the C terminus of NS5B and the 3′UTR of HCV, a ribozyme of hepatitis D virus, and a T7 terminator in these plasmids were excluded. For digoxigenin (DIG)-labeled RNA synthesis, 1 μg of DNA template, 5× reaction buffer (Promega), and 2 μl of 10× labeling mixture (ATP, CTP, GTP, 10 mM; UTP, 6.5 mM; DIG-labeled UTP, 3.5 mM; Boehringer GmbH, Mannheim, Germany) were incubated with 2 μl of T3 or T7 enzyme mix (Ambion, Austin, Tex.) at 37°C for 2 h. For nonlabeled RNA synthesis, 2 μl each of ATP, CTP, GTP, and UTP (7.5 mM; Ambion) was used instead of the 10× labeling mixture. For capped-RNA synthesis, 2 μl each of ATP, CTP, and UTP (7.5 mM), 1 μl of GTP (7.5 mM), and 1 μl of cap homologue m7G (5′) ppp (5′) G (7.5 mM; Ambion) were used instead of the 10× labeling mixture. After incubation, the mixtures were treated twice with 2 U of DNase I (Ambion) at 37°C for 20 min and precipitated with lithium chloride (3.75 M; Ambion) after the addition of EDTA (25 mM).
Immunoblotting.
Cells infected with the recombinant baculoviruses were harvested at 2 days after infection, washed twice with phosphate-buffered saline (PBS), and boiled for 10 min in 1× sodium dodecyl sulfate (SDS) sample buffer (10% glycerol, 2.3% SDS, 62.5 mM Tris-HCl [pH 6.8], 5% 2-mercaptoethanol, 0.1% bromophenol blue). Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on a 12.5% gel and were electrophoretically blotted onto a polyvinylidene difluoride membrane (Millipore, Tokyo, Japan). The filter was blocked with Block Ace (Snow Brand, Tokyo, Japan) for 1 h at 37°C, incubated with anticore monoclonal antibody (54) at room temperature (RT) for 1 h, washed twice with Tris-buffered saline containing 0.1% Tween 20 (TTBS), and incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G goat serum (Amersham, Little Chalfont, Buckinghamshire, United Kingdom) for 1 h at RT. After three washes with TTBS, the protein was detected by enhanced chemiluminescence Western detection reagents (Amersham) instructed by the manufacturer.
RNA transfection and luciferase assay.
Cells (2.5 × 105) in a 24-mm-diameter dish were washed twice with 500 μl of Opti-MEM (GIBCO BRL, Life Technologies, Gaithersburg, Md.), and the same volume of Opti-MEM was added. DIG-labeled or nonlabeled RNA (2 μg) and 5 μl of Lipofectine (GIBCO) or Tfx-20 (Promega) were mixed well in 100 μl of Opti-MEM and incubated at RT for 15 min. The RNA mixture was inoculated into the cells and incubated at 37°C for 2 h.
For the experiment to determine the RNA regions of HCV interacting with the core protein, cells in a 24-mm-diameter dish were transfected with 2 μg of DIG-labeled RNAs. For experiments to determine the effect of expression of core protein on translation, cells expressing HCV core protein were transfected with reporter RNA (0.5 of HCVLuc, 0.8 of HCV07Luc, 1.5 of HCV09Luc, or 0.08 μg of EMCVLuc/well), together with the capped RLuc RNA (0.34 μg/well) as an internal control to normalize the efficiency of transfection, and were incubated at 37°C for 6 h. Firefly and RLuc activities were determined by the Dual-Luciferase reporter assay system (Promega) as described previously (5). Relative light units (RLU) were measured with a luminometer (Berthold, Wildbad, Germany), and the activity of firefly luciferase was normalized to that of RLuc.
Fluorescent ELISA Immunoassay.
Expression of the core protein was quantified by a fluorescent enzyme-linked immunosorbent assay (ELISA) as described previously (21); 10−3 μl of the cell lysates used in the luciferase assay was processed as instructed by the manufacturer (International Reagents Co., Kobe, Japan). Relative fluorescence intensity was determined by an ELSIA-F3000 reader (International Reagents Co.).
Immunoprecipitation.
Two days after infection with AcCA39, the cells were washed twice with 500 μl of PBS, suspended in 400 μl of TNE buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 250 μg of yeast RNA extracts [Boehringer] per ml), and sonicated for 30 s. After centrifugation at 2 × 104 × g for 10 min, 390 μl of supernatants was incubated with 0.5 μg of the anticore monoclonal antibody and 20 μl of protein A-Sepharose (50% suspension [vol/vol] in TNE buffer; Pharmacia, Tokyo, Japan) for 1 h at 4°C with rotation. After centrifugation at 8 × 103 × g for 10 s at 4°C, the pellets were washed twice with TNE buffer. For the detection of core protein, the immunoprecipitates were suspended in 1× SDS sample buffer, boiled for 10 min, separated by SDS-PAGE, and analyzed by immunoblotting.
Detection of HCV RNA in the immunoprecipitates.
The immunoprecipitates with anticore antibody were suspended in 400 μl of RNAzolB (Tel-Test, Inc., Friendswood, Tex.) and extensively vortexed. After addition of 40 μl of chloroform, the samples were incubated at 4°C for 5 min and centrifuged at 2 × 104 × g for 15 min. The aqueous phase was collected, the RNA was precipitated with an equal volume of 2-propanol, washed with 75% ethanol, and dissolved in 1.7 μl of 0.1% diethylpyrocarbonate-treated water, and 5.8 μl of sample buffer (17.5% formaldehyde, 50% formamide in 1× morpholinepropanesulfonic acid [MOPS] buffer containing 20 mM MOPS, 5 mM sodium acetate, and 1 mM EDTA [pH 7.0]) was added. The solution was denatured at 65°C for 15 min and cooled in ice-cold water; 7.5 μl of 2× loading buffer (80% formamide, 0.1% bromophenol blue, 0.1% xylene cyanol, 2 mM EDTA) was then added. After separation on the formaldehyde-denatured agarose gel (18% formaldehyde, 1% agarose) in 1× MOPS buffer, the gel was washed twice for 15 min with 20× SSC (3 M NaCl, 0.3 M trisodium citrate dihydrate [pH 7.0]) and transferred onto a Hybond N+ membrane filter (Amersham) by the capillary method. The filter was dried, and RNAs were fixed on the membrane by irradiation with UV light (UV Crosslinker; Funakoshi, Tokyo). DIG-labeled RNAs were detected by using a DIG luminescent detection kit (Boehringer) according to the manufacturer’s protocol. For dot blot analysis, the RNAs dissolved in water were dotted onto a Hybond N+ membrane filter, dried, fixed by irradiation with UV light, and detected as described above.
RESULTS
System determining the interaction between HCV core protein and its RNA.
To examine the interaction between HCV core protein and its RNA and to determine which regions of the RNA interact with the core protein, we established an in vivo system. HCV core protein was transiently expressed in HepG2 cells by infection with a recombinant baculovirus and transfected with either viral sense or antiviral sense DIG-labeled HCV RNAs. Cells were lysed with TNE buffer and immunoprecipitated with the anticore antibody; then DIG-labeled RNAs were extracted from the immunoprecipitates and detected by dot blotting or Northern blottings. In this system, if HCV RNAs interact with the core protein within cells, they can be recovered from the immunoprecipitates. We first determined the expression of HCV core protein in HepG2 cells by infection with a recombinant baculovirus, AcCA39, expressing full-length HCV core protein, and then determined whether the core protein could be immunoprecipitated by anticore antibody. Two days after infection, the core protein of about 22 kDa on polyacrylamide gels was immunoprecipitated with the anticore antibody (Fig. 2B). The core protein expressed in cells by the infection was detected mainly in the cytoplasm (data not shown).
FIG. 2.

Specific interaction of HCV core protein with HCV RNA. HepG2 cells were infected with AcCA39 at an MOI of 50, transfected with +094 RNA (lane 1), −094 RNA (lane 2), or a transcript derived from pBlue (lane 3), and immunoprecipitated with anticore antibody. DIG-labeled RNAs were extracted from the immunoprecipitates and detected by dot blotting (A). HCV core protein was detected in the immunoprecipitates by Western blotting (B).
Specific interaction of core protein with viral sense HCV RNA.
Cells expressing HCV core protein were transfected with either viral sense or antiviral sense 094 (+094 or −094, respectively) RNA, which covers almost the full-length of HCV RNA (nt 62 to 9402 [Fig. 1]). We detected a clear positive signal in the immunoprecipitates of cells transfected with +094 RNA by dot blot analysis but not in those of cells transfected with −094 RNA or transcripts of pBlue (Fig. 2A).
To confirm that the same amount of HCV core protein was accumulated in each cell and immunoprecipitated with anticore antibody, the immunoprecipitates were analyzed by Western blotting. As shown in Fig. 2B, almost equal amounts of the core protein were detected. We also confirmed that almost the same amounts of viral or antiviral sense labeled RNAs were detected when the RNAs were extracted without immunoprecipitation from cells that had been transfected with the equal amount of +094 or −094 RNA, respectively (data not shown; see below). These results imply that the core protein specifically interacts with viral sense HCV RNAs but not with antiviral sense RNAs or RNAs irrelevant to HCV.
To further confirm the specific interaction of viral sense RNA with the core protein obtained by dot blotting, RNAs recovered from the immunoprecipitates were run on the denatured agarose gel. Although degraded, the DIG-labeled RNAs were detected in the case of +094 RNA (data not shown). The failure to detect RNA corresponding to the length of 094 RNA on the gel may be due to the lower efficiency of the transfection with the longer RNA or the easier degradation of longer RNA during the transfection and extraction procedures for RNAs. We therefore divided the full-length HCV RNA into smaller regions and determined the RNA regions that retain the ability to interact with the core protein.
Identification of RNA regions responsible for specific interaction with core protein.
To determine the RNA regions responsible for interaction with the core protein, various regions of both strands of HCV RNAs were prepared (Fig. 1). HepG2 cells expressing core protein by the infection were transfected with viral or antiviral sense DIG-labeled 03 (nt 1 to 334), 014 (nt 62 to 1358), 1123 (nt 1124 to 2327), 2333 (nt 2282 to 3313), 3247 (nt 3212 to 4739), 4763 (nt 4740 to 6345), 6275 (nt 6207 to 7476), 7486 (nt 7420 to 8648), or 8395 (nt 8280 to 9548) RNA. Specific interaction with core protein was observed only in the viral sense 03, 014, and 1123 RNAs, not in other regions of viral sense RNAs or antiviral sense RNAs (Fig. 3A).
FIG. 3.

Interaction of various regions of HCV RNA with core protein. HepG2 cells infected with AcCA39 were transfected with DIG-labeled 03, 014, 1123, 2333, 3247, 4763, 6275, 7486, or 8395 RNA. + and − indicate cells transfected with positive- and negative-sense RNAs, respectively. (A) Northern blots of RNAs extracted from the immunoprecipitates; (B) DIG-labeled RNAs recovered from the cells before immunoprecipitation.
To exclude the possibility that the positive-sense HCV RNAs are more stable than the negative-sense ones in the cells, RNAs were extracted without immunoprecipitation from cells transfected with equal amounts of sense- or antisense RNAs. As shown in Fig. 3B, almost the same amounts of both senses of RNAs were recovered from the cells. Furthermore, in cells infected with a control virus, AcCAG, and transfected with either sense of HCV RNAs, very faint RNAs were detected in the immunoprecipitates with anticore antibody (data not shown). Likewise, anti-NS3 antibody did not coimmunoprecipitate either sense of HCV 3247 RNA, including the NS3 coding region in cells expressing NS3 protein (data not shown). Therefore, these results indicate that HCV core protein specifically interacts with the positive-sense HCV RNAs within nt 1 to 2327.
In this region of the RNA (nt 1 to 2327), the overlapping regions, nt 62 to 334 and 1124 to 1358, were expected to interact with the core protein. To determine which regions in the RNA (nt 1 to 2327) are important for interaction with the core protein, three RNAs, 37 (nt 329 to 716), 714 (nt 708 to 1357), and 1423 (nt 1358 to 2327) (Fig. 1), were prepared. As shown in Fig. 4, significantly stronger signals were detected in all cells transfected with each of the positive-sense RNAs than in those transfected with the negative-sense RNAs. The stability of these RNAs in the cells was almost the same between viral and antiviral sense RNAs (data not shown). These results indicate that the core protein interacts with these regions independently.
FIG. 4.
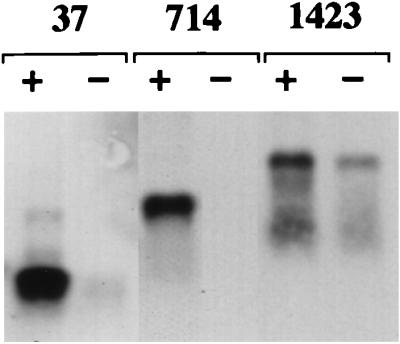
HCV RNA regions responsible for a specific interaction with core protein. HepG2 cells infected with AcCA39 were transfected with DIG-labeled 37, 714, or 1423 RNA. + and − indicate cells transfected with positive- and negative-sense RNAs, respectively. RNAs extracted from the immunoprecipitates were analyzed by Northern blotting.
Suppression of HCV RNA translation in cells expressing core protein.
The RNA regions responsible for specific interaction with the core protein include the IRES of HCV, which is important for the initiation of the cap-independent translation of HCV RNA (43, 57). In particular, the region from nt 40 to 370 of the HCV RNA including the sequence encoding the N-terminal portion of the core protein is required for efficient initiation of translation (43). This implies that core protein may have some effects on the translation of HCV. We therefore determined the translational efficiency of reporter RNA carrying nt 1 to 709 of HCV RNA followed by a firefly luciferase gene (HCV07Luc [Fig. 5]) in cells expressing the core protein or other proteins in vivo. Two days after infection with AcCA39 or AcCAG, HepG2 cells were cotransfected with both HCV07Luc and the capped RLuc RNAs. The latter was used as an internal control to normalize the efficiency of the transfection. The cells were lysed, and the activities of each luciferase were measured at 6 h posttransfection. The relative activity of luciferase in cells expressing core protein was lower than that in cells infected with AcCAG (Fig. 6A). To determine the specificity of the suppression of the translation of HCV07Luc RNA by the HCV core protein, we investigated the effect of the expression of other proteins on the translation of the reporter RNA. When cells that had been infected with recombinant baculovirus AcCA816 (expressing HCV E1 protein) or AcCAlacZ (expressing β-Gal) were transfected with HCV07Luc RNA, the expression of luciferase was not suppressed, as in cells infected with AcCAG (Fig. 6A). We also determined whether HCV core protein suppresses the translation of reporter RNA containing the IRES of EMCV (EMCVLuc RNA). Expression of HCV core protein did not affect the translation of EMCVLuc RNA, as shown in Fig. 6B. The activities of RLuc used as an internal control were almost the same among the cells infected with AcCA39, AcCA816, AcCAlacZ, and AcCAG (data not shown). These results indicate that the expression of HCV core protein but not E1 protein or β-Gal specifically suppresses its own translation.
FIG. 5.

Structures of reporter RNAs. HCVLuc consists of the 5′UTR of HCV followed by the luciferase gene. HCV07Luc is composed of the 5′UTR of HCV and a part of the coding region of the core protein (nt 1 to 709) followed by the luciferase gene. HCV09Luc consists of the 5′UTR of HCV, the whole core protein coding region, and part of the E1 protein coding region (nt 1 to 924) followed by the luciferase gene. EMCVLuc consist of the 5′UTR (nt 271 to 831) of EMCV followed by the luciferase gene.
FIG. 6.

Suppression of translation of HCV RNA by core protein expression. (A) HepG2 cells infected with AcCA39 (lane 1, gray bar), AcCA816 (lane 2), AcCAlacZ (lane 3), or AcCAG (lane 4) at an MOI of 20 were transfected with HCV07Luc together with the internal standard, capped RLuc RNA. (B) HepG2 cells infected with AcCA39 (gray bar) or AcCAG (open bar) at an MOI of 20 were transfected with EMCVLuc together with the capped RLuc RNA. The activities of both firefly and RLuc were measured by a luminometer. Relative luciferase activity (RLU) is shown after normalization with that of the RLuc, which was used as an internal standard. Relative activities were determined in at least three independent experiments, each conducted with triplicate samples. Standard deviations are represented by vertical lines.
Dose-dependent suppression of HCV RNA translation by core protein.
Luciferase activity was reduced in correlation with increases in the multiplicity of infection (MOI) of AcCA39, whereas no reduction in activity was observed in correlation with the MOI of AcCAG (Fig. 7A). The concentration of core protein in the lysates used in the luciferase assay increased in accordance with the MOI of AcCA39 (Fig. 7B). These results suggest that the translation of HCV RNA is suppressed by the expression of the core protein in a dose-dependent manner.
FIG. 7.

Dose-dependent suppression of HCV RNA translation by core protein. (A) HepG2 cells infected with AcCA39 (gray bars) or AcCAG (open bars) at MOIs of 5 to 50 were cotransfected with HCV07Luc and capped RLuc RNAs. Cells were lysed at 6 h posttransfection, and both firefly and RLuc activities (RLU) were measured. The hatched bar indicates mock-infected cells. Relative activities (RLU) were determined as described for Fig. 6. (B) The amount of core protein in the same sample used for the luciferase assay was measured by ELISA.
HCV RNA region responsible for suppression of its translation.
To determine the regions in HCV RNA responsible for suppression of its translation by the core protein, we synthesized two more reporter RNAs: (i) HCVLuc, containing only the 5′UTR of HCV (nt 1 to 344, in which nt 342–344 [AUG] was the common sequence for the initiation codon of luciferase) followed by a firefly luciferase gene (5); and (ii) HCV09Luc, possessing the 5′UTR, the coding sequence of the whole core protein and a part of the E1 protein (nt 1 to 924), followed by a firefly luciferase gene (Fig. 5) (61). HepG2 cells infected with AcCA39 or AcCAG were transfected with HCVLuc, HCV07Luc, or HCV09Luc RNA. Translational suppression of each of the reporter RNAs was observed in all cells expressing the core protein (Fig. 8). This result indicates that the 5′UTR (nt 1 to 344) of HCV RNA plays an important role in suppression of translation of its own RNA by interaction with the core protein.
FIG. 8.

HCV core protein suppresses the translation of RNA possessing the 5′UTR of HCV. HepG2 cells infected with AcCA39 (gray bars) or AcCAG (open bars) at an MOI of 20 were cotransfected with HCVLuc, HCV07Luc, or HCV09Luc RNA together with the internal standard, capped RLuc RNA. Relative activities (RLU) were determined as described for Fig. 6.
DISCUSSION
In this study, we analyzed the interaction of HCV core protein with its genomic RNA. We found (i) a specific interaction of HCV RNA with the core protein and (ii) suppression of HCV RNA translation in cells expressing HCV core protein.
Interaction of core protein with HCV RNA.
In this study, we developed a system to examine the interaction of HCV core protein with its RNA in vivo and succeeded in demonstrating specific interaction of the HCV core protein with viral sense RNAs. We found that the positive-strand RNA from the 5′ end to nt 2327 specifically interacts with the core protein in vivo compared with each of the negative-strand RNAs. Furthermore, each region of the RNAs, +03 (nt 1 to 334), +37 (nt 329 to 716), +714 (nt 708 to 1357), and +1423 (nt 1358 to 2327), interacts with the core protein independently. There is no significant sequence homology among these four regions; therefore, secondary or tertiary structure may be important for the specific interaction with the core protein. HCV core protein was shown to be specifically coimmunoprecipitated with the viral sense HCV RNA by anticore antibody. A similar phenomenon has previously been observed in the case of mouse hepatitis virus (MHV). Baric et al. have reported that a monoclonal antibody against the nucleocapsid protein of MHV specifically coimmunoprecipitates MHV genomic RNA as well as all six MHV subgenomic mRNAs in MHV-infected cells; they also have determined the region of the RNAs important for the interaction with the nucleocapsid protein (6). These results may support our own findings concerning the regions of HCV RNA interacting with the core protein.
The physical interaction of HCV core protein with HCV RNA of nt 1 to 341 or 1 to 73 has been reported based on the results of in vitro assays (19, 46). These results are not inconsistent with our in vivo results. However, no strand specificity of RNA in interaction with core protein has been shown. We likewise tested for interaction between core protein and genomic RNA in an in vitro system (gel mobility shift assay, Northwestern blotting, etc.) but could not detect any specific interaction. As mentioned above, we found a specific interaction of HCV RNA with its core protein in an in vivo system. The results obtained in the in vitro and in vivo conditions may differ because the core protein and/or RNAs retain their native conformations in our in vivo system, because some host factors in cells are involved in the specific interaction, or because HCV core protein is easily precipitated in the reaction buffer in vitro. We are now trying to determine the interaction of the core protein with the viral RNA by another in vitro method; which could provide clues to understanding the capsid formation of HCV.
It has recently been reported that (i) viruslike particles (VLP) are successfully produced in insect cells infected with a recombinant baculovirus possessing nt 259 to 2819, corresponding to one portion of the 5′UTR, and the coding sequence for the core, E1, and E2 proteins and (ii) these VLP contain the viral sense RNA (7). This RNA region, important for VLP formation, is almost the same as regions shown in this study to interact with core protein, indicating that these RNA regions are important in nucleocapsid formation.
We also examined the region in the core protein responsible for the specific interaction with HCV RNA. The core protein has four clusters of basic amino acid residues: 5 to 13 (PKPQRKTKR), 38 to 43 (PRRGPR), 58 to 71 (PRGRRQPIPKARRP), and 112 to 117 (PRRRSR) from the N terminus. These clusters are expected to bind with genomic RNAs. We therefore constructed four recombinant baculoviruses carrying a deletion in each one of the four clusters and then determined their interactions with HCV RNAs by the method described in this study. All of the four deletion mutants interacted with viral sense RNA (data not shown). This result indicates that none of the four basic amino acid clusters in the core protein are crucial for the specific interaction with viral sense RNA. Santolini et al. have reported that aa 1 to 75 of core protein interact with the 5′UTR of HCV RNA (46). This region includes three clusters of basic amino acid residues. These results suggest that more than two basic amino acid clusters may be involved in interaction with the viral RNA.
Suppression of HCV translation by HCV core protein.
We also demonstrated that the expression of core protein specifically suppresses the translation of reporter RNA possessing the 5′UTR of HCV (nt 1 to 344). These results indicate that HCV core protein suppresses its own translation by interacting with its 5′UTR. The effects of baculovirus infection on translation of the reporter RNA carrying the 5′UTR of HCV could be ruled out because the efficiency of translation of the HCV RNA was almost the same in cells infected with the recombinant baculovirus AcCA816, AcCAlacZ, or AcCAG (Fig. 6). Furthermore, the effects of core protein expression on host translational machinery could also be eliminated because the efficiency of the cap-dependent translation of RLuc RNA used as a internal standard was almost the same, irrespective of the expression of foreign proteins by infection with recombinant baculoviruses to the extent of our experiment (data not shown). These results strongly suggest that the expression of HCV core protein specifically suppress its own translation of HCV RNA.
Since the core protein interacts with not only the 5′UTR but also +37, 714, and 1423 RNAs, it is possible that the core protein suppresses the translation of RNAs containing the above regions other than the 5′UTR. It is of interest, for example, to examine whether the translational suppression by the core protein can be maintained by replacing the IRES of HCV in the HCV07Luc RNA with that of EMCV, whose translation is not suppressed by the HCV core protein as shown in Fig. 6B.
Cellular factors such as ribosome (46), La antigen (3), pyrimidine tract binding protein (2), eukaryotic initiation factor 3 (51), and p25 protein (14) were shown to bind to the 5′UTR of HCV. It is therefore possible that HCV core protein binds to these factors and interferes with their translational functions; in other words, there is indirect inhibition by the core protein. It is also possible that the core protein directly binds to the 5′UTR of HCV, inhibit the access of factor(s) and suppressing translation.
Rous sarcoma virus Pr76gag protein has been shown to regulate its own translation (52). At low concentrations of Pr76gag protein, RNA carrying the 5′ leader sequence followed by a coding sequence of Pr76gag is translated efficiently. On the other hand, the protein suppresses the translation of the RNA by inhibiting the ribosome from scanning on the RNA at high concentrations of the protein. The authors speculated that the translation of Pr76gag protein competes with the packaging (52). These findings may be helpful in understanding the regulation of the translation of HCV RNA by expression of core protein, or even in elucidating the mechanism of persistent HCV infection. It is not possible, however, to verify this hypothesis regarding HCV at present, since there is no cell culture system for the efficient replication of HCV.
HCV core protein has multiple functions; it trans regulates viral and host gene promoters (24, 40, 42), and it is involved in the cellular signal pathways (31, 41, 44, 62) and tumorigenicity (35, 39). These observations suggest that HCV core protein functions not only as a structural nucleocapsid protein but also as a regulator of gene expression. The downregulation of the translation of viral RNA by the core protein might be involved in the establishment or maintenance of viral persistence, which is a major characteristic of HCV infection.
ACKNOWLEDGMENTS
We thank T. Suzuki for helpful discussion and suggestions, H. Aizaki for helpful discussion and for constructing plasmids for the reporter RNAs, and H. S. Irvine, Jr., for critical review of the manuscript. We also thank T. Mizoguchi for secretarial work and Y. Hirama-Suzuki and S. Ogawa for technical assistance.
This work was supported in part by Second Term Comprehensive 10-year Strategy for Cancer Control, Health Sciences Research Grants of Ministry of Health & Welfare, and by the Program for Promotion of Fundamental Studies in Health Sciences of the Organization for Drug ADR Relief, R&D Promotion and Product Review of Japan. T.S. is a Science and Technology Agency fellow in Japan.
REFERENCES
- 1.Aizaki H, Aoki Y, Harada T, Ishii K, Suzuki T, Nagamori S, Toda G, Matsuura Y, Miyamura T. Full-length complementary DNA of hepatitis C virus genome from an infectious blood sample. Hepatology. 1998;27:621–627. doi: 10.1002/hep.510270242. [DOI] [PubMed] [Google Scholar]
- 2.Ali N, Siddiqui A. Interaction of polypyrimidine tract-binding protein with the 5′ noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J Virol. 1995;69:6367–6375. doi: 10.1128/jvi.69.10.6367-6375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali N, Siddiqui A. The La antigen binds 5′ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc Natl Acad Sci USA. 1997;94:2249–2254. doi: 10.1073/pnas.94.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alter M J, Margolis H S, Krawczynski K, Judson F N, Mares A, Alexander W J, Hu P Y, Miller J K, Gerber M A, Sampliner R E, Meeks E L, Beach M J. The natural history of community-acquired hepatitis C in the United States. N Engl J Med. 1992;327:1899–1905. doi: 10.1056/NEJM199212313272702. [DOI] [PubMed] [Google Scholar]
- 5.Aoki Y, Aizaki H, Shimoike T, Tani H, Ishii K, Saito I, Matsuura Y, Miyamura T. A human liver cell line exhibits efficient translation of HCV RNAs produced by a recombinant adenovirus expressing T7 RNA polymerase. Virology. 1998;250:140–150. doi: 10.1006/viro.1998.9361. [DOI] [PubMed] [Google Scholar]
- 6.Baric R, Nelson G, Fleming J, Deans R, Keck J, Casteel N, Stohlman S. Interactions between coronavirus nucleocapsid protein and viral RNAs: implications for viral transcription. J Virol. 1988;62:4280–4287. doi: 10.1128/jvi.62.11.4280-4287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumert T, Ito S, Wong D, Liang T. Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J Virol. 1998;72:3827–3836. doi: 10.1128/jvi.72.5.3827-3836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bukh J, Purcell R, Miller R. Sequence analysis of the 5′ noncoding region of hepatitis C virus. Proc Natl Acad Sci USA. 1992;89:4942–4946. doi: 10.1073/pnas.89.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, You L, Hwang L, Lee Y. Direct interaction of hepatitis C virus core protein with the cellular lymphotoxin-β receptor modulates the signal pathway of the lymphotoxin-β receptor. J Virol. 1997;71:9417–9426. doi: 10.1128/jvi.71.12.9417-9426.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choo Q-L, Kuo G, Weiner A J, Overby L R, Bradley D W, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 11.Choo Q-L, Richman K H, Han J H, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby A, Barr P J, Weiner A J, Bradley D W, Kuo G, Houghton M. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci USA. 1991;88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke B. Molecular virology of hepatitis C virus. J Gen Virol. 1997;78:2397–2410. doi: 10.1099/0022-1317-78-10-2397. [DOI] [PubMed] [Google Scholar]
- 13.Dash S, Halim A B, Tsuji H, Hiramatsu N, Gerber M. Transfection of Hep G2 cells with infectious hepatitis C virus genome. Am J Pathol. 1997;151:363–373. [PMC free article] [PubMed] [Google Scholar]
- 14.Fukushi S, Kurihara C, Ishiyama N, Hoshino F, Oya A, Katayama K. The sequence element of the internal ribosome entry site and a 25-kilodalton cellular protein contribute to efficient internal initiation of translation of hepatitis C virus RNA. J Virol. 1997;71:1662–1666. doi: 10.1128/jvi.71.2.1662-1666.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geigenmuller-Gnirke U, Nitschko H, Schlesinger S. Deletion analysis of the capsid protein of Sindbis virus: identification of the RNA binding region. J Virol. 1993;67:1620–1626. doi: 10.1128/jvi.67.3.1620-1626.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grakoui A, McCourt D W, Wychowski C, Feinstone S M, Rice C M. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J Virol. 1993;67:2832–2843. doi: 10.1128/jvi.67.5.2832-2843.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hijikata M, Kato N, Ootsuyama Y, Nakagawa M, Shimotohno K. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc Natl Acad Sci USA. 1991;88:5547–5551. doi: 10.1073/pnas.88.13.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Houghton M, Weiner A, Han J, Kuo G, Choo Q-L. Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral disease. Hepatology. 1991;14:381–388. [PubMed] [Google Scholar]
- 19.Hwang S B, Lo S Y, Lai M M C. Detection of cellular proteins and viral core protein interacting with the 5′ untranslated region of hepatitis C virus RNA. J Biomed Sci. 1995;2:227–236. doi: 10.1007/BF02253382. [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Mukaigawa J, Zuo J, Hirabayashi Y, Mitamura K, Yasui K. Cultivation of hepatitis C virus in primary hepatocyte culture from patients with chronic hepatitis C results in release of high titer infectious virus. J Gen Virol. 1996;77:1043–1054. doi: 10.1099/0022-1317-77-5-1043. [DOI] [PubMed] [Google Scholar]
- 21.Kashiwakuma T, Hasegawa A, Kajita T, Takata A, Mori H, Ohta Y, Tanaka E, Kiyosawa K, Tanaka T, Tanaka S, Hattori N, Kohara M. Detection of hepatitis C virus specific core protein in serum of patients by a sensitive fluorescence enzyme immunoassay (FEIA) J Immunol Methods. 1996;190:79–89. doi: 10.1016/0022-1759(95)00261-8. [DOI] [PubMed] [Google Scholar]
- 22.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T, Shimotohno K. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci USA. 1990;87:9524–9528. doi: 10.1073/pnas.87.24.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato N, Nakazawa T, Mizutani T, Shimotohno K. Susceptibility of human T-lymphotropic virus type I infected cell line MT-2 to hepatic C virus infection. Biochem Biophys Res Commun. 1995;206:863–869. doi: 10.1006/bbrc.1995.1123. [DOI] [PubMed] [Google Scholar]
- 24.Kim D W, Suzuki R, Harada T, Saito I, Miyamura T. Trans-suppression of gene expression by hepatitis C viral core protein. Jpn J Med Sci Biol. 1994;47:211–220. doi: 10.7883/yoken1952.47.211. [DOI] [PubMed] [Google Scholar]
- 25.Kolykhalov A A, Agapov E V, Blight K J, Mihalik K, Feinstone S M, Rice C M. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 26.Kuo G, Choo Q-L, Alter H J, Gitnick G L, Redeker A G, Purcell R H, Miyamura T, Dienstag J L, Alter M J, Stevens C E, Tegtmeier G E, Bonino F, Colombo M, Lee W-S, Kuo C, Berger K, Shuster F R, Overby L R, Bradley D W, Houghton M. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 27.Large M K, Kittlesen D J, Hahn Y S. Suppression of host immune response by the core protein of hepatitis C virus: implications for hepatitis C virus persistence. J Immunol. 1999;162:931–938. [PubMed] [Google Scholar]
- 28.Liu Z, Yang D, Qiu Z, Lim K, Chong P, Gillam S. Identification of domains in rubella virus genomic RNA and capsid protein necessary for specific interaction. J Virol. 1996;70:2184–2190. doi: 10.1128/jvi.70.4.2184-2190.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo S Y, Selby M J, Ou J H. Interaction between hepatitis C virus core protein and E1 envelope protein. J Virol. 1996;70:5177–5182. doi: 10.1128/jvi.70.8.5177-5182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masters P. Localization of an RNA-binding domain in the nucleocapsid protein of the coronavirus mouse hepatitis virus. Arch Virol. 1992;125:141–160. doi: 10.1007/BF01309634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumoto M, Hsieh T-Y, Zhu N, Van Arsdale T, Hwang S B, Jeng K-S, Gorbalenya A E, Lo S-Y, Ou J-H, Ware C F, Lai M M C. Hepatitis C virus core protein interacts with the cytoplasmic tail of lymphotoxin-β receptor. J Virol. 1997;71:1301–1309. doi: 10.1128/jvi.71.2.1301-1309.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsumoto M, Hwang S, Jeng K, Zhu N, Lai M M. Homotypic interaction and multimerization of hepatitis C virus core protein. Virology. 1996;218:43–51. doi: 10.1006/viro.1996.0164. [DOI] [PubMed] [Google Scholar]
- 33.Miller R H, Purcell R H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc Natl Acad Sci USA. 1990;87:2057–2061. doi: 10.1073/pnas.87.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moradpour D, Englert C, Wakita T, Wands J R. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology. 1996;222:51–63. doi: 10.1006/viro.1996.0397. [DOI] [PubMed] [Google Scholar]
- 35.Moriya K, Fujie H, Yotsuyanagi H, Shintani Y, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 36.Moriya K, Yotsuyanagi H, Ishibashi K, Shintani Y, Fujie H, Matsuura Y, Miyamura T, Koike K. Hepatitis C virus core protein induces steatosis in transgenic mice. J Gen Virol. 1997;78:1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- 37.Moss B, Elroy-Stein O, Mizukami T, Alexander W, Fuerst T. Product review. New mammalian expression vectors. Nature. 1990;348:91–92. doi: 10.1038/348091a0. [DOI] [PubMed] [Google Scholar]
- 38.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 39.Ray R B, Lagging L M, Meyer K, Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol. 1996;70:4438–4443. doi: 10.1128/jvi.70.7.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray R B, Lagging L M, Meyer K, Steele R, Ray R. Transcriptional regulation of cellular and viral promoters by the hepatitis C virus core protein. Virus Res. 1995;37:209–220. doi: 10.1016/0168-1702(95)00034-n. [DOI] [PubMed] [Google Scholar]
- 41.Ray R, Meyer K, Ray R. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology. 1996;226:176–182. doi: 10.1006/viro.1996.0644. [DOI] [PubMed] [Google Scholar]
- 42.Ray R, Steele R, Meyer K, Ray R. Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene. 1998;208:331–336. doi: 10.1016/s0378-1119(98)00030-4. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds J E, Kaminski A, Kettinen H, Grace K, Clarke B, Carroll A, Rowlands D, Jackson R. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 1995;14:6010–6020. doi: 10.1002/j.1460-2075.1995.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruggieri A, Harada T, Matsuura Y, Miyamura T. Sensitization to Fas-mediated apoptosis by hepatitis C virus core protein. Virology. 1997;229:68–76. doi: 10.1006/viro.1996.8420. [DOI] [PubMed] [Google Scholar]
- 45.Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe Y, Koi S, Onji M, Ohta Y, Choo Q-L, Houghton M, Kuo G. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87:6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santolini E, Migliaccio G, La Monica N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J Virol. 1994;68:3631–3641. doi: 10.1128/jvi.68.6.3631-3641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu Y K, Iwamoto A, Hijikata M, Purcell R H, Yoshikura H. Evidence for in vitro replication of hepatitis C virus genome in a human T-cell line. Proc Natl Acad Sci USA. 1992;89:5477–5481. doi: 10.1073/pnas.89.12.5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shih C-M, Lo S J, Miyamura T, Chen S-Y, Lee Y-H W. Suppression of hepatitis B virus expression and replication by hepatitis C virus core protein in HuH-7 cells. J Virol. 1993;67:5823–5832. doi: 10.1128/jvi.67.10.5823-5832.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shoji I, Aizaki H, Tani H, Ishii K, Chiba T, Saito I, Miyamura T, Matsuura Y. Efficient gene transfer into various mammalian cells, including non-hepatic cells, by baculovirus vectors. J Gen Virol. 1997;78:2657–2664. doi: 10.1099/0022-1317-78-10-2657. [DOI] [PubMed] [Google Scholar]
- 50.Shoji I, Suzuki T, Sato M, Aizaki H, Chiba T, Matsuura Y, Miyamura T. Internal processing of hepatitis C virus NS3 protein. Virology. 1999;254:315–323. doi: 10.1006/viro.1998.9540. [DOI] [PubMed] [Google Scholar]
- 51.Sizova D, Kolupaeva V, Pestova T, Shatsky I, Hellen C. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J Virol. 1998;72:4775–4782. doi: 10.1128/jvi.72.6.4775-4782.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sonstegard T, Hackett P. Autogenous regulation of RNA translation and packaging by Rous sarcoma virus Pr76gag. J Virol. 1996;70:6642–6652. [PMC free article] [PubMed] [Google Scholar]
- 53.Stohlman S, Baric R, Nelson G, Soe L, Welter L, Deans R. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J Virol. 1988;62:4288–4295. doi: 10.1128/jvi.62.11.4288-4295.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki R, Matsuura Y, Suzuki T, Ando A, Chiba J, Harada S, Saito I, Miyamura T. Nuclear localization of the truncated hepatitis C virus core protein with its hydrophobic C terminus deleted. J Gen Virol. 1995;76:53–61. doi: 10.1099/0022-1317-76-1-53. [DOI] [PubMed] [Google Scholar]
- 55.Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J Virol. 1991;65:1105–1113. doi: 10.1128/jvi.65.3.1105-1113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takeuchi K, Boonmar S, Kubo Y, Katayama T, Harada H, Ohbayashi A, Choo Q-L, Kuo G, Houghton M, Saito I, Miyamura T. Hepatitis C virus cDNA clones isolated from a healthy carrier donor implicated in post-transfusion non-A, non-B hepatitis. Gene. 1990;91:287–291. doi: 10.1016/0378-1119(90)90102-w. [DOI] [PubMed] [Google Scholar]
- 57.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiss B, Nitschko H, Ghattas I, Wright R, Schlesinger S. Evidence for specificity in the encapsidation of Sindbis virus RNAs. J Virol. 1989;63:5310–5318. doi: 10.1128/jvi.63.12.5310-5318.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weiss B, Geigenmuller-Gnirke U, Schlesinger S. Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res. 1994;22:780–786. doi: 10.1093/nar/22.5.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yanagi M, Purcell R H, Emerson S U, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yap C C, Ishii K, Aizaki H, Tani H, Aoki Y, Ueda Y, Matsuura Y, Miyamura T. Expression of target genes by coinfection with replication-deficient viral vectors. J Gen Virol. 1998;79:1879–1888. doi: 10.1099/0022-1317-79-8-1879. [DOI] [PubMed] [Google Scholar]
- 62.Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C, Lai M M. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J Virol. 1998;72:3691–3697. doi: 10.1128/jvi.72.5.3691-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]